

Abstract
Background and Objectives
Rituximab is used widely for relapse prevention in neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein (MOG)-IgG–associated disease (MOGAD); however, data regarding the effectiveness and safety of long-term rituximab use in these conditions are limited. In this study, we sought to evaluate long-term clinical outcomes in patients with aquaporin-4 IgG–seropositive (AQP4-IgG+) NMOSD and MOGAD treated with rituximab.
Methods
We performed a retrospective chart review of patients with AQP4-IgG+ NMOSD or MOGAD followed at the Johns Hopkins Neuromyelitis Optica Clinic and included patients who had received at least 1 dose of rituximab.
Results
We identified 111 patients with NMOSD and 23 patients with MOGAD who fulfilled the inclusion criteria. The median duration of rituximab treatment for the patients with NMOSD was 3.7 years (range: 0.5–13.2 years) and for the patients with MOGAD was 2.1 years (range: 0.5–7.0 years). The annualized relapse rate (ARR) decreased after rituximab initiation in both NMOSD (median ARR: pretreatment 1.1, posttreatment 0; p < 0.001) and MOGAD (median ARR: pretreatment 1.9, posttreatment 0.3; p = 0.002). Relapses on rituximab occurred in 31 patients with NMOSD (28%) and 14 patients with MOGAD (61%). The majority of NMOSD treatment failures (37/48 relapses; 77%) occurred either within the initial 6 months after starting rituximab (n = 13 relapses) or in the setting of delayed/missed rituximab doses and/or peripheral B-cell reconstitution (n = 24 relapses), whereas in MOGAD, these circumstances were present in a smaller proportion of treatment failures (19/35 relapses; 54%). The risk of relapse on rituximab was greater for patients with MOGAD compared with patients with NMOSD (hazard ratio: 2.8, 95% CI: 1.5–5.2, p = 0.001). Infections requiring hospitalization occurred in 13% and immunoglobulin G (IgG) hypogammaglobulinemia in 17% of patients. The median rituximab treatment duration before IgG hypogammaglobulinemia onset was 5.4 years (interquartile range: 3.8–7.7 years).
Discussion
Rituximab treatment is associated with the reduced annualized relapse rate in AQP4-IgG–seropositive NMOSD, especially in the absence of gaps in treatment and/or B-cell reconstitution. In MOGAD, although a reduction in relapses was observed after initiation of rituximab, this association appeared to be less robust than in AQP4-IgG–seropositive NMOSD. Severe infections and hypogammaglobulinemia occurred in a significant proportion of patients, highlighting the need for close monitoring of infectious complications.
Classification of Evidence
This study provides Class IV evidence that rituximab decreases the annualized relapse rate in AQP4-IgG–seropositive NMOSD and MOGAD.
Neuromyelitis optica spectrum disorder (NMOSD) is a relapsing inflammatory CNS disorder that primarily affects the optic nerves and spinal cord and is associated in the majority of cases with seropositivity for antibodies targeting aquaporin-4 (AQP4-IgG).1,2 In a subset of patients with AQP4-IgG–seronegative NMOSD, serum antibodies against myelin oligodendrocyte glycoprotein (MOG-IgG) may be detected. MOG-IgG–associated disease (MOGAD) has been recognized as a distinct CNS demyelinating disorder, with manifestations mainly including optic neuritis, transverse myelitis, and acute disseminated encephalomyelitis.3
Treatment for relapse prevention is recommended for all patients with AQP4-IgG+ NMOSD, given the high risk for relapse and severe neurologic disability.4,5 In contrast, the optimal treatment approach for MOGAD remains controversial because MOGAD may be monophasic (especially in children), and recovery from relapses is typically good; preventive treatment is generally recommended for patients with relapsing disease.3,6
Recently, phase 3 randomized placebo-controlled clinical trials demonstrated the efficacy of 4 biologic drugs (eculizumab, inebilizumab, satralizumab, and rituximab) for relapse prevention in AQP4-IgG+ NMOSD, leading to application submission and United States Food and Drug Administration approval for the former 3 for this indication.7-11 Off-label immunosuppressive treatments remain widely prescribed worldwide for relapse prevention in AQP4-IgG+ NMOSD, likely due to a variety of factors, including cost, availability/accessibility, and clinical experience.5 Rituximab, a chimeric monoclonal antibody that depletes circulating B cells by targeting the CD20 surface antigen, is a commonly used preventive treatment in AQP4-IgG+ NMOSD and shares a similar mechanism of action with inebilizumab. Although large-scale randomized controlled trials of rituximab are lacking, existing evidence from smaller randomized controlled trials (including a phase 3 randomized placebo-controlled trial) and observational studies supports that rituximab is highly effective for relapse prevention in AQP4-IgG+ NMOSD.11-15 However, reports regarding the long-term effectiveness and safety of rituximab in AQP4-IgG+ NMOSD are limited. Furthermore, rituximab has been used for relapse prevention in MOGAD, but it has been suggested that relapses may frequently occur in MOGAD, despite rituximab treatment and adequate B-cell depletion.16,17 In this retrospective observational study, we evaluated the long-term effectiveness and safety of rituximab in a large, real-world cohort of patients with AQP4-IgG+ NMOSD and MOGAD.
Methods
Study Design and Participants
We reviewed charts of patients followed at the Johns Hopkins Outpatient Clinic between January 2010 and June 2021 with AQP4-IgG+ NMOSD or MOGAD, according to the 2015 International Panel for Neuromyelitis Optica Diagnosis criteria or previously proposed MOGAD diagnostic criteria, respectively.1,18 Additional inclusion criteria included (1) treatment with at least 1 dose of rituximab, (2) verifiable diagnostic and therapeutic records, and (3) at least 1 clinic visit after starting rituximab. Data were extracted from the electronic medical record by reviewing all encounters using standardized data collection forms including medical comorbidities, rituximab infusion dates/dosing, infusion reactions, symptomatic infections, treating physician-determined relapses, and laboratory information.
An infusion cycle consisted of 1 set of paired infusions administered 14 days apart or a single infusion. Infusion reactions were rated according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0, from grade 1 (mild intervention not indicated) to 5 (death).19
We evaluated the annualized relapse rate (ARR) defined as the number of relapses divided by the total observation time, before rituximab initiation and during the rituximab treatment period. Similar to prior studies, the ARR calculation required at least 3 months of observation before rituximab initiation to avoid an artificially inflated pretreatment ARR.20
Ambulatory/micturition disability was classified per the Aminoff-Logue Disability Scale by reviewing available documentation at each clinic visit.21 Ambulatory function was graded as normal; 1: abnormal but no restricted activity; 2: unassisted but restricted activity; 3: requires unilateral assistance for walking; 4: requires bilateral assistance or walker; and 5: requires a wheelchair. Micturition was graded as normal; 1: hesitancy, urgency, or frequency; 2: occasional incontinence or retention; and 3: total incontinence or retention. Visual disability was classified using the Expanded Disability Status Scale (EDSS) visual function system score (0–6).22 The treatment period was defined as the time from the first rituximab infusion until 6 months after the last infusion. Information was also captured retrospectively from the first documented attack onward.
Infections were classified as mild if management was exclusively outpatient, moderate in cases requiring hospitalization but not intensive care unit (ICU) admission, and high in cases that necessitated ICU admission. Leukopenia was defined as white blood cell count (WBC) <3,000/μL and classified as mild (WBC <2,000–3,000/μL), moderate (1,000–2,000/μL), or severe (<1,000/μL). Lymphopenia was defined as absolute lymphocyte count (ALC) l <1,000/μL and classified as grade 1 (ALC: 800–999/μL), grade 2 (ALC: 500–799/μL), grade 3 (ALC: 200–499/μL), or grade 4 (ALC <200/ul). Neutropenia was defined as absolute neutrophil count (ANC) <1,500/μL and classified as mild (ANC: 1,000–1,500/μL), moderate (ANC: 500–999/μL), or severe (ANC <500/μL). Complete blood count values within 1 month of steroid use were omitted from the analysis. Immunoglobulin (Ig) levels below the reference range for the reference laboratory (Johns Hopkins Immunology Laboratory) when assessed by turbidimetry were considered low (IgG <600 mg/dL, IgM <35 mg/dL, and IgA <61 mg/dL). Mild IgG hypogammaglobulinemia was defined as 400–599 mg/dL, moderate 200–399 mg/dL, or severe <200 mg/dL.23 Ig levels that were captured within 6 months after intravenous Ig (IVIG) use or plasma exchange were not considered.
Statistical Analysis
To assess differences between groups, the Student t test for continuous variables and the Fisher exact test for categorical variables were used. The ARR before and after rituximab initiation was compared using the Wilcoxon signed-rank test. Kaplan-Meier curves were used to describe the time after starting rituximab to relapse, hypogammaglobulinemia, or infection requiring hospitalization. IgG levels were modeled using a linear mixed-effects model including time since rituximab initiation as a fixed effect. Assessment of potential factors associated with risk of relapse after rituximab initiation was performed using Cox proportional regression models where we considered the first documented clinical relapse as the outcome variable. The association of disability and age with risk of infection requiring hospitalization was assessed using Anderson-Gill models for recurrent events to account for multiple infection events. Statistical significance was considered p < 0.05. Statistical analyses were performed using Python v3.9, Stata v16 and GraphPad v9.1.1.
Data Availability
Anonymized data used for this study are available from the corresponding author on reasonable request, with the proper data sharing agreements in place.
Standard Protocol, Approvals, Registrations, and Patient Consents
The study protocol was approved by the Johns Hopkins Institutional Review Board (IRB00265758), and patient consent was waived for this retrospective chart review.
Results
Study Population
The study inclusion criteria were fulfilled for 111 participants with AQP4-IgG+ NMOSD and 23 participants with MOGAD treated with rituximab (Figure 1). The demographic and baseline clinical characteristics of the study population (at the time of rituximab initiation) are shown in Table 1. The median duration of treatment with rituximab for the AQP4-IgG+ NMOSD patients was 3.7 years (range: 0.5–13.2 years) and for the patients with MOGAD was 2.1 years (range: 0.5–7.0 years). The median age at rituximab initiation was 48.9 years for the AQP4-IgG+ NMOSD group (interquartile range [IQR]: 35.8–58.7) and 42.6 years for the MOGAD group (IQR: 32.3–51.2). Both groups were predominantly female (AQP4-IgG+ NMOSD: 88%, MOGAD: 74%), and 36% of patients with AQP4-IgG+ NMOSD and 65% of patients with MOGAD were White. Before rituximab initiation, the median ARR was 1.1 (IQR: 0.5–2.6) for AQP4-IgG+ NMOSD and 1.9 (IQR: 1.0–4.3) for MOGAD.
Figure 1. Study Population.

AQP4-IgG+ = aquaporin-4 IgG seropositive; MOGAD = myelin oligodendrocyte glycoprotein–IgG–associated disease; NMOSD = neuromyelitis optica spectrum disorder.
Table 1.
Demographics and Baseline Clinical Characteristics at the Time of Rituximab Initiation
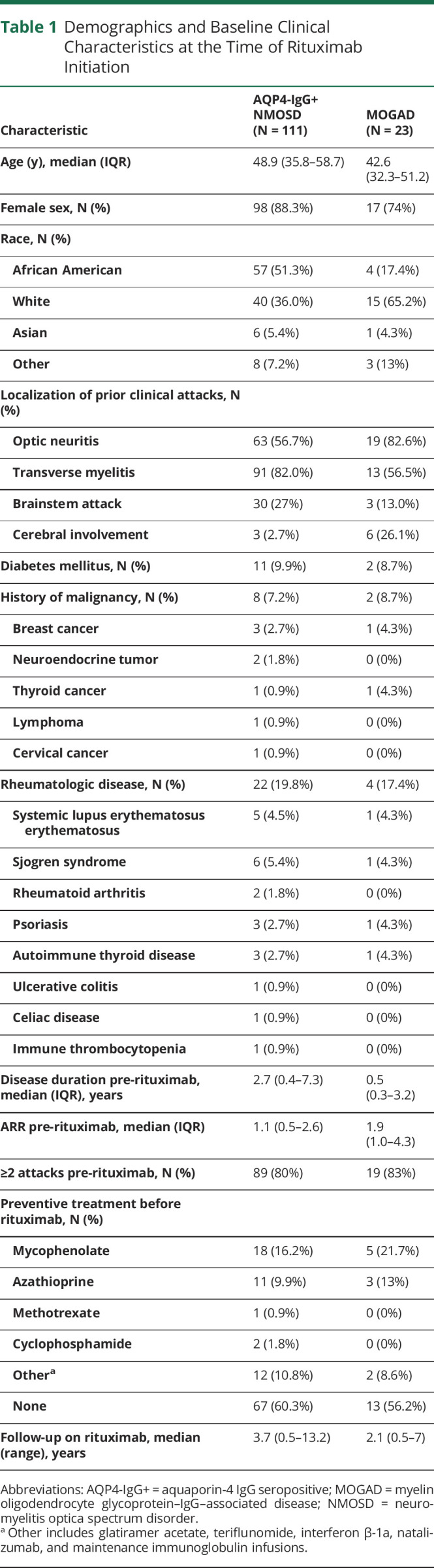
Rituximab was selected as the initial preventive therapy for the majority of patients in both groups (AQP4-IgG+ NMOSD: 60%; MOGAD: 56%). For most patients during the treatment period, rituximab was prescribed as a total of 2,000 mg divided in 2 doses 14 days apart, repeated every 6 months (AQP4-IgG+ NMOSD: 62%; MOGAD: 91%). In the rest of the cases, individualized dosing schemes were implemented with single infusions and/or variable intervals between treatment cycles by monitoring of CD19+ counts. Adherence issues also resulted in variable dosing intervals. The median interval between infusion cycles was 7.3 months (IQR: 6.4–8.9 months) for AQP4-IgG+ NMOSD and 6.7 months (IQR: 5.2–9.6 months) for MOGAD, and the median number of rituximab infusion cycles was 6 (IQR: 3–12) for AQP4-IgG+ NMOSD and 4 (IQR: 2–7) for MOGAD.
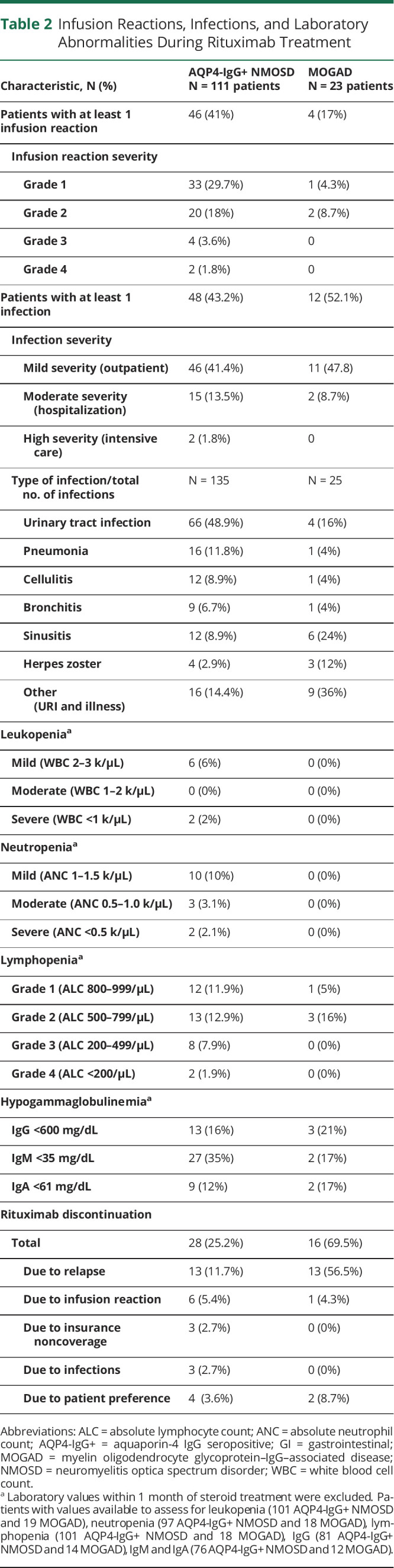
During follow-up, rituximab was discontinued in 28 (25%) of patients with AQP4-IgG+ NMOSD and 16 (69%) of patients with MOGAD. The most common reason for both AQP4-IgG+ NMOSD and MOGAD was a relapse while on therapy (Table 2).
Table 2.
Infusion Reactions, Infections, and Laboratory Abnormalities During Rituximab Treatment
Relapses and Disability
The distribution of relapses for all patients relative to rituximab initiation, cumulative incidence of relapses, and the annualized relapse rate is presented in Figures 2 and 3.
Figure 2. Clinical Attacks, Infections Requiring Hospitalization, and Hypogammaglobulinemia Over Time in Patients With AQP4-IgG+ NMOSD and MOGAD Before and After Initiation Rituximab Treatment.

Attacks are shown up to 15 years before rituximab initiation. From the AQP4-IgG+ NMOSD 11 patients (patients 9, 10, 17, 20, 23, 25, 29, 30, 49, 101, and 111) and from the MOGAD group 2 patients (patients 8 and 10) had experienced relapses before 15 years pre-rituximab. AQP4-IgG+ = aquaporin-4 IgG seropositive; MOGAD = myelin oligodendrocyte glycoprotein–IgG–associated disease; NMOSD = neuromyelitis optica spectrum disorder.
Figure 3. Characterization of Clinical Relapses of Patients With AQP4-IgG+ NMOSD and MOGAD on Rituximab.

(A) Flow diagram of type of relapses during treatment. (B) Kaplan-Meier curves showing the time to relapse after starting rituximab treatment (1 patient with NMOSD considered in 2 different epochs given prolonged treatment gap of 9 years). (C) Annualized relapse rate before and after rituximab treatment. ** p = 0.003; **** p < 0.001 AQP4-IgG+ = aquaporin-4 IgG seropositive; HR = hazard ratio MOGAD = myelin oligodendrocyte glycoprotein–IgG–associated disease; NMOSD = neuromyelitis optica spectrum disorder.
After starting rituximab, 31 (28%) patients with AQP4-IgG+ NMOSD and 14 (61%) patients with MOGAD experienced at least 1 relapse during treatment. The majority of AQP4-IgG+ NMOSD treatment failures (37/48 relapses; 77%) occurred either within the initial 6 months after starting rituximab (13 relapses; median time to relapse 1.8 months), in the setting of missed or delayed (≥1 month delay) rituximab infusions (12 relapses), peripheral B-cell reconstitution with CD19+ >0.5% less than 6 months after a rituximab infusion (4 relapses), or within 6 months of restarting rituximab after a prolonged gap in treatment (at least 1 year) accompanied by peripheral CD19+ reconstitution (6 relapses). Furthermore, no relapses were observed after continuous rituximab treatment for at least 2.5 years in AQP4-IgG+ NMOSD.
Of the 17 patients with AQP4-IgG+ NMOSD who experienced a relapse within 6 months of starting or restarting rituximab, 12 remained on rituximab for a median follow-up of 6.6 years (range: 1.1–10.7 years), with only 2 of these patients experiencing an additional relapse. In MOGAD, 10 relapses occurred within 6 months of rituximab initiation (median time to relapse 2.5 months) and 9 relapses in the setting of missed doses, whereas 16 relapses occurred in the absence of any of these circumstances or B-cell reconstitution. Of the 8 patients with MOGAD who experienced a relapse within 6 months of starting or restarting rituximab, 5 remained on rituximab, but all of them continued to experience relapses and were transitioned to different therapies. When considering only relapses occurring at least 6 months after starting rituximab and in the absence of reconstitution or missed doses, 93% of AQP4-IgG+ NMOSD and 57% of MOGAD were relapse-free.
The majority of patients in this study were not treated with prolonged corticosteroid tapers after rituximab initiation, as this was not standard of care at our center before 2020. Of note, 88% of the patients with AQP4-IgG+ NMOSD and 62% of the patients with MOGAD who relapsed within 6 months of starting or restarting rituximab after a prolonged treatment gap (>1 year) were not on oral corticosteroids at the time of the relapse.
The ARR decreased after initiation of rituximab, compared with the pretreatment period (Figure 2C), in both AQP4-IgG+ NMOSD (median ARR: pretreatment 1.1, posttreatment: 0; p < 0.001) and MOGAD (median ARR: pretreatment 1.9, posttreatment 0.28; p = 0.003). For AQP4-IgG+NMOSD the pretreatment compound ARR (total number of relapses per patient-years) was 0.55 and posttreatment was 0.09; for MOGAD pretreatment compound ARR was 0.68 and posttreatment 0.52. The risk of relapse on rituximab was greater for patients with MOGAD compared with patients with AQP4-IgG+ NMOSD (hazard ratio: 2.8, 95% CI: 1.5–5.2, p = 0.001).
Analyses examining associations of demographic variables (age, race/ethnicity, sex, and ARR before rituximab initiation) with risk of relapse on rituximab in each group revealed that older age was associated with a lower risk of relapse in MOGAD (hazard ratio: 0.70 per 10-year increment in age; 95% CI: 0.55–0.90, p = 0.006), but otherwise no significant associations were detected in the AQP4-IgG+ NMOSD or MOGAD groups, including with overall relapse risk during follow-up or risk of relapse in the first 6 months of rituximab treatment.
Disability outcomes are depicted in Figure 4. Ambulatory disability improved or was stable in 82% of patients with AQP4-IgG+ NMOSD and 72% of patients with MOGAD, micturition disability in 79% of patients with AQP4-IgG+ NMOSD and in 62% of patients with MOGAD, and visual disability in 91% of patients with AQP4-IgG+ NMOSD and in 66% of patients with MOGAD. Of the early relapses occurring in the AQP4-IgG+ NMOSD group within the first 6 months after rituximab initiation, 5 relapses were severe (defined as a ≥2-point change in the Aminoff-Logue score and/or EDSS visual score), 3 did not have sufficient information to describe severity, and 4 were mild with only a 1-point change. In the MOGAD group, of the 10 early relapses, only 1 was a severe relapse, for 2 relapses, severity could not be determined, and the other 7 were mild relapses.
Figure 4. Disability Outcomes.

Disability scores before rituximab treatment and at the last visit; data for pre- and post-treatment disability were available for 96 patients with AQP4-IgG+ NMOSD and 18 patients with MOGAD for gait, 84 patients with AQP4-IgG+ NMOSD and 16 patients with MOGAD for micturition, and 69 patients with AQP4-IgG+ NMOSD and 15 patients with MOGAD for vision. AQP4-IgG+ = aquaporin-4 IgG seropositive; MOGAD = myelin oligodendrocyte glycoprotein–IgG–associated disease; NMOSD = neuromyelitis optica spectrum disorder.
Adverse Events
The frequency of adverse events during the treatment period is described in Table 2. Infusion reactions were common in both groups, with 41% of patients with AQP4-IgG+ NMOSD and 17% of patients with MOGAD having at least 1 infusion reaction during the course of treatment. Most reactions were mild, with only 5% of patients with AQP4-IgG+ NMOSD and none of the patients with MOGAD having infusion reactions grade 3 or worse. Rituximab was discontinued due to infusion reactions in 5% of patients with AQP4-IgG+ NMOSD and 4% of patients with MOGAD.
Infections during the rituximab treatment period were common in both groups, with the majority of them being mild and managed in an outpatient setting (Table 2). For the AQP4-IgG+ NMOSD group, 15 patients (13.5%) had at least 1 infection that required hospitalization (2 of which necessitated ICU admission), of which 1 was fatal. Infections requiring hospitalization in the MOGAD group occurred in 2 patients (8.7%). The risk of an infection requiring hospitalization was higher in those with more severe disability (hazard ratio: 1.54 per 1-point increment in the Aminoff-Logue motor function scale; 95% CI: 1.28–1.86, p < 0.001). Age was not significantly associated with the risk of infection (hazard ratio: 1.14 per 10-year increment in age, 95% CI: 0.86–1.50, p = 0.35).
In the patients with AQP4-IgG+ NMOSD, the most frequent infection type was urinary tract infection (UTI), representing 49% of all infections. On the other hand, upper respiratory tract infections (36%) and sinusitis (24%) were the most common in the MOGAD group. Most infections requiring hospitalization were UTIs (15/29 severe infections), 8/29 severe infections were pneumonias, and 2/29 were cellulitis. Two patients had COVID-19 while on rituximab, with 1 of them requiring hospitalization (these infectious occurred before vaccines for SARS-CoV2 became available). The cumulative incidence of infections severe enough to require hospitalization is demonstrated in Figure 5A.
Figure 5. Adverse Events During Follow-up on Rituximab Treatment in Patients With AQP4-IgG+ NMOSD and MOGAD.
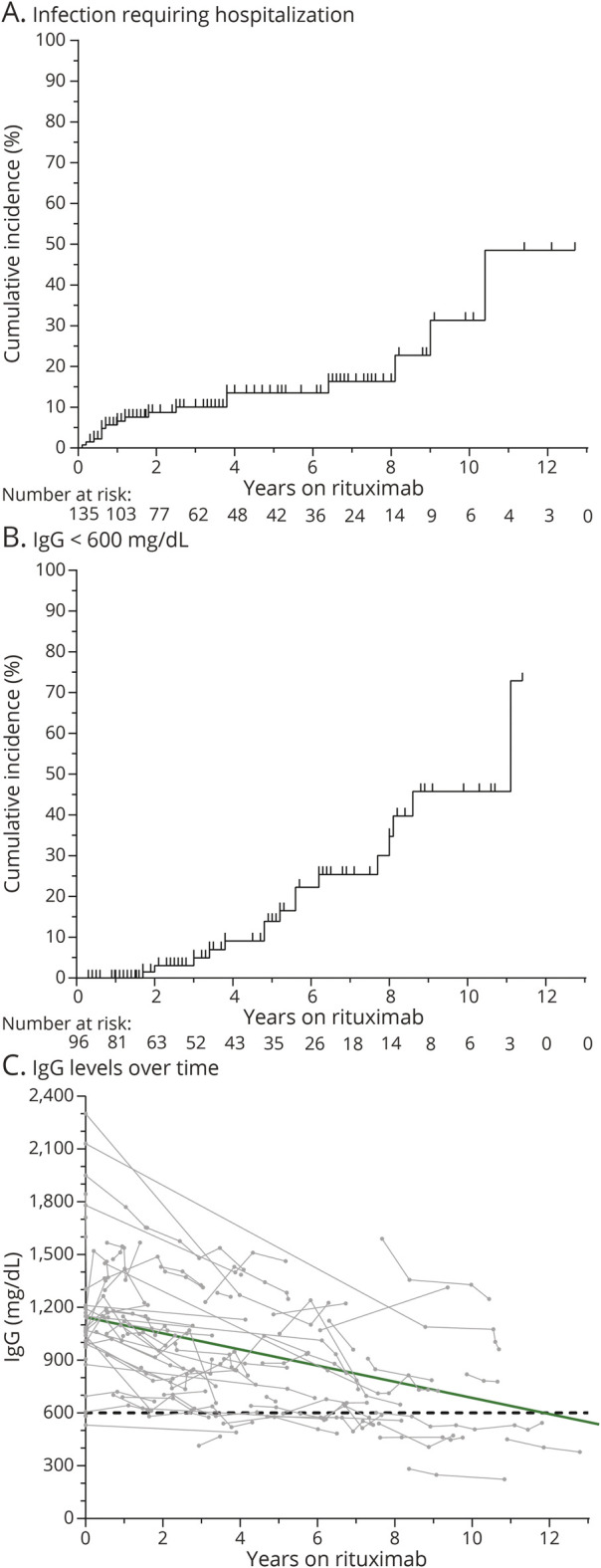
Infections requiring hospitalization (A) and hypogammaglobulinemia (B and C). The green line in C corresponds to the average trend of IgG levels, derived from a linear mixed-effects model. One patient with AQP4-IgG+ NMOSD was considered 2 different treatment epochs given prolonged treatment gap (9 years). AQP4-IgG+ = aquaporin-4 IgG seropositive; MOGAD = myelin oligodendrocyte glycoprotein–IgG–associated disease; NMOSD = neuromyelitis optica spectrum disorder.
Hypogammaglobulinemia (reduction of IgG, IgA, and/or IgA) was frequently observed in both groups (Table 2). IgG hypogammaglobulinemia (<600 mg/dL) was found in 16% of patients with AQP4-IgG+ NMOSD and 21% of patients with MOGAD. The cumulative incidence of hypogammaglobulinemia and trends of IgG levels over time are shown in Figure 5B and 5C, respectively. Only 2 patients had IgG levels within the 200–399 mg/dL range (moderate hypogammaglobulinemia), and no patients had IgG <200 mg/dL (severe hypogammaglobulinemia). Of the 16 patients who developed low IgG <600 mg/dL after rituximab, 13 had sustained low IgG in subsequent measurements, 1 was within the reference range on repeat testing, and 2 were not retested by the time the study ended. Four patients in the AQP4-IgG+ NMOSD group and 1 patient in the MOGAD group required IVIG administration due to persistent hypogammaglobulinemia and recurrent infections, and in all 5 patients, IVIG allowed for continuation of rituximab. Lymphopenia grade 3 or worse (ALC < 500/ul) developed in 8% of patients with AQP4-IgG+ NMOSD and none of the patients with MOGAD. Severe leukopenia and neutropenia were very rare in both groups.
A new diagnosis of malignancy during rituximab therapy was made in 7 patients with NMOSD: 3 had melanoma, 2 had breast cancer, 1 had thyroid cancer, and 1 had a neuroendocrine tumor. Five of these 7 malignancies were diagnosed within 2 years of NMOSD diagnosis. Two patients with MOGAD had a history of cancer that preceded rituximab treatment.
During follow-up (including after cessation of rituximab), there were 3 deaths in the AQP4-IgG+ NMOSD group. Two were related to severe infections attributable to complications from the underlying disease: 1 occurred 2 years after discontinuing rituximab in the setting of bacteremia/sepsis from infected sacral ulcers related to immobility and 1 while on rituximab due to urosepsis in the setting of an indwelling suprapubic catheter. One patient died due to complications of breast cancer that was diagnosed 10 months after NMOSD onset and 6 months after rituximab initiation (which was discontinued 2 years before death). None of the patients with MOGAD were diagnosed with malignancy or died during follow-up.
Classification of Evidence
This study provides Class IV evidence that rituximab decreases the annualized relapse rate in AQP4-IgG–seropositive AQP4-IgG+ NMOSD and MOGAD.
Discussion
In summary, we investigated the effectiveness and long-term safety of rituximab treatment in a large, observational single-center cohort of people with AQP4-IgG+ NMOSD and MOGAD. Rituximab treatment was associated with a significantly reduced relapse rate in both groups, but notably the risk of relapse on rituximab was substantially higher in MOGAD compared with AQP4-IgG+ NMOSD. Infections, hypogammaglobulinemia, and hematologic abnormalities were relatively common in both groups.
Rituximab has been empirically used for decades as a preventive therapy in AQP4-IgG+ NMOSD, due to the role B cells play in the disease pathophysiology, as AQP4-IgG autoantibodies have been clearly established to be pathogenic. Although large-scale, randomized placebo-controlled trials in AQP4-IgG+ NMOSD are lacking, the RIN-1 placebo-controlled trial that included 38 participants with AQP4-IgG+ NMOSD (19 per arm) demonstrated Class I evidence of rituximab's efficacy, with no relapses occurring in the rituximab arm compared with 7 relapses in the placebo arm, a finding that was statistically significant.11 The RIN-1 study findings are in line with the findings of meta-analyses of observational studies and results of an open-label, randomized controlled trial of rituximab vs azathioprine, however relapse rates while on rituximab treatment are variable across these studies, likely due to relatively small study sample sizes, and differences in the study designs and populations (including the proportion of patients with AQP4-IgG seropositivity).12,13,15 Furthermore, the efficacy of B cell–depleting therapies in NMOSD is supported by the efficacy of inebilizumab in a phase 2/3 randomized, placebo-controlled trial in NMOSD (N-MOmentum trial). Inebilizumab targets CD19, which is expressed more broadly on B-cell lineage cells than CD20, including on pro–B cells and plasmablasts/plasma cells.24
Our study builds on the existing literature regarding rituximab effectiveness in AQP4-IgG+ NMOSD, by reporting long-term effectiveness, with a treatment duration of up to 13 years.11,12,15 Notably, we observed that the majority of relapses occurring during rituximab treatment occurred either within the first 6 months after initiation, or in the setting of missed/delayed infusions and/or peripheral blood B-cell reconstitution. Importantly, no relapses were observed in patients who remained on rituximab and had been continuously treated for at least 2.5 years. This supports that, with optimal dosing/adherence and monitoring for peripheral B-cell reconstitution, rituximab treatment is associated with a high relapse-free rate in NMOSD. Furthermore, our observation that many relapses occurred early after treatment initiation (or reinitiation after a prolonged treatment gap) is consistent with prior reports that have supported that there may be a persistent and/or heightened risk of relapse during the first months after initiation of rituximab, which could relate to transient increases in AQP4-IgG titers in association with induction of B cell–activating factor.25-27 Of interest, a similar phenomenon was observed in the N-MOmentum trial of inebilizumab, with the majority of relapses occurring during the first year and a relatively stable attack-free probability through subsequent years of follow-up.28 Importantly, patients in the N-MOmentum trial were treated routinely with oral corticosteroids only for a 21-day period following inebilizumab initiation.9 In contrast, in the RIN-1 rituximab trial, although the sample size was small, all patients were treated with corticosteroids at baseline, which were slowly tapered according to a predefined protocol, with no relapses observed in the rituximab-treated arm.11 It has previously been recommended that steroids be overlapped with rituximab for at least 1 month following initiation of rituximab, followed by tapering.4,5 In our cohort, steroid use was variable, but in most cases were tapered rapidly within a month following rituximab initiation. Collectively, the above data support a delayed onset of effectiveness of B cell–depleting therapies in AQP4-IgG+ NMOSD and support the rationale for more prolonged duration of overlap of rituximab with corticosteroids (3–6 months) to mitigate early risk of relapse (similar to the approach implemented in the RIN-1 trial), including in patients reinitiating treatment after a prolonged treatment gap with B-cell reconstitution. Alternatively, it is conceivable that this observation could be related to a depletion of susceptibles phenomenon, in which those patients experiencing relapses early on are a subgroup that is less responsive to rituximab treatment.29 However, of the 12 patients who remained on rituximab despite experiencing a relapse within 6 months of starting or restarting rituximab, only 2 experienced an additional relapse during follow-up. This supports the notion that a relapse occurring in AQP4-IgG+ NMOSD early after rituximab initiation (or other B cell–depleting therapies) should not necessarily be considered a treatment failure.
Although the topic of specific regimens of oral corticosteroids overlapping with rituximab treatment in AQP4-IgG+ NMOSD warrants further investigation and is beyond the scope of the present study, the RIN-1 study steroid tapering protocol may help guide clinical practice. In the RIN-1 study, the initial dose of oral corticosteroids was 5–30 mg daily of prednisolone equivalents and fixed for the first 2 months of the trial and subsequently reduced by ∼10% every 4 weeks11 At our center, although the oral corticosteroid regimen may vary depending on individual patient characteristics, including comorbidities (e.g., diabetes and osteopenia) and tolerability of steroid treatment, we have recently implemented an approach of initiating oral prednisone at 1mg/kg (up to 60mg) after completion of an IV steroid pulse for treatment of an attack (which is the typical scenario that brings patients to attention and prompts initiation of long-term therapy with rituximab) and then tapering the daily dose by 10 mg every 1–2 weeks until reaching a dose of 20 mg daily, which we continue for the first 3 months after initiation of rituximab and then taper over the subsequent 3 months. Although more work is needed to inform recommendations for a specific tapering regimen, we feel that this approach has the potential to balance the risk of an early attack following initiation of rituximab with risks related to long-term exposure to high-dose corticosteroids.
In MOGAD, although we found that rituximab use was associated with a decrease in the ARR compared with the pre-rituximab period, the majority of patients with MOGAD experienced relapses on rituximab, and the risk of relapse was significantly greater than patients with AQP4-IgG+ NMOSD. In addition, a large proportion of relapses in MOGAD occurred in the absence of missed doses or peripheral blood B-cell reconstitution. These findings are consistent with prior observational studies reporting a reduction in relapse risk in rituximab-treated patients with MOGAD, with a substantial proportion of patients, however, continuing to relapse despite rituximab treatment and B-cell depletion.16,17,20,30-32 A recent systematic review and meta-analysis of rituximab treatment in patients with MOGAD including 238 patients found that rituximab was associated with a reduction in relapse risk in MOGAD, but only 55% remained relapse-free.33 Furthermore, all patients in our study with MOGAD who relapsed within the first 6 months of initiating treatment and then remained on rituximab continued to experience relapses while on treatment. This suggests that a low threshold should be maintained to switch from rituximab to another agent in patients with MOGAD experiencing relapses on rituximab, especially since maintenance immune globulin (IV or subcutaneous) has been reported to be associated with a markedly decreased risk of relapse in MOGAD.20,30,31,34
Regarding adverse events associated with rituximab treatment, although infusion reactions were common, most were minor and rarely resulted in treatment discontinuation. Lymphopenia and hypogammaglobulinemia were prevalent in both patients with MOGAD and AQP4-IgG+ NMOSD over time, with frequencies comparable to prior reports.23,35,36 Furthermore, the risk of infection during rituximab therapy was considerable in our cohort, with 13% of patients experiencing an infection requiring hospitalization, in line with findings from prior studies in individuals with multiple sclerosis and NMOSD.12,37 Higher ambulatory disability was strongly associated with risk of severe infection, likely due to factors including immobility and concomitant bladder dysfunction, which can increase the risk of UTIs, the most frequently observed infection in this cohort. Infections more classically associated with hypogammaglobulinemia (e.g., pneumonia and cellulitis) were less common but present in a relatively large proportion of the hospitalized cases. Furthermore, infections for which some protection may be provided by vaccination were observed, including pneumonia, herpes zoster, and COVID-19 (although the study period preceded the availability of SARS-CoV2 vaccines). Rituximab treatment is associated with decreased vaccination-induced humoral responses, but relatively preserved T-cell responses.38 This emphasizes the importance of administering vaccinations before the initiation of rituximab, if possible. In addition, for patients already on treatment, consideration should be given to the timing of vaccinations (we ideally aim for vaccinations to be administered 4 weeks before a rituximab infusion, consistent with consensus recommendations for SARS-CoV2 vaccination from the American College of Rheumatology and an expert panel convened by the National MS Society).39,40
There are several strengths of the current study. These include the large sample size and long-term follow-up (up to 13 years), especially for participants with AQP4-IgG+ NMOSD. Notably, the most recent meta-analysis of rituximab treatment in NMOSD included 435 patients with AQP4-IgG+ NMOSD from 26 different studies; thus, the present study increases the reported number of rituximab-treated patients with AQP4-IgG+ NMOSD in the literature by more than 25%. Moreover, our study included only participants from a single center, which reduces issues related to heterogeneity of treatment approaches, monitoring practices and outcome ascertainment. Furthermore, our study included a high proportion of African Americans in the AQP4-IgG+ NMOSD group (51%), a population that is often underrepresented in clinical trials, including in the recent phase 3 clinical trials in NMOSD, and which has been proposed to be predisposed to develop NMOSD and to experience worse clinical outcomes.7,8,10,11,41-43 In addition, our study included a large proportion of participants with concomitant autoimmune diseases, for which rituximab had the potential benefit of covering these conditions as well. Finally, we only included participants with AQP4-IgG or MOG-IgG seropositive disease, which improves the generalizability of our results, and avoids challenges with the interpretation of studies in the literature performing analyses of mixed populations of AQP4-IgG seropositive and seronegative participants, especially prior to the availability of MOG-IgG testing.
Our study has several limitations that warrant discussion. First, because this investigation was retrospective, it is limited to relapses and adverse events that were documented in the medical record, although we expect that lack of documentation would mainly bias toward reduced ascertainment of mild events such as minor infections. Relapses in AQP4-IgG+ NMOSD and MOGAD generally present with significant neurologic dysfunction, and thus, we would not expect such instances to be omitted from the medical record. Second, participants with MOGAD constituted a relatively small fraction of the cohort, somewhat limiting the interpretation of results in this subgroup, although our findings are consistent with prior reports. Finally, laboratory data were not routinely available for all patients.
In conclusion, our study supports that rituximab treatment is associated with a reduced annualized relapse rate in AQP4-IgG–seropositive NMOSD, especially in the absence of gaps in treatment and/or B-cell reconstitution, with the exception of a critical window of persistent relapse risk during the first months after rituximab initiation. Although a reduction in the relapse rate was observed in MOGAD after rituximab initiation, most patients experienced relapses, consistent with a less robust association of rituximab treatment with reduced relapse frequency in MOGAD compared with AQP4-IgG+ NMOSD. Given the relatively large proportions of patients experiencing hypogammaglobulinemia, lymphopenia, and infection requiring hospitalization, especially in patients with more severe disability, infection risk in patients treated with B cell–depleting therapies is an important consideration when counseling patients regarding treatment selection, and patients should be closely monitored during follow-up. The use of concurrent corticosteroids in the first 6-month of rituximab therapy to mitigate early relapse risk warrants further study.
Glossary
- ALC
absolute lymphocyte count
- ANC
absolute neutrophil count
- AQP4-IgG+
aquaporin-4 IgG seropositive
- ARR
annualized relapse rate
- EDSS
Expanded Disability Status Scale
- ICU
intensive care unit
- MOG
myelin oligodendrocyte glycoprotein
- Ig
Immunoglobulin
- IgG
immunoglobulin G
- IVIG
intravenous Ig
- MOGAD
myelin oligodendrocyte glycoprotein–IgG–associated disease
- NMOSD
neuromyelitis optica spectrum disorder
- UTI
urinary tract infection
- WBC
white blood cell count
Appendix. Authors
Footnotes
Class of Evidence: NPub.org/coe
CME Course: NPub.org/cmelist
Study Funding
Supported by the Caring Friends NMO Research fund, NIH/NINDS (K23NS117883 to E.S. Sotirchos), and National MS Society (TA-1904-33834 to E.S. Sotirchos).
Disclosure
P. Barreras, E.S. Vasileiou, A. Filippatou, and K. Fitzgerald report no disclosures relevant to the manuscript. M. Levy has received personal compensation from Alexion, Horizon, Genentech/Roche, Sanofi, and UCB for work on advisory boards, and his laboratory has received grant funding from Alexion, Horizon, Genentech/Roche, and Bluerock. C.A. Pardo reports no disclosures relevant to the manuscript. S.D. Newsome has received consultant fees for scientific advisory boards from Biogen, Genentech, Bristol Myers Squibb, Greenwich Biosciences, Novartis, Horizon Therapeutics, and EMD Serono; is an advisor for Autobahn; is a clinical adjudication committee member for a MedDay Pharmaceuticals clinical trial; is the study lead PI for a Roche clinical trial; and has received research funding (paid directly to his institution) from Biogen, Roche, and Genentech. E.M. Mowry has grants from and is site PI for studies sponsored by Biogen and Genentech; has received free medication for a clinical trial from Teva; and receives royalties for editorial duties from UpToDate. P.A. Calabresi has received consulting fees from Biogen, Nervgen, Avidea, and Disarm Therapeutics and is PI on grants from Principia and Genentech. E.S. Sotirchos has received speaker honoraria from Alexion, Viela Bio, and Biogen and has served on scientific advisory boards for Alexion, Viela Bio, Horizon Therapeutics, and Genentech. Go to Neurology.org/N for full disclosures.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. ; International Panel for NMO Diagnosis. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marignier R, Hacohen Y, Cobo-Calvo A, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021;20(9):762-772. [DOI] [PubMed] [Google Scholar]
- 4.Flanagan EP. Neuromyelitis optica spectrum disorder and other non-multiple sclerosis central nervous system inflammatory diseases. Continuum (Minneap Minn). 2019;25(3):815-844. [DOI] [PubMed] [Google Scholar]
- 5.Carnero Contentti E, Rojas JI, Cristiano E, et al. Latin American consensus recommendations for management and treatment of neuromyelitis optica spectrum disorders in clinical practice. Mult Scler Relat Disord. 2021;52:103026. [DOI] [PubMed] [Google Scholar]
- 6.Whittam DH, Karthikeayan V, Gibbons E, et al. Treatment of MOG antibody associated disorders: results of an international survey. J Neurol. 2020;267(12):3565-3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(22):2114-2124. [DOI] [PubMed] [Google Scholar]
- 8.Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cree BAC, Bennett JL, Kim HJ, et al. ; N-MOmentum Study Investigators. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352-1363. [DOI] [PubMed] [Google Scholar]
- 10.Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12(6):554-562. [DOI] [PubMed] [Google Scholar]
- 11.Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020;19(4):298-306. [DOI] [PubMed] [Google Scholar]
- 12.Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol. 2016;73(11):1342-1348. [DOI] [PubMed] [Google Scholar]
- 13.Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264(9):2003-2009. [DOI] [PubMed] [Google Scholar]
- 14.Paolilo RB, Hacohen Y, Yazbeck E, et al. Treatment and outcome of aquaporin-4 antibody-positive NMOSD: a multinational pediatric study. Neurol Neuroimmunol Neuroinflamm. 2020;7(5):e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao F, Chai B, Gu C, et al. Effectiveness of rituximab in neuromyelitis optica: a meta-analysis. BMC Neurol. 2019;19(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durozard P, Rico A, Boutiere C, et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol. 2020;87(2):256-266. [DOI] [PubMed] [Google Scholar]
- 17.Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS, et al. Treatment of MOG-IgG-associated disorder with rituximab: an international study of 121 patients. Mult Scler Relat Disord. 2020;44:102251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.López-Chiriboga AS, Majed M, Fryer J, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG–Associated disorders. JAMA Neurol. 2018;75(11):1355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.NIH Common Terminology Criteria for Adverse Events v3.0 (CTCAE). Accessed February 11, 2022. ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm. [Google Scholar]
- 20.Chen JJ, Flanagan EP, Bhatti MT, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. 2020;95(2):e111-e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aminoff MJ, Logue V. Clinical features of spinal vascular malformations. Brain. 1974;97(1):197-210. [DOI] [PubMed] [Google Scholar]
- 22.Kurtzke JF. Historical and clinical perspectives of the expanded disability status scale. Neuroepidemiology. 2008;31(1):1-9. [DOI] [PubMed] [Google Scholar]
- 23.Barmettler S, Ong MS, Farmer JR, Choi H, Walter J. Association of immunoglobulin levels, infectious risk, and mortality with rituximab and hypogammaglobulinemia. JAMA Netw Open. 2018;1(7):e184169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen D, Gallagher S, Monson NL, Herbst R, Wang Y. Inebilizumab, a B cell-depleting anti-CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med. 2016;5(12):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi B, Zhao M, Qiao L, et al. Relapses shortly after rituximab treatment in neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. 2021;54:103143. [DOI] [PubMed] [Google Scholar]
- 26.Perumal JS, Kister I, Howard J, Herbert J. Disease exacerbation after rituximab induction in neuromyelitis optica. Neurol Neuroimmunol Neuroinflam. 2015;2(1):e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakashima I, Takahashi T, Cree BAC, et al. Transient increases in anti-aquaporin-4 antibody titers following rituximab treatment in neuromyelitis optica, in association with elevated serum BAFF levels. J Clin Neurosci. 2011;18(7):997-998. [DOI] [PubMed] [Google Scholar]
- 28.Rensel M, Zabeti A, Mealy MA, et al. Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: analysis of aquaporin-4-immunoglobulin G-seropositive participants taking inebilizumab for 4 years in the N-MOmentum trial. Mult Scler. 2022;28(6):925-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Renoux C, Dell'Aniello S, Brenner B, Suissa S. Bias from depletion of susceptibles: the example of hormone replacement therapy and the risk of venous thromboembolism. Pharmacoepidemiol Drug Saf. 2017;26(5):554-560. [DOI] [PubMed] [Google Scholar]
- 30.Hacohen Y, Wong YY, Lechner C, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody–associated disease. JAMA Neurol. 2018;75(4):478-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanathan S, Mohammad S, Tantsis E, et al. ; Australasian and New Zealand MOG Study Group. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89(2):127-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cobo-Calvo A, Sepúlveda M, Rollot F, et al. Evaluation of treatment response in adults with relapsing MOG-ab-associated disease. J Neuroinflammation. 2019;16(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nepal G, Kharel S, Coghlan MA, Rayamajhi P, Ojha R. Safety and efficacy of rituximab for relapse prevention in myelin oligodendrocyte glycoprotein immunoglobulin G (MOG-IgG)-associated disorders (MOGAD): a systematic review and meta-analysis. J Neuroimmunol. 2022;364:577812. [DOI] [PubMed] [Google Scholar]
- 34.Sotirchos ES, Vasileiou ES, Salky R, et al. Treatment of myelin oligodendrocyte glycoprotein antibody associated disease with subcutaneous immune globulin. Mult Scler Relat Disord. 2022;57:103462. [DOI] [PubMed] [Google Scholar]
- 35.Avouac A, Maarouf A, Stellmann JP, et al. Rituximab-induced hypogammaglobulinemia and infections in AQP4 and MOG antibody-associated diseases. Neurol Neuroimmunol Neuroinflamm. 2021;8(3):e977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marcinnò A, Marnetto F, Valentino P, et al. Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. 2018;5(6):e498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luna G, Alping P, Burman J, et al. Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol. 2020;77(2):184-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gadani SP, Reyes-Mantilla M, Jank L, et al. Discordant humoral and T cell immune responses to SARS-CoV-2 vaccination in people with multiple sclerosis on anti-CD20 therapy. EBioMedicine. 2021;73:103636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The National Multiple Sclerosis Society. COVID-19 Vaccine Guidance for People Living With MS. Accessed June 10, 2022.
- 40.ACR COVID-19 Vaccine Clinical Guidance Task Force. COVID-19 Vaccine Clinical Guidance Summary for Patients With Rheumatic and Musculoskeletal Diseases. Accessed June 10, 2022.
- 41.Mealy MA, Kessler RA, Rimler Z, et al. Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol Neuroimmunol Neuroinflamm. 2018;5(4):e468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim SH, Mealy MA, Levy M, et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology. 2018;91(22):e2089-e2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hor JY, Asgari N, Nakashima I, et al. Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front Neurol. 2020;11:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data used for this study are available from the corresponding author on reasonable request, with the proper data sharing agreements in place.