

Graphical abstract
Keywords: MyD88, Proinflammatory, Stress, Depression, Coronavirus disease 2019
Abstract
Increasing evidence supports the pathogenic role of neuroinflammation in psychiatric diseases, including major depressive disorder (MDD) and neuropsychiatric symptoms of Coronavirus disease 2019 (COVID-19); however, the precise mechanism and therapeutic strategy are poorly understood. Here, we report that myeloid differentiation factor 88 (MyD88), a pivotal adaptor that bridges toll-like receptors to their downstream signaling by recruiting the signaling complex called ‘myddosome’, was up-regulated in the medial prefrontal cortex (mPFC) after exposure to chronic social defeat stress (CSDS) or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein. The inducible expression of MyD88 in the mPFC primed neuroinflammation and conferred stress susceptibility via amplifying immune danger signals, such as high-mobility group box 1 and SARS-CoV-2 spike protein. Overexpression of MyD88 aggravated, whereas knockout or pharmacological inhibition of MyD88 ameliorated CSDS-induced depressive-like behavior. Notably, TJ-M2010-5, a novel synthesized targeting inhibitor of MyD88 dimerization, alleviated both CSDS- and SARS-CoV-2 spike protein-induced depressive-like behavior. Taken together, our findings indicate that inhibiting MyD88 signaling represents a promising therapeutic strategy for stress-related mental disorders, such as MDD and COVID-19-related neuropsychiatric symptoms.
1. Introduction
A growing body of evidence indicates that neuroinflammation is critically associated with the pathophysiology of stress-related psychiatric diseases, such as major depressive disorder (MDD) (Leng et al., 2018, Norman et al., 2010). However, much less is known about the precise mechanism for neuroinflammation initiation underlying psychiatric diseases. It has been demonstrated that stress-induced release of immune danger signals, such as high-mobility group box 1 (HMGB1), initiate innate immune signaling by engaging toll-like receptors (TLRs) and the receptor for advanced glycation end-products (RAGE) (Frank et al., 2015, Franklin et al., 2018), followed by production of proinflammatory cytokines such as interleukin-1 beta (IL-1β), which increases stress susceptibility (Cheng et al., 2016). Except for endogenous danger-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) such as spike protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) also binds to TLRs and triggers innate immune response (Frank et al., 2021, Khan et al., 2021, Olajide et al., 2021, Zhao et al., 2021). Recent studies have revealed that COVID-19 increases blood–brain barrier (BBB) permeability (Soung et al., 2022), and spike protein of SARS-CoV-2 crosses the murine BBB by adsorptive transcytosis, entries the central nerve system (CNS) and induces neuroinflammation, which may lead to brain alterations and behavioral abnormalities (Frank et al., 2021, Rhea et al., 2021). Considering coronaviruses are neurotropic (Bauer et al., 2022, Kumari et al., 2021, Song et al., 2021), and neuropsychiatric symptoms (Huang et al., 2021a, Huang et al., 2021b) accompanied by the pandemic of coronavirus disease 2019 (COVID-19), TLRs-dependent neuroinflammation may increase mental risk in COVID-19 patients.
TLRs-dependent innate immune signaling requires the self-assembly of signaling proteins into oligomeric complexes (Deliz-Aguirre et al., 2021, Fitzgerald and Kagan, 2020), including inflammasome and myddosome (Gay et al., 2011, Lin et al., 2010), to coordinate signal transduction cascades in time and space. Nearly all TLRs interact with a central cytoplasmic signaling adaptor, myeloid differentiation primary response protein 88 (MyD88), which reorganize myddosome complex to govern immune signaling cross-talk and initiate downstream signal transduction pathways (Jang et al., 2013). Recent studies have revealed that MyD88 expression is associated with the severity of COVID-19 disease, and MyD88 is required for SARS-CoV-2-induced inflammatory response (Khan et al., 2021, Zheng et al., 2021). Notably, it has been recently reported that MyD88-deficient mice exhibit altered stress response (Hosoi et al., 2021), and the expression of MyD88 mRNA is increased in peripheral blood mononuclear cells of MDD patients (Hajebrahimi et al., 2014). However, the exact role of MyD88 in psychiatric disorders is unclear. Herein, we utilized behavioral, molecular, and genetic approaches to clarify that MyD88 as a novel pharmacological target for the treatment of MDD and neuroinflammation-related behavior abnormalities.
2. Materials and methods
2.1. Animals
Adult male C57BL/6J mice (6–7 weeks old, 18–20 g) and male CD-1 (6–8 months old) were purchased from Hunan SJA Laboratory Animal (Changsha, Hunan, China) and Vital River Laboratory Animal Technology (Beijing, China), respectively. MyD88−/− and their wild-type mice were kindly given from Professor Ping Zhou (Institute of Organ Transplantation, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China). Mice were housed in individual ventilation cage (IVC, ≤ 6 mice per cage) and maintained on a 12 h light–dark cycle (lights on at 8:00 a.m.) with free access to food and water all the time under standard laboratory conditions (ambient temperature: 22 ± 1 °C; humidity: 40 % ± 5 %). The study was conducted according to the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Welfare Committee of Huazhong University of Science and Technology [reference number: 2019-S1827]. All behavioral testing and tissue harvesting procedures were performed on male mice and investigators were blind to the treatments.
2.2. Chronic social defeat stress (CSDS) model
CSDS was conducted as described previously (Li et al., 2018). Briefly, a single invading C57BL/6J mice was physically attacked by different CD-1 residents for 10 min once a day for 10 consecutive days. The mice and the CD-1 were then kept overnight in a two-compartment mouse cage separated by a perforated plexiglass divider to provide the pressure of sensory contact. Control mice were placed in equivalent cages with a different littermate every day.
2.3. Subthreshold social defeat stress (SSDS) model
The SSDS paradigm was conducted as previously described (He et al., 2021). For SSDS protocol, mice were exposed to three social defeat sessions (5 min) with a novel CD-1 aggressor, and then the mice were placed on the other side of a perforated divider to provide psychological stress for 15 min. After SSDS session, the mice were single-housed and social interaction test (SIT) was tested at 24 h later.
2.4. BV2 cell culture
The mouse BV2 cell line, a model of microglia, was obtained from the Jennio Biotech Co. Ltd (Cat No. JNO-598; Guangzhou, China) and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco Laboratories, Cat No. C11995500BT; Grand Island, NY, USA) supplemented with 10 % fetal bovine serum (FBS) (Gibco Laboratories, Cat No. 10099141C; Grand Island, NY, USA) and 1 % penicillin/streptomycin (Gibco Laboratories, Cat No. 2441838; Grand Island, NY, USA) in a humidified incubator of 5 % CO2 at 37 °C. The cells were exposed to corticosterone (CORT, 5 nM, 50 nM and 100 nM) and SARS-CoV-2 spike receptor-binding domain (RBD, 0.1 nM, 1 nM and 10 nM) for 24 h before harvesting.
2.5. Primary microglial culture and virus transfection
The cortical tissue was separated from the brains of neonatal C57BL/6 mice under aseptic conditions, then rinsed and cut into pieces in precooled phosphate buffered saline (PBS). The cortical tissue was digested with 0.25 % trypsin (the final concentration is 0.125 %) for 15 min at 37 °C, and filtered through 70 µm cell strainer after termination of digestion. After centrifugation, the precipitated cells were resuspended in the DMEM/F12 intact medium with 10 % FBS and plated at a final density of 1 × 106 cells/flask in 75 cm2 flasks containing 0.1 g/ml of poly-l-lysine solution, then cultured at 37 °C for 7 to 9 d. Mechanical shaking (37 °C, 6 h) was applied for isolation and purification of glia from mixed glia and neurons culture, then collected supernatant to centrifugation and inoculation. Primary microglia were cultured in the DMEM medium containing 10 % FBS, 1 % penicillin/streptomycin at 37 °C in a 5 % CO2 incubator. The purity of these microglial cells was 99 % as determined by ionized calcium binding adaptor molecule 1 (Iba1) immunoreactivity. Primary microglia were resuspended by DMEM/F12 and seeded into 24-well plates pre-coated with poly-l-lysine at a density of 1.0 × 106 cells/well for overnight cultured in 5 % CO2 at 37 °C. Next, adeno-associated virus 9 (AAV9) vector delivery system containing green fluorescent protein (GFP) was used to overexpress MyD88 (AAV-MyD88, NCBI Reference Sequence: NM-010851.3), and the vector (AAV-GFP) was used as a control. After 8 h of transfection, serum-free transfer solution was replaced by complete medium to culture for 6 d.
2.6. Drugs preparation
Detailed information about drugs is described. CORT (HY-B1618), FPS-ZM1 (HY-19370), JSH-23 (HY-13982) and sparstolonin B (SSnB) (HY-116213) were purchased from MedCheExpress (Monmouth Junction, NJ, USA). MyD88 inhibitor peptide set (MIP) (NBP2-29328) was purchased from Novus Biologicals (Littleton, CO, USA). Recombinant Human HMGB1 protein (rHMGB1) (CF-1690-HMB-050) was purchased from RD systems (Minneapolis, MN, USA). Biotinylated SARS-CoV-2 spike RBD (SPD-C82E9, HEK293-expressed) was purchased from Acrobiosystems (Beijing, China). TJ-M2010-5 (PCT/CN2012/070811) was kindly given from Professor Feng-Chao Jiang (Department of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China).
CORT was dissolved in phosphate-buffered saline (PBS) to 5 mM concentration. For in vitro study, the final concentrations of CORT were 5 nM, 50 nM, 500 nM, respectively. Control inhibitor peptide (CP) and MIP were dissolved in PBS to 5 mM concentration. For in vivo or in vitro study, the final concentrations of CP and MIP were 50 µM. JSH-23 and SB203580 were dissolved in sodium l-ascorbate and dimethyl sulfoxide (DMSO) to 20 µM and 5 µM as final concentrations, respectively. rHMGB1 protein was dissolved in PBS to 40 µM concentration. For in vivo, the working concentrations of rHMGB1 were 0.2 µM and 2 µM. FPS-ZM1 and SSnB were dissolved in normal saline to 200 nM and 100 µM as final concentrations, respectively. SARS-CoV-2 spike RBD was dissolved in PBS to 7 µM concentration. For in vitro, the final concentrations of SARS-CoV-2 spike RBD were 0.1 nM, 1 nM and 10 nM, respectively. For in vivo, the working concentration of SARS-CoV-2 spike RBD was 10 nM. TJ-M2010-5 was dissolved in distilled deionized water to a stock solution of 5 mg/kg and further diluted to deliver doses of 1.25 mg/kg and 2.5 mg/kg in 0.1 ml. For in vitro, the final concentration of TJ-M2010-5 was 30 µM.
2.7. Behavioral experiments
For all behavioral tests, C57BL/6J mice were transported to behavioral room and adapted for at least 1 h before testing. Mice were assessed in SIT (He et al., 2021), sucrose preference test (SPT) (Luo et al., 2020), tail suspension test (TST) (Zhou et al., 2019), forced swim test (FST) (Deng et al., 2021), open field test (OFT), (Wu et al., 2021) and elevated plus maze test (EPM) (Shen et al., 2019) as previous reports.
2.7.1. SIT
Mice were placed in a novel and open area (42 cm × 42 cm × 42 cm) with a small-animal cage (10 cm × 6 cm) on one side to explore social interaction and corner zones for 2.5 min in the absence or presence of an unfamiliar CD-1 invader mouse, respectively. Movements were video-taped and recorded by the ANY-maze tracking system 5.3 (Stoelting Co., Wood Dale, IL, USA). The social interaction (SI) index was calculated as the total time spent by the mice in the interaction zone with target absent or present or as social interaction ratio (time spent in the interaction with social target present / social target absent) was classified mice as susceptible (ratio < 1) and resilient (ratio ≥ 1).
2.7.2. SPT
Mice was singly housed and adapted to two bottles (50 ml) with one containing 1 % sucrose solution and the other tap water for 48 h, and the position of the bottles was exchanged every 12 h to prevent position preference. After water deprivation for 12 h, mice were exposed to 1 % sucrose solution and tap water for 12 h in the dark phase. Sucrose preference was calculated by dividing the weight of sucrose intake consumed by the total weight of fluid intake (sucrose consumption + water consumption) × 100 %.
2.7.3. TST
Mice were suspended with tape to the suspension bar with the nose 20 cm above the ground. A 6-min test period was recorded by the ANY-maze tracking system 5.3 (Stoelting Co., Wood Dale, IL, USA) and the time of immobility was measured when they exhibited no body movement.
2.7.4. FST
Mice were individually placed into the clear plexiglass cylinder (height 35 cm; diameter 15 cm) filled with water (24 ± 1 °C, 10 cm-depth), and recorded with ANY-maze tracking system 5.3 (Stoelting Co., Wood Dale, IL, USA) for a 6-min session. The immobility time was recorded during the last 4 min. Immobility time was defined as the time spent by the mice floating without struggling in the water away from the wall of a cylinder.
2.7.5. OFT
Mice were placed in a large square open box (50 cm × 50 cm × 40 cm), which was divided into central area of 35 cm × 35 cm and the surrounding marginal zone and allowed to explore the area for 10 min. The movement were tracked with ANY-maze tracking system 5.3 (Stoelting Co., Wood Dale, IL, USA), including the total distance traveled in the open field, the time spent in the central area and the number of entries into the central area.
2.7.6. EPM
The apparatus consisted of two opposing open arms (30 cm × 5 cm × 0.5 cm) and two closed arms (30 cm × 5 cm × 15 cm) extending from a central platform (5 cm × 5 cm) and elevated 40 cm above the ground. Mice were placed in the central platform facing an open arm, and tracked the total distance traveled in the open and closed arms, the number of entries into open and closed arms and the time spent in open and closed arms during the 5-min test period using ANY-maze tracking system 5.3 (Stoelting Co., Wood Dale, IL, USA).
2.8. Western blotting
Cell or tissue samples were homogenized in ice-cold radioimmunoprecipitation (RIPA) lysis buffer with protease and phosphatase inhibitors (150 mM NaCl, 50 mM Tris, 1 % Triton X-100, 0.1 % sodium dodecyl sulfate (SDS), 1 % sodium deoxycholate, protease inhibitor mixture, pH 7.4). The samples were centrifuged at 4 °C with 12,000 g for 20 min and quantified by Bicinchoninic Acid Assay (BCA) protein assay kit (Beyotime Biotechnology, Shanghai, China). All protein samples were heated for 10 min at 95 °C in loading buffer. Equal amounts of protein (30 μg) were separated by 10 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Merck Millipore, Billerica, MA, USA). The membranes were blocked for 1 h at room temperature in 5 % bovine serum albumin (BSA) (Merck Millipore, Billerica, MA, USA) in Tris-buffered saline containing 0.1 % Tween-20 (TBST). Blots were incubated overnight at 4 °C with the different primary antibodies. anti-MyD88 (ab2064, RRID: AB_302807; 1:1000), anti-MAL (ab133332, 1:1000), anti-TLR2 (ab209216, 1:1000), anti-IL-1β (ab9722, RRID: AB_308765; 1:1000), anti-Iba1 (ab178846, RRID: AB_2636859; 1:1000), anti-GluA1 (ab183797, RRID: AB_2728702; 1:1000) and anti-GluA2 (ab52932, RRID: AB_880226; 1:1000) were purchased from Abcam (Cambridge, MA, USA). anti-IκBα (#4812, RRID: AB_10694416; 1:1000), anti-NF-κB p65 (#59674, RRID: AB_2799570; 1:1000), and anti-phospho-NF-κB p65 (#13346, RRID: AB_2798185; 1:1000) were obtained from Cell Signaling Company (San Francisco, CA, USA). Anti-Caspase-1 (06-503-1, 1:200) was purchased from Merck Millipore (Billerica, MA, USA). Anti-Caspase-1 p10 (sc-56036, RRID: AB_781816; 1:500) and anti-β-actin (sc-47778, RRID: AB_626632; 1:2000) were purchased from Santa Cruz Biotechnology (Waltham, MA, USA). Goat anti-rabbit 800 CW (926-32211, RRID: AB_621843; 1:10000) and Goat anti-mouse 800 CW (926-32210, RRID: AB_621842; 1:10000) were obtained from LI-COR (Nebraska, USA). After washing three times at room temperature with TBST, blots incubated with anti-NFκB p65 (HRP conjugate) were directly reacted with enhanced chemiluminescence substrate (Super Signal West Pico; Pierce Chemical Co., Rockford, IL) and captured by Micro Chemi (DNR Bio-imaging systems, Jerusalem, Israel), and the optical densities of the immunoblots were measured by ImageJ (NIH, Washington, USA) software. Other blots were incubated with secondary antibodies labeled by fluorescent dye in dark at room temperature for 1 h. Odyssey DLx imaging system (Lincoln, Nebraska, USA) and image studio software (NIH, Bethesda, MD, USA) were used to quantify the optical densities of bands. The results were presented as the percentage of control after normalization. All original of key western blots and information were presented in the Figs. S6–S9 and Table S2.
2.9. RNA preparation and real-time PCR
Total RNA was extracted from medial prefrontal cortex (mPFC) using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Then 1 µg total RNA was used for cDNA synthesis by using the RevertAid First Strand cDNA Synthesis kit (Fermentas, Thermo Scientific, Canada). Quantitative real-time PCR was performed in a StepOnePlusTM Real-Time PCR System (Applied Biosystems, Foster City, CA) with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). The protocol consisted of denaturation at 95 °C for 10 min followed by 39 cycles of 95 °C for 30 s, 95 °C for 5 s and 60 °C for 30 s. The gene expression was analyzed by ΔΔ Ct method and relative gene expression was normalized to housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers sequences were listed as follows (5′ to 3′): IL-1β, CCTGCAGCTGGAGAGTGTGGAT (forward), TGTGCTCTGCTTGTGAGGTGCT (reverse); GAPDH, ATGGTGAAGGTCGG TGTG (forward), CATTCTCGGCCTTGACTG (reverse).
2.10. Immunofluorescent staining
Mice were anesthetized with sodium pentobarbital (45 mg/kg, intraperitoneally) and perfused intracardially with 0.9 % saline followed by 4 % paraformaldehyde in PBS through the heart. The brains were post-fixed overnight in paraformaldehyde at 4 °C and placed in 10 %–30 % sucrose gradient at 4 °C for 3 d followed cut into 30 µm thick slices using a freezing microtome (CM1900, Leica Microsystems, Wetzlar, Germany). For immunofluorescence staining of cells, the cells were fixed in 4 % paraformaldehyde for 15 min. The cells or brain sections were washed with PBS and blocked with 5 % BSA containing 0.1 % Triton X-100 at room temperature for 2 h followed by antibodies against CD68 (MAC1957, 1:400; Biorad, California, USA), Iba1 (ab178846, 1:400; ab5076, 1:200; Abcam, Cambridge, MA, USA), TLR2 (ab209216, 1:200; Abcam, Cambridge, MA, USA), glial fibrillary acidic protein (GFAP) (#3670, 1:400; Cell Signaling Company, San Francisco, CA, USA) overnight at 4 °C, then the secondary antibodies labeled with Alexa dye (Invitrogen, Auckland, NEW ZEALAND) and 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (D9542, 1:8000; Sigma-Aldrich, St. Louis, MO, USA) were incubated. The argon laser lines of 405 nm, 488 nm, 559 nm and 635 nm were used for excitation. The fluorophores were captured with appropriate filters, and images were acquired under 20 × objective with a confocal laser scanning microscope (FV1000, Olympus, Tokyo, Japan). ImageJ (NIH, Washington, USA) was used for immunofluorescence analysis. Images were converted to 8-bit black-and-white images, and background was subtracted. A fixed threshold was set to acquire optimal representation for each staining group. The number of Iba1 positive cells was obtained using the “analyze particles” plugins in ImageJ, and divided by the total area of the acquired field to represent microglia density (microglia/mm2). A mean count per mouse was calculated and used for statistical analysis. For the immunofluorescent staining in vivo, the percentage of CD68-immunostained area (% area) was calculated for each field and each section. For the immunofluorescent staining in vitro, average intensities of the Iba1 or CD68 protein immunostaining were normalized with the number of cells. All results were presented as the percentage of control group following normalization. All analyses were conducted by someone blind to the groups of animals.
2.11. Stereotaxic surgery and cannula infusion
Mice were anesthetized with sodium pentobarbital (40 mg/kg, intraperitoneally) and immobilized in a stereotaxic apparatus (RWD Life Science Co., Shenzhen, Guangdong Province, China). For cannula implantation, mice were bilaterally implanted 22-gauge stainless steel guide cannulas into mPFC (AP = 2.0 mm, ML = ±0.4 mm, DV = −2.2 mm relative to the bregma; AP, ML, and DV denote anteroposterior, mediolateral and dorsoventral distances from bregma, respectively) and recovered for at least 5 d after surgery. Agents were microinjected into mPFC with the microsyringe pump and the injector remained for 2 min after infusion. For stereotaxic viral injection, AAV vector containing the MyD88 or GFP were bilaterally microinjection into mPFC (0.5 µl/side) at a slow rate of 50 nl/min followed by an additional 10 min of rest to diffusion. Behavioral test was commenced 3 weeks after viral injection.
2.12. Statistical analysis
Animals were randomly assigned to different groups. The experimenters were blinded to groups during experiments and quantitative analyses. Sample sizes were determined according to those used in similar studies from our group (He et al., 2021, Luo et al., 2020, Zhou et al., 2019) and justified by the power analyses (http://www.powerandsamplesize.com/). For power analyses, α was 0.05, the desired power (1-β) was 0.8, and the anticipated effect size and standard deviation were based on the result of our preliminary experiments. The information for power analyses were presented in Supplementary materials and methods. Data were expressed as the mean ± SEM and analyzed using GraphPad 8.0 software (GraphPad Software, Inc., USA). We performed statistical comparisons between two groups using Student’s t-tests. One or two-way analysis of variance (ANOVA) and Bonferroni post hoc analyses were used in analyses with multiple experimental groups, where appropriate. p < 0.05 was considered statistically significant. Exact p values, F values and other detailed statistical information for each figure are provided in the Table S1.
3. Results
3.1. Inducible expression of MyD88 by stress primes neuroinflammation in the mPFC
Glucocorticoid-dependent processes underlie the development of chronic stress-induced disorders, in particularly, MDD. We found that the expression of MyD88 (F(3, 53) = 4.452, p = 0.0073) in BV2 cells were elevated by ∼20 % after incubation with CORT (50 nM and 500 nM) for 24 h (Fig. 1 A). Then, primary microglia were pretreated with CP (50 µM) or MIP (50 µM) to interfere with the formation of MyD88 homodimer, followed by exposure to CORT (50 nM) for 24 h. Our results found that CORT (50 nM) changed morphology of primary microglia into ameboid-like shape with an increased immunostaining for CD68 (a marker of phagocytic activity) and Iba1 (a marker for microglia), which was abolished by MIP (CD68: F(1, 21) = 11.02, p = 0.00331, Vehicle + CP vs CORT + CP, p = 0.0002; CORT + CP vs CORT + MIP, p = 0.1486; Iba1: F(1, 21) = 9.492, p = 0.0057; Vehicle + CP vs CORT + CP, p = 0.0214; CORT + CP vs CORT + MIP, p = 0.0010) (Fig. 1B). Meanwhile, transfection of primary microglia with AAV-MyD88 increased the immunofluorescence intensity for CD68 (t = 2.487, p = 0.0418) and Iba-1 (t = 3.280, p = 0.0135) by 128.4 % and 109.8 % respectively. And MyD88-transfected microglia displayed a more ‘‘amoeboid’’ morphology, characterized by hypertrophic bodies, with fewer and shorter processes (Fig. 1C).
Fig. 1.

MyD88 promotes stress-induced microglial priming and neuroinflammation. (A) Incubation with CORT (50 nM, 500 nM) increased MyD88 expression in BV2 cells (n = 12–15 wells/group). (B) Representative images (left) of immunostaining for CD68 (red) and Iba1 (grey) in primary microglia. Quantitative analyses (right) showed that MIP (50 µM) significantly blocked CORT-induced microglial activation. Scale bar, 50 µm (n = 6–7 wells/group). (C) Representative images (left) of immunostaining for CD68 (red) and Iba1 (grey) in primary microglia. Quantitative analyses (right) showed that exposure of primary microglia to AAV-MyD88 resulted in microglial activation. Scale bar, 50 µm (n = 4–5 wells/group). (D) Representative images (left) of immunostaining for CD68 (green) and Iba1 (red) in the mPFC. Quantitative analyses (right) showed that CSDS increased the number of Iba1+ cell and immunofluorescence intensity of CD68 in microglial. Scale bar, 20 µm (n = 4 mice/group). (E) Representative western blots showing a decreased IκBα level (n = 13–14 mice/group), an increased phospho-NF-κB p65 (n = 13–14 mice/group) and IL-1β (n = 7–8 mice/group) expression in the mPFC of susceptible mice. (F) IL-1β mRNA was increased in the mPFC of susceptible mice (n = 8 mice/group). (G) The expression of MyD88 was upregulated in the mPFC (n = 8–13 mice/group), but not Hippo (n = 5–11 mice/group) and NAc (n = 4–11 mice/group) of susceptible mice. (H) The levels of TLR2 and MAL were elevated in the mPFC of susceptible mice (n = 6–12 mice/group). (I) Schematic timeline of CSDS, CP/MIP treatment and behavioral tests. (J) MIP reversed CSDS-induced upregulation of IL-1β expression in the mPFC (n = 5 mice/group). Data are presented as mean ± SEM, with each point representing data from an individual. *p < 0.05, **p < 0.01, ***p < 0.001, by one-way (A, G, H) or two-way (B, J) analysis of variance followed by Bonferroni’s post hoc test, Student’s t test (C–F). See also Fig. S1.
Then, we evaluated the effect of stress on microglia in vivo using a CSDS model (Fig. S1A). Immunostaining results showed that CSDS resulted in an approximately 46.1 % increase in the number of Iba1 + microglia (t = 4.255, p = 0.0054) and 46.7 % increase in the immunofluorescence intensity of CD68 (t = 2.969, p = 0.0250) in the mPFC, indicating that CSDS promotes microglial activation (Fig. 1D). In addition, CSDS increased neuroinflammation in the mPFC, as indicated by IκBα level reduced by 17.4 % (t = 2.880, p = 0.0080), with an 22.9 %, 36.0 % and 66.8 % increase in the level of NF-κB p65 phosphorylation (t = 2.099, p = 0.0461), pro-IL-1β and mat-IL-1β protein, respectively (pro-IL-1β, t = 2.484, p = 0.0274; mat-IL-1β, t = 5.065, p = 0.0002) (Fig. 1E). IL-1β mRNA expression was raised by ∼ 70 % (t = 5.324, p = 0.0001) in the mPFC of stressed mice (Fig. 1F).
We next examined the impact of CSDS on MyD88 expression in the mPFC. Exposure to CSDS resulted in an increase in MyD88 expression in the mPFC of susceptible mice, but not resilient mice (F(2, 30) = 22.15, p < 0.0001; Control vs Susceptible, p < 0.0001; Control vs Resilient, p = 0.1031). However, little effect of CSDS on MyD88 expression in the hippocampus (Hippo) (F(2, 22) = 2.021, p = 0.1564; Control vs Susceptible, p > 0.9999) and nucleus accumbens (NAc) (F(2, 22) = 0.8071, p = 0.4589; Control vs Susceptible, p > 0.9999) were observed (Fig. 1G). In addition, CSDS also increased the levels of other elements involved in myddosome signaling, including TLR2 (F(2, 26) = 3.509, p = 0.0447; Control vs Susceptible, p = 0.0450) and MyD88 adaptor-like protein (MAL) (F(2, 26) = 3.804, p = 0.0355; Control vs Susceptible, p = 0.0428) (Fig. 1H) in the mPFC of susceptible mice. Immunostaining results revealed that in the mPFC of both control and stressed mice, TLR2 was mainly expressed in the microglia, but not in the astrocyte (Fig. S1B), suggesting that innate immune responses of microglia may confer CSDS-induced neuroinflammation.
Considering that exposure of microglia to MIP significantly attenuated CORT-induced microglial activation, we next sought to determine the effect of MIP on stress-induced inflammation in vivo (Fig. 1I). We found that consecutive intracerebroventricular (i.c.v.) administration of MIP (50 µM, 1 µl/side) in the mPFC for 3 d reversed CSDS-induced increase in the expression of IL-1β (pro-IL-1β: F(1, 16) = 7.522, p = 0.0144; CSDS + CP vs CSDS + MIP, p = 0.0020; mat-IL-1β: F(1, 16) = 6.368, p = 0.0226; CSDS + CP vs CSDS + MIP, p = 0.0038) (Fig. 1J). Together, these results suggest that inducible expression of MyD88 promotes stress-induced microglial priming in the mPFC.
3.2. Targeting MyD88 in the mPFC blocks CSDS-induced depressive-like behaviors
We further used a viral expression approach to confirm whether inducible expression of MyD88 in the mPFC could generate a pro-depressive effect. Immunofluorescence staining confirmed that AAV-GFP and AAV-MyD88 was localized and distributed in the mPFC, and the expression of MyD88 protein was increased by 89.1 % in the MyD88-overexpressed mice compared with AAV-GFP group (t = 5.759, p < 0.0001) (Fig. S2A and Fig. 2 A). Overexpression of MyD88 in the mPFC was sufficient to increase the number of microglia (t = 4.988, p = 0.0076; AAV-GFP = 265 ± 19.74; AAV-MyD88 = 482.6.0 ± 38.91) and the expression of IL-1β protein (t = 5.500, p = 0.0002; AAV-GFP = 1.000 ± 0.04011; AAV-MyD88 = 1.605 ± 0.1099), which implicates an inflammatory activation of microglia (Fig. 2B-C). Furthermore, overexpression of MyD88 induced a significant increase in behavioral despair than that of AAV-GFP group, indicating by a prolonging duration of immobility in TST from 67.56 ± 9.802 s to 115.3 ± 10.08 s (t = 3.215, p = 0.0026) and FST from 52.66 ± 6.318 s to 92.37 ± 10.74 s (t = 2.760, p = 0.0088) (Fig. 2D). Meanwhile, MyD88-overexpressed mice developed anxiety-like behaviors in the EPM test, including a 35.4 % decrease in time (t = 2.111, p = 0.0416) and 42.9 % decrease in distance (t = 2.254, p = 0.0299) in the open arm (Fig. 2E), but exerted little effect on the time spent in the interaction zone with target (F(1, 40) = 0.2269, p = 0.6364; AAV-GFP vs AAV-MyD88, p > 0.9999; Fig. S2B), sucrose preference (t = 0.9311, p = 0.08757; AAV-GFP = 88.37 ± 2.441 %; AAV-MyD88 = 88.12 ± 1.543 %; Fig. S2C), as well as motor function in the OFT (t = 0.8054, p = 0.4255; AAV-GFP = 54.67 ± 2.577 m; AAV-MyD88 = 57.13 ± 1.793 m; Fig. S2D).
Fig. 2.

MyD88 in the mPFC mediates susceptibility to stress. (A) Representative photomicrographs (left) of injection site in the mPFC. Scale bars, 200 µm. Quantitative western blot assay (right) the expression of AAV-mediate MyD88 in the mPFC (n = 9 mice/group). (B) Quantitative analyses showed that MyD88 overexpression increased the number of Iba1+ cells in the mPFC. Scale bar, 50 µm (Left), 20 µm (Right) (n = 3 mice/group). (C) Representative western blots showing an increase in IL-1β expression in the mPFC of MyD88-overexpressed mice (n = 6–7 mice/group). (D) MyD88 overexpression increased the immobility time in TST and FST in mice (n = 16–25 mice/group). (E) MyD88 overexpression decreased the time (n = 16–23 mice/group) and distance (n = 16–25 mice/group) in open-arm in the EPM. (F) Schematic illustration of virus injection, SSDS and behavioral tests. (G-I) SSDS increased social avoidance (G), decreased sucrose preference (H) and prolonged the immobility time in TST and FST (I) in MyD88-overexpressed mice (n = 18–22 mice/group). (J) Schematic illustration of virus injection, CSDS and behavioral tests. (K-M) MyD88-overexpressed mice aggravated CSDS-induced decrease in the social interaction time (K) and sucrose preference (L), and prolonged the immobility time in TST (M) (n = 8–10 mice/group). (N and O) MyD88−/− mice prevented CSDS-induced decrease in the social interaction time (N) and social interaction ratio (O) (n = 3–6 mice/group). (P–Q) MIP (50 µM) reversed CSDS-induced social avoidance (P, n = 10–15 mice/group) and sucrose preference deficits (Q, n = 10–14 mice/group). Data are presented as mean ± SEM, with each point representing data from an individual. *p < 0.05, **p < 0.01, ***p < 0.001, by two-way (G, K–Q) analysis of variance followed by Bonferroni’s post hoc test, Student’s t test (A–E, H, I). See also Fig. S2.
We further explored directly the potential role of MyD88 in stress vulnerability (Fig. 2F). As expected, SSDS lead to depressive-like behaviors in MyD888-overexpressed mice, which was characterized by increased social avoidance (F(1, 76) = 4.724, p = 0.0329; AAV-GFP(Target) vs AAV-MyD88(Target), p = 0.0171; Fig. 2G), reduced sucrose preference from 81.74 ± 3.299 % to 57.37 ± 5.396 % (t = 3.649, p = 0.0008; Fig. 2H), and prolonged immobility time in TST from 97.64 ± 14.22 s to 144.5 ± 14.28 s (t = 2.300, p = 0.0271) and FST from 45.15 ± 6.611 s to 93.14 ± 9.073 s (t = 4.105, p = 0.0002; Fig. 2I), without changing the locomotor activity in the OFT (t = 1.675, p = 0.1042; AAV-GFP = 26.08 ± 2.298 m; AAV-MyD88 = 20.32 ± 2.513 m; Fig. S2E), suggesting that MyD88 may aggravate stress vulnerability. We further verified the effect of MyD88 on CSDS-induced depressive-like behaviors (Fig. 2J). Under the CSDS paradigm, MyD88-overexpressed mice showed less social interaction (F(1, 32) = 0.6651, p = 0.4208; CSDS + AAV-GFP vs CSDS + AAV-MyD88, p = 0.0035; Fig. 2K) and sucrose preference (F(1, 32) = 2.427, p = 0.1291; CSDS + AAV-GFP vs CSDS + AAV-MyD88, p = 0.0444; Fig. 2L) than that of AAV-GFP group, with an increase in about 22.6 % in the immobility time in TST (F(1, 32) = 0.7303, p = 0.3991; CSDS + AAV-GFP vs CSDS + AAV-MyD88, p = 0.1274; Fig. 2M). These results indicate that MyD88 in the mPFC determines the susceptibility to stress.
To further verify the role of MyD88 in stress susceptibility, MyD88 knockout (MyD88−/−) mice were treated by the CSDS paradigm. MyD88−/− mice were found to be resistant to stress-induced depressive-like behaviors under the SIT paradigm, such as increased social interaction time with target (F(1, 14) = 3.022, p = 0.1041; CSDS + WT vs CSDS + MyD88-/-, p = 0.0312; Fig. 2N) and social interaction ratio (F(1, 14) = 8.298, p = 0.0121; CSDS + WT vs CSDS + MyD88-/-, p = 0.0092; Fig. 2O). Furthermore, to examine the effect of MIP on depressive-like behaviors, we implanted cannulations into the mPFC of mice, and microinjected with CP (50 µM, 1 µl/side) or MIP (50 µM, 1 µl/side) for 3 d. We found that the local administration of MIP in the mPFC reversed CSDS-induced deficits in social interactions with target (F(1, 45) = 11.97, p = 0.0012; CSDS + CP vs CSDS + MIP, p < 0.0001; Fig. 2P) and sucrose preference (F(1, 43) = 5.237, p = 0.0271; CSDS + CP vs CSDS + MIP, p = 0.0234; Fig. 2Q). Taken together, these findings suggest that targeting MyD88 in the mPFC may serve as a potential strategy for treatment of depression.
3.3. Overexpression of MyD88 in the mPFC facilitates danger signal-associated behavioral abnormalities
Previous studies have shown that DAMPs, such as HMGB1, may contribute to neuroinflammatory priming (Fonken et al., 2016, Frank et al., 2015), and we found that microinjection with rHMGB1 (2 µM, 1 µl/side) into the mPFC followed by SSDS induced depression-related behaviors, including increased social avoidance (F(2, 23) = 3.728, p = 0.0396; Vehicle vs 2 μM, p = 0.0244; Fig. S3A) and prolonged immobility time in TST (F(2, 24) = 4.512, p = 0.0217; Vehicle vs 2 μM, p = 0.0171; Fig. S3B), but exerted little effect on the locomotor activity in the OFT (F(2, 24) = 1.242, p = 0.3068; Vehicle vs 2 μM, p = 0.2563; Fig. S3C). We further found that microinjection with rHMGB1 (2 µM, 1 µl/side) into the mPFC (Fig. 3 A) only increased the immobility time of FST in MyD88-overexpressed mice (F(1, 71) = 2.127, p = 0.1491; AAV-MyD88 + Vehicle vs AAV-MyD88 + rHMGB1, p = 0.0434; Fig. 3B), but not in GFP-injected mice, with no significant effect on locomotor activity measured by the OFT (F(1, 72) = 1.112, p = 0.2952; AAV-MyD88 + Vehicle vs AAV-MyD88 + rHMGB1, p > 0.9999; Fig. 3C). These results indicate that overexpression of MyD88 in the mPFC aggravates HMGB1-induced behavioral despair.
Fig. 3.

Overexpression of MyD88 in the mPFC facilitates DAMPs-induced behavioral abnormalities. (A) Schematic timeline of viral injection, cannula implantation, drugs treatment and behavioral tests. (B and C) Intra-mPFC infusion of rHMGB1 (2 µM) increased the immobility time in FST (B) with no changes total distance traveled (C) in the OFT in MyD88-overexpressed mice (n = 18–20 mice/group). (D-F) Exposure with FPS-ZM1 (200 nM) in the mPFC reduced the immobility time in TST (D) and FST (E) with no effect on total distance traveled (F) in the OFT in MyD88-overexpressed mice (n = 7–12 mice/group). (G-I) Intra-mPFC injection of SSnB (100 µM) blocked the prolongation of immobility time in TST (G, n = 7–10 mice/group) and FST (H, n = 16–21 mice/group) without changing total distance traveled (I, n = 8–10 mice/group) in the OFT in MyD88-overexpressed mice. Data are presented as mean ± SEM, with each point representing data from an individual. *p < 0.05, **p < 0.01, by two-way (B-I) analysis of variance followed by Bonferroni’s post hoc test. See also Fig. S3.
We next investigated whether MyD88 overexpression mediates neuroinflammation via engaging TLR2/4 and RAGE. It was found that intra-mPFC injection of FPS-ZM1 (200 nM, 1 µl/side), a RAGE inhibitor (Deane et al., 2012), for 3 d, prevented MyD88 overexpression-induced behavioral despair, indicated by reduced immobility time in TST (F(1, 34) = 5.205, p = 0.0289; AAV-MyD88 + Vehicle vs AAV-MyD88 + FPS-ZM1, p = 0.0399; Fig. 3D) and FST (F (1, 34) = 7.199, p = 0.0112; AAV-MyD88 + Vehicle vs AAV-MyD88 + FPS-ZM1, p = 0.0101; Fig. 3E), without changing motor function in the OFT (F(1, 34) = 0.1956, p = 0.6611; AAV-MyD88 + Vehicle vs AAV-MyD88 + FPS-ZM1, p > 0.9999; Fig. 3F). Next, intra-mPFC injection of SSnB (100 µM, 1 µl/side), a TLR2/4 inhibitor (Liang et al., 2011), for 3 d, also prevented MyD88 overexpression-induced behavioral despair, such as reduced immobility time in TST (F(1, 32) = 3.234, p = 0.0816; AAV-MyD88 + Vehicle vs AAV-MyD88 + SSnB, p = 0.0284; Fig. 3G) and FST (F(1,66) = 8.355, p = 0.0052; AAV-MyD88 + Vehicle vs AAV-MyD88 + SSnB, p = 0.0102; Fig. 3H), but exerted little effect on locomotor activity in the OFT (F(1, 33) = 0.1020, p = 0.7515; AAV-MyD88 + Vehicle vs AAV-MyD88 + SSnB, p > 0.9999; Fig. 3I). These findings further suggest that inhibition of DAMPs signaling may antagonize MyD88 overexpression-induced behavioral abnormalities.
3.4. MyD88 controls stress susceptibility via p38-MAPK/NF-κB signaling pathway
Our pervious study has reported that caspase-1-IL-1β signaling pathway underlies depression pathogenesis via regulating α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) (Li et al., 2018). In this study, we found that SSDS resulted in an approximately 32.7 % and 23.9 % increase in the level of activated caspase-1 (Casp 1 p10, t = 3.004, p = 0.0110) and IL-1β (pro-IL-1β: t = 2.735, p = 0.0097), respectively (Fig. 4 A), with a 36.6 % and 25.2 % decrease in the expression of AMPAR subunit glutamate receptor 1 (GluA1, t = 2.799, p = 0.0108) and AMPAR subunit glutamate receptor 2 protein (GluA2, t = 2.496, p = 0.0209) (Fig. 4B) in MyD88-overexpressed mice, respectively, prompting a possible mechanism underlying MyD88-promoted stress susceptibility.
Fig. 4.

MyD88/p38-MAPK/NF-κB signaling pathway mediates stress susceptibility. (A-B) Representative western blots showing the increased levels of activated Casp1 (A, n = 7 mice/group) and IL-1β (A, n = 17–20 mice/group), as well as decreased levels of GluA1 and GluA2 (B, n = 11–12 mice/group) in the mPFC of MyD88-overexpressed mice exposure to SSDS. (C) Experimental paradigms for viral injection, cannula implantation, drugs treatment and behavioral tests. (D) Representative heatmap of SI behavior from MyD88-overexpressed mice after JSH-23 or SB203580 pretreatment followed by stimulation with SSDS. (E-H) SB203580 (5 µM) or JSH-23 (20 µM) prevented SSDS-induced social avoidance (E), sucrose preference deficits (F), and prolongation of immobility time in TST (G) and FST (H) in MyD88-overexpressed mice (n = 10–12 mice/group). (I) Pretreatment of MyD88-overexpressed mice with JSH-23 or SB203580 followed by stimulation with SSDS produced little effect on total distance traveled in the OFT (n = 12 mice/group). (J and K) Representative western blots showed that not SB203580 (5 µM) (J) but JSH-23 (20 µM) (K) prevented SSDS-induced an increase in the expression of IL-1β in MyD88-overexpressed mice (n = 10 mice/group). Data are presented as mean ± SEM, with each point representing data from an individual. *p < 0.05, **p < 0.01, ***p < 0.001, by two-way (E-K) analysis of variance followed by Bonferroni’s post hoc test, Student’s t test (A and B).
MyD88 bridges TLRs to their downstream signaling pathways, such as the p38-mitogen-activated protein kinase (MAPK) and NF-κB signaling cascades, which have been involved in the regulation of IL-1β production (Lawrence, 2009, Liu et al., 2019b). We next sought to confirm the molecular pathway that cooperated MyD88 with depressive-like behaviors. MyD88-overexpressed mice were pretreated with SB203580 (i.c.v., 5 µM, 1 µl/side), a specific inhibitor of p38-MAPK (Liu et al., 2020), and JSH-23 (i.c.v., 20 µM, 1 µl/side), a NF-κB inhibitor (Koo et al., 2010), for 24 h, respectively, followed by exposure to SSDS (Fig. 4C). We found that pretreatment with SB203580 and JSH-23 prevented SSDS-induced depressive-like behaviors in MyD88-overexpressed mice, as evidenced by increased social interactions (F(2, 63) = 6.900, p = 0.0020; AAV-MyD88 + Vehicle vs AAV-MyD88 + SB203580, p = 0.0018; AAV-MyD88 + Vehicle vs AAV-MyD88 + JSH-23, p = 0.0054; Fig. 4D-E) and sucrose preference (F(2, 63) = 4.411, p = 0.0161; AAV-MyD88 + Vehicle vs AAV-MyD88 + SB203580, p = 0.0196; AAV-MyD88 + Vehicle vs AAV-MyD88 + JSH-23, p = 0.0473; Fig. 4F), as well as shortened immobility time in TST (F(2, 63) = 4.371, p = 0.0167; AAV-MyD88 + Vehicle vs AAV-MyD88 + SB203580, p = 0.0320; AAV-MyD88 + Vehicle vs AAV-MyD88 + JSH-23, p = 0.0135; Fig. 4G) and FST (F(2, 63) = 5.322, p = 0.0073; AAV-MyD88 + Vehicle vs AAV-MyD88 + SB203580, p = 0.0006; AAV-MyD88 + Vehicle vs AAV-MyD88 + JSH-23, p = 0.0262; Fig. 4H). Neither SB203580 nor JSH-23 affected locomotor activity in the OFT (F(2, 66) = 0.4539, p = 0.6371; AAV-MyD88 + Vehicle vs AAV-MyD88 + SB203580, p > 0.9999; AAV-MyD88 + Vehicle vs AAV-MyD88 + JSH-23, p = 0.6809) in MyD88-overexpressed mice after SSDS (Fig. 4I). Meanwhile, exposure of MyD88-overexpressed mice to SSDS also resulted in an increase in the expression of IL-1β, which was abolished by pretreatment with JSH-23, but not SB203580 (pro-IL-1β: F(1, 36) = 3.384, p = 0.0741; AAV-GFP + Vehicle vs AAV-MyD88 + Vehicle, p = 0.0177; AAV-MyD88 + Vehicle vs AAV-MyD88 + SB203580, p = 0.3367, Fig. 4J; pro-IL-1β: F(1, 36) = 5.550, p = 0.0240; AAV-GFP + Vehicle vs AAV-MyD88 + Vehicle, p = 0.0045; AAV-MyD88 + Vehicle vs AAV-MyD88 + JSH-23, p = 0.0375; Fig. 4K). These results suggest that p38-MAPK/NF-κB signaling pathway may underlie the increase in stress susceptibility in MyD88-overexpressed mice.
3.5. Injection of SARS-COV-2 spike RBD into mPFC induces depressive-like behaviors via MyD88-dependent proinflammatory signaling
Previous studies have reported that the binding of coronavirus spike protein to the TLRs induced the production of proinflammatory cytokine (Aboudounya and Heads, 2021, Lewis et al., 2021). Thus, we investigated whether SARS-CoV-2 spike protein induced microglial activation and depressive-like behaviors. We used SARS-CoV-2 spike RBD to mimic the effect of SARS-CoV-2 on microglia. It was shown that exposure of primary microglia to spike RBD (10 nM, but not 0.1 nM and 1 nM) for 24 h increased the immunofluorescence intensity of CD68 and Iba1 by 65.6 % and 52.0 %, respectively, together with more “amoeboid” morphology (CD68: t = 3.416, p = 0.0027; Iba1: t = 2.103, p = 0.0483; Fig. 5 A; and CD68: F(2, 12) = 0.5785, p = 0.5756; Iba1: F(2, 12) = 0.2968, p = 0.7485; Fig. S4A). Meanwhile, the expression of IL-1β and interleukin-6 (IL-6) in spike RBD (10 nM, but not 0.1 nM and 1 nM, 24 h)-exposed BV2 cells were elevated by about 20 % (pro-IL-1β: t = 2.342, p = 0.0286; IL-6: t = 2.844, p = 0.0174; Fig. 5B; and pro-IL-1β: F(2, 33) = 0.02688, p = 0.9735; mat-IL-1β: F(2, 33) = 0.4513, p = 0.6407; Fig. S4B), indicating that SARS-CoV-2 spike RBD triggers neuroinflammation. The final concentration of 10 nM (1 µl/side) of spike RBD was employed in the following animal experiments.
Fig. 5.

SARS-COV-2 spike RBD induces microglial activation and increases behavioral abnormalities by activating MyD88 signaling pathway. (A) Representative images (left) of immunostaining for CD68 (red) and Iba1 (grey) in primary microglia. Quantitative analyses (right) showing the activated microglia induced by Spike RBD (10 nM). Scale bar, 50 µm (Left), 20 µm (Right) (n = 11 wells/group). (B) Representative western blots showing increased expression of IL-1β (n = 12 wells/group) and IL-6 (n = 6 wells/group) in BV2 cells exposed to Spike RBD (10 nM). (C) Schematic timeline of cannula implantation, Spike RBD treatment and behavioral tests. (D) Spike RBD (10 nM) increased the immobility time in TST and FST in naïve mice (n = 17–19 mice/group). (E-F) Spike RBD (10 nM) did not alter sucrose preference (E, n = 7–8 mice/group) and total distance traveled (F, n = 17–19 mice/group) in mice. (G) Representative western blots showing the upregulated expression of MyD88 (n = 6–8 mice/group), TLR2 (n = 6–8 mice/group), phospho-NF-κB p65 (n = 9–10 mice/group) and Iba1 (n = 10 mice/group) in mice exposed to Spike RBD. (H) Representative western blots showing the increased IL-1β (n = 12 mice/group) production and reduced expression of GluA1 and GluA2 (n = 6–7 mice/group) in mice exposed to Spike RBD. (I) Schematic timeline of cannula implantation, MIP, Spike RBD treatment and behavioral tests. (J-K) MIP (50 µM) alleviated the increase in immobility time induced by Spike RBD in FST (J), without changing total distance traveled (K) in the OFT (n = 7–9 mice/group). Data are presented as mean ± SEM, with each point representing data from an individual. *p < 0.05, **p < 0.01, ***p < 0.001, by two-way (J-K) analysis of variance followed by Bonferroni’s post hoc test, Student’s t test (A, B, D-H). See also Fig. S4.
To confirm that spike RBD induced a pro-depressive effect, we infused spike RBD (10 nM) stereotactically in naive mice and tested them 24 h later in behavioral test (Fig. 5C). Intriguingly, spike RBD alone, without priming by SSDS was sufficient to induce behavioral despair of mice, indicating by prolonged immobility time in TST from 80.62 ± 8.285 s to 114.2 ± 13.64 s (t = 2.046, p = 0.0485) and FST from 50.84 ± 3.792 s to 78.60 ± 9.938 s (t = 2.498, p = 0.0175; Fig. 5D), but exerted little effect on sucrose preference (t = 0.2274, p = 0.8236; Vehicle = 87.21 ± 4.004 %; Spike RBD = 88.18 ± 1.855 %; Fig. 5E) and locomotor activity in the OFT (t = 0.4502, p = 0.6554; Vehicle = 40.81 ± 3.304 m; Spike RBD = 42.80 ± 2.965 m; Fig. 5F). Subsequent analysis showed that spike RBD exposure resulted in a 30.2 %, 131.4 %, 200.4 % and 26.8 % increase in the expression of MyD88 (t = 2.373, p = 0.0352), TLR2 (t = 3.032, p = 0.0104) and NF-κB p65 phosphorylation (t = 3.150, p = 0.0058), as well as Iba1 (t = 2.313, p = 0.0327), respectively (Fig. 5G). Furthermore, spike RBD also elevated the expression of pro-IL-1β and mat-IL-1β by 63.5 % and 39.7 % (pro-IL-1β: t = 3.150, p = 0.0046; mat-IL-1β: t = 2.280, p = 0.0326), and decreased the expression of GluA1 (t = 3.061, p = 0.0108) and GluA2 (t = 2.892, p = 0.0146) by about 30 % (Fig. 5H). We next assessed whether MyD88-dependent proinflammatory signaling mediated spike RBD-induced behavioral abnormalities (Fig. 5I). Pretreatment with MIP (i.c.v., 50 µM, 1 µl/side), a classic MyD88 antagonist, for 3 d prevented spike RBD-induced behavioral despair of mice, as indicated by reduced immobility time in FST (F(1, 28) = 11.58, p = 0.0020; Spike RBD + CP vs Spike RBD + MIP, p < 0.0001; Fig. 5J), without altering general locomotion measured the OFT (F(1, 29) = 0.003286, p = 0.9547; Spike RBD + CP vs Spike RBD + MIP, p > 0.9999; Fig. 5K). Taken together, these findings suggest that SARS-CoV-2 spike RBD may work as a precipitating factor that induces behavioral abnormalities.
3.6. Injection of SARS-COV-2 spike RBD into mPFC increases stress susceptibility via MyD88-dependent proinflammatory signaling
Thus, we asked whether injection of SARS-COV-2 spike RBD into the mPFC followed by exposure to SSDS would increase susceptibility to stress (Fig. 6 A). Our results found that spike RBD-treated mice are more susceptible to SSDS, as indicated by an approximately 38.5 % increase in social avoidance (F(1,66) = 11.34, p = 0.0013; Vehicle (Target) vs Spike RBD (Target), p = 0.0155; Fig. 6B), with a 43.2 % decrease in sucrose preference (t = 3.060, p = 0.0099; Fig. 6C), and prolonged immobility time in TST from 81.80 s ± 9.553 s to 126.1 ± 10.56 s (t = 3.061, p = 0.0044) and FST from 45.77 ± 7.804 s to 81.62 ± 9.735 s (t = 2.799, p = 0.0085; Fig. 6D), without altering spontaneous activity in the OFT (t = 1.810, p = 0.0794; Vehicle = 33.22 ± 3.240 m; Spike RBD = 26.84 ± 1.745 m; Fig. 6E). Notably, following SSDS exposure, the expression of MyD88 (t = 5.721, p < 0.0001), TLR2 (t = 2.717, p = 0.0137) and NF-κB p65 phosphorylation (t = 4.045, p = 0.0007) in the mPFC of spike RBD-treated mice were increased by 208.4 %, 86.6 % and 148.1 %, respectively (Fig. 6F). In addition, SSDS resulted in an approximately 221.9 % and 106.2 % increase in the expression of pro-IL-1β and mat-IL-1β (pro-1β: t = 3.229, p = 0.0044; mat-1β: t = 2.765, p = 0.0123), respectively, with a decrease in 54.1 % and 40.5 % in the total levels of GluA1 (t = 3.231, p = 0.0044) and GluA2 (t = 2.627, p = 0.0166) in spike RBD-treated mice (Fig. 6G). Next, we investigated whether MyD88 signaling pathway was required for SSDS-induced behavioral abnormalities in spike RBD-treated mice. Mice were pre-injected with MIP (i.c.v., 50 µM, 1 µl/side) for 3 d followed by treatment with spike RBD and SSDS (Fig. 6H). It was found that MIP prevented SSDS-induced behavioral abnormalities in spike RBD-exposed mice, such as deficits in social interactions (F(1, 29) = 11.44, p = 0.0021; Spike RBD + CP vs Spike RBD + MIP, p = 0.0026; Fig. 6I) and sucrose preference (F(1, 29) = 8.109, p = 0.0080; Spike RBD + CP vs Spike RBD + MIP, p = 0.0295; Fig. 6J), as well as behavioral despair (TST: F(1, 29) = 22.60, p < 0.0001; Spike RBD + CP vs Spike RBD + MIP, p < 0.0001; FST: F(1, 29) = 3.788, p = 0.0614; Spike RBD + CP vs Spike RBD + MIP, p = 0.0430; Fig. 6K-L), with no effect on locomotor activity (F(1, 29) = 0.3645, p = 0.5507; Spike RBD + CP vs Spike RBD + MIP, p > 0.9999; Fig. 6M). Together, these results suggest that spike RBD-increased stress susceptibility is mediated by activation of MyD88 signaling pathway in the mPFC.
Fig. 6.

MyD88 signaling pathway mediates SARS-CoV-2 spike RBD-increased stress susceptibility. (A) Schematic timeline of cannula implantation, Spike RBD treatment, SSDS and behavioral tests. (B–E) SSDS decreased the social interaction time (B, n = 16–19 mice/group) and sucrose preference (C, n = 6–8 mice/group), as well as increased immobility time in TST and FST (D, n = 16–19 mice/group), without altering total distance (E, n = 16–19 mice/group) in Spike RBD-treated mice. (F) Representative western blots showing the elevated levels of MyD88, TLR2 and phospho-NF-κB p65 in the mPFC of Spike RBD-treated mice after SSDS (n = 10–11 mice/group). (G) Representative western blots showing the increased IL-1β expression, and reduced expression of GluA1 and GluA2 in the mPFC of Spike RBD-treated mice after SSDS (n = 10–11 mice/group). (H) Schematic timeline of cannula implantation, MIP and Spike RBD treatment, SSDS and behavioral tests. (I-M) MIP (50 µM) reversed SSDS-induced social avoidance (I), sucrose preference deficits (J), and prolongation of immobility time in TST (K) and FST (L) in Spike RBD-treated mice, without changing total distance traveled (M) in the OFT (n = 8–9 mice/group). Data are presented as mean ± SEM, with each point representing data from an individual. *p < 0.05, **p < 0.01, ***p < 0.001, by two-way (B, I–M) analysis of variance followed by Bonferroni’s post hoc test, Student’s t test (C–G).
3.7. Targeting MyD88 dimerization by TJ-M2010-5 alleviates CSDS-/SARS-COV-2 spike RBD-induced depressive-like behaviors and proinflammatory signaling
In our previous studies (Ding et al., 2019, Liu et al., 2019a, Xie et al., 2016, Zou et al., 2020), we designed, synthesized and characterized a novel small MyD88 inhibitor, TJ-M2010-5, which specifically interacts with the MyD88 TIR domain and interferes with MyD88 dimerization. In vitro, we sought to validate the effect of TJ-M2010-5 on MyD88-related inflammatory pathways. It was shown that pretreatment with TJ-M2010-5 (30 µM) inhibited CORT (50 nM)-induced neuroinflammatory activation in BV2 cells, indicating by a reduced level of NF-κB p65 phosphorylation (F(1, 20) = 4.904, p = 0.0386; CORT + Vehicle vs CORT + TJ-M2010-5, p = 0.0301) and IL-1β protein (F(1, 20) = 8.442, p = 0.0087; CORT + Vehicle vs CORT + TJ-M2010-5, p = 0.0048) (Fig. 7 A). Next, we sought to determine the effect of TJ-M2010-5 on behavioral despairin mice and screened the effective dosage of TJ-M2010-5 using TST. We found that exposure of mice to TJ-M2010-5 at 1.25, 2.5 and 5 mg/kg (i.p., once daily) for 1 d (F(3, 32) = 0.6535, p = 0.5867) or 2 d (F(3, 32) = 0.2472, p = 0.8627) exerted little effect on the immobility time in TST (Fig. S5A, B). The results showed that intraperitoneal injection of TJ-M2010-5 (2.5 mg/kg, twice daily) for 2 d reduced the immobility time in TST (F(3, 32) = 4.075, p = 0.0147; Vehicle vs 1.25 mg/kg, p = 0.0295; Vehicle vs 2.5 mg/kg, p = 0.0082; Fig. 7B). Additionally, further analysis indicated that injection of TJ-M2010-5 (2.5 mg/kg, i.p., twice daily) for at least two consecutive days reduced the immobility time in TST (F(2, 35) = 15.50, p < 0.0001; 0 d vs 1 d, p = 0.2385; 0 d vs 2 d, p < 0.0001; Fig. 7C, Fig. S5C) and FST (F(2, 36) = 8.847, p = 0.0007; 0 d vs 1 d, p = 0.8170; 0 d vs 2 d, p = 0.0005; Fig. 7D), indicating a two-day administration was required for the antidepressant effects of TJ-M2010-5.
Fig. 7.

TJ-M2010-5, a small molecular MyD88 inhibitor, prevents CSDS-/SARS-COV-2 Spike RBD-induced depressive-like behaviors and neuroinflammation in mice. (A) TJ-M2010-5 (30 µM) blocked CORT-induced increase in phospho-NF-κB p65 and IL-1β level (n = 5–7 wells/group). (B) TJ-M2010-5 (1.25, 2.5 and 5 mg/kg, intraperitoneally, twice daily) for 2 d reduced the immobility time in TST in naïve mice (n = 9 mice/group). (C and D) Time-dependent effects of TJ-M2010-5 (2.5 mg/kg, intraperitoneally, twice daily) on the immobility time in TST (C, n = 9–19 mice/group) and FST (D, n = 9–20 mice/group). (E) Experimental timeline of CSDS, TJ-M2010-5 administration and behavioral tests. (F-G) TJ-M2010-5 (2.5 mg/kg, intraperitoneally, twice daily) for 2 d rescued CSDS-induced social avoidance (F, n = 11–12 mice/group) and sucrose preference deficits (G, n = 10–12 mice/group). (H and I) TJ-M2010-5 (2.5 mg/kg, intraperitoneally, twice daily) for 2 d blocked CSDS-induced the increase in the phosphorylation expression of NF-κB p65 (H, n = 11–13 mice/group) and IL-1β production (I, n = 12–14 mice/group) in the mPFC. (J–L) TJ-M2010-5 (2.5 mg/kg, intraperitoneally, twice daily) blocked the effect of Spike RBD on immobility time in TST (J, n = 19 mice/group) and FST (K, n = 18–19 mice/group) without affecting total distance traveled (L, n = 19 mice/group) in the OFT. Data are presented as mean ± SEM, with each point representing data from an individual. *p < 0.05, **p < 0.01, ***p < 0.001, by one-way (B-D) or two-way (A, F-L) analysis of variance followed by Bonferroni’s post hoc test. See also Fig. S5.
Then, the mice exposed to CSDS were used to further evaluate the beneficial effects of TJ-M2010-5 (Fig. 7E). We found that intraperitoneal injection of TJ-M2010-5 (2.5 mg/kg, twice daily) for 1 d failed to reverse the social avoidance in susceptible mice (F (1, 17) = 0.5569, p = 0.4657; Fig. S5D), while exposure of susceptible mice to TJ-M2010-5 (2.5 mg/kg, intraperitoneally, twice daily) for 2 d rescued CSDS-induced deficits in social interactions (F(1, 42) = 1.736, p = 0.1947; CSDS + Vehicle vs CSDS + TJ-M2010-5, p = 0.0401; Fig. 7F) and sucrose preference (F(1, 41) = 2.319, p = 0.1355; CSDS + Vehicle vs CSDS + TJ-M2010-5, p = 0.0330; Fig. 7G). TJ-M2010-5 (2.5 mg/kg, intraperitoneally, twice daily) for 2 d also blocked CSDS-induced NF-κB overactivation (F(1, 42) = 4.235, p = 0.0458; CSDS + Vehicle vs CSDS + TJ-M2010-5, p = 0.0131; Fig. 7H) and IL-1β overproduction (pro-IL-1β: F(1, 47) = 8.487, p = 0.0055; CSDS + Vehicle vs CSDS + TJ-M2010-5, p = 0.0043; mat-IL-1β: F(1, 47) = 9.331, p = 0.0037; CSDS + Vehicle vs CSDS + TJ-M2010-5, p = 0.0419; Fig. 7I).
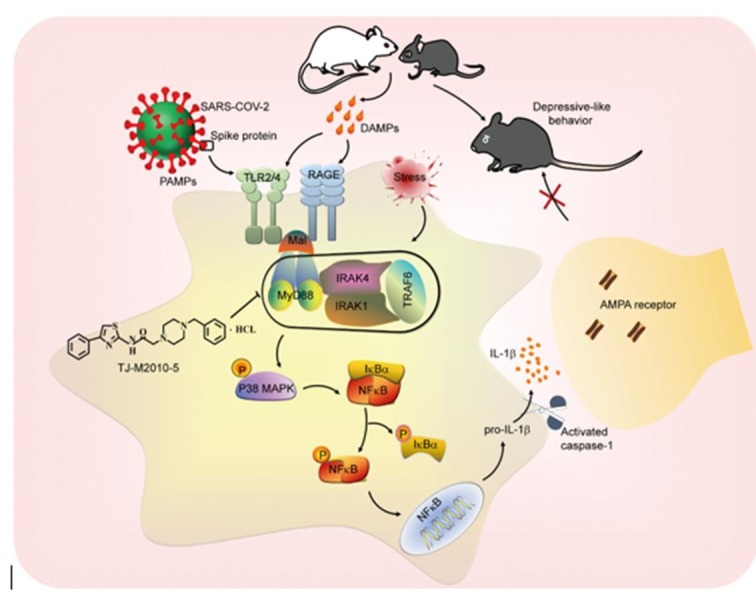
We next assessed whether TJ-M2010-5 prevented depression-related behaviors in mice exposed to spike RBD (Fig. 7J). Pretreatment of mice with TJ-M2010-5 (2.5 mg/kg, intraperitoneally, twice daily) for 2 d completely prevented the increase in immobility time in TST (F(1, 72) = 11.30, p = 0.0012; Spike RBD + Vehicle vs Spike RBD + TJ-M2010-5, p < 0.0001; Fig. 7K) and FST (F(1, 71) = 10.55, p = 0.0018; Spike RBD + Vehicle vs Spike RBD + TJ-M2010-5, p < 0.0001; Fig. 7L) caused by spike RBD, without altering locomotor activity in the OFT (F(1, 72) = 0.8654, p = 0.3553; Fig. 7M and Fig. S5E). Thus, TJ-M2010-5 may produce antidepressant effects via disrupting MyD88-dependent p38-MAPK/NF-κB/IL-1β signaling in the mPFC, and ameliorate stress- or SARS-CoV-2 spike protein-induced depressive-type behaviors (Fig. 8 ), thereby offering a potential therapy against MDD and neuroinflammation-related behavior abnormalities.
Fig. 8.

TJ-M2010-5 alleviates CSDS-/SARS-COV-2 Spike protein-related behavior abnormalities via targeting MyD88-dependent neuroinflammation. Stress-triggered DAMPs or SARS-COV-2 Spike protein as PAMPs bind to TLR2/4 or RAGE, leading to assembly of Myddosome, which heightens neuroinflammation by activating p38 MAPK/NF-kB signaling pathways. TJ-M2010-5 specifically interferes with MyD88 dimerization and thus interrupts the assembly of Myddosome, which may alleviate the development of depression by inhibiting neuroinflammation in vivo.
4. Discussion
Evidence from rodents and humans supports that depressive-like behaviors are associated with neuroinflammatory priming (Fenn et al., 2014, Fonken et al., 2018). Emerging evidence also indicates a role of neuroinflammation underlying COVID-19 neuropathology (Costanza et al., 2022, Matschke et al., 2020, Mingoti et al., 2022, Ribeiro et al., 2021). Our results revealed that MyD88, a pivotal adaptor that forms innate immune signaling complex called the ‘myddosome’, controls stress susceptibility via amplifying proinflammatory signaling. Inducible MyD88 expression by stress or SARS-CoV-2 spike protein induced behavioral abnormalities, which may be blocked by a small molecular MyD88 inhibitor, and highlighted MyD88 as a novel potential therapeutic target for depression, especially inflammation-related depression.
Our findings strongly support the emerging view that innate immune signaling contributes to the development of psychiatric disorders. Clinical findings have shown that the levels of the serum proinflammatory cytokines in MDD patients were significantly higher in comparison with controls (Dantzer et al., 2008, Stewart et al., 2009). However, as a noninfectious disease, less is known about how inflammation is activated in MDD. Previous reports have indicated that chronic stress-induced inflammatory responses occur at least partly via DAMPs signaling, and the receptors underlying stress-related DAMP signaling have recently been identified, including RAGE and TLR4 (Gong et al., 2020, Heijmans et al., 2012, Huebener et al., 2015). Except for DAMPs signaling, several lines of evidence have indicated that stress increases the permeability of the BBB by reducing the expression of tight junction proteins, and proinflammatory cytokines from peripheral circulation may entry central nervous system and activate TLRs in the brain of stressed individuals (Dion-Albert et al., 2022, Menard et al., 2017, Welcome and Mastorakis, 2020). Emerging roles of adaptor proteins in the function of TLRs have been clarified (Brown et al., 2011, Chen and Jiang, 2013). MyD88 is a key adaptor of TLRs and RAGE, which initiates key signal transduction pathways to elicit critical inflammatory immune responses by inducing the assembly of signaling complexes termed myddosomes (Deliz-Aguirre et al., 2021). In our study, rHMGB1 was used to mimic the effect of DAMPs, and we found that rHMGB1 only induced behavioral despair in MyD88-overexpressed mice. Meanwhile, both FPS-ZM1, a RAGE inhibitor, and SSnB, a TLR2/4 inhibitor, abolished MyD88 overexpression-induced behavioral despair, highlighting the role of innate immune signaling in stress susceptibility.
Increasing evidence has confirmed the role of inflammation in depression, whereas only a handful of studies have explored mechanisms to mediate neuroinflammation-induced depression-related behaviors. A very late study revealed that neuroinflammation promotes the development of depression-like symptoms during adolescence via excessive microglial engulfment of neuronal spines (Cao et al., 2021). Our previous study demonstrates that activation of IL-1β signaling pathway may generate depressive-like behaviors by downregulating the surface level of AMPARs (Li et al., 2018), a key glutamate receptor involved in depression and antidepressant therapy. Other studies have proved that lipopolysaccharide administration significantly increased IL-1β production and decreased GluA1 expression, which induced depressive-like behaviors (Li et al., 2019, Zhang et al., 2017). Our results suggested that chronic stress and SARS-CoV-2 infection elevated MyD88 expression, which drove IL-1β production via p38-MAPK/NF-κB pathway. It has been demonstrated that the increase in IL-1β led to the downregulation of AMPAR in the mPFC, which may directly generate depressive-like behaviors. In this study, we clarified the upstream signaling of IL-1β-AMPAR and revealed that inducible expression of MyD88 by stress facilitated the pattern recognition to trigger AMPAR deficits, which may represent a basis for inflammation-induced depression.
Our results also indicated that spike-MyD88 signaling may induce behavior abnormalities via amplifying neuroinflammation. The number of reported COVID-19 infection cases had surpassed 601 million, and the number of deaths caused by COVID-19 was more than 6.47 million (WHO operational update). COVID-19 is a multifaceted disease with multi-organ complications, including neurological symptoms. COVID-19-related mood disorders can occur during acute infection and persist or even emerge within 6 months after symptom onset (Huang et al., 2021a). It has been reported that SARS-CoV-2 infects brain vascular endothelial cells and crosses the blood–brain barrier, consequently activates the microglia, leading to a hyperinflammatory response, which may further induce neurological symptoms (Goncalves de Andrade et al., 2021, Zhang et al., 2021). Previous studies indicated that the coronavirus spike protein binds to TLRs and activates proinflammatory signaling (Olajide et al., 2021, Shirato and Kizaki, 2021, Zhao et al., 2021). Our results indicated that the SARS-CoV-2 spike protein generated behavioral abnormalities in vivo, and we proposed that the induction of neuroinflammation by spike protein was mediated through MyD88-dependent proinflammatory signaling, possibly as a result of TLRs activation. However, it should be noted that spike protein-induced behavioral abnormality is a highly limited model system to evaluate how SARS-CoV-2 induces behavior abnormalities. The mechanism involved in the COVID-19-related neurological manifestations are complicated. Based on our results, we hypothesized that MyD88 contributed to neuroinflammation-related depression via two mechanisms: Firstly, inducible MyD88 expression by SARS-CoV-2 spike protein in the mPFC may directly activate neuroinflammation and generate behavioral abnormalities, indicating by prolonged immobility time in TST and FST caused by spike RBD alone, without priming by SSDS (Fig. 5H). Secondly, as a stressful event, COVID-19 has an alarming impact on mental health (Amsalem et al., 2021). Our findings indicate that inducible MyD88 expression increases stress susceptibility. COVID-19 patients might be more vulnerable to mental health risk; thus, MyD88-increased stress susceptibility may be involved. Therefore, MyD88 may serve as a promising therapeutic target for neuroinflammation-related mental disorders.
A greater understanding of the role of neuroinflammation in stress responses and mood disorders may facilitate the development of antidepressants by targeting innate immune signaling. To date, however, no anti-inflammatory drugs have been used in clinical practice to directly improve MDD symptoms. Several peptide MyD88 inhibitors are commercially available, including ST2825 (Wang et al., 2019), and Pepinh-MYD (Sulaiman et al., 2017); however, due to their large molecular weights, pharmacokinetic disadvantages limit their application in neurological diseases. TJ-M2010-5 was one member of a series of 2-aminothiazole-derived MyD88 inhibitors (TJ-M2010 series; WIPO patent application number: PCT/CN2012/070811) in our laboratory. TJ-M2010-5 can bind to the TIR domain of MyD88 and inhibit MyD88 dimerization (Xie et al., 2016), thus disrupting myddosome formation. TJ-M2010-5 has been demonstrated to be effective in the treatment of acute liver injury (Ding et al., 2019), transplant rejection (Li et al., 2017), and colitis-associated colorectal cancer (Xie et al., 2016). Here, we found that TJ-M2010-5 at a moderate dose, but not the highest dose, exerted a better antidepressant effect, indicating that the highest dose of TJ-M2010-5 may produce side effects that counteracted the behavior benefits. Additionally, TJ-M2010-5 ameliorated both stress- and coronavirus spike protein-induced depressive-like behaviors, illustrating the potential value of TJ-M2010-5 in the treatment of psychiatric disorders. Considering the delayed onset of action, limited therapeutic efficacy and modest response rates of currently available antidepressants, and few drugs with demonstrated clinical efficacy for COVID-19-related mental disorders are available, targeting MyD88 dimerization by TJ-M2010-5 may provide a new avenue for stress-related disorders.
It should be noted that our study still has limitations that need to be addressed. First, although an increase in MyD88 expression was observed in CORT-treated BV2 cells and CSDS-treated mice, our current results cannot clarify the mechanism underlying the inducible expression of MyD88. More-detailed mechanisms required further investigation. Second, although overexpression of MyD88 may underlie the behavior effects of spike protein, some inconsistency also existed. For instance, MyD88 overexpression in the mPFC decreased sucrose preference but microinjection of spike protein exerted little effect. Third, although some evidence indicates that a possible role of immune activation in mood disorders due to COVID-19 (Al-Jassas et al., 2022), the level of neuroinflammation in the COVID-19 patients are difficult to evaluate. It should be noted that the spike protein-induced behavioral abnormality was induced by directly injecting the spike protein into the brain, not by natural infection. Although it was very useful to assess how MyD88 signaling contributes to neuroinflammation-induced behaviors, the limitations were obvious for its use in investigations about how SARS-CoV-2 affects behaviors. Additionally, some pattern recognition receptors such as TLR2 could be as well expressed in reactive astrocytes. Although our immunohistological data indicated that in the mPFC, the expression of TLR2 was mainly expressed in microglia, but not astrocyte, both in the normal and stressed mice, the effect of TLR2 and MyD88-related neuroinflammation signaling in astrocyte could not be fully excluded. Thus, the role of reactive astrocytes in inflammation-related depression should be investigated in the next study.
In summary, our results reveal a distinct role for MyD88 in regulating stress and spike protein-induced depressive-like behaviors, indicating that MyD88 may be an attractive therapeutic target for stress- and COVID-19-related mental disorders. We also show that MDD patients or COVID19 patients with mental disorders may benefit from MyD88-targeted therapeutics.
CRediT authorship contribution statement
Xia-Ping Yao: . Jian Ye: . Ting Feng: . Feng-Chao Jiang: . Ping Zhou: . Fang Wang: Funding acquisition. Jian-Guo Chen: Funding acquisition. Peng-Fei Wu: Methodology.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Foundation for National Key R&D Program of China (Grant No. 2021ZD0202900 to J-G.C.), National Natural Science Foundation of China (Grant No. 82130110 to J-G.C. and Grant No. U21A20363 to F.W.), Innovative Research Groups of National Natural Science Foundation of China (Grant No. 81721005 to J-G.C. and F.W.), National Natural Science Foundation of China (No. 82073834 to P.F.W., No. 81971279 to F.W., No. 81973310 to J.G.C.), and PCSIRT (No. IRT13016 to J.G.C).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbi.2022.12.007.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- Aboudounya M.M., Heads R.J. COVID-19 and Toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediators Inflamm. 2021;2021:8874339. doi: 10.1155/2021/8874339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jassas H.K., Al-Hakeim H.K., Maes M. Intersections between pneumonia, lowered oxygen saturation percentage and immune activation mediate depression, anxiety, and chronic fatigue syndrome-like symptoms due to COVID-19: A nomothetic network approach. J. Affect. Disord. 2022;297:233–245. doi: 10.1016/j.jad.2021.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsalem D., Dixon L.B., Neria Y. The coronavirus disease 2019 (COVID-19) outbreak and mental health: Current risks and recommended actions. JAMA Psychiatry. 2021;78:9–10. doi: 10.1001/jamapsychiatry.2020.1730. [DOI] [PubMed] [Google Scholar]
- Bauer L., Laksono B.M., de Vrij F.M.S., Kushner S.A., Harschnitz O., van Riel D. The neuroinvasiveness, neurotropism, and neurovirulence of SARS-CoV-2. Trends Neurosci. 2022;45:358–368. doi: 10.1016/j.tins.2022.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J., Wang, H., Hajishengallis, G.N., Martin, M., 2011. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 90, 417–427. https://doi.org/10.1177/0022034510381264. [DOI] [PMC free article] [PubMed]
- Cao P., Chen C., Liu A., Shan Q., Zhu X., Jia C., Peng X., Zhang M., Farzinpour Z., Zhou W., Wang H., Zhou J.N., Song X., Wang L., Tao W., Zheng C., Zhang Y., Ding Y.Q., Jin Y., Xu L., Zhang Z. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron. 2021;109:2573–2589.e9. doi: 10.1016/j.neuron.2021.06.012. [DOI] [PubMed] [Google Scholar]
- Chen H., Jiang Z. The essential adaptors of innate immune signaling. Protein Cell. 2013;4:27–39. doi: 10.1007/s13238-012-2063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Pardo M., Armini R.S., Martinez A., Mouhsine H., Zagury J.F., Jope R.S., Beurel E. Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav. Immun. 2016;53:207–222. doi: 10.1016/j.bbi.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanza A., Amerio A., Aguglia A., Serafini G., Amore M., Hasler R., Ambrosetti J., Bondolfi G., Sampogna G., Berardelli I., Fiorillo A., Pompili M., Nguyen K.D. Hyper/neuroinflammation in COVID-19 and suicide etiopathogenesis: Hypothesis for a nefarious collision? Neurosci. Biobehav. Rev. 2022;136 doi: 10.1016/j.neubiorev.2022.104606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R., O'Connor J.C., Freund G.G., Johnson R.W., Kelley K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R., Singh I., Sagare A.P., Bell R.D., Ross N.T., LaRue B., Love R., Perry S., Paquette N., Deane R.J., Thiyagarajan M., Zarcone T., Fritz G., Friedman A.E., Miller B.L., Zlokovic B.V. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deliz-Aguirre R., Cao F., Gerpott F.H.U., Auevechanichkul N., Chupanova M., Mun Y., Ziska E., Taylor M.J. MyD88 oligomer size functions as a physical threshold to trigger IL1R Myddosome signaling. J. Cell Biol. 2021;220:e202012071. doi: 10.1083/jcb.202012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S.L., Hu Z.L., Mao L., Gao B., Yang Q., Wang F., Chen J.G. The effects of Kctd12, an auxiliary subunit of GABAB receptor in dentate gyrus on behavioral response to chronic social defeat stress in mice. Pharmacol. Res. 2021;163 doi: 10.1016/j.phrs.2020.105355. [DOI] [PubMed] [Google Scholar]
- Ding Z., Du D., Yang Y., Yang M., Miao Y., Zou Z., Zhang X., Li Z., Zhang X., Zhang L., Wang X., Zhao Y., Jiang J., Jiang F., Zhou P. Short-term use of MyD88 inhibitor TJ-M2010-5 prevents d-galactosamine/lipopolysaccharide-induced acute liver injury in mice. Int. Immunopharmacol. 2019;67:356–365. doi: 10.1016/j.intimp.2018.11.051. [DOI] [PubMed] [Google Scholar]
- Dion-Albert L., Cadoret A., Doney E., Kaufmann F.N., Dudek K.A., Daigle B., Parise L.F., Cathomas F., Samba N., Hudson N., Lebel M., Signature C., Campbell M., Turecki G., Mechawar N., Menard C. Vascular and blood-brain barrier-related changes underlie stress responses and resilience in female mice and depression in human tissue. Nat. Commun. 2022;13:164. doi: 10.1038/s41467-021-27604-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn A.M., Gensel J.C., Huang Y., Popovich P.G., Lifshitz J., Godbout J.P. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol. Psychiatry. 2014;76:575–584. doi: 10.1016/j.biopsych.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald K.A., Kagan J.C. Toll-like receptors and the control of immunity. Cell. 2020;180:1044–1066. doi: 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken L.K., Frank M.G., Kitt M.M., D'Angelo H.M., Norden D.M., Weber M.D., Barrientos R.M., Godbout J.P., Watkins L.R., Maier S.F. The alarmin HMGB1 mediates age-induced neuroinflammatory priming. J. Neurosci. 2016;36:7946–7956. doi: 10.1523/JNEUROSCI.1161-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken L.K., Frank M.G., Gaudet A.D., D'Angelo H.M., Daut R.A., Hampson E.C., Ayala M.T., Watkins L.R., Maier S.F. Neuroinflammatory priming to stress is differentially regulated in male and female rats. Brain Behav. Immun. 2018;70:257–267. doi: 10.1016/j.bbi.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M.G., Weber M.D., Watkins L.R., Maier S.F. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav. Immun. 2015;48:1–7. doi: 10.1016/j.bbi.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M.G., Nguyen K.H., Ball J.B., Hopkins S., Kelley T., Baratta M.V., Fleshner M., Maier S.F. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav. Immun. 2021;100:267–277. doi: 10.1016/j.bbi.2021.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin T.C., Wohleb E.S., Zhang Y., Fogaca M., Hare B., Duman R.S. Persistent increase in microglial RAGE contributes to chronic stress-induced priming of depressive-like behavior. Biol. Psychiatry. 2018;83:50–60. doi: 10.1016/j.biopsych.2017.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay N.J., Gangloff M., O'Neill L.A. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 2011;32:104–109. doi: 10.1016/j.it.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Goncalves de Andrade E., Simoncicova E., Carrier M., Vecchiarelli H.A., Robert M.E., Tremblay M.E. Microglia fighting for neurological and mental health: on the central nervous system frontline of COVID-19 pandemic. Front. Cell. Neurosci. 2021;15 doi: 10.3389/fncel.2021.647378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong T., Liu L., Jiang W., Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020;20:95–112. doi: 10.1038/s41577-019-0215-7. [DOI] [PubMed] [Google Scholar]
- Hajebrahimi B., Bagheri M., Hassanshahi G., Nazari M., Bidaki R., Khodadadi H., Arababadi M.K., Kennedy D. The adapter proteins of TLRs, TRIF and MYD88, are upregulated in depressed individuals. Int. J. Psychiatry Clin. Pract. 2014;18:41–44. doi: 10.3109/13651501.2013.859708. [DOI] [PubMed] [Google Scholar]
- He J.G., Zhou H.Y., Xue S.G., Lu J.J., Xu J.F., Zhou B., Hu Z.L., Wu P.F., Long L.H., Ni L., Jin Y., Wang F., Chen J.G. Transcription factor TWIST1 integrates dendritic remodeling and chronic stress to promote depressive-like behaviors. Biol. Psychiatry. 2021;89:615–626. doi: 10.1016/j.biopsych.2020.09.003. [DOI] [PubMed] [Google Scholar]
- Heijmans J., Buller N.V., Muncan V., van den Brink G.R. Rage mediated DAMP signaling in intestinal tumorigenesis. Oncoimmunology. 2012;1:1165–1166. doi: 10.4161/onci.20929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi T., Yamawaki Y., Kimura H., Honda S., Ozawa K. Possible involvement of MyD88 in regulating stress response in mice. Front. Neurosci. 2021;15 doi: 10.3389/fnins.2021.621446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Huang L., Wang Y., Li X., Ren L., Gu X., Kang L., Guo L., Liu M., Zhou X., Luo J., Huang Z., Tu S., Zhao Y., Chen L., Xu D., Li Y., Li C., Peng L., Li Y., Xie W., Cui D., Shang L., Fan G., Xu J., Wang G., Wang Y., Zhong J., Wang C., Wang J., Zhang D., Cao B. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet. 2021;397:220–232. doi: 10.1016/s0140-6736(20)32656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Yao Q., Gu X., Wang Q., Ren L., Wang Y., Hu P., Guo L., Liu M., Xu J., Zhang X., Qu Y., Fan Y., Li X., Li C., Yu T., Xia J., Wei M., Chen L., Li Y., Xiao F., Liu D., Wang J., Wang X., Cao B. 1-year outcomes in hospital survivors with COVID-19: a longitudinal cohort study. Lancet. 2021;398:747–758. doi: 10.1016/s0140-6736(21)01755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebener P., Pradere J.P., Hernandez C., Gwak G.Y., Caviglia J.M., Mu X., Loike J.D., Schwabe R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Invest. 2015;125:539–550. doi: 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S.E., Hyam S.R., Jeong J.J., Han M.J., Kim D.H. Penta-O-galloyl-beta-D-glucose ameliorates inflammation by inhibiting MyD88/NF-kappaB and MyD88/MAPK signalling pathways. Br. J. Pharmacol. 2013;170:1078–1091. doi: 10.1111/bph.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S., Shafiei M.S., Longoria C., Schoggins J., Savani R.C., Zaki H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. eLife. 2021;10:e68563. doi: 10.7554/eLife.68563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo J.W., Russo S.J., Ferguson D., Nestler E.J., Duman R.S. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. U.S.A. 2010;107:2669–2674. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari P., Rothan H.A., Natekar J.P., Stone S., Pathak H., Strate P.G., Arora K., Brinton M.A., Kumar M. Neuroinvasion and encephalitis following intranasal inoculation of SARS-CoV-2 in K18-hACE2 mice. Viruses. 2021;13:132. doi: 10.3390/v13010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009;1 doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng L., Zhuang K., Liu Z., Huang C., Gao Y., Chen G., Lin H., Hu Y., Wu D., Shi M., Xie W., Sun H., Shao Z., Li H., Zhang K., Mo W., Huang T.Y., Xue M., Yuan Z., Zhang X., Bu G., Xu H., Xu Q., Zhang J. Menin deficiency leads to depressive-like behaviors in mice by modulating astrocyte-mediated neuroinflammation. Neuron. 2018;100(551–563):e7. doi: 10.1016/j.neuron.2018.08.031. [DOI] [PubMed] [Google Scholar]
- Lewis S.A., Sureshchandra S., Zulu M.Z., Doratt B., Jankeel A., Ibraim I.C., Pinski A.N., Rhoades N.S., Curtis M., Jiang X., Tifrea D., Zaldivar F., Shen W., Edwards R.A., Chow D., Cooper D., Amin A., Messaoudi I. Differential dynamics of peripheral immune responses to acute SARS-CoV-2 infection in older adults. Nat. Aging. 2021;1:1038–1052. doi: 10.1038/s43587-021-00127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.M., Liu L.L., Su W.J., Wang B., Zhang T., Zhang Y., Jiang C.L. Ketamine may exert antidepressant effects via suppressing NLRP3 inflammasome to upregulate AMPA receptors. Neuropharmacology. 2019;146:149–153. doi: 10.1016/j.neuropharm.2018.11.022. [DOI] [PubMed] [Google Scholar]
- Li C., Zhang L.M., Zhang X., Huang X., Liu Y., Li M.Q., Xing S., Yang T., Xie L., Jiang F.C., Jiang H.Y., He W.T., Zhou P. Short-term pharmacological inhibition of MyD88 homodimerization by a novel inhibitor promotes robust allograft tolerance in mouse cardiac and skin transplantation. Transplantation. 2017;101:284–293. doi: 10.1097/TP.0000000000001471. [DOI] [PubMed] [Google Scholar]
- Li M.X., Zheng H.L., Luo Y., He J.G., Wang W., Han J., Zhang L., Wang X., Ni L., Zhou H.Y., Hu Z.L., Wu P.F., Jin Y., Long L.H., Zhang H., Hu G., Chen J.G., Wang F. Gene deficiency and pharmacological inhibition of caspase-1 confers resilience to chronic social defeat stress via regulating the stability of surface AMPARs. Mol. Psychiatry. 2018;23:556–568. doi: 10.1038/mp.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q., Wu Q., Jiang J., Duan J., Wang C., Smith M.D., Lu H., Wang Q., Nagarkatti P., Fan D. Characterization of sparstolonin B, a Chinese herb-derived compound, as a selective Toll-like receptor antagonist with potent anti-inflammatory properties. J. Biol. Chem. 2011;286:26470–26479. doi: 10.1074/jbc.M111.227934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.C., Lo Y.C., Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Gao Q., Guan L., Sheng W., Hu Y., Gao T., Jiang J., Xu Y., Qiao H., Xue X., Liu S., Li T. Atorvastatin attenuates isoflurane-induced activation of ROS-p38MAPK/ATF2 pathway, neuronal degeneration, and cognitive impairment of the aged mice. Front. Aging Neurosci. 2020;12 doi: 10.3389/fnagi.2020.620946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Zhang Y., Liu S., Liu Y., Yang X., Liu G., Shimizu T., Ikenaka K., Fan K., Ma J. Cathepsin C promotes microglia M1 polarization and aggravates neuroinflammation via activation of Ca(2+)-dependent PKC/p38MAPK/NF-kappaB pathway. J. Neuroinflammation. 2019;16:10. doi: 10.1186/s12974-019-1398-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Zhang X., Wang H., Zhang M., Peng Y., Li M., Xie L., Jiang F., Gong Y., Zhao Q., Zhou P. Implication of myeloid differentiation factor 88 inhibitor TJ-M2010-5 for therapeutic intervention of hepatocellular carcinoma. Hepatol. Res. 2019;49:1182–1194. doi: 10.1111/hepr.13359. [DOI] [PubMed] [Google Scholar]
- Luo H., Wu P.F., Cao Y., Jin M., Shen T.T., Wang J., Huang J.G., Han Q.Q., He J.G., Deng S.L., Ni L., Hu Z.L., Long L.H., Wang F., Chen J.G. Angiotensin-converting enzyme inhibitor rapidly ameliorates depressive-type behaviors via bradykinin-dependent activation of mammalian target of rapamycin complex 1. Biol. Psychiatry. 2020;88:415–425. doi: 10.1016/j.biopsych.2020.02.005. [DOI] [PubMed] [Google Scholar]
- Matschke J., Lütgehetmann M., Hagel C., Sperhake J.P., Schröder A.S., Edler C., Mushumba H., Fitzek A., Allweiss L., Dandri M., Dottermusch M., Heinemann A., Pfefferle S., Schwabenland M., Sumner Magruder D., Bonn S., Prinz M., Gerloff C., Püschel K., Krasemann S., Aepfelbacher M., Glatzel M. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet. Neurol. 2020;19:919–929. doi: 10.1016/s1474-4422(20)30308-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard C., Pfau M.L., Hodes G.E., Kana V., Wang V.X., Bouchard S., Takahashi A., Flanigan M.E., Aleyasin H., LeClair K.B., Janssen W.G., Labonte B., Parise E.M., Lorsch Z.S., Golden S.A., Heshmati M., Tamminga C., Turecki G., Campbell M., Fayad Z.A., Tang C.Y., Merad M., Russo S.J. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017;20:1752–1760. doi: 10.1038/s41593-017-0010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingoti M.E.D., Bertollo A.G., Simoes J.L.B., Francisco G.R., Bagatini M.D., Ignacio Z.M. COVID-19, oxidative stress, and neuroinflammation in the depression route. J. Mol. Neurosci. 2022;23:1–16. doi: 10.1007/s12031-022-02004-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman G.J., Karelina K., Zhang N., Walton J.C., Morris J.S., Devries A.C. Stress and IL-1beta contribute to the development of depressive-like behavior following peripheral nerve injury. Mol. Psychiatry. 2010;15:404–414. doi: 10.1038/mp.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olajide O.A., Iwuanyanwu V.U., Adegbola O.D., Al-Hindawi A.A. SARS-CoV-2 spike glycoprotein S1 induces neuroinflammation in BV-2 microglia. Mol. Neurobiol. 2021;59:445–458. doi: 10.1007/s12035-021-02593-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea E.M., Logsdon A.F., Hansen K.M., Williams L.M., Reed M.J., Baumann K.K., Holden S.J., Raber J., Banks W.A., Erickson M.A. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat. Neurosci. 2021;24:368–378. doi: 10.1038/s41593-020-00771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro D.E., Oliveira-Giacomelli A., Glaser T., Arnaud-Sampaio V.F., Andrejew R., Dieckmann L., Baranova J., Lameu C., Ratajczak M.Z., Ulrich H. Hyperactivation of P2X7 receptors as a culprit of COVID-19 neuropathology. Mol. Psychiatry. 2021;26:1044–1059. doi: 10.1038/s41380-020-00965-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z.C., Wu P.F., Wang F., Xia Z.X., Deng Q., Nie T.L., Zhang S.Q., Zheng H.L., Liu W.H., Lu J.J., Gao S.Q., Yao X.P., Long L.H., Hu Z.L., Chen J.G. Gephyrin palmitoylation in basolateral amygdala mediates the anxiolytic action of benzodiazepine. Biol. Psychiatry. 2019;85:202–213. doi: 10.1016/j.biopsych.2018.09.024. [DOI] [PubMed] [Google Scholar]
- Shirato K., Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. 2021;7:e06187. doi: 10.1016/j.heliyon.2021.e06187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E., Zhang C., Israelow B., Lu-Culligan A., Prado A.V., Skriabine S., Lu P., Weizman O.E., Liu F., Dai Y., Szigeti-Buck K., Yasumoto Y., Wang G., Castaldi C., Heltke J., Ng E., Wheeler J., Alfajaro M.M., Levavasseur E., Fontes B., Ravindra N.G., Van Dijk D., Mane S., Gunel M., Ring A., Kazmi S.A.J., Zhang K., Wilen C.B., Horvath T.L., Plu I., Haik S., Thomas J.L., Louvi A., Farhadian S.F., Huttner A., Seilhean D., Renier N., Bilguvar K., Iwasaki A. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021;218:e20202135. doi: 10.1084/jem.20202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soung A.L., Vanderheiden A., Nordvig A.S., Sissoko C.A., Canoll P., Mariani M.B., Jiang X., Bricker T., Rosoklija G.B., Arango V., Underwood M., Mann J.J., Dwork A.J., Goldman J.E., Boon A.C.M., Boldrini M., Klein R.S. COVID-19 induces CNS cytokine expression and loss of hippocampal neurogenesis. Brain. 2022;awac270 doi: 10.1093/brain/awac270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, J.C., Rand, K.L., Muldoon, M.F., Kamarck, T.W., 2009. A prospective evaluation of the directionality of the depression-inflammation relationship. Brain Behav. Immun. 23, 936-944. https://doi.org/10.1016/j.bbi.2009.04.011. [DOI] [PMC free article] [PubMed]
- Sulaiman G., Cooke A., Ffrench B., Gasch C., Abdullai O.A., O'Connor K., Elbaruni S., Blackshields G., Spillane C., Keegan H., McEneaney V., Knittel R., Rogers A., Jeffery I.B., Doyle B., Bates M., d'Adhemar C., Lee M.Y., Campbell E.L., Moynagh P.N., Higgins D.G., O'Toole S., O'Neill L., O'Leary J.J., Gallagher M.F. MyD88 is an essential component of retinoic acid-induced differentiation in human pluripotent embryonal carcinoma cells. Cell Death Differ. 2017;24:1975–1986. doi: 10.1038/cdd.2017.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Tan Y., Huang Z., Huang N., Gao M., Zhou F., Hu J., Feng W. Disrupting myddosome assembly in diffuse large Bcell lymphoma cells using the MYD88 dimerization inhibitor ST2825. Oncol. Rep. 2019;42:1755–1766. doi: 10.3892/or.2019.7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcome M.O., Mastorakis N.E. Stress-induced blood brain barrier disruption: Molecular mechanisms and signaling pathways. Pharmacol. Res. 2020;157 doi: 10.1016/j.phrs.2020.104769. [DOI] [PubMed] [Google Scholar]
- Wu P.F., Han Q.Q., Chen F.F., Shen T.T., Li Y.H., Cao Y., Chen J.G., Wang F. Erasing m(6)A-dependent transcription signature of stress-sensitive genes triggers antidepressant actions. Neurobiol. Stress. 2021;15 doi: 10.1016/j.ynstr.2021.100390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L., Jiang F.C., Zhang L.M., He W.T., Liu J.H., Li M.Q., Zhang X., Xing S., Guo H., Zhou P. Targeting of MyD88 homodimerization by novel synthetic inhibitor TJ-M2010-5 in preventing colitis-associated colorectal Cancer. J. Natl. Cancer Inst. 2016;108:djv364. doi: 10.1093/jnci/djv364. [DOI] [PubMed] [Google Scholar]
- Zhang J.C., Yao W., Dong C., Yang C., Ren Q., Ma M., Han M., Wu J., Ushida Y., Suganuma H., Hashimoto K. Prophylactic effects of sulforaphane on depression-like behavior and dendritic changes in mice after inflammation. J. Nutr. Biochem. 2017;39:134–144. doi: 10.1016/j.jnutbio.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Zhang L., Zhou L., Bao L., Liu J., Zhu H., Lv Q., Liu R., Chen W., Tong W., Wei Q., Xu Y., Deng W., Gao H., Xue J., Song Z., Yu P., Han Y., Zhang Y., Sun X., Yu X., Qin C. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target. Ther. 2021;6:337. doi: 10.1038/s41392-021-00719-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Kuang M., Li J., Zhu L., Jia Z., Guo X., Hu Y., Kong J., Yin H., Wang X., You F. SARS-CoV-2 spike protein interacts with and activates TLR4. Cell Res. 2021;31:818–820. doi: 10.1038/s41422-021-00495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M., Karki R., Williams E.P., Yang D., Fitzpatrick E., Vogel P., Jonsson C.B., Kanneganti T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021;22:829–838. doi: 10.1038/s41590-021-00937-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.Y., He J.G., Hu Z.L., Xue S.G., Xu J.F., Cui Q.Q., Gao S.Q., Zhou B., Wu P.F., Long L.H., Wang F., Chen J.G. A-kinase anchoring protein 150 and protein kinase a complex in the basolateral amygdala contributes to depressive-like behaviors induced by chronic restraint stress. Biol. Psychiatry. 2019;86:131–142. doi: 10.1016/j.biopsych.2019.03.967. [DOI] [PubMed] [Google Scholar]
- Zou Z., Du D., Miao Y., Yang Y., Xie Y., Li Z., Zhou L., Zhang L., Zhou P., Jiang F. TJ-M2010-5, a novel MyD88 inhibitor, corrects R848-induced lupus-like immune disorders of B cells in vitro. Int. Immunopharmacol. 2020;85 doi: 10.1016/j.intimp.2020.106648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.