

Abstract
Liver cancer is the sixth most prevalent type of cancer worldwide and accounts for the third most frequent cause of cancer-associated mortality. Conventional anticancer drugs display limited efficacy owing to their short half-life, poor solubility and inefficient drug delivery. Despite advancements being made in drug discovery and development for the treatment of hepatocellular carcinoma (HCC), drug inefficacy and drug continue to pose significant obstacles to effective treatment. Therefore, it is imperative that novel treatment strategies be developed with the aim of developing anticancer treatments without any side-effects and with long-term durability. Extracellular vesicles, such as exosomes, intercellular communication agents which have the ability to carry heterogenous molecules with high penetrability, low immunogenicity and longer durability, may provide a versatile natural delivery system. The present review article illustrates the innovative treatment strategy using exosomes as a delivery agent for two distinct anticancer candidates, i.e., tumor necrosis factor-related apoptosis-inducing ligand and microRNA-335. The aim of the present review was to present a unique strategy for the development of an exceptional anticancer treatment therapy exploiting exosomes as a delivery vehicle which may be used for HCC.
Keywords: hepatocellular carcinoma, miR335, tumor necrosis factor-related apoptosis-inducing ligand, anticancer therapy, exosomes, drug resistance
1. Introduction
Liver cancer is the sixth most prevalent form of cancer worldwide and accounts for the third most frequent cause of cancer-associated mortality (1,2) (Fig. 1). Hepatocellular carcinoma (HCC) is the most predominant form of malignant liver cancer, which is responsible for ~90% of all non-metastatic liver cancers and has been reported to be associated with cirrhosis and hepatitis B or C virus (HBV/HCV) infection (3,4). Other risk factors include obesity, iron overload, alcohol consumption, diabetes, fatty liver disease and smoking (5).
Figure 1.

Worldwide Epidemiology of Liver Cancer in 2022 (International Agency for Research on Cancer) Cancer Today; https://gco.iarc.fr/today/home; accessed October 21, 2022). (A) The estimated incidences of different types of cancer worldwide in 2022. (B) Histogram representation of the estimated number of incident cases and deaths worldwide. (C) Number of deaths from liver cancer from 2020 to 2040 as estimated by the World Health Organization. (D) Current treatment approaches for HCC. HCC, hepatocellular carcinoma; TACE, transarterial chemoembolization; HAIC, hepatic arterial infusion chemotherapy; TKI, tyrosine kinase inhibitor; ICI, immune checkpoint inhibitor.
However, according to statistics, only 25% of patients with HCC are diagnosed in the initial stages at the onset of the disease (6). A probable reason for this may be the absence of initial symptoms and frequent overlap with other diseases, thus making it difficult to distinguish HCC from other clinical conditions. The survival rates of cancer patients may be markedly enhanced by timely and precise diagnosis at the initial stages (7). As with the late detection of advanced-stage HCC, the diagnosis of cancer at a late stage suggests that the patient has reached a stage that is non-curative. Henceforth, the chances of survival are greatly minimized, and such patients are placed under palliative care due to the high rate of metastasis and relapse, as occurs in patients with late-stage HCC (8,9).
At present, the most common strategies for the treatment of HCC include ablation, surgical resection, liver transplantation, chemotherapy, transarterial chemoembolization (TACE), radiotherapy and combination therapy depending on the disease staging and patient's profile (Fig. 1) (10). However, these conventional therapies have multiple limitations that compromise the quality of life of patients receiving these therapies. For example, undesired effects of radiotherapy and the development of resistance to chemotherapy due to long-term treatment and transplantation lead to long-term immunosuppressive therapy (11). Although substantial advancements have been made over the past decade in the management and treatment of HCC, including liver resection or transplantation and ablation, only ~15% of patients with early-stage HCC without cirrhosis are eligible for surgical removal. TACE is another available treatment option for patients with intermediate-stage HCC that results in a 23% increase in the 2-year survival rate when compared to traditional treatment therapies (12).
Most HCC cases are diagnosed predominantly in the late stages of the disease, which renders both surgical (resection and transplantation) and locoregional treatment (chemoembolization) inadequate for the overall survival of patients. As a result, there is an urgent need for the development of an effective therapy for patients with advanced-stage HCC.
In the SHARP randomized controlled trial, sorafenib as a monotherapy was shown to be efficacious for advanced HCC. With a high safety profile, the sorafenib-treated group exhibited an overall survival rate of 10.7 months compared to 7.9 months for the placebo group (13) (Fig. 2). Sorafenib is currently the only approved prescribed option for the treatment of patients with advanced-stage HCC. Patients with HCC who have not responded to earlier treatments are recommended to use sorafenib, which received authorization by the US Food and Drug Administration (FDA) in 2007 (14). The anticancer effect of sorafenib is based on its ability to obstruct cell proliferation and angiogenesis, which inhibits tumor growth (15). However, sorafenib treatment is beneficial to only a limited number of patients and is often accompanied by drug resistance within 6 months of commencing the treatment. Moreover, the use of sorafenib is also associated with side-effects, such as nausea, alopecia and hypertension (14).
Figure 2.

Timeline of FDA-approved drugs for HCC. The SHARP trial (13) demonstrated the effectiveness of sorafenib against HCC. Compared to the placebo group, the sorafenib-treated group exhibited a markedly longer OS (mOS 10.7 vs. 7.9 months; HR, 0.69; 95% CI, 0.55-0.87; P<0.001). In the REFLECT trial (16), lenvatinib displayed non-inferiority in OS corresponding to sorafenib monotherapy (mOS, 13.6 vs. 12.3 months; HR, 0.92; 95% CI, 0.79-1.06). In the IMbrave150 trial (17), the efficacy of bevacizumab combined with atezolizumab was compared with that of sorafenib. The combination treatment resulted in a markedly improved outcome than sorafenib monotherapy, exhibiting prolonged OS and PFS (mOS 19.2 vs. 13.4 months; HR, 0.66; 95% CI, 0.52-0.85; P=0.0009). FDA, Food and Drug Administration; HCC, hepatocellular carcinoma; OS, overall survival; HR, hazard ratio; Mos, median OS; CI, confidence interval; ORR, objective response rate; PFS, progress free survival; AEs, adverse effects.
It took >10 years following the approval of sorafenib before a second first-line targeted drug for HCC was developed. As per the outcome from REFLECT trial (16), a randomized phase III non-inferiority trial reported by Kudo et al (16), led to the approval of lenvatinib to be used as the first-line treatment for advanced HCC.
In terms of overall survival, this randomized phase III trial in 2018 demonstrated that lenvatinib was not inferior to sorafenib showing overall survival of 13.6 months compared to 12.6 months for the sorafenib-treated group (16). A recent milestone in the development of HCC first-line drug development was achieved in 2020 when the FDA approved bevacizumab plus atezolizumab, an antibody combination strategy, as a first-line treatment for patients with unresectable HCC on the basis of safety and efficacy determined in the IMbrave150 trial (17). In this phase III study, 501 patients with HCC who had not previously received systemic treatment were compared to the effectiveness of bevacizumab coupled with atezolizumab against sorafenib. By significantly improving the overall survival by 12.6% at 12 months, the combination treatment significantly outperformed sorafenib monotherapy (17). Although all three drugs approved by the FDA for first-line therapy had led to an improved response and survival rate, they are all associated with multiple adverse effects (Fig. 2).
Despite advancements being made in several first- and second-line drugs for the treatment of HCC, the use of these drugs still presents the issue of a compromised lifestyle with the provision of less benefit overall. The quality of life of patients receiving therapy with these drugs does not appear to be improving, and there is also the issue of the high costs of these drugs. Moreover, these targeted therapies are associated with the drawback of an inadequate objective response rate (ORR) and adaptive/acquired resistance (18). Furthermore, the long-term usage of such chemotherapeutic drugs may pose the issue of toxicity, as well as drug inefficacy. Therefore, it is imperative that a novel treatment strategy be developed with the aim of developing targeted therapy that can be applied to patients with HCC at any stage of treatment without posing any side-effects with longer durability.
The present review article illustrates the innovative exosome-based therapy as a delivery agent of two potential anticancer candidates, i.e., tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and microRNA (miRNA/miR)-335. The summary and discussion of scientific investigations highlights the immense potential of harnessing the ability of exosomes for developing an effective anticancer drug therapy for patients with liver cancer and their role in addressing the issue of drug resistance development conferred by existing chemotherapeutics.
2. MicroRNA-335: A novel candidate for anticancer treatment
miRNAs are non-coding RNAs whose function is to perform post-transcriptional gene regulation. They play a key role in tumor development by modulating the expression of various oncogenes and tumor suppressor genes (19). miRNAs are poorly regulated in multiple types of cancers and, as per their function, they function either as oncogenes or tumor suppressors. Oncomirs, such as miR-21 stimulate tumor growth by impeding tumor suppressor genes (20). On the contrary, tumor suppressor miRNAs, such as miR-145 restrict the progression of tumors by blocking oncogenes (21). Furthermore, miRNAs are involved in several biological processes, including cell proliferation, apoptosis, metastasis and angiogenesis (22,23), which are the key features in cancer progression.
miRNAs as a novel class of regulatory factors are crucial to the biological and pathological processes in several cases of human solid tumors (24). According to numerous recent studies, as a tumor suppressor gene, miR-335-5p has been linked to the emergence and growth of several tumors, including colorectal cancer (25), non-small cell lung cancer (26) and HCC (27). Furthermore, hepatic stellate cell-derived miR-335-5p exosomes may be used as prospective miRNA biomarkers in HBV-related HCC (28) and thus have potential therapeutic significance in HCC (29).
Previous research has demonstrated that miR-335 is a suppressor of tumor formation, invasion and metastasis, and is responsible for the regulation of apoptosis and thus has prognostic value in HCC (30). miR-335-3p, sometimes referred to as miR-335*, is generated concurrently with miR-335. Notably, these miRNAs have also been shown to be responsible for inducing the activation of the p53 tumor suppressor pathway to prevent cellular proliferation and neoplastic cell transformation (31). Emerging evidence has depicted the role of miR-335 in the majority of oncogenic signaling pathways responsible for cell growth and survival. It has been discovered that the downregulation of miR-335 in lung cancer enhances cell proliferation by activating the AKT/mTOR signaling pathway, which is one of the most commonly dysregulated signaling pathways in human malignancies (32). This characteristic of miR-335 could be exploited in therapeutic applications for restricting HCC progression and controlling metastasis.
miR-335-5p reportedly targets downstream genes to modulate the biological activity of cancer cells. For example, miR-335-5p overexpression has been shown to suppress the proliferation, migration and invasion of non-small cell lung cancer by targeting CPNE1 (33). In the case of HCC, miR-335-5p has been reported to significantly attenuate the development of this type of cancer. For example, a previous study demonstrated how the circ_0009910/miR-335-5p/Rho-associated coiled-coil-containing protein kinase 1 (ROCK1) axis is crucial to the onset and development of HCC (34). In that study, miR-335-5p inhibited HCC cell proliferation, migration and invasion by targeting ROCK1. By enhancing the inhibitory effects of miR-335-5p on the expression of ROCK1 in HCC, circ_0009910 knockdown exhibited anticancer properties. In addition to that, it was found that the invasiveness of cancer cells was positively associated with ROCK1 (34). In a similar context, Liu et al (35) demonstrated that miR-335 was involved in suppressing HCC cell proliferation, migration and invasion by downregulating ROCK1 expression. Their research investigating molecular mechanisms revealed that ROCK1, which is associated with cell movement and invasion in various cancer types, was a target gene for miR-335 for modulating the proliferation and metastasis of HCC cells (35). Reportedly, ROCK1 functions as an oncogene in HCC and promotes the development of HCC (36).
The significance of miR-335 was further validated by another study which demonstrated that miR-335 restoration inhibited hepatic stellate cell (HSC) migration (37). The findings of that study concluded that miR-335 considerably decreased during HSC activation. Restoring miR-335 expression markedly decreased collagen type I and α-smooth muscle actin levels and prevented cell migration, at least in part through the downregulation of tenascin-C, an extracellular matrix glycoprotein involved in cell migration. The overexpression of miR-335 in HSC may thus provide a novel strategy for the treatment of hepatic fibrosis (37). Another recent study by Yang et al (38) revealed that a newly characterized circular RNA, circ_0005075, promoted HCC progression by suppressing the function of miR-335. Circ_0005075 was discovered to be upregulated in HCC tissues where it was found that the downregulation of circ_0005075 inhibited HCC progression (38). As per their study mitogen-activated protein kinase 1 (MAPK1) was shown to be regulated by miR-335, divulging it as one of the downstream regulatory targets of circ_0005075. It was also shown that the upregulation of circ_0005075 may be responsible for the elevated level of MAPK1 (38). As a crucial member of the MAPK family, the main function of MAPK1 involves cell proliferation, gene expression, differentiation, mitosis, cell survival and apoptosis (39). Likewise, their role is also imperative in tumorigenesis. With the overexpression of MAPK1 in multiple types of cancer, including breast cancer (40), they could be the likely candidates to be used for the prognosis of patients with HCC (38). To sum up, Yang et al (38) demonstrated that miR-335 may target and inhibit MAPK1 (38). Moreover, Ji et al (41) identified octamer-binding transcription factor 4 (OCT4) as another target gene of miR-335-5p. miR-335-5p was demonstrated to prevent OCT4 gene expression to restrict the downstream activation of the Akt signaling pathway, which was demonstrated to be a canonical regulator in the development of liver cancer (41).
Notably, a number of scientific investigations have also discovered a pertinent link between miR-335 expression and the survival of cancer patients (26,42). Furthermore, several findings have indicated that miR-335 influences the chemotherapeutic response in patients receiving standard treatment (43,44). For example, the study by Cui et al (45) demonstrated that serum miR-335 levels can be utilized as a marker to identify the status of disease progression, apart from clinical outcome in patients receiving TACE therapy. TACE therapy is the standard of care treatment for patients with large or multinodular HCC whose treatment response varies and still lacks any prognostic marker. However, the study by Cui et al (45) indicated that low levels of miR-335 in patient serum were associated with a low survival rate with a poor treatment response. Chen and Xia (46) demonstrated the role of miR-335 as a biomarker for HCC treatment and demonstrated its function responsible for regulating sensitivity to sorafenib in HCC. They depicted the fact that miR-335 regulates sorafenib sensitivity in HCC cells by inhibiting the AKT pathway ia targeting C-MET, which is a tyrosine kinase protein involved in the development of cancer (46).
Dohi et al (47) first reported that miR-335, which is located within the intron of its protein-coding host gene, MEST, was downregulated due to aberrant promoter hypermethylation. Primary HCC tissues exhibited considerably higher levels of miR-335/MEST methylation and miR-335 expression was much lower in tumors compared to the non-tumor tissue counterparts (47). Their finding suggested that aberrant DNA methylation in primary HCC was the cause of the decreased miR-335 expression. Furthermore, their findings suggested that a decreased expression of miR-335 may be linked to distant metastases in HCC (47). Similar findings were also provided on the expression of miR-335 in tumor tissues, which was reported to be much lower than in non-tumor tissues compare to HCC patient samples (48). The recent study by Nie et al (49) demonstrated ROCK1 as a target gene of miR-335-5p, where circ_0064288 enhanced ROCK1 expression by competitively binding with miR-335-5p. They suggested that circ_0064288, which is highly expressed in HCC, functions as an oncogene by inhibiting miR-335-5p expression and promoting ROCK1 expression, which is responsible for regulating cell motility. A list of various studies investigating the clinical significance of miR-335 in HCC and their molecular mechanisms is presented in Table I. Thus, potential diagnostic and treatment options for HCC may be provided via the modulation of miR-335.
Table I.
Clinical function and mechanisms of action of miRNA-335 in HCC.
| Clinical significance | Mechanisms | Recipient cells | (Refs.) |
|---|---|---|---|
| Suppresses HCC cell proliferation, migration and invasion | By enhancing the inhibitory effects of miR-335-5p on the expression of ROCK1 in HCC | HepG2, Hep3B, HCCLM3, MHCC97 (human HCC cell lines) | (34) |
| Restricts the proliferation, migration and invasion of HCC cells | Via downregulating the Rho-associated coiled-coil-containing protein kinase 1 | HuH7 cells and HepG2 | (35) |
| circ_0005075 promotes HCC proliferation, migration, invasion, anti-apoptosis, and chemotherapeutic resistance | Via repressing the function of miR-335 | HepG2 and SMMC-7721 cells | (38) |
| Restricts the proliferation of Huh-7 liver cancer cells | Via targeting the OCT4/Akt pathway | Huh7 human liver cancer cells | (41) |
| miR-335 contributes to the sensitizing effects of anticancer drugs | SIAH2 is the target of miR-335 by enhancing the expression of HDAC3 | SNU387R, Malme3MR, SNU387Rtaxol, Malme3MR-Taxol, SNU387-R Vinblastine | (43) |
| Prognostic marker | Via aberrant promoter hypermethylation | Patients with HCC | (45) |
| Modulates sorafenib resistance | Via suppressing the c-Met-Akt pathway through lncRNA NEAT1 | HepG2/Bel7404 | (46) |
| Diminishes the expression of miR-335, which may be associated with distant metastasis in HCC | DNA hypermethylation of CpG islands within promoter regions of protein-coding host gene, MEST | Huh1, Huh7, HLE, HLF and HepG2 | (48) |
| miR-335-5p overexpression partly counteracts the effect of circ_0064288 responsible for HCC cell growth and migration | Circ_0064288 facilitates HCC cell growth and migration by regulating the miR-335-5p/ROCK1 axis | Huh7, Hep3B, HCCLM3, and MHCC97-L | (49) |
| Obstructs the proliferation and invasion, and increases the apoptosis of HCC cells | Shuttle between hepatoma cells and HSCs, downregulate mRNA targets for miR-335 | MHCC97L, MHCC97H, Huh7 and HepG2 cells | (137) |
HCC, hepatocellular carcinoma; OCT4, octamer-binding transcription factor 4; SIAH2, Siah E3 ubiquitin protein ligase 2; HDAC3, histone deacetylase 3; ROCK1, Rho-associated coiled-coil-containing protein kinase 1.
3. Significance of TRAIL in HCC treatment
TRAIL is a pro-apoptotic ligand that has received increasing attention owing to its property of inducing apoptosis in multiple types of cancer cells without affecting normal cells (50). This unique feature of TRAIL has allowed several researchers to investigate the development of TRAIL-receptor agonists as a form of anticancer therapy (51,52). Additionally, TRAIL-based therapeutics are independent of p53 in tumor cells, unlike other chemotherapeutic drugs, which renders them unique in terms of the cell death pathway (53,54).
TRAIL is a transmembrane protein reported to be found on natural killer (NK) cells and cytotoxic T-cell surfaces with a predominant expression in tissues, including the prostate, lungs and spleen (55). TRAIL can be secreted in the soluble form, which is non-toxic to normal cells, while healthy adult plasma contains a trace quantity of endogenous TRAIL (100 pg/ml) (56). Multiple types of cancer cells overexpress the death receptors (DRs), DR4 and DR5 (57). Upon secretion from NK cells, TRAIL binds to DR4 and DR5 (58) and upon binding, it leads to the recruitment of caspase-8 to the Fas-associated death domain adaptor protein. Following activation, it culminates in apoptotic signaling via caspase-3 activation, ultimately leading to cell death (Fig. 3) (59).
Figure 3.

Schematic representation of the exosomal delivery of TRAIL and TRAIL-mediated cell death in recipient HCC cells. TRAIL binds to its cognate DR4 or DR5 on target cancer cells. Upon binding, it leads to the recruitment of caspase-8 to the FADD adaptor protein. Following activation, it culminates in apoptosis signaling via caspase-3-activation, ultimately leading to cell death. The figure was created using BioRender.com. TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; DR, death receptor; FADD, Fas-associated death domain; HCC, hepatocellular carcinoma; Casp, caspase.
The ability of TRAIL to induce tumor-specific cell death renders it a promising candidate for antitumor therapy. In numerous in vitro studies, TRAIL protein in its soluble recombinant form has been widely explored as an anticancer agent (57,60-62). A number of previous publications with the aim of developing antitumor therapy have centered their findings on the in vitro and in vivo tumoricidal activity of TRAIL protein; they demonstrated a peculiar feature of apoptosis induction by recombinant, soluble TRAIL protein in a broad range of cancer cell lines, although they demonstrated no activity against normal cells (63-65). The safe use of TRAIL as a ligand for therapeutic purposes, displaying no noticeable cytotoxicity to normal tissues, has also been validated in mouse models (57) and in humans (66).
The study by Grisendi et al (67) on adipose-derived (AD)-mesenchymal stem cells (MSCs) producing TRAIL revealed that when AD-MSCs loaded with TRAIL were injected into mice, they localized to the tumor site and induced apoptosis without causing any significant toxicity to normal tissues. Those authors also proposed that using stably genetically modified AD-MSCs to deliver TRAIL alone or in conjunction with sensitizing drugs may provide new treatment options for malignancies that remain incurable (67). Furthermore, El-Shemi et al (61) revealed TRAIL and inhibitor of growth 4 (ING4) as potent apoptosis-inducing genes in an orthotopic mouse model of human HCC bearing utilizing oncolytic adenoviruses as a gene delivery agent. Their study found that the combination of these drugs significantly reduced tumor-driven angiogenesis and neovascularization, and also triggered apoptosis and immune responses, without exhibiting any overlapping toxicities (61). Another study by Liu et al (65) revealed that TRAIL plasmid DNA delivered via HCC-targeted lipid/calcium/phosphate/protamine nanoparticles in conjunction with traditional sorafenib therapy decreased HCC development, as well as liver fibrosis in a mouse model of HCC. Overall, these findings provide a promising treatment strategy for cancer based on TRAIL that may be applied in clinical settings.
Another study recently demonstrated the underlying mechanisms of action of TRAIL, which involved causing substantial cytotoxicity to tumor cells only, but seldom affecting non-transformed cells (68). That research revealed an interaction between TRAIL and immediate early response gene (IER3), which is expressed in a number of human tissues, and appears to be downregulated in cancer cells, and its overexpression can stimulate the apoptosis of cancer cells and enhance their sensitivity to chemotherapeutic drugs (68). It was demonstrated that these two proteins may be responsible for directing HCC cells to undergo apoptosis and interrupt with their capacity to proliferate and migrate. These findings demonstrate that TRAIL can partly influence the pathogenesis of HCC by interacting with IER3 to reduce Wnt/-catenin signaling (68). A number of TRAIL-based therapeutics for the treatment of HCC are currently undergoing or have undergone clinical trials, as demonstrated in Table II.
Table II.
List of clinical trials conducted on TRAIL-based therapy against various types of cancer.
| Cancer type | Mechanism | Settings | Clinical trial/status |
|---|---|---|---|
| Advanced-stage HCC | Monoclonal antibody targeting TRAIL-R1 (mapatumumab) | Combination therapy (sorafenib) | Phase II completed (143) (NCT01258608) |
| Advanced non-small cell lung cancer | Apoptosis-inducing recombinant TRAIL via DR4 and DR5 activation (mapatumumab,) | Combination therapy (Paclitaxel and Carboplatin) | Phase III completed in 2018 (144) (NCT00583830) |
| Relapsed and refractory multiple myeloma | Recombinant TRAIL triggering apoptosis via the activation of DR4 and DR5 (CPT) | Combination therapy (thalidomide) | Phase III completed in 2014 (ChiCTRONC-1200206) |
| Advanced-stage HCC | TRAIL receptor agonists against TRAIL-R1(DR4) | Combination of mapatumumab with sorafenib | Phase II (completed in 2013) (NCT01258608) |
| Metastatic triple-negative breast cancer | Monoclonal antibody targeting TRAIL-R2 (tigatuzumab) | Combination therapy (abraxane) | Phase II (completed in 2017) (NCT01307891) |
| Non-small cell lung cancer | Targeted stem cells expressing TRAIL (MSC TRAIL) | Combination therapy (pemetrexed/cisplatin chemotherapy) | Phase III clinical trial estimated to be completed in September, 2025 (NCT03298763) |
| B-cell non-Hodgkin's lymphoma | Recombinant TRAIL triggering apoptosis via activation of DR4 and DR5 (dulanermin) | Combination therapy (rituximab) | Phase II (completed in 2010) (145) (NCT00118209) |
| Advanced solid tumors | TRAIL receptor agonists against DR5 (DS-8273a) | Monotherapy | Phase I (completed in 2017) (146) (NCT02076451) |
| Colorectal cancer non-small cell lung cancer, triple-negative breast cancer, renal cell carcinoma, gastric cancer, pancreatic cancer | Equal mixture of two humanized non-competing DR5-specific monoclonal antibodies (GEN1029) | Monotherapy | Phase III clinical trial estimated to be completed in March, 2022 (NCT03576131) (terminated) |
HCC, hepatocellular carcinoma; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; DR, death receptor.
Challenges with TRAIL therapy
Unfortunately, despite being such a prominent feature of being specific to tumor cells, TRAIL-based therapy still has a long way to go for successful clinical translation. TRAIL-based therapy, including recombinant or agonistic monoclonal antibodies against DR4/DR5 (69,70), has exhibited limited efficacy in a number of clinical trials of different stages due to the short plasma half-life of recombinant TRAIL, limited bioavailability and undesirable systemic toxicity (69). Moreover, TRAIL-based therapy is associated with several challenges, including the development of TRAIL-mediated cell death resistance that leads to TRAIL-induced apoptosis being ineffective in HCC. The root cause of the development of therapeutic resistance may be intrinsic resistance in some highly malignant tumors and acquired resistance post-frequent exposure to TRAIL (71). Other factors may include the activation of anti-apoptotic molecules and multiple receptors of various signaling pathways (71-73).
Developing agonist monoclonal antibodies (mAbs) targeting DR4 or DR5 receptors has been the most prevailing approach due to their long serum half-lives in vivo. A number of clinical trials, have been conducted on several agonists, such as mapatumumab (NCT01258608), lexatumumab (NCT00428272) and tigatuzumab (NCT01307891); however, none of them displayed any antitumor response rates in cancer patients (69). Similarly, in preclinical tumor xenograft mouse models, mapatumumab and lexatumumab have failed to eradicate tumors (74,75). The dimeric structure of the antibody is most likely to blame for the failure of all agonist mAb clinical trials. Since the binding of ligand on DR4 and DR5 induces receptor trimerization following the activation of the extrinsic pathway, maintaining its trimeric structure will be required in the future to enable its functional mechanism of apoptosis induction.
The strategy employing recombinant human native TRAIL appears to be more feasible as it allows the preservation of the original trimeric structure with full functional efficacy. Dulanermin, which is Amgen's version of TRAIL, was not found to be effective against cancer in human clinical trials, even though it was effective in preclinical tumor xenograft models (76). The probable reason for the inefficacy of dulanermin may be related to its poor pharmacokinetic profile due to its very short half-life in mammals. Given its low molecular weight and its non-covalently linked trimeric structure instability, which may cause rapid renal elimination. All these facts illustrate the requirement of a novel approach to tackle this challenge.
To obtain such biologically active TRAIL, many expression systems, such as His, Flag tag or the incorporation of trimerization domains, such as leucine zipper or isoleucine zipper and the stabilization of trimers with cations, as zinc were identified. It has been validated that to assemble and maintain a functionally folded ligand trimer of TRAIL, zinc chelation is critical (77,78). However, it appears to have its own set of drawbacks. For example, both leucine zipper-fused TRAIL and an N-terminus His-tagged TRAIL stabilized by insertion mutation are likely to be immunogenic in humans (57). In addition, particularly the His-tagged version, has been linked to hepatotoxicity not observed with native TRAIL (79,80). Other strategies for prolonging the half-life of TRAIL, which include albumin-conjugated TRAIL nanoparticles or liposome conjugated TRAIL, present the issue of production limitation (81,82).
The recent study by Naval et al (83) demonstrated the significance of the oligomerization of TRAIL receptors in TRAIL-induced apoptosis. They stated that TRAIL, as a transmembrane protein, exhibited more potent pro-apoptotic properties than its soluble form (83). In immune system cells, TRAIL is expressed as a type II membrane protein in the plasma membrane (84,85) or is enclosed within microvesicles (86,87). DR5 is solely triggered by the membrane-bound form of TRAIL, whereas DR4 can be activated by both the soluble and membrane-bound forms of the ligand (88). Given the property of TRAIL of being naturally secreted as a membrane protein in exosomes, a number of nanocarriers, such as liposomes, whose lipid composition replicates natural exosomes with surface-bound TRAIL, have been investigated in several pre-clinical studies on its anticancer properties. Various in vitro and in vivo studies have demonstrated that this membrane-bound form of TRAIL has greater antitumor activity than the soluble form against hematological and solid tumors (89-91). In comparison to soluble TRAIL, this liposomal formulation with TRAIL attached to the liposome surface produces improved DR5 clustering and increased DISC recruitment, resulting in a greater apoptotic signal (90). As TRAIL produces high-order TRAIL oligomers on the lipid nanoparticle surface, improved DR5 clustering and higher DISC recruitment is accomplished (92). Furthermore, TRAIL-encapsulated liposomes and nanoparticles face hurdles with agent release from carriers (93,94). In summary, the therapeutic benefits of TRAIL therapy have been limited, possibly due to the resistance displayed by HCC cells and poor pharmacokinetics. Thus, an effective delivery agent is required for the delivery of TRAIL, which can increase its circulation time in the human body with optimal encapsulation.
4. Exosomes: A novel approach for drug delivery
Conventional anticancer drugs display limited efficacy owing to their short half-life, poor solubility and inefficacious delivery, resulting in the development of drug resistance and substantial systemic toxicity (95). Apart from this, poor drug delivery is also a major contributor to treatment failure. After entering the blood circulation by injection, the drug faces a variety of hurdles before reaching and acting on the target site (96). To improve the efficacy of HCC chemotherapeutics, a drug delivery system with active targeting and local, controlled, and continuous drug release is urgently required.
Extracellular vesicles (EV)-based therapeutics are a promising drug delivery system, since they can penetrate tissues and even cross the blood-brain barrier as natural nanoscale agents (97,98). Exosomes in particular, offer numerous benefits as drug delivery vehicles, which include a small size, low cytotoxicity, long half-life in the circulation, and the ability to load various cargoes with high a biocompatibility (Fig. 4) (99,100). Conventional drug delivery strategies frequently fall short of the intended results for several reasons, including the rapid in vivo degradation of miRNAs, the loss of native structure in proteins and the potential for severe toxicity in normal cells. However, these issues may be resolved by using exosomes as carriers and thus, by deploying exosomes to deliver drugs to tumor sites, an effective and promising approach may be made available for targeted cancer therapy.
Figure 4.
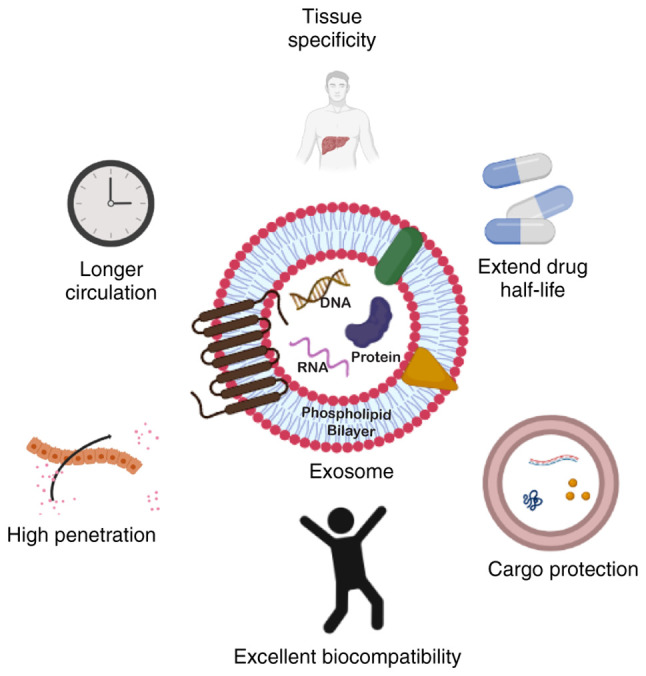
Structure of exosomes and benefits of using exosomes as a drug delivery system compared to other existing agents. The figure was created using BioRender.com.
Exosomes are endosomal-derived EVs with a size of 30-200 nm that have been reported to be released by a multitude of cell types. They have the characteristic cargo-loading capacity of carrying heterogenous biomolecules, such as DNA, RNA, proteins and lipids, and transferring them to recipient cells, thus acting as intercellular messengers. Exosomes are formed by the intraluminal budding of multivesicular bodies (MVBs) of an intact cell and are released into the extracellular environment when these MVBs fuse with the plasma membrane of the recipient cell (101).
Exosomes have the potential to mirror the intricacy of the parental cell with the innate capacity to regulate multitude of roles in crucial biological activities (102). Consequently, this characteristic led to exosome-based applications in cancer treatment and diagnoses being more feasible. Exosomes contain molecules with a wide range of functions, but lack the complexity of cells and organs; as a result, exosomes are regarded as excellent tools for use in the treatment of a variety of disorders, including cancer. Exosomes also have a number of advantages in terms of biocompatibility, stability, cellular uptake mechanism, biodistribution, pharmacokinetics and immunogenicity, rendering them promising anticancer candidates. These characteristics can raise the therapeutic index of exosome-based cancer treatments by preferentially targeting tumor cells, while reducing undesirable side-effects. Below is a brief illustration of key facts of the therapeutic potential of exosomes over present drug delivery platforms.
Source and safety
MSCs have been reported to be the most favorable selection for the commercial production of exosomes due to the ease of availability and are reported to produce an excessive number of exosomes (EVs) with consistent sustainability and reproducibility compared to other cell lines (103). Most importantly, applying MSC-exosomes as an agent to deliver drugs has been reported to have no safety concerns, including the chances of inducing tumorigenicity (104,105). MSC-derived EVs exhibit significant flexibility for in vitro and in vivo modifications (106), as well as a high stability in human plasma and at storage at 20°C (107,108).
Furthermore, MSC-derived EVs have been demonstrated to be well-tolerated in a variety of animal models, aside from possessing therapeutic benefits as proven in the treatment of myocardial infarction, chronic kidney disease, wound healing and liver injury in mouse models (109,110). Previous studies have employed EVs as an efficient systemic natural gene carrier for transporting anticancer miRNAs and proteins (111,112). A number of phase I clinical trials have validated the safety of EV administration; no reports of grade II toxicity were reported with the determination of the maximal tolerated dose (113-115).
Drug delivery and cellular uptake
As aforementioned, exosomes, being a natural cellular messenger, provide the benefit of a heterogeneous cargo-loading capacity and specificity. Exosomes are known to possess homing properties that can deliver cargo even to distant targets and in between cells with suitable biocompatibility, and to regulate the functions of targeted cells transiently (116). Reportedly, the interaction of exosomes with target cells involves multiple mechanisms, as exosomes can directly bind to membrane receptors of the recipient cell for content internalization, or they can transport bioactive cargo by fusing with the plasma membrane of the target cell.
Presently, a number of drugs face the issue of not being able to cross the blood-brain barrier, limiting the efficacy of several therapies, including cancer therapies. However, exosomes can cross the blood-brain barrier to increase intracranial drug concentration (117). For example, exosomes have been shown to carry medicines or siRNA to the brains of mice with Alzheimer's disease (111,118). Unlike the traditional approach of drug administration, delivery using exosomes does not have the drawback of drug toxicity, intracranial infection and imprecise absorption (118).
Cargo protection and improved durability
The ideal delivery agent does not only perform the site-specific transportation of enclosed therapeutics, but should also be capable of protecting the enclosed material and avoiding premature degradation by the body's immune system. Exosomes have a lipid bilayer structure that not only aids in transport efficiency and supports the load of hydrophobic or hydrophilic drugs, but also protects the encapsulated material (119). Furthermore, being able to have a reduced clearance rate can sustain the drugs in the body's circulation. Exosomes, being natural products of the body, do not invoke any immune response and possess a longer circulation half-life, which can prevent therapeutic cargo from degrading too rapidly (118).
Stability
Exosomes are well-known for their stability, as they preserve the identity of their parental cells, while maintaining their long-term innate integrity (107). Multiple freeze-thaw cycles have been shown to have no effect on their size, indicating that freezing has no effect on the quality of exosomes stored (107). Kalra et al (108) demonstrated the stability of colon cancer-derived exosomes and noted that the majority of samples retained their integrity even without protease inhibitors for 3 months, and that the highest stability was found at 80°C. This property of being stable for a long period of time in storage at 80°C suggests another advantage of exosomes over existing anticancer drugs (120).
Furthermore, a previous study found that therapeutic exosomes maintained antitumor activity even after being frozen for at least 5 months (121). Exosomes can shield therapeutic nucleic acids and proteins from RNases and proteinase degradation as they contain fragile bioactive molecules within a lipid bilayer membrane (122). In addition, under both physiological and pathological conditions, exosomes appear to display an enhanced stability in the blood, that facilitates long-distance travel within the body. Thus, exosome stability covers not just the human body, but also storage in the field. In addition to this, they have also been shown to have improved stability in the blood, allowing them to cover long distances throughout the body under both normal and pathological conditions (123).
Therapeutic significance and drug resistance
It has been demonstrated that drugs encapsulated with exosomes lead to an enhanced chemotherapeutic efficacy (124). Exosomes appear to have a much higher potential (>10-fold) in targeting cancer cells compared to liposomes of similar size (125). Furthermore, currently, the major barrier to an effective therapy or complete cure is multi-drug resistance (MDR). This resistance is commonly shown by all cancer patients undergoing long-term chemotherapy. The exosome is one such natural nanocarrier which has proven to be efficient in overcoming the issue of MDR in tumors. Kim et al (126) confirmed this by integrating paclitaxel (PTX) into exosomes released from macrophages for the treatment of MDR cancer. They determined that exosomes augmented cytotoxicity in drug-resistant cells by >50-fold when compared to exosome-free drugs (126). Furthermore, when doxorubicin (DOX)-loaded exosomes were administered intranasally to animals with pulmonary metastasis, confocal fluorescence microscopy revealed an almost perfect co-localization with cancer cells. These findings suggest that PTX-loaded exosomes inhibit MDR tumors and pulmonary metastasis growth more effectively (126).
5. Challenges, synergism, and strategies against liver cancer
Several drug delivery strategies have been developed over the years for the treatment of cancer; however, only a few of these have obtained clinical approval. The likelihood of an efficacious drug delivery to cancer tissues following in vivo administration is <0.7% (127). The expected accomplishments in the case of HCC drug development may not be as satisfactory as in the case of other types of cancer. Furthermore, based upon the outcome of recent clinical trials, a single drug therapy appears to be inadequate in the case of treatment for advanced-stage HCC (128). Thus, combination therapy is a major area of research for the treatment of advanced-stage HCC.
Surprisingly, formulations containing recombinant TRAIL have encountered considerable difficulties in being evaluated for human application due to its undesirable systemic toxicity, the short plasma half-life of recombinant TRAIL, or the activation of anti-apoptotic proteins (62,69,94,129). However, the exosome-based encapsulation of TRAIL protein can convey better pharmacokinetic characteristics, greater bioavailability and the ability to cross target tissues to the enclosed protein/drug (130). For example, in a previous study, TRAIL was transduced into leukemia K562 cells with human membrane TRAIL and produced TRAIL secreted exosomes, which were shown to trigger the apoptosis of melanoma and lymphoma cell lines in vitro (131). These findings reveal that cells that have been genetically engineered to express TRAIL can secrete exosomes that contain the pro-apoptotic ligand in an active form in their membranes. Although therapeutic success varied in the different tumor models studied, TRAIL exosomes exhibited potent killing activity in vitro and in vivo, in both local and systemic therapy modalities (131).
Another study by Yuan et al (132) demonstrated that TRAIL-loaded exosomes were more efficient in inducing cell death than recombinant soluble TRAIL. It was shown that the fluidic nature of the lipid bilayer membrane in exosomes harboring TRAIL may allow higher order TRAIL oligomerization and, as a result, the stronger clustering of its receptors, which is a crucial signal for effective extrinsic death pathway activation (132). It was demonstrated that the limited bioavailability of TRAIL, the low activity and cell resistance to TRAIL ligand can be overcome by TRAIL-expressing EVs derived from MSCs, thereby improving the clinical efficacy of TRAIL. This EV-loaded TRAIL effectively induced apoptosis in a variety of cancer cell lines, including lung (A549, NCI-H460 and NCI-H727), neuroblastoma (SHEP-TET), breast (M231), kidney (RCC10) and malignant pleural mesothelioma lines (H2795). While there was no toxicity to control healthy cells, TRAIL+ exosomes were capable of triggering apoptosis in TRAIL-resistant cancer cells (132).
The TRAIL receptor binding to target cells, which activates the caspase cascade and results in death, was suggested as the therapeutic mechanism of TRAIL-MSC-EVs. Furthermore, TRAIL-MSC-EVs have exhibited therapeutic efficacy in TRAIL-resistant cancer cell lines, which is noteworthy (132). The study by Shamili et al (133) also demonstrated the anti-tumor activity of TRAIL-transfected MSC-derived exosomes in a mouse model of melanoma. Their findings suggested that when TRAIL-expressing exosomes were injected into mice, they delayed the appearance of tumors and attenuated tumor growth (133). Of note, they proposed that a combination of TRAIL-exosomes with another chemotherapeutic may be explored as a promising therapeutic tool (133). The diagrammatic representation of how TRAIL-expressing exosomes would lead to cell death in recipient HCC cells is presented in Fig. 3. A list of studies demonstrating the exosomal delivery by TRAIL and its clinical significance is presented Table III.
Table III.
List of recent studies on exosomal delivery of TRAIL for cancer treatment.
| Cancer types | Donor cells | Results | (Refs.) |
|---|---|---|---|
| Melanoma and lymphoma | K562 cells (lymphoblasts) | Induction of apoptosis in cancer cells and control tumor progression in vivo | (131) |
| Lung cancer (in vitro), pleural mesothelioma (in vitro), renal cancer (in vitro), breast adenocarcinoma (in vitro), neuroblastoma (in vitro) | MSCs | Highly efficient at selectively inducing apoptosis in cancer cells and TRAIL delivery by MSC-EVs at least partially overcomes TRAIL resistance in cancer cells | (132) |
| Melanoma | MSCs | Delay in the appearance of tumors and attenuation of tumor growth | (133) |
| Lymphoma | Myeloid leukemia cells | Increased apoptosis of leukemia cells | (147) |
| Human lung adenocarcinoma | 293T cells | Dinaciclib and TRAIL exert synergistic effects on TRAIL-mediated apoptosis | (148) |
| Lung cancer | MSCs | EV-encapsulated TRAIL and dinaciclib can overcome the drug-resistance of lung cancer cells and are highly efficient for inducing the apoptosis of the TRAIL-resistant A549 cell line | (149) |
| Malignant melanoma | RAW 264.7 (macrophage cell line) | TRAIL-Exo/triptolide improved tumor targetability, enhanced cellular uptake, inhibited theproliferation, invasion, and migration, and induced the apoptosis of A375 cells | (150) |
MSCs, mesenchymal stem cells; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; DR, death receptor; EVs, extracellular vesicles.
Several studies have found a synergistic effect between TRAIL and sorafenib, suggesting that combined treatment with these agents may lead to the development of effective therapy for overcoming TRAIL resistance in cancer cells. For example, a previous study demonstrated the synergistic effects of sorafenib and TRAIL, where Sorafenib considerably increased the cytotoxicity TRAIL to HCC cells (134). The enhancement in cytotoxicity may be obtained from the downregulation of anti-apoptotic proteins by sorafenib. Similarly, another study by Chen et al (135) revealed that sorafenib sensitized TRAIL-resistant HCC cells to TRAIL-induced apoptosis by inhibiting STAT3 (135).
Moreover, the application of Sorafenib enclosed within exosomes into the target site offers a number of benefits as opposed to oral administration. As depicted by a previous study, exosome-encapsulated DOX delivery increased the therapeutic index in breast and ovarian cancer mouse models compared to exosome-free DOX (124). It was also demonstrated by in vitro and in vivo experiments that exosomes loaded with DOX limited heart toxicity by partially reducing the passage of DOX through cardiac endothelial cells (124).
The major issue with the safe, specific and efficient delivery of miRNAs is their property of being easily degradable before reaching the target organ. The lipid bilayer membrane of EVs protects the enclosed miRNA, preventing it from degradation and facilitating its effective delivery to the target site (136). Previous research has demonstrated that exosomes can safely enclose and carry miRNA to target cells of multiple types of cancer. For example, Almanza et al (137) demonstrated that EVs containing miR-335 effectively and long-lastingly restored the endogenous miR-335 pool in human triple-negative breast cancer cells, suppressing the expression of the miR-335 target gene SOX4 transcription factor, and significantly reducing tumor development in vivo.
Previously, another group (138) reported utilizing EVs as a delivery agent for miR-335 both in vivo and in vitro. They were successful in demonstrating that the safe administration of fibroblast-derived EVs that were loaded with miR-195 may concentrate inside the tumor, reduce the size of tumors, and increase the longevity of treated rats in a rat model of cholangiocarcinoma (138). Wang et al (139) demonstrated that miR335-5p could be successfully supplied to hepatoma cells by utilizing exosomes as a delivery agent both in vivo and in vitro. They observed the progression of HCC cell development when stellate cells were co-cultured with HCC cells due to exosomal transfer and noted the downregulated expression of miR-3355p in both cells and exosomes (139). The target genes identified in terms of HCC are CDC42, NRG1, EIF5, CDK2, EIF2C2, LIMK1, PLK2, RGS19, THBS1, YBX1 and TCF3. However, upregulating the expression of miR-335-5p in stellate cell-derived exosomes has been shown to restrict HCC cell proliferation and invasion in vitro, and cause tumor shrinkage in mouse models (139). A schematic diagram of the exosomal delivery of miR-335 into recipient HCC cells and the mode of action based on the afore-mentioned investigations is presented in Fig. 5.
Figure 5.

Graphical overview of the exosomal delivery of miR-335 into recipient HCC cells and mode of action. miR-335 inhibits the expression of MAPK1, OCT4 and ROCK1 to suppress the malignancy of cancer cells, inhibiting cell proliferation, metastasis, and tumorigenesis. The figure was created using BioRender.com. HCC, hepatocellular carcinoma; MAPK1, mitogen-activated protein kinase 1; OCT4, octamer-binding transcription factor 4; ROCK1, Rho-associated coiled-coil-containing protein kinase 1.
It is also noteworthy that miR-335 modulates the sensitivity of sorafenib against HCC cells. As shown in the study by Kim et al (43), Siah E3 ubiquitin protein ligase 2 (SIAH2) is the target of miR-335, where miR-335 contributes to the sensitizing effect of anticancer drugs via the expression enhancement of histone deacetylase 3 (HDAC3). SIAH2 overexpression was shown to result in anticancer drug resistance due to its effect on HDAC3 expression and ubiquitination (43). These findings suggest that miR-335 may be a promising anticancer agent. When combined with sorafenib, encapsulating it in exosomes increases its sensitivity to the drug, thus enhancing the therapeutic efficacy and preventing degradation.
6. Development of exosome-based TRAIL + miR-335 therapy
Although exosome-based cancer therapy displays exceptional therapeutic potential, there are still a number of substantial challenges that need to be resolved in order for its use to be feasible in clinical applications. The technology of exosome mass production is not yet standardized. Although small-scale GMP exosome production has been shown to be viable, there are still numerous obstacles in large-scale production (121,140). Numerous companies are still struggling to produce exosome on mass scale level. However, few companies, such as CK Exogene, a Korean biotechnology firm, have managed to overcome the issue of low exosome yield and have acquired the patented technology (10-2020-0062365) for exosome mass production (141) and this company is currently developing exosome-based anticancer drug for patients with liver cancer using the aforementioned candidates i.e., TRAIL and miR-335. A schematic overview of anticancer candidates, including dorafenib encapsulated in exosomed as a delivery agent is presented in Fig. 6).
Figure 6.

Overall strategy of exosome-based targeted therapy encapsulating TRAIL, miR-335 and sorafenib. The figure was created using BioRender.com. HCC, hepatocellular carcinoma.
To the best of our knowledge, the present review is the first of its kind, exploring and combining the cutting-edge feature of exosome with novel anticancer candidates (TRAIL and miR-335). The aim of the present review article was to present the compiled investigations of TRAIL and miR-335, both of which have been extensively explored in the past, along with the added benefit of utilizing exosomes as a carrier. Exosomes encapsulating TRAIL have the potential to overcome the challenging issue of resistance among HCC cells towards TRAIL-induced apoptosis, thus rendering TRAIL more effective in killing cancer cells. Such a strategy not only provides long-term and effective anticancer treatment for patients with HCC, but it has also been reported to overcome the issue of drug resistance, which is the major challenge to current drug therapy for liver cancer.
Additionally, to receive the successful outcome of any drug treatment, the accessibility of the drug to the target organ is imperative and necessitates the requirement of an effective delivery route. Even though drugs targeted against HCC comprise various delivery routes, including the direct injection into the liver, intra-arterial drug delivery is an effective technique for targeting the tumor site with multiple agents. Compared to intravenous delivery, intra-arterial drug administration expedites the systemic clearance and enhances the intra-tumor drug concentration (142). Owing to such high magnitude of benefits conferred by exosome-based TRAIL-miR-335 delivery from preventing metastasis to inducing cytotoxicity in cancer cells specifically, this approach has the prospects to be provided to patients with all stages of liver cancer from stages 0 to 4.
7. Conclusions
Exosomes offer the versatile characteristics of an efficient delivery system for both TRAIL and miR-335 as anticancer candidates. The incorporation of the benefits of exosomes, with them being a natural cellular carrier and the combination of these novel candidates with a standard drug, such as sorafenib would have a sensitizing effect and has been proven to yield a synergistic anti-cytotoxicity effect on TRAIL-resistant cancer cells. Moreover, this strategy also has the potential to overcome resistance to sorafenib, the most prevalent issue of the current drug treatment program among patients with HCC. The most prominent significance of exosome-based technology lies in the fact that this approach can be applied to all types of cancer and encompasses the benefit of overcoming drug resistance, which is the most prevalent issue in current drug treatment regimen. The present review thus provides an insight into the development of exosome-based therapy and the possibility of its bench-to-bed translation for providing an exceptional anticancer treatment.
Acknowledgments
Not applicable.
Funding Statement
No funding was received.
Availability of data and materials
Data sharing is not applicable to this article, as no data sets were generated or analyzed during the current study.
Authors' contributions
All authors (NT, YJC, KHY, TBW, DK, DC and JK) were involved in the drafting and revision of the manuscript, and in critically revising the manuscript for important intellectual content. All authors have read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The purification strategy for the mass production of highly purified and concentrated exosomes is subject to Korean patent application no. 10-2020-0062365, associated with CK-Exogene, Inc. JK and NT are employees of CK-Exogene, Inc. The other authors (YJC, KHY, TBW, DK and DC) are not associated with CK-Exogene, Inc. and declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Singal AG, El-Serag HB. Hepatocellular carcinoma from epidemiology to prevention: Translating knowledge into practice. Clin Gastroenterol Hepatol. 2015;13:2140–2151. doi: 10.1016/j.cgh.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dasgupta P, Henshaw C, Youlden DR, Clark PJ, Aitken JF, Baade PD. Global trends in incidence rates of primary adult liver cancers: A systematic review and meta-analysis. Front Oncol. 2020;10:171. doi: 10.3389/fonc.2020.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomes MA, Priolli DG, Tralhão JG, Botelho MF. Hepatocellular carcinoma: Epidemiology, biology, diagnosis, and therapies. Rev Assoc Med Bras (1992) 2013;59:514–524. doi: 10.1016/j.ramb.2013.03.005. In English, Portuguese. [DOI] [PubMed] [Google Scholar]
- 5.Center MM, Jemal A. International trends in liver cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2011;20:2362–2368. doi: 10.1158/1055-9965.EPI-11-0643. [DOI] [PubMed] [Google Scholar]
- 6.Farinati F, Sergio A, Baldan A, Giacomin A, Di Nolfo MA, Del Poggio P, Benvegnu L, Rapaccini G, Zoli M, Borzio F, et al. Early and very early hepatocellular carcinoma: When and how much do staging and choice of treatment really matter? A multi-center study. BMC Cancer. 2009;9:33. doi: 10.1186/1471-2407-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kakushadze Z, Raghubanshi R, Yu W. Estimating cost savings from early cancer diagnosis. Data. 2017;2:30. doi: 10.3390/data2030030. [DOI] [Google Scholar]
- 8.Finn RS. Emerging targeted strategies in advanced hepatocellular carcinoma. Semin Liver Dis. 2013;33(Suppl 1):S11–S19. doi: 10.1055/s-0033-1333632. [DOI] [PubMed] [Google Scholar]
- 9.WHO's International Agency for Research on Cancer (IARC) In: World Cancer Report 2014. Stewart BW, Kleihues P, editors. IARC Press; Lyon: 2014. [Google Scholar]
- 10.Dimitroulis D, Damaskos C, Valsami S, Davakis S, Garmpis N, Spartalis E, Athanasiou A, Moris D, Sakellariou S, Kykalos S, et al. From diagnosis to treatment of hepatocellular carcinoma: An epidemic problem for both developed and developing world. World J Gastroenterol. 2017;23:5282–5294. doi: 10.3748/wjg.v23.i29.5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arruebo M, Vilaboa N, Saez-Gutierrez B, Lambea J, Tres A, Valladares M, González-Fernández A. Assessment of the evolution of cancer treatment therapies. Cancers (Basel) 2011;3:3279–3330. doi: 10.3390/cancers3033279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–1763. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 13.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 14.Kane RC, Farrell AT, Madabushi R, Booth B, Chattopadhyay S, Sridhara R, Justice R, Pazdur R. Sorafenib for the treatment of unresectable hepatocellular carcinoma. Oncologist. 2009;14:95–100. doi: 10.1634/theoncologist.2008-0185. [DOI] [PubMed] [Google Scholar]
- 15.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 16.Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, Baron A, Park JW, Han G, Jassem J, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 17.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda K, Kudo M, Kawazoe S, Osaki Y, Ikeda M, Okusaka T, Tamai T, Suzuki T, Hisai T, Hayato S, et al. Phase 2 study of lenvatinib in patients with advanced hepatocellular carcinoma. J Gastroenterol. 2017;52:512–519. doi: 10.1007/s00535-016-1263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 20.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin C, Wang A, Liu L, Wang G, Li G, Han Z. miR-145-5p inhibits tumor occurrence and metastasis through the NF-κB signaling pathway by targeting TLR4 in malignant melanoma. J Cell Biochem. 2019 Jan 30; doi: 10.1002/jcb.28388. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 22.Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget. 2015;6:8474–8490. doi: 10.18632/oncotarget.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hydbring P, Wang Y, Fassl A, Li X, Matia V, Otto T, Choi YJ, Sweeney KE, Suski JM, Yin H, et al. Cell-cycle-targeting MicroRNAs as therapeutic tools against refractory cancers. Cancer Cell. 2017;31:576–590.e8. doi: 10.1016/j.ccell.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8:9227. doi: 10.1038/s41598-018-27521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Zhao Y, Xu M, Zhou F, Yan J. Serum miR-1301-3p, miR-335-5p, miR-28-5p and their target B7-H3 may serve as novel biomarkers for colorectal cancer. J BUON. 2019;24:1120–1127. [PubMed] [Google Scholar]
- 26.Du W, Tang H, Lei Z, Zhu J, Zeng Y, Liu Z, Huang JA. miR-335-5p inhibits TGF-β1-induced epithelial-mesenchymal transition in non-small cell lung cancer via ROCK1. Respir Res. 2019;20:225. doi: 10.1186/s12931-019-1184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Tao Y, Shan L, Chen R, Jiang H, Qian Z, Cai F, Ma L, Yu Y. The role of MicroRNAs in hepatocellular carcinoma. J Cancer. 2018;9:3557–3569. doi: 10.7150/jca.26350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang G, Dong F, Xu Z, Sharma S, Hu X, Chen D, Zhang L, Zhang J, Dong Q. MicroRNA profile in HBV-induced infection and hepatocellular carcinoma. BMC Cancer. 2017;17:805. doi: 10.1186/s12885-017-3816-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gougelet A. Exosomal microRNAs as a potential therapeutic strategy in hepatocellular carcinoma. World J Hepatol. 2018;10:785–789. doi: 10.4254/wjh.v10.i11.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye L, Wang F, Wu H, Yang H, Yang Y, Ma Y, Xue A, Zhu J, Chen M, Wang J, Zhang QA. Functions and targets of miR-335 in cancer. Onco Targets Ther. 2021;14:3335–3349. doi: 10.2147/OTT.S305098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scarola M, Schoeftner S, Schneider C, Benetti R. miR-335 directly targets Rb1 (pRb/p105) in a proximal connection to p53-dependent stress response. Cancer Res. 2010;70:6925–6933. doi: 10.1158/0008-5472.CAN-10-0141. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Bian T, Feng J, Qian L, Zhang J, Jiang D, Zhang Q, Li X, Liu Y, Shi J. miR-335 inhibited cell proliferation of lung cancer cells by target Tra2β. Cancer Sci. 2018;109:289–296. doi: 10.1111/cas.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang H, Zhu J, Du W, Liu S, Zeng Y, Ding Z, Zhang Y, Wang X, Liu Z, Huang J. CPNE1 is a target of miR-335-5p and plays an important role in the pathogenesis of non-small cell lung cancer. J Exp Clin Cancer Res. 2018;37:131. doi: 10.1186/s13046-018-0811-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li HW, Liu J. Circ_0009910 promotes proliferation and metastasis of hepatocellular carcinoma cells through miR-335-5p/ROCK1 axis. Eur Rev Med Pharmacol Sci. 2020;24:1725–1735. doi: 10.26355/eurrev_202002_20349. [DOI] [PubMed] [Google Scholar]
- 35.Liu H, Li W, Chen C, Pei Y, Long X. MiR-335 acts as a potential tumor suppressor miRNA via downregulating ROCK1 expression in hepatocellular carcinoma. Tumour Biol. 2015;36:6313–6319. doi: 10.1007/s13277-015-3317-2. [DOI] [PubMed] [Google Scholar]
- 36.Chen K, Zhang L. LINC00339 regulates ROCK1 by miR-152 to promote cell proliferation and migration in hepatocellular carcinoma. J Cell Biochem. 2019;120:14431–14443. doi: 10.1002/jcb.28701. [DOI] [PubMed] [Google Scholar]
- 37.Chen C, Wu CQ, Zhang ZQ, Yao DK, Zhu L. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp Cell Res. 2011;317:1714–1725. doi: 10.1016/j.yexcr.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Yang X, Song H, Zi Z, Kou J, Chen S, Dai Y, Wang J, Yuan L, Gao K. Circ_0005075 promotes hepatocellular carcinoma progression by suppression of microRNA-335. J Cell Physiol. 2019;234:21937–21946. doi: 10.1002/jcp.28757. [DOI] [PubMed] [Google Scholar]
- 39.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 40.Zhang D, Li X, Yao Z, Wei C, Ning N, Li J. GABAergic signaling facilitates breast cancer metastasis by promoting ERK1/2-dependent phosphorylation. Cancer Lett. 2014;348:100–108. doi: 10.1016/j.canlet.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Ji YY, Song Y, Wang AN. MiR-335-5p inhibits proliferation of Huh-7 liver cancer cells via targeting the Oct4/Akt pathway. Eur Rev Med Pharmacol Sci. 2021;25:1853–1860. doi: 10.26355/eurrev_202102_25080. [DOI] [PubMed] [Google Scholar]
- 42.Zhang BJ, Gong HY, Zheng F, Liu DJ, Liu HX. Up-regulation of miR-335 predicts a favorable prognosis in esophageal squamous cell carcinoma. Int J Clin Exp Pathol. 2014;7:6213–6218. [PMC free article] [PubMed] [Google Scholar]
- 43.Kim Y, Kim H, Park D, Jeoung D. miR-335 targets SIAH2 and confers sensitivity to anti-cancer drugs by increasing the expression of HDAC3. Mol Cells. 2015;38:562–572. doi: 10.14348/molcells.2015.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng Y, Shen P. miR-335 acts as a tumor suppressor and enhances ionizing radiation-induced tumor regression by targeting ROCK1. Front Oncol. 2020;10:278. doi: 10.3389/fonc.2020.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui L, Hu Y, Bai B, Zhang S. Serum miR-335 level is associated with the treatment response to trans-arterial chemoembolization and prognosis in patients with hepatocellular carcinoma. Cell Physiol Biochem. 2015;37:276–283. doi: 10.1159/000430352. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Xia X. Long noncoding RNA NEAT1 suppresses sorafenib sensitivity of hepatocellular carcinoma cells via regulating miR-335-c-Met. J Cell Physiol. 2019 Apr 1; doi: 10.1002/jcp.27567. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 47.Dohi O, Yasui K, Gen Y, Takada H, Endo M, Tsuji K, Konishi C, Yamada N, Mitsuyoshi H, Yagi N, et al. Epigenetic silencing of miR-335 and its host gene MEST in hepatocellular carcinoma. Int J Oncol. 2013;42:411–418. doi: 10.3892/ijo.2012.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shang X, Li G, Liu H, Li T, Liu J, Zhao Q, Wang C. Comprehensive circular RNA profiling reveals that hsa_ circ_0005075, a new circular RNA biomarker, is involved in hepatocellular crcinoma development. Medicine (Baltimore) 2016;95:e3811. doi: 10.1097/MD.0000000000003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nie Y, Zhu X, Bu N, Jiang Y, Su Y, Pan K, Li S. Circ_0064288 acts as an oncogene of hepatocellular carcinoma cells by inhibiting miR-335-5p expression and promoting ROCK1 expression. BMC Cancer. 2022;22:265. doi: 10.1186/s12885-022-09323-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol. 1999;11:255–260. doi: 10.1016/S0955-0674(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 51.Wajant H. Molecular mode of action of TRAIL receptor agonists-common principles and their translational exploitation. Cancers (Basel) 2019;11:954. doi: 10.3390/cancers11070954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amarante-Mendes GP, Griffith TS. Therapeutic applications of TRAIL receptor agonists in cancer and beyond. Pharmacol Ther. 2015;155:117–131. doi: 10.1016/j.pharmthera.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willms A, Schittek H, Rahn S, Sosna J, Mert U, Adam D, Trauzold A. Impact of p53 status on TRAIL-mediated apoptotic and non-apoptotic signaling in cancer cells. PLoS One. 2019;14:e0214847. doi: 10.1371/journal.pone.0214847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol. 2013;169:1723–1744. doi: 10.1111/bph.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim B, Allen JE, Prabhu VV, Talekar MK, Finnberg NK, El-Deiry WS. Targeting TRAIL in the treatment of cancer: New developments. Expert Opin Ther Targets. 2015;19:1171–1185. doi: 10.1517/14728222.2015.1049838. [DOI] [PubMed] [Google Scholar]
- 56.Graves JD, Kordich JJ, Huang TH, Piasecki J, Bush TL, Sullivan T, Foltz IN, Chang W, Douangpanya H, Dang T, et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell. 2014;26:177–189. doi: 10.1016/j.ccr.2014.04.028. [DOI] [PubMed] [Google Scholar]
- 57.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 58.Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. 1998;188:2375–2380. doi: 10.1084/jem.188.12.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang Y, Yang X, Xu T, Kong Q, Zhang Y, Shen Y, Wei Y, Wang G, Chang KJ. Overcoming resistance to TRAIL-induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int J Oncol. 2016;49:153–163. doi: 10.3892/ijo.2016.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Galal El-Shemi A, Mohammed Ashshi A, Oh E, Jung BK, Basalamah M, Alsaegh A, Yun CO. Efficacy of combining ING4 and TRAIL genes in cancer-targeting gene virotherapy strategy: First evidence in preclinical hepatocellular carcinoma. Gene Ther. 2018;25:54–65. doi: 10.1038/gt.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28:2839–2846. doi: 10.1200/JCO.2009.25.1991. [DOI] [PubMed] [Google Scholar]
- 63.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 64.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–12690. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 65.Liu CH, Chern GJ, Hsu FF, Huang KW, Sung YC, Huang HC, Qiu JT, Wang SK, Lin CC, Wu CH, et al. A multifunctional nanocarrier for efficient TRAIL-based gene therapy against hepatocellular carcinoma with desmoplasia in mice. Hepatology. 2018;67:899–913. doi: 10.1002/hep.29513. [DOI] [PubMed] [Google Scholar]
- 66.Kim CY, Jeong M, Mushiake H, Kim BM, Kim WB, Ko JP, Kim MH, Kim M, Kim TH, Robbins PD, et al. Cancer gene therapy using a novel secretable trimeric TRAIL. Gene Ther. 2006;13:330–338. doi: 10.1038/sj.gt.3302658. [DOI] [PubMed] [Google Scholar]
- 67.Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E, De Santis G, Spano C, Tagliazzucchi M, Barti-Juhasz H, et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res. 2010;70:3718–3729. doi: 10.1158/0008-5472.CAN-09-1865. [DOI] [PubMed] [Google Scholar]
- 68.Liu S, Qiu J, He G, He W, Liu C, Cai D, Pan H. TRAIL promotes hepatocellular carcinoma apoptosis and inhibits proliferation and migration via interacting with IER3. Cancer Cell Int. 2021;21:63. doi: 10.1186/s12935-020-01724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21:1350–1364. doi: 10.1038/cdd.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holland PM. Death receptor agonist therapies for cancer, which is the right TRAIL? Cytokine Growth Factor Rev. 2014;25:185–193. doi: 10.1016/j.cytogfr.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 71.Zhang L, Gu J, Lin T, Huang X, Roth JA, Fang B. Mechanisms involved in development of resistance to adenovirus-mediated proapoptotic gene therapy in DLD1 human colon cancer cell line. Gene Ther. 2002;9:1262–1270. doi: 10.1038/sj.gt.3301797. [DOI] [PubMed] [Google Scholar]
- 72.Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, Bayer E, Walczak H, Kalthoff H, Ungefroren H. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene. 2000;19:5477–5486. doi: 10.1038/sj.onc.1203936. [DOI] [PubMed] [Google Scholar]
- 73.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–1319. [PubMed] [Google Scholar]
- 74.Marini P, Denzinger S, Schiller D, Kauder S, Welz S, Humphreys R, Daniel PT, Jendrossek V, Budach W, Belka C. Combined treatment of colorectal tumours with agonistic TRAIL receptor antibodies HGS-ETR1 and HGS-ETR2 and radiotherapy: Enhanced effects in vitro and dose-dependent growth delay in vivo. Oncogene. 2006;25:5145–5154. doi: 10.1038/sj.onc.1209516. [DOI] [PubMed] [Google Scholar]
- 75.Pukac L, Kanakaraj P, Humphreys R, Alderson R, Bloom M, Sung C, Riccobene T, Johnson R, Fiscella M, Mahoney A, et al. HGS-ETR1, a fully human TRAIL-receptor 1 monoclonal antibody, induces cell death in multiple tumour types in vitro and in vivo. Br J Cancer. 2005;92:1430–1441. doi: 10.1038/sj.bjc.6602487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J, Fox JA. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efcacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001;299:31–38. [PubMed] [Google Scholar]
- 77.Hymowitz SG, O'Connell MP, Ultsch MH, Hurst A, Totpal K, Ashkenazi A, de Vos AM, Kelley RF. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry. 2000;39:633–640. doi: 10.1021/bi992242l. [DOI] [PubMed] [Google Scholar]
- 78.Mérino D, Lalaoui N, Morizot A, Solary E, Micheau O. TRAIL in cancer therapy: Present and future challenges. Expert Opin Ther Targets. 2007;11:1299–1314. doi: 10.1517/14728222.11.10.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med. 2001;7:383–385. doi: 10.1038/86397. [DOI] [PubMed] [Google Scholar]
- 80.Ashley DM, Riffkin CD, Lovric MM, Mikeska T, Dobrovic A, Maxwell JA, Friedman HS, Drummond KJ, Kaye AH, Gan HK, et al. In vitro sensitivity testing of minimally passaged and uncultured gliomas with TRAIL and/or chemotherapy drugs. Br J Cancer. 2008;99:294–304. doi: 10.1038/sj.bjc.6604459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bae S, Ma K, Kim TH, Lee ES, Oh KT, Park ES, Lee KC, Youn YS. Doxorubicin-loaded human serum albumin nanoparticles surface-modified with TNF-related apoptosis-inducing ligand and transferrin for targeting multiple tumor types. Biomaterials. 2012;33:1536–1546. doi: 10.1016/j.biomaterials.2011.10.050. [DOI] [PubMed] [Google Scholar]
- 82.Mitchell MJ, Wayne E, Rana K, Schaffer CB, King MR. TRAIL-coated leukocytes that kill cancer cells in the circulation. Proc Natl Acad Sci USA. 2014;111:930–935. doi: 10.1073/pnas.1316312111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naval J, de Miguel D, Gallego-Lleyda A, Anel A, Martinez-Lostao L. Importance of TRAIL molecular anatomy in receptor oligomerization and signaling Implications for cancer therapy. Cancers (Basel) 2019;11:444. doi: 10.3390/cancers11040444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Griffith T, Wiley SR, Kubin MZ, Sedger LM, Maliszewski CR, Fanger NA. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J Exp Med. 1999;189:1343–1354. doi: 10.1084/jem.189.8.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kayagaki N, Yamaguchi N, Nakayama M, Kawasaki A, Akiba H, Okumura K, Yagita H. Involvement of TNF-related apoptosis-inducing ligand in human CD4+ T cell-mediated cytotoxicity. J Immunol. 1999;162:2639–2647. [PubMed] [Google Scholar]
- 86.Monleón I, Martínez-Lorenzo MJ, Anel A, Lasierra P, Larrad L, Pineiro A, Naval J, Alava MA. CD59 cross-linking induces secretion of APO2 ligand in overactivated human T cells. Eur J Immunol. 2000;30:1078–1087. doi: 10.1002/(SICI)1521-4141(200004)30:4<1078::AID-IMMU1078>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 87.Monleón I, Martinez-Lorenzo MJ, Monteagudo L, Lasierra P, Taulés M, Iturralde M, Piñeiro A, Larrad L, Alava MA, Naval J, Anel A. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J Immunol. 2001;167:6736–6744. doi: 10.4049/jimmunol.167.12.6736. [DOI] [PubMed] [Google Scholar]
- 88.Wajant H, Moosmayer D, Wüest T, Bartke T, Gerlach E, Schönherr U, Peters N, Scheurich P, Pfizenmaier K. Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene. 2001;20:4101–4106. doi: 10.1038/sj.onc.1204558. [DOI] [PubMed] [Google Scholar]
- 89.De Miguel D, Basáñez G, Sánchez D, Malo PG, Marzo I, Larrad L, Naval J, Pardo J, Anel A, Martinez-Lostao L. Liposomes decorated with Apo2L/TRAIL overcome chemoresistance of human hematologic tumor cells. Mol Pharm. 2013;10:893–904. doi: 10.1021/mp300258c. [DOI] [PubMed] [Google Scholar]
- 90.De Miguel D, Gallego-Lleyda A, Anel A, Martinez-Lostao L. Liposome-bound TRAIL induces superior DR5 clustering and enhanced DISC recruitment in histiocytic lymphoma U937 cells. Leuk Res. 2015;39:657–666. doi: 10.1016/j.leukres.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 91.De Miguel D, Gallego-Lleyda A, Ayuso JM, Erviti-Ardanaz S, Pazo-Cid R, del Agua C, Fernández LJ, Ochoa I, Anel A, Martinez-Lostao L. TRAIL-coated lipid-nanoparticles overcome resistance to soluble recombinant TRAIL in non-small cell lung cancer cells. Nanotechnology. 2016;27:185101. doi: 10.1088/0957-4484/27/18/185101. [DOI] [PubMed] [Google Scholar]
- 92.De Miguel D, Gallego-Lleyda A, Ayuso JM, Pejenaute-Ochoa D, Jarauta V, Marzo I, Fernández LJ, Ochoa I, Conde B, Anel A, Martinez-Lostao L. High-order TRAIL oligomer formation in TRAIL-coated lipid nanoparticles enhances DR5 cross-linking and increases antitumour effect against colon cancer. Cancer Lett. 2016;383:250–260. doi: 10.1016/j.canlet.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 93.Kim TH, Jiang HH, Park CW, Youn YS, Lee S, Chen X, Lee KC. PEGylated TNF-related apoptosis-inducing ligand (TRAIL)-loaded sustained release PLGA microspheres for enhanced stability and antitumor activity. J Control Release. 2011;150:63–69. doi: 10.1016/j.jconrel.2010.10.037. [DOI] [PubMed] [Google Scholar]
- 94.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 95.Chinnappan M, Srivastava A, Amreddy N, Razaq M, Pareek V, Ahmed R, Mehta M, Peterson JE, Munshi A, Ramesh R. Exosomes as drug delivery vehicle and contributor of resistance to anticancer drugs. Cancer Lett. 2020;486:18–28. doi: 10.1016/j.canlet.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J, Yeung BZ, Cui M, Peer CJ, Lu Z, Figg WD, Guillaume Wientjes M, Woo S, Au JL. Exosome is a mechanism of inter-cellular drug transfer: Application of quantitative pharmacology. J Control Release. 2017;268:147–158. doi: 10.1016/j.jconrel.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012;21:R125–R134. doi: 10.1093/hmg/dds317. [DOI] [PubMed] [Google Scholar]
- 98.Lee JK, Park SR, Jung BK, Jeon YK, Lee YS, Kim MK, Kim YG, Jang JY, Kim CW. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One. 2013;8:e84256. doi: 10.1371/journal.pone.0084256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Syn NL, Wang L, Chow EK, Lim CT, Goh BC. Exosomes in cancer nanomedicine and immunotherapy: Prospects and challenges. Trends Biotechnol. 2017;35:665–676. doi: 10.1016/j.tibtech.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 100.Ha D, Yang N, Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm Sin B. 2016;6:287–296. doi: 10.1016/j.apsb.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Farooqi AA, Desai NN, Qureshi MZ, Librelotto DRN, Gasparri ML, Bishayee A, Nabavi SM, Curti V, Daglia M. Exosome biogenesis, bioactivities and functions as new delivery systems of natural compounds. Biotechnol Adv. 2018;36:328–334. doi: 10.1016/j.biotechadv.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 102.Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. 2018;6:18. doi: 10.3389/fcell.2018.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yeo RW, Lai RC, Zhang B, Tan SS, Yin Y, The BJ, Lim SK. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65:336–341. doi: 10.1016/j.addr.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 104.Lou G, Chen Z, Zheng M, Liu Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp Mol Med. 2017;49:e346. doi: 10.1038/emm.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cho BS, Kim JO, Ha DH, Yi YW. Exosomes derived from human adipose tissue-derived mesenchymal stem cells alleviate atopic dermatitis. Stem Cell Res Ther. 2018;9:187. doi: 10.1186/s13287-018-0939-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, Alvarez-Erviti L, Sargent IL, Wood MJ. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7:2112–2126. doi: 10.1038/nprot.2012.131. [DOI] [PubMed] [Google Scholar]
- 107.Sokolova V, Ludwig AK, Hornung S, Rotan O, Horn PA, Epple M, Giebel B. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf B Biointerfaces. 2011;87:146–150. doi: 10.1016/j.colsurfb.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 108.Kalra H, Adda CG, Liem M, Ang CS, Mechler A, Simpson RJ, Hulett MD, Mathivanan S. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics. 2013;13:3354–3364. doi: 10.1002/pmic.201300282. [DOI] [PubMed] [Google Scholar]
- 109.Rani S, Ryan AE, Griffin MD, Ritter T. Mesenchymal stem cell-derived extracellular vesicles: Toward cell-free therapeutic applications. Mol Ther. 2015;23:812–823. doi: 10.1038/mt.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sun L, Xu R, Sun X, Duan Y, Han Y, Zhao Y, Qian H, Zhu W, Xu W. Safety evaluation of exosomes derived from human umbilical cord mesenchymal stromal cell. Cytotherapy. 2016;18:413–422. doi: 10.1016/j.jcyt.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 111.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 112.El Andaloussi S, Lakhal S, Mäger I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. 2013;65:391–397. doi: 10.1016/j.addr.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 113.Escudier B, Dorval T, Chaput N, André F, Caby MP, Novault S, Flament C, Leboulaire C, Borg C, Amigorena S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J Transl Med. 2005;3:10. doi: 10.1186/1479-5876-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, Valente N, Shreeniwas R, Sutton MA, Delcayre A, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3:9. doi: 10.1186/1479-5876-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, Li G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther. 2008;16:782–790. doi: 10.1038/mt.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iranifar E, Seresht BM, Momeni F, Fadaei E, Mehr MH, Ebrahimi Z, Rahmati M, Kharazinejad E, Mirzaei H. Exosomes and microRNAs: New potential therapeutic candidates in Alzheimer disease therapy. J Cell Physiol. 2019;234:2296–2305. doi: 10.1002/jcp.27214. [DOI] [PubMed] [Google Scholar]
- 117.Qu M, Lin Q, Huang L, Fu Y, Wang L, He S, Fu Y, Yang S, Zhang Z, Zhang L, Sun X. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson's disease. J Control Release. 2018;287:156–166. doi: 10.1016/j.jconrel.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 118.Wang H, Sui H, Zheng Y, Jiang Y, Shi Y, Liang J, Zhao L. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3β pathway. Nanoscale. 2019;11:7481–7496. doi: 10.1039/C9NR01255A. [DOI] [PubMed] [Google Scholar]
- 119.Kim MS, Haney MJ, Zhao Y, Yuan D, Deygen I, Klyachko NL, Kabanov AV, Batrakova EV. Engineering macro-phage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine. 2018;14:195–204. doi: 10.1016/j.nano.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 120.Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013;32:623–642. doi: 10.1007/s10555-013-9441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mendt M, Kamerkar S, Sugimoto H, McAndrews KM, Wu CC, Gagea M, Yang S, Blanko EVR, Peng Q, Ma X, et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight. 2018;3:e99263. doi: 10.1172/jci.insight.99263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang Y, Balaji V, Kaniyappan S, Krüger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E, Mandelkow EM. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener. 2017;12:5. doi: 10.1186/s13024-016-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jiang XC, Gao JQ. Exosomes as novel bio-carriers for gene and drug delivery. Int J Pharm. 2017;521:167–175. doi: 10.1016/j.ijpharm.2017.02.038. [DOI] [PubMed] [Google Scholar]
- 124.Hadla M, Palazzolo S, Corona G, Caligiuri I, Canzonieri V, Toffoli G, Rizzolio F. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine (Lond) 2016;11:2431–2441. doi: 10.2217/nnm-2016-0154. [DOI] [PubMed] [Google Scholar]
- 125.Smyth TJ, Redzic JS, Graner MW, Anchordoquy TJ. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim Biophys Acta. 2014;1838:2954–2965. doi: 10.1016/j.bbamem.2014.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, Inskoe E, Piroyan A, Sokolsky M, Okolie O, et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine. 2016;12:655–664. doi: 10.1016/j.nano.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014. doi: 10.1038/natrevmats.2016.14. [DOI] [Google Scholar]
- 128.Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: Looking outside the box. J Hepatol. 2020;72:342–352. doi: 10.1016/j.jhep.2019.09.010. [DOI] [PubMed] [Google Scholar]
- 129.Trivedi R, Mishra DP. Trailing TRAIL resistance: Novel targets for TRAIL sensitization in cancer cells. Front Oncol. 2015;5:69. doi: 10.3389/fonc.2015.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sterzenbach U, Putz U, Low LH, Silke J, Tan SS, Howitt J. Engineered exosomes as vehicles for biologically active proteins. Mol Ther. 2017;25:1269–1278. doi: 10.1016/j.ymthe.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rivoltini L, Chiodoni C, Squarcina P, Tortoreto M, Villa A, Vergani B, Bürdek M, Botti L, Arioli I, Cova A, et al. TNF-related apoptosis-inducing ligand (TRAIL)-armed exosomes deliver proapoptotic signals to tumor site. Clin Cancer Res. 2016;22:3499–3512. doi: 10.1158/1078-0432.CCR-15-2170. [DOI] [PubMed] [Google Scholar]
- 132.Yuan Z, Kolluri KK, Gowers KH, Janes SM. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J Extracell Vesicles. 2017;6:1265291. doi: 10.1080/20013078.2017.1265291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shamili FH, Bayegi HR, Salmasi Z, Sadri K, Mahmoudi M, Kalantari M, Ramezani M, Abnous K. Exosomes derived from TRAIL-engineered mesenchymal stem cells with effective anti-tumor activity in a mouse melanoma model. Int J Pharm. 2018;549:218–229. doi: 10.1016/j.ijpharm.2018.07.067. [DOI] [PubMed] [Google Scholar]
- 134.Nojiri K, Sugimoto K, Shiraki K, Tameda M, Inagaki Y, Ogura S, Kasai C, Kusagawa S, Yoneda M, Yamamoto N, et al. Sorafenib and TRAIL have synergistic effect on hepatocellular carcinoma. Int J Oncol. 2013;42:101–108. doi: 10.3892/ijo.2012.1676. [DOI] [PubMed] [Google Scholar]
- 135.Chen KF, Tai WT, Liu TH, Huang HP, Lin YC, Shiau CW, Li PK, Chen PJ, Cheng AL. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res. 2010;16:5189–5199. doi: 10.1158/1078-0432.CCR-09-3389. [DOI] [PubMed] [Google Scholar]
- 136.Munir J, Yoon JK, Ryu S. Therapeutic miRNA-enriched extracellular vesicles: Current approaches and future prospects. Cells. 2020;9:2271. doi: 10.3390/cells9102271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Almanza G, Rodvold JJ, Tsui B, Jepsen K, Carter H, Zanetti M. Extracellular vesicles produced in B cells deliver tumor suppressor miR-335 to breast cancer cells disrupting oncogenic programming in vitro and in vivo. Sci Rep. 2018;8:17581. doi: 10.1038/s41598-018-35968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li L, Piontek K, Ishida M, Fausther M, Dranoff JA, Fu R, Mezey E, Gould SJ, Fordjour FK, Meltzer SJ, et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology. 2017;65:501–514. doi: 10.1002/hep.28735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang F, Li L, Piontek K, Sakaguchi M, Selaru FM. Exosome miR-335 as a novel therapeutic strategy in hepatocellular carcinoma. Hepatology. 2018;67:940–954. doi: 10.1002/hep.29586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Andriolo G, Provasi E, Lo Cicero V, Brambilla A, Soncin S, Torre T, Milano G, Biemmi V, Vassalli G, Turchetto L, et al. Exosomes from human cardiac progenitor cells for therapeutic applications: Development of a GMP-grade manufacturing method. Front Physiol. 2018;9:1169. doi: 10.3389/fphys.2018.01169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yoo KH, Thapa N, Kim BJ, Lee JO, Jang YN, Chwae YJ, Kim J. Possibility of exosome-based coronavirus disease 2019 vaccine (review) Mol Med Rep. 2022;25:26. doi: 10.3892/mmr.2021.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cooke JN, Ellis JA, Hossain S, Nguyen J, Bruce JN, Joshi S. Computational pharmacokinetic rationale for intra-arterial delivery to the brain. Drug Deliv Transl Res. 2016;6:622–629. doi: 10.1007/s13346-016-0319-6. [DOI] [PubMed] [Google Scholar]
- 143.Ciuleanu T, Bazin I, Lungulescu D, Miron L, Bondarenko I, Deptala A, Rodriguez-Torres M, Giantonio B, Fox NL, Wissel P, et al. A randomized, double-blind, placebo-controlled phase II study to assess the efficacy and safety of mapatumumab with sorafenib in patients with advanced hepatocellular carcinoma. Ann Oncol. 2016;27:680–687. doi: 10.1093/annonc/mdw004. [DOI] [PubMed] [Google Scholar]
- 144.von Pawel J, Harvey JH, Spigel DR, Dediu M, Reck M, Cebotaru CL, Humphreys RC, Gribbin MJ, Fox NL, Camidge DR. Phase II trial of mapatumumab, a fully human agonist monoclonal anti-body to tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1), in combination with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Clin Lung Cancer. 2014;15:188–196.e2. doi: 10.1016/j.cllc.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 145.Davies A, Sage B, Kolluri K, Alrifai D, Graham R, Weil B, Rego R, Bain O, Patrick PS, Champion K, et al. TACTICAL: A phase I/II trial to assess the safety and efficacy of MSCTRAIL in the treatment of metastatic lung adenocarcinoma. J Clin Oncol. 2019;37:TPS9116. doi: 10.1200/JCO.2019.37.15_suppl.TPS9116. [DOI] [Google Scholar]
- 146.Forero A, Bendell JC, Kumar P, Janisch L, Rosen M, Wang Q, Copigneaux C, Desai M, Senaldi G, Maitland ML. First-in-human study of the antibody DR5 agonist DS-8273a in patients with advanced solid tumors. Invest New Drugs. 2017;35:298–306. doi: 10.1007/s10637-016-0420-1. [DOI] [PubMed] [Google Scholar]
- 147.Xie Y, Bai O, Zhang H, Yuan J, Zong S, Chibbar R, Slattery K, Qureshi M, Wei Y, Deng Y, Xiang J. Membrane-bound HSP70-engineered myeloma cell-derived exosomes stimulate more efficient CD8(+) CTL- and NK-mediated antitumour immunity than exosomes released from heat-shocked tumour cells expressing cytoplasmic HSP70. J Cell Mol Med. 2010;14:2655–2666. doi: 10.1111/j.1582-4934.2009.00851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ke C, Hou H, Li J, Su K, Huang C, Lin Y, Lu Z, Du Z, Tan W, Yuan Z. Extracellular vesicle delivery of TRAIL eradicates resistant tumor growth in combination with CDK inhibition by dinaciclib. Cancers (Basel) 2020;12:1157. doi: 10.3390/cancers12051157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yuan Q, Su K, Li S, Long X, Liu L, Yang M, Yuan X, Sun J, Hu J, Li Q, et al. Pulmonary delivery of extracellular vesicle-encapsulated dinaciclib as an effective lung cancer therapy. Cancers (Basel) 2022;14:3550. doi: 10.3390/cancers14143550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jiang L, Gu Y, Du Y, Tang X, Wu X, Liu J. Engineering exosomes endowed with targeted delivery of triptolide for malignant melanoma therapy. ACS Appl Mater Interfaces. 2021;13:42411–42428. doi: 10.1021/acsami.1c10325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article, as no data sets were generated or analyzed during the current study.