

ABSTRACT
In this 14th installment of the annual Antibodies to Watch article series, we discuss key events in commercial monoclonal antibody therapeutics development that occurred in 2022 and forecast events that might occur in 2023. As of mid-November, 12 antibody therapeutics had been granted first approvals in either the United States or European Union (tebentafusp (Kimmtrak), faricimab (Vabysmo), sutimlimab (Enjaymo), relatlimab (Opdualag), tixagevimab/cilgavimab (Evusheld), mosunetuzumab (Lunsumio), teclistamab (TECVAYLI), spesolimab (SPEVIGO), tremelimumab (Imjudo; combo with durvalumab), nirsevimab (Beyfortus), mirvetuximab soravtansine (ELAHERE™), and teplizumab (TZIELD)), including 4 bispecific antibodies and 1 ADC. Based on FDA action dates, several additional product candidates could be approved by the end of 2022. An additional seven were first approved in China or Japan in 2022, including two bispecific antibodies (cadonilimab and ozoralizumab). Globally, at least 24 investigational antibody therapeutics are undergoing review by regulatory agencies as of mid-November 2022. Our data show that, with antibodies for COVID-19 excluded, the late-stage commercial clinical pipeline grew by ~20% in the past year to include nearly 140 investigational antibody therapeutics that were designed using a wide variety of formats and engineering techniques. Of those in late-stage development, marketing application submissions for at least 23 may occur by the end of 2023, of which 5 are bispecific (odronextamab, erfonrilimab, linvoseltamab, zanidatamab, and talquetamab) and 2 are ADCs (datopotamab deruxtecan, and tusamitamab ravtansine).
KEYWORDS: Antibody therapeutics, cancer, COVID-19, food and drug administration, european medicines agency, immune-mediated disorders, SARS-CoV-2
Introduction
Each year since 2010, the Antibodies to Watch article series has endeavored to capture a snapshot of all commercially sponsored monoclonal antibody therapeutics in late-stage clinical development, regulatory review, and those recently approved.1–13 The data presented in each report is derived from a dataset that now includes nearly 1200 antibody therapeutics currently in clinical studies and ~175 that are in regulatory review or approved. We define an antibody therapeutic as a protein molecule that includes at least one binding site derived from an antibody gene. We have thus included molecules such as tebentafusp (Kimmtrak®), which is a recently approved product comprising a high-affinity T cell receptor specific to a peptide sequence fused to an anti-CD3 single-chain antibody fragment, but exclude fusion proteins in which the antibody component is an Fc incorporated solely to extend the half-life of the molecule. Within the current dataset, we identified ~140 antibody therapeutics undergoing evaluation in pivotal Phase 2, Phase 2/3 or Phase 3 studies, referred to collectively as ‘late-stage’ because data derived from them may be used to support submission of a marketing application in the United States (US), European Union (EU),r other regions of the world. Extensive data for this late-stage commercial pipeline are found in Supplemental Table S1 and S2.
The majority of our data were collected during August 1 to November 1, 2022, with only major changes such as approvals that occurred during November 2022 included. We briefly describe relevant details for 19 antibody therapeutics granted a first approval in 2022, and 24 product candidates for which marketing applications are under consideration in at least one country or region. Possible regulatory submissions for 23 investigational antibody therapeutics are forecast based on company disclosures. We also discuss the status of antibody-based COVID-19 interventions as the pandemic wanes in 2022. While we aimed to cite appropriate sources, due to the large volume of literature for the molecules, we focused on publications and other disclosures made public during January 1 to November 1, 2022.
COVID-19 interventions
As the third year of the COVID-19 pandemic concludes, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continues to cause global disruption as variants of concern such as Omicron persist in circulation. As of November 2022, the COVID-19 Dashboard, maintained by the Center for Systems Science and Engineering at Johns Hopkins University (coronavirus.jhu.edu/map), shows that total cases and deaths world-wide have exceeded 630 million and 6.6 million, respectively. Cases and deaths now occur, however, at much lower levels than during 2021 and early 2022 due to a combination of public health measures (e.g., use of masks), wide-spread availability of vaccines, and development of drugs for the disease.
Changing pandemic conditions, in particular the emergence of the Omicron variant, substantially altered the anti-SARS-CoV-2 antibody development landscape during 2022. Due to the high frequency of the Omicron variant, emergency use authorizations (EUAs) granted in 2020 and 2021 by the US Food and Drug Administration (FDA) for casirivimab and imdevimab (REGN-COV), bamlanivimab and etesevimab, and sotrovimab were paused during 2022.14 In addition, clinical development of numerous investigational anti-SARS-CoV-2 antibodies was paused or abandoned. For example, in April 2022 Adagio Therapeutics (now Invivyd) announced they paused their plans to submit an EUA request for anti-SARS-CoV-2 adintrevimab based on feedback from the FDA regarding the antibody’s lack of neutralizing activity against the BA.2 Omicron variant.
As of early November 2022, only two anti-SARS-CoV-2 antibody product candidates, bebtelovimab and Evusheld (tixagevimab co-packaged with cilgavimab) retained an EUA from FDA. Bebtelovimab was first issued an EUA on February 11, 2022, for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 y of age and older weighing at least 40 kilograms) with a positive COVID-19 test, and who are at high risk for progression to severe COVID-19, including hospitalization or death, and for whom alternative COVID-19 treatment options approved or authorized by the FDA are not accessible or clinically appropriate. Evusheld was first authorized on December 8, 2021, for emergency use as pre-exposure prophylaxis for prevention of COVID-19 in adults and pediatric individuals who meet certain conditions. EUAs for bebtelovimab and Evusheld were reissued on October 27, 2022,14 although a preprint released online in September 2022 reported that Evusheld does not neutralize Omicron variants BA.4.6, BA.4.7, and BA.5.9 in pseudovirus neutralization assays.15
Several antibodies that do not target the virus itself, but rather target antigens relevant to symptoms of COVID-19, are either under consideration for an EUA (vilobelimab) or had EUAs reissued (Actemra®, tocilizumab) in 2022. In September 2022, InflaRx N.V. announced that they had submitted a request for an EUA for vilobelimab, which was previously granted FDA’s Fast Track designation for the treatment of critically ill, intubated, mechanically ventilated COVID-19 patients.16 Vilobelimab, a chimeric IgG4κ antibody targeting complement 5a, is being developed for various diseases, including pyoderma gangrenosum, but is not currently approved for any indication. Actemra® was first issued an EUA in June 2021 for treatment of COVID-19 in hospitalized adults and pediatric patients (2 y of age and older) who are receiving systemic corticosteroids and require supplemental oxygen, noninvasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation, and this EUA was reissued on October 27, 2022.14 Actemra®, a humanized anti-IL-6 receptor IgG1κ antibody, has been commercially available since 2005, and is currently marketed in the US for rheumatoid arthritis (RA), giant cell arteritis, systemic sclerosis-associated interstitial lung disease, polyarticular juvenile idiopathic arthritis, systemic juvenile idiopathic arthritis, and cytokine release syndrome.
Antibody therapeutics granted a first approval in the US or EU in 2022
Despite the ongoing pandemic, the annual number of antibody therapeutics granted a first approval in either the US or EU by mid-November 2022 was near the record of 13 products, which was achieved in both 2018 and 2021,17 and has the potential to exceed the record by the end of the year. As detailed in Table 1 and the summaries below, the 12 new antibody-based therapeutics granted first approvals in these two regions as of mid-November 2022 are: tebentafusp (Kimmtrak), faricimab (Vabysmo), sutimlimab (Enjaymo), relatlimab (Opdualag), tixagevimab/cilgavimab (Evusheld), mosunetuzumab (Lunsumio), teclistamab (TECVAYLI), spesolimab (SPEVIGO), tremelimumab (Imjudo; combo with durvalumab), nirsevimab (Beyfortus), mirvetuximab soravtansine (ELAHERE™), and teplizumab (TZIELD). An equal number of the 12 newly approved drugs are for cancer (6 products) and non-cancer (6 products) indications. Notably, the 2022 approvals include 4 bispecifics (tebentafusp, faricimab, mosunetuzumab, and teclistamab) and mirvetuximab soravtansine, which is an antibody–drug conjugate (ADC). All four bispecifics target combinations of antigens that are unique among the currently approved antibody therapeutics. Mirvetuximab soravtansine and the immune checkpoint modulatory antibody relatlimab also target unique antigens (folate receptor (FR) α and LAG3, respectively) compared to antibody therapeutics now on the market. The BLAs for two product candidates, toripalimab, and ublituximab, have FDA action dates before the end of the year, and an FDA action date for penpulimab has not been verified, which allows for the possibility of at least three possible additional FDA approvals by the end of 2022.
Table 1.
Commercially sponsored monoclonal antibody therapeutics granted first approvals in the European Union or United States during 2022. Table includes information publicly available as of November 18, 2022. Abbreviations: ADC, antibody–drug conjugate; BCMA, B cell maturation antigen; CTLA-4, Cytotoxic T-lymphocyte associated protein 4; EU, European Union; EUA, Emergency use authorization; FR, folate receptor; IgG, immunoglobulin; IL, interleukin; LAG-3, Lymphocyte-activation gene 3; NA, not applicable; PD-1, programmed cell death protein 1; PDUFA, Prescription Drug User Fee Act; RSV, Respiratory syncytial virus; SARS-CoV-2, Severe acute respiratory syndrome-coronavirus-2; VEGF, vascular endothelial growth factor.
| INN (Brand name) | Target; Format | Indication first approved | Date of first EU approval | Date of first US approval |
|---|---|---|---|---|
| Tebentafusp (Kimmtrak) | gp100, CD3; Bispecific immunoconjugate | Metastatic uveal melanoma | 4/1/2022 | 1/25/2022 |
| Faricimab (Vabysmo) | VEGF-A, Ang-2; Human/humanized IgG1 κ/λ bispecific | Diabetic macular edema and neovascular age-related macular degeneration | 9/15/2022 | 1/28/2022 |
| Sutimlimab (Enjaymo) | C1s; Humanized IgG4κ | Cold agglutinin disease | 11/15/2022 | 2/4/2022 |
| Relatlimab (Opdualag) | LAG-3; Human IgG4κ | Melanoma | 9/15/2022 | 3/18/2022 |
| Tixagevimab/cilgavimab (Evusheld) | SARS-CoV-2; Human IgG1κ | COVID-19 | 3/25/2022 | NA (EUA) |
| Mosunetuzumab (Lunsumio) | CD20, CD3; Humanized IgG1κ bispecific | Follicular lymphoma | 6/3/2022 | In review (PDUFA date 12/29/2022) |
| Teclistamab (TECVAYLI) | BCMA, CD3; Humanized/human IgG4λ bispecific | Multiple myeloma | 8/23/2022 | 10/25/2022 |
| Spesolimab (SPEVIGO) | IL-36 receptor; Humanized IgG1κ | Generalized pustular psoriasis | In review | 9/01/2022 |
| Tremelimumab (Imjudo; combo with durvalumab) | CTLA-4; Human IgG2κ | Hepatocellular carcinoma | In review | 10/21/2022 |
| Nirsevimab (Beyfortus) | RSV; Human IgG1κ | Prevention of RSV infection | 10/31/2022 | NA |
| Mirvetuximab soravtansine (ELAHERE™) | FRα; Humanized IgG1κ ADC | Ovarian cancer | NA | 11/14/2022 |
| Teplizumab (TZIELD) | CD3; Humanized IgG1κ | Delay of onset of Stage 3 Type 1 diabetes | NA | 11/17/2022 |
Tebentafusp (Immunocore Holdings plc)
Tebentafusp (Tebentafusp-tebn, Kimmtrak®) is a bispecific gp100 peptide-HLA-directed CD3 T cell engager indicated for the treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma. Tebentafusp is composed of an affinity-enhanced T-cell receptor (TCR) fused to an anti-CD3 antibody single-chain variable fragment (scFv) that can redirect T cells to target glycoprotein 100-positive cells. The TCR targets a human leukocyte antigen (HLA)-A*02:01 complexed with gp100 peptide, a lineage antigen expressed in melanocytes and melanoma. First approved in the US, Kimmtrak® has now been approved for marketing throughout the EU and in several other countries.
On January 25, 2022, the FDA approved Kimmtrak® for the treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma (mUM), which is a rare type of cancer that originates in the eye.18 The recommended dosage of Kimmtrak® is 20 μg on Day 1, 30 μg on Day 8, 68 μg on Day 15, and 68 μg once every week thereafter in unresectable or metastatic uveal melanoma patients who are HLA‐A*02:01‐positive; the product is administered via intravenous (IV) infusion. Tebentafusp was granted Breakthrough Therapy, Fast Track, and Orphan Drug designations by the FDA. The European Commission (EC) approved Kimmtrak® for the treatment of HLA-A*02:01-positive adult patients with mUM in April 2022; Kimmtrak® is also approved in the UK, Australia, and Canada for this indication.
The marketing approvals of Kimmtrak® are based on the results of an open-label, randomized Phase 3 clinical trial (NCT03070392), which compared tebentafusp with the investigator’s choice of treatment as first-line systemic therapy in patients with mUM. The study, which included 392 patients randomized 2:1 to receive tebentafusp or the control treatment, demonstrated a statistically and clinically meaningful overall survival (OS) benefit (hazard ratio 0.51), with median OS of almost 22 months in the tebentafusp group and 16 months in the control group.19 Cytokine release syndrome (CRS), which can be serious and life threatening, occurred in 89% of the patients in the tebentafusp group. The US prescribing information for Kimmtrak® includes a black box warning for CRS occurring in patients who receive the product.
Faricimab (Genentech)
Faricimab (faricimab-svoa, Vabysmo®) is a bispecific antibody inhibiting vascular endothelial growth factor-A (VEGF-A) and angiopoietin-2 (Ang-2) developed to treat ophthalmic disorders. The Fc domain of faricimab was engineered to abolish binding to Fcγ receptors and the neonatal Fc receptor (FcRn), which removes effector functions and causes faster systemic clearance, respectively, of the molecule. On January 28, 2022, the FDA approved the use of Vabysmo® to treat neovascular (wet) aged-related macular degeneration (AMD) and diabetic macular edema (DME).20 For both indications, the recommended dose for Vabysmo® is 6 mg administered by intravitreal injection every 4 weeks for the first four doses, after which the patient’s response is evaluated to determine the suitable dosage and frequency of additional doses. Vabysmo was also approved in the EU for AMD and DME in September 2022.
The approvals were based on positive results across two identical Phase 3 studies in wet AMD (TENAYA and LUCERNE) and two identical Phase 3 studies in DME (YOSEMITE and RHINE). In these studies, after four initial monthly doses, patients treated with Vabysmo given at intervals of up to 4 months achieved non-inferior vision gains versus aflibercept given every 2 months in the first year.21,22 In July 2022 Genentech released two-year data from the TENAYA and LUCERNE studies confirming Vabysmo treatment improved vision with fewer treatments for people with wet AMD.23 In particular, the new 2-year data showed that more than 60% of patients administered Vabysmo® could be treated every 4 months, which is an increase from 45% at year one. A longer interval between treatments is expected to be more convenient for patients.
Sutimlimab (Sanofi)
Sutimlimab (sutimlimab-jome, Enjaymo®) is a humanized IgG4ҡ monoclonal antibody that inhibits the classical complement pathway and specifically binds to complement protein component 1 s subcomponent (C1s), a serine protease that cleaves C4. The Fc domain of sutimlimab is engineered to stabilize the hinge and reduce binding to Fcγ receptors and C1q, thereby removing effector functions.
On February 4, 2022, the FDA approved the use of Enjaymo® as the first ever and only treatment for patients with cold agglutinin disease (CAD), a rare autoimmune blood disorder.24 The recommended dose for Enjaymo® is 6.5–7.5 g (patient weight-based dosage) administered via IV injection weekly for the first 2 weeks with administration every other week thereafter to decrease the need for red blood cell transfusion due to hemolysis in patients with CAD. Sutimlimab received FDA’s Breakthrough Therapy and Orphan Drug designations for CAD, and Orphan Drug designation in the EU for this indication. A marketing authorization application (MAA) for sutimlimab for the treatment of hemolytic anemia in adult patients with cold agglutinin disease submitted to the European Medicines Agency (EMA) received a positive opinion in September 2022 and was approved by the EC in November 2022.
The FDA’s approval in February 2022 was based on data from the single-arm Phase 3 CARDINAL study (NCT03347396), which included 24 patients with CAD who had a recent history of blood transfusion. In Part A of the study, patients received a fixed weight-based dose (6.5 g or 7.5 g) of Enjaymo via IV infusion on Day 0, Day 7, and then once every other week up to Week 26. Part B of the study evaluated the long-term safety and durability of response to Enjaymo in patients with CAD over a 2-year follow-up. Treatment with sutimlimab resulted in increased hemoglobin levels and reduced bilirubin levels, and patients remained transfusion-free after Week 5 and did not use other CAD-related therapies.25
The EC’s approval in November 2022 was based on results from the CARDINAL and CADENZA (NCT03347422) studies. In the placebo-controlled Phase 3 CADENZA study, 42 participants with primary CAD without a recent history of blood transfusion were randomized 1:1 to receive a fixed weight-based dose (6.5 g or 7.5 g) of Enjaymo or placebo via IV infusion on Day 0, Day 7, and then once every other week up to Week 26. The open-label Part B of the study assessed long-term safety and durability of response to Enjaymo in patients with CAD. Results from the study showed treatment with sutimlimab reduced hemolysis, anemia, and fatigue in CAD patients without transfusion requirement, and statistically significant and clinically meaningful differences were demonstrated between sutimlimab and placebo.26
Relatlimab (Bristol Myers Squibb, Ono Pharmaceutical Co., Ltd.)
Relatlimab (relatlimab-rmbw) is a human lymphocyte activation gene-3 (LAG-3) blocking IgG4ҡ antibody. Opdualag™, a combination of relatlimab-rmbw, and anti-programmed cell death protein 1 (PD-1) nivolumab (Opdivo®), was developed by Bristol Myers Squibb (BMS) and Ono Pharmaceutical Co., Ltd. for the treatment of adult and pediatric patients 12 y of age or older with unresectable or metastatic melanoma. The companies have a strategic collaboration agreement to jointly develop and commercialize multiple immunotherapies, either as single agents or combination regimens, for cancer in Japan, South Korea, and Taiwan. Opdualag™ was approved by FDA on March 18, 2022, for the treatment of adult and pediatric patients 12 y of age or older with unresectable or metastatic melanoma.27 The product was subsequently approved in the EU on September 15, 2022, for first-line treatment of advanced (unresectable or metastatic) melanoma in adults and adolescents 12 y of age and older with tumor cell programmed cell death protein ligand-1 (PD-L1) expression <1%.
Opdualag™ is a first-in-class, fixed-dose combination of nivolumab (480 mg) and relatlimab (160 mg), administered as a single IV infusion every 4 weeks in patients 12 y or older who weight at least 40 kg. Approvals of the drug are based on the randomized, double-blind Phase 2/3 RELATIVITY-047 trial (NCT03470922), which compared Opdualag™ to nivolumab alone in a total of 714 melanoma patients. Opdualag more than doubled the median progression-free survival (PFS) when compared to nivolumab monotherapy (10.1 months versus 4.6 months, respectively). PFS at 12 months was 47.7% with relatlimab–nivolumab vs. 36.0% with nivolumab alone. Moreover, PFS across key subgroups favored relatlimab–nivolumab over nivolumab. Grade 3 or 4 treatment-related adverse events occurred in 18.9% of patients in the relatlimab–nivolumab group and in 9.7% of patients in the nivolumab group.28
BMS is recruiting an estimated 1050 patients for the randomized, double-blind Phase 3 RELATIVITY-098 study (NCT05002569) of adjuvant immunotherapy with relatlimab and nivolumab fixed-dose combination versus nivolumab monotherapy after complete resection of Stage III–IV melanoma. The primary outcome measure is recurrence-free survival time, and the estimated primary completion date is December 15, 2025. The company is also evaluating the relatlimab-nivolumab fixed-dose combination versus regorafenib or trifluridine + tipiracil for patients with later-lines of metastatic colorectal cancer in a Phase 3, randomized, open-label study (RELATIVITY-098; NCT05328908), which will include an estimated 700 patients and has an estimated primary completion date in January 2025.
Tixagevimab and cilgavimab (AstraZeneca)
Evusheld, (AZD7442) is a combination of two half-life extended antibodies, tixagevimab (AZD8895) and cilgavimab (AZD1061). Derived from B cells donated by convalescent patients after SARS-CoV-2 infection, these two human anti-SARS-CoV-2 IgG1ҡ antibodies were discovered at Vanderbilt University Medical Center and licensed to AstraZeneca in June 2020. Tixagevimab and cilgavimab bind to distinct epitopes on the SARS-CoV-2 spike protein and were optimized by AstraZeneca to have extended half-life and reduced Fc receptor and complement C1q binding via mutations to the Fc domain of the antibodies.
On December 8, 2021, Evusheld received an EUA from the FDA for pre-exposure prophylaxis of COVID-19 in adults and pediatric individuals (12 y or older weighing at least 40 kg) who may be immune compromised due to a medical condition or immunosuppressive medications and who may not mount an adequate immune response to COVID-19 vaccination, as well as those individuals for whom COVID-19 vaccination is not recommended.29 The primary data supporting Evusheld’s EUA are from the randomized, double-blind, placebo-controlled, PROVENT Phase 3 pre-exposure prevention trial (NCT04625725), with 5197 enrolled participants, which showed a statistically significant reduction (77% at primary analysis, 83% at median six-month analysis) in the risk of developing symptomatic COVID-19 compared to placebo, with protection from the virus continuing for at least 6 months.
The initial FDA-recommended dose was intramuscular administration of 150 mg tixagevimab and 150 mg cilgavimab administered in two separate, consecutive injections. In June 2022, the FDA revised the Evusheld dosing to recommend repeat dosing every 6 months with a dose of 300 mg of tixagevimab and 300 mg cilgavimab if patients need ongoing protection. The revisions were based on nonclinical data and pharmacokinetic modeling that suggested activity against the Omicron BA.2, BA.2.12.1, BA.4, and BA.5 subvariants may be retained for 6 months at drug concentrations achieved following an Evusheld dose of 300 mg of tixagevimab and 300 mg cilgavimab.30
Marketing approvals were subsequently granted for pre-exposure use of Evusheld in the UK and EU in early 2022. On March 17, 2022, AstraZeneca announced that Evusheld received a Conditional Marketing Authorization from the UK’s Medicines and Healthcare products Regulatory Agency, thereby becoming the first antibody combination for pre-exposure prophylaxis against COVID-19 licensed in Great Britain.31 On March 24, 2022, AstraZeneca announced that Evusheld was recommended for marketing authorization by EMA based on the PROVENT Phase 3 trial (NCT04625725);32 the product was granted an EU-wide approval for SARS-CoV-2 pre-exposure prophylaxis by the EC on March 25, 2022.
AstraZeneca has also pursued marketing authorizations for use of Evusheld as a treatment for SARS-CoV-2 infections. In August 2022, Evusheld was approved in Japan for both prevention and treatment of symptomatic disease caused by SARS-CoV-2 infection.33 In September 2022, Evusheld was approved in the EU for the treatment of adults and adolescents (aged 12 y and older weighing at least 40 kg) with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19. The approvals in Japan and the EU for Evusheld as a treatment for COVID-19 were based on data from the randomized, placebo-controlled Phase 3 TACKLE study (NCT04723394), which showed reduced risk of severe COVID-19 or death in patients who received Evusheld (18/407 (4%)) compared to those administered placebo (37/415 (9%); relative risk reduction 50.5% [95% CI 14 · 6–71 · 3]; p = 0 · 0096).34
Mosunetuzumab (Genentech, Roche)
Mosunetuzumab (Lunsumio®) is a humanized IgG1ҡ bispecific antibody, generated using knobs-in-holes technology, that targets CD20 and CD3, thereby redirecting T cells to eliminate malignant B cells while avoiding the destruction of the engaged T cells. The modified aglycosylated Fc domain of this antibody results in ablated effector functions. The FDA granted Breakthrough Therapy designation to mosunetuzumab for the treatment of adults with relapsed or refractory (r/r) follicular lymphoma (FL) who have received at least two prior systemic therapies in June 2020 and Orphan Drug designation in December 2018.
On June 3, 2022, the EC granted conditional marketing authorization for Lunsumio® for the treatment of adult patients with r/r FL who have received at least two prior systemic therapies.35 FL is a type of non-Hodgkin’s lymphoma (NHL) that is typically slow-growing (i.e., indolent). Mosunetuzumab was granted an Orphan Drug designation by EMA. Lunsumio is administered via IV injection, normally in cycles of 21 d. The recommended treatment duration is at least 8 treatment cycles, and up to 17 cycles. In cycle 1, 3 doses of Lunsumio® are administered (Day 1: 1 mg; Day 8: 2 mg; Day 15: 60 mg), but only 1 dose is administered in cycle 2 (Day 1: 60 mg) and cycles 3 to 17 (Day 1: 30 mg).
The marketing authorization granted in the EU was based on positive results from the pivotal Phase 1/2 GO29781 study (NCT02500407), which is evaluating escalating doses of mosunetuzumab as a single agent and in combination with anti-PD-L1 atezolizumab in patients with relapsed or refractory B-cell NHL and chronic lymphocytic leukemia (CLL). In heavily pre-treated FL patients included in this study, Lunsumio® demonstrated high complete response rates, with most of the responders maintaining responses for at least 18 months, and favorable tolerability. After a median follow-up of 18.3 months, the median duration of response (DOR) among responders was 22.8 months (95% CI: 9.7-not estimable), the complete response rate was 60% (n = 54/90), the objective response rate was 80% (n = 72/90).35
On July 5, 2022, Genentech announced that the FDA had accepted a BLA and granted priority review for mosunetuzumab, for the treatment of adults with r/r FL who have received at least two prior systemic therapies.36 The application is also based on results from the Phase 1/2 GO29781 study. FDA’s first action on the BLA is expected by December 29, 2022.
Teclistamab (Janssen Research & Development, LLC)
Teclistamab (TECVAYLI) is a T-cell redirecting IgG4λ bispecific antibody recognizing B-cell maturation antigen (BCMA) on target cells and CD3ε on T cells. Generated from Ligand’s transgenic mouse (OmniAb) and Genmab’s DuoBody technology, the Fc was engineered with the stabilizing S228P mutation and L234A/L235A mutations to minimize its effector functions. Teclistamab was granted Orphan Drug designations for the treatment of multiple myeloma (MM) in both the US and EU, and received the Breakthrough Therapy designation for the treatment of r/r MM by the FDA, and a PRIority MEdicines (PRIME) designation by the EMA for treatment of adult patients with r/r MM who previously received ≥3 prior lines of therapy. In August 2022, TECVAYLI (teclistamab) was granted conditional marketing authorization in the EU as monotherapy for the treatment of adult r/r MM patients who previously received ≥3 prior lines of therapy.37 In October 2022, FDA granted TECVAYLI an accelerated approval for adult r/r MM patients who received at least four prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
The authorizations for marketing were based on the results from the multicohort, open-label, Phase 1 and Phase 2 MajesTEC-1 studies (NCT03145181, NCT04557098, respectively), evaluating the safety and efficacy of teclistamab in adults with r/r MM. The ongoing first-in-human dose escalation and dose expansion clinical study (NCT03145181) is assessing the efficacy of teclistamab in patients with r/r MM, with the antibody administered IV (range: 0.3 − 19.2 μg/kg [once every 2 weeks] or 19.2 − 720 μg/kg [once per week]) or subcutaneously (SC) (range: 80 − 3000 μg/kg [once per week]) in different cohorts, with step-up dosing for 38.4 μg/kg or higher doses. Based on the dose escalation data, in the Phase 2 portion of the study patients received a weekly SC dose of teclistamab (1.5 mg/kg), after receiving step-up doses of 0.06 mg/kg and 0.3 mg/kg. Results of the MajesTEC-1 study showed that teclistamab induced durable responses that deepened over time in patients with triple-class exposed r/r MM (n = 165), with an overall response rate of 63%, including a complete response in 39.4% of the patients. The median DOR and duration of PFS were 18.4 months (95% confidence interval [CI], 14.9 to not estimable) and 11.3 months (95% CI, 8.8 to 17.1), respectively. Adverse events were consistent with this patient population and toxicities consistent with T-cell redirection were mostly Grade 1/2.38
Teclistamab is being investigated also in combination studies, including the Phase 3 MajesTEC-3 (NCT05083169), MajesTEC-4 (NCT05243797), and MajesTEC-7 (NCT05552222) studies. Of these, the MajesTEC-3 study is scheduled to be completed soonest. MajesTEC-3 study is comparing the effects of the teclistamab–daratumumab combination administered SC with SC daratumumab in combination with pomalidomide and dexamethasone or SC daratumumab in combination with bortezomib and dexamethasone. The estimate enrollment is 560 patients, and the primary outcome measure is PFS up to 5 y and 2 months. Initiated in October 2021, the estimated primary completion date of the study is in July 2024.
Spesolimab (Boehringer Ingelheim)
Spesolimab (SPEVIGO®), a humanized anti-IL-36 IgG1κ antibody developed by Boehringer Ingelheim, was approved by the FDA as a treatment option for generalized pustular psoriasis (GPP) flares in adults on September 1, 2022. GPP is a rare and potentially life-threatening neutrophilic skin disease characterized by episodes of widespread eruptions of painful, sterile pustules. The FDA had previously granted spesolimab Breakthrough Therapy and Orphan Drug designations for the treatment of GPP, and the BLA for spesolimab received a Priority review. In addition, spesolimab has received Breakthrough Therapy Designation in China and Taiwan, Priority Review in the China, Orphan Drug Designation in Korea, Switzerland and Australia, and Rare Disease designation and fast track in Taiwan for the treatment of GPP flares. An MAA for use of spesolimab as a treatment of flares in GPP is undergoing evaluation by the EMA.39
The approval by FDA was based in part on results from the 12-week pivotal Phase 2 Effisayil™ 1 clinical trial (NCT03782792), which evaluated the efficacy, safety, and tolerability of a single 900 mg dose of IV administered spesolimab, with the option of a second dose if symptoms persisted on Day 8, vs placebo in 53 patients experiencing a GPP flare. After 1 week, 54% of patients treated with SPEVIGO showed no visible pustules compared to 6% of those who received placebo.40 A 3-arm, 5-year Phase 2 study (NCT03886246) to evaluate spesolimab in GPP patients who took part in previous studies with spesolimab is currently recruiting an estimated 155 participants. Patients will be administered SPEVIGO® at 4-, 6-, or 12-week intervals. The primary outcome measure of the study is the occurrence of treatment emergent adverse events (TEAEs) up to Week 252 of maintenance treatment; secondary outcome measures relate to the efficacy of the drug.
Tremelimumab (AstraZeneca)
Tremelimumab (CP-675,206), originally developed by Pfizer using Abgenix’s XenoMouse™ technology, is a human IgG2ҡ antibody targeting cytotoxic T lymphocyte-associated antigen-4 (CTLA-4). In 2011, MedImmune (now AstraZeneca) gained tremelimumab’s global development rights, while Pfizer retained the rights for use in certain combination therapies. Tremelimumab blocks the activity of the immune checkpoint CTLA-4, contributing to T-cell activation, fostering antitumor immune responses and cancer cell death. Tremelimumab and anti-PD-L1 durvalumab (Imfinzi) were granted Orphan Drug designation in the US for the treatment of hepatocellular carcinoma (HCC), and tremelimumab was also granted Orphan Drug designation for HCC in the EU. On October 21, 2022, FDA approved the combination of tremelimumab with Imfinzi for unresectable advanced liver cancer based on the results of the Phase 3 HIMALAYA trial.41 On November 10, 2022, FDA granted a supplemental approval for the combination of tremelimumab with Imfinzi and chemotherapy for first-line metastatic non-small cell lung cancer (NSCLC) based in results of the Phase 3 POSEIDON study.42
Marketing applications for the tremelimumab with Imfinzi combination for liver cancer and for the combination of tremelimumab with Imfinzi and chemotherapy for first-line NSCLC are under review by regulatory authorities in other countries and regions.
HIMALAYA (NCT03298451) is a randomized, open-label, global Phase 3 trial evaluating the safety and efficacy of durvalumab monotherapy and the combination of durvalumab and tremelimumab versus sorafenib, a standard-of-care multi-kinase inhibitor, as first-line treatment in patients with unresectable HCC who had not received prior systemic therapy and were not eligible for localized treatment. The combination of durvalumab and tremelimumab, called the STRIDE regimen (Single Tremelimumab Regular Interval Durvalumab), comprises a single priming dose of 300 mg of tremelimumab added to 1500 mg of durvalumab followed by durvalumab every 4 weeks. Patients were randomized to STRIDE (n = 393), durvalumab (n = 389), or sorafenib (n = 389). The primary outcome measure was OS. Results of the HIMALAYA trial were presented at the American Society of Clinical Oncology Gastrointestinal Cancers Symposium held January 20–22, 2022, in San Francisco. At data cutoff, the primary objective was met: OS was significantly improved for STRIDE vs sorafenib (hazard ratio [HR], 0.78; 96% confidence interval [CI], 0.65–0.92; p = .0035. 3). In addition, the objective response rates were higher for STRIDE and durvalumab (20.1% and 17.0%, respectively) than for sorafenib (5.1%).43
The 3-arm, randomized, Phase 3 POSEIDON study (NCT03164616) evaluated the efficacy of durvalumab or durvalumab and tremelimumab, both administered via IV infusion, in combination with platinum-based chemotherapy as first-line treatment in stage 4 NSCLC patients. In the experimental arms, treatment-naïve patients with EGFR/ALK wild-type metastatic NSCLC were randomized (1:1:1) to receive: (a) 1500 mg of durvalumab with up to 4 cycles of platinum-based chemotherapy every 3 weeks, followed by a maintenance treatment of 1500 mg of durvalumab every 4 weeks until progression; (b) 1500 mg of durvalumab and 75 mg of tremelimumab with up to 4 cycles of platinum-based chemotherapy every 3 weeks, followed by a maintenance treatment of 1500 mg of durvalumab every 4 weeks until progression with one additional 5th dose of 75 mg of tremelimumab post chemotherapy at Week 16; or (c) up to six cycles of chemotherapy alone. Updated results after approximately 4 y follow-up, showing a sustained improvement in OS of tremelimumab and durvalumab with chemotherapy compared to chemotherapy alone, as well as exploratory analyses, showing trends for OS improvement in subgroups with high unmet need, were presented at the European Society of Medical Oncology Congress held September 9–13, 2022, in Paris, France.44
Tremelimumab is being evaluated in combination with durvalumab in studies that include patients with other types of cancer, including small cell lung cancer (SCLC) (ADRIATIC, NCT03703297), bladder cancer (VOLGA, NCT04960709; NILE, NCT03682068) and renal cell carcinoma (RAMPART, NCT03288532). AstraZeneca anticipates regulatory submission acceptances for tremelimumab combined with durvalumab and standard of care for treatment of first-line urothelial cancer in the second half of 2023.
Nirsevimab (AstraZeneca, Sanofi)
Nirsevimab (MEDI8897, Beyfortus) is human IgG1ҡ antibody targeting the respiratory syncytial virus (RSV). The Fc domain was engineered using AstraZeneca’s proprietary YTE half-life extension technology. Developed by AstraZeneca and Sanofi, nirsevimab is designed to offer newborns and infants direct protection against RSV and help prevent RSV-related lower respiratory tract infections. Nirsevimab has received regulatory designations to facilitate development, including a Promising Innovative Medicine designation from the UK Medicines and Healthcare Products Regulatory Agency; Breakthrough Therapy designation from China’s National Medical Products Administration (NMPA); Breakthrough Therapy designation from FDA; and PRIME designation from EMA. In addition, it was named “a medicine for prioritized development” under the Project for Drug Selection to Promote New Drug Development in Pediatrics by Japan’s Agency for Medical Research and Development. Based on results of the Phase 3 MELODY (NCT03979313), Phase 2/3 MEDLEY (NCT03959488), and Phase 2b (NCT02878330) clinical trials, nirsevimab was evaluated under an accelerated assessment procedure and received a positive opinion by EMA in September 2022, and was approved by the EC in early November 2022.45
The Phase 2b trial is a randomized, placebo-controlled trial designed to measure the efficacy of nirsevimab in preventing medically attended RSV-related lower respiratory tract infections through 150 d post-dose. The study was conducted on healthy preterm infants (29–35 weeks’ gestation) who were randomized (2:1) to receive a single intramuscular injection of nirsevimab (50 mg) or placebo. The primary endpoint of the study was met, with a reduction of the incidence of medically attended RSV-related lower respiratory tract infections by 70.1% (95% CI: 52.3, 81.2) compared to placebo.45
MELODY is a randomized, placebo-controlled Phase 3 trial evaluating the safety and efficacy of nirsevimab for the prevention of medically attended lower respiratory tract infections in healthy late preterm and term infants (i.e., born at 35 weeks’ gestation or later). Participants (n = 1490) up to 1 y of age, were randomized (2:1) to receive a single intramuscular injection of nirsevimab (50 mg if <5 kg or 100 mg if >5 kg body weight) or placebo. The primary endpoint of this study was met, with a reduction in the incidence of medically attended lower respiratory tract infections caused by RSV of 74.5% (95% CI 49.6, 87.1; P < .001) compared to placebo.46
The randomized, double-blind, palivizumab-controlled Phase 2/3 MEDLEY study evaluated the safety and pharmacokinetics (PK) of nirsevimab compared to palivizumab in preterm infants and infants with congenital heart disease and/or chronic lung disease of prematurity eligible to receive palivizumab entering their first RSV season. Participants (n = 925) up to 1 year of age entering their first RSV season were randomized to receive a single intramuscular injection of nirsevimab or palivizumab (50 mg if <5 kg or 100 mg if >5 kg body weight). Safety was assessed by monitoring the occurrence of treatment-emergent adverse events or treatment-emergent serious adverse events through 360 d post-dose. Serum levels of nirsevimab following dosing (on Day 151) in this trial were comparable with those observed in the MELODY study, suggesting a similar protection in this population to that in the healthy term and late preterm infants.47
Mirvetuximab soravtansine (Immunogen, Inc.)
Mirvetuximab soravtansine (mirvetuximab soravtansine-gynx, ELAHERE™), developed by ImmunoGen as a treatment for epithelial malignancies such as ovarian adenocarcinoma, is an ADC targeting FRα. The cytotoxic warhead, the tubulin-targeting maytansinoid drug DM4, is conjugated to the humanized IgG1ҡ antibody via a cleavable disulfide linker. The ADC has been granted Orphan Drug designations for ovarian cancer in the US and EU, and FDA’s Fast Track designation for a specific subset of ovarian cancer patients with medium to high FRα-positive platinum-resistant lesions who received between one and three prior systemic treatments, and for whom single-agent chemotherapy is appropriate as the next line of therapy. FDA granted an accelerated approval for mirvetuximab soravtansine for the treatment of adult patients with FRα-positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, who have received one to three prior systemic treatment regimens, on November 14, 2022. FDA also approved a companion diagnostic, VENTANA FOLR1 (FOLR1-2.1) RxDx Assay, developed by Roche.48
FDA’s approval was based on positive results of the Phase 3 SORAYA study (NCT04296890), which evaluated the efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant advanced high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancer, whose tumors express a high-level of FRα. A total of 106 platinum-resistant ovarian cancer patients with high FRα expression previously treated with at least one, but less than three prior systemic treatments, at least one of which included bevacizumab, received mirvetuximab soravtansine (6 mg/kg adjusted ideal body weight) administered on Day 1 of every 3-week cycle. Results from the SORAYA trial were presented at the Society of Gynecologic Oncology (SGO) annual meeting held in March 2022. Additional efficacy analyses based on a 120-day cutoff date showing tumor reduction in 71.4% of patients, an objective response rate of 32.4% as assessed by the investigator, and a preliminary median OS of 13.8 months were presented at the American Society of Clinical Oncology (ASCO) Annual Meeting held June 3–7, 2022. A retrospective safety analysis based on 464 patients with FRα positive, recurrent ovarian cancer pooled across three studies (a Phase 1 first-in-human trial and the Phase 3 FORWARD I and SORAYA trials) demonstrating a differentiated and consistent safety profile was also presented at the 2022 ASCO meeting.49
Mirvetuximab soravtansine was also evaluated in the randomized Phase 3 FORWARD I trial (NCT02631876), which enrolled 366 patients with platinum-resistant ovarian cancer, randomized 2:1 to receive either the ADC or the physician’s choice of pegylated liposomal doxorubicin, topotecan, or weekly paclitaxel. Improved patient-reported outcomes associated with mirvetuximab compared with chemotherapy were presented at the European Society for Medical Oncology (ESMO) in held in Paris, France in September 2022.50 In addition, ImmunoGen continues to enroll patients in the randomized, open-label Phase 3 MIRASOL study (NCT04209855), which is evaluating mirvetuximab soravtansine vs. investigator’s choice of chemotherapy in platinum-resistant, advanced high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancers with high FRα expression. Top-line data from the confirmatory MIRASOL study are expected to be announced in early 2023. If positive, the results may support a full approval by FDA.48
Teplizumab (Provention Bio, Inc.)
Teplizumab (PRV-031, MGA-031) is a humanized, anti-CD3e IgG1κ antibody originally developed at Tolerance Therapeutics, Inc. and the University of California. The antibody Fc region was mutated (L234A; L235A) to reduce effector functions. Teplizumab binds CD3 expressed on mature T cells and may induce expansion and/or regulatory function in T cell subsets. In 2005, teplizumab was licensed to MacroGenics. In 2018, Provention Bio acquired all rights to teplizumab and subsequently continued its development for the prevention and treatment on type I diabetes (T1D). The FDA granted teplizumab Orphan Drug designation for the treatment of recent-onset T1D. Teplizumab was also granted FDA’s Breakthrough Therapy designation for the prevention or delay of clinical T1D in at-risk individuals and EMA’s PRIME designation for the same indication. As of October 2022, Provention Bio and Sanofi had entered into a co-promotion agreement for teplizumab.
Provention Bio submitted a BLA for teplizumab in November 2020, but FDA issued a complete response letter in July 2021. In the letter, the FDA stated that a single, low-dose PK/ pharmacodynamics (PD) study in healthy volunteers had failed to show PK comparability between the intended commercial product and the clinical trial product. The FDA also cited additional considerations related to product quality. Provention Bio announced in March 2022 that the BLA had been resubmitted, and on November 17, 2022, FDA approved TZIELD™ (teplizumab-mzwv) to delay the onset of Stage 3 T1D in adult and pediatric patients aged 8 y and older with Stage 2 T1D.51
FDA’s approval was based in part on a clinical trial in Stage 2 T1D patients in which TZIELD delayed the median onset of Stage 3 T1D by 25 months, or approximately 2 y, compared to placebo. Tzield is administered by IV infusion once daily for 14 consecutive days.52
Provention Bio is currently evaluating teplizumab in patients with newly diagnosed insulin-dependent T1D in the global PROTECT (PROvention T1D trial Evaluating C-peptide with Teplizumab) Phase 3 study (NCT03875729). This randomized, double-blind, placebo-controlled, multicenter trial will enroll 300 patients with recent onset T1D who will be randomized 2:1 to either two 12-day cycles of teplizumab (IV) or placebo. The primary efficacy endpoint is C-peptide change. Secondary endpoints include insulin use, HbA1c, hypoglycemic episodes, and safety. The company expects top-line data from PROTECT Phase 3 study in the second half of 2023.
Antibody therapeutics first approved or undergoing regulatory review outside the US or EU in 2022
While uncommon a decade ago, first approvals of antibody therapeutics in regions outside the US or EU are now expected each year. As detailed in Table 2, this trend continued in 2022, with 5 first approvals granted in China (ormutivimab, serplulimab (HANSIZHUANG), cadonilimab (开坦尼®), pucotenlimab (Puyouheng), ripertamab (Anpingxi®)), and 2 granted in Japan (nemolizumab (Mitchga®), ozoralizumab (Nanozora®)). Notable among the seven approvals, nemolizumab targets a unique antigen (interleukin-31 receptor (IL-31 R)α), cadonilimab is a bispecific antibody that targets a unique antigen combination (PD-1, CTLA-4) compared to currently marketed products, and ozoralizumab is the first approved bispecific nanobody. Further first approvals of antibody therapeutics outside the US or EU are expected in 2023, as marketing applications for at least 7 antibody candidates are undergoing review in China (Table 2). An NDA for concizumab is under review by Japan’s Pharmaceuticals and Medical Devices Agency (PMDA), but FDA may approve a BLA submitted in the US before PMDA approves concizumab in Japan. Relevant details about the products approved in 2022 and those in review are summarized below.
Table 2.
Commercially sponsored monoclonal antibody therapeutics first approved or undergoing regulatory review outside the European Union or United States in 2022. Table includes information publicly available as of November 1, 2022. Abbreviations: dMMR, mismatch repair deficient; IL, interleukin; MSI-H, microsatellite instability-high; PCSK, proprotein convertase substilisin/kexin type; PD-1, programmed cell death protein 1; PD-L1, programmed cell death protein ligand 1; RANKL, receptor activator of nuclear factor-κB ligand; TNF, tumor necrosis factor.
| INN (Brand name) | Target(s); Format | Indication first approved or in review | Status |
|---|---|---|---|
| Ormutivimab | Rabies virus; Human IgG1λ | Rabies infection | Approved in China |
| Serplulimab (HANSIZHUANG) | PD-1; Humanized IgG4κ | MSI-high/dMMR solid tumors | Approved in China |
| Cadonilimab (开坦尼®) |
PD-1, CTLA-4; Humanized IgG1κ bispecific | Cervical cancer | Approved in China |
| Pucotenlimab (Puyouheng) | PD-1; Humanized IgG4κ | MSI-high/dMMR solid tumors | Approved in China |
| Ripertamab (Anpingxi®) | CD20; Chimeric IgG1κ | Diffuse large B-cell lymphoma | Approved in China |
| Nemolizumab (Mitchga®) |
IL-31 Rα; Humanized IgG2κ | Atopic dermatitis | Approved in Japan |
| Ozoralizumab (Nanozora®) | TNF, albumin; Humanized bispecific nanobody | Rheumatoid arthritis | Approved in Japan |
| Geptanolimab (Aibining 艾比寧®) |
PD-1; Humanized IgG4κ | Peripheral T-cell lymphoma | Regulatory review in China |
| Socazolimab | PD-L1; Human IgG1λ2 | Cervical cancer | Regulatory review in China |
| Adebrelimab | PD-L1; Humanized IgG4κ | Small cell lung cancer | Regulatory review in China |
| Tagitanlimab | PD-L1; Humanized IgG1κ | Solid tumor indications | Regulatory review in China |
| Crovalimab | Complement C5; Humanized IgG1κ | Paroxysmal nocturnal hemoglobinuria | Regulatory review in China |
| Narlumosbart | RANKL; Human IgG4κ | Unresectable or surgically difficult giant cell tumor of bone | Regulatory review in China |
| Tafolecimab | PCSK9; Human IgG2κ | Primary hypercholesterolemia and mixed dyslipidemia | Regulatory review in China |
| Concizumab | Tissue factor pathway inhibitor; Humanized IgG4κ | Hemophilia A or B with inhibitors | Regulatory review in Japan |
Ormutivimab (Molecular Targeting Technologies Inc., North China Pharmaceutical Co., Ltd)
Ormutivimab is a recombinant human antibody with rabies virus neutralizing function for post-exposure prophylaxis. Ormutivimab is a human IgG1λ antibody that targets the rabies virus surface glycoprotein 4 epitope 1. Created at Thomas Jefferson University, ormutivimab was licensed to Molecular Targeting Technologies Inc., which has conducted trials in China in partnership with the North China Pharmaceutical Co., Ltd. On January 26, 2022, Molecular Targeting Technologies, Inc., announced that China’s NMPA had approved ormutivimab for post-exposure prophylaxis of rabies.53
In post-exposure prophylaxis animal models, ormutivimab was able to neutralize a broad panel of Chinese-prevalent street rabies viruses and had comparable protection to that achieved with polyclonal human rabies immunoglobulin (HRIG) up to a high dose level (200 IU/kg).54 The effects of ormutivimab or HRIG administered with or without rabies vaccine in healthy adults were evaluated in a Phase 2 study (NCT02559921). The study enrolled 300 patients and had 3 arms in which patients received antibody only (ormutivimab (20 IU·kg−1), ormutivimab (40 IU·kg−1), or HRIG (20 IU·kg−1)), and 4 arms in which participants received antibody in combination with a purified Vero cell rabies vaccine (ormutivimab (20 IU·kg−1), ormutivimab (40 IU·kg−1), HRIG (20 IU·kg−1), or placebo. Population PD modeling was used to compare the activity, reaction characteristics, and impacting factors of the neutralizing antibodies induced by the co-administration of ormutivimab or HRIG with the rabies vaccine, allowing the determination of the target dose of 20 IU·kg−1 ormutivimab in a Phase 3 study.55 Further clinical study has shown ormutivimab to be safe and effective, achieving its main and secondary efficacy endpoints after a single injection in exposed individuals.53
Serplulimab (Shanghai Henlius Biotech Inc.)
Serplulimab (HANSIZHUANG), a humanized IgG4κ antibody targeting PD-1, was developed by Shanghai Henlius Biotech, Inc. The antibody’s hinge region was mutated (S228P) to stabilize the molecule. On March 25, 2022, Shanghai Henlius Biotech, Inc. announced serplulimab had been approved by NMPA for the treatment of adult patients with advanced unresectable or metastatic microsatellite instability-high (MSI-H) solid tumors that have failed to respond to previous standard treatments.56 On November 1, 2022, the company announced that NMPA granted a supplemental approval for serplulimab, in combination with carboplatin and albumin-bound paclitaxel for the first-line treatment of unresectable locally advanced or metastatic squamous NSCLC.57
The NMPA’s first approval was based on a single-arm, multi-center, pivotal Phase 2 clinical study that enrolled 108 patients with unresectable or metastatic MSI-H/dMMR solid tumors who had progressed on or been intolerant to standard therapies. The primary efficacy endpoint, the objective response rate, was assessed by the independent radiology review committee (IRRC) per RECIST v1.1. For the 68 patients with confirmed MSI-H that comprised the main efficacy analysis population, the IRRC- and investigator-assessed objective response rates were 39.7% (95% CI: 28.03, 52.30; 3 CR, 24 PR) and 35.3% (95% CI: 24.1–47.8%), respectively.56
The NMPA is reviewing supplemental new drug applications (NDAs) for serplulimab for extensive-stage (ES) SCLC and, in combination with chemotherapy, first-line treatment of patients with esophageal squamous cell carcinoma (ESCC). Shanghai Henlius Biotech, Inc. plans to submit an MAA for serplilimab for ES-SCLC in the EU in 2022. Results for a Phase 3 study (NCT04063163) of serplulimab in combination with chemotherapy (carboplatin-etoposide) in previously untreated patients with ES-SCLC were published in September 2022.58 In this study, patients were randomized 2:1 to receive either 4.5 mg/kg of serplulimab (n = 389) or placebo (n = 196) IV every 3 weeks, and all patients received IV carboplatin and etoposide every 3 weeks for up to 12 weeks. The median duration of follow-up was 12.3 months (range, 0.2–24.8 months). The median OS was significantly longer in the serplulimab group (15.4 months [95% CI, 13.3 months-not evaluable]) than in the placebo group (10.9 months [95% CI, 10.0–14.3 months]) (hazard ratio, 0.63 [95% CI, 0.49–0.82]; P < .001). The median PFS (assessed by an IRRC) also was longer in the serplulimab group (5.7 months [95% CI, 5.5–6.9 months]) than in the placebo group (4.3 months [95% CI, 4.2–4.5 months]) (hazard ratio, 0.48 [95% CI, 0.38–0.59]).58 Late-stage clinical studies evaluating the safety and efficacy of serplulimab in other cancers, including gastric, colorectal, triple-negative breast cancer, are planned or ongoing.
Cadonilimab (Akeso, Inc.)
Cadonilimab (开坦尼®) is an Fc-silenced, humanized tetravalent IgG1κ bispecific antibody that targets PD-1 and CTLA-4. Developed by Akesobio, cadonilimab was approved by China’s NMPA in June 2022 for the treatment of relapsed or metastatic cervical cancer patients who progressed on or after platinum-based chemotherapy.59 The NMPA previously granted cadonilimab Breakthrough Therapy designation for this indication.
The NMPA’s approval of 开坦尼® is based on positive results from a pivotal Phase 2 study of cadonilimab for treatment of relapsed or metastatic cervical cancer patients who progressed on or after platinum-based chemotherapy. Of the 111 patients enrolled, 100 were evaluable with tumor assessment. As confirmed by the IRRC, the objective response rate and complete response were 33.0% and 12.0%, respectively, and the DOR rates at 6 and 12 months were 77.6% and 52.9%, respectively. The median PFS and OS were 3.75 and 17.51 months, respectively. Among the 64 patients with a combined positive score ≥1 for PD-L1 staining, the objective response rate was 43.8%, median PFS was 6.34 months and median OS was not reached.59
A Phase 3 trial (NCT04982237) evaluating cadonilimab plus platinum-containing chemotherapy with or without bevacizumab as first-line treatment for persistent, recurrent, or metastatic cervical cancer has completed enrollment, and a Phase 3 trial (NCT05235516) of cadonilimab combined with chemoradiotherapy for locally advanced CC is ongoing. Akeso is also evaluating cadonilimab in late-stage clinical trials of patients with early-stage HCC and gastric or gastroesophageal junction cancer (GEJ).
Pucotenlimab (LEPU BIOPHARMA)
Pucotenlimab (Puyouheng) is a humanized, hinge-stabilized IgG4κ anti-PD-1 monoclonal antibody with an Fc domain engineered for half-life extension (S254T, V308P, N434A mutations). Pucotenlimab binds human PD-1 with high affinity and suppresses the interaction of PD-1 with its ligands PD-L1 and PD-L2.60 On July 22, 2022, LEPU BIOPHARMA announced that Puyouheng had been conditionally approved for marketing by the NMPA. The drug is indicated for patients with unresectable or metastatic MSI-H or mismatch repair deficient (dMMR) advanced solid tumors, including advanced colorectal cancers that have progressed following previous treatment with a fluoropyrimidine, oxaliplatin and irinotecan, and other advanced solid tumors that have progressed following at least previous first-line therapy with no satisfactory alternative treatment option.61 LEPU BIOPHARMA’s marketing application for Puyouheng as second-line monotherapy for advanced melanoma was accepted by NMPA in July 2021.
Puyouheng’s approval for MSI-H/dMMR solid tumors was based on a multi-center, open-label, Phase 2 clinical study (NCT03704246) with the objective response rate assessed by the Independent Review Committee (IRC) according to the RECIST1.1 as the primary outcome measure. A total of 100 patients with histologically confirmed advanced solid tumors that were identified as having MSI-H/dMMR had enrolled in the study as of December 4, 2021. Patients were administered 200 mg of pucotenlimab IV every 3 weeks until disease progression, unacceptable toxicity, or patient withdrawal. The median follow-up duration for the intention to treat (ITT) population was 22.5 months. The objective response rate for the ITT population was 49.0% (95% CI: 38.86%, 59.20%), with 9 cases of complete response and 40 cases of partial response. The objective response rate was 50.0% (95% CI: 31.30%, 68.70%) in the subgroup of patients with colorectal cancers who failed previous triplet therapy (a fluoropyrimidine, oxaliplatin, and irinotecan).61
Ripertamab (SinoCellTech Ltd., CSPC Pharmaceutical Group Ltd.)
Ripertamab (Anpingxi®) is a chimeric anti-CD20 IgG1κ antibody that differs from rituximab by only one amino acid (V219A in the CH1 domain of the heavy chain). SinoCellTech created and developed ripertamab as a treatment for hematological malignancies as part of a licensing agreement with CSPC Pharmaceutical Group Ltd. In August 2022, ripertamab was approved by NMPA for the treatment of newly diagnosed CD20-positive diffuse large B-cell lymphoma (DLBCL).62
The approval for DLBCL was based on a randomized, single-blind Phase 3 study (NCT02772822) that compared the efficiency and safety of ripertamab plus CHOP (cyclophosphamide, hydroxydaunomycin, oncovin, and prednisone) with rituximab plus CHOP in untreated CD20-positive DLBCL patients. The aim of the study was to show non-inferiority of first-line treatment with ripertamab plus CHOP compared to rituximab plus CHOP. A total of 364 participants were randomized 2:1 to receive up to six cycles of either ripertamab or rituximab plus CHOP. The primary endpoint was the objective response rate as assessed by the Independent Review Committee. The objective response rate difference between the two groups was −0.4% (95% CI: −5.5%, 4.8%), which met the pre-specified non-inferiority margin of −12%. There were no significant differences between ripertamab plus CHOP compared to rituximab plus CHOP in 1-year PFS rates (81.1% vs. 83.2%, p = .8283), 1 year event-free survival rates (56.2% vs. 58.1%, p = .8005), and 3-year OS rates (81.0% vs. 82.8%, p = .7183).63
Nemolizumab (Maruho Co., Ltd, Chugai Pharmaceutical Co. Ltd.)
Nemolizumab (Mitchga®) is an anti-IL-31 Rα humanized IgG2κ monoclonal created by Chugai. In July 2016, Chugai entered into a global license agreement granting Galderma S.A. exclusive rights for the development and marketing of nemolizumab worldwide, with the exception of Japan and Taiwan. In September 2016, Chugai granted Maruho Co., Ltd., the rights in Japan for the development and marketing of nemolizumab for the skin diseases such as atopic dermatitis.
On March 28, 2022, Maruho Co., Ltd. obtained regulatory approval from the Ministry of Health, Labor and Welfare for Mitchga® SC Injection 60 mg Syringes for the treatment of itching (pruritus) associated with atopic dermatitis when existing treatment is insufficiently effective.64 The approval was based on results from Phase 3 clinical studies conducted in Japan, including the JapicCTI-173740 and JapicCTI‐183894 studies in patients with moderate-to-severe atopic dermatitis (13 y or older) who were tolerant to existing treatments. In both studies, nemolizumab 60 mg was administered SC every 4 weeks along with topical corticosteroids, topical calcineurin inhibitors, or oral antihistamines. Treatment with nemolizumab resulted in continuous improvement in pruritus, signs of atopic dermatitis, and quality of life for up to 68 weeks.65
Galderma, which was founded in 1981 as a joint venture between L’Oréal and Nestlé, is sponsoring numerous late-stage clinical studies of nemolizumab for moderate-to-severe atopic dermatitis, as well as prurigo nodularis. Prurigo nodularis is a rare, potentially debilitating, inflammatory skin disease characterized by disfiguring skin nodules often covering extensive areas of the body and an intense and chronic itch. There are currently no approved therapeutic options for patients with this disease. Nemolizumab was granted Breakthrough Therapy designation by the FDA for pruritus associated with prurigo nodularis.
In June 2022, Galderma announced that the randomized, placebo-controlled Phase 3 OLYMPIA 2 (NCT04501679) trial met all primary and key secondary endpoints, showing nemolizumab as monotherapy significantly improved skin lesions and pruritus compared with placebo in adult patients with moderate-to-severe prurigo nodularis. A total of 274 patients were enrolled in the study. For patients in the nemolizumab arm of the study, dose was based on body weight. Patients weighing less than (<) 90 kilogram received two SC injections of 30 mg nemolizumab (60 mg loading dose) at baseline, then one SC injection once every 4 weeks. Patients weighing greater than or equal to (≥) 90 kg received two SC injections of 60 mg nemolizumab at baseline (no loading dose) and two SC injections every 4 weeks throughout the treatment period of 16 weeks. Of the nemolizumab-treated patients, 38% reached clearance or almost-clearance of skin lesions, assessed using the investigator’s global assessment score vs 11% in the placebo group (p < .0001). In addition, 56% of nemolizumab-treated patients achieved an at least four-point reduction in itch, as measured by the peak-pruritus numerical rating scale score, compared to 21% in the placebo group (p < .0001).66 The confirmatory Phase 3 OLYMPIA 1 trial, which has a similar design but longer treatment period (up to 24 weeks), is currently enrolling an estimated 270 patients.
Ozoralizumab (Taisho Pharmaceutical Co., Ltd.)
Ozoralizumab (TS-152) is a humanized antibody composed of three NANOBODY®VHHs, of which two target the disease-relevant antigen tumor necrosis factor (TNF) and one targets human serum albumin, which extends the serum half-life of the molecule. Discovered by Ablynx, ozoralizumab was licensed to Taisho Pharmaceutical in 2015. Taisho applied for approval to manufacture and market ozoralizumab in Japan for RA in March 2021, and the company announced in September 2022 that it received approval to manufacture and market Nanozora® 30 mg Syringes for SC Injection from the Ministry of Health, Labor and Welfare for the indication of RA that is inadequately managed by currently available treatments.67
The results of a multicenter, double-blind, parallel-group, placebo-controlled Phase 2/3 study (JapicCTI-184029) that assessed the efficacy and safety of SC administration of ozoralizumab patients with active RA despite methotrexate (MTX) therapy were published in June 2022.68 The study included a total of 381 patients who were randomized to receive 30 mg ozoralizumab, 80 mg ozoralizumab, or placebo via SC injection every 4 weeks, along with MTX, for 24 weeks. The primary endpoints were a 20% improvement rate in the American College of Rheumatology criteria (ACR20 response rate) at Week 16 and change in the baseline modified total Sharp score (ΔmTSS) at Week 24. At Week 16, the ACR20 response was significantly higher (p < .001) in both ozoralizumab groups (30 mg, 79.6%; 80 mg, 75.3%), compared with placebo (37.3%). Similar results were observed at Week 24. Structural non-progression (ΔmTSS ≤ 0) was significantly higher in both ozoralizumab groups compared to the placebo group. Taisho also sponsored a smaller Phase 3 study of ozoralizumab in patients with active RA without MTX (JapicCTI-184031). The target sample size of this study was 135 patients; first enrollment was in February 2018.
Geptanolimab (Genor Biopharma Co., Inc.)
Geptanolimab (Aibining 艾比寧®; GB226, APL-501, CBT 501) is a hinge-stabilized, humanized IgG4κ antibody targeting PD-1. The antibody was developed by Genor BioPharma Co. Ltd., which owns development and commercialization rights in China. Apollomics, Inc. (formerly CBT Pharmaceuticals, Inc.) holds the rest of the world rights.
An NDA for geptanolimab for r/r peripheral T-cell lymphomas submitted by Genor was accepted by the NMPA and granted priority review. The company anticipates approval in China in 2022. Genor is also evaluating geptanolimab’s safety and efficacy in a pivotal clinical trial as second-line or later treatment of cervical cancer, and in early-stage clinical trials, either as monotherapy or in combination with other drugs, in patients with various other cancers.69
Socazolimab (Lee’s Pharmaceutical Holdings Limited)
Socazolimab (ZKAB001, STI-A1014) is a recombinant human IgG1λ2 antibody that targets PD-L1. The antibody was licensed from Sorrento Therapeutics, Inc. by China Oncology Focus Limited, a Lee’s Pharmaceutical Holdings Limited subsidiary, for development in Greater China, including Mainland China, Hong Kong, Macau, and Taiwan. In October 2021, an NDA for socazolimab to treat recurrent or metastatic cervical cancer was accepted by the Center for Drug Evaluation of the NMPA for review. Socazolimab was previously granted Breakthrough designation by the NMPA.
Results for an open-label, dose-escalation, and dose-expansion Phase 1 study (NCT03676959) investigating the safety and efficacy of the socazolimab for recurrent or metastatic cervical cancer were recently reported. In this study, 5, 10, or 15 mg/kg doses of socazolimab were administered via IV injection biweekly in the dose escalation phase, which included 12 patients. In the dose-expansion phase, 92 patients, with 54 patients (59.3%) expressing baseline PD-L1-positive tumors, were administered the 5 mg/kg dose of socazolimab. The overall response rate was 15.4% (95% CI, 8.7% to 24.5%), and the median PFS was 4.44 months (95% CI, 2.37 to 5.75 months). The median OS was 14.72 months (95% CI, 9.59 to NE months) and the 12-month OS was 58.2% (45.4%, 69.0%). The overall response rates for PD-L1-positive and PD-L1-negative patients were 16.7% and 17.9%, respectively.70
Socazolimab with or without carboplatin plus etoposide is being evaluated in a Phase 3 multicenter, randomized, double blinded, placebo-controlled clinical trial (NCT04878016) as first-line treatment of patients with ES-SCLC. Enrollment for this study, estimated as 498 patients, has been completed.71
Adebrelimab (Jiangsu Hengrui Medicine Co., Ltd.)
Adebrelimab (SHR-1316, HTI-1088) is a humanized anti-PD-L1 IgG4κ antibody developed by Jiangsu Hengrui Medicine Co. Ltd. An NDA for adebrelimab in combination with chemotherapy as a treatment for SCLC has been submitted to the NMPA.72
The results of the randomized, double-blind, placebo-controlled Phase 3 CAPSTONE-1 study (NCT03711305) evaluating the safety and efficacy of adebrelimab as first-line treatment in patients with ES-SCLC were recently published in Lancet Oncology.73 Patients were randomized in a 1:1 ratio to receive either adebrelimab (20 mg/kg, Day 1 of each 21-day cycle) + carboplatin + etoposide or placebo + carboplatin + etoposide for 4–6 cycles in the induction phase followed by maintenance with adebrelimab or placebo until progressive disease as assessed by the investigator using Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST v1.1). A total of 462 patients were enrolled, with 230 receiving adebrelimab plus chemotherapy (adebrelimab group) and 232 receiving placebo plus chemotherapy (placebo group). All treatments were IV administered. At data cutoff (Oct 8, 2021), median follow-up was 13 · 5 months (IQR 8 · 9–20 · 1). Median OS was significantly improved in the adebrelimab group (median 15.3 months [95% CI 13 · 2–17 · 5]) vs. the placebo group (12.8 months [11 · 3–13 · 7]; hazard ratio 0 · 72 [95% CI 0 · 58–0 · 90]; one-sided p = 0 · 0017). Treatment-related serious adverse events occurred in 39% and 28% of the patients in the adebrelimab and placebo groups, respectively.
Adebrelimab in combination with chemo-radiotherapy is being evaluated in a placebo-controlled, multi-center, randomized, double-blinded, Phase 3 trial (NCT04691063) that is enrolling patients with limited-stage SCLC by invitation. In addition, a randomized, double-blind, multicenter, Phase 1b/3 study (NCT04316364) is evaluating adebrelimab or placebo in combination with chemotherapy as perioperative treatment of resectable Stage II or III NSCLC.
Tagitanlimab (Sichuan Kelun Pharmaceutical Co., Ltd.)
Tagitanlimab (A167, KL-A167, KLA-167, HBM9167) is a humanized IgG1κ antibody that targets PD-L1. Developed by KLUS Pharma, a subsidiary of Kelun-Biotech, which is a holding subsidiary of Sichuan Kelun Pharmaceutical Co., Ltd., the antibody includes several mutations (L234A L235A G237A) to reduce effector functions. Klus Pharma’s pipeline indicates the BLA has been filed to NMPA in China for two solid tumor indications.74 Tagitanlimab was licensed to Harbor BioMed worldwide, excluding China, Hong Kong, Macau, and Taiwan, in 2018, and has received Orphan Drug designation from the FDA for nasopharyngeal carcinoma (NPC).
A single arm, Phase 2 study (NCT03848286) to evaluate the efficacy and safety of tagitanlimab in patients with recurrent or metastatic NPC sponsored by Sichuan Kelun Pharmaceutical Research Institute Co., Ltd. was completed in January 2022. Tagitanlimab (900 mg, IV) was administered every 2 weeks to 153 patients diagnosed with histopathologically confirmed recurrent or metastatic nonkeratinizing NPC and had received ≥ two lines of chemotherapy until disease progression, intolerable toxicity, or withdrawal of consent. The primary endpoint was overall response rate evaluated by the independent review committee (IRC) according to RECIST v1.1.
Sichuan Kelun Pharmaceutical Research Institute Co., Ltd. is the sponsor of additional clinical studies that are not yet recruiting patients. A Phase 3 study (NCT05294172) that will evaluate the efficacy of tagitanlimab combined with cisplatin and gemcitabine vs placebo combined with cisplatin and gemcitabine in the treatment of recurrent or metastatic NPC is not yet recruiting patients as of the last record update on April 4, 2022. In addition, several Phase 2 studies of tagitanlimab in triple-negative breast cancer (NCT05445908) and NSCLC (NCT05351788) patients are also not yet recruiting when accessed in mid-November 2022.
Crovalimab (Chugai Pharmaceuticals, F. Hoffmann-La Roche Ltd.)
Crovalimab (SKY59, RG6107, RO7112689), a humanized IgG1κ antibody targeting complement C5, was developed by Chugai Pharmaceuticals. The antibody includes mutations that reduce effector functions (L235R G236R S239K A327G A330S P331S) and enhance half-life (M428L N434A). It was also designed to have pH-dependent binding to the target.75 In August 2022, Chugai announced that the NMPA accepted an application for regulatory approval of crovalimab for paroxysmal nocturnal hemoglobinuria (PNH) and granted priority review. Crovalimab had previously been designated as a Breakthrough Therapy for PNH by NMPA. F. Hoffmann-La Roche Ltd. is responsible for the development of crovalimab outside Japan and Taiwan, and so the regulatory application was filed by a China affiliate of Roche.76
The application is based on primary analysis of the multicenter, single-arm COMMODORE 3 study (NCT04654468), which evaluated the efficacy, safety, pharmacokinetics, and pharmacodynamics of crovalimab in patients with PNH, not previously treated with complement inhibitors. Participants (n = 51) received a loading series of crovalimab comprising an IV dose (1000 or 1500 mg based on body weight) on Day 1, followed by weekly crovalimab SC doses (340 mg) for 4 weeks on Week 1 Day 2, then on Weeks 2, 3, and 4. For Week 5 and Q4W thereafter, the crovalimab dose was either 680 mg SC or 1020 mg SC, depending on body weight. The primary endpoints of the study were the percentage of patients who achieved predetermined hemolytic control based on LDH level between Week 5 through Week 25, and the change in the percentage of patients achieving predetermined transfusion avoidance from baseline at Week 25. Results are to be presented at a future medical meeting.76
Two randomized, active-controlled Phase 3 clinical trials evaluating crovalimab for PHN are ongoing. COMMODORE 1 (NCT04432584) and COMMODORE 2 (NCT04434092) trials are evaluating the efficacy and safety of crovalimab versus eculizumab (SOLIRIS®) in children or adults with PNH that is currently treated or not previously treated with complement inhibitors, respectively. For COMMODORE 1, the estimated enrollment is 250 patients and the primary study completion date is in July 2025. For COMMODORE 2, the estimated enrollment is 200 patients and the primary study completion date is in January 2023.
Narlumosbart (CSPC Pharmaceutical Group Limited)
Narlumosbart (JMT103) is a human IgG4κ monoclonal antibody that binds and inhibits the activity of receptor activator of nuclear factor-κB ligand (RANKL). The antibody was developed by Shanghai JMT-BioTechnology Co., Ltd., a subsidiary of CSPC Pharmaceutical Group Limited. As announced in June 2022, a BLA for narlumosbart for the treatment of unresectable or surgically difficult giant cell tumor of bone (GCTB) was accepted for priority review by the NMPA. The BLA for narlumosbart includes data from two pivotal clinical studies that demonstrated narlumosbart had a better clinical efficacy in the treatment of unresectable or surgically difficult GCTB, with a tumor response rate of 93.5%, and a trend higher than that of the denosumab group.77
Liang et al. recently reported the results of a Phase 1 dose escalation and expansion study (NCT03550508) that evaluated the safety, tolerability, and preliminary PK/PD of narlumosbart in 59 patients with bone metastases from tumors.78 In the dose expansion phase of the study, patients (n = 39) received narlumosbart at 1.0, 2.0, or 3.0 mg/kg on Day 1, Day 29, and Day 57. Based on the study data, the authors concluded that narlumosbart had a good safety profile and potential clinical activity in patients with bone metastases from solid tumors, and that narlumosbart’s PK/PD profiles appeared to be similar to those of the marketed anti-RANKL product denosumab.
In May 2021, Niu et al. reported results for 38 patients enrolled June 3 through December 24, 2020 in a multicenter, single-arm, open-label Phase 1b/2 study that evaluated the efficacy and safety of narlumosbart in surgically unsalvageable or refractory GCTB.79 Patients were administered 2 mg/kg narlumosbart SC every 4 weeks with a loading dose on Days 8 and Day 15 of the first 4 weeks of therapy. Among 32 patients with at least 1 efficacy evaluation within 12 weeks, 26 (81.3%, 95% CI: 63.6–92.8) had a tumor response by at least one response criteria. A real-world clinical study (NCT05402865) of denosumab and non-denosumab therapies in the treatment of Chinese patients with unresectable GCTB conducted during 2013–2021 served as the external control for the single-arm Phase 1b/2 trial of narlumosbart treatment in GCTB patients.
Tafolecimab (Innovent Biologics, Inc.)
Tafolecimab (IBI306) is a human IgG2κ antibody that binds proprotein convertase substilisin/kexin type 9 (PCSK9), preventing PCSK9ʹs interaction with its receptor low-density lipoprotein cholesterol receptor (LDL-R), thereby restoring LDL-R recycling and low-density lipoprotein cholesterol (LDL-C) uptake. In June 2022, Innovent Biologics, Inc. announced that the NMPA accepted the NDA for tafolecimab for the treatment of primary hypercholesterolemia, including non-familial hypercholesterolemia (non-FH) and heterozygous familial hypercholesterolemia (HeFH), and mixed dyslipidemia.80
The NDA includes data from three randomized, double-blind Phase 3 clinical studies conducted in China, CREDIT-1 (NCT04289285), CREDIT-2 (NCT04179669), and CREDIT-4 (NCT04709536), that evaluated the safety and efficacy of tafolecimab for primary hypercholesterolemia. In these studies, treatment with tafolecimab reduced LDL-C levels by ~57-67% compared to placebo.
The 4-arm, placebo-controlled CREDIT-1 study (NCT04289285) enrolled 804 non-FH patients at high or very high cardiovascular risk. Patients received SC doses of either tafolecimab 450 mg every 4 weeks or 600 mg every 6 weeks, or they were administered placebo every 4 or 6 weeks. After 48 weeks of continuous treatment with tafolecimab, LDL-C levels of the tafolecimab arms were significantly reduced compared with the placebo arms (LS mean difference of 450 mg Q4W = −65.04%, 97.5%CI: −70.22% ~ −59.86%, p < .0001; LS mean difference of 600 mg Q6W = −57.31%, 97.5%CI: −63.95% ~ −50.68%, p < .0001).81
The randomized, double-blind, placebo-controlled CREDIT-2 study (NCT04179669) evaluated the efficacy and safety of tafolecimab in 148 Chinese patients with HeFH. During the 12-week double-blind treatment period, patients who had previously received a stable lipid-lowering therapy for at least 4 weeks were randomized 2:1:2:1 to receive SC injection of tafolecimab or placebo 150 mg every 2 weeks, or tafolecimab or placebo 450 mg every 4 weeks, respectively. In Week 12–24, patients in the tafolecimab groups continued to receive the drug according to the previous regimens, whereas patients in placebo groups were crossed over to receive open-label tafolecimab 150 mg Q2W or 450 mg Q4W, respectively. The primary endpoint was the percent change in LDL-C levels from baseline to Week 12. Study results showed that tafolecimab significantly reduced LDL-C levels (treatment difference versus placebo: −57.4% [97.5% CI, −69.2% to −45.5%] for 150 mg every 2 weeks; −61.9% [−73.4% to −50.4%] for 450 mg every 4 weeks; P < 0 · 0001 for both comparisons). The results for key secondary endpoints were also positive.82
The randomized, double-blind, placebo-controlled CREDIT-4 (NCT04709536) study recruited an estimated 300 patients with hypercholesterolemia, including non-FH and HeFH, in China. Results after 12 weeks of continuous treatment indicate that the LDL-C levels of control arm were significantly reduced compared with the placebo arm (LS mean difference of 450 mg Q4W = −63.02%, 95%CI: −66.48% ~ −59.56%, p < .0001).81
Concizumab (Novo Nordisk)
Concizumab (NNC172-2021, NN7415) is a hinge-stabilized (S228P mutation), humanized IgG4κ antibody that targets the Kunitz-2 domain tissue factor pathway inhibitor (TFPI) and downregulates its activity. By binding TFPI, the antibody prevents TFPI from binding Factor Xa, thereby enabling Factor Xa to contribute to the maintenance of hemostasis. Concizumab was discovered and developed for prophylactic treatment of hemophilia A and B by Novo Nordisk. FDA granted concizumab Breakthrough Therapy designation for prophylaxis treatment of people with hemophilia B with inhibitors toward Factor VIII or Factor IX, and Orphan Drug designation for hemophilia A and B. Concizumab was granted Orphan Drug designation in the EU for treatment of hemophilia B. Pharma Japan reported in August 2022 that Novo Nordisk filed an NDA in Japan for concizumab for the prophylactic treatment of hemophilia A or B.83 Novo Nordisk reported that they will submit a BLA to FDA for concizumab in this indication by the end of 2022. Based on historical trends in approval times for PMDA vs FDA, this BLA may be approved in the US before the NDA is approved in Japan.
Novo Nordisk’s ongoing Explorer clinical trial program is evaluating the efficacy and safety of concizumab administered as a once-daily SC prophylactic treatment in people with hemophilia A or B with or without inhibitors. The program includes the Phase 3 explorer7 (NCT04083781), explorer8 (NCT04082429), and explorer10 (NCT05135559) studies. The explorer7 and explorer8 studies were initiated in October and November 2019, respectively, but paused between March and August 2020 due to non-fatal thrombotic events in three patients enrolled in the Phase 3 program. Both studies are active but not recruiting as of September 2022.
Results of the explorer7 study were reported at the International Society on Thrombosis and Hemostasis meeting held in July 2022.84 In this study, males aged 12 y and over were randomized 1:2 to either a no prophylaxis (n = 19; arm one; ≥24 weeks) or concizumab prophylaxis (n = 33; arm two; ≥32 weeks) or assigned to concizumab prophylaxis (n = 81; arm three and four). After treatment restart following the study pause due to thromboembolic events, patients received a 1.0 mg/kg concizumab loading dose, followed by an initial 0.20 mg/kg daily dose, with potential adjustment to 0.15 or 0.25 mg/kg based on plasma concizumab concentration at Week 4. The primary outcome measure was the number of treated spontaneous and traumatic bleeding episodes, measured as the annualized bleeding rate (ABR), between arms one and two. The estimated mean ABR was 1.7 (95% CI, 1.0–2.9) for concizumab versus 11.8 (95% CI, 7.0–19.9) for no prophylaxis (ABR ratio, 0.14 [95% CI, 0.07–0.29]; P < .001). At 24 weeks, 21 (63.6%) concizumab patients, including those who discontinued before 24 weeks, and 2 (10.5%) of patients on no prophylaxis had zero treated bleeds. No thromboembolic events were reported after treatment restart.84
Antibody therapeutics undergoing first regulatory review in the US or EU
As of mid-November 2022, BLAs or MAAs for 16 antibody therapeutics are undergoing review by either FDA or EMA, respectively (Table 3). The BLAs for toripalimab and ublituximab have first action dates of December 23 and December 28, respectively, and FDA’s first action date for penpulimab’s BLA is not confirmed. If at least 2 of these are approved by the end of the year, then the annual number of first antibody therapeutics approved in 2022 would reach a new record. Of the antibodies in regulatory review in the US or EU, 2 (glofitamab, epcoritamab) are bispecific antibodies targeting CD20 and CD3, and 1 (trastuzumab duocarmazine) is an ADC. In addition, BLAs for 2 product candidates intended as treatments for early Alzheimer’s disease, lecanemab, and donanemab, are being evaluated by FDA. Considering the substantial unmet medical need in this area, approval of these mAbs has the potential to improve the lives of many patients, as well as their caregivers.
Table 3.
Commercially sponsored investigational monoclonal antibody therapeutics in regulatory review in the European Union or United States. Table includes information publicly available as of November 18, 2022. Abbreviations: ADC, antibody–drug conjugate; CNS, central nervous system; CRL, complete response letter; HER2, human epidermal growth factor receptor 2; IL, interleukin; MASP, mannan-binding lectin-associated serine protease; NA, Not applicable or available; PD-1, programmed cell death protein 1; PDUFA, Prescription Drug User Fee Act.
| International non- proprietary name | Target; Format | Indication(s) under review | Status in EU | Status in US (estimated decision date) |
|---|---|---|---|---|
| Toripalimab | PD-1; Humanized IgG4κ | Nasopharyngeal carcinoma, esophageal squamous cell carcinoma | In review | In review (PDUFA date 12/23/2022) |
| Ublituximab | CD20; Chimeric IgG1κ | Multiple sclerosis | In review | In review (PDUFA date 12/28/2022) |
| Lecanemab | Amyloid beta protofibrils; Humanized IgG1κ | Early Alzheimer’s disease | NA | In review (PDUFA date 1/6/2023) |
| Trastuzumab duocarmazine | HER2; Humanized IgG1κ ADC |
HER2+ unresectable locally advanced or metastatic breast cancer | In review | In review (PDUFA date 5/12/2023) |
| Mirikizumab | IL-23p19; Humanized IgG4ҡ | Ulcerative colitis | In review | In review (2023) |
| Glofitamab | CD20, CD3e; IgG1λ/ҡ bispecific | Diffuse large B-cell lymphoma | In review | NA |
| Penpulimab | PD-1; Humanized IgG1ҡ | Metastatic nasopharyngeal carcinoma | NA | In review |
| Inolimomab | CD25; Murine IgG1ҡ | Acute graft-vs-host disease | NA | In review |
| Donanemab | Amyloid β; Humanized IgG1ҡ | Early Alzheimer’s disease | NA | In review (Q1 2023) |
| Lebrikizumab | IL-13; Humanized IgG4κ | Atopic dermatitis | In review | NA |
| Epcoritamab | CD20, CD3; Humanized IgG1κ/λ bispecific | Large B-cell lymphoma | In review | In review (PDUFA date 5/21/2023) |
| Tislelizumab | PD-1; Humanized IgG4ҡ | Esophageal squamous cell carcinoma | In review | In review (2nd cycle) |
| Retifanlimab | PD-1; Humanized IgG4ҡ | Carcinoma of the anal canal | Application withdrawn | In review (2nd cycle) |
| Narsoplimab | MASP-2; Human IgG4λ | Hematopoietic stem cell transplant-associated thrombotic microangiopathy | NA | In review (CRL appealed) |
| Sintilimab | PD-1; Human IgG4ҡ | Non-small cell lung cancer | NA | In review (2nd cycle) |
| 131I-Omburtamab | B7-H3; Murine IgG1κ; radiolabeled | CNS/leptomeningeal metastasis from neuroblastoma | In review | In review |
Toripalimab (Shanghai Junshi Bioscience Co., Ltd, Coherus BioSciences, Inc.)
Toripalimab (marketed as Tuoyi® in China) is an IgG4κ anti-PD-1 monoclonal antibody developed by Shanghai Junshi Bioscience Co., Ltd. Coherus has partnered with the company to co-develop toripalimab, with Coherus responsible for the development and commercialization of toripalimab in the US and Canada. In 2018, toripalimab became the first anti-PD1 approved in China, and the product is now approved there for multiple types of cancer, including melanoma, NPC, urothelial carcinoma, NSCLC, and ESCC.
Toripalimab was granted Orphan Drug designations by the FDA for the treatment of NPC, mucosal melanoma, soft tissue sarcoma, esophageal cancer, and SCLC. FDA also granted Breakthrough Therapy designation to toripalimab for the treatment of recurrent or metastatic NPC with disease progression on or after platinum-containing chemotherapy and in combination with gemcitabine and cisplatin as a first-line treatment for patients with recurrent or metastatic NPC. The EMA has granted Orphan Drug designation to toripalimab for the treatment of NPC.
In September 2021, Junshi Biosciences and Coherus Biosciences announced the completion of a rolling BLA submission to the FDA for toripalimab in combination with gemcitabine and cisplatin as first-line treatment for patients with advanced recurrent or metastatic NPC and toripalimab monotherapy for the second-line or later treatment of patients with recurrent or metastatic NPC after platinum-based chemotherapy. The FDA issued a complete response letter requesting a quality process change in May 2022, and in July 2022, the companies announced FDA’s acceptance of the BLA resubmission. FDA’s first action on the BLA is expected by December 23, 2022.85 As of November 2022, an MAA had been submitted to EMA for toripalimab combined with cisplatin and gemcitabine for the first-line treatment of patients with locally recurrent or metastatic NPC and toripalimab combined with paclitaxel and cisplatin for the first-line treatment of patients with unresectable locally advanced/recurrent or metastatic ESCC.
The BLA submission included data from the Phase 3 JUPITER-02 (NCT03581786) study of patients with recurrent or metastatic NPC and no previous chemotherapy for recurrent or metastatic disease. Patients (n = 289) were randomized (1:1) to receive either toripalimab (240 mg) or placebo in combination with gemcitabine-cisplatin therapy every 3 weeks for up to six cycles, followed by monotherapy with toripalimab or placebo. The primary endpoint was PFS as assessed by a blinded independent review committee according to RECIST v.1.1. The combination of toripalimab and gemcitabine-cisplatin improved the median PFS compared to the chemotherapy arm (11.7 vs 8 months, respectively), the overall response rate (77.4% vs. 66.4% (P = .033), respectively) and the median DOR (10.0 vs. 5.7 months, respectively).86
Ublituximab (TG Therapeutics, Inc)
Ublituximab (TG-1101) is a chimeric anti-CD20 IgG1κ antibody glycoengineered for enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) that was originally developed by LFB Biotechnology and licensed to TG Therapeutics as a treatment for CLL and multiple sclerosis (MS).
After the BLA acceptance by the FDA in May 2021 for ublituximab in combination with umbralisib (Ukoniq®) as a treatment for patients with CLL and small lymphocytic lymphoma, TG therapeutics withdrew this BLA based on data from the UNITY-CLL Phase 3 study (NCT02612311) showing an increasing imbalance in OS in the control arm. In June 2022, the FDA withdrew its approval of Ukoniq® for marginal zone lymphoma and FL.87 A separate BLA submitted in 2021 for ublituximab monotherapy as a treatment for patients with relapsing forms of MS is undergoing review at the FDA and a first action by FDA is expected by December 28, 2022.88
The BLA for ublituximab for MS included results of the randomized, double-blinded, active-controlled Phase 3 trials ULTIMATE I (NCT03277261) and ULTIMATE II (NCT03277248) evaluating ublituximab (450 mg dose IV every 6 months, following a Day 1 infusion of 150 mg over four hours and a Day 15 infusion over 1 hour) compared to teriflunomide (14 mg oral tablets taken once daily) in a total of 1,094 patients with relapsing MS for both studies. The primary endpoint was the annualized relapse rate (ARR). Results from the ULTIMATE I and II Phase 3 trials showed that the primary endpoint was met, with a significant reduction of the ARR for ublituximab vs. teriflunomide over a period of 96 weeks. In the ULTIMATE I trial, which included 549 patients, the ARRs were 0.08 vs. 0.19 for those who received ublituximab vs teriflunomide, respectively, (rate ratio, 0.41; 95% confidence interval [CI], 0.27 to 0.62; P < .001), while in the ULTIMATE II trial (n = 545 patients) the ARRs were 0.09 and 0.18, respectively (rate ratio, 0.51; 95% CI, 0.33 to 0.78; P = .002).89
Lecanemab (Eisai Co., Ltd., Biogen, Inc.)
Lecanemab (BAN2401) is a humanized anti-amyloid beta protofibril IgG1κ antibody initially developed by BioArctic Neuroscience. BAN2401 was licensed to Eisai in a collaboration agreement, allowing the jointly development of lecanemab as a treatment for Alzheimer disease (AD). Under an agreement with Biogen, Eisai and Biogen co-commercialize and co-promote lecanemab, with Eisai having final decision-making authority. The FDA granted Breakthrough Therapy designation for lecanemab for the treatment of AD. In March 2022, Eisai initiated the submission of application data to Japan’s PMDA under the prior assessment consultation system with the aim of obtaining early approval for lecanemab. In May 2022, the companies announced completion of a rolling BLA submission to the FDA for lecanemab in the treatment of mild cognitive impairment due to AD. In July 2022, the FDA accepted the BLA for lecanemab under the accelerated approval pathway and granted Priority Review. The FDA’s target date for a first action on the BLA is January 6, 2023.90
The BLA submission was supported by data from Study 201 (NCT01767311) and the Phase 3 Clarity AD study (NCT03887455). Study 201 is a multicenter, double-blind, placebo-controlled study assessing the clinical efficacy of lecanemab at multiple doses (2.5 mg/kg biweekly, 5 mg/kg monthly, 5 mg/kg biweekly, 10 mg/kg monthly, 10 mg/kg biweekly) or placebo and exploring the dose response of lecanemab using a composite clinical score (ADCOMS) in 856 patients with mild cognitive impairment due to AD and mild AD with confirmed presence of amyloid pathology. The primary outcome measure was the change from baseline in the AD Composite Score (ADCOMS) at 12 months, and key secondary endpoints included amyloid pathophysiology as measured by amyloid positron emission tomography at 18 months.
Clarity AD Phase 3 is a placebo-controlled, double-blind, 18-month study designed to evaluate the efficacy, long-term safety, and tolerability of 10 mg/kg IV lecanemab administered every 2 weeks in 1795 patients with early AD. The primary outcome measurement was the change from baseline in the Clinical Dementia Rating-Sum of Boxes (CDR-SB) at 18 months of treatment. In September 2022, Eisai Co and Biogen Inc announced positive topline results from Clarity AD Phase 3 trial where lecanemab met the primary endpoint by reducing by 27% the clinical decline on the CDR-SB compared with placebo at 18 months. All key secondary endpoints were also met. Based on results of the study, Eisai aims to file for traditional approval in the US and for marketing authorization applications in Japan and Europe by the end of Eisai’s 2022 fiscal year, which is March 31, 2023.91
Trastuzumab duocarmazine (Byondis, medac GmbH)
[vic-]trastuzumab duocarmazine (SYD985) is an ADC composed of the IgG1ҡ anti- human epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab and the cleavable linker-drug called valine-citrulline-seco-DUocarmycin-hydroxyBenzamide-Azaindole (vc-seco-DUBA). This ADC has been designed to bind HER2-expressing cancer cells. After internalization, the linker is cleaved by proteolysis and the free drug is activated to become a minor groove DNA-damaging cytotoxin. The selectively cleavable linker connecting the antibody to the payload confers high stability in circulation and efficient release of the cytotoxin in the tumor. FDA granted the therapy Fast Track designation based on data from a Phase 1 study that included heavily pre-treated last-line HER2-positive metastatic breast cancer patients. In May 2022 Byondis and medac GmbH agreed to partner to commercialize trastuzumab duocarmazine in Europe.92 In July 2022, Byondis announced that FDA and EMA accepted the company’s BLA and MAA submissions, respectively, for [vic-]trastuzumab duocarmazine in patients with HER2-positive unresectable locally advanced or metastatic breast cancer.93,94 FDA’s target first action date for the BLA is May 12, 2023.93
The marketing applications are supported by data from the pivotal Phase 3 TULIP clinical trial (NCT03262935), a multi-center, open-label, randomized study comparing [vic-]trastuzumab duocarmazine (administered via IV injection every 3 weeks) to physician’s choice (PC) of treatment in patients with pre-treated HER2-positive unresectable locally advanced or metastatic breast cancer. Patients (n = 437) were randomly assigned 2:1 between [vic-]trastuzumab duocarmazine (1.2 mg/kg every 3 weeks) and PC therapy. The study met its primary endpoint of PFS, demonstrating a statistically significant improvement of 2.1 months in [vic-]trastuzumab duocarmazine treated patients over those who received PC therapy. TULIP also demonstrated supportive OS results.91,92
Mirikizumab (Eli Lilly and Company)
Mirikizumab (LY3074828), developed by Eli Lilly and Company, is a humanized IgG4ҡ monoclonal antibody that blocks the activity of interleukin 23 by targeting the p19 subunit the cytokine. The antibody is engineered with the following mutations: S228P for hinge stabilization, F234A and L235A to abrogate effector function, and K447> del to reduce IgG4 C-terminal heterogeneity. Mirikizumab is being investigated for the treatment of immune-mediated diseases, including ulcerative colitis and Crohn’s disease. In the first quarter of 2022, Lilly submitted marketing applications in the US, EU, and Japan for the approval of mirikizumab for ulcerative colitis, and regulatory decisions are expected in 2023. If approved, mirikizumab would become the first and only anti-IL23p19 treatment for people with ulcerative colitis.
Mirikizumab is being evaluated for the treatment of ulcerative colitis in the LUCENT Phase 3 clinical program, which includes LUCENT-1 (NCT03518086), LUCENT-2 (NCT03524092), and LUCENT-3 (NCT03519945). LUCENT-1 is a multicenter, randomized, double-blind, placebo-controlled induction study in over 1,000 patients with moderately-to-severely active ulcerative colitis who have previously failed conventional and/or biologic therapies and/or JAK inhibitors. Patients enrolled in the trial receive IV infusions of placebo or 300 mg mirikizumab every 4 weeks on Weeks 0, 4, 8 for 12 weeks. The primary endpoint is clinical remission, achieved when inflammation of the colon is controlled or resolved, leading to normalization or near-normalization of symptoms. Positive results from the LUCENT-1 study, presented at the 17th Congress of the European Crohn’s and Colitis Organization (ECCO) in February 2022, show that patients with moderately-to-severely active ulcerative colitis who took mirikizumab achieved significant superior rates of clinical remission at 12 weeks compared to patients taking placebo (45.5%, n = 395/868 vs. 27.9%, n = 82/294, respectively; p < .001). Patients who received mirikizumab also achieved statistically significant improvements across key secondary endpoints compared to those taking placebo.95
LUCENT-2 (NCT03524092) is a 3-arm, randomized, double-blind, placebo-controlled maintenance study in patients who completed the 12-week induction study (LUCENT-1). In the LUCENT-2 study, patients who achieved clinical response with mirikizumab induction therapy in LUCENT-1 were re-randomized to receive SC administration of mirikizumab at one of 2 dose levels or placebo for an additional 40 weeks. Positive results from the pivotal Phase 3 LUCENT-2 study demonstrate that patients with ulcerative colitis who responded to mirikizumab at 12 weeks achieve and maintain statistically superior and clinically meaningful improvements at 1 year compared to placebo in terms of the clinical remission primary endpoint (49.9%, n = 182/365 vs. 25.1%, n = 45/179, respectively; p < .001) and all key secondary endpoints.96
LUCENT-3 (NCT03519945) is a single-arm, open label extension study to investigate the long-term efficacy and safety of mirikizumab administered SC in patients with moderately to severely active ulcerative colitis who participated in mirikizumab ulcerative colitis trials. The primary outcome measure is the percentage of participants in clinical remission in a time frame of 52 weeks. The study is recruiting an estimated 960 patients and has an estimated primary completion date in June 2025.
Lilly is also evaluating mirikizumab for Crohn’s disease in two large Phase 3 trials. The randomized, placebo- and active-controlled VIVID-1 (NCT03926130) is evaluating of the safety and efficacy of mirikizumab given IV and SC vs. either ustekinumab or placebo in patients with moderately to severely active Crohn’s disease. The VIVID-1 study is recruiting an estimated 1100 patients and has an estimated primary completion date in August 2023. The open-label VIVID-2 (NCT04232553) study will evaluate the long-term efficacy and safety of mirikizumab given IV and SC in an estimated 778 Crohn’s disease patients. The VIVID-2 has an estimated primary completion date in January 2025.
Glofitamab (Hoffmann-La Roche)
Glofitamab (RO7082859, CD20-TCB, RG6026) is a full-length IgG1λ/ҡ bispecific T cell redirecting antibody targeting CD20 on malignant B cells and CD3 on T cells. This bispecific antibody was developed by Roche using the 2:1 CrossMab technology, characterized by 3 antigen-binding fragment (Fab) arms enabling monovalent binding to CD3ɛ and bivalent binding to CD20, with the second CD20 arm fused to the CD3ɛ-binding arms via a flexible linker. Glofitamab also features a heterodimeric Fc region engineered with PG LALA mutations to abolish binding to FcɣRs and C1q. An MAA containing data from the Phase 1/2 NP30179 study (NCT03075696) evaluating glofitamab for NHL was submitted to the EMA, and submissions to additional health authorities in 2022 are planned.97
New pivotal results of the NP30179 study were presented at the 2022 American Society of Clinical Oncology Annual Meeting and at the European Hematology Association Congress held in June 2022. The NP30179 study is a multicenter, open-label, dose-escalation study evaluating the efficacy, safety, tolerability, and PK of glofitamab as a single agent and in combination with obinutuzumab (Gazyva®/Gazyvaro™). Glofitamab is administered by IV infusion as a single agent and in combination with obinutuzumab (up to 25 mg), following pretreatment with a single dose of obinutuzumab (1 g), in patients with relapsed/refractory B-cell NHL who had received a median of three prior therapies. After a median follow-up of 12.6 months, 39.4% of patients achieved a complete response (primary efficacy endpoint) and half of them achieved an overall response (the percentage of patients with a partial or complete response; secondary efficacy endpoint), as assessed by an independent review committee. The majority (77.6%) of complete responses were durable and ongoing at 12 months and the median duration of complete response had not yet been reached (not evaluable). The most common adverse event was cytokine release syndrome occurring in 63.0% of patients.97
Glofitamab is under investigation in a randomized, open-label, multicenter Phase 3 study (STARGLO, NCT04408638) where patients with relapsed or refractory DLBCL receive glofitamab or rituximab in combination with gemcitabine + oxaliplatin (GemOx). Patients will receive up to 8 cycles of glofitamab IV or rituximab IV in combination with GemOx IV followed by up to 4 cycles of glofitamab monotherapy. The primary outcome measure is OS. The estimated primary completion date is November 2022.
Penpulimab (Akesobio, Chia Tai-Tianqin Pharmaceutical Group Co, Ltd.)
Penpulimab (AK105) is a humanized, Fc-engineered (L234A L235A G237A mutations), anti-PD-1 IgG1κ antibody with reduced effector functions, which may contribute to a better safety profile. Penpulimab has demonstrated a slower antigen binding off rate compared with current marketed PD-1 antibodies that resulting in higher receptor occupancy that may improve antitumor activity.98 Akesobio and Chia Tai-Tianqin Pharmaceutical Group Co, Ltd, a subsidiary of Sino Biopharmaceutical Limited, are developing penpulimab as a treatment for various tumor types. In 2021, the NMPA granted penpulimab its first approval, for treatment of patients with relapsed or refractory classic Hodgkin’s lymphoma after at least second-line systemic chemotherapy treatment in China.
Penpulimab was granted Breakthrough Therapy, Orphan Drug and Fast Track designations by the FDA for the treatment of metastatic NPC. In May 2021, Akeso, Inc. and Sino Biopharmaceutical Limited jointly announced that a BLA for penpulimab was submitted to the FDA for third-line treatment of mNPC, and that the FDA will review the BLA under the new policy of Real-Time Oncology Review, which aims to accelerate the process of drug approval. The companies, however, have not provided further updates regarding this BLA.
Inolimomab (ElsaLys Biotech)
Inolimomab (Leukotac) is a murine IgG1κ anti-IL2 receptor alpha (CD25) antibody originally developed by Jazz Pharmaceuticals. ElsaLys Biotech licensed the worldwide rights to Leukotac from Jazz Pharmaceuticals in 2017. Mediolanum Farmaceutici Spa acquired ElsaLys Biotech in 2020. Inolimomab has been granted orphan designation by the EMA and the FDA for the treatment of graft versus host disease (GvHD).
In July 2020, ElsaLys Biotech announced that FDA had accepted a BLA under the Real time Oncology Review program for inolimomab as a potential treatment for adult patients with steroid-refractory acute GvHD based on data from Phase 3 study INO107 (EUDRACT 2007–005009-24) that compared inolimomab vs antithymocyte globulin. Since then, the company has provided no further information. The status of the BLA and any of its modules is thus unknown. Inolimomab is available in France under a cohort Temporary Authorization for Use in adult and pediatric patients with steroid-refractory or steroid-dependent acute GvHD.
Donanemab (Eli Lilly and Company)
Donanemab (LY3002813) is a humanized IgG1k monoclonal antibody that targets a form of Aβ, N3pG, found in cerebral amyloid plaques. The FDA granted Breakthrough Therapy designation for donanemab for treatment of AD. By the end of the second quarter of 2022, Lilly’s BLA for an accelerated approval of donanemab for early AD was accepted by FDA and granted priority review,99 and a decision is anticipated in early 2023.
The BLA includes data from the randomized, placebo-controlled Phase 2 TRAILBLAZER-ALZ trial (NCT03367403) of donanemab as monotherapy or in combination with LY3202626 vs. placebo in 272 patients with early symptomatic AD. In this study, patients were randomized to receive donanemab 700 mg IV every 4 weeks for the first 3 doses, then 1400 mg donanemab IV every 4 weeks for up to 72 weeks; donanemab 700 mg IV every 4 weeks for the first 3 doses, then 1400 mg donanemab IV every 4 weeks in combination with 12 mg of LY3202626 orally for up to 72 weeks; or IV placebo. The study outcomes and measures included the change in amyloid, tau, and clinical decline after donanemab treatment. The change from baseline to 76 weeks in the integrated AD rating scale (iADRS) score as a primary objective was achieved, with a 32% difference in slowing decline for the donanemab group.100 A post-hoc analysis that included use of a disease-progression model found donanemab slowed tau accumulation in a region-dependent manner and there was a significant association between percentage amyloid reduction and change on the iADRS only in apolipoprotein E (APOE) ε4 carriers (95% CI, 24%–59%; P < .001).101
Data from the Phase 3 TRAILBLAZER-ALZ4 (NCT05108922) and TRAILBLAZER-ALZ2 (NCT04437511) studies of donanemab are expected by the end of 2022 and mid-2023, respectively. The TRAILBLAZER-ALZ4 study is evaluating the effects of donanemab compared to aducanumab (Aduhelm) in 200 participants with early symptomatic AD. The TRAILBLAZER-ALZ2 study is assessing the safety, tolerability, and efficacy of donanemab in an estimated 1800 patients with early symptomatic AD.
Lebrikizumab (Almirall S.A., Eli Lilly and Company)
Lebrikizumab (TNX-650, PRO301444, MILR1444A, RG3637) is a humanized anti-IL-13 IgG4κ antibody with a hinge region that is stabilized via an S228P mutation. IL-13 is a key mediator of the proinflammatory processes in atopic dermatitis and potent enhancer of neuronal responses to the persistent itch stimuli that characterize the disease. Originally developed by F. Hoffmann-La Roche Ltd and Genentech, Inc., a member of the Roche Group, in 2017 Dermira obtained exclusive, worldwide rights to develop and commercialize lebrikizumab for atopic dermatitis and all other indications, except Roche retained certain rights, including exclusive rights to develop and promote lebrikizumab for interstitial lung diseases. In 2019, Almirall acquired and exercised an option from Dermira to exclusively license rights to develop and commercialize lebrikizumab for the treatment of atopic dermatitis and certain other indications in Europe. In 2020, Dermira was acquired by Eli Lilly and Company. Almirall announced in October 2022 that EMA accepted the filing of the MAA for lebrikizumab in atopic dermatitis and approval in Europe is expected in the second half of 2023.102
Lebrikizumab is being evaluated in a comprehensive clinical development program in atopic dermatitis that includes at least 10 Phase 3 studies and more than 2,000 patients in total. The ongoing Phase 3 ADjoin study (NCT04392154), which is the largest, will evaluate the long-term effects of the drug in an estimated 1000 patients who completed participation in a Dermira- or Lilly-sponsored lebrikizumab study (parent study), DRM06-AD04, DRM06-AD05, DRM06-AD06, DRM06-AD17, or DRM06- AD18.
Almirall recently announced results from two randomized, 52-week, double-blind, placebo-controlled Phase 3 studies, ADvocate 1 (NCT04146363), and ADvocate 2 (NCT04178967) evaluated lebrikizumab as monotherapy in adult and adolescent patients (aged 12 to less than 18 y of age and weighing at least 40 kg) with moderate-to-severe atopic dermatitis.103 During the 16-week treatment period, patients received SC lebrikizumab 500-mg initially and at 2 weeks, followed by lebrikizumab 250-mg or placebo every 2 weeks. In the maintenance period, patients with moderate-to-severe atopic dermatitis who achieved a clinical response after 16 weeks of lebrikizumab treatment were re-randomized to receive lebrikizumab every 2 weeks or every 4 weeks or placebo for an additional 36 weeks. Patients who required rescue treatment during the induction period or who did not achieve clinical response at 16 weeks received lebrikizumab every 2 weeks for an additional 36 weeks. The primary endpoints were measured by an Investigator Global Assessment (IGA) score of clear or almost clear skin with a reduction of at least two points from baseline and at least 75% change in baseline in the Eczema Area and Severity Index (EASI-75) score at 16 weeks. Results from the Advocate 1 (n = 424 patients) and Advocate 2 (n = 445 patients) studies showed 79% and 85% of patients who received lebrikizumab every 4 weeks, and 79% and 77% of patients who received lebrikizumab every 2 weeks, maintained 75% or greater skin improvement at 1 year of treatment, respectively.103
Epcoritamab (Genmab, AbbVie)
Epcoritamab (GEN3013, DuoBody®-CD3xCD20), a T cell-engaging bispecific IgG1κ/λ antibody targeting CD20 and CD3, created using Genmab’s DuoBody® technology, is jointly owned by Genmab and AbbVie. It was designed to bind to CD3 on T cells and CD20 on B cells to induce T cell-mediated killing of malignant B cells. In November 2022, Genmab announced that the FDA accepted for Priority Review the BLA for subcutaneous epcoritamab for the treatment of patients with relapsed/refractory large B-cell lymphoma (LBCL) after two or more lines of systemic therapy, and set a target action date of May 21, 2023.104
In April 2022, Genmab and AbbVie presented top-line data from the first cohort of the Phase 1/2 EPCORE NHL-1 trial (NCT03625037) studying epcoritamab in 157 patients with relapsed or refractory large B-cell lymphoma who had received at least two prior systemic therapies, including some who had received prior treatments with CAR-T cell therapy. The dose escalation findings from the Phase 1 part identified a dose of 48 mg as the recommended Phase 2 dose.105 Thus, in the Phase 2 trial, patients received 48 mg of epcoritamab as 1 ml SC injections in 28-day cycles, with weekly dosing in Cycles 1–2, dosing every second week in Cycles 3–6, and dosing every 4 weeks from Cycle 7 onward. An independent review committee confirmed an overall response rate of 63.1%, exceeding the prespecified threshold for efficacy. Patients naïve to CAR-T cell therapy achieved a 69% overall response rate, while patients who received prior CAR-T cell therapy achieved a 54% overall response rate. After a median follow-up of 10.7 months, the median DOR was estimated to be 12 months, while the median DOR among patients achieving a complete response was not reached.106
Epcoritamab is also being investigated in multiple ongoing clinical studies across different settings and histologies. The most advanced of these is the randomized, open-label Phase 3 EPCORE™DLBCL-1 trial (NCT04628494) of epcoritamab vs investigator’s choice chemotherapy in patients with relapsed or refractory DLBCL. The study is recruiting an estimated 480 patients and has an estimated primary completion date in June 2023.
Tislelizumab (BeiGene, Novartis Pharmaceuticals Corporation)
Tislelizumab (BGB-A317) is an anti-PD-1 hinge-stabilized, humanized IgG4κ monoclonal antibody Fc-engineered (E233P/F234V/L235A/D265A) to reduce Fc receptor binding on macrophages, which has been shown to compromise the anti-tumor activity of PD-1 antibodies. Developed by BeiGene, tislelizumab is currently approved in China as a treatment for nine indications. Novartis acquired the rights to develop, manufacture, and commercialize tislelizumab in North America, Europe, and Japan through a license and collaboration agreement with BeiGene. Novartis is evaluating tislelizumab in 14 pivotal clinical trials in a broad array of solid tumors, with more than 8800 patients enrolled to date in 35 countries.
The NMPA is evaluating a regulatory submission for tislelizumab in combination with chemotherapy as first-line treatment in patients with unresectable, locally advanced, recurrent, or metastatic ESCC. The application is based on data from an interim analysis of the global Phase 3 RATIONALE 306 trial (NCT03783442), in which 649 patients with advanced or metastatic ESCC were randomized to receive 200 mg tislelizumab every 3 weeks or matching placebo plus chemotherapy. An improvement of median OS from 10.6 months with chemotherapy alone to 17.2 months with the addition of tislelizumab was observed, and an OS benefit was observed regardless of baseline PD-L1 expression.107
In September 2021, the FDA accepted a BLA for tislelizumab for second-line treatment of unresectable advanced or metastatic ESCC, but in July 2022 BeiGene announced that the FDA has deferred action on the application due to the inability to complete site inspections because of COVID-19 travel restrictions in China.108
In April 2022, Novartis announced that the EMA validated MAAs for tislelizumab for two indications: 1) monotherapy for unresectable, recurrent, locally advanced, metastatic ESCC after prior chemotherapy, and 2) first-line treatment as a monotherapy or in combination with chemotherapy for locally advanced or metastatic squamous or non-squamous NSCLC.109 The MAAs, which included data from the Phase 3 RATIONALE 302 trial in ESCC, and the Phase 3 RATIONALE 303, RATIONALE 304 and 307 trials in NSCLC, remain in review by the EMA as of November 2022.
RATIONALE 302 (NCT03430843) is an open-label Phase 3 study involving 512 patients with advanced unresectable or metastatic ESCC who had received prior systemic therapy. Patients were randomized 1:1 to receive 200 mg tislelizumab IV every 3 weeks or investigator-chosen chemotherapy (paclitaxel, docetaxel, or irinotecan). In this study, OS was significantly longer with tislelizumab versus chemotherapy in all patients (median, 8.6 v 6.3 months; hazard ratio [HR], 0.70 [95% CI, 0.57 to 0.85]; one-sided P = .0001), and in patients with tumor area positivity scores ≥ 10% (median, 10.3 months v 6.8 months; HR, 0.54 [95% CI, 0.36 to 0.79]; one-sided P = .0006). Treatment with tislelizumab was associated with a greater overall response rate (20.3% vs 9.8%), a higher median DOR (7.1 months vs 4.0 months), and a favorable safety profile compared with chemotherapy.110
The open-label randomized RATIONALE 303 (NCT03358875) study compared treatment with 200 mg IV tislelizumab every 3 weeks to docetaxel in 805 patients with locally advanced or metastatic NSCLC who had progressed on a prior platinum-containing treatment. OS was increased by 6 months, from 11.9 months (95% CI: 10.18–13.93 months) for patients receiving docetaxel treatment to 17.2 months (95% CI: 15.28–20.04 months) for patients in the tislelizumab treatment arm.111
RATIONALE 304 (NCT03663205) is an open-label, multicenter, randomized Phase 3 study of tislelizumab plus chemotherapy versus chemotherapy alone in 332 patients with untreated advanced non-squamous NSCLC who were randomized 1:1 to receive either 200 mg tislelizumab plus chemotherapy or chemotherapy. Patients treated with tislelizumab plus chemotherapy had a significantly longer PFS compared to chemotherapy alone (median PFS: 9.7 months vs 7.6 months; hazard ratio 0.645, 95% CI: 0.462–0.902).112
The open-label Phase 3 trial RATIONALE 307 (NCT03594747) of tislelizumab plus chemotherapy versus chemotherapy randomized 355 patients with untreated advanced squamous NSCLC to receive 200 mg tislelizumab plus paclitaxel and carboplatin, or 200 mg tislelizumab plus nab-paclitaxel and carboplatin, or paclitaxel and carboplatin on a 21-day cycle. After a median study follow-up of 8.6 months, PFS, objective response rate and DOR were improved by the addition of tislelizumab to both chemotherapy regimens, compared to chemotherapy alone.113
Retifanlimab (Incyte Corporation, Macrogenics, Zai Lab)
Retifanlimab (INCMGA00012, MGA012, ZL-1306) is a humanized, hinge-stabilized IgG4κ anti-PD-1 antibody in development as a treatment for various tumor types, including squamous cell carcinoma of the anal canal (SCAC), NSCLC, and gastric cancer. Incyte obtained the exclusive worldwide rights for the development and commercialization of retifanlimab from MacroGenics in 2017, although MacroGenics retains rights to develop pipeline assets in combination with retifanlimab. In July 2019, Zai Lab obtained the rights to develop and exclusively commercialize retifanlimab in hematology and oncology in Greater China. The FDA granted Fast Track and Orphan Drug designations for retifanlimab for the treatment of anal cancer.
In January 2021, Incyte submitted a BLA for accelerated approval of retifanlimab for the treatment of locally advanced or metastatic SCAC who have progressed on, or who are intolerant of, platinum-based chemotherapy. The BLA was based on data from a single-arm Phase 2 POD1IUM-202 trial (NCT03597295) that enrolled 94 patients with locally advanced or metastatic SCAC. In this study, the objective response rate of 13.8% (95% CI, 7.6–22.5), with the best overall response being a complete response in 1.1% of the patient population. In June 2021, the BLA was discussed at a meeting of FDA’s Oncologic Drugs Advisory Committee,114 and in July 2021, the FDA issued a complete response letter stating that the FDA cannot approve the application in its present form, and that additional data are needed to demonstrate the clinical benefit of retifanlimab for the treatment of patients with advanced or metastatic SCAC.115
Retifanlimab is currently being investigated in the randomized, double-blind Phase 3 POD1UM-303/InterAACT 2 trial (NCT04472429) in combination with carboplatin-paclitaxel chemotherapy in patients with inoperable locally recurrent or metastatic SCAC. Retifanlimab (500 mg) or placebo will be IV administered on Day 1 of each 28-day cycle. Patients in both arms of the study will receive up to six induction cycles (24 weeks) of carboplatin (area-under-the-curve 5 on Day 1) and paclitaxel (80 mg/m2 on Days 1, 8, and 15) every 28 d.116 The estimated enrollment is 300 patients and the estimated study primary completion date is in October 2024.
Retifanlimab is also being evaluated for gastric cancer or GEJ cancer and NSCLC in late-stage clinical studies sponsored by Macrogenics/Zai Lab and Incyte/Zai Lab, respectively. The Phase 2/3 MAHOGANY study (NCT04082364) is evaluating margetuximab in combination with retifanlimab and chemotherapy or tebotelimab (MGD013) and chemotherapy in 82 patients with metastatic or locally advanced, treatment-naïve, HER2-positive gastric or GEJ cancer. Initiated in September 2019, the study has an estimated primary completion date in December 2023. The randomized, double-blind Phase 3 study POD1UM-304 study (NCT04205812) will investigate the efficacy and safety of retifanlimab in combination with platinum-based chemotherapy for first-line treatment of metastatic NSCLC. Approximately 530 patients will be randomized 2:1 to receive platinum-based chemotherapy plus 375 mg retifanlimab or placebo every 3 weeks. This study has an estimated primary completion date in August 2025.
Narsoplimab (Omeros Corporation)
Narsoplimab (OMS721) is a hinge-stabilized (S228P mutation) human IgG4λ monoclonal antibody targeting mannan-binding lectin-associated serine protease-2 (MASP-2) being developed by Omeros Corporation for hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA). Narsoplimab was granted Breakthrough Therapy and Orphan drug designations by the FDA for HSCT-TMA and IgA nephropathy. The FDA also granted Fast track designation to narsoplimab for the treatment of patients with atypical hemolytic uremic syndrome.
In January 2021, the FDA accepted and granted priority review to a BLA for narsoplimab for the treatment of HSCT-TMA. The BLA was based on data from a single-arm, open-label pivotal Phase 2 trial (NCT02222545) in which patients treated with at least one dose (n = 28) of narsoplimab has a response rate of 61%.117 In October 2021, the company announced that it had received a complete response letter from the FDA that requested additional information to estimate narsoplimab’s effects on patients with HSCT-TMA to support regulatory approval. In June 2022, Omeros requested Formal Dispute Resolution regarding the complete response letter, which was denied. According to the company, the FDA’s decision proposes a path forward for the resubmission of the BLA based on survival data from the completed pivotal trial versus an historical control group.118
Sintilimab (Innovent Biologics, Eli Lilly and Company)
Sintilimab (marketed as TYVYT® in China) is an anti-PD-1 monoclonal IgG4κ antibody jointly developed by Innovent and Eli Lilly. In China, sintilimab has been approved in six indications with the first four indications included in the National Reimbursement Drug List. The six approvals are for: 1) treatment of relapsed or refractory classic Hodgkin’s lymphoma after two lines or later of systemic chemotherapy (approved in December 2018); 2) first-line treatment of unresectable locally advanced or metastatic nonsquamous NSCLC lacking EGFR or ALK driver gene mutations in combination with pemetrexed and platinum chemotherapy (approved in Feb 2021); 3) first-line treatment of unresectable locally advanced or metastatic squamous NSCLC in combination with gemcitabine and platinum chemotherapy (approved in June 2021); 4) first-line treatment of unresectable locally advanced or metastatic HCC in combination with a bevacizumab biosimilar (approved in June 2021); 5) first-line treatment of unresectable locally advanced, recurrent or metastatic ESCC in combination with cisplatin plus paclitaxel or cisplatin plus 5-fluorouracil (approved in June 2022); and 6) first-line treatment of unresectable locally advanced, recurrent, or metastatic gastric or GEJ adenocarcinoma in combination with fluorouracil and platinum-based chemotherapy (approved in June 2022). Innovent also currently has a regulatory submission under review in China for sintilimab for the treatment of EGFR-mutated nonsquamous NSCLC following EGFR-TKI treatment in combination with a bevacizumab biosimilar and pemetrexed and cisplatin chemotherapy.
A BLA for sintilimab injection in combination with pemetrexed and platinum chemotherapy for the first-line treatment of people with nonsquamous NSCLC was submitted in March 2021 by Innovent Biologics, Inc. and their partner Eli Lilly and Company, and accepted for review by FDA in May 2021. The BLA was primarily based on the results of the Phase 3 ORIENT-11 trial (NCT03607539), in which a total of 397 patients were enrolled and randomized 2:1 to receive either sintilimab 200 mg or placebo in combination with pemetrexed and platinum chemotherapy every three weeks for up to four cycles, followed by either sintilimab or placebo plus pemetrexed maintenance therapy. In the ORIENT-11 trial, sintilimab in combination with pemetrexed and platinum-based chemotherapy demonstrated clinically meaningful treatment effect across all endpoints. The study, however, was intended to support registration in China, and thus the companies were relying on foreign data as the sole basis for marketing approval in the US.119 The FDA’s Oncologic Drugs Advisory Committee met to discuss the BLA in February 2022. In March 2022, the FDA issued a complete response letter for the BLA.120 The letter indicates that the FDA is unable to approve the application in its current form, and recommends an additional clinical study, specifically a multiregional clinical trial comparing standard of care therapy for first-line metastatic NSCLC to sintilimab with chemotherapy.
131I-Omburtamab (Y-mAbs Therapeutics, Inc.)
131I-Omburtamab (Omblastys®) is a monoclonal radioimmunoconjugate targeting B7-H3, which is expressed in several tumor types. Developed at Memorial Sloan Kettering Cancer Center and exclusively licensed to Y-mAbs therapeutics, the 131I-labeled, murine IgG1κ antibody is being developed as a treatment for pediatric patients with central nervous system (CNS)/leptomeningeal metastases from neuroblastoma. 131I-omburtamab was granted Breakthrough Therapy designation by FDA for this indication.
A rolling BLA seeking FDA’s approval of 131I-omburtamab for the treatment of pediatric patients with CNS/leptomeningeal metastases from neuroblastoma was complete by August 2020, but the FDA issued a refusal to file letter in October 2020 because the Chemistry, Manufacturing, and Control and Clinical modules required additional detail. The BLA was resubmitted to the FDA in April 2022 and subsequently accepted for priority review. In October 2022, FDA’s Oncologic Drugs Advisory Committee voted 16 to 0 that Y-mAbs had not provided sufficient evidence to conclude that 131I-omburtamab improves OS. The FDA’s target date for a first action on the application is November 30, 2022.
The BLA included data from the pivotal 101 (NCT03275402) and 03–133 (NCT00089245) studies. Interim results for 32 patients enrolled in the 101 multicenter study of 131I-omburtamab were presented at the International Society of Pediatric Oncology Annual Congress held September 28 through October 1, 2022, in Barcelona, Spain. Patients received up to two cycles of intracerebroventricular 131I-omburtamab, with one treatment cycle consisting of 1 dose at 50 millicurie (mCi) at Week 1. For Japan only one treatment cycle of 131I-omburtamab consists of 2 doses: 2 mCi at Week 1 and 50 mCi at Week 2. With a median follow-up of 25 months, the 12-month OS was 73.5% and the objective response rate in patients with measurable disease after central review based on Response Assessment in Neuro-Oncology criteria and European Association of Neuro-Oncology/European Society for Medical Oncology criteria was 31.3%. A total of 75.0% of patients with measurable disease achieved disease control.121
Y-mAbs therapeutics is currently also evaluating omburtamab for patients with desmoplastic small round cell tumors and peritoneal cancers in Phase 2 (NCT04022213), recurrent medulloblastoma, recurrent ependymoma (NCT04743661).
Late-stage commercial clinical pipeline
Since the first Antibodies to Watch article13 was published in 2010, the number of antibody therapeutics in late-stage clinical studies has dramatically increased, from 26 included in the first article to nearly 140 included here (Figure 1; Supplemental Tables S1 and S2). The increase in those for non-cancer indications (Figure 1a; Supplemental Table S1) outpaced those for cancer indications (Figure 1b; Supplemental Table S2) during 2013–2017. This trend reversed in 2018, although the difference in the annual numbers for each group is not substantial. A key difference between the non-cancer and cancer groups is the application of novel formats that enhance the biological activity of the antibody. In Figure 1, each molecule was included in only one format category, which was selected based in how greatly the modification altered molecular properties. Using this stratification method, antibodies linked to a small-molecule drug would be categorized as ADCs, and excluded from the full length or antibody fragment categories. Since 2010, the use of enhanced antibodies, such as ADCs and bispecifics, increased in both groups, but the increase has been greater among the antibodies for cancer. Since 2017, approximately half of the antibodies for cancer in late-stage clinical studies in any given year were not canonical full-length molecules, whereas most antibodies for non-cancer indications were full-length molecules in each of the years (range 83% to 100%).
Figure 1.
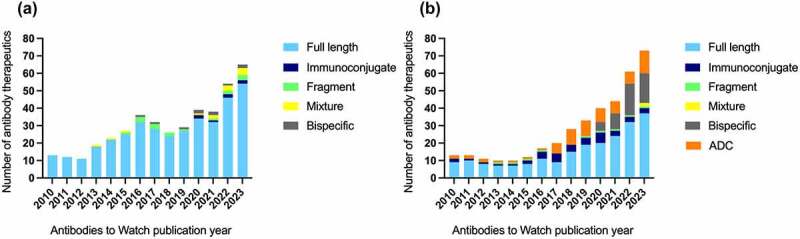
Format and number of antibody therapeutics in late-stage clinical studies from 2010–2023*. (a) Format and number of antibodies in late-stage clinical studies for diseases other than cancer (i.e., all non-cancer indications). Antibodies that entered late-stage studies for COVID-19 during 2020–2022 were excluded to enable accurate comparisons to data from previous years. (b) Format and number of antibodies in late-stage clinical studies for cancer. As defined here, ADCs are antibody-derived proteins conjugated to a small molecule drug through a linker, while immunoconjugates are antibody-derived proteins fused or conjugated to any other biologically relevant modality, e.g., protein, radioisotope. Data were derived from the ‘Antibodies to Watch’ articles;1–13 data for each article are collected at the end of the previous year. *Data for 2023 is as of October 1, 2022. Abbreviations: ADC, antibody–drug conjugate.
We further evaluated the formats and engineering methods applied to the design of the antibody therapeutics in the current late-stage pipeline to illustrate their diversity (Figure 2). The majority of antibodies in late-stage clinical trials for both non-cancer and cancer indications are based on a full-length IgG1 backbone, while approximately a third of antibodies in both indications are formatted onto the less immuno-active subclasses, IgG2 and IgG4 (Figure 2a, b). As described below, however, the IgG1 Fc can be engineered to reduce effector functions. While the details of all formats have not yet been divulged, there are at least 8 antibody fragments in late-stage clinical trials, 3 for non-cancer indications (Figure 2a) and 5 for cancer indications (Figure 2b), which is a broad category including scFv and derivatives, TandAb, VHH homodimers, VH-VH’, and Fabs. Antibody fragments may have beneficial properties over full-length IgGs, such as better tissue penetration, access to difficult to reach targets, and minimal undesirable immune activation due to the lack of Fc domain. However, one of the main drawbacks of antibody fragments is the greatly reduced half-life, which has led to engineering approaches to improve this feature while retaining the beneficial properties of their small size.
Figure 2.
Antibody format and engineering strategies in late-stage clinical studies*. (a) Formats for antibodies in late-stage clinical studies for diseases other than cancer (i.e., all non-cancer indications), and (b) cancer indications. (c) Engineering of antibodies in late-stage clinical studies for diseases other than cancer (i.e., all non-cancer indications), and (d) cancer indications. As defined here, ADCs are antibody-derived proteins conjugated to a small molecule drug through a linker, while immunoconjugates are antibody-derived proteins fused or conjugated to any other biologically relevant modality, e.g., protein, radioisotope. Antibodies were included in more than one category if more than one engineering strategy was used in the design of the molecule. *Data as of October 1, 2022. Antibody engineering and format data are included in Supplemental Tables S1 and S2. Abbreviations: Ab, antibody.
When all engineering approaches for each molecule are included, our data show that over two-thirds have been engineered to improve their therapeutic effect in some way (Figure 2c, d). Engineering the Fc domain to increase binding to FcRn, and therefore increase serum half-life, is used more in non-cancer indications, especially in antibodies against infectious diseases caused by agents such as SARS-CoV-2, Staphylococcus aureus, and RSV (Figure 2c). Engineering that increases affinity for Fc receptors, such as improving ADCC via afucosylation, is commonly used in cancer indications (Figure 2d), but some antibodies engineered for increased affinity for Fc receptors for a different purpose have been developed for non-cancer indications. An example of this approach involves “sweeping” or potent reduction of antigen via enhanced FcγRIIb and FcRn binding, or increased neutralization of lipopolysaccharide signaling via toll-like receptor-4 by enhanced FcγRIIa binding. ADCs and immunoconjugates use the specificity of the antibodies to deliver another functional moiety, such as toxic drugs, cytokines, or cytokine-binding domains directly to the target. This form of engineering is much more common for cancer indications compared to non-cancer indications. Reduction of immune effector functions by the introduction of mutations into the Fc domain is being used both for cancer indications, such as in checkpoint inhibitors, and for non-cancer indications when blocking ligand–receptor interactions for which an immune response is unwanted or detrimental.122
We also assessed the main mechanisms of action (MOA) for the antibody therapeutics in late-stage studies (Figure 3), which are diverse as a consequence of their molecular design. The data are presented as Euler diagrams to illustrate overlaps, indicating antibodies with more than one mechanism. Most of the antibodies for non-cancer indication are blocking antibodies, with 11 and 4 functioning via antigen clearance and cell depletion properties, respectively (Figure 3a). The single agonist antibody is NNC0365-3769 (Mim8), a bispecific antibody targeting FIXa and FX for the treatment of hemophilia. Four antibodies bentracimab, ligelizumab, birtamimab, and E2814, which target tricagrelor, IgE, amyloid, and Tau, respectively, have both blocking and antigen clearance properties. Two antibodies, rocatinlimab, and inalumab targeting OX40 and BLyS/BAFF/TACI/BCMA receptor, respectively, have both blocking and cell depletion properties.
Figure 3.

Mechanisms of action for antibody therapeutics in late-stage clinical studies*. Area proportional Euler diagrams representing the main mechanisms of action of antibodies in late-stage clinical studies. (a) Antibodies for non-cancer indications. (b) Antibodies for cancer indications. *Data as of October 1, 2022; n = 65 antibodies for non-cancer indications, n = 73 antibodies for cancer indications. Abbreviations: CDC, complement-dependent cytotoxicity; ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody dependent cell phagocytosis. Figure created using euler: Area-Proportional Euler and Venn Diagrams with Ellipses, R package version 6.1.1, available at https://eulerr.co/.
The MOA diversity is greater among the antibodies for cancer indications (Figure 3b). Antibodies that block a target also comprise the majority, but 16 are designed for payload delivery, 11 act via antigen clearance, 7 are cell engagers, and 7 act via complement-dependent cytotoxicity (CDC), ADCC, and antibody-dependent cell phagocytosis (ADCP). Of the blocking antibodies, 31 are immunomodulatory, 9 target the tumor and 3 target the tumor microenvironment. Only two antibodies are agonistic, INBRX-109, and ivuxolimab targeting DR5 and OX40, respectively. Of the antibodies acting via cytotoxic or radioactive payload delivery, 13 are ADCs, of which 6 are tubulin inhibitors, 4 are DNA binding agents, and 3 are topoisomerase 1 inhibitors. Only 2 are radio-immunoconjugates and 1 is an immunocytokine. Of the seven immune cell engager antibodies, six are designed to recruit T cells via CD3 and 1, AFM13, is designed to engage natural killer (NK) cells via CD16a. Apart from tarlatamab, which redirects T cells toward a solid tumor via Delta-like ligand 3 (DLL3), these immune cell engagers are designed to redirect T cells or NK cells toward hematological malignancies via CD20, BCMA, G protein-coupled receptor 5D or CD30.
In contrast to the cell engaging antibodies, which have a single mechanism of action, other antibodies for cancer indications function through multiple mechanisms. Two antibodies, telisotuzumab vedotin and zilovertamab vedotin, act via cytotoxic payload delivery and by blocking receptor signaling (cMET and ROR1, respectively). Seven antibodies have a dual mechanism of action, acting as blocking antibodies and promoting antigen clearance. Of these, 1 is a mixture of two antibodies targeting non-overlapping epitopes of EGFR, 1 is a CD73-targeting antibody, and 5 are bispecific proteins (two targeting PD-L1 and transforming growth factor-β, and three targeting VEGF and either DLL3 (two antibodies) or PD-1 (one antibody)). Two antibodies, bemarituzumab and nadunolimab, targeting fibroblast growth factor receptor 2b and IL1 receptor accessory protein, respectively, function both via CDC, ADCC, ADCP mechanisms and as blocking agents. One antibody, the anti-HER2 antibody zanidatamab, has a triple mechanism of action, acting as a blocking antibody, promoting antigen internalization and clearance, and triggering ADCC.
Of the nearly 140 antibody therapeutics currently in late-stage clinical studies, at least 23 have progressed sufficiently that companies may submit marketing applications for them by the end of 2023. These molecules are described below in the sections on Antibodies to Watch in 2023.
Antibodies to watch in 2023: Non-cancer indications
Of the antibody therapeutics in late-stage clinical studies that include patients with any disease other than cancer, companies sponsoring six of these (bentracimab, pozelimab, garadacimab, suciraslimab, tarcocimab tedromer, and axatilimab) have indicated that they may submit marketing applications to regulatory authorities during the fourth quarter of 2022 or in 2023 (Table 4). Relevant details for these 6 molecules are summarized below, with the summaries approximately in chronological order according to when a marketing application may be submitted.
Table 4.
Commercially sponsored investigational monoclonal antibodies in late-stage clinical studies for non-cancer indications, with regulatory submission anticipated during 2022–2023. *Indication for which a regulatory submission is anticipated. See Supplemental Table S1 for more details about each antibody. #First marketing application dates are estimates; table includes information publicly available as of November 15, 2022. Abbreviations: BLA, biologics license application (US); Fab, antigen-binding fragment; MAA, marketing authorization application (EU); NDA, new drug application (China); VEGF, vascular endothelial growth factor.
| INN | Target; Format | Indication of relevant late-stage study* | Status# |
|---|---|---|---|
| Bentracimab | Ticagrelor; Human IgG1 Fab | Reversal of the antiplatelet effects of ticagrelor | Phase 3 (BLA, Q4 2022) |
| Pozelimab | Complement 5; Human IgG4κ | CD55-deficient protein-losing enteropathy | Phase 3 (BLA, H2 2022) |
| Garadacimab | Factor XIIa; Human IgG4λ | Hereditary angioedema | Phase 3 (BLA, 2023) |
| Suciraslimab | CD22; chimeric IgG1κ | Rheumatoid arthritis | Phase 3 (NDA, 2023) |
| Tarcocimab tedromer | VEGF; Humanized IgG1κ antibody-biopolymer conjugate | Retinal vein occlusion | Phase 3 (BLA, 2023) |
| Axatilimab | Colony stimulating factor 1 receptor; Humanized IgG4κ | Graft vs. host disease | Phase 3 (BLA, 2023) |
Bentracimab (PhaseBio Pharmaceuticals, Inc.)
Bentracimab (PB2452; formerly MEDI2452) is a human antibody Fab that neutralizes the effects of the platelet inhibitor Brilinta®/Brilique® (ticagrelor), which is an oral anticoagulant given to patients undergoing surgery and to patients with certain cardiovascular- and hemostasis-related disorders. The Fab binds to ticagrelor with high affinity (Ki 20 pM), whereas ticagrelor binds to P2Y12 on platelets with lower affinity (Ki 2 nM). Created by MedImmune, world-wide rights to bentracimab were licensed to PhaseBio in 2018, and PhaseBio subsequently licensed rights in the EU and European Economic Area, United Kingdom, Russia, Ukraine, and other countries within the Commonwealth of Independent States to Alfasigma S.p.A. PhaseBio’s global clinical development of bentracimab is supported by SFJ Pharmaceuticals, which will lead development and regulatory activities in China and Japan. Bentracimab has received Breakthrough Therapy and PRIME designations from the FDA and EMA, respectively. PhaseBio is focused on clinical development and regulatory efforts to support a planned BLA submission for bentracimab in the fourth quarter of 2022.123
The safety and efficacy of bentracimab are being evaluated in the open-label, prospective single-arm Phase 3 REVERSE-IT study (NCT04286438) of reversal of the antiplatelet effects of ticagrelor with bentracimab in patients who present with uncontrolled major or life-threatening bleeding or who require urgent surgery or invasive procedure. Eligible patients received an initial IV bolus of 6 g bentracimab infused over 10 minutes for rapid reversal, followed immediately by a 6 g IV loading infusion over 4 hours and then a 6 g IV maintenance infusion over 12 hours. An alternative regimen comprising administration of 36 g over an active treatment period of 24 hours and 10 min was available to patients with potential drug interaction from recent concomitant use of moderate or strong CYP3A inhibitors with ticagrelor. A prespecified interim analysis that included study data for 150 patients, of which 142 required urgent surgery or invasive procedures and 8 had major bleeding, was reported in December 2021.124 For the 129 patients with analyzable platelet data, administration of bentracimab reversed the effects of ticagrelor within 5 to 10 minutes, and this reversal was sustained for more than 24 hours. Adjudicated hemostasis was achieved for more than 90% of 122 patients for which these data were available (P < .001). The study will enroll an estimated 200 patients and has a primary completion date in December 2023.
Pozelimab (Regeneron Pharmaceuticals, Inc.)
Pozelimab (REGN3918) is a human IgG4κ antibody targeting complement component 5 (C5) developed by Regeneron for C5-related disorders, including CD55-deficient protein-losing enteropathy (PLE), PNH, and myasthenia gravis. FDA has granted pozelimab Orphan Drug designations for the treatment of CD55-deficient PLE and PNH. Regeneron anticipates submission of a BLA for pozelimab for CD55-deficient PLE in the second half of 2022.125
The efficacy and safety study of pozelimab in patients with CD55-deficient PLE were evaluated in an open-label Phase 2/3 study (NCT04209634). The study included 10 patients aged 1 year and older with a clinical diagnosis of CD55-deficient PLE disease who received a single loading IV dose on Day 1, then fixed SC doses based on body weight) every week (±2 d) over the treatment period. The primary outcome measure of the study was the proportion of patients with active disease at baseline achieving both normalization of serum albumin and clinical outcome improvement within up to 24 weeks. The primary study completion date was November 9, 2021.
A Phase 3 study open-label extension study (NCT04162470) to evaluate the long-term safety, tolerability, and efficacy of pozelimab in patients with PNH was completed in April 2022. The study included 24 participants who completed 1 of the 2 parent studies (R3918-PNH-1852 [NCT03946748] or R3918-PNH-1853). Participants were administered SC pozelimab every week over the treatment period. The study’s primary outcome measures were the incidence and severity of treatment-emergent adverse events up to Week 104 and the proportion of patients achieving lactate dehydrogenase less than or equal to 1.5 of the upper limit of normal up to Week 26.
Two additional Phase 3 studies, ACCESS-1 (NCT05133531) and ACCESS-2 (NCT05131204) started in the first half of 2022 are evaluating the effects of pozelimab in combination with cemdisiran in PNH patients. The ACCESS-1 study is a randomized, open-label, ravulizumab-controlled study to evaluate the efficacy and safety of pozelimab and cemdisiran combination therapy in patients with PNH who are complement inhibitor treatment-naive or have not recently received complement inhibitor therapy. The study is recruiting an estimated 124 patients and has a primary completion date in May 2026. The ACCESS-2 study is a randomized, open-label, eculizumab- and ravulizumab-controlled study to evaluate the efficacy and safety of pozelimab and cemdisiran combination therapy in patients with PNH who are currently treated with eculizumab or ravulizumab. The study is recruiting an estimated 140 patients and has a primary completion date in June 2025.
The combination of pozelimab with cemdisiran is also being evaluated in a Phase 3 study (NCT05070858) of patients with symptomatic generalized myasthenia gravis. Initiated in December 2021, this study will enroll and estimated 235 patients and has a primary completion date in August 2024.
Garadacimab (CSL Limited)
Garadacimab (CSL312) is a hinge-stabilized (S228P mutation), human anti-Factor XIIa IgG4λ antibody developed by CSL for hereditary angioedema, a rare disorder characterized potentially life-threatening, recurrent episodes of severe swelling. Factor XII is the principal initiator of the plasma contact system, a protease cascade that results in inflammation, increased vascular permeability, vasodilation, and chemotaxis, as well as activation of the intrinsic coagulation pathway. Garadacimab was granted Orphan Drug designations in the EU for hereditary angioedema and in the US for the prevention of bradykinin-mediated angioedema and for idiopathic pulmonary fibrosis. CSL aims to begin filing with global health authorities at the end of their current fiscal year, i.e., by July 2023, for full approval.126
Results of a Phase 1 study of garadacimab (single‐dose IV and SC) in healthy male volunteers showed that the drug was well tolerated. In this Phase 1 study, subjects were randomized to eight cohorts, with IV doses of 0.1, 0.3, 1, 3, and 10 mg/kg and SC doses of 1, 3, and 10 mg/kg. The follow-up for safety lasted 85 d after dosing. A total of 48 subjects were enrolled; 32 received garadacimab and 16 received placebo. Mean garadacimab half‐life ranged between 14 and 20 d and 18 and 20 d in the cohorts that received the drug via IV and SC administration, respectively.127
CSL Behring sponsored a double-blind, randomized, placebo-controlled, parallel-arm Phase 3 study (NCT04656418) to investigate the efficacy and safety of SC administration of garadacimab in the prophylactic treatment of hereditary angioedema. Initiated in January 2021, the study enrolled 64 patients and was completed in June 2022. The study met its primary and secondary efficacy objectives and also demonstrated favorable safety and tolerability.126 The company is also sponsoring an open-label Phase 3 study (NCT04739059) to Evaluate the Long-term Safety and Efficacy of garadacimab in the Prophylactic Treatment of Hereditary Angioedema. Started in March 2021, the study has an actual enrollment of 171 participants and primary completion date in November 2025.
Suciraslimab (SinoMab BioScience Limited)
Suciraslimab (SM03), a chimeric IgG1κ antibody targeting CD22 on human B lymphocytes, is in development by SinoMab BioScience Limited for immune-mediated diseases, including RA. Wong et al. recently proposed that, upon binding to CD22, suciraslimab converts the configuration of CD22 from cis-binding to trans-binding. This conversion of the CD22 configuration allows the B cell to differentiate self from nonself and modulates B cells that trigger autoimmune attacks on autologous tissues, thereby alleviating the symptoms of immune-mediated diseases such as RA.128 SinoMab BioScience plans to file an NDA for suciraslimab for RA with the NMPA of the People’s Republic of China in the first half of 2023 and expects to commercialize suciraslimab by the second half of 2023 at the earliest.129
The efficacy and safety of suciraslimab were evaluated in a randomized, placebo-controlled Phase 2 study (NCT04192617) of 156 moderate-to-severely active RA patients in China who were receiving background MTX. Patients were randomized in a 1:1:1 ratio to receive a cumulative dose of 3600 mg (high dose, 600 mg × 6 infusions at Weeks 0, 2, 4, 12, 14 and 16) or 2400 mg suciraslimab (low dose, 600 mg × 4 infusions at Weeks 0, 2, 12 and 14) or placebo. Patients in all three arms of the study also received MTX. The primary outcome measure was the 24-week ACR 20% improvement criteria (ACR20) response rate. The 24-Week ACR20 response rate was significantly higher in both groups that received suciraslimab (high-dose (65.3%, P = .002) and low-dose suciraslimab (56.9%, P = .024)) compared to the placebo group (34.0%), and were comparable between the high- and low-dose groups.130
As of the end of 2021, a 2-arm, randomized, placebo-controlled Phase 3 clinical trial (NCT04312815) evaluating the safety and efficacy of suciraslimab in moderate-to-severely RA patients with an inadequate response to MTX had completed its enrollment of 530 patients. Patients received 600 mg IV suciraslimab on Week 0, 2, 4 and 12, 14, 16. Participants with inadequate response were rescued with open label IV suciraslimab (600 mg dose). The primary outcome was the 24-week ACR20 response rate. The preliminary result for the primary endpoint at Week 24 is expected in the third quarter of 2022 and the readout of the final study result for both safety and efficacy at Week 52 is expected in the first quarter of 2023.
SinoMab BioScience plans to evaluate suciraslimab for other immune-mediated disorders, including systemic lupus erythematosus, Alzheimer’s disease, and Sjogren’s syndrome.129
Tarcocimab tedromer (Kodiak Sciences Inc.)
Tarcocimab tedromer (KSI-301) is a humanized, Fc-silenced (L234A L235A G237A mutations) anti-VEGF IgG1κ antibody site-specifically (L443C) conjugated to an optically clear, high molecular weight phosphorylcholine polymer. The overall molecular weight of tarcocimab tedromer is 950 kDa, with the antibody and biopolymer contributing ~150 kDa and 800 kDa, respectively. Developed by Kodiak Sciences Inc., tarcocimab tedromer is designed to provide improved bioavailability, biocompatability, and stability compared to conventional biologic treatments for ocular disorders. Tarcocimab tedromer is being evaluated as an intravitreally administered treatment for retinal vein occlusion (RVO), DME, wet AMD, and non-proliferative diabetic retinopathy (NPDR). Based on positive results of the Phase 3 BEACON study of tarcocimab tedromer in RVO, and anticipated readouts of Phase 3 studies in DME (GLEAM and GLIMMER), wet AMD (DAYLIGHT), NPDR (GLOW) in 2023, Kodiak may submit a BLA for tarcocimab tedromer in all four indications in 2023.131,132
The randomized Phase 3 BEACON (NCT04592419) non-inferiority study is evaluating 5 mg tarcocimab tedromer administered every 2 months after two monthly loading doses vs 2 mg aflibercept every 1 month in treatment-naïve RVO patients. A total of 568 patients were randomized (1:1) to the two treatment arms. The study met its primary efficacy endpoint of non-inferior visual acuity gains at Week 24 for patients who were administered tarcocimab tedromer compared to those given aflibercept according to the study protocol. In the BEACON study, tarcocimab tedromer had a favorable safety profile with low rates of intraocular inflammation and no cases of intraocular inflammation with vasculitis or vascular occlusion.133
The paired, non-inferiority Phase 3 studies GLIMMER (NCT04603937) and GLEAM ((NCT04611152) are expected to report topline data in mid-2023. These studies are evaluating the effects of 5 mg tarcocimab tedromer every 2–6 months after three monthly loading doses vs. 2 mg aflibercept every two months after 5 monthly doses in in DME patients. The estimated primary completion dates of the GLIMMER and GLEAM studies are in April and May 2023, respectively. The short-interval, non-inferiority Phase 3 DAYLIGHT study (NCT04964089) in patients with treatment-naïve wet AMD is expected to report topline results in mid-2023. In the DAYLIGHT study, patients are administered tarcocimab tedromer (5 mg) once a month or aflibercept (2 mg) once a month for 3 monthly doses followed aflibercept (2 mg) once every 2 months. The estimated primary completion date of the DAYLIGHT study is in March 2023. The Phase 3 GLOW study (NCT05066230) of tarcocimab versus sham in NPDR without DME has completed enrollment. The estimated primary completion date of the GLOW study is in November 2023.
Axatilimab (Syndax Pharmaceuticals, Inc., Incyte Corporation)
Axatilimab (SNDX-6352) is a humanized hinge-stabilized (S228P) anti-colony stimulating factor 1 receptor (CSF-1R) IgG4κ antibody in clinical development by Syndax Pharmaceuticals and their partner Incyte as a treatment for chronic GvHD. CSF-1R signaling is involved in the expansion and infiltration of donor-derived macrophages that mediate chronic GVHD-related disease processes. Syndax licensed axatilimab from UCB during its preclinical development. Under Syndax and Incyte’s global partnership and license agreement, Incyte will oversee the international (outside US) commercialization activities of axatilimab for all indications. Syndax expects to report topline data from the pivotal AGAVE-201 study evaluating axatilimab in patients with cGVHD in mid-2023, with the expectation for a BLA filing later in 2023.134
The randomized Phase 2 AGAVE-201 study is evaluating the safety and efficacy of three dosing regimens of axatilimab in 241 patients with cGVHD following two or more prior lines of therapy. Patients are IV administered 0.3, 1 or 3 mg/kg axatilimab every 2 weeks for up to 2 y. The primary endpoint will assess objective response rate based on the 2014 National Institutes of Health consensus criteria for cGVHD. The study’s estimated primary completion date is in December 2023.
Antibodies to watch in 2023: Cancer indications
Of the antibody therapeutics in late-stage clinical studies that include patients with cancer, companies sponsoring 17 of these (IBI310, odronextamab, cosibelimab, apamistamab- iodine-131, datopotamab deruxtecan, zolbetuximab, camrelizumab, sugemalimab, felzartamab, tusamitamab ravtansine, tiragolumab, elranatamab, nofazinlimab, erfonrilimab, linvoseltamab, zanidatamab, and talquetamab) have indicated that they may submit marketing applications to regulatory authorities during the fourth quarter of 2022 or in 2023 (Table 5). Notably, 5 of these (odronextamab, erfonrilimab, linvoseltamab, zanidatamab, and talquetamab) are bispecific and 2 are ADCs (datopotamab deruxtecan and tusamitamab ravtansine), which suggests that these formats will become increasingly critical to medical care if they are approved in the future. Relevant details for the 17 molecules are summarized below, with the summaries approximately in chronological order according to when a marketing application may be submitted.
Table 5.
Commercially sponsored investigational monoclonal antibodies in late-stage clinical studies for cancer indications, with regulatory submission anticipated during 2022–2023. *Indication for which a regulatory submission is anticipated. See Supplemental Table S2 for more details about each antibody. #First marketing application dates are estimates; table includes information publicly available as of November 15, 2022. Abbreviations: ADC, antibody–drug conjugate; BCMA, B-cell maturation antigen; BLA, biologics license application (US); CEACAM5, carcinoembryonic antigen-related cell adhesion molecule-5; CTLA-4, cytotoxic T lymphocyte antigen-4; GPRC5D, G protein-coupled receptor class C group 5 member D; HER2, human epidermal growth factor receptor 2; MAA, marketing authorization application (EU); NDA, new drug application; PD-L1, programmed cell death protein ligand 1; TIGIT, T-cell Immunoreceptor with Ig and ITIM domains.
| INN | Target(s); Format | Indication of relevant* late-stage study | Status# |
|---|---|---|---|
| IBI310 | CTLA-4; Human mAb | Cervical cancer | Phase 3 (NDA, 2022) |
| Odronextamab | CD20, CD3; Human IgG4κ bispecific | Non-Hodgkin’s lymphoma | Pivotal Phase 2 (BLA, H2 2022) |
| Cosibelimab | PD-L1; Human IgG1λ | Squamous cell carcinoma | Phase 3 (MAA, Q2 2023) |
| Apamistamab- Iodine-131 | CD45; Murine IgG1κ, radiolabeled | Acute myeloid leukemia | Phase 3 (BLA, H1 2023) |
| Datopotamab deruxtecan | TROP-2; Humanized IgG1κ ADC | Non-small cell lung cancer | Phase 3 (H1 2023) |
| Zolbetuximab | Claudin-18.2; Chimeric IgG1κ | Gastric and gastro-esophageal junction adenocarcinoma | Phase 3 (BLA, 2023) |
| Camrelizumab | PD-1; Humanized IgG4κ | Hepatocellular carcinoma | Phase 3 (NDA, 2023) |
| Sugemalimab | PD-L1; Human IgG4λ | Relapsed or refractory extranodal natural killer/T-cell lymphoma | Phase 3 (BLA, 2023) |
| Felzartamab | CD38; Human IgG1κ | Multiple myeloma | Phase 3 (BLA, 2023) |
| Tusamitamab ravtansine | CEACAM5; Humanized IgG1κ ADC | Non-small cell lung cancer | Phase 3 (2023) |
| Tiragolumab | TIGIT; Human IgG1κ | Non-small cell lung cancer, esophageal cancer | Phase 3 (2023) |
| Elranatamab | BCMA, CD3; Humanized IgG2a | Multiple myeloma | Phase 3 (2023) |
| Nofazinlimab | PD-1; humanized IgG4κ | Hepatocellular carcinoma | Phase 3 (NDA, 2023) |
| Erfonrilimab | PD-L1, CTLA-4; Humanized/chimeric IgG1 bispecific VH-VH-h-CH2-CH3 dimer | Non-small cell lung cancer | Phase 3 (NDA) |
| Linvoseltamab | BCMA, CD3; Human IgG4κ bispecific | Multiple myeloma | Pivotal Phase 2 (BLA, 2023) |
| Zanidatamab | HER2, HER2; Humanized IgG1 bispecific | Biliary tract cancer | Pivotal Phase 2 (2023) |
| Talquetamab | GPRC5D, CD3; Humanized IgG4κ/λ bispecific | Multiple myeloma | Pivotal Phase 2 (2023–25) |
IBI310 (Innovent Biologics, Inc)
IBI310 is a human anti-CTLA-4 antibody developed by Innovent Biologics. The NMPA has granted Breakthrough Therapy designation for IBI310 in combination with sintilimab for the treatment of patients with recurrent or metastatic cervical cancer. Innovent plans to submit an NDA with the NMPA for IBI310 in combination with sintilimab for second-line cervical cancer by end 2022.135
The NDA will include results from a randomized, placebo-controlled Phase 2 trial evaluating the efficacy and safety of IBI310 versus placebo, in combination with Tyvyt (sintilimab), in 205 advanced cervical cancer patients who failed first and later line, or are intolerant of, platinum-based chemotherapy.
Odronextamab (Regeneron Pharmaceuticals, Inc.)
Odronextamab (REGN1979) is a human IgG4κ T cell-engaging bispecific antibody targeting CD20 and CD3, with Fc modifications to stabilize the hinge (S228P) and reduce Fc effector functions (E234P, F234V, L235A). Odronextamab was invented using Regeneron’s proprietary VelocImmune® technology and Veloci-Bi® platform. Odronextamab is in development by Regeneron Pharmaceuticals for the treatment of relapsed or refractory B-cell NHL. The FDA has granted Fast Track designation for odronextamab in relapsed or refractory FL and DLBCL. In April 2020, Zai Labs obtained exclusive rights from Regeneron to develop and commercialize odronextamab in Greater China. Regeneron anticipates BLA submission in the second half of 2022.136
Results from the single-arm, non-randomized, dose-escalation, and dose-expansion Phase 1 ELM-1 trial (NCT02290951) of 154 patients over 18 y old with CD20 + r/r B cell malignancies who had previously received CD20-directed antibody therapy showed promising preliminary activity and a manageable safety profile.137 Patients received IV injections of odronextamab in 21-day cycles according to a step-up dosing schedule in cycle 1, and then treatment weekly at doses ranging from 0.1 mg to 320 mg during cycles 2–4, followed by treatment every 2 weeks until disease progression or unacceptable toxicity. Efficacy results showed in r/r FL patients treated with >5 mg (n = 28), the objective response rate was 92.9% and complete response rate was 75%, while the objective response rate was 60% and 33.3% in r/r DLBLC patients who had not (n = 10) or who had (n = 21) received prior CAR-T cell therapy treated with odronextamab at doses of at least 80 mg.137
Results from the potentially pivotal Phase 2 ELM-2 study (NCT03888105) are expected in the second half of 2022.136 This study is made up of five arms investigating the safety and efficacy of IV-administered odronextamab in patients with FL, DLBLC, mantle cell lymphoma, marginal zone lymphoma, or B-cell non-Hodgkin lymphoma. The estimated enrollment is 512 patients, and the estimated study primary completion date is in December 2025.
Cosibelimab (Checkpoint Therapeutics, Inc.)
Cosibelimab (CK-301) is a human anti-PD-L1 IgG1λ antibody licensed from the Dana-Farber Cancer Institute in 2015 by Checkpoint Therapeutics, Inc. The company successfully completed two pre-BLA meetings with the FDA in July 2022, and a BLA submission for cosibelimab for metastatic and locally advanced cutaneous squamous cell carcinoma (cSCC) is expected by the end of 2022.138 An MAA submission for cosibelimab is expected in the second quarter of 2023.
Checkpoint Therapeutics announced the results from a registration-enabling Phase 1 (NCT03212404) trial of patients with metastatic or locally advanced cSCC in June 2022.139 The top-line results from the 78 metastatic cSCC patients demonstrated cosibelimab, at a fixed dosage of 800 mg every 2 weeks, met its primary endpoint with a confirmed objective response rate of 47.4% (95% CI, 36–59%) by independent central review using RECIST 1.1 criteria. Positive interim results were also announced from 31 patients with locally advanced cSCC with the objective response rate being 54.8% (95% CI, 36–73%).
Checkpoint Therapeutics was also evaluating cosibelimab as a treatment for NSCLC. However, the Phase 3 CONTERNO study (NCT04786964) of cosibelimab in combination with pemetrexed and platinum chemotherapy for first-line treatment of patients with metastatic NSCLC has been discontinued due to the substantially longer enrollment period than expected caused by the ongoing conflict in Ukraine.139
Apamistamab-I-131 (Actinium Pharmaceuticals, Inc.)
Apamistamab-I-131 (Iomab-B) is an iodine-131 radiolabelled murine IgG1κ antibody that targets CD45, an immune cell marker expressed on normal immune cells as well as on leukemia and lymphoma cells, including those in the bone marrow. Actinium Pharmaceuticals, Inc. is developing this antibody for targeted radiation to the bone marrow as a conditioning therapy to be used prior to an allogenic bone marrow transplant (BMT) for patients with active, relapsed or refractory acute myeloid leukemia (AML). Both the FDA and the EMA have granted Iomab-B Orphan Drug designations for this indication. Actinium has secured Immedica AB as their Europe, Middle East, and North Africa commercial partner, and anticipates BLA submission in the first half of 2023.140
In October 2022, Actinium announced that the pivotal Phase 3 SIERRA trial (NCT02665065) met its primary endpoint of durable complete remission of 6 months post initial remission after a BMT in Iomab-B arm compared to conventional care arm, demonstrating statistical significance (p < .0001).141 This study evaluated 153 patients aged over 55 y old with active, relapsed or refractory AML receiving Iomab-B compared to those receiving conventional care prior to BMT. Actinium reported a consistent 100% rate of BMT engraftment in all patients receiving Iomab-B compared to 17% of patients in the control arm, and an approximate 5x increase in patients potentially evaluable for the durable Complete Remission primary endpoint at 100 d post-BMT in the Iomab-B arm compared to the control arm.142
Datopotamab deruxtecan (Daiichi Sankyo, AstraZeneca)
Datopotamab deruxtecan (DS1062) is an anti-trophoblast cell surface antigen 2 (TROP2) humanized IgG1κ antibody conjugated to DXd, a derivative of exatecan, a topoisomerase I inhibitor. After binding to TROP2, DS1062 is internalized into tumor cells and DXd is released, inducing DNA damage and apoptosis in TROP2-expressing tumor cells. In July 2020, Daiichi Sankyo and AstraZeneca entered into a global collaboration to jointly develop datopotamab deruxtecan, except in Japan where Daiichi Sankyo maintains exclusive rights to the ADC. AstraZeneca has indicated that they anticipate regulatory submissions for datopotamab deruxtecan in second-line NSCLC with and without actionable alterations genomic alterations (AGAs) in the first half of 2023 and potentially in second-line or third-line hormone receptor-positive, HER2-negative breast cancer and in first-line triple-negative breast cancer in the second half of 2023.143
Regulatory submissions would be based on the Phase 3 TROPION-Lung01, TROPIONBreast01, and TROPIONBreast02 studies. Initiated in December 2020, the TROPION-Lung01 (NCT04656652) is evaluating the efficacy, safety, and pharmacokinetics of IV DS-1062a at 6.0 mg/kg vs IV docetaxel at 75 mg/m2 every 3 weeks in 590 participants with advanced or metastatic NSCLC with or without AGAs. The PFS and OS are the primary outcome measures. The estimated primary completion date of the study is in September 2023.
The TROPION-Breast01 (NCT05104866) study is evaluating the safety and efficacy of datopotamab deruxtecan vs investigator’s choice of standard of care single-agent chemotherapy (eribulin, capecitabine, vinorelbine, or gemcitabine) in 700 patients with inoperable or metastatic HR-positive, HER2-negative breast cancer who have been treated with one or two prior lines of systemic chemotherapy. Initiated in October 2021, the estimated primary completion date of the study is in July 2025. In the randomized, open-label, multicenter TROPION-Breast02 study (NCT05374512), the efficacy and safety of datopotamab deruxtecan is being compared with investigator’s choice chemotherapy in approximately 600 locally recurrent inoperable or metastatic triple-negative breast cancer patients who are not candidates for PD-1/PD-L1 inhibitor therapy. Initiated in May 2022, the estimated primary completion date of the study is in December 2025. PFS and OS are the primary outcome measures for the TROPIONBreast01 and TROPIONBreast02 studies.
Zolbetuximab (Astellas)
Zolbetuximab (IMAB362) is an anti-Claudin 18.2 (CLDN18.2) chimeric IgG1κ antibody, originally developed by Ganymed Pharmaceuticals AG, which was acquired by Astellas in 2016. CLDN18.2 expression is confined to tight junctions of the gastric mucosa in healthy tissues, but due to changes in cell polarity in malignant cells, the epitopes of CLDN18.2 are exposed on the cancer cell surface. Orphan Drug designation was granted to zolbetuximab for gastric cancer in the EU. Astellas aims to file for regulatory approval of zolbetuximab for gastric and GEJ adenocarcinoma in their fiscal year 2023 (April 1, 2023 – March 31, 2024).144
Results from two randomized, double-blind, placebo-controlled Phase 3 studies, SPOTLIGHT (NCT03504397) and GLOW (NCT03653507), investigating zolbetuximab in gastric and GEJ adenocarcinoma patients may support regulatory submissions. The SPOTLIGHT trial is investigating the safety and efficacy of zolbetuximab plus modified FOLFOX6 (folinic acid, fluorouracil, oxaliplatin) chemotherapy compared to placebo plus the same chemotherapy in 566 patients with unresectable or metastatic CLDN18.2-positive, HER2-negative gastric or GEJ adenocarcinoma. Patients receive 800 mg/m2 zolbetuximab administered as a minimum 2-hour IV infusion on Day 1 of Cycle 1 as a loading dose, and then 600 mg/m2 IV zolbetuximab every 3 weeks. The study met its primary endpoint showing statistical significance in PFS for patients in the zolbetuximab study arm vs those in the placebo study arm. The study also met the OS secondary endpoint, showing statistical significance for patients treated with zolbetuximab plus mFOLFOX6 compared to placebo plus mFOLFOX6.145
The GLOW trial has a similar design, but the study is evaluating the combination of zolbetumab with oxaliplatin and capecitabine (CAPOX) as the chemotherapy drug instead of modified FOLFOX6. Patients with CLDN18.2-positive, HER2-negative, locally advanced unresectable or metastatic G/GEJ adenocarcinoma enrolled in the GLOW study were treated with the same zolbetuximab dosing regimen used in the SPOTLIGHT trial. Enrollment (n = 507) for the GLOW study is complete.
Camrelizumab (Jiangsu HengRui Medicine Co., Ltd, Elevar Therapeutics, Inc.)
Camrelizumab (INCSHR1210, SHR-1210, AiRuiKa) is a hinge-stabilized (S228P mutation) IgG4κ antibody that targets the immune system checkpoint protein PD-1. Jiangsu Hengrui Pharmaceuticals Co., Ltd. (Hengrui Pharma) markets camrelizumab in China under the brand name AiRuiKa as a treatment for numerous tumor types. Hengrui Pharma and Elevar Therapeutics (formerly LSK BioPharma) have a global clinical collaboration to evaluate the safety and efficacy of Elevar’s rivoceranib (also known as apatinib) in combination with camrelizumab in patients with advanced HCC. Following a pre-NDA meeting with FDA, Elevar plans to file an NDA for the combination of rivoceranib plus camrelizumab as early as is feasible in 2023.146
A randomized, open-label, Phase 3 trial NCT03764293) evaluated the efficacy and safety of camrelizumab plus rivoceranib versus sorafenib as first-line therapy in patients with incurable, locally advanced, or metastatic HCC who had not received previous systematic treatment. The primary endpoints were OS and PFS as assessed by the blinded independent review committee according to RECIST v1.1. Patients (n = 543) were randomized 1:1 to receive camrelizumab (200 mg IV once every 2 weeks) plus rivoceranib (250 mg orally once daily) or sorafenib (400 mg orally twice daily). Camrelizumab plus rivoceranib significantly prolonged OS and PFS, and improved the overall response rate versus sorafenib. The median OS for camrelizumab + rivoceranib was 22.1 mos. [95% CI 19.1–27.2] vs. 15.2 mos. [13.0–18.5]; hazard ratio (HR) 0.62 [95% CI 0.49–0.80]; 1-sided p < .0001, and the median PFS for camrelizumab + rivoceranib was 5.6 mos. [95% CI 5.5–6.3] vs. 3.7 mos. [2.8–3.7]; HR 0.52 [95% CI 0.41–0.65]); 1-sided p < .0001. The confirmed overall response rate for camrelizumab + rivoceranib was 25.4% (95% CI 20.3–31.0), compared to 5.9% (3.4–9.4) for sorafenib.147
Sugemalimab (CStone Pharmaceuticals, Pfizer)
Sugemalimab (Cejemly®, 择捷美®) is a human IgG4λ anti-PD-L1 monoclonal antibody discovered using Ligand’s OmniAb platform and developed by CStone Pharmaceuticals. This antibody therapeutic is approved in China for first-line treatment of metastatic (stage IV) NSCLC in combination with chemotherapy and for the treatment of patients with unresectable stage III NSCLC whose disease has not progressed following concurrent or sequential platinum-based chemoradiotherapy. CStone formed a strategic partnership with Pfizer that includes the development and commercialization of sugemalimab in mainland China.
Sugemalimab was also evaluated as a treatment for relapsed or refractory extranodal natural killer/T-cell lymphoma (r/r ENKTL). The NMPA has accepted and granted priority review to the supplemental NDA for sugemalimab for this indication. CStone anticipates regulatory submissions for indications in stage III NSCLC, stage IV NSCLC, and r/r ENKTL in multiple countries and regions. The FDA granted sugemalimab received Breakthrough Therapy designation for r/r ENKTL, and CStone expects to submit a BLA to FDA for this indication in 2023.148
The single-arm Phase 2 GEMSTONE-201 study (NCT03595657) evaluated the efficacy and safety of sugemalimab monotherapy in 80 patients with r/r-ENKTL after prior asparaginase-based chemotherapy or chemo-radiotherapy. Patients were administered 1200 mg sugemalimab by IV administration every 3 weeks for up to 24 months until progression, death, or withdrawal from study. The primary endpoint of the study was the objective response rate (Complete Responses + Partial Responses) as assessed by the IRRC per Lugano 2014 criteria. Among the 78 evaluable patients as per IRRC, the objective response rate was 46.2% (95% CI: 34.8%, 57.8%), 29 (37.2%) patients achieved CR, the median DOR was not reached, and the 12-mo DOR rate was 86%. The 1- and 2-year OS rates were 68.6% and 54.6%, respectively.149
Felzartamab (I-Mab Biopharma, MorphoSys)
Felzartamab (TJ202, MOR202) is a human IgG1κ antibody directed against CD38, which is highly expressed on the surface of plasma cells. The antibody induces the depletion of such cells via ADCC and ADCP mechanisms. Felzartamab is being developed in MM, but also in autoimmune disorders, including glomerulonephritis and IgA nephropathy. Originally developed by MorphoSys, I-Mab Biopharma has exclusive rights for the development and commercialization of felzartamab in Greater China. In June 2022, MorphoSys granted Human Immunology Biosciences, Inc. exclusive rights to develop and commercialize felzartamab across all indications worldwide, excluding Greater China.150 As of October 2022, I-Mab Biopharma anticipates submission of a BLA in China for felzartamab for third-line MM in 2023–2025.151
Preset primary and secondary endpoints were met in a Phase 3 study of felzartamab for third-line MM. In this study, lower infusion-related reaction rates and a shorter infusion time allowed out-patient administration. I-Mab has also completed enrollment for the registrational randomized, open-label, parallel-controlled Phase 3 trial assessing the efficacy and safety of the combination of felzartamab with lenalidomide for second-line MM patients. The topline data is expected to be released in 2023 and could support a potential BLA submission in China.151
Tusamitamab ravtansine (Sanofi)
Tusamitamab ravtansine (SAR408701) is a humanized IgG1κ antibody (SAR408377) targeting carcinoembryonic antigen-related cell adhesion molecule-5 (CEACAM5) conjugated to DM4, an anti-tubulin maytansinoid agent. Initially developed by ImmunoGen, Sanofi was granted an exclusive license to tusamitamab ravtansine in 2017. In August 2022, Sanofi and Innovent Biologics announced a collaboration in which Innovent will be responsible for developing and exclusively commercializing tusamitamab ravtansine in multiple oncology-based indications in China, and Sanofi will be entitled to receive a milestone payment and royalties on the net sales of the product in China upon approval.152 Sanofi anticipates a regulatory submission for tusamitamab ravtansine for 2nd-3rd line NSCLC in 2023.153
The randomized, open-label Phase 3 trial (NCT04154956; CARMEN-LC03) is evaluating the safety and efficacy of tusamitamab ravtansine in metastatic non-squamous NSCLC patients with CEACAM-5-positive tumors who were previously treated with standard-of-care platinum-based chemotherapy and an immune checkpoint inhibitor. An estimated 554 participants ae being enrolled and randomly assigned to receive SAR408701 IV every 2 weeks or docetaxel IV every 3 weeks. The primary outcome measures are the PFS and OS. Initiated in February 2020, the study’s estimated primary completion date is in January 2025.
Two Phase 2 studies in non-squamous NSCLC with CEACAM5-positive expression are also ongoing. The CARMEN-LC04 study (NCT04394624) is evaluating the activity of tusamitamab ravtansine in combination with ramucirumab in 36 pretreated patients who will receive ramucirumab priori to IV tusamitamab ravtansine every 2 weeks. Part 1 of the study will assess the tolerability and confirm the recommended dose of the combination. Part 2 will investigate the antitumor activity of the combination. Initiated in August 2020, the study’s estimated primary completion date is in October 2022.
In the CARMEN-LC05 trial (NCT04524689), an estimated 120 patients with CEACAM positive-expression non-squamous NSCLC will be enrolled and assigned to 4 arms: tusamitamab ravtansine + pembrolizumab, tusamitamab ravtansine + pembrolizumab plus carboplatin, tusamitamab ravtansine + pembrolizumab plus cisplatin or tusamitamab ravtansine + pembrolizumab + carboplatin plus pemetrexed. The primary objective of the trial is to assess each combination’s tolerability and to determine the recommended dose of tusamitamab ravtansine in each combination. Secondary objectives include evaluating the safety, antitumor activity, pharmacokinetics, and immunogenicity of each combination. Initiated in October 2020, the study’s estimated primary completion date is in July 2023.
Tiragolumab (Genentech, a member of the Roche Group)
Tiragolumab (MTIG7192A, RO7092284, RG6058) is a human IgG1κ mAb targeting T-cell Immunoreceptor with Ig and ITIM domains (TIGIT), an inhibitory immune checkpoint expressed on T cells and NK cells. Roche is developing tiragolumab to synergistically enable the re-activation of T cells and enhance NK cell-mediated anti-tumor activity with anti-PD-1/anti-PD-L1 therapies. The FDA granted Breakthrough Therapy designation for tiragolumab in combination with anti-PD-L1 atezolizumab for first-line treatment of people with metastatic NSCLC whose tumors have high PD-L1 expression with no EGFR or ALK genomic tumor aberrations, based on the promising results from the Phase 2 CITYSCAPE trial. Roche anticipates that marketing applications may be submitted in 2023 for tiragolumab in combination with atezolizumab (Tecentriq) for first-line PD-L1-positive NSCLC and for tiragolumab in combination with Tecentriq and chemotherapy (cisplatin and paclitaxel) for first-line esophageal cancer (China only).154
Tiragolumab is being evaluated as a treatment for several types of solid tumors in the Phase 3 SKYSCRAPER program, which to date includes nine clinical studies (SKYSCRAPER-01 through −09). The randomized, double-blind, placebo-controlled Phase 3 SKYSCRAPER-01 study (NCT04294810), investigating the combination of tiragolumab with atezolizumab in 534 patients with PD-L1-high, locally advanced or metastatic NSCLC, did not meet the co-primary endpoint of PFS, and the other co-primary endpoint of OS was immature.155 Patients received 1200 mg atezolizumab plus 600 mg tiragolumab, or atezolizumab alone in the placebo arm, via IV injection on Day 1 of every 21-day cycle. The study is ongoing until the next analysis, as numerical improvement was observed for both endpoints. The next set of results is expected in 2023.
The randomized, placebo-controlled Phase 3 SKYSCRAPER-08 study (NCT04540211) is evaluating atezolizumab plus tiragolumab in combination with paclitaxel and cisplatin compared with paclitaxel and cisplatin as first-line treatment in 461 patients with unresectable locally advanced, unresectable recurrent, or metastatic ESCC. Patients in the investigational drug arm of the study receive atezolizumab at a fixed dose of 1200 mg and tiragolumab at a fixed dose of 600 mg, each administered by IV infusion every 3 weeks on Day 1 of each 21-day cycle, in combination with paclitaxel and cisplatin. The study primary endpoints are OS and PFS. All study locations are in Asia (China, Hong Kong, Republic of Korea, Taiwan, and Thailand). Recruitment for the study was completed in the fourth quarter of 2021.154
Elranatamab (Pfizer Inc.)
Elranatamab (PF-06863135) is a humanized IgG2a T-cell engaging bispecific antibody designed to target BCMA, which is highly expressed on tumor cells, and CD3 found on the T cell surface. Developed by Pfizer for the treatment of patients with r/r MM, elranatamab is formulated for SC, rather than IV, administration to allow higher doses without increasing adverse events. Elranatamab was granted Orphan Drug designations by the FDA and the EMA for the treatment of MM. In addition, FDA granted elranatamab Fast Track and Breakthrough Therapy, and EMA granted elranatamab PRIME designation, for r/r MM.156 Pfizer has indicated that key pivotal study readouts, which could support potential regulatory filings, are expected on the second half of 2022.157
Pfizer reported interim analysis of the open label, single-arm, pivotal Phase 2 MagnetisMM-3 study (NCT04649359) assessing the safety and efficacy of elranatamab monotherapy in r/r MM patients whose disease is refractory to at least one agent in each of three major classes of medications (proteasome inhibitor, anti-CD38 antibody, immunomodulatory drug) approved for the disease. Patients are enrolled in two cohorts depending on whether they had previously received treatment with a BCMA-directed ADC or CAR-T therapy. Following a series of 2 priming doses (12 and 32 mg) administered the first week, patients then receive 76 mg SC elranatamab weekly. The primary endpoint is objective response rate as assessed by blinded independent central review. For 123 patients naïve to BCMA-directed therapies (Cohort A), the overall response rate after a median follow-up of 6.8 months was 61.0%, and the probability of maintaining a response was 90.4% among responders. The results also suggested elranatamab may have a manageable safety profile in patients with triple-class r/r MM.156
Safety and efficacy of elranatamab in MM patients is being studied in two Phase 3 trials. The open label 3-arm MagnetisMM-5 trial (NCT05020236) is enrolling an estimated 589 participants. The safety lead-in dose escalation part of the study will evaluate the safety and activity of different doses of elranatamab in combination with daratumumab in r/r MM patients. Part 2 includes 3 randomized arms that will compare the safety and activity of SC elranatamab as monotherapy vs SC elranatamab plus SC daratumumab or SC daratumumab + pomalidomide + dexamethasone. The primary outcome measure related to efficacy is progression-free survival per the International Myeloma Working Group criteria from the date of randomization to the date of progressive disease, discontinuation from the study, death, or censoring, whichever occurs first, assessed up to 32 months. Initiated in October 2021, the estimated primary completion date of the study is in October 2024.
Initiated in March 2022, the 2-arm Phase 3 MagnetisMM-7 study (NCT05317416) is evaluating elranatamab monotherapy versus lenalidomide for newly diagnosed MM patients who are positive for minimum residual disease after undergoing autologous stem cell transplant. In this randomized study, an estimated 366 enrolled participants will receive SC elranatamab every week or oral lenalidomide once daily. Primary outcome measures include the incidence of dose limiting toxicities and PFS. The estimated primary completion date of the study is in June 2027.
Nofazinlimab (CStone Pharmaceuticals)
Nofazinlimab (EQ176, CS1003) is a humanized IgG4κ monoclonal antibody targeting PD-1 developed by CStone Pharmaceuticals for the treatment of HCC. CStone retains rights to nofazinlimab in Greater China and licensed the anti-PD1 to US firm EQRx for development and commercialization outside of China. The FDA has granted nofazinlimab Orphan Drug designation for the treatment of patients with HCC. CStone expects to file an NDA filing for first line treatment of HCC in China in 2023.158
Results from a single-arm Phase 1 (NCT03809767, CS1003-102) study conducted in China of nofazinlimab in HCC were presented at the 2022 American Society of Clinical Oncology annual meeting, which was held June 3–7, 2022, in Chicago, IL. In this open-label, multi-center, dose escalation trial, the combination of nofazinlimab with lenvatinib (LEN) was assessed as first-line treatment in patients with unresectable HCC. A total of 107 patients received 200 mg nofazinlimab IV once every 3 weeks and LEN orally (body weight ≥60 kg: 12 mg; <60 kg: 8 mg) daily as first-line treatment. The primary endpoint was objective response rate assessed by investigators per RECIST v1.1. As of the data cutoff for the analysis presented, 20 patients had been treated. The objective response rate for these patients was 45.0% (95% CI: 23.06%, 68.47%) and the median PFS was 10.4 months. The DOR range was 4.2 to 18.7 months, but the median DOR and OS had not yet reached as of the data cutoff date. Nofazinlimab was well tolerated, with a manageable safety profile.159
An international randomized Phase 3 study (NCT04194775, CS1003-305) is underway to evaluate the efficacy and safety of IV nofazinlimab in combination with lenvatinib compared with placebo in combination with lenvatinib as first-line treatment in patients with unresectable advanced HCC. The estimated enrollment is 525 patients. The primary endpoints of the study are OS and PFS evaluated by a blinded independent central review committee based on RECIST v1.1. Initiated in December 2019, the primary completion date of the study is in June 2023.
Erfonrilimab (Alphamab Oncology)
Erfonrilimab (KN046) is a humanized bispecific single‐domain antibody‐Fc fusion protein (VH-VH-h-CH2-CH3 dimer) targeting immune checkpoints PD-L1 and CTLA-4 developed by Jiangsu Alphamab Biopharmaceuticals Co., Ltd., a wholly owned subsidiary of Alphamab Oncology. Orphan drug designations were granted by the FDA for KN046 in thymic epithelial tumors and in combination with the anti-HER2 bispecific antibody KN026 in gastric cancers. The company plans to submit an NDA for erfonrilimab in squamous NSCLC.160
The efficacy and the safety of KN046 in combination with paclitaxel and carboplatin is being assessed in squamous NSCLC patients in the randomized, double blind, placebo-controlled Phase 3 ENREACH-LUNG-01 study (NCT04474119). The 482 patients enrolled in the study were randomized to receive either KN046 administered IV at 5 mg/kg every 3 weeks plus chemotherapy (carboplatin and paclitaxel) or placebo plus chemotherapy as first-line treatment. In March 2022, the company reported that the study met its prespecified endpoint of improving PFS. OS data will be reported at a later date.160
Two-year follow-up data from the Phase 2 KN046-202 study (NCT04054531), which evaluated the efficacy, safety, and tolerability of erfonrilimab combined with chemotherapy as first-line therapy in NSCLC patients, were reported at the 2022 ESMO Congress held September 9–13, 2022, in Paris, France. Patients with non-squamous NSCLC received KN046 (5 mg/kg IV every 3 weeks) and chemotherapy (carboplatin (area under the free carboplatin plasma concentration versus time curve (AUC) = 5), and pemetrexed (500 mg/m2)), and those with squamous NSCLC received KN046 (5 mg/kg IV every 3 weeks) in combination with carboplatin (AUC = 5), and paclitaxel (175 mg/m2). As of March 15, 2022 data cutoff date, the median follow-up was 23.1 months. In all 87 efficacy evaluable patients, the confirmed objective response rate was 46% (95% CI: 35.2, 57.0). For patients with non-squamous (cohort 1, n = 51) and squamous disease (cohort 2, n = 36), the confirmed objective response rates were 43.1% (95% CI: 29.3, 57.8) and 50% (95% CI: 32.9, 67.1), respectively, and the DORs were 9.7 months (95% CI:4.01, 20.73) and 7.3 months (95% CI:3.52, -), respectively. The median PFS of cohort 1 and cohort 2 were 5.8 months (95% CI: 4.80, 7.16) and 5.7 m (95% CI: 4.17, 8.71), and median OS were 27.2 months (95% CI: 15.18, -) and 26.6 months (95% CI: 12.19, -), respectively.161
In total, the company is conducting ~20 clinical trials of erfonrilimab, including pivotal Phase 2/3 or Phase 3 studies in NSCLC (NCT05001724, NCT04474119) and in pancreatic cancer in combination with gemcitabine and nab-paclitaxel (NCT05149326), as well as Phase 2 studies in thymic carcinoma (NCT04925947; NCT04469725).
Linvoseltamab (Regeneron Pharmaceuticals, Inc.)
Linvoseltamab (REGN5458) is an IgG4κ bispecific T-cell engaging antibody that targets BCMA and CD3. It was derived from Regeneron’s proprietary VelocImmune® technology and created using the company’s Veloci-Bi® platform. The company plans to complete enrollment in a potentially pivotal Phase 2 study of linvoseltamab in MM, initiate studies with an SC formulation, initiate Phase 3 studies in earlier lines of therapy, and potentially submit a BLA for linvoseltamab in r/r MM to FDA in 2023.162
Linvoseltamab is under investigation in the single-arm Phase 1/2 LINKER-MM1 trial (NCT03761108) enrolling an estimated 291 participants with r/r MM who had received at least three prior lines of therapy proteasome inhibitors, immunomodulatory drugs and CD38 antibody treatments or were double refractory. In Part 1 of the study, the safety, tolerability, dose-limiting toxicities, and recommended Phase 2 dose of linvoseltamab as monotherapy are being assessed. Interim results for 73 patients treated with linvoseltamab doses ranging from 3 to 800 mg for up to 21 months have been reported.163 Patients had received a median of five prior lines of therapy, with 38% (n = 28) being penta-refractory and 90% (n = 66) being refractory to the last line of therapy. The overall response rate was 75% at the highest dose levels (200–800 mg, n = 18/24), and 51% among all enrolled patients (n = 37/73). Part 2 of the study will assess the objective response rate of linvoseltamab in triple-class refractory patients as measured using the International Myeloma Working Group criteria within a time frame of up to 5 y. Initiated in January 2019, the estimated primary completion date of the study is May 2023.
Zanidatamab (Zymeworks Inc., Jazz Pharmaceuticals, BeiGene)
Zanidatamab (ZW25) is a humanized anti-HER2 bispecific antibody, with an asymmetric IgG1-like format; one chain is IgG1κ and the other chain is an scFvκ linked to the IgG1 hinge, CH2, and CH3 domains, developed by Zymeworks from the proprietary Azymetric platform. Zanidatamab is biparatopic, simultaneously binding two distinct, non-overlapping epitopes of HER2, the ECD4 domain (trastuzumab-binding domain) and the ECD2 domain (pertuzumab-binding domain), improving binding, clustering, and receptor internalization. FDA granted Breakthrough Therapy designation for zanidatamab for previously treated HER2 gene-amplified biliary tract cancer (BTC), in addition to two Fast Track designations, one for previously treated or recurrent HER2-positive BTC and another for first-line gastroesophageal adenocarcinoma (GEA) in combination with standard of care chemotherapy. Zanidatamab has also received Orphan Drug designation for the treatment of biliary tract and gastric cancers from the FDA and for gastric cancer from the EMA. In 2018, BeiGene was granted the exclusive rights for the research, development, and commercialization of zanidatamab in Asia-Pacific, excluding Japan and India. In October 2022, Jazz Pharmaceuticals announced it would acquire development and commercialization rights to zanidatamab across all indications in the United States, Europe, Japan, and all other territories except for those Asia/Pacific territories previously licensed by Zymeworks. Top-line clinical data from the pivotal Phase 2 HERIZON-BTC-01 study evaluating zanidatamab in BTC that may support first global regulatory filings is expected by end of 2022. Positive data may allow marketing application submissions in 2023.164
As of April 2022, enrollment was completed in the open-label, single-arm HERIZON-BTC-01 study (NCT04466891) investigating the anti-tumor efficacy of zanidatamab monotherapy in 100 patients with previously treated advanced or metastatic HER2-amplified BTCs, including gallbladder cancer and bile duct cancer.165 Zanidatamab was administered via IV injection at 20 mg/kg in 28-day cycles on Day 1 and Day 15. Early results from 20 patients first reported in January 2021 showed a disease control rate of 65% (n = 13) and a confirmed partial response in 40% (n = 8), with a median DOR of 7.4 months (95% CI; 3.2 not estimable).166
Zanidatamab is also being investigated in the randomized Phase 3 HERIZON-GEA-01 trial (NCT05152147; EudraCT 2021–000296-36) of patients with advanced or metastatic HER2-positive GEA. This study is recruiting an estimated 714 patients who are randomized into three treatment arms: trastuzumab plus chemotherapy, zanidatamab plus chemotherapy, or zanidatamab and tislelizumab plus chemotherapy. Trastuzumab (6 mg/kg), zanidatamab (1800 mg for patients <70 kg, or 2400 mg for patients >70 kg) and tislelizumab (200 mg) are administered via IV injection every 3 weeks concurrently to the investigator’s choice of chemotherapy. The primary completion date is in June 2024, with study completion anticipated by July 2025.167
Talquetamab (Janssen Research & Development, LLC)
Talquetamab (JNJ-64407564) is a humanized IgG4κ/λ T-cell engaging bispecific antibody developed by Janssen. The antibody targets G protein-coupled receptor class C group 5 member D (GPRC5D), which is highly expressed in malignant plasma cells, and CD3 on T cells. Talquetamab was granted Breakthrough Therapy designation by the FDA for use in patients with r/r MM based on results of the MonumenTAL-1 study,168 in addition to the PRIME designation received from the EMA in the same indication earlier.
Preliminary results from the MonumenTAL-1 Phase 1 first-in-human dose escalation study of talquetamab (NCT03399799) determining the recommended Phase 2 dose(s) and dosing schedule in subjects with r/r MM were presented at the European Hematology Association’s 2022 meeting.169 In this study, participants received either SC talquetamab every week at 405 µg/kg (n = 30) or SC talquetamab every 2 weeks at 800 µg/kg (n = 44). The overall response rate in the 405 μg/kg every week group was 70%, and 64% in the 800 μg/kg every 2 weeks group. Partial responses were seen in 57% of patients in both groups. The median time to first confirmed response among patients in the 405 μg/kg every week group was 0.9 months (range = 0.2–3.8) and 1.2 months (range = 0.3–6.8) in the 800 μg/kg every 2 weeks group. These results are based on median patient follow-ups that were longer (13.2 months; range = 1.1–24.0) in the 405 μg/kg every week group than that of the 800 μg/kg every 2 weeks group (7.7 months; range = 0.7–16.0).169 The study’s estimated enrollment is 260 patients, and the actual primary completion date is July 7, 2022.
Talquetamab is being evaluated in the MonumenTAL-5 Phase 3 study (NCT05461209) to compare its efficacy with BLENREP (anti-BCMA belantamab mafodotin) based on primary outcome measures (overall response rate and PFS) in r/r MM patients who have received at least four prior therapies, Including an immunomodulatory drug, a proteasome inhibitor, and an anti-CD38 antibody. Participants are randomized to receive SC talquetamab or IV BLENREP. Initiated in November 2022, the estimated primary completion date of the study is in February 2024. Positive study results could allow regulatory submissions in 2023, although submission in 2024 or later may be more realistic.
Notable set-backs
Companies prune their pipelines each year by discontinuing development of molecules for reasons that include lack of sufficient efficacy or the presence of safety signals, as well as business considerations, such as priority of other assets, changes in the competitive landscape or medical practice, and availability of funds for further development. When anti-SARS- CoV-2 antibody therapeutics are excluded, the number of antibody therapeutics discontinued while in late-stage development in 2022 was small, and similar in number to past years. Examples include Regeneron’s discontinuation of clinical development of fasinumab and REGN1908-1909.170 Anti-nerve growth factor fasinuamb was being studied in osteoarthritis pain of the knee or hip in collaboration with Teva and Mitsubishi Tanabe Pharma. REGN1908-1909 was a mixture of two antibodies that targeted Fel d 1, which is a causative agent of cat allergy. Both fasinumab and REGN1908-1909 had reached Phase 3 studies, but development was discontinued due to futility.
Like Schrödinger’s cat, however, molecules may have uncertain fates following the announcement of disappointing late-stage clinical study results, business decisions, or regulatory actions. In 2022, examples of molecules in this situation include lilotomab satetraxetan (Nordic Nanovector), ARX788 (Ambrx Biopharma Inc., NovoCodex), gantenerumab (Roche), and oportuzumab monatox (Sesen Bio). While the molecules have encountered setbacks, they may in fact not be dead. Nordic Nanovector announced in July 2022 that the company discontinued the pivotal PARADIGME study of radiolabeled anti-CD37 antibody lilotomab satetraxetan (Betalutin®) in r/r FL for lack of efficacy, and in November 2022 announced that the company had entered into a definitive merger agreement to combine with APIM Therapeutics.171 This action suggests a change in priorities, but a partnership that may allow continued development of Betalutin® is (remotely) possible. Ambrex announced strategic reprioritization of its pipeline in October 2022, noting that it would no longer directly pursue anti-HER2 ADC ARX788 as its lead clinical asset due to changes in the HER2 competitive landscape. Development of ARX788 by licensee Zhejiang Medicine Co., Ltd.’s subsidiary Novocodex may continue, and Ambrx may seek a development partner(s) ex-China to progress ARX788.172 In November 2022, Roche announced that the GRADUATE I and II studies (NCT03444870 and NCT03443973, respectively) evaluating anti-amyloid β gantenerumab in people with mild cognitive impairment due to Alzheimer’s and mild Alzheimer’s dementia, collectively called early Alzheimer’s disease, did not meet their primary endpoint of slowing clinical decline, but did not specifically state that further studies would be halted.173
Discontinuation of an antibody therapeutic after a marketing application has been submitted to a regulatory agency is highly unusual, with fewer than a half-dozen cases occurring in the past 20 y. Oportuzumab monatox (Vysyneum™, Vicineum), an immunotoxin composed of a humanized anti-epithelial cell adhesion molecule scFv linked to ETA(252–608) Pseudomonas exotoxin, seems poised to join this select group, but its final fate is still uncertain. Sesen Bio, Inc. developed oportuzumab monatox for the treatment of BCG-unresponsive non-muscle invasive bladder cancer, and submitted a BLA and MAA to FDA and EMA, respectively, for this indication. In August 2021, however, the company received a complete response letter from the FDA that recommended additional clinical/statistical data and analyses, and then withdrew the MAA. In 2022, Sesen announced a voluntary pause on further development of the asset, underwent a company restructuring, and then entered into a definitive merger agreement with Carisma Therapeutics Inc, which develops cell and gene therapies.174 Sesen had previously licensed certain rights to oportuzumab monatox in Greater China, the Middle East and North Africa, and Turkey to Qilu Pharmaceutical Co., Hikma Pharmaceuticals Plc Ltd., and Eczacibasi Pharmaceuticals Marketing Co., respectively. We await further news of progress (or the lack of it) from these companies.
Outlook for the future
As documented here, the commercial clinical pipeline of antibody therapeutics has shown substantial growth over the past year, although at a somewhat slower pace (~20%) compared to the annual growth (over 30%) noted in the Antibodies to Watch in 2022 article.1 The annual number of approvals in the US or US appears poised to meet or exceed the previous record, annual approvals in regions outside the US and EU are increasing, and marketing applications for a substantial number of antibody therapeutics are being evaluated by regulatory authorities (Tables 2 and 3). Notably, more highly engineered (e.g., bispecific) and conjugated (e.g., ADC) molecules are successfully navigating late-stage clinical studies and regulatory review, and entering the market.
For the past several years, however, BLAs for some antibody therapeutics have required second cycles of review, which delays, but does not necessarily derail, their approval. Each BLA is unique, and the reasons for second cycles vary, but FDA has recently started placing increased emphasis on confirmatory clinical studies for drugs in development by companies that aim for an accelerated approval, with a particular focus on cancer drugs. The accelerated approval pathway allows FDA to approve products intended for serious or life-threatening disease based on the drug’s effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit to patients, but a required post-approval trial is needed to verify that the drug provides the expected clinical benefit. If no benefit is confirmed, then the product may be removed from the market either voluntarily by the sponsoring company or by FDA.
In 2022, the cancer drugs teclistamab (TECVAYLI) and mirvetuximab soravtansine ((ELAHERE™) were granted accelerated approvals, but submission of a BLA for camidanlumab tesirine (ADCT-301; ADC Therapeutics SA) was delayed due to FDA’s guidance that, for it to consider an accelerated approval path, a randomized confirmatory Phase 3 study must be well underway and ideally fully enrolled at the time of any BLA submission. ADC Therapeutics had conducted a single-arm, registrational Phase 2 Hodgkin lymphoma trial (NCT04052997) that included 117 patients with relapsed or refractory Hodgkin lymphoma.175 After receiving FDA’s guidance, ADC Therapeutics tersely noted in a November 8, 2022 press release that they will not submit a BLA for camidanlumab tesirine in 2023, and they are pausing any material investments in the Hodgkin’s lymphoma program and will evaluate options for camidanlumab tesirine in this indication with a disciplined and strategic approach to resource allocation.176
FDA’s current thinking regarding accelerated approvals for cancer drugs was articulated in a New England Journal of Medicine article published on October 20, 2022.177 In that article, Fashoyin-Aje and coauthors at FDA’s Oncology Center of Excellence argue for the use of approaches that will decrease the time from an accelerated approval to confirmation that the drug does, or does not, provide a benefit. They provide data showing that the median time to withdrawal is 3.8 y if a confirmatory study was ongoing, vs. 7.3 y if such a study had not started, when the drug was granted accelerated approval. During this period, patients are exposed to the risks of the drug, but do not derive the expected benefits. In the antibody therapeutics space, gemtuzumab ozogamicin (Mylotarg) offers an example of this. Mylotarg was granted an accelerated approval by FDA in 2000 and was marketed for over a decade before it was voluntarily withdrawn after confirmatory trials failed to verify clinical benefit and demonstrated safety concerns, including a high number of early deaths. Subsequent studies ultimately allowed the drug to return to the US market in 2017. In a more recent example, in November 2022 GSK voluntarily initiated the process for withdrawal of the US marketing authorization for Blenrep (anti-BCMA belantamab mafodotin-blmf) following a request from FDA.178 Blenrep was granted an accelerated approval in 2020 is a monotherapy treatment for adult patients with r/r MM who have received at least four prior therapies, but the confirmatory Phase 3 DREAMM-3 trial did not meet the requirements of the accelerated approval regulations. Other ongoing potentially confirmatory trials (DREAMM-7, DREAMM-8) of belantamab mafodotin may allow the drug to return to the US market in the future.
FDA’s more stringent approach may prove challenging to small companies that are relying on accelerated approvals to enable partnering or fund-raising, in particular if these are needed to initiate confirmatory clinical studies. We are currently tracking nearly 800 antibody therapeutics in clinical development for cancer, which comprise over 60% of the commercial clinical pipeline. We look forward to documenting the progress of these molecules, as well as antibody therapeutics for other indications, as they enter late-stage clinical studies and become ‘Antibodies to Watch’ in the future.
Supplementary Material
Acknowledgments
The authors thank Vandna Prasad Rath and Andy Cook at Hanson Wade for providing access to the Beacon Targeted Therapies database.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
List of Abbreviations
Aβ, amyloid beta; ACR20, American College of Rheumatology 20%; AD, Alzheimer’s disease; ADC, antibody–drug conjugate; ADCC, antibody-dependent cell-mediated cytotoxicity; ADP, adenosine diphosphate; ALK, anaplastic large-cell lymphoma kinase; AMD, age-related macular degeneration; AML, acute myeloid leukemia; Ang-2, angiopoietin-2; ANGPTL3, angiopoietin-like protein 3; ASCO, American Society of Clinical Oncology; BCG, bacillus Calmette-Guérin; BCMA, B cell maturation antigen; BCVA, Best-corrected visual acuity; BLA, biologics license application; BMS, Bristol Myers Squibb; BMT, bone marrow transplant; BTC, biliary tract cancer; CAD, cold agglutinin disease; CAPOX, capecitabine/oxaliplatin; CAR-T, Chimeric Antigenic Receptor – T cell; CDC, complement-dependent cytotoxicity; CDR-SB, Clinical Dementia Rating Scale – Sum of Boxes; CHMP, Committee for Medicinal Products for Human Use; CI, confidence interval; CLDN18.2, claudin 18.2; CLL, chronic lymphocytic leukemia; CMC, Chemistry, Manufacturing and Controls; CNS, central nervous system; COVID-19, coronavirus disease 2019; CR, complete response; CRS, cytokine release syndrome, CSCC, cutaneous squamous cell carcinoma; CSU, chronic spontaneous urticaria; CTLA-4, cytotoxic T lymphocyte antigen-4; DCR, disease control rate; DLBCL, diffuse large B-cell lymphoma; DM4, N2’-deacetyl-N2’-(4-mercapto-4-methyl-1-oxopentyl) maytansine; DME, diabetic macular edema; dMMR, deficient mismatch repair; EC, European Commission; ECMO, extracorporeal membrane oxygenation; EGFR, epidermal growth factor receptor; EMA, European Medicines Agency; ENKTL, extranodal natural killer/T-cell lymphoma; EpCAM, epithelial cell adhesion molecule; ESCC, esophageal squamous cell carcinoma; ESMO, European Society for Medical Oncology; EU, European Union; EUA, Emergency use authorization; Fab, antigen-binding fragment; Fc, crystallizable fragment; FcyR, Receptors for IgG Fc; FcRn, neonatal Fc receptor; FDA, US Food and Drug Administration; FL, follicular lymphoma; FRα, folate receptor alpha; GEA, gastroesophageal adenocarcinoma; GEJ, gastroesophageal junction; GM-CSF, granulocyte-macrophage colony stimulating factor; GPP, generalized pustular psoriasis; GPRC5D, G Protein-Coupled Receptor Class C Group 5 Member D; GvHD, graft-vs-host disease; HCC, hepatocellular carcinoma; HER2, human epidermal growth factor receptor 2; HLA, human leukocyte antigen; HoFH, homozygous familial hypercholesterolemia; HR, hazard ratio; HS, hidradenitis suppurativa; HSCT, hematopoietic stem cell transplant; hTfR, human transferrin receptor; iADRS, Integrated AD Rating Scale; IDS, iduronate-2-sulfatase; IFN, interferon; IFNAR1, interferon alpha receptor 1; IGA, Investigator’s Global Assessment; IgE, immunoglobulin E; IgG, immunoglobulin G; IL, interleukin; IM, intramuscular; INN, International Nonproprietary Names; IRRC, independent radiology review committee; IV, intravenous; LAG-3, lymphocyte-activation gene 3; LDH, lactate dehydrogenase; LDL, low-density lipoprotein; LM, leptomeningeal metastases; MAA, marketing authorization application; mAb, monoclonal antibody; MASP-2, mannan-binding lectin-associated serine protease-2; MDS, myelodysplastic syndrome; MET, mesenchymal epithelial transition factor; MHLW, Ministry of Health, Labor and Welfare; MM, multiple myeloma; MMAE, monomethyl auristatin E; MMR, mismatch repair; MPS-II, mucopolysaccharidosis II; MS, multiple sclerosis; MSI, microsatellite instability; MTX, methotrexate; NDA, new drug application; NHL, non-Hodgkin’s lymphoma; NIH, National Institutes of Health; NK, natural killer cells; NMIBC, non-muscle invasive bladder cancer; NPDR, non-proliferative diabetic retinopathy; NMPA, National Medical Products Administration; NSCLC, non-small cell lung cancer; OA, osteoarthritis; OR, overall response; OS, overall survival; PD, pharmacodynamics; PD-1, programmed cell death protein 1; PD-L1, programmed cell death protein ligand 1; PD-L2, programmed death ligand 2; PDUFA, Prescription Drug User Fee Act; PFS, progression-free survival; PHN, paroxysmal nocturnal hemoglobinuria; PK, pharmacokinetics; PMDA, Pharmaceuticals and Medical Devices Agency; PR, partial response; PRIME, Priority Medicines; PTCL, peripheral T cell lymphoma; PTI, personalized treatment intervals; RA, rheumatoid arthritis; RECIST, Response Evaluation Criteria in Solid Tumors; RSV, respiratory syncytial virus, RT-qPCR, Quantitative reverse transcription PCR; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SC, subcutaneous; SCAC, squamous cell carcinoma of the anal canal; scFv, single-chain variable fragment; SLE, systemic lupus erythematosus; T1D, type 1 diabetes; TCR, T cell receptor; TGA, Australian Therapeutic Goods Administration; TIGIT, T-cell Immunoreceptor with Ig and ITIM domains; TIM-3, T-cell immunoglobulin and mucin-domain domain-containing molecule-3; TMAs, thrombotic microangiopathies; TNF, tumor necrosis factor; UK, United Kingdom; US, United States; VEGF, human vascular endothelial growth factor; VHH, variable heavy chain single-domain antibodies
Disclosure statement
HK and JV are employed by companies that develop antibody therapeutics. JMR is employed by The Antibody Society, a non-profit trade association funded by corporate sponsors that develop antibody therapeutics or provide services to companies that develop antibody therapeutics, and she is Editor-in-Chief of mAbs, a biomedical journal focused on topics relevant to antibody therapeutics development. Data in this publication were collected from publicly available sources.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2153410
References
- 1.Kaplon H, Chenoweth A, Crescioli S, Reichert JM.. Antibodies to watch in 2022. MAbs. 2022;14(1):2014296. doi: 10.1080/19420862.2021.2014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1):1860476. doi: 10.1080/19420862.2020.1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs. 2020;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11(2):219–42. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs. 2018;10(2):183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9(2):167–81. doi: 10.1080/19420862.2016.1269580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reichert JM. Antibodies to watch in 2016. MAbs. 2016;8(2):197–204. doi: 10.1080/19420862.2015.1125583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichert JM. Antibodies to watch in 2015. MAbs. 2015;7(1):1–8. doi: 10.4161/19420862.2015.988944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichert JM. Antibodies to watch in 2014. MAbs. 2014;6(1):5–14. doi: 10.4161/mabs.27333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reichert JM. Which are the antibodies to watch in 2013? MAbs. 2013;5(1):1–4. doi: 10.4161/mabs.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reichert JM. Which are the antibodies to watch in 2012? MAbs. 2012;4(1):1–3. doi: 10.4161/mabs.4.1.18719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs. 2011;3(1):76–99. doi: 10.4161/mabs.3.1.13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reichert JM. Antibodies to watch in 2010. MAbs. 2010;2(1):84–100. doi: 10.4161/mabs.2.1.10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.U.S. Food and Drug Administration. Emergency Use Authorization . [accessed 2022 Nov 19]. https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization.
- 15.Wang Q, Li Z, Ho J, Guo Y, Yanchen A, Liu M, Wang M, Yu J, Sheng Z, Huang Y, et al. Resistance of SARS-CoV-2 Omicron subvariant BA.4.6 to antibody neutralization. bioRxiv preprint posted September 6, 2022 accessed 2022 September 6. 10.1101/2022.09.05.506628 [DOI] [Google Scholar]
- 16.InflaRx NV InflaRx submits request for Emergency Use Authorization to US FDA for vilobelimab for treatment of critically ill COVID-19 patients. September 29, 2022 press release [accessed 2022 Nov 12]. https://www.inflarx.de/Home/Investors/Press-Releases/09-2022-InflaRx-Submits-Request-for-Emergency-Use-Authorization-to-US-FDA-for-Vilobelimab-for-Treatment-of-Critically-Ill-COVID-19-Patients-.html.
- 17.The Antibody Society, Inc. Antibody therapeutics approved or in regulatory review in the EU or US. [accessed 2022 Nov 18]. https://www.antibodysociety.org/resources/approved-antibodies/.
- 18.Immunocore Holdings pl . Immunocore announces FDA approval of KIMMTRAK® (tebentafusp-tebn) for the treatment of unresectable or metastatic uveal melanoma. January 26, 2022, press release [accessed 2022 August 25]. https://ir.immunocore.com/news-releases/news-release-details/immunocore-announces-fda-approval-kimmtrakr-tebentafusp-tebn.
- 19.Nathan P, Hassel JC, Rutkowski P, Baurain JF, Butler MO, Schlaak M, Sullivan RJ, Ochsenreither S, Dummer R, Kirkwood JM, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385(13):1196–206. doi: 10.1056/NEJMoa2103485. [DOI] [PubMed] [Google Scholar]
- 20.Genentech . FDA approves Genentech’s Vabysmo, the first bispecific antibody for the eye, to treat two leading causes of vision loss. January 28, 2022, press release [accessed 2022 Aug 23]. https://www.gene.com/media/press-releases/14943/2022-01-28/fda-approves-genentechs-vabysmo-the-firs.
- 21.Heier JS, Khanani AM, Quezada Ruiz C, Basu K, Ferrone PJ, Brittain C, Figueroa MS, Lin H, Holz FG, Patel V, et al. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): two randomised, double-masked, phase 3, non-inferiority trials. Lancet. 2022;399(10326):729–40. doi: 10.1016/S0140-6736(22)00010-1. [DOI] [PubMed] [Google Scholar]
- 22.Wykoff CC, Abreu F, Adamis AP, Basu K, Eichenbaum DA, Haskova Z, Lin H, Loewenstein A, Mohan S, Pearce IA, et al. Efficacy, durability, and safety of intravitreal faricimab with extended dosing up to every 16 weeks in patients with diabetic macular oedema (YOSEMITE and RHINE): two andomized, double-masked, phase 3 trials. Lancet. 2022;399(10326):741–55. doi: 10.1016/S0140-6736(22)00018-6. [DOI] [PubMed] [Google Scholar]
- 23.Genentech . New two-year data confirm Genentech’s Vabysmo improves vision with fewer treatments for people with wet-age-related macular degeneration. July 14, 2022, press release [accessed 2022 Aug 23]. https://www.gene.com/media/press-releases/14960/2022-07-14/new-two-year-data-confirm-genentechs-vab.
- 24.Sanofi . FDA approves Enjaymo™ (sutimlimab-jome), first treatment for use in patients with cold agglutinin disease. February 4, 2022, press release [accessed 2022 Aug 23]. https://www.sanofi.com/en/media-room/press-releases/2022/2022-02-04-22-00-00-2379517.
- 25.Sanofi. European commission approves enjaymo® (sutimlimab) for treatment of hemolytic anemia in adult patients with cold agglutinin disease. Nov 17, 2022. press release [accessed 2022 Nov 18]. https://www.sanofi.com/en/media-room/press-releases/2022/2022-11-17-17-50-00-2558583.
- 26.Röth A, Berentsen S, Barcellini W, D’Sa S, Jilma B, Michel M, Weitz IC, Yamaguchi M, Nishimura J-I, Vos JMI, et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood. 2022;blood.2021014955. doi: 10.1182/blood.2021014955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bristol Myers Squibb US Food and Drug Administration approves first LAG-3-blocking antibody combination, Opdualag™ (nivolumab and relatlimab-rmbw), as treatment for patients with unresectable or metastatic melanoma. March 18, 2022, press release [accessed 2022 Aug 23]. https://news.bms.com/news/corporate-financial/2022/U.S.-Food-and-Drug-Administration-Approves-First-LAG-3-Blocking-Antibody-Combination-Opdualag-nivolumab-and-relatlimab-rmbw-as-Treatment-for-Patients-with-Unresectable-or-Metastatic-Melanoma/default.aspx.
- 28.Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, Rutkowski P, Gogas HJ, Lao CD, De Menezes JJ, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24–34. doi: 10.1056/NEJMoa2109970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.AstraZeneca . Evusheld (formerly AZD7442) long-acting antibody combination authorised for emergency use in the US for pre-exposure prophylaxis (prevention) of COVID-19. December 8, 2021 press release [accessed 2022 Aug 29]. https://www.astrazeneca.com/content/astraz/media-centre/press-releases/2021/evusheld-long-acting-antibody-combination-authorised-for-emergency-use-in-the-us-for-pre-exposure-prophylaxis-prevention-of-covid-19.html.
- 30.U.S. Food and Drug Administration . FDA authorizes revisions to Evusheld dosing. June 29, 2022 [accessed 2022 Aug 30]. https://www.fda.gov/drugs/drug-safety-and-availability/fda-authorizes-revisions-evusheld-dosing.
- 31.AstraZeneca . AstraZeneca’s antibody combination, Evusheld (tixagevimab co-packaged with cilgavimab) authorised for use in Great Britain for pre-exposure prophylaxis (prevention) of COVID-19. March 17, 2022 [accessed 2022 Aug 30] https://www.astrazeneca.com/media-centre/press-releases/2022/astrazenecas-antibody-combination-evusheld-authorised-for-use-in-great-britain-for-pre-exposure-prophylaxis-prevention-of-covid-19.html.
- 32.AstraZeneca . Evusheld long-acting antibody combination recommended for approval in the EU for the pre-exposure prophylaxis (prevention) of COVID-19. March 24, 2022 [2022. Aug 30] https://www.astrazeneca.com/content/astraz/media-centre/press-releases/2022/evusheld-long-acting-antibody-combination-recommended-for-approval-in-the-eu-for-the-pre-exposure-prophylaxis-prevention-of-covid-19.html.
- 33.AstraZeneca . Evusheld long-acting antibody combination approved for prevention and treatment of COVID-19 in Japan. August 30, 2022 press release [accessed 2022 Aug 30]. https://www.astrazeneca.com/media-centre/press-releases/2022/evusheld-approved-for-covid-19-in-japan.html#!.
- 34.Montgomery H, Hobbs FDR, Padilla F, Arbetter D, Templeton A, Seth Seegobin S, Kim K, Campos JAS, Arends RH, Brodek BH, et al. Efficacy and safety of intramuscular administration of tixagevimab–cilgavimab for early outpatient treatment of COVID-19 (TACKLE): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Resp Med. 2022;10(10):985–96. doi: 10.1016/S2213-2600(22)00180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roche . European Commission approves Roche’s first-in-class bispecific antibody Lunsumio for people with relapsed or refractory follicular lymphoma. June 8, 2022, press release [accessed 2022 Aug 30]. https://www.roche.com/media/releases/med-cor-2022-06-08.
- 36.Genentech . FDA grants priority review to Genentech’s mosunetuzumab for people with relapsed or refractory follicular lymphoma. July 5, 2022, press release [accessed 2022 Aug 30]. https://www.gene.com/media/press-releases/14958/2022-07-05/fda-grants-priority-review-to-genentechs.
- 37.The Janssen Pharmaceutical Companies of Johnson & Johnson . Janssen marks first approval worldwide for TECVAYLI® (teclistamab) with EC authorisation of first-in-class bispecific antibody for the treatment of patients with multiple myeloma. August 24, 2022 press release [accessed 2022 Aug 30]. https://www.jnj.com/janssen-marks-first-approval-worldwide-for-tecvayli-teclistamab-with-ec-authorisation-of-first-in-class-bispecific-antibody-for-the-treatment-of-patients-with-multiple-myeloma.
- 38.Moreau P, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, Nooka AK, Martin T, Rosinol L, Chari A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495–505. doi: 10.1056/NEJMoa2203478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boehringer Ingelheim. U.S . FDA approves first treatment option for generalized pustular psoriasis flares in adults. September 2, 2022 press release [accessed 2022 Sep 2]. https://www.boehringer-ingelheim.com/human-health/skin-diseases/fda-approves-first-gpp-flare-treatment.
- 40.Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, Navarini AA, Zheng M, Xu J, Turki H, et al. Trial of spesolimab for generalized pustular psoriasis. N Engl J Med. 2021;385(26):2431–40. doi: 10.1056/NEJMoa2111563. [DOI] [PubMed] [Google Scholar]
- 41.AstraZeneca . Imjudo (tremelimumab) in combination with Imfinzi approved in the US for patients with unresectable liver cancer. October 24, 2022 press release [accessed 2022 Nov 19]. https://www.astrazeneca.com/media-centre/press-releases/2022/imfinzi-and-imjudo-approved-in-advanced-liver-cancer.html.
- 42.AstraZeneca . Imfinzi and Imjudo with chemotherapy approved in the US for patients with metastatic non-small cell lung cancer. November 11, 2022 press release [accessed 2022 Nov 19]. https://www.astrazeneca.com/media-centre/press-releases/2022/imfinzi-and-imjudo-approved-in-us-for-lung-cancer.html.
- 43.Abou-Alfa GK, Chan SL, Kudo M, Lau G, Kelley RK, Furuse J, Sukeepaisarnjaroen W, Kang Y, Dao TV, De Toni EN, et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. Journal of Clinical Oncology. 2022;40(4_suppl):379–379. doi: 10.1200/JCO.2022.40.4_suppl.379. [DOI] [Google Scholar]
- 44.AstraZeneca . Imfinzi and tremelimumab with chemotherapy demonstrated sustained survival benefit in metastatic non-small cell lung cancer, nearly doubling the number of patients alive after three years vs. chemotherapy. September 11, 2022. press release [accessed 2022 Sep 25]. https://www.astrazeneca.com/content/astraz/media-centre/press-releases/2022/imfinzi-and-tremelimumab-with-chemotherapy-demonstrated-sustained-survival-benefit-in-metastatic-non-small-cell-lung-cancer.html.
- 45.Sanofi . European Commission grants first approval worldwide of Beyfortus® (nirsevimab) for prevention of RSV disease in infants. November 4, 2022 press release [accessed 2022 Nov 19]. https://www.sanofi.com/en/media-room/press-releases/2022/2022-11-04-07-00-00-2548492.
- 46.Hammitt LL, Dagan R, Yuan Y, Baca Cots M, Bosheva M, Madhi SA, Muller WJ, Zar HJ, Brooks D, Grenham A, et al. Nirsevimab for prevention of RSV in healthy late-preterm and term infants. N Engl J Med. 2022 Mar 3;3869:837–46. 10.1056/NEJMoa2110275 [DOI] [PubMed] [Google Scholar]
- 47.Domachowske J, Madhi SA, Simões EAF, Atanasova V, Cabañas F, Furuno K, Garcia-Garcia ML, Grantina I, Nguyen KA, Brooks D, et al. Safety of nirsevimab for RSV in infants with heart or lung disease or prematurity. N Engl J Med. 2022;386(9):892–94. doi: 10.1056/NEJMc2112186. [DOI] [PubMed] [Google Scholar]
- 48.ImmunoGen, Inc . ImmunoGen announces FDA accelerated approval of ELAHERE™ (mirvetuximab soravtansine-gynx) for the treatment of platinum-resistant ovarian cancer. November 14, 2022 press release [accessed 2022 Nov 19]. https://investor.immunogen.com/news-releases/news-release-details/immunogen-announces-fda-accelerated-approval-elaheretm.
- 49.ImmunoGen, Inc . ImmunoGen presents additional efficacy and safety analyses evaluating mirvetuximab soravtansine in ovarian cancer at ASCO. May 26, 2022 press release [accessed 2022 Sept 19]. https://investor.immunogen.com/news-releases/news-release-details/immunogen-presents-additional-efficacy-and-safety-analyses.
- 50.ImmunoGen, Inc . ImmunoGen presents additional analyses evaluating mirvetuximab soravtansine in ovarian cancer at ESMO. September 11, 2022 press release [accessed 2022 Sept 19]. https://investor.immunogen.com/news-releases/news-release-details/immunogen-presents-additional-analyses-evaluating-mirvetuximab.
- 51.Provention Bio , Inc. TZIELD™ (teplizumab-mzwv) approved by FDA as the first and only treatment indicated to delay the onset of Stage 3 type 1 diabetes (T1D) in adult and pediatric patients aged 8 years and older with Stage 2 T1D. November 17, 2022 press release [accessed 2022 Nov 19]. https://investors.proventionbio.com/2022-11-17-TZIELD-TM-teplizumab-mzwv-approved-by-FDA-as-the-first-and-only-treatment-indicated-to-delay-the-onset-of-Stage-3-type-1-diabetes-T1D-in-adult-and-pediatric-patients-aged-8-years-and-older-with-Stage-2-T1D.
- 52.U.S. Food and Drug Administration . FDA briefing document, Endocrinologic and Metabolic Drugs Advisory Committee Meeting, May 27, 2021: Teplizumab BLA761183. [accessed 2022 Nov 19]. https://www.fda.gov/media/149388/download. [Google Scholar]
- 53.Molecular Targeting Technologies, Inc . Molecular Targeting Technologies, Inc., Thomas Jefferson University and North China Pharmaceutical Co., Ltd announced the Chinese approval for a rabies drug. January 26, 2022 press release [accessed 2022 Sept 19]. https://www.businesswire.com/news/home/20220126005104/en/Molecular-Targeting-Technologies-Inc.-Thomas-Jefferson-University-and-North-China-Pharmaceutical-Co.-Ltd-Announced-the-Chinese-Approval-for-a-Rabies-Drug.
- 54.Zhai LL, Wang H, Zhao W, Zhang SF, Miao FM, Cao Y, Chen C, Li YF, Gao J, Lv RY, et al. Efficacy of ormutivimab, a novel recombinant human anti-rabies monoclonal antibody, in post-exposure prophylaxis animal models. Travel Med Infect Dis. 2022;46:102267. doi: 10.1016/j.tmaid.2022.102267. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Shi N, Li G, Li L, Bai Y, Yang L, Zhao W, Gao J, Wei J, Zhao W, et al. Population pharmacodynamic analyses of human anti-rabies virus monoclonal antibody (ormutivimab) in healthy adult subjects. Vaccines (Basel). 2022;10(8):1218. doi: 10.3390/vaccines10081218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shanghai Henlius Biotech, Inc . Henlius receives NMPA approval for its first innovative monoclonal antibody HANSIZHUANG. March 25, 2022 press release [accessed 2022 Sept 19]. https://www.henlius.com/en/NewsDetails-3512-26.html.
- 57.Shanghai Henlius Biotech, Inc . Henlius’ novel anti-PD-1 mAb HANSIZHUANG (Serplulimab) receives NMPA approval for the treatment of sqNSCLC. November 1, 2022 press release [accessed 2022 Nov 10]. https://www.henlius.com/en/NewsDetails-3837-26.html.
- 58.Cheng Y, Han L, Wu L, Chen J, Sun H, Wen G, Ji Y, Dvorkin M, Shi J, Pan Z, et al. Effect of first-line serplulimab vs placebo added to chemotherapy on survival in patients with extensive-stage small cell lung cancer: the ASTRUM-005 randomized clinical trial. JAMA. 2022;328(12):1223–32. doi: 10.1001/jama.2022.16464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akeso, Inc. Akeso’s cadonilimab (PD-1/CTLA-4), the first dual immune checkpoint inhibitor to treat cancer, approved for marketing in China. June 29, 2022 press release [accessed 2022 Sept 19]. https://www.akesobio.com/en/media/akeso-news/20220629/.
- 60.Zhang J, Huang Y, Xi G, Zhang F. HX008: a humanized PD-1 blocking antibody with potent antitumor activity and superior pharmacologic properties. MAbs. 2020;12(1):1724751. doi: 10.1080/19420862.2020.1724751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biopharma LEPU. Anti-PD-1 monoclonal antibody Puyouheng (pucotenlimab injection) developed by LEPU BIOPHARMA was approved for marketing by the national medical products administration. July 22, 2022 press release [accessed 2022 Sept 19]. https://en.lepubiopharma.com/new/187.html.
- 62.SinoCellTech Ltd . China Cell’s commercialization process adds new recruits: anpingxi® - Repertuzumab approved for marketing. August 30, 2022 press release [translated from Chinese, accessed 2022 Sept 19]. http://www.sinocelltech.com/Detail/desc/pid/23/sid/363.html.
- 63.Shi Y, Zhang Q, Hong X, Wang Z, Gao Y, Zou L, Cen H, Gui L, Li Y, Feng J, et al. Comparison of efficacy and safety of ripertamab (SCT400) versus rituximab (Mabthera ®) in combination with CHOP in patients with previously untreated CD20-positive diffuse large B-cell lymphoma: a randomized, single-blind, phase III clinical trial. Hematol Oncol. 2022 Jul 20; 10.1002/hon.3054. [DOI] [PubMed] [Google Scholar]
- 64.Maruho Co., Ltd . Maruho acquires manufacturing and marketing approval in Japan for “Mitchga subcutaneous injection 60mg syringes”, a new treatment targeting itch associated with atopic dermatitis. March 28, 2022 press release [accessed 2022 Sept 19]. https://www.maruho.co.jp/english/information/20220328.html.
- 65.Kabashima K, Matsumura T, Komazaki H, Kawashima M; Nemolizumab JP01 andJP02 Study Group . Nemolizumab plus topical agents in patients with atopic dermatitis (AD) and moderate-to-severe pruritus provide improvement in pruritus and signs of AD for up to 68 weeks: results from two phase III, long-term studies. Br J Dermatol. 2022;186(4):642–51. doi: 10.1111/bjd.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Galderma . Galderma announces positive data from phase III trial, demonstrating efficacy and safety of nemolizumab in patients with prurigo nodularis. June 22, 2022 press release [accessed 2022 Sept 19]. https://www.galderma.com/news/galderma-announces-positive-data-phase-iii-trial-demonstrating-efficacy-and-safety-nemolizumab.
- 67.Taisho Pharmaceutical Co., Ltd . Notification of approval to manufacture and market Nanozora® 30mg syringes for S.C. injection, a therapy for rheumatoid arthritis, in Japan: Japan’s first NANOBODY® therapeutic. September 26, 2022 press release [accessed 2022 Nov 10]. https://www.taisho.co.jp/global/news/2022/20220926001109.html.
- 68.Takeuchi T, Kawanishi M, Nakanishi M, Yamasaki H, Tanaka Y. Phase II/III results of the anti-TNF multivalent NANOBODY® compound ‘ozoralizumab’ in patient with rheumatoid arthritis (OHZORA trial). Arthritis Rheumatol. 2022;74(11):1776–85. doi: 10.1002/art.42273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Genor Biopharma Co., Inc . 1H 2022 Interim results presentation. August 2022. presentation [accessed 2022 Sept 19]. https://www.genorbio.com/media/1300/1h22-interim-results-presentation.pdf.
- 70.An J, Guiling L, Tang J, Li BX, Xiong HH, Qiu H, Luo L, Wang L, Wang D, Zhou Q, et al. Efficacy and safety of the anti–PD-L1 monoclonal antibody socazolimab for recurrent or metastatic cervical cancer: results from the phase I dose-escalation and expansion study. J. Clin. Oncol. 2022;40(16_suppl):5526–5526.https://ascopubs.org/doi/abs/10.1200/JCO.2022.40.16_suppl.5526. [DOI] [PubMed] [Google Scholar]
- 71.Sorrento Therapeutics, Inc . Sorrento’s license partner, Lee’s Pharm, announces full enrollment of 498 patients in a Phase III trial of socazolimab (anti-PD-L1 antibody) for first-line treatment of extensive-stage small-cell lung cancer (ES-SCLC). May 17, 2022 press release [accessed 2022 Sept 19]. https://investors.sorrentotherapeutics.com/news-releases/news-release-details/sorrentos-license-partner-lees-pharm-announces-full-enrollment.
- 72.Jiangsu Hengrui Pharmaceuticals Co., Ltd . Pipeline highlights as of September 2022. [accessed 2022 Nov 19]. www.hengrui.com/en/pipeline.html.
- 73.Wang J, Zhou C, Yao W, Wang Q, Min X, Chen G, Xu X, Li X, Xu F, Fang Y, et al. Adebrelimab or placebo plus carboplatin and etoposide as first-line treatment for extensive-stage small-cell lung cancer (CAPSTONE-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022;23(6):739–47. doi: 10.1016/S1470-2045(22)00224-8. [DOI] [PubMed] [Google Scholar]
- 74.Pharma K. Pipeline. [accessed 2022 Nov 19]. www.kluspharma.com/pipeline.
- 75.Sampei Z, Haraya K, Tachibana T, Fukuzawa T, Shida-Kawazoe M, Gan SW, Shimizu Y, Ruike Y, Feng S, Kuramochi T. Antibody engineering to generate SKY59, a long-acting anti-C5 recycling antibody. PLoS One. 2018;13(12):e0209509. doi: 10.1371/journal.pone.0209509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chugai Pharmaceutical Co ., Ltd. Anti-C5 recycling antibody crovalimab obtains priority review in China for the treatment of paroxysmal nocturnal hemoglobinuria. August 10, 2022 press release [accessed 2022 Aug 30]. www.chugai-pharm.co.jp/english/news/detail/20220810160000_942.html.
- 77.CSPC Pharmaceutical Group Limited . Biologics license application for JMT103 accepted by the NMPA. June 22, 2022 press release [accessed 2022 Aug 30]. https://doc.irasia.com/listco/hk/cspc/announcement/a220622.pdf.
- 78.Liang X, Xue J, Ge X, Li J, Li H, Xue L, Di L, Tang W, Song G, Li Q, et al. Safety, tolerability, and pharmacokinetics/pharmacodynamics of JMT103 in patients with bone metastases from solid tumors. Front Oncol. 2022. Aug 5;12:971594. 10.3389/fonc.2022.971594eCollection 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Niu X, Wei F, Tu C, Huang G, Bi W, Li J, Yao W, Zhang X, Shen J, Zhang G, et al. Efficacy and safety of JMT103 in patients with giant cell tumor of bone: a multicenter, single-arm, open-label, phase Ib/II study. J Clin Oncol. 2021;39(15_suppl):11526. doi: 10.1200/JCO.2021.39.15_suppl.11526. [DOI] [Google Scholar]
- 80.Innovent Biologics, Inc . Innovent announces the NMPA acceptance of the new drug application for tafolecimab injection (anti-PCSK-9 antibody). June 14, 2022 press release [accessed 2022 Aug 30]. https://www.innoventbio.com/InvestorsAndMedia/PressReleaseDetail?key=332.
- 81.Innovent Biologics, Inc . Innovent announces two registration studies of IBI306 (anti-PCSK-9 antibody) met primary endpoint. February 17, 2022 press release [accessed 2022 Aug 30]. https://www.innoventbio.com/InvestorsAndMedia/PressReleaseDetail?key=306.
- 82.Innovent Biologics, Inc . Innovent releases results of a Phase 3 clinical study of IBI306 (PCSK-9 inhibitor) in Chinese patients with heterozygous familial hypercholesterolemia at the American College of Cardiology Annual Congress 2022. April 4, 2022. press release [accessed 2022 Sept 19]. https://www.innoventbio.com/InvestorsAndMedia/PressReleaseDetail?key=314. [Google Scholar]
- 83.Pharma Japan . Novo seeks Japan approval of hemophilia drug concizumab. August 31, 2022 business news [accessed 2022 Nov 25]. https://pj.jiho.jp/article/247369.
- 84.Jiménez-Yuste V, Angchaisuksiri P, Castaman G, Cepo K, Haaning J, Jacobsen S, Mahlangu J, Matsushita T, Nogami K, Shapiro A. Concizumab prophylaxis in patients with haemophilia A or B with inhibitors: efficacy and safety results from the primary analysis of the Phase 3 explorer7 trial. Abstract Number: LB 01.2; International Society on Thrombosis and Haemostasis, London, UK; July 9-13, 2022. [accessed 2022 July 9-13]. https://abstracts.isth.org/abstract/concizumab-prophylaxis-in-patients-with-haemophilia-a-or-b-with-inhibitors-efficacy-and-safety-results-from-the-primary-analysis-of-the-phase-3-explorer7-trial/. [Google Scholar]
- 85.Coherus BioSciences, Inc., Shanghai Junshi Biosciences Co., Ltd . Coherus and Junshi Biosciences announce FDA acceptance of resubmission of BLA for toripalimab for the treatment of nasopharyngeal carcinoma. July 6, 2022 press release [accessed 2022 Oct 15]. https://investors.coherus.com/news-releases/news-release-details/coherus-and-junshi-biosciences-announce-fda-acceptance.
- 86.Mai HQ, Chen QY, Chen D, Hu C, Yang K, Wen J, Li J, Shi YR, Jin F, Xu R, et al. Toripalimab or placebo plus chemotherapy as first-line treatment in advanced nasopharyngeal carcinoma: a multicenter randomized phase 3 trial. Nat Med. 2021;27(9):1536–43. doi: 10.1038/s41591-021-01444-0. [DOI] [PubMed] [Google Scholar]
- 87.U.S. Food and Drug Administration . FDA approval of lymphoma medicine Ukoniq (umbralisib) is withdrawn due to safety concerns. June 1, 2022 FDA Drug Safety Communication; [accessed 2022 Oct 15]. https://www.fda.gov/media/158834/download. [Google Scholar]
- 88.TG Therapeutics Inc . TG Therapeutics announces FDA extension of BLA PDUFA date for ublituximab to treat patients with RMS. May 31, 2022 press release [accessed 2022 Oct 15]. https://ir.tgtherapeutics.com/news-releases/news-release-details/tg-therapeutics-announces-fda-extension-bla-pdufa-date.
- 89.Steinman L, Fox E, Hartung H-P, Alvarez E, Qian P, Wray S, Robertson D, Huang D, Selmaj K, Wynn D, et al. Ublituximab versus teriflunomide in relapsing multiple sclerosis. N Engl J Med. 2022;387(8):704–14. doi: 10.1056/NEJMoa2201904. [DOI] [PubMed] [Google Scholar]
- 90.Eisai Co., Ltd., Biogen , Inc. The U.S. FDA accepts and grants priority review For Eisai’s biologics license application of lecanemab for early Alzheimer’s disease under the accelerated approval pathway. July 6, 2022 press release [accessed 2022 Oct 15]. https://www.eisai.com/news/2022/news202254.html.
- 91.Eisai Co., Ltd., Biogen , Inc. Lecanemab confirmatory Phase 3 clarity ad study met primary endpoint, showing highly statistically significant reduction of clinical decline in large global clinical study of 1,795 participants with early Alzheimer’s disease. September 27, 2022 press release [accessed 2022 Oct 15]. https://investors.biogen.com/news-releases/news-release-details/lecanemab-confirmatory-phase-3-clarity-ad-study-met-primary.
- 92.Byondis . Byondis and medac enter into license and collaboration and supply agreements for anti-HER2 ADC trastuzumab duocarmazine (SYD985). May 3, 2022 press release [accessed 2022 Sept 19]. https://www.byondis.com/media/press-releases/byondis-and-medac-enter-into-license-and-collaboration-and-supply-agreements-for-anti-her2-adc-trastuzumab-duocarmazine.
- 93.Byondis . FDA accepts Byondis’ biologics license application for [vic-]trastuzumab duocarmazine (SYD985) in HER2-positive metastatic breast cancer. July 12, 2022 press release [accessed 2022 Sept 19]. https://www.byondis.com/media/press-releases/fda-accepts-byondis-biologics-license-application-for-vic-trastuzumab-duocarmazine-syd985-in-her2-positive-metastatic-breast-cancer.
- 94.Byondis . EMA validates marketing authorization application for trastuzumab duocarmazine (SYD985) in HER2-positive metastatic breast cancer. July 18, 2022 press release [accessed 2022 Sept 19]. https://www.byondis.com/media/press-releases/ema-validates-marketing-authorization-application-for-trastuzumab-duocarmazine-syd985-in-her2-positive-metastatic-breast-cancer.
- 95.Eli Lilly and Company . Nearly two-thirds of patients respond to mirikizumab treatment at 12 weeks in Lilly’s first-in-class ulcerative colitis Phase 3 LUCENT-1 study. February 18, 2022 press release [accessed 2022 Sep 25]. https://investor.lilly.com/news-releases/news-release-details/nearly-two-thirds-patients-respond-mirikizumab-treatment-12.
- 96.Eli Lilly and Company . Fifty percent of patients with ulcerative colitis treated with mirikizumab achieved clinical remission at one year in Lilly’s pivotal Phase 3 study. May 24, 2022. press release [accessed 2022 Sep 25]. https://investor.lilly.com/news-releases/news-release-details/fifty-percent-patients-ulcerative-colitis-treated-mirikizumab.
- 97.Genentech . New pivotal data demonstrate clinical benefit of Genentech’s glofitamab, a potential first-in-class bispecific antibody for people with aggressive lymphoma. May 26, 2022. press release [accessed 2022 Sept 19]. https://www.gene.com/media/press-releases/14954/2022-05-26/new-pivotal-data-demonstrate-clinical-be.
- 98.Huang Z, Pang X, Zhong T, Qu T, Chen N, Ma S, He X, Xia D, Wang M, Xia M, et al. Penpulimab, an Fc-engineered IgG1 anti-PD-1 antibody, with improved efficacy and low incidence of immune-related adverse events. Front Immunol. 2022;13:924542. doi: 10.3389/fimmu.2022.924542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eli Lilly and Company . Lilly reports second-quarter financial results, highlights momentum of new medicines and pipeline advancements. August 4, 2022 press release [accessed 2022 Oct 15]. https://investor.lilly.com/news-releases/news-release-details/lilly-reports-second-quarter-financial-results-highlights.
- 100.Eli Lilly and Company . Lilly’s donanemab slows clinical decline of Alzheimer’s disease in positive Phase 2 trial. January 11, 2021. press release [accessed 2022 Oct 15]. https://investor.lilly.com/news-releases/news-release-details/lillys-donanemab-slows-clinical-decline-alzheimers-disease.
- 101.Shcherbinin S, Evans CD, Lu M, Andersen SW, Pontecorvo MJ, Willis BA, Gueorguieva I, Hauck PM, Brooks DA, Mintun MA, et al. Association of amyloid reduction after donanemab treatment with tau pathology and clinical outcomes: the TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022;79(10):1015–24. doi: 10.1001/jamaneurol.2022.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Almirall SA Almirall announces EMA acceptance for filing of Marketing Authorization Application (MAA) for lebrikizumab in atopic dermatitis. October 28, 2022 press release [accessed 2022 Nov 25]. https://www.almirall.com/newsroom/news/almirall-announces-ema-acceptance-for-filing-of-marketing-authorization-application-maa-for-lebrikizumab-in-atopic-dermatitis.
- 103.Almirall SA. Almirall: eight out of ten patients maintained skin clearance at one year in lebrikizumab atopic dermatitis monotherapy trials. June 7, 2022 press release [accessed 2022 Sep 25]. https://www.almirall.com/newsroom/news/almirall-eight-out-of-ten-patients-maintained-skin-clearance-at-one-year-in-lebrikizumab-atopic-dermatitis-monotherapy-trials-1.
- 104.Genmab, Inc . Genmab announces U.S. Food and Drug Administration accepts for priority review biologics license application (BLA) for Epcoritamab (DuoBody®-CD3xCD20) for the treatment of relapsed/refractory large B-cell lymphoma (LBCL). 2022 Nov 21 press release [accessed 2022 Nov 21]. https://ir.genmab.com/news-releases/news-release-details/genmab-announces-us-food-and-drug-administration-accepts.
- 105.Hutchings M, Mous R, Clausen MR, Johnson P, Linton KM, Chamuleau MED, Lewis DJ, Sureda Balari A, Cunningham D, Oliveri RS, et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet. 2021;398(10306):1157–69. doi: 10.1016/S0140-6736(21)00889-8. [DOI] [PubMed] [Google Scholar]
- 106.AbbVie, Inc . AbbVie announces late-breaking results from Phase 2 trial of investigational epcoritamab (DuoBody®-CD3xCD20) in patients with relapsed/refractory large B-cell lymphoma (LBCL) at the European Hematology Association (EHA) Annual Congress. June 11, 2022 Press Release [accessed 2022 Sep 25]. https://news.abbvie.com/news/press-releases/abbvie-announces-late-breaking-results-from-phase-2-trial-investigational-epcoritamab-duobody-cd3xcd20-in-patients-with-relapsedrefractory-large-b-cell-lymphoma-lbcl-at-european-hematology-association-eha-annual-congress.htm. [Google Scholar]
- 107.BeiGene LTD BeiGene announces acceptance of 11th regulatory submission for PD-1 inhibitor tislelizumab in China. August 23, 2022 press release [accessed 2022 Sep 25]. https://ir.beigene.com/news/beigene-announces-acceptance-of-11th-regulatory-submission-for-pd-1-inhibitor-tislelizumab-in-china/2c7d63b6-7abe-441b-b23d-8d850c8f7ba4/.
- 108.BeiGene LTD BeiGene provides regulatory update on the U.S. biologics license application (BLA) for PD-1 inhibitor tislelizumab in 2L ESCC. July 14, 2022 press release [accessed 2022 Sep 25]. https://ir.beigene.com/news/beigene-provides-regulatory-update-on-the-u-s-biologics-license-application-bla-for-pd-1-inhibitor-tislelizumab/09127f95-fd94-4a1b-b215-3e356e5fcfd9/.
- 109.Novartis . Novartis announces anti-PD-1 tislelizumab accepted by EMA for regulatory review in esophageal and lung cancers. April 6, 2022 press release [accessed 2022 Sep 25]. https://www.novartis.com/news/media-releases/novartis-announces-anti-pd-1-tislelizumab-accepted-ema-regulatory-review-esophageal-and-lung-cancers.
- 110.Shen L, Kato K, Kim S-B, Ajani JA, Zhao K, He Z, Yu X, Shu Y, Luo Q, Wang J, et al. Tislelizumab versus chemotherapy as second-line treatment for advanced or metastatic esophageal squamous cell carcinoma (RATIONALE-302): a randomized phase iii study. J Clin Oncol. 2022 Sep 10;40(26):3065–76. 10.1200/JCO.21.01926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.BeiGene LTD Results from RATIONALE 303: a global Phase 3 study of tislelizumab vs docetaxel as second- or third-line therapy for patients with locally advanced or metastatic NSCLC. BeiGene presentation [accessed 2022 Oct 5]. https://www.beigenemedical.com/CongressDocuments/Zhou_BGB-A317-303_AACR_presentation_2021.pdf.
- 112.Lu S, Wang J, Yu Y, Yu X, Hu Y, Ai X, Ma Z, Li X, Zhuang W, Liu Y, et al. Tislelizumab plus chemotherapy as first-line treatment for locally advanced or metastatic nonsquamous NSCLC (RATIONALE 304): a Randomized Phase 3 Trial. J Thorac Oncol. 2021;16(9):1512–22. doi: 10.1016/j.jtho.2021.05.005. [DOI] [PubMed] [Google Scholar]
- 113.Wang J, Lu S, Yu X, Hu Y, Sun Y, Wang Z, Zhao J, Yu Y, Hu C, Yang K, et al. Tislelizumab plus chemotherapy vs chemotherapy alone as first-line treatment for advanced squamous non–small-cell lung cancer: a Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021;7(5):709–17. doi: 10.1001/jamaoncol.2021.0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee Meeting , June 24, 2021: BLA 761209 – retifanlimab. [accessed 2022 Sep 25]. https://www.fda.gov/media/150325/download.
- 115.Incyte Corporation . Incyte provides regulatory update on retifanlimab for the treatment of certain patients with squamous cell carcinoma of the anal canal (SCAC). July 23, 2021 press release [accessed 2022 Sep 25]. https://investor.incyte.com/news-releases/news-release-details/incyte-provides-regulatory-update-retifanlimab-treatment-certain.
- 116.Rao S, Jones M, Bowman J, Tian C, Spano JP. POD1UM-303/InterAACT 2: a phase III, global, randomized, double-blind study of retifanlimab or placebo plus carboplatin–paclitaxel in patients with locally advanced or metastatic squamous cell anal carcinoma. Front Oncol. 2022;12:935383. doi: 10.3389/fonc.2022.935383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Khaled SK, Claes K, Goh YT, Kwong YL, Leung N, Mendrek W, Nakamura R, Sathar J, Ng E, Nangia N, et al. Narsoplimab, a mannan-binding lectin-associated serine protease-2 inhibitor, for the treatment of adult hematopoietic stem-cell transplantation–associated thrombotic microangiopathy. J Clin Oncol. 2022;40(22):2447–57. doi: 10.1200/JCO.21.02389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Omeros Corporation . Omeros Corporation receives decision from FDA on formal dispute resolution request for narsoplimab. November 8, 2022 press release [accessed 2022 Nov 10]. https://investor.omeros.com/news-releases/news-release-details/omeros-corporation-receives-decision-fda-formal-dispute.
- 119.Innovent Biologics, Inc., Eli Lilly and Company . BLA761222 ODAC - Sintilimab BLA in non-squamous NSCLC. February 10, 2022 presentation [accessed 2022 Oct 15]. https://www.fda.gov/media/156090/download.
- 120.Eli Lilly and Company . Lilly announces complete response letter for sintilimab in combination with pemetrexed and platinum chemotherapy for the first-line treatment of people with nonsquamous non-small cell lung cancer. March 24, 2022 press release [2022. Sep 25]. https://investor.lilly.com/news-releases/news-release-details/lilly-announces-complete-response-letter-sintilimab-combination.
- 121.Y-mAbs Therapeutics , Inc. Y-mAbs announces pivotal data for omburtamab. October 3, 2022 press release [accessed 2022 Oct 15]. https://ir.ymabs.com/news-releases/news-release-details/y-mabs-announces-pivotal-data-omburtamab.
- 122.Wilkinson I, Hale G. Systematic analysis of the varied designs of 819 therapeutic antibodies and Fc fusion proteins assigned international nonproprietary names. MAbs. 2022;14(1):2123299. doi: 10.1080/19420862.2022.2123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.PhaseBio Pharmaceuticals, Inc . PhaseBio Pharmaceuticals reports second quarter 2022 financial results and recent business highlights. August 12, 2022 press release [accessed 2022 Sep 25]. https://investors.phasebio.com/news-releases/news-release-details/phasebio-pharmaceuticals-reports-second-quarter-2022-financial.
- 124.Bhatt DL, Pollack CV, Mazer CD, Angiolillo DJ, Steg PG, James SK, Weitz JI, Ramnath R, Arnold SE, Mays MC, et al. Bentracimab for ticagrelor reversal in patients undergoing urgent surgery. NEJM Evid. 2022;1(3):3. doi: 10.1056/EVIDoa2100047. [DOI] [PubMed] [Google Scholar]
- 125.Regeneron Pharmaceuticals, Inc . Regeneron corporate presentation. August 2022. [accessed 2022 Sep 25]. https://investor.regeneron.com/static-files/2bed4561-b43f-4fe6-be95-021f223cd7cb.
- 126.CSL Limited . CSL announces positive top-line Phase 3 results for garadacimab as preventive treatment in patients with hereditary angioedema (HAE). August 17, 2022 press release [accessed 2022 Sep 25]. https://www.csl.com/news/2022/20220817-csl-announces-positive-results-for-garadacimab.
- 127.McKenzie A, Roberts A, Malandkar S, Feuersenger H, Panousis C, Pawaskar D. A phase I, first-in-human, randomized dose-escalation study of anti-activated factor XII monoclonal antibody garadacimab. Clin Transl Sci. 2022;15(3):626–37. doi: 10.1111/cts.13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wong KL, Li Z, Ma F, Wang D, Song N, Chong CH, Luk KK, Leung SO. SM03, an anti-CD22 antibody, converts cis-to-trans ligand binding of CD22 against α2,6-linked sialic acid glycans and immunomodulates systemic autoimmune diseases. J Immunol. 2022 Jun 15;208(12):2726–37. doi: 10.4049/jimmunol.2100820 [DOI] [PubMed] [Google Scholar]
- 129.SinoMab BioScience Limited . Interim report 2022. [accessed 2022 Oct 15]. https://files.services/files/517/2022/0927/20220927164502_35278113_en.pdf
- 130.Li J, Li M, Wu D, Zhou J, Leung S-O, Zhang F. SM03, an anti-human CD22 monoclonal antibody, for active rheumatoid arthritis: a phase II, randomized, double-blind, placebo-controlled study. Rheumatology (Oxford). 2022. 5;61(5):1841–48. doi: 10.1093/rheumatology/keab699. [DOI] [PubMed] [Google Scholar]
- 131.Kodiak Sciences Inc . Kodiak Sciences announces second quarter 2022 financial results and recent business highlights. August 9, 2022 press release [accessed 2022 Nov 10]. https://ir.kodiak.com/news-releases/news-release-details/kodiak-sciences-announces-second-quarter-2022-financial-results.
- 132.Kodiak Sciences Inc . Corporate presentation, August 2022. [accessed 2022 Nov 19]. https://ir.kodiak.com/static-files/687f81e2-520a-4b6e-a91b-cc06da614bb4.
- 133.Kodiak Sciences Inc . KSI-301 anti-VEGF antibody biopolymer conjugate for retinal vein occlusion: primary 24-week efficacy and safety outcomes of the beacon phase 3 pivotal study, September 30, 2022 presentation. [accessed 2022 Sep 25]. https://ir.kodiak.com/static-files/f3a30a82-ed03-4313-acb1-73f22e2a0c4b.
- 134.Syndax Pharmaceuticals, Inc . Syndax Pharmaceuticals reports third quarter 2022 financial results and provides clinical and business update. November 3, 2022 pess release [accessed 2022 Nov 10]. https://ir.syndax.com/news-releases/news-release-details/syndax-pharmaceuticals-reports-third-quarter-2022-financial.
- 135.Innovent Biologics, Inc . Innovent 2022 interim results, August 2022. [accessed 2022 Oct 31]. https://img.innoventbio.com/file1.pdf.
- 136.Regeneron Pharmaceuticals, Inc . Regeneron corporate presentation, August 2022. [accessed 2022 Oct 15]. https://investor.regeneron.com/static-files/2bed4561-b43f-4fe6-be95-021f223cd7cb.
- 137.Bannerji R, Arnason JE, Advani RH, Brown JR, Allan JN, Ansell SM, Barnes JA, O’Brien SM, Chávez JC, Duell J, et al. Odronextamab, a human CD20×CD3 bispecific antibody in patients with CD20-positive B-cell malignancies (ELM-1): results from the relapsed or refractory non-Hodgkin lymphoma cohort in a single-arm, multicentre, phase 1 trial. Lancet Haematol. 2022;9(5):e327–e339. doi: 10.1016/S2352-3026(22)00072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Checkpoint Therapeutics, Inc . Checkpoint Therapeutics corporate presentation September 2022. [accessed 2022 Sep 25]. https://ir.checkpointtx.com/company-information/presentations.
- 139.Checkpoint Therapeutics, Inc . Checkpoint Therapeutics reports second quarter 2022 financial results and recent corporate highlights. August 12, 2022 press release [accessed 2022 Sep 25]. https://ir.checkpointtx.com/news-events/press-releases/detail/87/checkpoint-therapeutics-reports-second-quarter-2022.
- 140.Actinium Pharmaceuticals, Inc. Actinium Investor Presentation September 2022. [accessed 2022 Sep 25]. https://d1io3yog0oux5.cloudfront.net/_f3b741a645f0b39c25728ef872153297/actiniumpharma/db/206/944/pdf/Actinium_Investor+Presentation_September+2022.pdf
- 141.Actinium Pharmaceuticals, Inc . Actinium announces positive top-line results from pivotal Phase 3 SIERRA trial of Iomab-B in patients with active relapsed or refractory acute myeloid leukemia. October 31, 2022 press release [accessed 2022 Oct 31]. https://ir.actiniumpharma.com/press-releases/detail/428/actinium-announces-positive-top-line-results-from-pivotal.
- 142.Actinium Pharmaceuticals, Inc. Actinium Pharmaceuticals, Inc . announces 82% of control arm patients did not receive a bone marrow transplant by conventional means but 100% of patients who received Iomab-B, including crossover patients, in the Phase 3 SIERRA trial successfully engrafted at the Transplantation & Cellular Therapy Tandem Meetings of ASTCT and CIBMTR. April 25, 2022 press release [accessed 2022 Sep 25]. https://ir.actiniumpharma.com/press-releases/detail/421/actinium-pharmaceuticals-inc-announces-82-of-control-arm. [Google Scholar]
- 143.AstraZeneca . Clinical trials appendix H1 2022 results update [accessed 2022 Sep 25]. https://www.astrazeneca.com/content/dam/az/PDF/2022/h1-2022/H1-2022-results-clinical-trials-appendix.pdf.
- 144.Astellas Pharma , Inc. Integrated report 2022 for the year ending March 31, 2022. [accessed 2022 October 5]. https://www.astellas.com/en/system/files/astellas_ir2022_en_20221004.pdf.
- 145.Astellas Pharma, Inc . Astellas announces zolbetuximab meets primary endpoint in Phase 3 SPOTLIGHT trial as first-line treatment in Claudin 18.2 positive, HER2-negative locally advanced or metastatic gastric and gastroesophageal junction (GEJ) cancers. November 17, 2022 press release [accessed 2022 Nov 17]. https://www.astellas.com/en/news/26821.
- 146.Elevar Therapeutics, Inc . Elevar Therapeutics announces positive pre-NDA meeting for rivoceranib combination with camrelizumab as hepatocellular carcinoma treatment option. October 11, 2022. press release [accessed 2022 Oct 31]. https://elevartherapeutics.com/2022/10/11/elevar-therapeutics-announces-positive-pre-nda-meeting-for-rivoceranib-combination-with-camrelizumab-as-hepatocellular-carcinoma-treatment-option/.
- 147.Jiangsu Hengrui Pharmaceuticals Co., Ltd . Camrelizumab in combination with rivoceranib (apatinib) significantly prolonged overall survival and progression-free survival vs sorafenib in patients with unresectable hepatocellular carcinoma in a multinational Phase 3 trial, a joint program with Elevar Therapeutics. September 10, 2022 press release [2022 Oct 31]. https://www.hengrui.com/en/media/detail-254.html.
- 148.CStone Pharmaceuticals . CStone Pharmaceuticals reports 2022 interim results and business updates. August 25, 2022 press release [accessed 2022 Oct 31]. https://www.cstonepharma.com/en/html/news/2808.html.
- 149.Huang H, Tao R, Yang Y, Cen H, Zhou H, Guo Y, Zou L, Cao J, Huang Y, Jin J, et al. GEMSTONE-201: preplanned primary analysis of a multicenter, single-arm, phase 2 study of sugemalimab (suge) in patients (pts) with relapsed or refractory extranodal natural killer/T cell lymphoma (R/R ENKTL). J Clin Oncol. 2022;40(16):7501–7501. doi: 10.1200/JCO.2022.40.16_suppl.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.I-Mab Biopharma . I-Mab partner MorphoSys announces new license agreements for felzartamab and TJ210. June 15, 2022 press release [accessed 2022 Sep 25]. https://ir.i-mabbiopharma.com/news-releases/news-release-details/i-mab-partner-morphosys-announces-new-license-agreements.
- 151.I-Mab Biopharma . Pioneering the Next-Generation of Immuno-Oncology. October 2022. corporate presentation [accessed 2022 Nov 25]. https://ir.i-mabbiopharma.com/system/files-encrypted/nasdaq_kms/assets/2022/10/19/7-56-59/Corporate%20Presentation_Oct%202022_v2.pdf.
- 152.Sanofi . Sanofi and Innovent Biologics enter strategic collaboration to accelerate development of oncology medicines and expand presence in China. August 4, 2022 press release [accessed 2022 Sep 25]. https://www.sanofi.com/en/media-room/press-releases/2022/2022-08-04-15-30-00-2492626.
- 153.Sanofi . Sanofi Q2 2022 results. July 28, 2022 presentation. [accessed 2022 Sep 25]. www.sanofi.com/dam/jcr:2c6b06a7-37bd-4a6a-b243-3ac5cee2752c/2022_07_28_Sanofi_Q2_2022_Results.pdf [Google Scholar]
- 154.Roche Group . Roche YTD September 2022 sales. October 18, 2022 presentation with appendix [accessed 2022 Nov 10]. https://www.roche.com/investors/events/q3-2022.
- 155.Roche Group . Roche reports interim results for phase III SKYSCRAPER-01 study in PD-L1-high metastatic non-small cell lung cancer. May 11, 2022 Roche press release [accessed 26 Sep 2022]. https://www.roche.com/media/releases/med-cor-2022-05-11.
- 156.Pfizer Inc . Pfizer’s elranatamab granted FDA Breakthrough Therapy designation for relapsed or refractory multiple myeloma. November 3, 2022 press release [accessed 2022 Nov 10]. https://www.pfizer.com/news/press-release/press-release-detail/pfizers-elranatamab-granted-fda-breakthrough-therapy.
- 157.Pfizer Inc . Second quarter 2022 earnings teleconference. July 28, 2022 [accessed 2022 Sep 25]. https://s28.q4cdn.com/781576035/files/doc_financials/2022/q2/Q2-2022-Earnings-Charts-FINAL.pdf.
- 158.CStone Pharmaceuticals . Investor presentation, July 2022. [accessed 2022 Sep 25]. https://www.cstonepharma.com/en/uploads/2022/07/165846866935356.pdf.
- 159.Shen L, Guo Y, Zhang Y, Li W, Gong J, Li Q, Ma Z, Wang N, Su R, Cai Z, et al. Updated efficacy and safety results from a phase 1b study of the PD-1 antagonist CS1003 combined with lenvatinib (LEN) as first-line (1L) treatment in Chinese patients (pts) with unresectable hepatocellular carcinoma (uHCC). Journal of Clinical Oncology. (June 01, 2022);40(16_suppl):e16191–e16191. 10.1200/JCO.2022.40.16_suppl.e16191 [DOI] [Google Scholar]
- 160.Alphamab Oncology . The first interim analysis of KN046 Phase III trial successfully reached prespecified PFS endpoint. March 31, 2022 press release [accessed 2022 Sep 25]. https://www.alphamabonc.com/en/html/news/2414.html.
- 161.Zhao Y, Chen G, Li X, Wu J, Chang B, Hu S, Yang S, Liu Y, Wang N, Huang Y, et al. Two-year follow-up from KN046 in combination with platinum doublet chemotherapy as first-line (1L) treatment for NSCLC: an open-label, multi-center phase II trial. European Society for Medical Oncology Congress, September 10, 2022, Paris, France. Poster session 14; abstract 1029P. [accessed 2022 Sep 25]. https://oncologypro.esmo.org/meeting-resources/esmo-congress/two-year-follow-up-from-kn046-in-combination-with-platinum-doublet-chemotherapy-as-first-line-1l-treatment-for-nsclc-an-open-label-multi-center. [Google Scholar]
- 162.Regeneron Pharmaceuticals, Inc . Regeneron corporate presentation, August 2022. [accessed 2022 Sep 25]. https://investor.regeneron.com/static-files/2bed4561-b43f-4fe6-be95-021f223cd7cb.
- 163.Regeneron Pharmaceuticals, Inc . New REGN5458 (BCMAXCD3) Phase 1 data show 75% response rate at highest dose levels studied in patients with heavily pretreated multiple myeloma. December 11, 2021 press release [accessed 2022 Sep 25]. https://newsroom.regeneron.com/news-releases/news-release-details/new-regn5458-bcmaxcd3-phase-1-data-show-75-response-rate-highest.
- 164.Jazz Pharmaceuticals plc . Jazz Pharmaceuticals and Zymeworks announce exclusive license agreement to develop and commercialize zanidatamab, a HER2-targeted bispecific antibody. October 19, 2022 press release [accessed 2022 Oct 31]. http://investor.jazzpharma.com/news-releases/news-release-details/jazz-pharmaceuticals-and-zymeworks-announce-exclusive-license.
- 165.Zymeworks, Inc. Zymeworks reports last patient enrolled in pivotal study of zanidatamab in treatment of HER2-expressing late-line biliary tract cancer. April 28, 2022. press release [accessed 2022 Sep 25]. https://ir.zymeworks.com/news-releases/news-release-details/zymeworks-reports-last-patient-enrolled-pivotal-study.
- 166.Zymeworks, Inc. Corporate presentation, September 2022. [accessed 2022 Oct 31]. https://ir.zymeworks.com/static-files/efbb3a4f-99d3-4eb2-9c9f-681abd2e1b6c.
- 167.Tabernero J, Shen L, Elimova E, Ku G, Liu T, Shitara K, Lin X, Boyken L, Li H, Grim J, et al. HERIZON-GEA-01: zanidatamab + chemo ± tislelizumab for 1L treatment of HER2-positive gastroesophageal adenocarcinoma. Future Oncol. 2022;18. doi: 10.2217/fon-2022-0595. [DOI] [PubMed] [Google Scholar]
- 168.The Janssen Pharmaceutical Companies of Johnson & Johnson . Janssen announces U.S. FDA Breakthrough Therapy designation granted for talquetamab for the treatment of relapsed or refractory multiple myeloma. June 29, 2022 press release [accessed 2022 Nov 17]. https://www.janssen.com/us/sites/www_janssen_com_usa/files/janssen_announces_us_fda_breakthrough_therapy_designation_granted_for_talquetamab_for_the_treatment_of_relapsed_or_refractory_multiple_myeloma.pdf.
- 169.Minnema MC, Krishnan A, Berdeja JG, Oriol A, van de Donk NWCJ, Rodríguez-Otero P, Morillo D, Mateos M-V, Costa LJ, Caers J, et al. Talquetamab, a G protein-coupled receptor family C Group 5 member D x CD3 bispecific antibody, in relapsed/refractory multiple myeloma: updated efficacy and safety results from MONUMENTAL-1. Abstract release date: 05/26/22 EHA Library [accessed 2022 Nov 17]. https://library.ehaweb.org/eha/2022/eha2022-congress/357046/monique.c.minnema.talquetamab.a.g.protein-coupled.receptor.family.c.group.5.html.
- 170.Regeneron Pharmaceuticals, Inc . Regeneron reports third quarter 2022 financial and operating results. November 3, 2022 press release [accessed 2022 Nov 17]. https://investor.regeneron.com/news-releases/news-release-details/regeneron-reports-third-quarter-2022-financial-and-operating.
- 171.Nordic Nanovector ASA and APIM Therapeutics . Nordic Nanovector and APIM Therapeutics announce merger agreement. November 9, 2022 press release [accessed 2022 Nov 19]. https://www.prnewswire.com/news-releases/nordic-nanovector-and-apim-therapeutics-announce-merger-agreement-301673852.html.
- 172.Ambrx Biopharma Inc. Ambrx Biopharma Inc . announces strategic reprioritization and provides corporate update. October 18, 2022 press release [accessed 2022 Nov 17]. https://ir.ambrx.com/news/news-details/2022/Ambrx-Biopharma-Inc.-Announces-Strategic-Reprioritization-and-Provides-Corporate-Update/default.aspx.
- 173.Roche . Roche provides update on Phase III GRADUATE programme evaluating gantenerumab in early Alzheimer’s disease. November 14, 2022 press release [accessed 2022 Nov 19]. https://www.roche.com/media/releases/med-cor-2022-11-14
- 174.Sesen Bio, Inc., Carisma Therapeutics Inc . Sesen Bio and Carisma Therapeutics announce merger agreement. September 21, 2022 press release [accessed 2022 Nov 19]. https://carismatx.com/sesen-bio-and-carisma-therapeutics-announce-merger-agreement/
- 175.Carlo-Stella C, Ansell S, Zinzani PL, Radford J, Maddocks K, Pinto A, Collins GP, Bachanova V, Bartlett N, Bence-Bruckler I, et al. Camidanlumab tesirine: updated efficacy and safety in an open-label, multicenter, Phase 2 study of patients with relapsed or refractory classical Hodgkin lymphoma (r/r CHL). European Hematology Association Hybrid Congress, June 10, 2022, Vienna (Austria). https://library.ehaweb.org/eha/2022/eha2022-congress/357065/carmelo.carlo-stella.camidanlumab.tesirine.updated.efficacy.and.safety.in.an.html. [Google Scholar]
- 176.ADC Therapeutics SA . ADC Therapeutics reports third quarter 2022 financial results and provides business updates. November 8, 2022 press release [accessed 2022 Nov 19]. https://ir.adctherapeutics.com/press-releases/press-release-details/2022/ADC-Therapeutics-Reports-Third-Quarter-2022-Financial-Results-and-Provides-Business-Updates/default.aspx.
- 177.Fashoyin-Aje LA, Mehta GU, Beaver JA, Pazdur R. The on- and off-ramps of oncology accelerated approval. N Engl J Med. 2022;387(16):1439–42. doi: 10.1056/NEJMp2208954. [DOI] [PubMed] [Google Scholar]
- 178.GSK . GSK provides an update on Blenrep (belantamab mafodotin-blmf) US marketing authorization. November 22, 2022 press release [accessed 2022 Nov 22]. https://www.gsk.com/en-gb/media/press-releases/gsk-provides-update-on-blenrep-us-marketing-authorisation/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.