

Abstract
The high mobility group (HMG) transcription factor TCF-1 is essential for early T cell development. Although in vitro biochemical assays suggest that HMG proteins can serve as architectural elements in the assembly of higher-order nuclear organization, the contribution of TCF-1 on the control of three-dimensional (3D) genome structures during T cell development remains unknown. Here, we investigated the role of TCF-1 in 3D genome reconfiguration. Using gain and loss of function experiments, we discovered that the co-occupancy of TCF-1 and the architectural protein CTCF altered the structure of topologically associating domains in T cell progenitors, leading to interactions between previously insulated regulatory elements and target genes at late stages of T cell development. The TCF-1 dependent gain in long-range interactions was linked to deposition of active enhancer mark H3K27ac and recruitment of the cohesin-loading factor NIPBL at active enhancers. These data indicate that TCF-1 has a role in controlling the global genome organization during T cell development.
The mammalian genome is folded into higher-order 3D structures with globular interaction domains1. These structures, variously called topologically associating domains (TADs) or insulated neighborhoods2–6, sequester regulatory elements, insulating them from genomic regions outside such domains. The ubiquitously-expressed protein CTCF has a prominent role in creating insulated neighborhoods through its convergent binding events, which can block cohesin-mediated loop extrusion7,8. A small number of lineage-determining transcription factors (LDTFs) which can endow competence to new developmental programs have been shown to control 3D genome structure8–12 but the underlying mechanisms of this control are not fully understood.
The T cell developmental pathway represents a leading exemplar to understand global genome organization13. T cell development starts when bone marrow-derived precursors seed the thymus and give rise to early thymic progenitors (ETP)14. TCF-1, encoded by Tcf7, is upregulated at ETP by Notch1 and remains highly expressed until maturation. This protein can control the expression of Gata3 and Bcl11b, which are necessary for T lineage commitment15,16. TCF-1 can also bind to nucleosomal DNA and create novel accessible regulatory elements17,18. Biochemical studies have indicated that the HMG proteins like TCF-1 and LEF1 can bend DNA19,20. Moreover, TCF-1 is linked to 3D genome organization of peripheral CD8+ T cells21 and T leukemic cells22,23. Yet, the role of TCF-1 on genome folding during T cell development is unknown.
Here we assessed whether the early and continuous expression of TCF-1 in the thymus had a role in chromatin folding. Employing gain and loss of function experiments, we observed that TCF-1 targeted boundaries of insulated neighborhoods in T cell progenitors, weakened the insulation between adjacent neighborhoods, and enhanced long-range interactions between regulatory elements and target genes located on previously insulated domains. TCF-1-dependent long-range interactions were linked to the recruitment of the cohesin-loading factor NIPBL to active enhancers. Our finding of TCF-1-dependent chromatin interactions across insulated neighborhoods demonstrates an LDTF dismantling insulation during a developmental trajectory.
Results
TCF-1 occupancy correlates with intra-TAD interactions
To study the relationship between TCF-1 occupancy and 3D genome folding during T cell development, we employed publicly available ChIP-seq measurements of TCF-1 binding events in thymocytes24 and Hi-C measurements of 3D genome interactions at different T cell developmental stages, including pre-commitment (CLP, ETP, and DN2) and post-commitment (DN3, DN4, and DP) stages25. We defined TADs in DPs using the insulation score strategy26, grouped TADs based on TCF-1 binding density and evaluated the extent of intra-TAD interactions. The increase in intra-TAD interactions in DPs compared with ETPs-DN4s was significantly correlated with the density of TCF-1 binding events within TADs (Fig. 1a), suggesting that genomic domains with the strongest TCF-1 occupancy acquired the largest extent of de novo 3D interactions in DPs.
Fig. 1. TCF-1 is associated with intra-TAD interactions and cohesin loops.
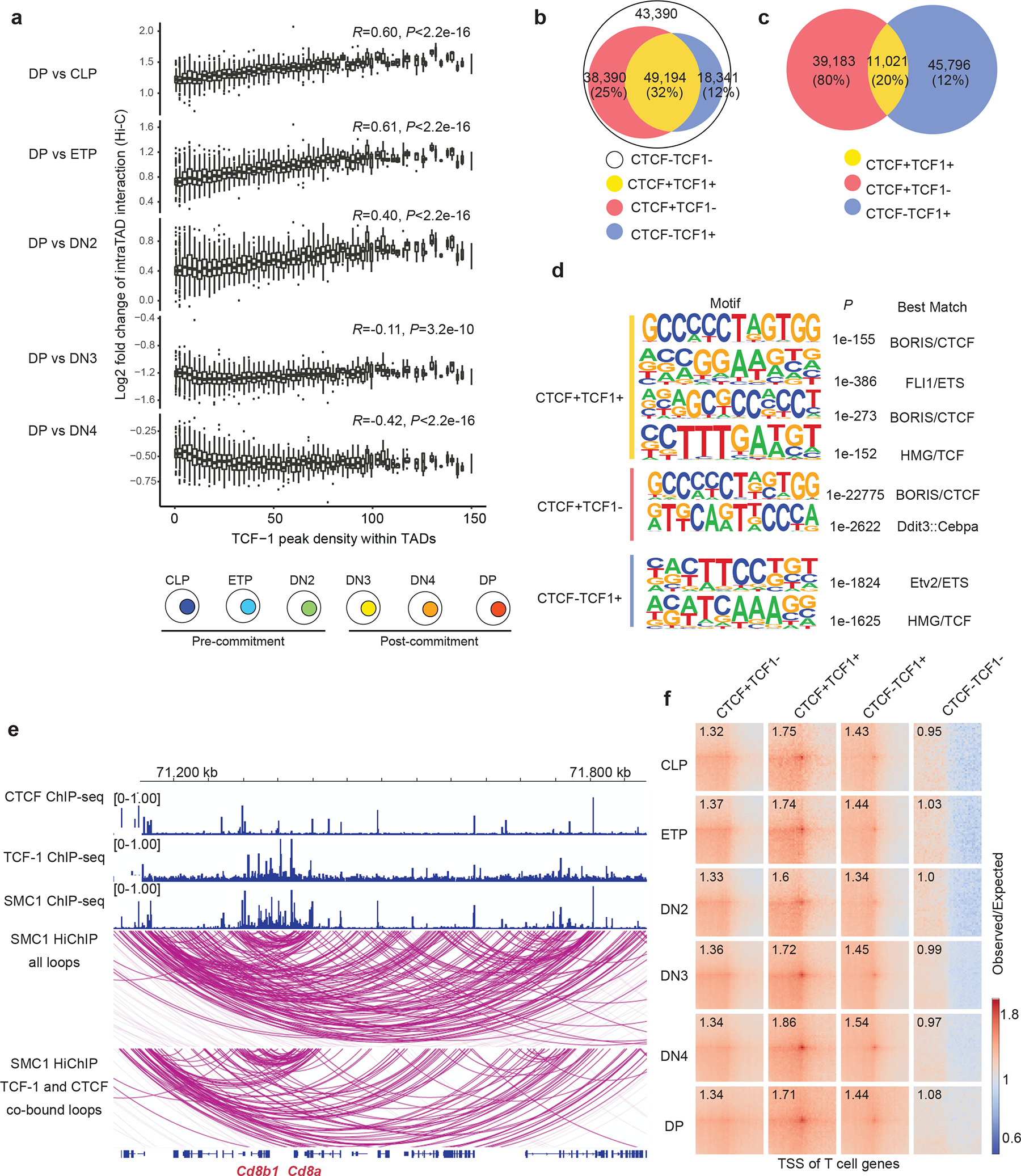
a. Box plots showing the association between TCF-1 binding density and log2 fold change difference in intra-TAD interactions between DP and CLP-to-DN4 (bottom) and schematic of T cell developmental stages (top). Long range interactions at various T cell developmental stages were measured using Hi-C26. Intra-TAD interactions were calculated based on TADs detected in DP T cells (n=3023), and peak density were calculated with number of TCF-1 peaks per 1 Mbp region. TCF-1 peaks were defined from TCF-1 ChIP-seq in thymocytes25. Data are shown as boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers). P value was calculated with two-sided Spearman correlation. b. Venn diagram showing the total number of cohesin loops measured by SMC1 HiChIP and the proportion of them co-bound by CTCF and/or TCF-1 at least on one anchor. The four classes of peaks were defined using CTCF ChIP-seq in DPs29 and publicly available TCF-1 ChIP-seq in thymocytes25. c. Venn diagram showing the number of TCF-1 and CTCF peaks, and the overlap between them in DPs. d. Seq-logos demonstrating motif enrichment using random background based on homer analysis at TCF-1+CTCF, CTCF-only and TCF-1-only sites in DPs. P values are calculated using hypergenometric test. e. Genome browser view showing the association of CTCF and TCF-1 peaks with cohesin loops at the Cd8a and Cd8b1 locus. Loops co-bound by CTCF and TCF-1 at least on one anchor are shown in the bottom panel. f. Pileups of interactions between pairs of T cell developmental genes and CTCF only, TCF-1+CTCF, TCF-1-only peaks and random regions within 100kbp windows. T cell genes were defined from single-cell RNA-seq analysis of the thymus30 (“T Cell Subset Markers”, Table S2 in ref30). The number in each box indicates the strength of interaction at the center pairing TSS of T cell genes and different sets of peaks.
To explore the association between TCF-1 binding and finer scale 3D structures, we identified cohesin-associated loops using SMC1 HiChIP in DPs27. Of the 149,315 loops detected in DPs, more than 40% were occupied by TCF-1 at least on one anchor, which was comparable to CTCF binding pattern (Fig. 1b) while 32% of loops were co-bound by TCF-1 and CTCF (Fig. 1b). To evaluate the genome-wide co-occupancy of these two proteins, we grouped CTCF and TCF-1 binding events in DPs into CTCF alone, TCF-1 alone, and regions co-occupied by both proteins (Fig. 1c, Supplementary Table 1). CTCF and TCF-1 co-occupancy accounted for ~20% of TCF-1 bound regions (Fig. 1c). The HMG motif was enriched at CTCF and TCF-1 co-bound sites (Fig. 1d), implying the direct binding of TCF-1 protein at these loci. The occupancy of CTCF and TCF-1 was associated with dense cohesin loops at the Cd8a (Fig. 1e), Ets1 (Extended Data Fig. 1a) and Tcf7 (Extended Data Fig. 1b) loci.
We next assessed the extent of long-range interactions between the regulatory elements in each TCF-1 binding group and genes that are selectively regulated during T cell development using single-cell RNA-seq profiling of thymocytes28 (Supplementary Table 2) and found that TCF-1 and CTCF co-bound sites had the strongest interactions with stage-specific genes (Fig. 1f). Thus, TCF-1 and CTCF co-occupancy might be a structurally salient feature of T cell development.
TCF-1 and CTCF co-binding weakens insulation at boundaries
We next measured the average 3D interactions anchored at the CTCF, TCF-1 or TCF-1+CTCF peaks, and a set of randomly sampled regions that lacked either protein as control. In CLPs, ETPs, and DN2s, TCF-1+CTCF sites had limited interactions between their upstream and downstream regions (Fig. 2a). The insulation at TCF-1+CTCF sites was gradually lost starting in DN3s (Fig. 2a), suggesting interactions between upstream and downstream insulated domains centered at TCF-1+CTCF sites. The interactions between the two sides of TCF-1-only regions also started to increase in DN3s (Fig. 2a), while the CTCF-occupied sites remained insulated throughout T cell development (Fig. 2a). To quantitatively assess changes in insulation at TCF-1+CTCF sites, we calculated genome-wide insulation scores26. We found that insulation at TCF-1+CTCF sites, but not CTCF-only sites, gradually declined during T cell development starting from DN3s (Fig. 2b and Extended Data Fig. 2a). The increase in insulation score in DPs compared to CLPs was significantly larger at TCF-1+CTCF sites compared with TCF-1 or CTCF regions (Extended Data Fig. 2b), indicating that co-binding of TCF-1 and CTCF could increase interactions across insulated neighborhoods, leading to a loss of insulation as T cells become more mature. Co-bound sites were preferentially enriched closer to genes expressed at DN3 and DN4 stages28 compared to genes associated with other stages (Fig. 2c, Supplementary Table 3). Thus, insulation at TCF-1+CTCF sites declined post T cell commitment due to de novo long-range interactions at T cell commitment genes.
Fig. 2. TCF-1+CTCF sites earmark weakening of insulated neighborhoods.
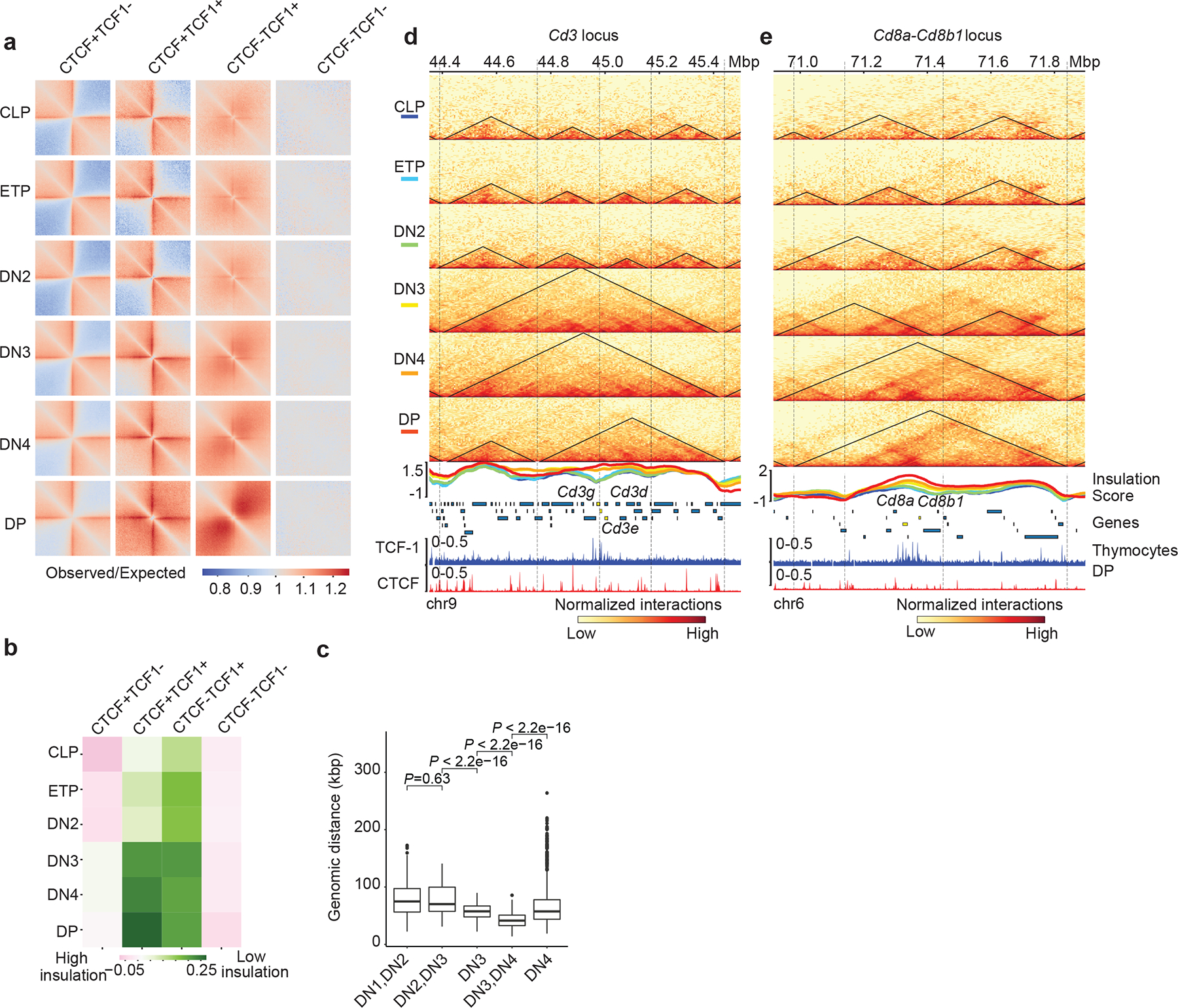
a. Heatmap showing local pileup interactions using Hi-C data at CTCF-only, CTCF+TCF-1, TCF-1-only peaks and random regions as control. The horizontal line and the vertical line of each plot are the 500 kb region centered on different peak groups.
b. Heatmap showing average insulation score during T cell development at the CTCF only, CTCF+TCF-1 co-bound, TCF-1 only peaks and random regions as control.
c. Boxplots showing distance of marker genes of T cell developmental stages to TCF-1+CTCF peaks. Data are shown as boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers). The comparison between different stages was done with two-sided Wilcoxon signed-rank test. The distance between these markder genes and TCF-1+CTCF peaks significantly (P < 2.22 × 10−16) changed except between DN1 and DN2. Selection of marker genes as described in Fig. 1g.
d-e Genome browser view showing the interactions at Cd3d-e (d) and CD8a,b1 (e) loci during T cell development, as well as TCF-1 and CTCF binding events in DPs. Triangles in the heatmap represent TADs called with cooltools. Boundaries are marked as vertical lines. Insulation score in each T cell developmental stage is shown as a line plot and colored according to their developmental stage as indicated in the heatmap legend. Heatmaps are normalized with sequencing depth and ChIP-seq tracks are normalized with count per million (CPM).
Based on the quantitative definition of TAD boundaries, the number of boundaries steadily decreased from ETP to DP (Extended Data Fig. 2c), suggesting the reorganization of TADs during T cell development. We further classified the boundaries into different groups according to their quantitative detection at each stage. We detected boundaries with mixed patterns of gain or loss (group I), boundaries that remained invariant during development (group H), boundaries that formed after T cell commitment (groups A and B), and boundaries that were lost at different stages (groups C, D, E, F and G) (Extended Data Fig. 2d, Supplementary Table 4). Examination of the long-range contacts across the nine groups indicated increased across boundary interactions at DN3s-DPs compared with CLPs to DN3s at lost boundaries and fewer across boundary interactions in DN3s to DPs at gained boundaries (Extended Data Fig. 2e,f). The interactions across domains separated by dynamic boundaries were confirmed by insulation score and directionality index analyses (Extended Data Fig. 3a,b). The percentage of group C boundaries that were in compartment B (PC1 <0) increased markedly between CLPs and DPs (Extended Data Fig. 3c). Lost boundaries in groups E and F were largely in compartment A in both CLPs and DPs (Extended Data Fig. 3c,d), suggesting no compartmental switching as thymocytes matured.
Next, we calculated the average occurrence of peaks for each binding group within 500 kb of dynamic boundaries. While CTCF-only sites were enriched at most boundaries (Extended Data Fig. 3e–h), TCF-1+CTCF as well as TCF-1-only sites were enriched at lost boundaries in groups E and F (Extended Data Fig. 3f–i). Analysis of histone acetylation and chromatin accessibility29 indicated that boundaries lost at DN3s and DN4s (group E and F) were highly associated with active enhancer marks, as well as transcriptionally active and accessible chromatin states across T cell development (Extended Data Fig. 3h,i), suggesting that TCF-1 binding events, especially TCF-1+CTCF regions, were abundant in transcriptionally active and open chromatin domains.
We found multiple boundaries in the Cd3d-g and Cd8a-b loci in CLPs and ETPs that weakened in DPs (Fig. 2d,e), suggesting that boundary weakening between two adjacent domains at loci encompassing T cell development genes corresponded to novel interactions between previously insulated domains. Dense TCF-1 occupancy was accompanied by increased expression of the Cd3d-g and Cd8a-b genes in DN3s-DPs (Fig. 2d,e). A cluster of TCF-1 binding events at a boundary encompassing the Cd3 locus corresponded to loss of insulation and gain in long-range interactions across the boundary at DN3s to DPs (Fig. 2d). Moreover, multiple TCF-1 binding events that occurred near the boundary of Cd8a-b locus, were also associated with loss of insulation and extensive long-range interactions (Fig. 2e). New boundaries were also detected at select loci such as a cluster of TCF-1 binding events at the Rag1-Rag2 locus (Extended Data Fig. 3j), corresponding to increased expression of these genes in mature T cells. Thus, TCF-1+CTCF co-binding was associated with dynamic domain boundaries encompassing T cell identity genes.
TCF-1+CTCF sites are evolutionarily conserved
We next examined the conservation scores of DNA sequences centered on different classes of CTCF and TCF-1 binding events. We found a sharp increase in sequence conservation at genomic regions with CTCF only or TCF-1 only peaks and an even broader and stronger degree of sequence conservation at TCF-1+CTCF sites (Extended Data Fig. 4a). To further explore the insulation status of conserved binding sites in human T cells, we employed Hi-C data from T cell acute lymphoblastic leukemia (T-ALL) and ETP leukemia (ETP-ALL)30, which arise from malignant transformation of T cell precursors. Moreover, we generated Hi-C maps from mature lymph node CD8+ T cells from one healthy organ donor (Extended Data Fig. 4b). We evaluated the extent of insulation and long-range 3D interactions at homologous CTCF and TCF-1 binding sites in the human genome and found loss of insulation at TCF-1+CTCF sites in mature CD8+ T cells compared to T-ALLs and ETP-ALLs (Extended Data Fig. 4b). We further analyzed the average local interactions at TCF-1+CTCF sites in multiple mouse cell types (Extended Data Fig. 4c). While the CTCF sites appeared insulated in all examined cell types, TCF-1 sites showed increased interactions only in naïve CD4+ T cells and DPs (Extended Data Fig. 4c). Based on analysis of public Hi-C data from 4D Nucleome project31, the TCF-1+CTCF sites exhibited the same level of insulation as CTCF sites in all non-T cell lines examined (Extended Data Fig. 4d). Thus, the highly conserved TCF-1+CTCF sites were insulated in non-T cells and early T cell progenitors in both humans and mice.
Overexpression of TCF-1 in fibroblasts creates de novo 3D interactions
To ask whether TCF-1 expression was required for long-range interactions across co-bound TCF-1+CTCF boundaries, we used fibroblasts because T cell lineage collaborators of TCF-1 are not expressed in this cell type. We overexpressed TCF-1 in the fibroblast cell line NIH-3T3 using lentiviral transduction with a doxycycline-inducible gene expression system (hereafter 3T3TCF-1). We measured TCF-1 and CTCF binding events and transcriptional outputs before and 72h post-TCF-1 induction by doxycycline in biological replicates (Supplementary Fig. 1), using ChIP-seq and RNA-seq, respectively. Transcriptomic measurements indicated the increased expression of 273 genes and decreased expression of 194 genes in 3T3TCF-1 compared to non-induced fibroblasts (Extended Data Fig. 5a, Supplementary Table 5). 1,872 CTCF binding events were predominantly stronger and 431 CTCF binding events were weaker in 3T3TCF-1 fibroblasts (Extended Data Fig. 5b). Thus, TCF-1 expression can interfere with the transcriptional control of fibroblasts17.
Next, we measured cohesin-mediated 3D genome interactions using SMC1 HiChIP before and after TCF-1 induction. Only a small number of regions switched compartments between 3T3TCF-1 and non-induced fibroblasts (Extended Data Fig. 5c). The overall compartmentalization, measured by the compartment strength (Extended Data Fig. 5d) or the extent of BB, AA and AB interactions (Extended Data Fig. 5e), was similar in 3T3TCF1 and non-induced fibroblasts, suggesting that TCF-1 expression did not alter the compartmentalization of the fibroblast genome 72h post induction. Most TAD boundaries were conserved (Extended Data Fig. 5f,g); however, we observed a significant increase in intra-TAD interactions within invariant TADs in 3T3TCF-1 compared to non-induced fibrobalsts (Fig. 3a). Measurements of the connectivity within a TAD, referred to as “domain score”32, indicated significantly larger domain scores in 3T3TCF-1 compared with non-induced fibroblasts (Fig. 3b), suggesting that TCF-1 can promote intra-TAD interactions. The density of TCF-1 binding correlated significantly with changes in domain score after TCF-1 induction (Extended Data Fig. 5h), suggesting that dense TCF-1 binding corresponded to a substantial increase in intra-TAD interactions. TCF-1+CTCF sites had the largest increase in 3D interactions in 3T3TCF-1 (Fig. 3c), suggesting their cooperativity in mediating 3D interactions. The increase in interactions originating from each genomic region was positively correlated with the number of TCF-1 peaks in the neighborhood (Extended Data Fig. 5i,j). The greatest increase in 3D interactions occurred when TCF-1 occupied both anchors and had high density at neighboring regions (Extended Data Fig. 6k,l). Thus, as exemplified at the Irf2bp2 locus (Fig. 3d), TCF-1 could increase intra-TAD interactions in fibroblasts at many loci.
Fig. 3. Gain and loss of TCF-1 reshapes long-range interactions.
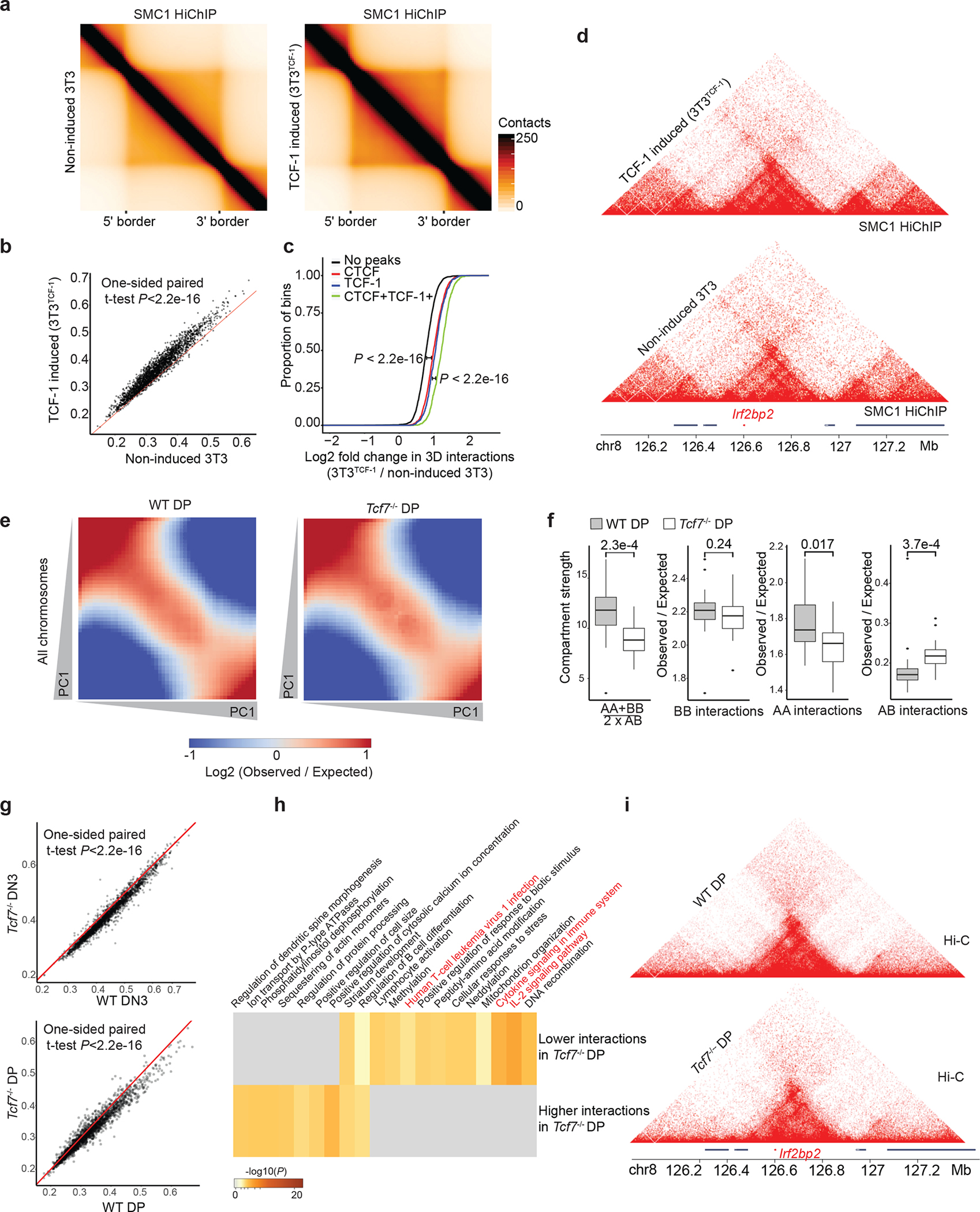
a. Heatmaps showing the aggregate of interactions across invariant TADs using SMC1 HiChIP in non-induced 3T3 and 3T3TCF-1. TADs are rescaled and normalized using GENOVA. One HiChIP experiment was performed per experimental condition.
b. Scatter plot showing domain scores in non-induced 3T3 and 3T3TCF-1. One-sided paired Student’s t-test assesses that domain scores are significantly (P<2.21e-16) higher in 3T3TCF-1 compared with non-induced 3T3.
c. Cumulative distribution plot showing the increase of interactions in genomic bins with TCF-1+CTCF or other groups. Comparisons between different groups using two-sided Kolmogorov–Smirnov test evaluates the significance of the difference. TCF-1+CTCF sites gained significantly (P < 2.2 × 10−16) more interactions than other groups.
d. Contact matrix and genome browser view of the TAD encompassing the Irf2bp2 locus in 3T3.
e. Saddle plot showing the compartmentalization across all chromosomes in wildtype and Tcf7−/− DPs based on Hi-C measurements. One Hi-C experiment was performed per genotype.
f. Data are shown as boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers) showing the compartment strength, BB interactions, AA interactions and AB interactions in wildtype and Tcf7−/− DPs (n=20 mouse chromosomes). The statistical test was done using two-sided Student’s t-test. (ns: not significant, *P< 0.05, ***P<0.001).
g. Scatter plot showing domains scores in DN3s and DPs. The red line represents equal domain scores in two conditions. One-sided paired Student’s t-test shows that domain score is significantly different in wildtype DN3s and DPs compared with Tcf7−/− cells (P<2.21e-16). One Hi-C experiment was performed in the wildtype condition and two Hi-C experiments were performed in two distinct Tcf7−/− DN3 clones.
h. Gene-ontology analysis of genes in the top 50 TADs that gain domain score and the top 50 TADs that lose domain score after TCF-1 deletion in DP T cells using metaScape.
i. Contact matrix and genome browser view of the TAD encompassing Irf2bp2 that lost substantial intra-TAD interactions in Tcf7−/− DPs.
TCF-1 controls the 3D chromatin conformation of T cells
T cells undergo a remarkable genome reorganization during the DN2-to-DN3 transition25. To explore whether TCF-1 was required for the 3D genome organization during T cell development, we genetically ablated TCF-1 expression using CRISPR/Cas9 (Extended Data Fig. 6a) in a DN3-like T cell line, also called Scid.adh33. In addition, we generated Vav-CreTcf7fl/fl mice, in which TCF-1 is conditionally ablated in all hematopoietic cells (Extended Data Fig. 6b), as indicated by immunoblot experiments (Extended Data Fig. 6c) and flow cytometry in the thymus (Extended Data Fig. 6d). We next mapped the global chromatin conformation using ultra-deep Hi-C measurements in DN3 cell lines and primary DPs in wildtype and TCF-1 deficient T cells (hereafter Tcf7−/− DN3s and DPs). We detected overall modest changes of PC1 values between wild-type and Tcf7−/− cells (Extended Data Fig. 6e,f). However, we observed weakening of compartment strengths in Tcf7−/− DPs (Figs. 3e). There was also a significant decrease of interactions in A compartment in both Tcf7−/− DN3s and DPs, a significant decrease in compartment strength in Tcf7−/− DPs, and a significant increase of interactions between A and B compartments in Tcf7−/− DPs (Fig. 3f and Extended Data Fig. 6h) compared to their wild-type counterparts. These data suggest TCF-1 was required for the establishment and maintenance of long-range interactions at the compartment level in DN3s and DPs. Most TAD boundaries were conserved between wild-type and Tcf7−/− cells (Extended Data Fig. 6i,j), but there was a significant reduction in intra-TAD interactions and hence, reduced domain scores in Tcf7−/− cells compared with wild-type DN3s and DPs (Fig. 3g), positing that TCF-1 was required for the increase in de novo 3D interactions at these stages.
In Tcf7−/− DN3s (or Tcf7−/− DPs), 196 (or 525) genes were downregulated, while 374 (or 664) genes were upregulated (Supplementary Tables 6–9). To ascertain whether TCF-1-dependent changes in intra-TAD interactions related to changes in transcriptional outputs, we investigated the expression of genes located in the top 50 TADs, which had the greatest change in their domain scores in Tcf7−/− cells. Genes positioned in TADs whose domain scores increased in Tcf7−/− DN3s and DPs were upregulated in Tcf7−/− DN3s and DPs (Extended Data Fig. 6k,l, Supplementary Table 10), while genes whose domain scores decreased after TCF-1 deletion were downregulated in Tcf7−/− DN3s and DPs (Extended Data Fig. 6k,l, Supplementary Table 10). Gene-ontology analysis indicated that genes positioned in TADs whose domain scores decreased in Tcf7−/− DPs were enriched in ontologies including ‘lymphocyte activation’, ‘cytokine signaling in immune system’ and ‘IL-2 signaling’ (Fig. 3h,i and Supplementary Table 11), while genes whose domain scores decreased in Tcf7−/− DN3 were enriched in ‘cellular response to IFN-beta’ (Extended Data Fig. 6m, Supplementary Table 11), indicating a potential regulatory role of TCF-1 on IFN-induced genes by directly reorganizing 3D genome folding. These findings indicated the requirement of TCF-1 for intra-TAD interactions, which are necessary for the transcriptional control of genes with developmental roles in T cells.
A dense cluster of TCF-1 binding events on IFN-induced genes corresponded to TCF-1 dependent SMC1 occupancy and 3D interactions (Extended Data Fig. 7a), suggesting the potential role of TCF-1 protein in recruiting the cohesin subunit. To further investigate whether sequence variation at this TCF-1 binding cluster can alter the 3D genome interactions and thus the expression of IFN-induced genes within this domain, we utilized the SMC1 HiChIP data in DPs in C57BL/6 and NOD27, which showed differences in the intra-TAD interactions (Extended Data Fig. 7b). A cluster of TCF-1 binding events coincided with stronger interactions in NOD compared with C57BL/6 DPs (Extended Data Fig. 7b). Consequently, most genes had higher expression in NOD compared with C57BL/6 DPs (Extended Data Fig. 7c). Long-range interactions at this domain were also TCF-1 dependent in fibroblasts and DPs (Extended Data Fig. 7d,e). Thus, TCF-1 was required to maintain long-range interactions at various length scales.
TCF-1 is required to diminish insulation
To investigate the extent to which TCF-1 acted on insulated neighborhoods, we examined interactions at TCF-1+CTCF sites in 3T3TCF-1 fibroblasts in addition to Tcf7−/− DN3s and DPs. We first quantified the interactions at a 500kbp region centered around different classes of TCF-1 and CTCF binding events in 3T3TCF-1 fibroblasts. Among 12,143 TCF-1+CTCF sites, interactions between upstream and downstream domains centered at 2,435 co-bound sites increased in 3T3TCF-1 fibroblasts compared with non-induced fibroblasts (Fig. 4a, Supplementary Table 12). Among 27,353 TCF-1 peaks, the interaction and insulation changed at 6,868 sites (Fig. 4a,b).
Fig. 4. TCF-1 is required to dismantle boundaries.
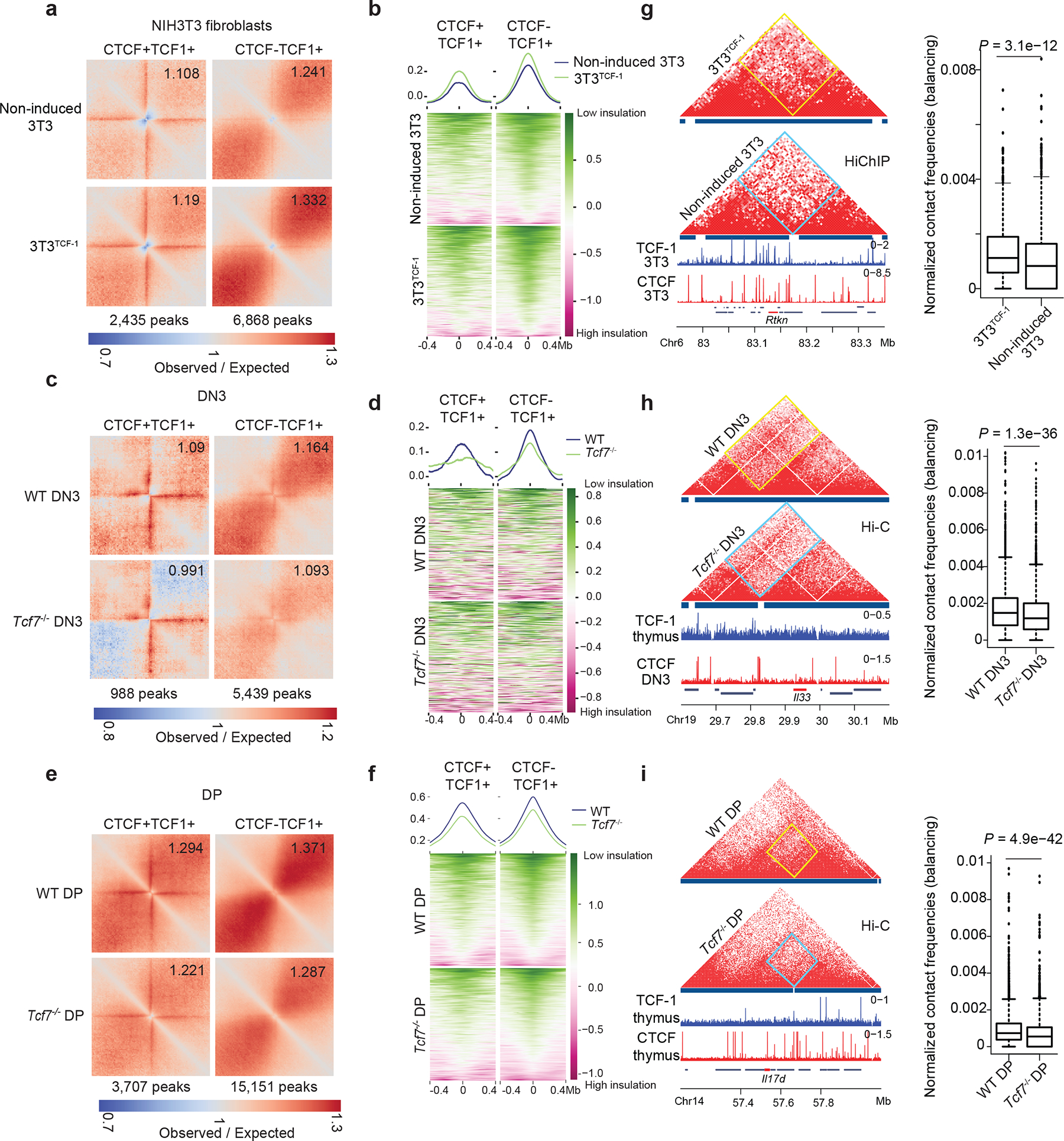
a. Local pileup plot of long-range interactions using SMC1 HiChIP in non-induced 3T3 and 3T3TCF-1 at a subset of CTCF+TCF-1 and TCF-1 only peaks which lost insulation in 3T3TCF-1. The two classes of peaks were defined using TCF-1 and CTCF ChIP-seq in 3T3TCF-1.
b. Summary plot and heatmap of insulation score in non-induced 3T3 and 3T3TCF-1.
c. Local pileup plot of long-range interactions using ultra-deep Hi-C in wildtype and Tcf7−/− DN3 at a subset of CTCF+TCF-1 and TCF-1 only peaks that gain insulation in Tcf7−/− DN3. The two classes of peaks were defined using TCF-1 and CTCF ChIP-seq in thymocytes and DPs, respectively.
d. Summary plot and heatmap showing insulation score in wildtype and Tcf7−/− DN3.
e. Local pileup plot of long-range interactions using ultra-deep Hi-C in wildtype and Tcf7−/− DPs at a subset of CTCF+TCF-1 and TCF-1 only peaks that gained insulation in Tcf7−/− DPs.
f. Summary plot and heatmap showing insulation score in wildtype and Tcf7−/− DPs at a subset of CTCF+TCF-1 and TCF-1 only peaks that gained insulation after TCF-1 deletion in DPs.
g-i. Contact matrix of HiChIP and Hi-C data at the Rtkn, Il22, and Il17d loci in NIH3T3 (g) and DN3 (h), and DPs (i) respectively. Inter-domain interactions are quantified by boxplots (n=1080 (g), 3404 (h) and 3300 genomic interactions (i) for 3T3TCF-1, DN3 and DPs, respectively; centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers). Two sided Paired wilcoxon ranksum test was performed.
To test whether TCF-1 was required to reshape the insulated neighborhoods, we quantified 3D interactions in wildtype, Tcf7−/− DN3s and DPs using our ultra-deep Hi-C measurements. We mapped TCF-1 binding events in DN3s using TCF-1 ChIP-seq and compared TCF-1 occupancy in DN3s with the genome-wide profile based on the public TCF-1 ChIP-seq data in thymocytes24 (Extended Data Fig. 8a). Based on the number of peaks, the public TCF-1 ChIP-seq data in thymocytes was of higher quality (Extended Data Fig. 8a), so we employed them in analyses of DN3s and DPs. Loss of TCF-1 in DN3s led to reduced interactions at 988 TCF-1+CTCF sites (Fig. 4c,d, Supplementary Table 13) and 5,439 TCF-1-only sites (Fig. 4c,d), with a corresponding increase in insulation. These effects were reproduced in another Tcf7−/− DN3 clone (Extended Data Fig. 8b). The effect of TCF-1 deletion in DPs on long-range interactions centered at TCF-1+CTCF sites was even more prominent than that in DN3s, and led to reduced interactions at 3,707 TCF-1 and CTCF co-bound peaks, while 15,151 TCF-1-only sites showed loss of 3D interactions (Fig. 4e,f, Supplementary Table 14). Hi-C data from wild-type and Bcl11b-deficient naïve CD4+ T cells25 indicated no effect of Bcl11b on insulation (Extended Data Fig. 8c). As representative examples, the Rtkn (Fig. 4g), Il33 (Fig. 4h) and Il17d (Fig. 4i) loci showed statistically significant changes in chromatin interactions between 3T3TCF-1 and non-induced 3T3 in addition to wildtype and Tcf7−/− DN3s and DPs. Thus, TCF-1 binding could change local interactions between insulated domains.
Oligopaint 3D FISH corroborates genomic measurements
A robust identification of TADs is still lacking due to the relatively low concordance of TAD definitions34,35. We used alternative methodologies to quantify interaction frequencies across TCF-1 dependent boundaries. We tested how chromatin interactions measured by Hi-C related to spatial localization of adjacent TADs using high-resolution Oligopaint DNA fluorescence in situ hybridization (FISH) with confocal imaging in 3D36,37. Because we detected TCF-1-dependent chromatin interactions across domains at the Cd8a-Cd8b1 locus in fibroblasts, DN3s and DPs (Fig. 5a and Extended Data Fig. 9a,b) we tiled Oligopaint probes along two domains of this locus, referred to as TAD1 (~284 kbp) and TAD2 (~437 kbp) (Fig. 5a), which were insulated in ETP-DN2 and combined into one domain after DN3. Because the largest extent of interactions across boundaries was detected at DPs, we performed Oligopaint FISH experiments in wild-type and Tcf7−/− DPs. To assess the spatial proximity of TADs, we measured the distance between centroids of each domain across individual alleles and used a 3D segmentation strategy38 to trace the edges of each domain’s signal, generating a distribution of domain volumes across ~300 alleles per condition (Fig. 5b). To report the fraction of overlap between two domains across single alleles, the overlap volume of TAD1 and TAD2 per allele was normalized to the volume of TAD1. If chromatin interactions across boundaries at the Cd8a-Cd8b1 locus in DPs was TCF-1-dependent, that would be reflected in a reduction in overlap between TAD1 and TAD2 volumes and an increase in distance between TAD1 and TAD2 centroids in Tcf7−/− compared with wildtype DPs. Representative examples of alleles at the Cd8 locus in wildtype and Tcf7−/− DPs and 3D rendering of TAD1 and TAD2 at the single-cell level corroborated the TCF-1-dependent spatial localization of two adjacent TADs (Fig. 5b,c). This finding was also confirmed for hundreds of DPs across biological replicates (Fig. 5d,e and Extended Data Fig. 9c,d). As such, high-resolution imaging quantified the TCF-1-dependent chromatin interactions across boundaries, independent of sequencing techniques at the single-allele level.
Fig. 5. 3D FISH reveals TCF-1 dependent TAD intermingling at the CD8a locus in DPs.
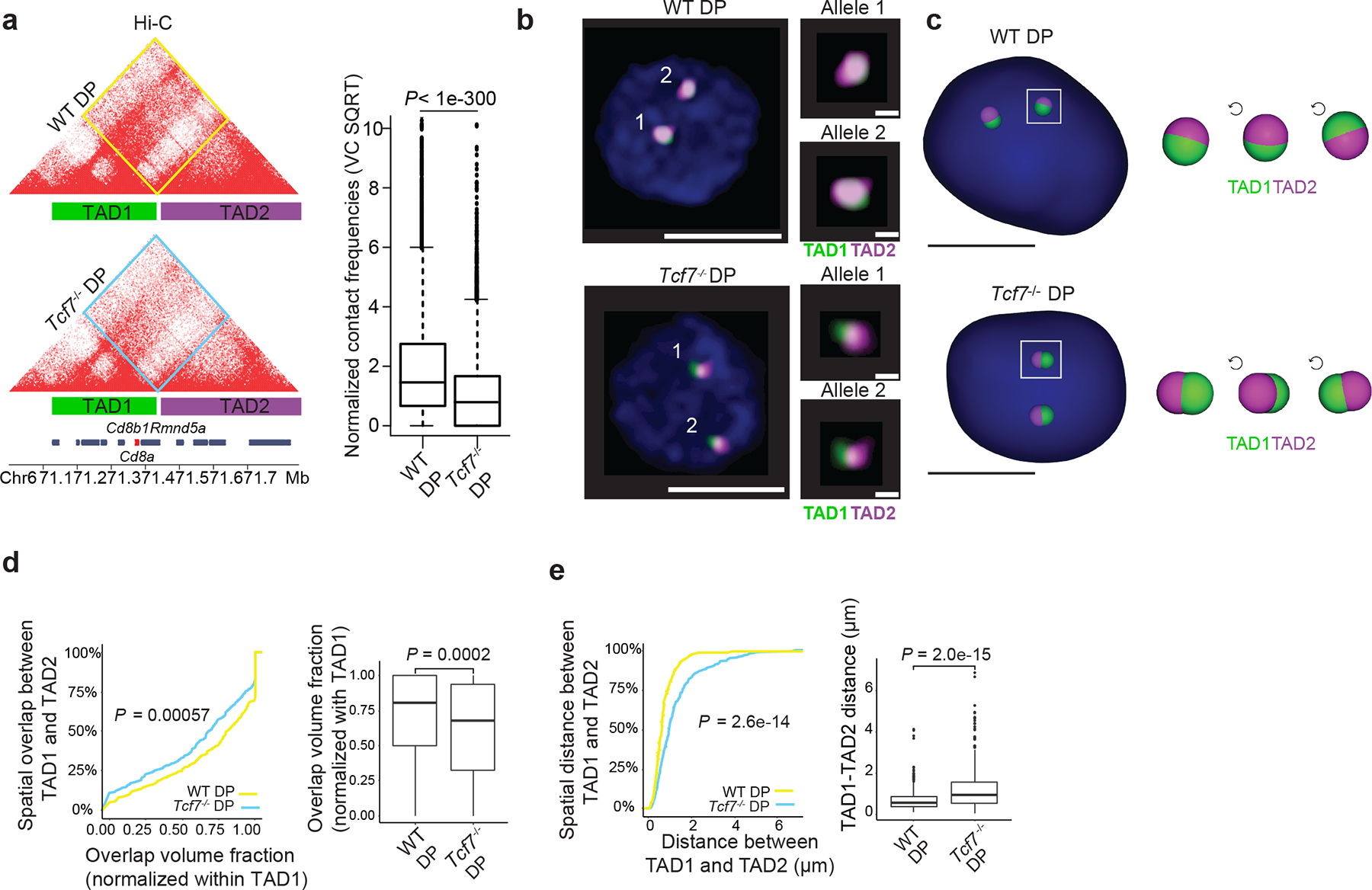
a. Contact matrix of Hi-C data at the Cd8a-Cd8b1 locus which includes the location of Oligopaint probes for TAD1 and TAD2 depicted as green and magenta bars. Inter-domain interactions are quantified by boxplot in wildtype and Tcf7−/− DPs (n=7221 genomic interactions; boxplot centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers). Two sided Paired wilcoxon ranksum test was performed. TAD1 spans chr6:71163477-71432963 while TAD2 spans chr6:71440321-71825773 in mm10.
b. A representative image of Oligopaint FISH probes in wildtype and Tcf7−/− DPs with magnification of each allele. Scale bar for full cell is 5μm and for magnified allele 1 μm, nuclei are stained with DAPI (blue).
c. 3D rendering of TAD1 and TAD2 in wildtype and TCF-1 deficient single DP T cells. Zoomed view of one allele per cell is shown in 3 rotations of 90°, scale bar is 5μm.
d-e. Cumulative distribution plot (left) and corresponding boxplot (right) of overlap volume (d) and distance (e) between TAD1 and TAD2 across 312 individual wildtype DPs and 367 individual Tcf7−/− DPs. Kolmogorov-Smirnov test P-values and two-sided Wilcoxon rank-sum test P-values are shown for cumulative distribution plots and corresponding boxplots, respectively. Cells were pooled from two to three mice per genotype. Overlap volume was defined using a 3D segmentation strategy48 across a minimum of 300 alleles per condition. The overlap volume of TAD1 and TAD2 per allele was normalized to the volume of TAD1 (d). Distance was measured between the centroids of each domain across individual alleles (e). Data are shown as boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers)
TCF-1 recruits NIPBL to reconstruct 3D genome
TADs are formed by cohesin-mediated extrusion of DNA1. Subunits of the cohesin complex are loaded onto chromatin by NIPBL and MAU239–41 and once loaded, they extrude DNA until convergent CTCF sites are recognized42–45. Although TCF-1 overexpression in fibroblasts or TCF-1 deletion in DN3s changed CTCF occupancy (Extended Data Figs. 5b and 10a), CTCF binding at TCF-1-dependent boundaries remained unchanged (Extended Data Fig. 7). We mapped the genome-wide binding patterns of NIPBL and SMC1 and measured the chromatin signature of active enhancers using H3K27ac ChIP-seq in wild-type and Tcf7−/− DN3s in biological replicates (Supplementary Figs. 2–3). We created a binding atlas of cohesin occupancy in DN3s by combining SMC1 and NIPBL peaks across various conditions and evaluated genomic regions with gain or loss of SMC1 and NIPBL, in addition to deposition of H3K27ac after TCF-1 deletion in DN3s. H3K27ac was lost in Tcf7−/− DN3s at more than 2,000 genomic regions, concomitant with an evident diminution of NIPBL and SMC1 binding at these sites (Fig. 6a, Supplementary Table 15). Notably, the overt chromatin accessibility at these regions, as measured by ATAC-seq, did not change commensurately (Fig. 6a). As a representative example, H3K27ac, SMC1 and NIPBL binding events were concordantly reduced in Tcf7−/− DN3s at a cluster of interferon-induced transmembrane genes (Fig. 6b). Gain of H3K27ac and co-occupancy of NIPBL and SMC1 occurred at more than 1,300 genomic regions in Tcf7−/− DN3s (Fig. 6c,d, Supplementary Table 16). ETS, RUNX, and TCF recognition motifs were enriched at sites where cohesin complex occupancy was lost in Tcf7−/− DN3s (Fig. 6e), while CTCF-like recognition motifs were enriched at genomic regions that gained cohesin occupancy in Tcf7−/− DN3s (Fig. 6f) compared to wild-type DN3s. SMC1 ChIP-seq indicated a similar motif enrichment lost in Tcf7−/− DN3s (Extended Data Fig. 10c, Supplementary Table 8). The long-range interactions anchored at TCF-1-dependent SMC1+NIPBL co-occupied regions that carried the H3K27ac signature were also TCF-1-dependent (Fig. 6g,h). Moreover, the TCF-1-dependent SMC1+NIPBL co-bound enhancers were enriched near weakened boundaries bound by TCF-1+CTCF (Extended Data Fig. 10d). Together, TCF-1 established the active enhancer repertoire and sequestered the cohesin machinery to these enhancers, a process which can create de novo long-range interactions and cause weakening of the previously insulated neighborhoods.
Fig. 6. TCF-1 promotes NIPBL recruitment to active enhancers to reconstruct genome organization.
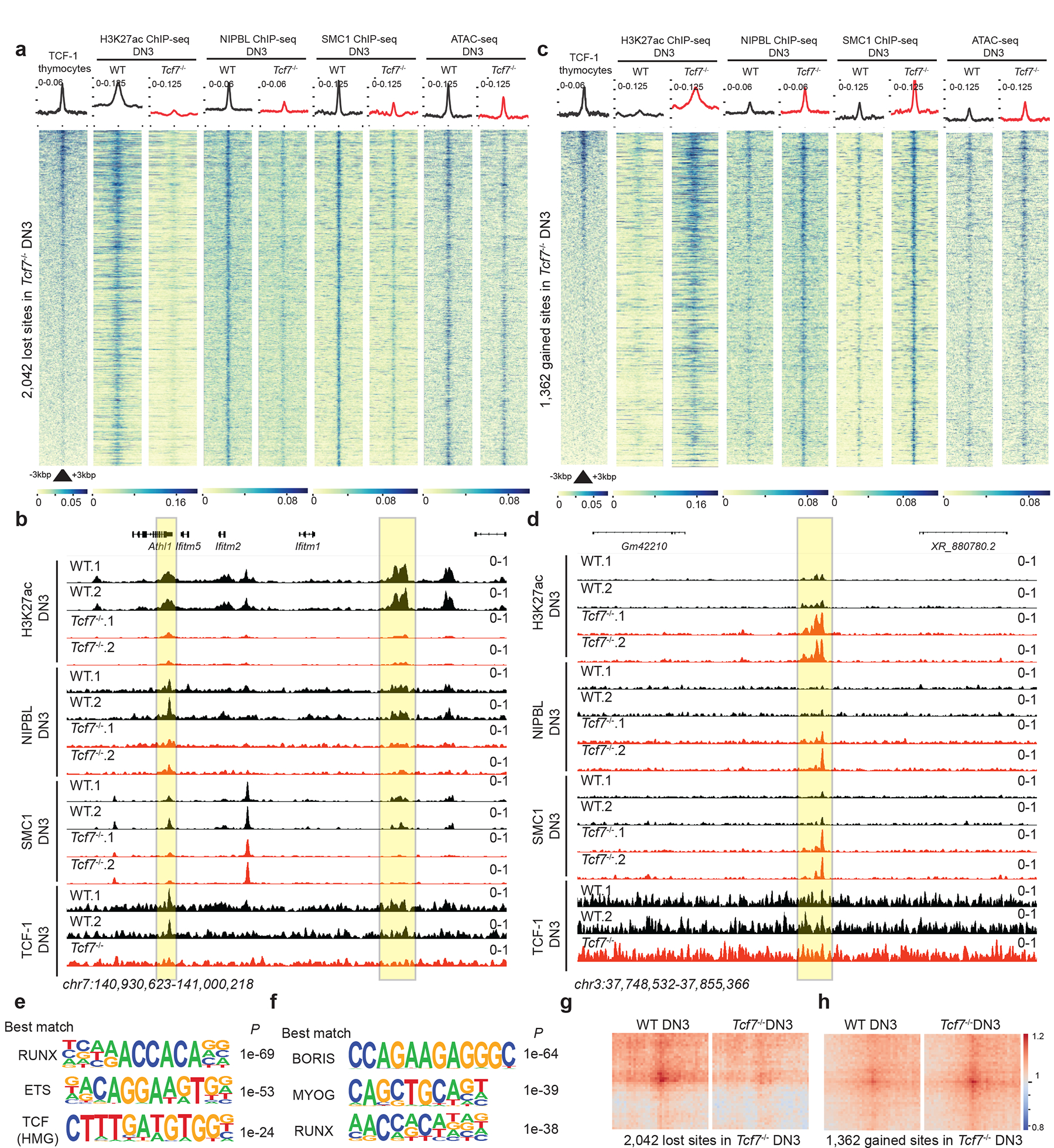
a,c. Heatmap depicting genomic regions where TCF-1 led to a loss (a) or a gain (c) on the binding events of these proteins in addition to deposition of H3K27ac in DN3s. A binding atlas of cohesin occupancy was created by combining SMC1 and NIPBL peaks across various conditions. DESeq2 was used to determine TCF-1 dependent set of peaks between wildtype and Tcf7−/− cells. Heatmaps show +/− 3kb around peak center. Pubicly available TCF-1 ChIP-seq from thymocytes is used25. H3K27Ac, NIPBL, and SMC1 ChIP-seq and ATAC-seq experiments were generated in DN3s.
b. Genome browser view at a cluster of interferon-induced transmembrane genes. Yellow highlight shows binding of TCF-1 at genomic loci where H3K27ac, SMC1, and NIPBL binding events were concordantly reduced in Tcf7−/− DN3s.
d. Genome browser view of a representative example locus, yellow highlight shows genomic loci where H3K27ac, SMC1, and NIPBL binding events were concordantly increased in Tcf7−/− DN3s.
e-f. Seq-logos demonstrating top motif enrichment based on Homer analysis of genomic regions in which occupancy of cohesin was lost (e) or gained (f) in Tcf7−/− DN3s compared to random background regions. P values are calculated using hypergenometric test.
g-h. Local pileup of long-range interactions anchored at genomic regions described in (a) and (c) with lost (g) and gained (h) occupancy. Hi-C data in DN3s were used.
Discussion
Here, we showed that co-binding of TCF-1 and CTCF earmarked TAD boundaries which reorganized during T cell development. The disruption of TAD boundaries, such as domains encompassing the Cd8 and Cd3 clusters, was accompanied by interactions of regulatory elements previously located in different insulated neighborhoods. Key regulatory elements and their target genes were spatially distanced on different TADs during early developmental stages when these target genes were transcriptionally silent. Our data indicated that TCF-1 endowed competence to the T cell developmental program by minimizing the spatial distance between regulatory elements required for the proper expression of T cell genes, even when located within insulated neighborhoods in progenitors.
The ability of TCF-1 to recruit NIPBL and the cohesin complex at active enhancers implied a possible mechanism through which TCF-1 could diminish insulation at TAD boundaries. Degrading the histone acetyltransferases P300 leads to loss of NIPBL loading, suggesting histone acetylation may represent a signal for cohesin loading46. We speculate that TCF-1-mediated histone acetylatation can lead to accumulation of NIPBL and recruitment of the cohesin complex, which in turn would lead to chromatin interactions across TCF-1 bound loci. Considering that TCF-1 expression is induced at ETP, it is puzzling why it takes so long for the boundary dynamics to occur after DN3s. We speculate that the cohesin-complex-mediated genome reorganization during T cell development might be rate limiting. This hypothesis is supported by the increased expression of NIBPL at DPs (ImmGen data), but further experiments are required. Whether the HMG domain of TCF-1 bends DNA to assist the cohesin complex during genome reorganization remains to be shown.
Although our work established an effect of TCF-1 on genome folding, it remains unclear to which extent the TCF-1-dependent global genome organization relates to transcriptional control. The emerging view is that transcription factors can alter genome organization prior to mediating transcriptional control. Pre-existing loops and the requirement of stage-specific histone acetylation for gene expression has been reported47,48. It is enticing to speculate that TCF-1 sets the thymocytes 3D genome landscape up for transcriptional control once T cells specialize and expand in the periphery. Additional work is needed to elucidate if CTCF and TCF-1 co-binding events enable spatial contacts between regulatory elements of genes essential in stem-like or memory CD8+ T cells. Despite these new questions, our study describes an LDTF with the ability to eliminate TAD-enforced spatial distance between regulatory elements and their target genes in progenitors. We speculate that other LDTFs might be proficient to enable interactions between regulatory elements and target genes in other developmental pathways.
METHODS
Resource availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Golnaz Vahedi (vahedi@pennmedicine.upenn.edu).
Materials availability
This study did not generate new unique reagents.
Cell culture
NIH 3T3 cells were purchased from ATCC as described 17 and maintained at a low passage number (< 12) using high glucose 1× DMEM medium with L-glutamine (GIBCO) with 100 U mL-1 penicillin and 100 mg mL-1 streptomycin (GIBCO) and 10% bovine serum (GIBCO). 293T (ATCC) cells were maintained in high glucose DMEM 1× medium with L-glutamine (GIBCO), and 100 U mL-1 penicillin and 100 mg mL-1 streptomycin (GIBCO) with 10% fetal bovine serum (FisherScientific). Scid.adh cell line, a pro-T cell line derived from spontaneous thymic lymphomas 64, was a kind gift from Warren Pear’s lab at the University of Pennsylvania. These cells were grown in RPMI 1640 medium (Invitrogen), supplemented with 10% fetal bovine serum (FisherScientific), 1mM sodium pyruvate (Gibco), 1% non-essential amino acids (Gibco), 2 mM L-Glutamine (Lonza), 1% penicillin-streptomycin and 0.1% 2-Mercaptoethanol (Gibco). All cells were grown at 37°C and 5% CO2.
Mice
Female and Male breeder Vav-iCre transgenic mice (Strain #008610)65–67 and Tcf7eGFP Mice (strain # 030909)68 were purchased from Jackson Laboratory. “Tcf7−/−” mice were generated by breeding Tcf7eGFP mice, in which 2 loxP sites are inserted on either side of exon 2 of the Tcf7 gene, with Vav-iCre mice. The F1 generation was backcrossed to Tcf7eGFP mice to reach homozygous floxed Cre+ experimental mice. Vav-iCre+ control mice were generated by crossing Vav-iCre mice with C57BL6/J (strain # #000664) and used as controls in all experiments. Experimental and control mice were 6–10 weeks old of either sex. All mice were bred and housed in an American Association for the Accreditation of Laboratory Animal Care (AAALAC) accredited vivarium at the University of Pennsylvania. All husbandry and experimental procedures were performed according to the protocol reviewed and approved by the Institutional Animal Care and Use Committee (IACUC).
Cell preparation
Single-cell suspensions were prepared from thymi of mice by dissociation of tissue through 70 mM mesh filters (Falcon) in RPMI 1640 (Corning) +1% FBS (Gemini). DP T cells were isolated utilizing serial magnetic bead separation. First EasySep™ Release Mouse Biotin Positive Selection Kit was utilized with biotin anti-mouse CD8a (53–6.7), followed by isolation with EasySep™ Mouse CD4 Positive Selection Kit II. Purity of DP cells post isolation was ≥ 90% as assessed by flow cytometry.
METHOD DETAILS
Lentiviral Packaging and Transduction for NIH 3T3 cells
For TCF-1 overexpression in 3T3 cells, the cDNA encoding the short isoform of TCF-1 (p33)17 was cloned into Tet-inducible lentiviral vector pINDUCER-20 (Addgene) using Gateway cloning strategy (Gateway Clonase II, Invitrogen). Lentiviral vectors were packaged in HEK293T cells. Briefly, 4 × 106 HEK293T cells were plated in 5 mL media in 10 cm dishes on the day prior to transfection. During transfection, 3.9 μg of pINDUCER-20-TCF-1 plasmid was co-transfected with packaging plasmids, 2.6 μg of pCMVdelta and 1.3 μg of VSV-G, using 26 μL FuGene HD (Promega). The cells were returned to the incubator for 6 hours. Subsequently, the medium was changed to fresh media. Virions were collected 24 and 48 hrs after transfection, snap-frozen, and stored at −80°C for future use. NIH 3T3 cells were transduced by addition of virions to culture media supplemented with polybrene (Sigma-Aldrich, cat# H9268) at 8 mg mL−1 followed by centrifugation at room temperature for 20 mins at 2000 rpm. Transduced cells were selected using 1 mg/ml G418 (Gibco) for 7 days. TCF-1 expression was induced using 500 ng/ml of doxycycline for 72 hrs. Cells which have been transduced with the same vector but have not been treated with doxycycline are referred to as ‘untreated NIH-3T3’ while doxycycline-treated cells are referred to as ‘TCF-1 induced NIH-3T3’.
Retroviral Packaging and Transduction for TCF-1 KO in Scid.adh cell line
CRISPR/Cas9 system was used to delete TCF-1 in Scid.adh cells. sgRNAs targeting the DNA-binding domain (HMG-box) were designed using CRISPR Targets tracks of UCSC genome browser. ACCGCAACCAGATCCTGGGTCGCA and AAACTGCGACCCAGGATCTGGTTG were used as sgRNA. sgRNA was cloned into pSL21-vex (Addgene 158230)69, MSCV-Cas9-puro plasmid (Addgene 65655) was used for retroviral introduction of Cas9 into the cells. For retroviral packaging, 4 × 106 293T cells were plated in 4 mL DMEM media in 10 cm dishes on the day prior to transfection. Immediately before transfection, chloroquine was added to the media to a final concentration of 25 mM. The retroviral construct/empty vector and the pCL-Eco plasmid were transiently co-transfected using Lipofectamine 3000 (Invitrogen). The cells were returned to the incubator for 6 hours. Subsequently, the medium was changed to fresh media. Virions were collected 24 and 48 hrs after transfection, snap-frozen, and stored at −80°C for future use. Scid.adh cells were transduced by addition of Cas9- and sgRNA-virions to culture media supplemented with polybrene (Sigma-Aldrich, cat# H9268) at 8 mg mL−1 and spinfected at 1750 rpm for 25 mins. Cas9-expressing cells were selected 4 days after spin infection with 1 μg/mL puromycin for 7 days, followed by checking Cas9 expression on the western blot. To obtain single cell clones of the TCF-1-knockout cells, we diluted bulk transduced cells to obtain a dilution of 1 cell in 100 μL media and plated them in 96-well plates. Knockout clones G9 and E10 were selected after performing western blot to check TCF-1 expression. Hi-C data in both clones are used to evaluate the effect of TCF-1 deletion in DN3 cell lines.
Western Blot
Western blotting was performed on whole cell lysates from Vav-Cre+, heterozygous and homozygous Tcf7−/− thymocytes as well as cells derived from Scid.adh Tcf7 KO single cell clones. Cells were lysed with 1X RIPA buffer supplemented with proteinase inhibitor cocktail. Equal numbers of cells per condition were utilized and equal volumes of lysate were loaded on a NuPAGE™ 4–12% Bis-Tris gel and transferred using the iBlot™ 2 Gel Transfer Device. Membranes were blocked with 5% non fat dry milk in 1X TBST buffer followed by incubation with primary rabbit-anti mouse and HRP conjugated secondary antibodies. Blots were visualized with SuperSignal™ West Femto Maximum Sensitivity Substrate on the ChemiDoc™ Imaging system.
Flow Cytometry
Single-cell suspensions were stained following standard protocols. The fluorochrome-conjugated, anti-mouse antibodies were as follows: PE CD4 (RM4-4), APC CD8a (53–6.7), BV785 CD44 (IM7), PECy7 CD25 (PC61) and APC Streptavidin. Antibodies used in the lineage cocktail (Lin) include biotinylated antibodies against GR-1 (RB6-8C5), NK1.1 (PK136), CD11b (M1/70), Ter119 (TER-119), and B220 (RA3-6B2). Cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit for discrimination of live cells. Data were collected on an LSRII running DIVA software (BD Biosciences) and were analyzed with FlowJo software v10.6.1.
ChIP-seq
ChIP-seq was performed as previously described 27. Cells were fixed for 30 min in RPMI 1640 (Gibco) with 1% FBS (Sigma-Aldrich) with 1.5 mM ethylene glycol-bis (succinic acid N-hydroxysuccinimideester) (EGS) (Thermo 21565) in DMSO followed by 15 min with 1% formaldehyde (Formaldehyde solution 16% Fisher 28906) and quenched for 10 min with 0.125 M glycine. Cells were washed 2× with PBS and frozen at −80°C. Cross-linked cells were lysed and then sonicated for 10 cycles at 10 sec each with 50 sec between cycles. Triton X-100 was added to a final concentration of 1% to centrifuge cleared lysates. Lysates were incubated overnight with Protein G Dynabeads (ThermoFisher 10003D) conjugated to 10 μg of anti-SMC1 Antibody (Bethyl A300-055A), 10 μg of anti-CTCF antibody (Millipore 07–729), 10 μg of anti-NIPBL antibody (Bethyl A301-779A), 10 μg of anti-H3K27ac antibody (Abcam ab4729) or 10 μg of anti-TCF-1 antibody (CST 2206S). Beads were washed and complexes were eluted for 30 min at 65°C with shaking. After reversal of cross-linking, RNase and proteinase K treatment were performed and DNA was purified and quantified. Two biological replicates were generated for each experiment. Two separate aliquot of fixed cells per condition were used as replicates for each experiment. Library preparation was carried out using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) and were paired-end sequenced (38bp+38bp) on a NextSeq 550 (Illumina).
RNA-seq
Cells were washed once with 1× PBS before resuspending pellet in 350 μL Buffer RLT Plus (QIAGEN) with 1% 2-Mercaptoethanol (Sigma), vortexed briefly, and stored at −80°C. Subsequently, total RNA was isolated using the RNeasy Plus Micro Kit (QIAGEN). RNA integrity numbers were determined using a TapeStation 2200 (Agilent), and all samples used for RNA-seq library preparation had RIN numbers greater than 9. Libraries were prepared using the SMARTer® Stranded Total RNA-seq Kit v2- Pico Input Mammalian kit (Takara). Two biological replicates were generated for each experiment. Two separate aliquot of fixed cells per condition were used as replicates for each experiment. Libraries were validated for quality and size distribution using a TapeStation 2200 (Agilent). Libraries were paired-end sequenced (38bp+38bp) on a NextSeq 550 (Illumina).
ATAC-seq
ATAC-seq was performed as previously described with minor modifications 27,70. Fifty thousand cells were pelleted at 550 × g and washed with 50 μL ice-cold 1× PBS, followed by treatment with 50 μL lysis buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). Nuclei pellets were resuspended in 50 μL transposition reaction containing 2.5 μL Tn5 transposase (FC-121-1030; Illumina). The reaction was incubated in a 37°C heat block for 45 min. Tagmented DNA was purified using a MinElute Reaction Cleanup Kit (QIAGEN) and amplified with varying cycles, depending on the side reaction results. Libraries were purified using a QIAQuick PCR Purification Kit (QIAGEN). Libraries were validated for quality and size distribution using a TapeStation 2200 (Agilent). Libraries were paired-end sequenced (38bp+38bp) on a NextSeq 550 (Illumina).
HiChIP
HiChIP was performed as described 71 using antibody against SMC1 (Bethyl A300-055A). Briefly, 10 × 106 cells were crosslinked with 1% formaldehyde (Thermo Fisher Scientific, cat# 28908) for 10 min and subsequently quenched with 0.125 M glycine (Invitrogen, cat# 15527–013). Chromatin was digested using MboI restriction enzyme (NEB, cat# R0147), followed by biotin incorporation with Biotin-14-dATP (Invitrogen, cat# 19524–016) during repair, ligation, and sonication. Sheared chromatin was 4-fold diluted with ChIP dilution buffer (16.7mM Tris pH 7.5, 167mM NaCl, 1.2mM EDTA, 0.01% SDS, 1.1% Triton X-100), pre-cleared, and then incubated with SMC1 antibody at 4°C overnight. Chromatin-antibody complexes were captured by Protein-A magnetic beads (Pierce, cat# 88846) and subsequently washed with Low Salt Wash Buffer, High Salt Wash Buffer, LiCl Wash Buffer and eluted. DNA was purified with MinElute PCR Purification Kit (QIAGEN, cat# 28004) and quantified using Qubit dsDNA HS Assay Kit (Invitrogen, cat# Q32851). 50–150ng was used for capture with Dynabeads MyOne Streptavidin C-1 (Invitrogen, cat# 65001) and an appropriate amount of Tn5 enzyme (Illumina, cat# FC-121-1030) was added to captured DNA to generate the sequencing library. Libraries were validated for quality and size distribution using a TapeStation 2200 (Agilent). Paired-end sequencing (38 bp+38 bp) was performed on a NextSeq 550.
Hi-C
Hi-C libraries were generated on 106 cells using with Arima-HiC+ kit (Arima Genomics) and Accel-NGS @S Plus DNA Library kit (21024 Swift Biosciences), according to the manufacturer’s recommendations. Libraries were validated for quality and size distribution using Qubit dsDNA HS Assay Kit (Invitrogen, cat# Q32851) and TapeStation 2200 (Agilent). Libraries were paired-end sequenced (66bp+66bp) on NovaSeq 6000 (Illumina).
Oligopaint Probe designing
The TADs and boundaries at CD8a locus were identified using insulation score 26 (as described in more details subsequently). The OligoMiner design pipeline 72 was then applied to design DNA FISH probes to the respective coordinates, chr6: 71163477–71432963 (TAD1) and chr6: 71440321–71825773 (TAD2). Oligopaint probes were designed to have 80 bases of homology with an average of 5 probes per kb and were purchased from Twist Bioscience. Oligopaint probes were synthesized as described previously 73,74.
Oligopaint DNA FISH
Cells were incubated on 75 × 25 mm polysine adhesion glass slides (Electron Microscopy Sciences, cat #63412-01) with silicone isolators (Electron Microscopy Sciences, cat #70339-05) for 1 hour at RT in a humidified chamber. Cells were then fixed to the slide for 10 min with 4% formaldehyde in phosphate-buffered saline (PBS) and rinsed briefly in PBS. Slides were stored in PBS at 4°C until use.
For DNA FISH primary probe hybridization, slides were warmed to RT in PBS for 10 min. Cells were permeabilized in 0.5% Triton-PBS for 15 min. Cells were then dehydrated in an ethanol row, consisting of 2-min consecutive incubations in 70%, 90%, and 100% ethanol. The slides were then allowed to air dry for about 2 min at RT. Cells were then washed in 4XSSCT/50% formamide (0.6M NaCl, 0.06M sodium citrate, 0.2% Tween-20) for 5 min, pre-denatured in 4XSSCT/50% formamide at 92°C for 2.5 min, and then in 4XSSCT/50% formamide at 60°C for 20 min. 50 pmol of primary Oligopaint probes in hybridization buffer (10% dextran sulfate/2xSSCT/50% formamide/4% polyvinylsulfonic acid (PVSA)/1.4 mM dNTPs) were then added to the cells, covered with a 24 × 50 mm glass coverslip (Fisher Scientific, cat #22037170), and sealed with no-wrinkle rubber cement (Elmer’s). Cells were denatured by placing slides on a heat block in a water bath set to 92°C for 2.5 minutes, after which slides were transferred to a humidified chamber and incubated overnight at 37°C. Approximately 16–18 hours later, coverslips were removed with a razor blade, and slides were washed in 2XSSCT (0.3M NaCl, 0.03M sodium citrate, 0.1% Tween-20) at 60°C for 15 min, 2XSSCT at RT for 10 min, and 0.2XSSC at RT for 10 min.
Secondary probes (10 pmol/25 μL) containing fluorophores Atto-565 and Alexa-647 (IDT) were added to the slides, again resuspended in hybridization buffer containing only 10% formamide and covered with a coverslip sealed with rubber cement. Slides were incubated at RT for 2 hours in a dark, humidified chamber, followed by washes in 2XSSCT at 60°C for 5 min, 2XSSCT at RT for 5 min. All slides were stained in PBS with DAPI (0.1 mg/mL) and then washed in 2XSSC. Slides were mounted with Slowfade Gold Antifade Reagent (Invitrogen, cat# S36936) and sealed with clear fingernail polish.
Image acquisition and processing
Imaging was performed on a Leica TCS SP8 Multiphoton Confocal microscope using a 1.3 NA ×40 oil immersion objective with pixels of 541.6 nm × 541.6 nm. Fields of view were selected, such that confluence was balanced to provide maximum data points and to ensure proper cell segmentation during downstream analysis. Z-stacks were determined, such that both homologs were within the imaged space and represented 10 μm in total axial thickness. Localizations were then recorded in 0.3 μm steps.
Representative images were also obtained on a Leica TCS SP8 Multiphoton Confocal microscope, this time using a 1.4 NA ×63 oil immersion objective with pixels of 343.9 × 343.9 nm. Z-stacks were determined and recorded as with the 1.3 NA ×40 oil immersion objective. Each cell, allele, and locus for each strain were individually processed using FIJI software to generate orthogonal projections of the Z-stacks and to split channels into individual TIFs.
3D reconstructions of representative cells were rendered using IMARIS v.7.4.2 software (Bitplane AG, Switzerland). DNA FISH dots were generated using the Spots tool with a 0.8 μm diameter, created at the intensity mass center of the fluorescent probe signal. Nuclear volume was created using the Surfaces tool with automatic settings based on the fluorescent signal from the DAPI stain.
ChIP-seq data analysis
Bowtie2 50 was used for alignment of ChIP-seq data. Reads aligned to the mitochondrial genome or chrY as well as reads mapping to multiple genomic loci were discarded from downstream analyses. Bigwig files were generated by bedtools 75 genomecov and wigToBigWig normalizing tracks to tags-per-million. For peak calling, macs2 54 with “macs2 callpeak -c inputfile–nolambda–nomodel–keep-dup all -p 0.00001” was used. Input sample was prepared by the same approach without immunoprecipitation and used as input control for peak calling. ChIP-seq peaks from two conditions and both replicates were merged, and the number of fragments in each peak were counted with bedtools. The count data of each peak was then fed to DEseq2 for differential analysis.
RNA-seq data analysis
RNA-seq samples were aligned by STAR_2.5.0a_alpha 51 with parameters ‘– readFilesCommand zcat–outFilterMultimapNmax 1–outSAMtype SAM–alignEndsType Local–outReadsUnmapped Fastx–outFilterMismatchNmax 1–alignMatesGapMax 400000–sjdbGTFfile’. HTSeq v0.6.1 facilitated counting RNA-seq reads on Gencode vM11 gene models with parameters ‘-s yes -t exon -m intersection-nonempty’. DESeq2 was subsequently applied on gene counts to identify genes differentially expressed.
ATAC-seq data analysis
Bowtie2 was used for alignment (bowtie2 -p 20 -X2000 -t). Reads aligned to the mitochondrial genome or chrY as well as reads mapping to multiple genomic loci were discarded from downstream analyses. Additionally, Picard was used to mark and remove duplicates. Furthermore, for each ATAC-seq library the insert size was calculated by Picard. The insert size distribution of sequenced fragments had clear periodicity of approximately 200 bp, suggesting many fragments are protected by integer multiples of nucleosomes. Bigwig files were generated by bedtools genomecov and wigToBigWig normalizing tracks to tags-per-million. For peak calling, macs2 with “macs2 callpeak–nomodel -B–keep-dup all–broad–broad-cutoff 0.1 -q 0.1” was used.
HiChIP and Hi-C data analysis
HiChIP data processing
Raw reads for HiChIP sample were processed with HiC-Pro (version 2.11.1) 56 to obtain putative interactions with default parameters except LIGATION_SITE = GATCGATC and GENOME_FRAGMENT generated for MboI restriction enzyme. For the purpose of downstream analysis, ValidPairs were converted to cool files and hic files using the “hicpro2higlass.sh” and “hicpro2juicebox.sh” in utils of HiC-Pro, respectively.
Hi-C data processing
For the 3e Hi-C data during T cell development 25, we downloaded the raw data from GSE79422 and processed it with HiC-Pro without setting the LIGATION_SITE and GENOME_FRAGMENT. For the Hi-C data we generated in wild type and TCF-1 knock out DN3 cell line, we processed the data with HiC-Pro using parameters LIGATION_SITE = GATCGATC and GENOME_FRAGMENT generated for MboI restriction enzyme, keeping other parameters as default. ValidPairs generated by HiC-Pro were further converted to cool and hic files as described above.
Compartment analysis
We did the compartment analysis of HiChIP and Hi-C data following the tutorial on cooltools github page (https://github.com/open2c/cooltools) and further visualized the compartment strength utilizing saddle plot implemented in cooltools. Scatter plot of PC1 values between the two different conditions was done with ggplot2 in R, of which the correlation was calculated with stat_cor() function in ggpubr package. The compartment strength was calculated with the top 30% PC1 regions representing A compartment and bottom 30% PC1 value representing B compartment. We calculated the compartment strength, AA interactions, BB interactions and AB interactions of each chromosome, and used boxplot to compare the two conditions implementing Student’s t-test for statistical test.
TAD and boundary analysis
TADs and boundaries were identified with insulation score26, which was implemented in cooltools “diamond-insulation” function. We calculated the genome-wide insulation score at 20 kb resolution with 500 kb window size utilizing cooltools diamond-insulation. The insulation score result was converted to bedgraph file, which was further converted to bigwig files for visualization purpose. Boundaries were identified with the bins whose boundary strength > 0.1 and were further used to define TADs with custom python script.
Common and unique boundaries between conditions and among T cell developmental stages were identified with custom python script, in which we clustered the boundaries whose minimum distance is less than 100 kb and the maximum distance between the boundaries should be within 200 kb. We generated a set of consensus boundaries based on the boundary clusters by taking the two ends of each cluster as boundary coordinates. In this way, we generated the common and unique boundaries between conditions and identified the boundaries that are shared between different T cell developmental stages. The boundaries that are shared or unique to the six T cell developmental stages were visualized with UpSetR 60. Common and unique TADs between conditions were identified with bedtools intersect (-wao -f 0.8 -r) 75 and followed with filtering the TADs by only keeping TADs whose distances of starts and ends in two conditions are less than 100 kb. Only TADs that are larger than 200 kb were kept for downstream analysis. We did domain score analysis based on these common TADs between two conditions. Cooler was used to dump the contact matrix from cool files, and custom script was used to calculate the PETs within the TAD, which was divided by PETs between the TAD and the chromosome to generate the domain score.
Directional index
Matrix was generated from cool files using cooltools dump-cworld with parameter “--balancing-type IC_unity”. Then directional index (DI) was calculated with script from dekker-cworld (https://github.com/dekkerlab/cworld-dekker). The results were further converted to bedgraph and bigwig files for visualization.
TCF-1 association with interaction change
In the TCF-1 association with increased intra-TAD interactions during T cell development, we identified the common TADs between five early stages and DP T cells. Then we calculated the log2 fold change of PETs within the common TADs from early stages to DP, as well as TCF-1 densities in the TADs. The relationship between TCF-1 density and increase of intra-TAD interactions was visualized with boxplot in ggplot2, and the correlation was calculated with stat_cor() function.
In the association of TCF-1 binding and domain score change in NIH3T3, we subtracted the domain scores of common TADs between TCF-1 induced and normal NIH3T3 and calculated the TCF-1 densities in the TADs. Then we plotted the relationship between these two features with boxplot in ggplot2. For the bin based analysis, we calculated the number of TCF-1 peaks within 50 kb to each bin, and further classified it according to whether there is TCF-1 binding in that bin. Then we calculate the total PETs between the bin and other regions of the TAD in both TCF-1 induced and normal NIH3T3, which was used to determine log2 fold change of interactions of the bin. We plotted the relationship between number of TCF-1 peaks within 50 kb and log2 fold change of interactions of the bins using boxplot in ggplot2, and further separated the bins to with or without TCF-1 binding. We used the classified bins, and aggregated the PETs between bin pairs with same TCF-1 peaks and binding status. Log2 fold change of the pairwise interaction between the bins was calculated and visualized with python script.
Loop analysis
ValidPairs from HiC-Pro and SMC1 ChIP-seq peaks were fed to hichipper 62 to call significant interactions. Only loops with PETs >= 2 were kept for downstream analysis. The comparison of increased loops after TCF-1 induction in fibroblast between different peaks was done using violin plot and boxplot in addition to stat_compare_means() for statistical tests in ggplot2.
We used loops from mango to do the association analysis of loops with TCF-1 and CTCF ChIP-seq peaks utilizing bedtools pairtobed function. The TCF-1 and CTCF peaks were extended 5 kb at both sides. Then we plotted the loops associated with TCF-1 and CTCF peaks with venn diagram in python (https://pypi.org/project/matplotlib-venn/). Genome browser view of cohesin loops and ChIP-seq signals on TCF-1, CTCF and SMC1 were shown using IGV.
3D Pileup analysis
Local pileup analysis at different sets of peaks was done with coolpup.py 63 using parameters “--pad 250 --local”. The average interaction of the upright corner of pileup plots is quantified with custom python script, by parsing the results from coolpup.py. Interactions between regions in bedfiles were done with parameters “--mindist 200000 --maxdist 2000000”. Pileup of loops was also plot with coolpup.py. To identify the regions that have insulation change, we quantified the average interactions between upstream and downstream of each peak. The peaks whose difference of average interactions between two conditions greater than 0.05 will be considered change of insulation. To do multiple pileup analysis parallelly, we used GNU parallel to run the shell script.
Stripe analysis
We used Stripenn76 to quantify stripiness of the stripe originating from the TCF1 cluster between NOD and C57BL/6.
1D pileup analysis
The pileup analysis of 1D features including insulation score, directional index, ChIP-seq and phastCons score was done with deeptools61.
Visualization of 1D and 3D features at selected regions
We used HiCExplorer 59 to visualize the interactions, insulation score, genes and ChIP-seq signals of selected regions. The cool files were converted to h5 format with hicConvertFormat using parameter “--load_raw_values”. The color scales of the interaction heatmaps were normalized with total PETs.
Triangle heatmaps
Triangle heatmaps for 3D chromatin conformation data and corresponding tracks were generated using Sushi R package (version 1.28.0)
Quantification of inter-domain interactions
The inter-domain interactions marked yellow and sky blue boxes in the Sushi plots were quantified using matrix.fetch function in the cooler package77. The square root of vanilla coverage method (VC SQRT)78 or matrix balancing normalization79 was used.
Detection of regions which gained or lost insulation at TCF-1 binding sites
We used custom script following the tutorial on cooltools GitHub page (https://github.com/open2c/cooltools) to calculate the interactions in a 500 kb window centered on each peak, for both untreated and TCF-1 induced 3T3, and for WT and TCF-1 KO DN3 cells. Those peaks that have an increase or decrease of interactions between upstream and downstream of the peaks by greater than 0.05 were selected for TCF-1 induced 3T3 and TCF-1 KO DN3 cells, respectively. The pileup at these peaks were plotted using coolpup.py.
Distance to T cell development genes
We downloaded the signature genes for each T cell development stage from single cell studies of thymus in mouse 28. Then calculated the distance of these genes to the closest TCF-1 and CTCF co-bound peak with bedtools closest.
Oligopaint FISH imaging analysis
The TANGO80 plug-in of FIJI software81 was utilized for the oligopaint FISH imaging analysis. An image with LIF file extension was used as an input. The imaging analysis is composed of two steps: (1) cell/probe segmentations and (2) the measurement of the overlapping volume and minimum distance between two probes. Before the cell segmentation, Fast Filter 3D (median filter) and Misc 3D Filters (Gaussian 3D filter) were applied to the image. Then, OTSU method was used for cell segmentation. After cell segmentation, Morphological Filters 3D (Fill Holes 3D and Fill holes 2D) was applied for cells as post-filtering. For probe channels, Fast Filters 3D (Mean filter), Subtract Background 2D (radius: 5 pixels, rolling ball method) and Misc 3D filters (Gaussian 3D filter) were applied before segmentation. For segmentation, Percentage of Bright Pixels was set as 3.5%. As post-filtering, Morphological filters 3D and Size and Edge filter (minimum volume=5) were applied. After cell segmentation, those including more than one cell were eliminated from the analysis. To measure the overlapping volume and the distance between two probes, object colocalization and minimal distance functions were utilized.
Extended Data
Extended Data Fig. 1. Loops associated with CTCF and TCF-1.
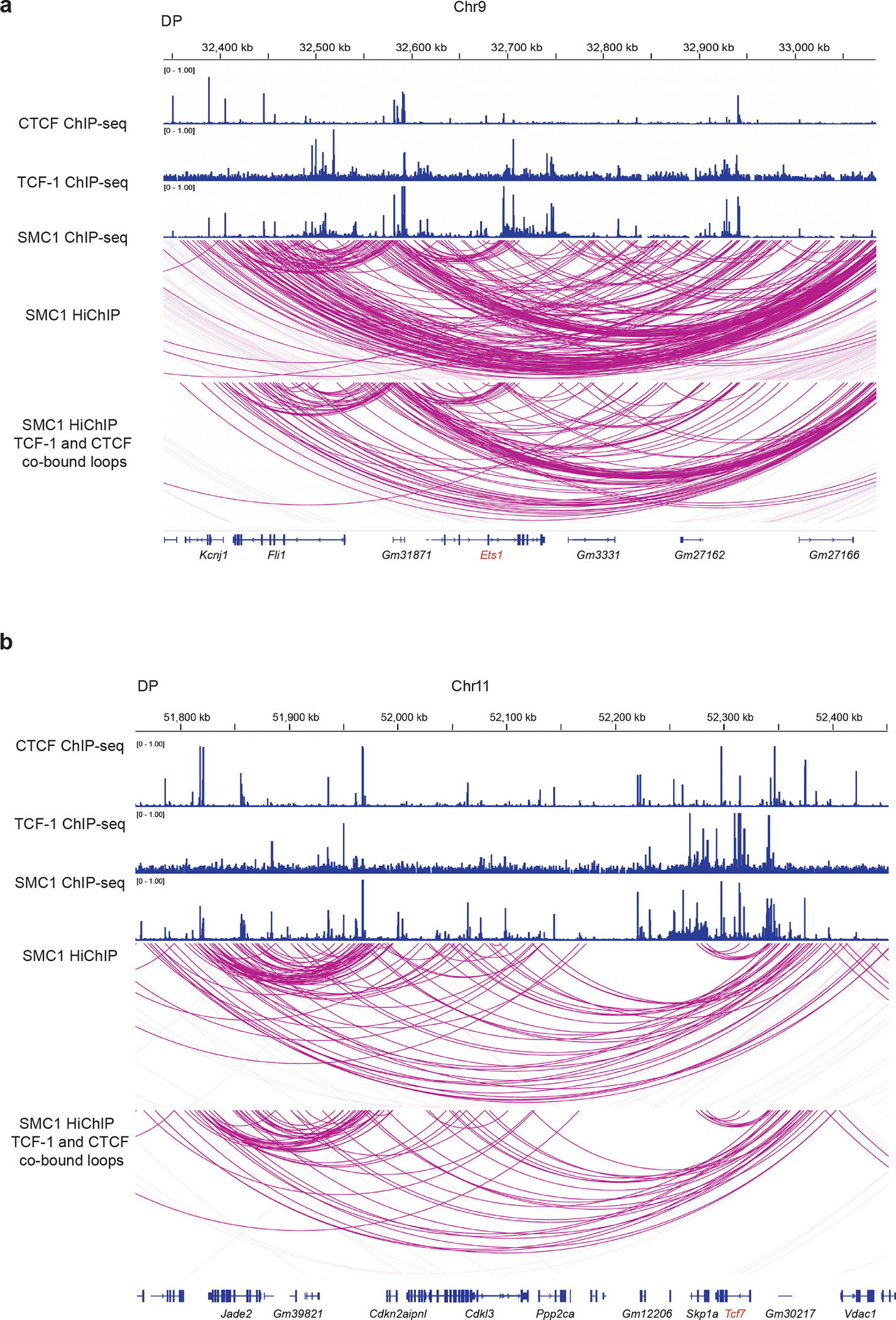
Genome browser view showing cohesin loops and CTCF, TCF-1, SMC1 binding at the Ets1 (a) and Tcf7 (b) loci. The loops associated with both TCF-1 and CTCF are shown in the bottom panel.
Extended Data Fig. 2. Loss of insulation and boundaries during T cell development.
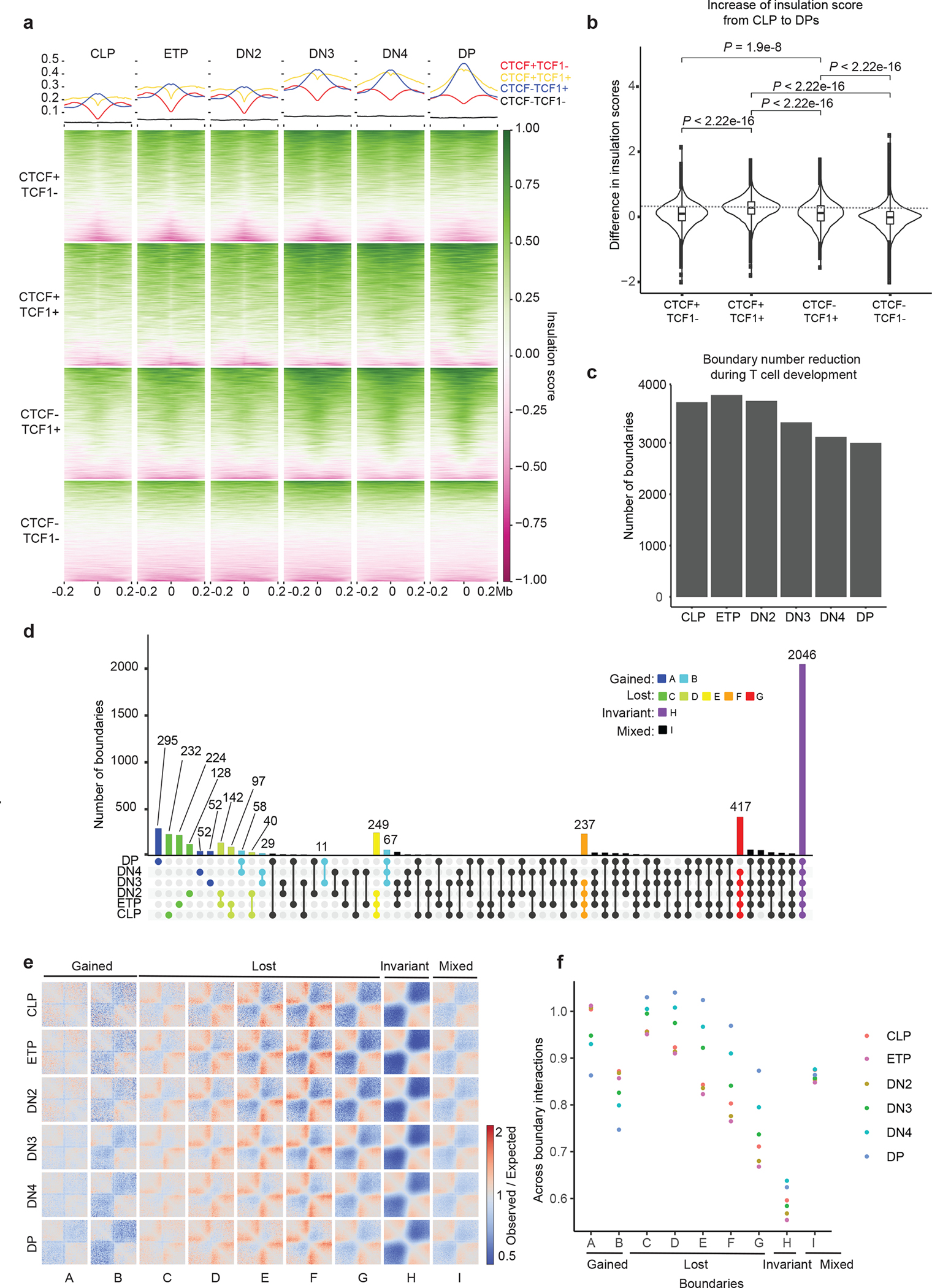
a. Summary plot and heatmap of insulation score in six T cell developmental stages at CTCF-only (red), CTCF+TCF-1 (yellow), TCF-1-only (blue) peaks and random regions as control (black), where lower value indicates higher insulation. Peaks defined in Fig 1c. b. Violin plot and boxplot (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers) showing differences in insulation score from CLP to DP at the four different sets of peaks. The comparison between different groups was done with one-sided Student’s t-test. The insulation score change is significantly (P < 2.2 × 10−16) different between different sets of peaks. c. Bar plot showing the total number of boundaries in six T cell developmental stages. d. Upset plot showing unique and shared boundaries among the six T cell developmental stages. The color shows different groups of boundaries that are lost or gained during T cell development. e. Local pileup in six T cell developmental stages at the nine groups of boundaries classified in (d). f. Quantification of interactions in six T cell developmental stages between upstream and downstream regions of the nine groups of boundaries.
Extended Data Fig. 3. Chromatin features at dynamic boundaries.
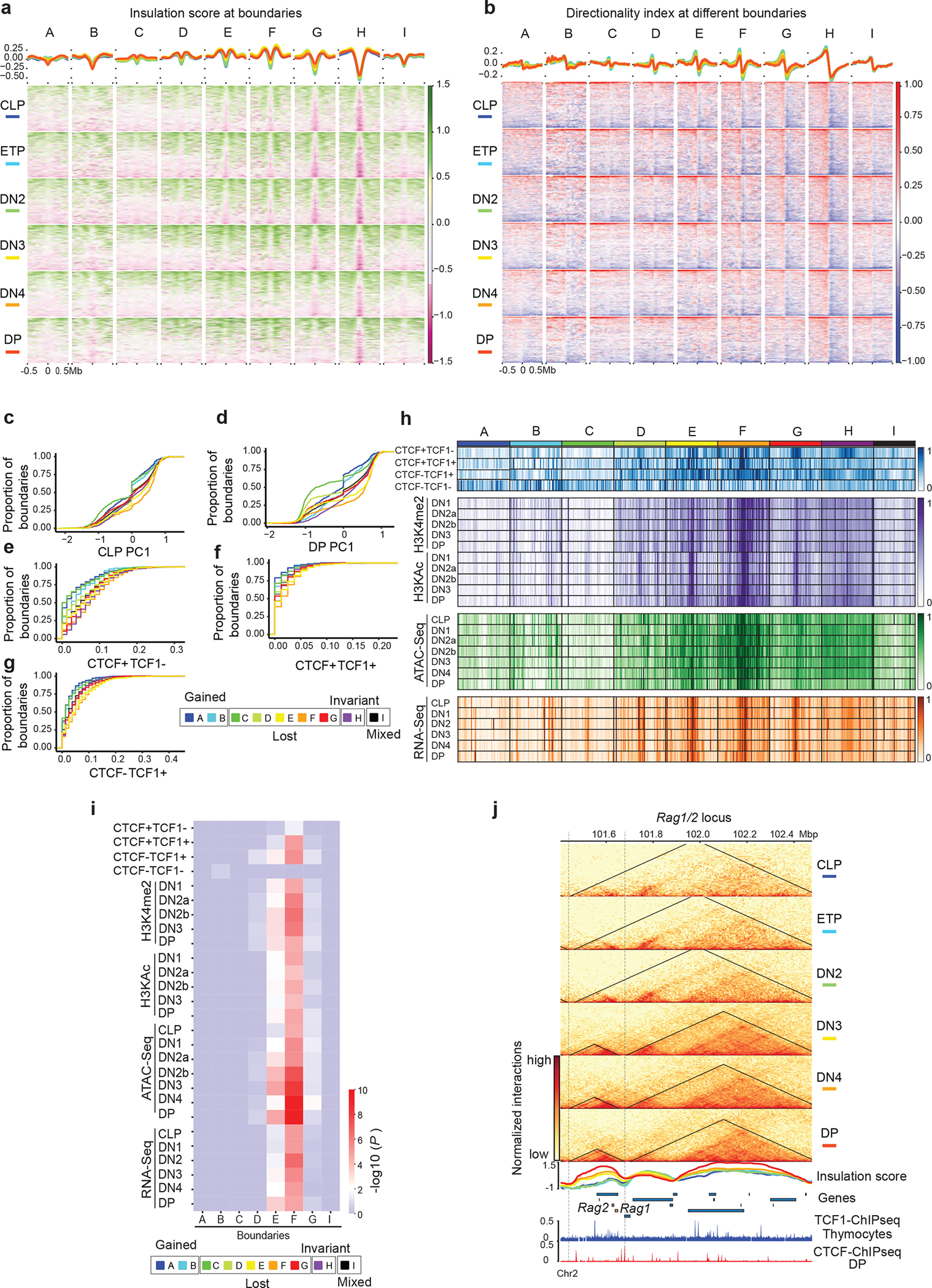
a. Summary plot and heatmap of insulation score in 1 Mbp regions centered at the nine groups of boundaries in six T cell developmental stages. b. Summary plot and heatmap of directionality index at the nine groups of boundaries in six T cell developmental stages. Cumulative distribution of PC1 values in CLP cells (c) and DP cells (d) at nine boundary groups. Cumulative distribution of CTCF only peak density (e), CTCF+TCF-1 peaks density (f), and TCF-1-only peak density (g) as defined in Fig. 1c at nine boundary groups h. Heatmap showing enrichment of CTCF only, CTCF+TCF-1, TCF-1 only peaks and random regions at nine boundary groups, as well as the H3K4me2, H3KAc, ATAC-seq and RNA-seq signals at T cell developmental stages. Signals in each row are normalized between 0 and 1. i. Heatmap showing the statistical test of enrichment of epigenetic features at each group of boundaries compared with conserved boundaries using Kolmogorov–Smirnov test.
j. Genome browser view and contact matrix at the Rag1/2 locus. Contact matrix is normalized according to the sequencing depth and triangles show the identified TADs in this region. Insulation score depicted as a line plot and colored according to their developmental stage as indicated in the figure legend. Genes in this region are shown with Rag1 and Rag2 genes highlighted in orange.
Extended Data Fig. 4. TCF-1+CTCF sites are evolutionarily conserved and insulated in non-T cells.
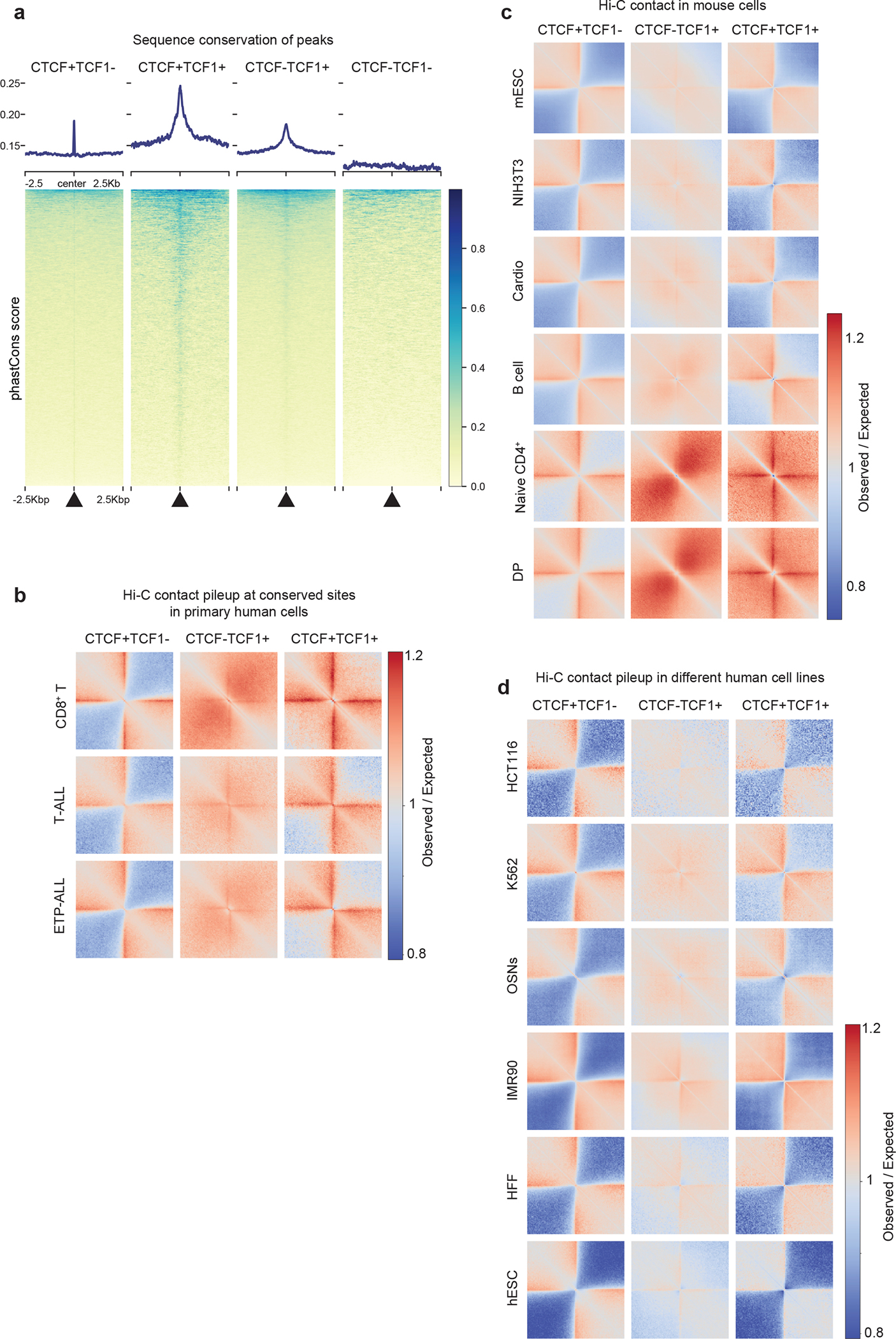
a. Summary plot and heatmap of sequence conservation using phastCons score at CTCF-only, CTCF+TCF-1, TCF-1-only peaks and random regions as control.
b. Local pileup plot of interactions using Hi-C measurements in progenitor and mature T cells in humans at homologues CTCF and TCF-1 peaks. Hi-C measurements were from human publicly available T-ALL, ETP T-ALL, and ultra-deep Hi-C in CD8+ T cells from a healthy donor generated for this study. Human homologous regions for CTCF only, TCF-1 only as well as TCF-1+CTCF peaks in mice were found using liftOver.
c. Local pileup plot of interactions at CTCF only, TCF-1+CTCF, TCF-1 only peaks in different mouse cell types, including mouse embryonic stem cell (mESC), fibroblast (NIH3T3), cardiomyocyte, B cells, naïve CD4+, and DP T cells.
d. Local pileup plot of interactions in different human cell types from 4D Nucleome repository at the liftOver sites of CTCF only, TCF-1 only as well as CTCF+TCF-1 peaks from mice.
Extended Data Fig. 5. Differential gene expression and CTCF binding after TCF-1 overexpression in fibroblast.
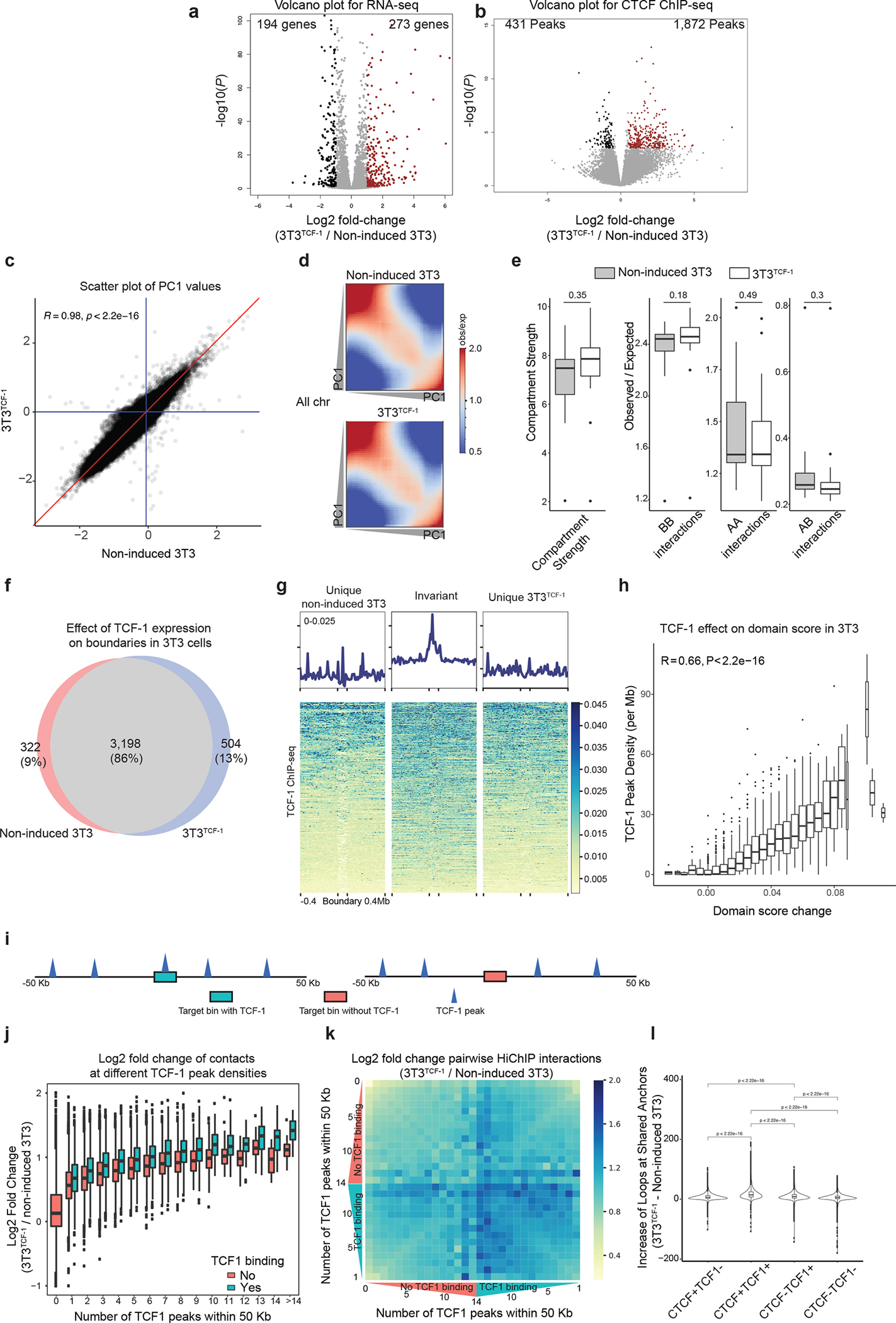
a. Volcano plot of differentially expressed genes in non-induced 3T3 and 3T3TCF-1 fibroblasts. Log2 fold change and P values are calculated with DEseq2. P depicts adjusted pvalue calculated by DESeq2. b. Volcano plot of differential CTCF binding in non-induced 3T3 and 3T3TCF-1 fibroblasts. Log2 fold change and P values are calculated with DEseq2. P depicts adjusted pvalue calculated by DESeq2. c. Scatter plot of PC1 values in non-induced 3T3 and 3T3TCF-1 fibroblasts. The blue lines are where PC1 values are equal to zero, and the red line is where PC1 values are equal in the two conditions. Pearson correlation and the two-sided significance level are shown, the PC1 values are significantly (P < 2.2 × 10−16) correlated. d. Saddle plot showing the compartmentalization in non-induced 3T3 and 3T3TCF-1 fibroblasts. The regions are sorted by PC1 value from B to A compartment, and interactions between different regions are shown in the heatmap. e. Boxplot (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers) shows the compartment strength, BB interactions, AA interactions and AB interactions in wildtype and Tcf7−/− DN3s on all chromosomes (n=20). The statistical test was done using two-sided Student’s t-test. (ns: not significant). f. Venn diagram showing the unique and shared boundaries non-induced 3T3 and 3T3TCF-1 fibroblasts. g. Summary plot and heatmap of TCF-1 ChIP-seq performed in NIH3T3 cells at unique and common boundaries between and after TCF-1 induction. The three different sets of boundaries are normalized to the same height. h. Boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers) showing the correlation of TCF-1 binding density in NIH3T3 cells and increase in domain score after TCF-1 overexpression. TADs (n=2573) are grouped based on the increase of domain score and the boxplots show TCF-1 peak density in each group. R and P values are calculated with two-sided Pearson correlation. The results shows TCF-1 binding is significantly (P < 2.2 × 10−16) associated with domain score increase. i. Illustration of our computational strategy to classify TCF-1-dependent 3D genome interactions originating from TCF-1 binding. Genomic regions were divided into bins and classified based on the number of TCF-1 peaks within 50 kb, as well as TCF-1 binding status in the bin. For every genomic bin, we therefore calculated the number of TCF-1 peaks in the 50 kbp neighborhood of the bin and additionally classified these bins into TCF-1 bound and unbound regions. j. Boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers) show correlation of interactions at bins with different number of TCF-1 binding events within their 50 kb neighborhood. Each bin itself is further divided based on TCF-1 occupancy. k. The greatest increase in 3D interactions after TCF-1 expression in fibroblasts occurs when TCF-1 occupies both anchors and has high density at anchors’ neighboring regions. We calculated the extent of increase in interactions between 10kbp bin pairs which were grouped based on the status of TCF-1 binding and the number of TCF-1 binding events in their 50kbp neighborhood. l. Violin plot and boxplot (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers) showing the number of loops after TCF-1 overexpression in 3T3 at anchors that are bound by CTCF only, CTCF+TCF-1, TCF-1 only and random regions. The statistical test was done with two-sided Student’s t-test. The results show that the gain of loops after TCF-1 overexpression are significantly (P < 2.2 × 10−16) different between different sets of peaks.
Extended Data Fig. 6. 3D genome reorganization after TCF-1 deletion in DN3 and DPs.
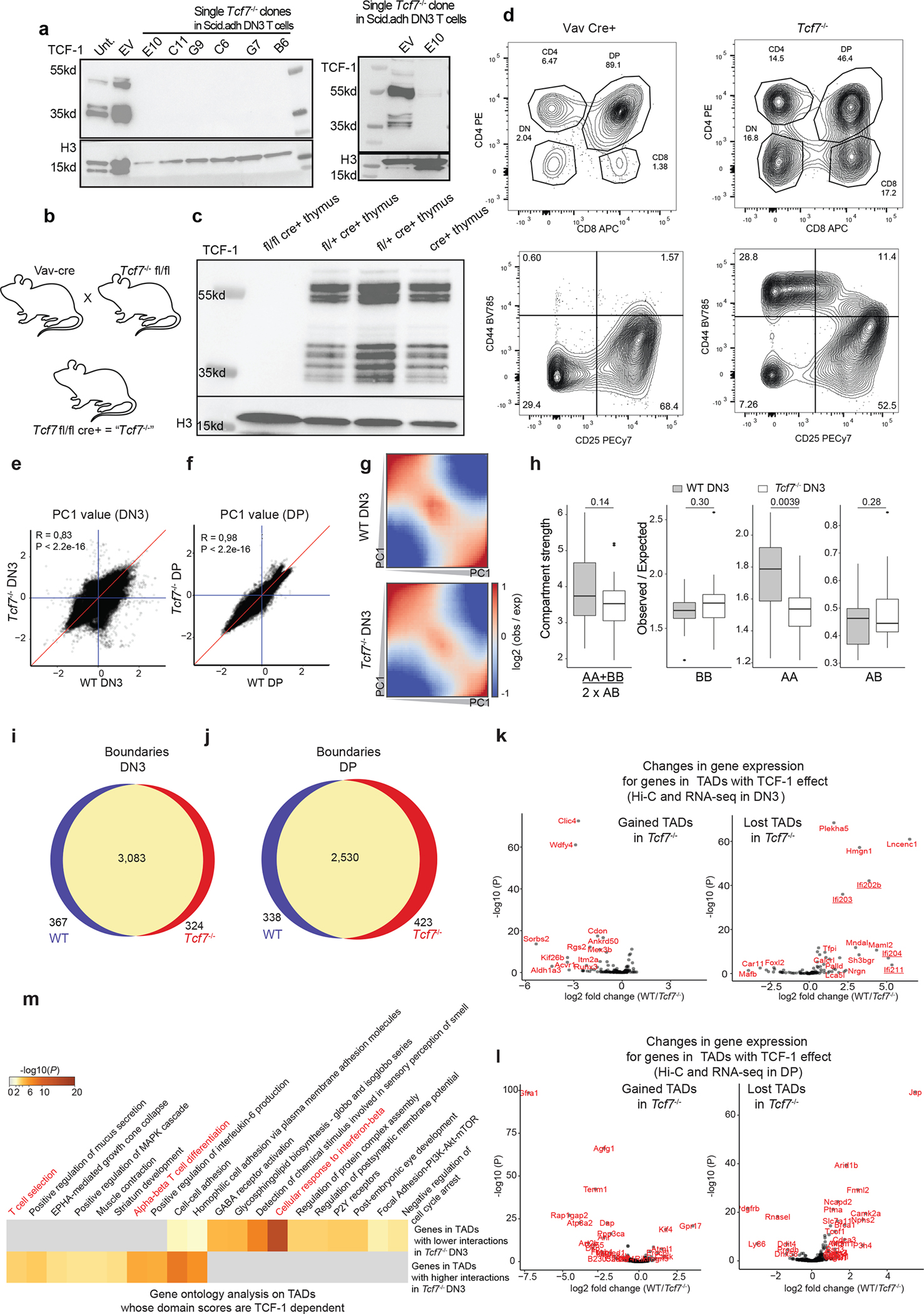
a. Western blot of TCF-1 in DN3s in which TCF-1 was disrupted using CRISPR/Cas9. Single cell clones in bold indicate those utilized for further experiments. Experiments were carried out twice and similar results were obtained.
b. Illustration of breeding strategy for disruption of Tcf7 in mice by crossing Vav-Cre with Tcf7 floxed mice, generating mice in which TCF-1 is conditionally ablated in all hematopoietic cells.
c-d. Western blot of TCF-1 in thymocytes from homozygous fl/fl cre+ (Tcf7−/−), heterozygous fl/+ cre+ (N=2), and cre+ mice. (c) Flow cytometric analysis of thymocytes from Vav cre+ control and Tcf7−/− experimental mice. Top panel shows CD4+ and CD8+ populations and is pre-gated on lymphocytes, live, and single cells. Bottom panel shows DN populations DN1-DN4 (upper left to lower left quadrant clockwise) as measured based on CD44 and CD25 expression and is pre-gated on lymphocytes, live, single cells and lineage negative cells (d).
e-f. Scatter plot showing A/B compartment distribution using correlation of PC1 values between wildtype and Tcf7−/− DN3s (e) and DPs (f). Blue lines are PC1 equals zero, and red line is where PC1 are equal in two conditions. Pearson correlation coefficients and P-values are shown.
g. Saddle plot shows the compartmentalization in wildtype and Tcf7−/− DN3s. The regions are sorted by PC1 value from B to A compartment, and interactions between different regions are shown in the heatmap. One Hi-C experiment was performed in the wildtype condition and two Hi-C experiments were performed in two distinct Tcf7−/− DN3 clones.
h. Data are shown as boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers) showing the compartment strength, BB interactions, AA interactions and AB interactions in wildtype and Tcf7−/− DN3s (n=20 mouse chromosomes). The statistical test was done using two-sided Student’s t-test. (ns: not significant, *: P< 0.05).
i-j. Venn diagram showing the unique and shared boundaries between wildtype and Tcf7−/− DN3s (i) and DP (j).
k-l. Scatter plot showing changes in gene expression located in the top 50 TADs that lost domain score in DN3 (k) and DP (l) T cells. Genes that were significantly (P < 0.05, abs(log2FoldChange) > 0.5) differentially expressed were shown in red. m. Gene-ontology analysis of genes in the top 50 TADs that gained domain score and the top 50 TADs that lost domain score after TCF-1 deletion in DN3s using metaScape. T cell-specific ontology terms are marked in red.
Extended Data Fig. 7. Reorganization of interferon genes by TCF-1.
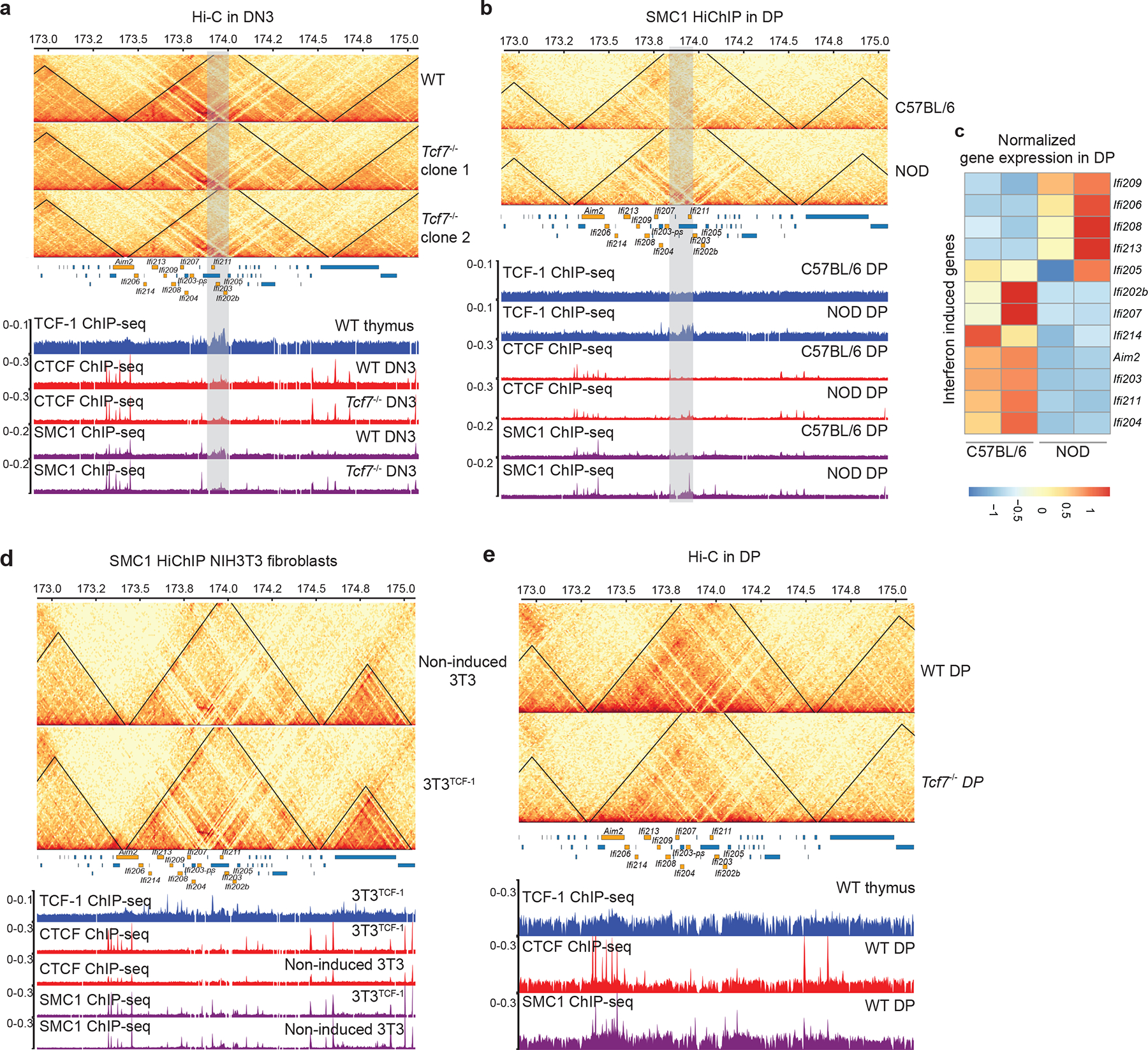
a,b,d,e. Contact matrix and genome browser views of ultra-deep Hi-C measurements at interferon-induced gene cluster (genes marked in orange) using two independent clones for deleting TCF-1 using CRISPR/Cas9 in DN3 (a), comparing SMC1 HiChIP in DPs of C57BL/6 and NOD mice (b). We used Stripenn to quantify stripiness of the stripe originating from the TCF1 cluster, which showed that NOD has a stripe score of 2.14, while C57BL/6 is -2.3. SMC1 HiChIP in TCF-1 induction in fibroblasts (d), and ultra-deep Hi-C in wildtype and Tcf7−/− DPs (e). The browser view focuses on a TAD that lose intra-TAD interactions after TCF-1 deletion in both replicates, which also have a cluster of TCF-1 binding (grey highlighted) in wildtype DN3s. The CTCF and SMC1 binding in both conditions are also shown. c. Heatmap showing the row normalized gene expression level from DPs in NOD and C57BL/6 of the interferon induced genes in the TAD with different intra-TAD interactions in C57BL/6 and NOD mice. Orange bars represent genomic coordinates of interferon genes.
Extended Data Fig. 8. TCF-1 ChIP-seq comparison in DP T and DN3 T cells.
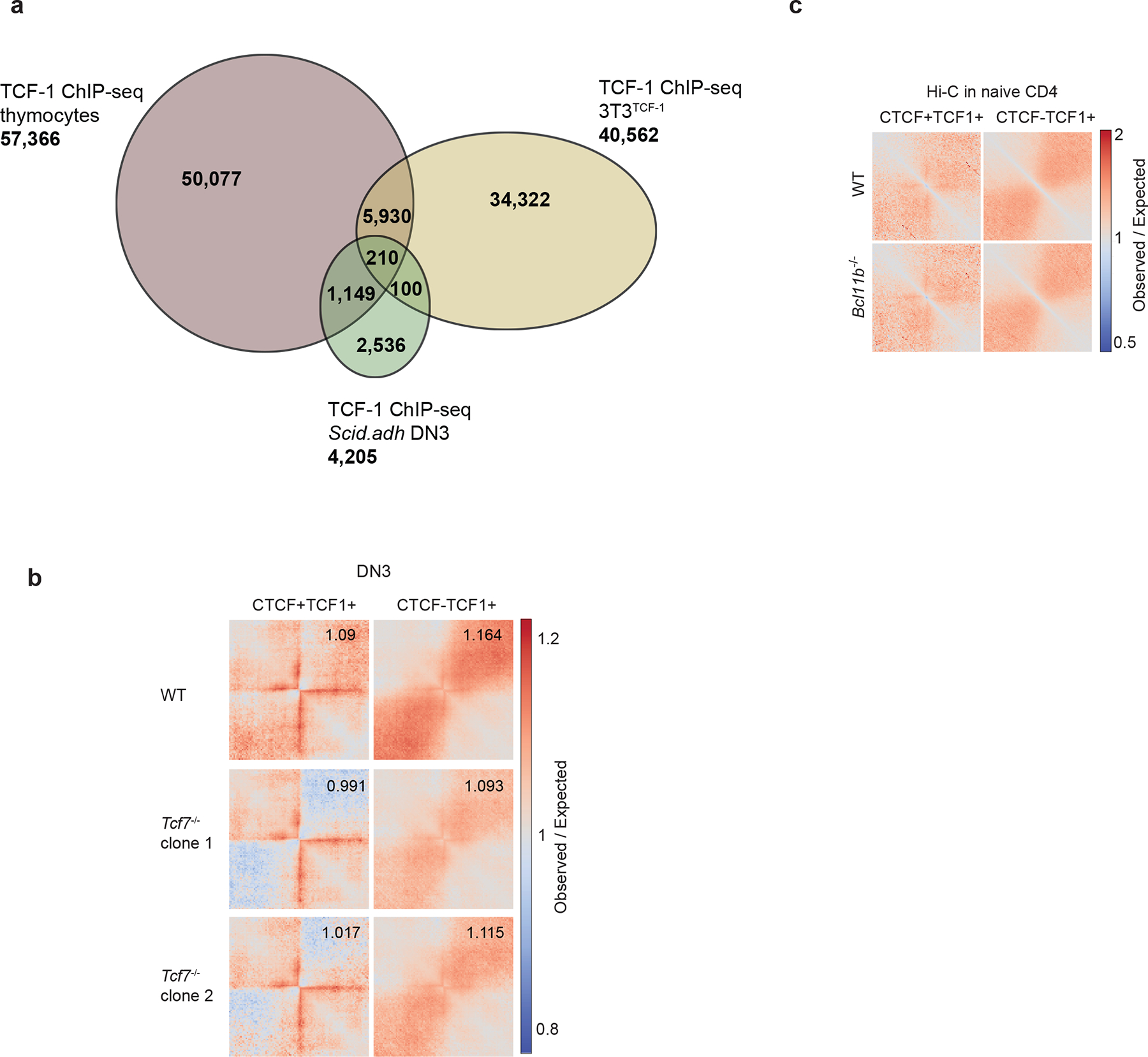
a. Venn diagram of overlapping and unique bound regions by TCF-1 ChIP-seq thymocytes, DN3s and 3T3TCF-1. b. Local pileup plot of long-range interactions using ultra-deep Hi-C in wildtype and two Tcf7−/− DN3 clones at a subset of CTCF+TCF-1 co-bound, as well as TCF-1-only peaks that gain insulation in Tcf7−/− DN3s. The two classes of peaks were defined using TCF-1 and CTCF ChIP-seq in thymocytes and DPs, respectively as in Fig. 1. These peaks were further selected based on the decrease of average interactions between the upstream and downstream of the peaks by at least 0.05 after TCF-1 deletion. The numbers are the average observed/expected interactions in the upper right square, which is the interactions between the upstream and downstream of the peaks.
c. Local pileup interactions in wildtype and Bcl11b deficient naïve CD4 T+ cells at the CTCF and TCF-1 co-bound, as well as TCF-1 only peaks.
Extended Data Fig. 9. Oligopaint 3D FISH corroborates the role of TCF-1 on chromatin interactions at the Cd8 locus.
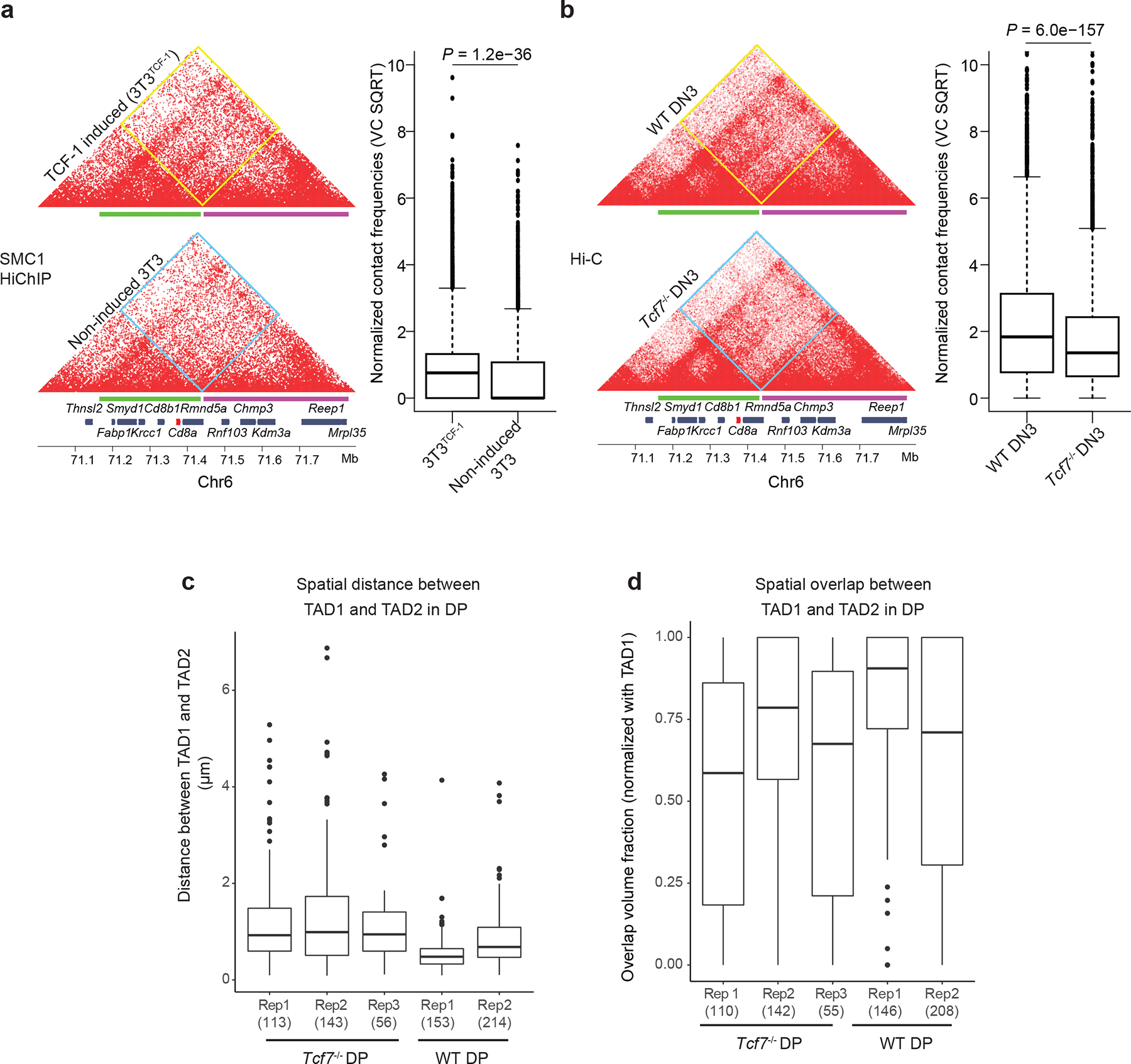
a-b. Contact matrix plot of Hi-C data sets at the Cd8a-Cd8b1 locus in NIH3T3 fibroblasts (a) and DN3s (b) (n=7221 genomic interactions). Inter-domain interactions were quantified by boxplots. Two sided Paired wilcoxon ranksum test was performed. Oligopaint probes for TAD1 (green) and TAD2 (magenta) are depicted. Two-sided Wilcoxon rank-sum test P-values are shown for boxplots.
c-d. Box plot showing spatial distance (c) and spatial overlap (d) between TAD1 and TAD2 compared between wildtype (N=2) and TCF-1-deficient (N=3) of DPs in biological mouse replicates. Spatial distance and overlap were calculated as in (Fig. 5d–e). Number of cells used for imaging analysis per mouse is depicted in parenthesis. a-d Data are shown as boxplots (centre, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5× interquartile range; points, outliers)
Extended Data Fig. 10. Recruitment of cohesin at TCF-1 binding sites.
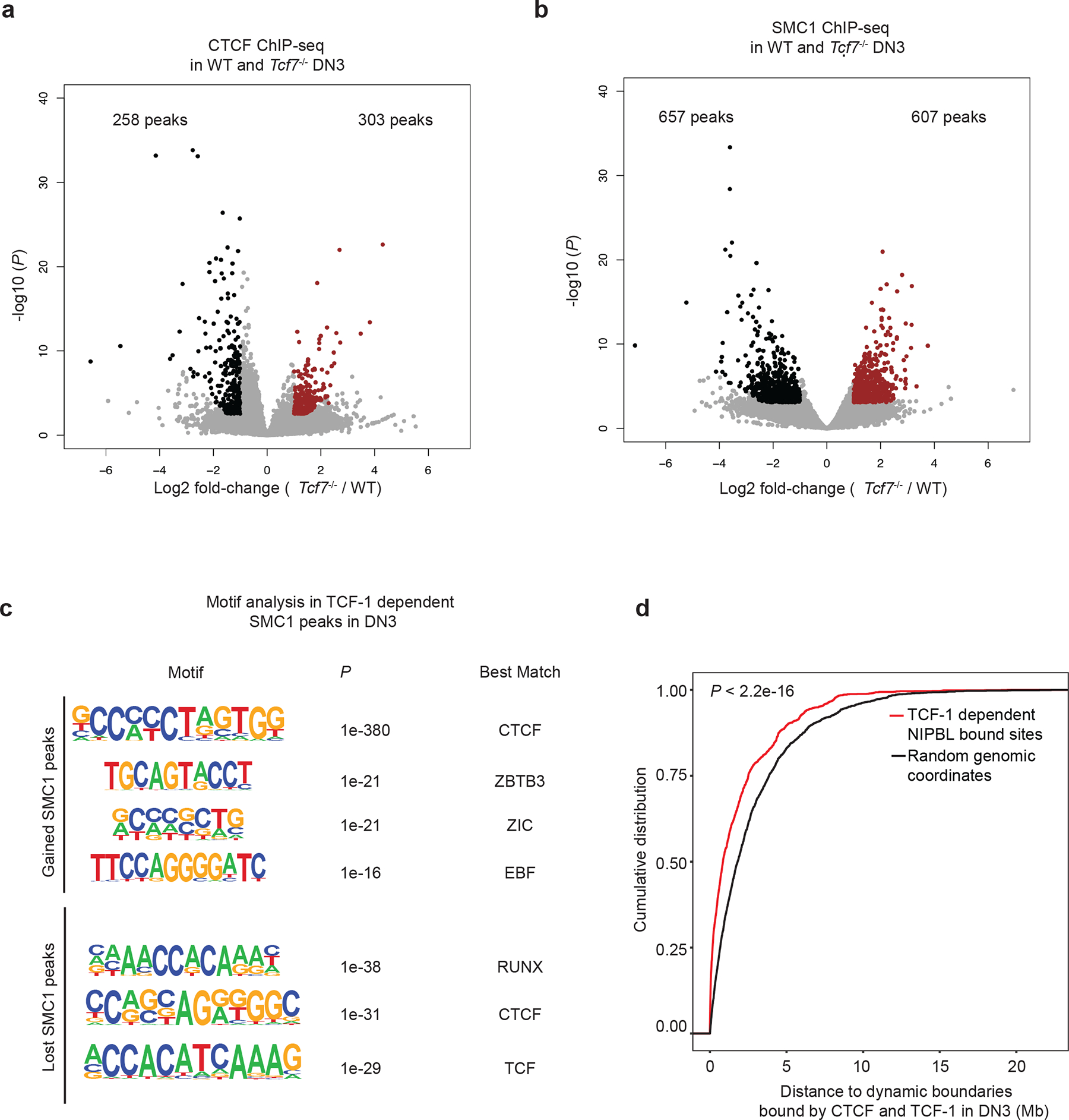
a. Volcano plot shows the differential CTCF binding in wildtype and Tcf7−/− DN3s. Red indicates stronger binding after TCF-1 deletion. Log2 fold change and P values are calculated with DEseq2. P depicts adjusted pvalue calculated by DESeq2.
b. Volcano plot showing the differential SMC1 binding in wildtype and Tcf7−/− DN3s. Log2 fold change and P values are calculated with DEseq2. P depicts adjusted pvalue calculated by DESeq2. Red indicates stronger binding after TCF-1 deletion. c. Enriched motifs from HOMER at the gained or lost SMC1 peaks after TCF-1 deletion in DN3 cells using random background. The most significantly enriched motifs and associated P values are shown. P values are calculated using hypergenometric test.
d. Cumulative distribution of genomic distance between 2,042 TCF-1 dependent SMC1 and NIPBL co-bound enhancers or 2,042 randomly selected genomic regions to 988 dynamic boundaries with TCF-1 and CTCF co-binding. Kolmogorov-Smirnov test P-value is shown for cumulative distribution plot.
Supplementary Material
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CTCF | Millipore | Cat# 07-729; RRID:AB_441965 |
| Anti-SMC1 | Bethyl | Cat# A300-055A; RRID:AB_2192467 |
| Normal Rabbit IgG | CST | Cat# #2729; RRID:AB_1031062 |
| Anti-TCF-1 | CST | Cat# 2206S; RRID:AB_2199300 |
| Biotin anti-mouse CD8a (53-6.7) | BioLegend | Cat# 100703, RRID:AB_312742 |
| Anti-NIPBL | Bethyl | A301-779A; RRID:AB_1211232 |
| Histone H3 (acetyl K27) antibody | Abcam | ab4729; RRID:AB_2118291 |
| Anti-Histone H3 Antibody, Unconjugated | CST | Cat# 9715, RRID:AB_331563 |
| PE anti-mouse CD4 (RM4-4) | BioLegend | Cat# 116005; RRID:AB_313690 |
| APC anti-mouse CD8 (53-6.7) | BioLegend | Cat# 100711; RRID:AB_312750 |
| PeCy7 anti-mouse CD25 (PC61) | BioLegend | Cat# 102015; RRID:AB_312864 |
| Brilliant Violet 785™ anti-mouse/human CD44 (IM7) | BioLegend | Cat# 103041; RRID:AB_11218802 |
| APC Streptavidin | BioLegend | Cat# 405207 |
| Biotin anti-mouse Ly-6G/Ly-6C (Gr-1) (RB6-8C5) | BioLegend | Cat# 108403; RRID:AB_313368 |
| Biotin anti-mouse NK1.1 (PK136) | BioLegend | Cat# 108703; RRID:AB_313390 |
| Biotin anti-mouse CD11b (M1/70) | BioLegend | Cat# 101203, RRID:AB_312786 |
| Biotin anti-mouse Ter119 (TER-119) | BioLegend | Cat# 116203, RRID:AB_313704 |
| Biotin anti-mouse B220 (RA3-6B2) | BioLegend | Cat# 103203, RRID:AB_312988 |
| Anti-rabbit IgG, HRP-linked Antibody | CST | Cat# 7074, RRID:AB_2099233 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E. coli | Thermo | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo | Cat# 11965084 |
| FBS (HyClone) | Cytiva | Cat# SH30910.03 |
| Bovine Serum, heat inactivated | Thermo | Cat# 26170035 |
| RPMI 1640 medium | Invitrogen | Cat# 11875085 |
| L-Glutamine 200 mM | Lonza | Cat# 17-605E |
| MEM Non-Essential Amino Acids Solution (100X) | Invitrogen | Cat# 11140050 |
| Sodium Pyruvate (100 mM) | Invitrogen | Cat# 11360070 |
| Penicillin-Streptomycin | Gibco | Cat# 15140122 |
| Gateway Clonase II LR | Thermo | Cat# 11791020 |
| Gateway Clonase II BP | Thermo | Cat# 11789020 |
| Polybrene | Sigma | Cat# TR-1003-G |
| Lipofectamine 3000 | Thermo | Cat# L3000008 |
| Fugene HD | Promega | Cat# E2311 |
| Chloroquine | Sigma | Cat# C6628-25G |
| G418 | GIBCO | Cat# 11811-023 |
| Doxycycline | Sigma | Cat# D9891 |
| Dimethylformamide | Sigma-Aldrich | Cat# D4551 |
| Puromycin | Takara | Cat# 631305 |
| Ethylene glycol bis(succinic acid N hydroxysuccinimideester) (EGS) | Thermo | Cat# 21565 |
| DMSO | Fisher | Cat# BP231-1 |
| Formaldehyde solution 16% | Thermo | Cat# PI28908 |
| Glycine | Thermo | Cat# 15527013 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| MboI | NEB | Cat# R0147 |
| Biotin-14-dATP | Invitrogen | Biotin-14-dATP |
| dCTP | Invitrogen | Cat# 18253-013 |
| dTTP | Invitrogen | Cat# 18255-018 |
| dGTP | Invitrogen | Cat# 18254-011 |
| Tris HCl, pH 7.5, 1M | Thermo | Cat# 15567027 |
| NaCl, 5M | Thermo | Cat# AM9759 |
| MgCl2 1M | Thermo | Cat# AM9530G |
| IGEPAL CA-630 | Sigma | Cat# I8896-50ML |
| EDTA | Invitrogen | Cat# 15575-038 |
| Protein A Magnetic beads | Pierce | Cat# 88846 |
| Lithium Chloride, 8M | Sigma-Aldrich | Cat# L7026 |
| Sodium bicarbonate | Sigma-Aldrich | Cat#144-55-8 |
| T4 DNA Ligase | NEB | Cat# M0202L |
| BSA, 50mg/mL | Invitrogen | Cat# AM2616 |
| Tween 20 | BIO-RAD | Cat# 170-6531 |
| DNA Polymerase I, Large (Klenow) Fragment | NEB | Cat# M0210S |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| Dynabeads MyOne Streptavidin C-1 | Invitrogen | Cat# 65001 |
| Phusion PCR Master Mix | NEB | Cat# M0531 |
| Nextera XT Index Kit | Illumina | Cat# FC-131-1001 |
| SPRIselect | Beckman Coulter | Cat# B23318 |
| MinElute Reaction Cleanup Kit | QIAGEN | Cat# 28204 |
| QIAQuick PCR Purification Kit | QIAGEN | Cat# 28104 |
| Phosphate-Buffered Saline, 1X | Thermo | Cat# 10010031 |
| RNase | Thermo | Cat# EN0531 |
| Proteinase K | Thermo | Cat# AM2546 |
| Protein G Dynabeads | Thermo | Cat# 10003D |
| 2-Mercaptoethanol | Thermo | Cat# 21985023 |
| Buffer RLT Plus | QIAGEN | Cat# 1053393 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| Trypsin-EDTA (0.05%) | Gibco | Cat# 25300054 |
| RIPA Buffer (10X) | CST | Cat# 9806 |
| NuPAGE LDS Sample Buffer (4X) | Thermo | Cat# NP0007 |
| NuPAGE™ 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well | Thermo | Cat# NP0321BOX |
| iBlot™ 2 Transfer Stacks, nitrocellulose, mini | Thermo | Cat# IB23002 |
| Tris Buffered Saline with Tween® 20 | CST | Cat# 9997S |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo | Cat# 34095 |
| Polysine adhesion glass slides | Electron Microscopy Sciences | Cat# 63412-01 |
| Silicone Isolators | Electron Microscopy Sciences | Cat# 70339-05 |
| Ethanol | Decon Laboratories | 2716 |
| Dimethylformamide | Sigma-Aldrich | Cat# D4551 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787-250ML |
| Polyvinylsulfonic acid (PVSA) | Sigma-Aldrich | Cat# 278424 |
| Fisherbrand™ Premium Cover Glasses | Fisher Scientific | Cat# 12-548-5M |
| Nowrinkle rubber cement | Elmer’s | Cat# 34633 |
| Slowfade Gold Antifade Reagent | Invitrogen by Thermo Fisher Scientific | Cat# S36936 |
| Secondary Oligopaint probe NDB_1279_Atto565: /5ATTO565N/ AGC GAA TCC GAC GCA CCG CTA /3ATTO565N/ | ||
| Secondary Oligopaint probe NDB_1281_Alexa647: /5Alex647N/ AGG ACC CTG TTC GGC TAA CCA /3AlexF647N/ | ||
| Dextran sulfate | Sigma Aldrich | Cat# D8906-50G |
| 20X SSC buffer | Corning | Cat# 46-020-CM |
| Phosphate-Buffered Saline, 10X | Stemcell Technologies | Cat# 37354 |
| Formamide, molecular grade | Promega | Cat# H5052 |
| Formamide, > 99.5% (4 × 4L) | Sigma Aldrich | Cat# 221198-4X4L |
| dNTP mixture, 100mM (25mM of each) | Thomas Scientific | Cat# C788T68 |
| RNase A, DNase and protease-free (10 mg/mL) | Thermo Fisher | Cat# EN0531 |
| Defy & Inspire™ clear nail polish | Target Corporation | Cat# 052-06-3573 |
| Critical commercial assays | ||
| Illumina Tagment DNA Enzyme and Buffer Large Kit | Illumina | Cat# 20034198 |
| SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian | Takara | Cat# 634413 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | NEB | Cat# E7645S |
| Arima HiC+ kit | Arima Genomics | |
| Accel-NGS 2S Plus DNA Library Kit | Swift Biosciences | Cat# 21024 |
| 2S SET A INDEXING KIT | Swift Biosciences | Cat# 26148 |
| KAPA Library Amplification Kit with Primer Mix | Roche | KK2620 |
| Kapa library Quantification kit | Roche | 7960140001 |
| D1000 ScreenTape | Agilent | Cat# 5067-5582 |
| D1000 Reagents | Agilent | Cat# 5067-5583 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067-5584 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067-5585 |
| Genomic DNA ScreenTape | Agilent | Cat# 5067-5365 |
| Genomic DNA Reagents | Agilent | Cat# 5067-5366 |
| RNA ScreenTape | Agilent | Cat# 5067-5576 |
| RNA ScreenTape Ladder | Agilent | Cat# 5067-5578 |
| RNA ScreenTape Sample Buffer | Agilent | Cat# 5067-5577 |
| EasySep™ Release Mouse Biotin Positive Selection Kit | Stemcell | Cat# 17655 |
| EasySep™ Mouse CD4 Positive Selection Kit II | Stemcell | Cat# 18952 |
| Recombinant DNA | ||
| Plasmid: pInducer20 | Addgene | Cat# 44012; RRID:Addgene_44012 |
| Plasmid: pSL21-vex | Addgene | Cat# 158230 |
| Plasmid: MSCV-Cas9_Puro | Addgene | Cat# 65655 |
| Plasmid: pCL-Eco | (Naviaux et al., 1996) | |
| Deposited data | ||
| HiC, HiChIP, ChIPseq, RNAseq | This study | GSE178348 |
| Publicly accessible data | ||
| TCF-1 ChIPseq in thymus | 24 | GSE46662 |
| CTCF ChIPseq in DP T cells | 27 | GSE141853 |
| SMC1 HiChIP in DP T cells | 27 | GSE141853 |
| 3e Hi-C during T cell development | 25 | GSE79422 |
| Histone modifications during T cell development | 29 | GSE31235 |
| ATAC-seq during T cell development | 49 | GSE100738 |
| RNA-seq during T cell development | 25 | GSE79422 |
| Mouse B cell hic | 42 | 4DNFISA93XFU |
| Mouse cardiomyocyte hic | (Rosa-Garrido et al., 2017) | GSE96693 |
| Mouse mESC hic | (Bonev et al., 2017) | 4DNFIC21MG3U |
| Human hESC hic | (Oksuz et al., 2020) | 4DNFI7JNCNFB |
| Human IMR90 hic | 45 | 4DNFIJTOIGOI |
| Human olfactory sensory neurons hic | (Zazhytska et al., 2021) | 4DNFI7V61PXA |
| Human HCT116 hic | 42 | 4DNFIV3PIEQJ |
| Human K562 hic | (Ray et al., 2019) | 4DNFI244AS29 |
| Human HFF hic | (Oksuz et al., 2020) | 4DNFIWMLVHWX |
| Experimental models: Cell lines | ||
| NIH 3T3 | ATCC | Cat# CRL-1658 RRID:CVCL_0594 |
| HEK293T | ATCC | Cat# CRL-3216 RRID:CVCL_0063 |
| Scid.adh | Warren Pear lab at UPENN | |
| Experimental models: Organisms and Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: B6(Cg)-Tcf7tm1Hhx/J | Jackson Laboratory | RRID:IMSR_JAX:030909 |
| Mouse: B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J | Jackson Laboratory | RRID:IMSR_JAX:008610 |
| Software and algorithms | ||
| FastQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; RRID:SCR_014583 | |
| Trim Galore | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/; RRID:SCR_011847 | |
| Bowtie2 | 50 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | 51 | https://github.com/alexdobin/STAR |
| Picard | https://broadinstitute.github.io/picard/ | |
| HTSeq | 52 | https://htseq.readthedocs.io/en/master/; RRID:SCR_005514 |
| DESeq2 | 53 | https://github.com/Bioconductor-mirror/DESeq2; RRID:SCR_015687 |
| R | https://cran.r-project.org; RRID:SCR_001905 | |
| MACS2 | 54 | https://github.com/taoliu/MACS |
| Homer | 55 | http://homer.ucsd.edu/homer; RRID:SCR_010881 |
| bedtools | https://bedtools.readthedocs.io/en/latest/#; RRID:SCR_006646 | |
| wigToBigWig | http://hgdownload.cse.ucsc.edu/admin/exe/ | |
| HiC-Pro | 56 | https://github.com/nservant/HiC-Pro; RRID:SCR_017643 |
| ggplot2 | https://ggplot2.tidvverse.org/ | |
| Metascape | 57 | https://metascape.org/gp/index.html#/main/step1; RRID:SCR_016620 |
| Juicer | 58 | https://github.com/aidenlab/juicer |
| Cooler | https://github.com/open2c/cooler | |
| cooltools | https://github.com/open2c/cooltools | |
| HiCExplorer | 59 | https://hicexplorer.readthedocs.io/en/latest/ |
| UpSetR | 60 | https://cran.r-project.org/web/packages/UpSetR/index.html |
| deeptools | 61 | https://deeptools.readthedocs.io/en/develop/ |
| dekker-cworld | https://github.com/dekkerlab/cworld-dekker | |
| hichipper | 62 | https://github.com/aryeelab/hichipper |
| Coolpup.py | 63 | https://github.com/open2c/coolpuppy |
| TANGO | ||
| Fiji | ||
| IMARIS | ||
| Sushi | ||
| OligoMiner | ||
| Flowjo v.10.6.1 | ||
Acknowledgments
We thank helpful discussions with R. B. Faryabi, K. Zaret, A. Karnay, E.J.Wherry, and R. Jain. We would like to thank J. Shi and his lab for help and discussion about the CRISPR/Cas9 plasmids. This work was supported by NIH grants UC4 DK112217, U01DK11221702A1, R01HL145754, U01DK127768, U01DA052715, the Burroughs Wellcome Fund, the Chan Zuckerberg Initiative, W. W. Smith Charitable Trust, the Penn Epigenetics Institute pilot and the Sloan Foundation awards to G.V.
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Code availability
The codes for Stripenn and data analysis are available from the author’s Github page (https://github.com/VahediLab/TCF13D_code).
Data availability
Genomics data generated in this study is publicly available on GEO: GSE178348. Oligopaint FISH data generated in this study are provided as Source Data.
References
- 1.Beagan JA & Phillips-Cremins JE On the existence and functionality of topologically associating domains. Nat Genet 52, 8–16, doi: 10.1038/s41588-019-0561-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon JR et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336, doi: 10.1038/nature14222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dowen JM et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387, doi: 10.1016/j.cell.2014.09.030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hnisz D, Shrinivas K, Young RA, Chakraborty AK & Sharp PA A Phase Separation Model for Transcriptional Control. Cell 169, 13–23, doi: 10.1016/j.cell.2017.02.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385, doi: 10.1038/nature11049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380, doi: 10.1038/nature11082 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen AS CTCF as a boundary factor for cohesin-mediated loop extrusion: evidence for a multi-step mechanism. Nucleus 11, 132–148, doi: 10.1080/19491034.2020.1782024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stadhouders R, Filion GJ & Graf T Transcription factors and 3D genome conformation in cell-fate decisions. Nature 569, 345–354, doi: 10.1038/s41586-019-1182-7 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Stadhouders R et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 50, 238–249, doi: 10.1038/s41588-017-0030-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weintraub AS et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588 e1528, doi: 10.1016/j.cell.2017.11.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Giammartino DC et al. KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat Cell Biol 21, 1179–1190, doi: 10.1038/s41556-019-0390-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johanson TM et al. Transcription-factor-mediated supervision of global genome architecture maintains B cell identity. Nat Immunol 19, 1257–1264, doi: 10.1038/s41590-018-0234-8 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Singh H, Khan AA & Dinner AR Gene regulatory networks in the immune system. Trends Immunol 35, 211–218, doi: 10.1016/j.it.2014.03.006 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Allman D et al. Thymopoiesis independent of common lymphoid progenitors. Nat Immunol 4, 168–174, doi: 10.1038/ni878 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Li L, Leid M & Rothenberg EV An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science 329, 89–93, doi: 10.1126/science.1188989 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kueh HY et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol 17, 956–965, doi: 10.1038/ni.3514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson JL et al. Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cells. Immunity 48, 243–257 e210, doi: 10.1016/j.immuni.2018.01.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emmanuel AO et al. TCF-1 and HEB cooperate to establish the epigenetic and transcription profiles of CD4(+)CD8(+) thymocytes. Nat Immunol 19, 1366–1378, doi: 10.1038/s41590-018-0254-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giese K, Cox J & Grosschedl R The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell 69, 185–195, doi: 10.1016/0092-8674(92)90129-z (1992). [DOI] [PubMed] [Google Scholar]
- 20.Love JJ et al. Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature 376, 791–795, doi: 10.1038/376791a0 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Shan Q et al. Tcf1 and Lef1 provide constant supervision to mature CD8(+) T cell identity and function by organizing genomic architecture. Nat Commun 12, 5863, doi: 10.1038/s41467-021-26159-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Y et al. EBF1 nuclear repositioning instructs chromatin refolding to promote therapy resistance in T leukemic cells. Mol Cell 82, 1003–1020 e1015, doi: 10.1016/j.molcel.2022.01.015 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antoszewski M et al. Tcf1 is essential for initiation of oncogenic Notch1-driven chromatin topology in T-ALL. Blood 139, 2483–2498, doi: 10.1182/blood.2021012077 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dose M et al. beta-Catenin induces T-cell transformation by promoting genomic instability. Proc Natl Acad Sci U S A 111, 391–396, doi: 10.1073/pnas.1315752111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu G et al. Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity 48, 227–242 e228, doi: 10.1016/j.immuni.2018.01.013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crane E et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244, doi: 10.1038/nature14450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fasolino M et al. Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity 52, 257–274 e211, doi: 10.1016/j.immuni.2020.01.003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kernfeld EM et al. A Single-Cell Transcriptomic Atlas of Thymus Organogenesis Resolves Cell Types and Developmental Maturation. Immunity 48, 1258–1270 e1256, doi: 10.1016/j.immuni.2018.04.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang JA, Mortazavi A, Williams BA, Wold BJ & Rothenberg EV Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell 149, 467–482, doi: 10.1016/j.cell.2012.01.056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kloetgen A et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet 52, 388–400, doi: 10.1038/s41588-020-0602-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dekker J et al. The 4D nucleome project. Nature 549, 219–226, doi: 10.1038/nature23884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krijger PH et al. Cell-of-Origin-Specific 3D Genome Structure Acquired during Somatic Cell Reprogramming. Cell Stem Cell 18, 597–610, doi: 10.1016/j.stem.2016.01.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dionne CJ et al. Subversion of T lineage commitment by PU.1 in a clonal cell line system. Dev Biol 280, 448–466, doi: 10.1016/j.ydbio.2005.01.027 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Forcato M et al. Comparison of computational methods for Hi-C data analysis. Nat Methods 14, 679–685, doi: 10.1038/nmeth.4325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eres IE & Gilad Y A TAD Skeptic: Is 3D Genome Topology Conserved? Trends Genet 37, 216–223, doi: 10.1016/j.tig.2020.10.009 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beliveau BJ et al. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat Commun 6, 7147, doi: 10.1038/ncomms8147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beliveau BJ et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc Natl Acad Sci U S A 109, 21301–21306, doi: 10.1073/pnas.1213818110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ollion J, Cochennec J, Loll F, Escude C & Boudier T TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29, 1840–1841, doi: 10.1093/bioinformatics/btt276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciosk R et al. Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell 5, 243–254, doi: 10.1016/s1097-2765(00)80420-7 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Murayama Y & Uhlmann F Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 505, 367–371, doi: 10.1038/nature12867 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petela NJ et al. Scc2 Is a Potent Activator of Cohesin’s ATPase that Promotes Loading by Binding Scc1 without Pds5. Mol Cell 70, 1134–1148 e1137, doi: 10.1016/j.molcel.2018.05.022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vian L et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178 e1120, doi: 10.1016/j.cell.2018.03.072 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fudenberg G et al. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep 15, 2038–2049, doi: 10.1016/j.celrep.2016.04.085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kueng S et al. Wapl controls the dynamic association of cohesin with chromatin. Cell 127, 955–967, doi: 10.1016/j.cell.2006.09.040 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680, doi: 10.1016/j.cell.2014.11.021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Y, Denholtz M, Lu H & Murre C Calcium signaling instructs NIPBL recruitment at active enhancers and promoters via distinct mechanisms to reconstruct genome compartmentalization. Genes Dev 35, 65–81, doi: 10.1101/gad.343475.120 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beagan JA et al. Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat Neurosci 23, 707–717, doi: 10.1038/s41593-020-0634-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrovic J et al. Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol Cell 73, 1174–1190 e1112, doi: 10.1016/j.molcel.2019.01.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida H et al. The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912 e820, doi: 10.1016/j.cell.2018.12.036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, doi: 10.1093/bioinformatics/btu638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137, doi: 10.1186/gb-2008-9-9-r137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589, doi: 10.1016/j.molcel.2010.05.004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Servant N et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259, doi: 10.1186/s13059-015-0831-x (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523, doi: 10.1038/s41467-019-09234-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durand NC et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98, doi: 10.1016/j.cels.2016.07.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolff J et al. Galaxy HiCExplorer: a web server for reproducible Hi-C data analysis, quality control and visualization. Nucleic Acids Res 46, W11–W16, doi: 10.1093/nar/gky504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Conway JR, Lex A & Gehlenborg N UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940, doi: 10.1093/bioinformatics/btx364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramirez F, Dundar F, Diehl S, Gruning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191, doi: 10.1093/nar/gku365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lareau CA & Aryee MJ hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat Methods 15, 155–156, doi: 10.1038/nmeth.4583 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flyamer IM, Illingworth RS & Bickmore WA Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinformatics 36, 2980–2985, doi: 10.1093/bioinformatics/btaa073 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carleton M et al. Signals transduced by CD3epsilon, but not by surface pre-TCR complexes, are able to induce maturation of an early thymic lymphoma in vitro. J Immunol 163, 2576–2585 (1999). [PubMed] [Google Scholar]
- 65.de Boer J et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33, 314–325, doi: 10.1002/immu.200310005 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Ogilvy S et al. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94, 1855–1863 (1999). [PubMed] [Google Scholar]
- 67.Shimshek DR et al. Codon-improved Cre recombinase (iCre) expression in the mouse. Genesis 32, 19–26, doi: 10.1002/gene.10023 (2002). [DOI] [PubMed] [Google Scholar]
- 68.Yang Q et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat Immunol 16, 1044–1050, doi: 10.1038/ni.3248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Z et al. In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262–1280 e1222, doi: 10.1016/j.cell.2021.02.019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218, doi: 10.1038/nmeth.2688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13, 919–922, doi: 10.1038/nmeth.3999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beliveau BJ et al. OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc Natl Acad Sci U S A 115, E2183–E2192, doi: 10.1073/pnas.1714530115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moffitt JR & Zhuang X RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). Methods Enzymol 572, 1–49, doi: 10.1016/bs.mie.2016.03.020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosin LF, Nguyen SC & Joyce EF Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLoS Genet 14, e1007393, doi: 10.1371/journal.pgen.1007393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoon S, Chandra A and Vahedi G Stripenn detects architectural stripes from chromatin conformation data using computer vision. Nature Communications 3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abdennur N & Mirny LA Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316, doi: 10.1093/bioinformatics/btz540 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rao SSP et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680, doi: 10.1016/j.cell.2014.11.021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Imakaev M et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9, 999–+, doi: 10.1038/Nmeth.2148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ollion J, Cochennec J, Loll F, Escude C & Boudier T TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29, 1840–1841, doi: 10.1093/bioinformatics/btt276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682, doi: 10.1038/Nmeth.2019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomics data generated in this study is publicly available on GEO: GSE178348. Oligopaint FISH data generated in this study are provided as Source Data.