

Abstract
Structural Variations, including recurrent Copy Number Variants (CNVs) at specific genomic loci, have been found to be associated with increased risk of several diseases and syndromes. CNV carrier status can be determined in large collections of samples using SNP-arrays and, more recently, sequencing data. Although there is some consensus among researchers about the essential steps required in such analysis (i.e., CNV Calling, Filtering of putative carriers, and Visual Validation using intensity data plots of the genomic region), standard methodologies and processes to control the quality and consistency of the results are lacking. Here, we present a comprehensive and user-friendly protocol that we have refined from our extensive research experience in the field. We cover every aspect of the analysis, from input data curation to final results. For each step we highlight which parameters affect the analysis the most and how different settings may lead to different results. We provide a pipeline to run the complete analysis with effective (but customizable) pre-sets. We present software that we developed to better handle and filter putative CNV carriers and perform visual inspection to validate selected candidates. Finally, we describe methods to evaluate the critical sections and actions to counterbalance potential problems. The current implementation is focused on Illumina SNP-array data. All the presented software is freely available and provided in a ready-to-use docker container.
Basic protocol 1:
From intensity data files to CNV calls. Perform SNP filtering, PennCNV calling pipeline, apply a light post-processing, tabix index intensity files.
Basic protocol 2:
From CNVs calls to validated CNV carriers. Filter putative CNV carriers in the selected loci, perform visual validation, and export results.
Support protocol 1:
Quality control. Run a comprehensive series of tests and guidance on which parameters to change if any problem should arise.
Support protocol 2:
Install the necessary software.
Keywords: CNVs, Structural Variation, SNPs, bioinformatics pipeline
INTRODUCTION
Structural Variants are genomic rearrangements involving a sequence of base pairs. They can be categorised into multiple subgroups including indels, tandem repeats, Copy Number Variants (deletions and duplications; CNVs), and large chromosomal abnormalities (Sudmant et al., 2015). Recurrent CNVs constitute a structural variant class of particular interest, as many of them have been shown to be associated with disease traits(Malhotra & Sebat, 2012; Weischenfeldt, Symmons, Spitz, & Korbel, 2013). Recurrent CNVs usually consist of duplications and deletions occurring in specific loci of the genome interspersed by so-called low-copy repeats (LCR). The LCR are typically relatively large (>10kb) sequence elements with high (>95%) sequence homology and during cell replication their proximity can facilitate a non-allelic homologous recombination (NaHR) between chromosomes/chromatids, resulting in the deletion and duplication of the genetic material in between the two LCR copies.(Sharp et al., 2005; Turner et al., 2008) Although such events occur sporadically, the breakpoints of the resulting CNVs will always be dictated by the genomic position of the LCRs, hence the term “recurrent”. Most recurrent disease-associated CNVs are rare (<0.1%) and rather large (>300kb) (Crawford et al., 2019; Stankiewicz & Lupski, 2002), they often occur de-novo in the carrier and tend to not segregate over many generations (Stefansson et al., 2008), although this varies widely across loci and CNV types depending on the severity and frequency of the pathogenic symptoms associated with them. Some examples include 1q21.1, 15q11.2, 15q13.3 and 22q11.2(Driscoll et al., 1992; Stefansson et al., 2008).
Large chromosomal rearrangements were traditionally detected via microscopic karyotype inspection, while most recurrent CNVs are too small for such detection (hence sometimes referred to as “submicroscopic”) and must be detected through inferential molecular genetics methods. Currently, most recurrent CNVs are routinely identified (“called”) from SNP-arrays and/or sequencing technology. Of these, SNP-arrays are more commonly applied in very large studies, as SNP-array genotyping is less expensive than whole genome sequencing, whereas the latter typically return more accurate and precise results but is more complex to analyse. The most commonly applied method to “call” CNVs from SNP-array data is PennCNV (Wang et al., 2007), a hidden Markov model (HMM) based approach which evaluates deviations from expected patterns in two key derived measures of the raw A and B allele intensities from which SNP-array genotypes are determined. These derived measures are the log-R-ratio (LRR), which measures the overall relative intensity of each probe compared to all other probes, and the B-allele-frequency (BAF), which measures the relative intensity of the B-allele to the total intensity for each probe. PennCNV is considered the current standard for CNV calling, however, it has some shortcomings. PennCNV internal HMM only considers successive SNPs at any step, making it extremely sensitive to local noise. Due to this intrinsic limitation of the model, false positives (calls only due to noise), over-segmentation (a true CNV erroneously split into smaller calls), and imprecise boundaries in general are common problems affecting PennCNV calls. These issues especially affect results obtained from noisy datasets (the main sources of noise are discussed in the main text). Of note, complete false negatives are not generally considered a concern.
Prompted by our own analyses of recurrent CNVs in several large SNP-array genotype datasets, and a perceived lack of standardised procedures to deal with the most commonly encountered issues in such analyses, we have developed a set of protocols that in our opinion will facilitate accurate and efficient calling of recurrent CNVs (or CNVs at other fixed loci) in any large SNP-array dataset. In protocol 1, we describe how to filter uninformative SNPs from intensity data in order to improve accuracy of CNV calls (for simplicity, here and in following protocols we consider Illumina “final report”, containing the key LRR and BAF measures, as raw data - see following sections, “Necessary resources” and “Basic protocol 1”, for a formal data description). Moreover, we show how to use the provided PennCNV pipeline (including some light processing), as well as the tabix-indexing of the raw data for the following steps. In protocol 2, we illustrate how to effectively filter putative carriers in selected loci as well as to visually validate the CNVs calls. This section is implemented using the software packages we provide. Figure 1 illustrates the overall protocol schematics. In support protocol 1, we illustrate how to run and interpret an extensive battery of tests in order to assess the quality of the results. Finally, we describe which parameters in which steps should be modified in order to improve the overall quality of the results, if any. In support protocol 2, we detail the install process of all necessary software.
Figure 1:
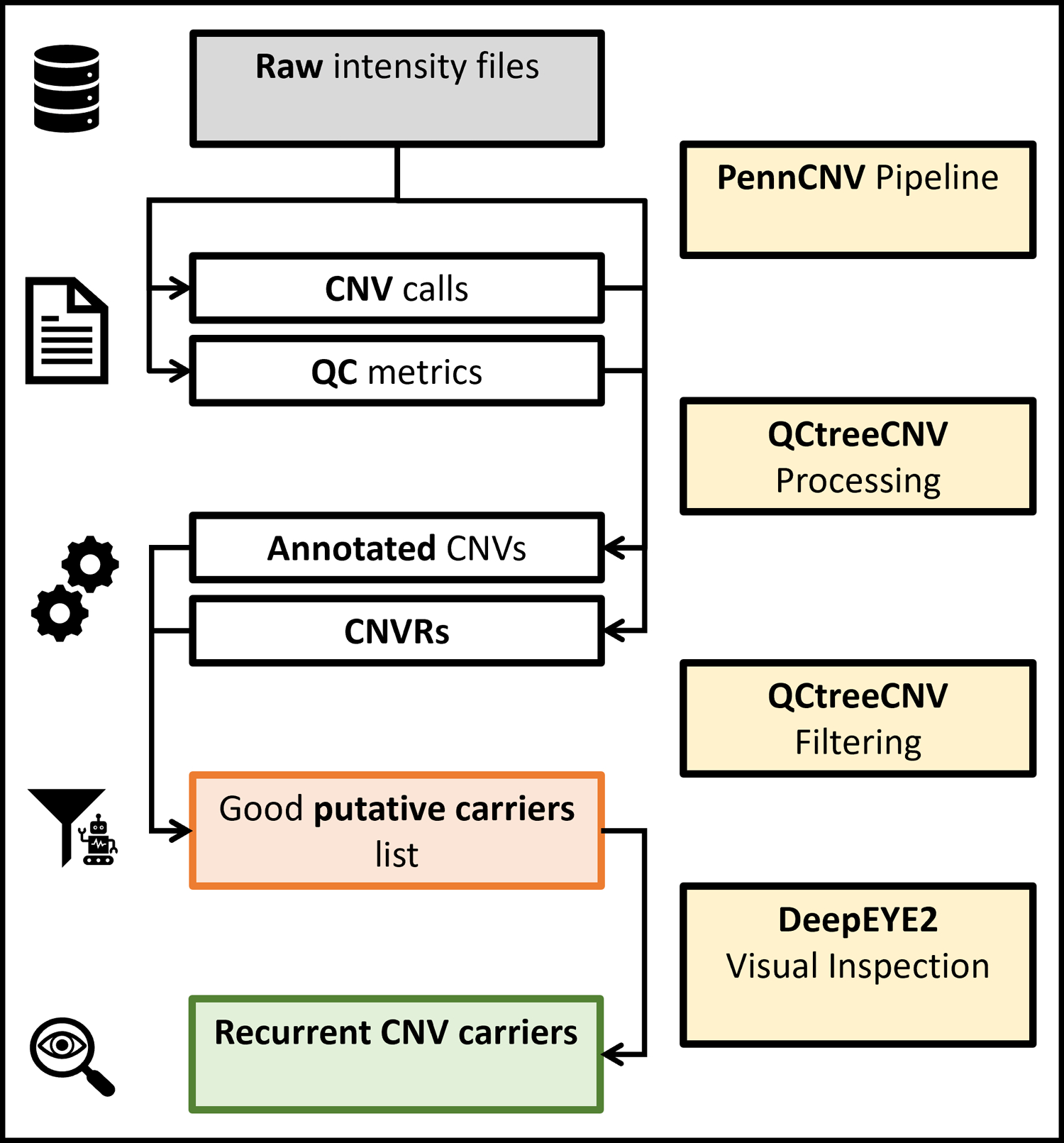
Schematic representation of the whole protocol. The four major processing blocks are shown in yellow, the main inputs (intensity files) in grey, intermediary outputs in red, and the final results in green.
As discussed in the commentary, the protocol is primarily designed for processing large datasets (e.g., > 50,000 samples). However, we also tried to code and describe most steps so that the whole protocol, or at least sections of it, can be as useful also in smaller studies and in ones not strictly focused on recurrent CNVs.
Necessary resources
Hardware:
The protocol is designed for large collections of genotyping data. For this reason, the user will need at least a modern workstation, but a high-performance computing cluster is preferred. While the RAM and disk space requirements are not high compared to, e.g., programs that handle next generation sequencing data, several steps of the pipeline need to read raw data, thus a fast storage solution is advised.
Software:
We provide a docker/singularity image containing all necessary software to run the protocol. Refer to support protocol 2 for instructions on how to use the image or how to install our software separately. The initial PennCNV calling pipeline is designed to take advantage of the job scheduler SLURM and to run all commands via the singularity container. Only Linux environments are supported.
Files:
The protocol is focused on Illumina genotyping data; raw data for each sample of the cohort is basically the only necessary prerequisite. There must be one file per sample. Additionally, a key file linking each sample to its raw data file is needed. The raw data format is Illumina “intensity file”/”final report” format, that is a text file containing at least the following columns: “Name”, “Log R Ratio”, “B Allele Freq”. Expected format is detailed in the following section. Please note that while intensity files are considered raw data, they are not the most primitive form in which Illumina SNP-array data can exist. If the raw data is in IDAT format, it needs to be processed into intensity files before proceeding with the protocol. IDAT files can be converted to GTC files using the Illumina software IAAP (available here https://support.illumina.com/array/array_software/illumina-array-analysis-platform.html), then GTCs can be converted to intensity file using the python library IlluminaBeadArrayFiles (available here https://github.com/Illumina/BeadArrayFiles). All required inputs are discussed in more detail at the beginning of basic protocols 1 and 2.
BASIC PROTOCOL 1
From raw intensity files to CNV calls
PennCNV (Wang et al., 2007) has been the de facto standard for CNV calling since its initial release fifteen years ago. While it is fairly easy to use, implementing a pipeline to process tens or hundreds of thousands of samples can be challenging. Moreover, there are many checks the users need to perform in order to ensure all commands run as intended, as well as some files that are not straightforward to generate or obtain. The pipeline we refined takes care of most of these issues. This protocol details how to run the pipeline and obtain the raw CNV calls and sample QC files to use for further processing.
Required files
To run the protocol three main files plus the intensity files are required, and they must be in the correct format. We provide GC content file (requirement 4), however, the user needs to generate the samples list (requirement 3) and the SNPs position file (requirement 2).
Intensity files. These files store the raw information regarding each Illumina SNP probe. Each file should contain at least the following three columns: “Name”, “Log R Ratio”, “B Allele Freq”. Column names must be exact; other columns can be present as long as these are the first three. These columns contain, namely: the name of the SNP markers, the value of Log2 R ratio (a relative measure of the light intensity), and B Allele Frequency (a measure of the allelic composition). Intensity files are also called “Final Reports” and may contain a multi-line header. Moreover, it is possible that multiple samples are stored in the same file (e.g., an entire genotyping batch) and that the files are compressed. In all cases PennCNV will not work. All these problems can be very different from case to case, but they can be solved using standard GNU utilities tools (included in every major linux distribution) such as sed or the program awk. To solve the second problem, the PennCNV script split_illumina_report.pl can also be used. Note that in this case the user may need to regenerate a correct samples_list.txt file. Note that it is important to know in which build of the human genome the intensity data is (e.g., “hg19”), and that all samples in the same project must be on the same version.
snppos.txt. This is a TAB separated text file that contains at least the three following columns: “Name”, “Chr” and “Position”, that are the name and genomic location of each SNP marker. The first column must use the same names as in the intensity files. In projects where each intensity file contains this information, any sample can be used to generate the SNP position file. Otherwise, there should be a file called “PFB” (Population Frequency of the B allele) that contains all the columns required by the snppos.txt format.
samples_list.txt. This must be a two columns TAB separated text file with a header. The two columns must be “sample_ID” and “file_path”. “sample_ID” needs to be the identifier for the specific sample and “file_path” needs to be the complete path (thus starting from root, “/”, avoid using links and the home directory, “~/”, in the path) to the intensity file for that sample. Again, this protocol assumes there is one intensity file per sample. If this is not the case, see next point. In the case that samples were genotyped in waves or batches, these can be used; otherwise, batches of approximately 2000 samples will be created. In the first instance, an additional column called “batch” must be present, and the content must be integers indicating the specific batch.
GC content file. This file is used by the PennCNV script cal_gc_snp.pl. This file is not straightforward to generate but we provide the hg38, hg19 and hg18 version as part of the IBPcnv repository. The hg38 version is available from the PennCNV repository, https://github.com/WGLab/PennCNV/blob/master/gc_file/. It is also included in the IBPcnv repository for user convenience. Finally, the original hg18 version can also be obtained directly from UCSC Genome Browser at http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/gc5Base.txt.gz.
Protocol steps with step annotations
Setup. We provide all required software and scripts in a docker/singularity container and a GitHub repository. Excluding the raw data, all files and subdirectories need to be in the same directory ($workingdir hereafter). Support protocol 2 details the installation process, as well as some suggestions on how to set up directories and the environment for the analysis.
- Initial checks and required files generation:
- This will do the following. First, it will run a series of tests to check that all intensity files are present and in the correct format. Then, it will check if samples were already divided in batches and that this is in the correct format. If not, it will create the batches and separate the samples into groups of approximately 2000 each, the actual number may vary in order to not have the last batch significantly smaller than the rest.
- To complete this step, move to the main working directory (cd $workingdir) and run
singularity exec ibpcnv.simg Rscript \
IBPcnv/penncnv_pipeline/01_preprocess.R \
$workingdir 1 2000 $tabix_folder - If the script fails checking the batches but no errors are found in the intensity files, the second parameter can be set to 0 to skip the initial checks that constitute the slower part of the testing.
- If all checks are successfully completed, the script will write the per-batch sample list files needed by PennCNV in $workingdir/listfile/. The script will also print the number of batches that will be used.
- Select the SNP markers:
- This step will do the following. First it will extract the marker names and position from an intensity file. It will then download the HRC SNP list, selecting only SNPs that are strictly biallelic, known (with a name in dbSNP142, no “.”) and with a minor allele frequency of at least $minMAF (MAF values for each SNP are obtained from the HRC), we suggest 0.001 (0.1%). Then, it will merge the two tables, remove markers with duplicated “SNP_ID”, remove markers that map to the same position and finally it will create the snppos.txt file needed by PennCNV.
- To complete this step, move to the main working directory and run
singularity exec ibpcnv.simg Rscript \
IBPcnv/penncnv_pipeline/02_select_SNPs.R \
$workingdir $minMAF $hgversion TRUE
- PennCNV calling pipeline:
- First, run the calling pipeline in parallel on each wave. To complete this step, move to the main working directory and run the command
bash IBPcnv/penncnv_pipeline/03_penncnv_pipeline.sh \
$workingdir $ibpcnvdir $n_batches hg19 - PennCNV calling parameters (minimum number of SNPs and minimum length of each call) can be changed in the script $ibpcnvdir/penncnv_pipeline/03_2_cnv_calling.sh, before launching the previous step. By default, these are set to 5 and 1000 bp respectively.
- Check that all batches are completed successfully. Catching any PennCNV error is quite straightforward, the following command can be used
cd $workingdir && grep ‘ERROR’ pennlogs/* if [ $(wc -l samples_list.txt | cut -d ‘ ‘ -f1) == \
$(wc -l results/autosome.qc | cut -d ‘ ‘ -f1) ]; then
echo “All good”; fijoin -v2 −1 1 −2 2 <(LANG=C tail -n+2 1 results/autosome.qc | sort -k) \
<(LANG=C tail -n+2 samples_list.txt | sort -k 2)sbatch IBPcnv/penncnv_pipeline/03_1_per_wave.sh $workingdir \
$ibpcnvdir $n_wave - After the calling pipeline is completed, the batches can be combined into two single files (CNV calls and QC) running (for PBS systems, use qsub instead of sbatch)
sbatch IBPcnv/penncnv_pipeline/04_combine_results.sh \
$workingdir $ibpcnvdir $maxgap - This will also perform a “soft stitching” step, meaning that for each sample, close calls with the same copy number will be stitched together. This is controlled via the $maxgap parameter; we recommend a value of 0.2 and not higher than 0.4 as it could alter the raw call-set. A stronger stitching will be performed in the basic protocol 2 when selecting putative CNVs in a specific locus.
- Tabix indexing the intensity files. In basic protocol 2, we take advantage of the speed of tabix indexed files as well as of the GC waviness-adjusted LRR values.
- The GC model file for the specific array in use should have already been generated. If the user is interested only in the second half of the protocol the following command can be used
bash IBPcnv/misc/create_gcmodel.sh $workingdir $ibpcnvdir - To perform the indexing run
bash IBPcnv/penncnv_pipeline/05_launch_tabix.sh $workingdir $ibpcnvdir
BASIC PROTOCOL 2
From CNVs calls to validated CNVs carriers
CNV calling using SNP-array data is an intrinsically imprecise process, and strongly depends on the quality of the initial SNP-array raw data. In particular, it is prone to pick up noise as signal, as well as to “over-segment”, that is, incorrectly splitting a (often large) CNV call into several smaller ones. This ultimately may lead to unreliable results. Moreover, precise CNV boundaries (at the level of 1–2 SNP probes) are very difficult to obtain. Different research groups have developed different strategies to overcome these problems. To counter over-segmentation, it is common practice to “stitch” close, consecutive calls with the same Copy Number (CN). To reduce noise, some kind of filtering is almost always included. At the CNV call level, the filtering can act on the call length, the number of SNP probes or, more rarely, on the confidence score. Some filtering is usually performed also at sample level, this may include removing samples with a too high LRR standard deviation (LRRSD), extreme BAF drift, or too many calls. Few studies, such as this protocol, also perform an initial filtering on the SNPs in order to reduce the general noise of the raw data. Notably, these approaches tend to lead to false positive calls being a larger issue than false negative ones. Changing the type and strength of the CNV calls level filtering enables researchers to balance between false negatives and the number of calls to validate (i.e., false positives to manually screen). In order to reduce the number of false positives two main strategies have typically been used. The first is to visually validate all putative CNV calls. This approach is time consuming and prone to human error, however, when done correctly, it is the overall best approach, and it is commonly used when calling CNV in a limited set of specific genomic loci, such as recurrent CNV loci (Stefansson et al., 2014; Calle Sánchez et al., 2021). This approach should always be preferred when the CNVs of interest are rare, and thus few false positives or negatives could significantly affect estimated prevalence and downstream analysis. The second approach is to use multiple algorithms to perform the CNV calling and intersect the resulting callsets (often using a form of reciprocal overlap in terms of base pairs or probes) and filter the results. An example is using more than one program (such as PennCNV (Wang et al., 2007), QuantiSNP (Colella et al., 2007) or others) and considering “validated” only calls where 2 or more programs made the call. In this case the reasoning is that even if some false positives are introduced, with a large sample size, these errors will be equally distributed among, e.g., cases and controls. Such studies usually also perform some kind of grouping before doing any analysis (e.g., deletions affecting at least one gene) and rarely consider individual CNVs on their own.
In this protocol, we follow the first approach (i.e., always relying on visual inspection as the final validation step) and we integrate it with newly developed filters that do not depend on the PennCNV output but directly on the raw data. By doing so we are able to reduce the number of putative carriers to inspect, while also minimising the number of false negatives as much as possible. This approach is particularly suited for large cohorts where the number of false positives can be high. In smaller datasets it may not be equally useful to perform the filtering step and the user can simply use our package to standardise the files and then use the graphical interface to validate all putative carriers. It is important to notice that, as stated in the main introduction, this protocol has been designed with a strong focus on recurrent CNV, meaning CNV with relatively fixed and precise start and end positions. As an example, a researcher interested in all recurrent CNVs in the larger 15q region should specify each specific locus individually in the loci.txt file (see required files), while a researcher interested in all CNVs overlapping the NRXN1 gene should use a very low minoverlap value (see step 3c) and avoid the advanced filtering (see step 4d).
Figure 2 shows a schematic representation of the basic protocol 2.
Figure 2:
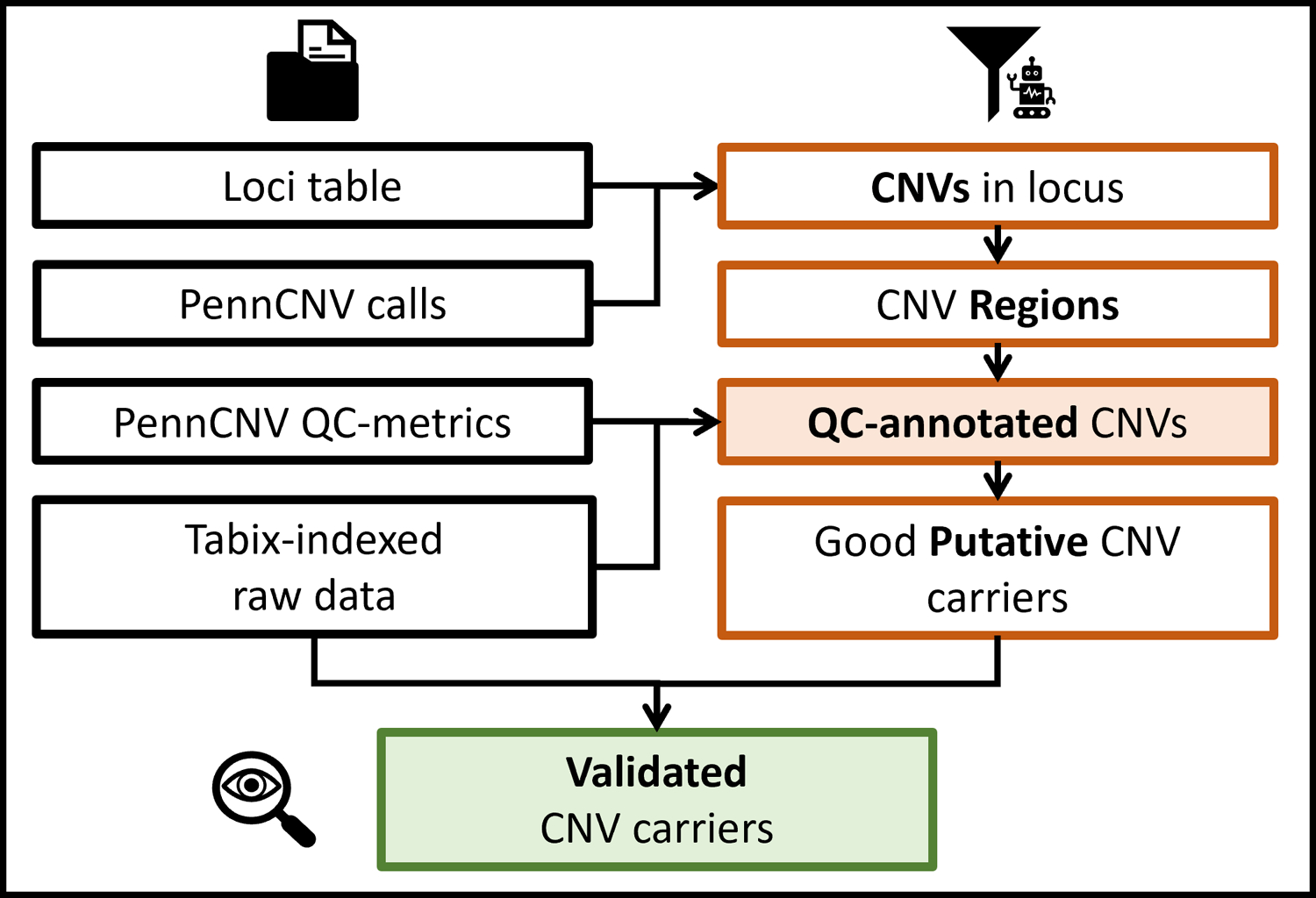
More detailed schematics of basic protocol 2. Colours and icons are kept consistent with figure 1, indicating starting data files, QCtreeCNV and DeepEYE respectively.
Required files
The only additional file needed to run the second part of the protocol is a list of the loci of interest. The file loci.txt must be a four column TAB separated text file with a header. The columns must be called “locus”, “chr”, “start”, “end”. The chromosome must be in integer format. This format is used in all the results and intermediate R objects, as well as the tabix indexed intensity files. It simply consists of integers from 1 to 22 that are used for autosomes, plus 23 for X, 24 for Y, 25 for XY and 26 for MT.
Protocol steps with step annotations
- Setup. All software and scripts required are provided in a docker/singularity container and a GitHub repository. Support protocol 2 details the installation process. It is assumed the user successfully completed basic protocol 1. Throughout this protocol all files are loaded in R as data.table objects. Please note that they behave slightly differently than data.frame in certain situations. If the user prefers using data.frame to explore the results, after the protocol completion the main objects can be converted by running
objectA_df <- as.data.frame(objectA_dt) QCtreeCNV filtering pipeline. This step is meant to be run interactively in an R session. The user can use the provided commands in an R script; however, we suggest exploring at least a couple of loci interactively before setting the final parameters.
- Preparation. All code lines are shown in code block 1.
- Load data into R. To launch an interactive R session using the provided singularity image, run
singularity exec ibpcnv.simg R - Check formats and select the calls in the loci of interest. These steps can be performed with a single function, qctree_pre(). It takes the four main objects from the previous step and the parameters for stitching close calls. Line 7 shows the code for default values. If there is any problem with the inputs the function will fail with an error message explaining the specific problem. By default, the function will also take care of multiple calls in a locus from a single locus, keeping only the largest call, regardless of the copy number. The user can avoid this by setting rm_dup = F. Note however that the downstream steps do not support multiple calls per sample in a single locus and will throw an error if any is found.
- The stitching function takes three main parameters, minimum SNPs number for calls to be considered, maximum gap between two consecutive calls with same CN in order to stitch them together, and minimum overlap between a call and a locus for the call to be selected as a putative CNV. Default values are respectively 20, 0.5 and 0.2. These values can be changed with the following parameters minsnp, maxgap, minoverlap, e.g. pre <- qctree_pre(loci, cnvs, qc, samples, minsnp = 15, maxgap = 0.4, minoverlap = 0.5).
- Compute CNV Regions. We provide two different functions to compute CNVRs (CNV Regions), cnvr_fast() and cnvrs_create(). Additionally, the user is free to use a different method as long as the results are in the correct format. CNVRs are used here to separate groups of largely overlapping calls within a certain locus, in particular groups with different lengths. CNVRs and their computation are further discussed in the commentary and in the package manuals and vignette. The suggested function can be run as shown in line 9.
1> library(data.table); setDTthreads(2); library(QCtreeCNV)
2> cnvs <- fread(“results/autosome.cnv”)
3> qc <- fread(“results/autosome.qc”)
4> loci <- fread(“loci.txt”)
5> samples <- fread(“samples_list.txt”)
6>
7> put_cnvs <- qctree_pre(loci, cnvs, qc, samples)
8>
9> cnvrs <- cnvr_fast(put_cnvs)
Code block 1.
- qctree() filtering:
- The filtering function is structured as a decision-making tree consisting of five main steps, and each step has multiple parameters the user can change. More technical details on each step and how the main parameters are connected with the outputs of support protocol 1 (quality control) is further discussed in the commentary. To run the function with default values type
cnvs_out <- qctree(cnvrs[[1]], cnvrs[[2]], loci) - Step 1 of the filtering tree is removing samples-wise QC outliers and this is the most accessible step to customise. By default, samples will be removed if they have LRRSD > 0.35, BAF drift > 0.01 or GCWF outside of the window −0.02 to 0.02. These values can be changed with the following parameters maxLRRSD, maxBAFdrift, maxGCWF, minGCWF.
- The resulting table will contain the column “excl” with the value 0, meaning the line is a good putative CNV call, or 1, meaning the line is a bad putative CNV and can be skipped in the visual inspection step. This table can be exported in the correct format for the visual inspection interface with
export_cnvs(cnvs_out[excl == 0, ], “putative_cnvs.txt”) export_cnvs(cnvs_out, “putative_cnvs.txt”) - If no advanced filtering is needed, the user can simply apply the standard filters (LRRSD, BAFdrift, GCWF) when exporting the table. Using the data.table syntax, run
export_cnvs(put_cnvs[LRRSD <= 0.35 & BAFdrift <= 0.01 &
between(GCWF, −0.02, 0.02, incbounds=T), ],
“putative_cnvs.txt”)
- Visual inspection.
- Visual inspection of the putative CNVs is necessary to validate the true CNV carriers with precision. The in-house program DeepEYE (included in the provided container) provides the user with a graphical interface to assign a label (true, false, unknown) to each CNV candidate. The results are stored in an extra column in the putative CNVs file.
- DeepEYE requires three main inputs, the samples and loci lists, plus the putative CNVs table. It can be run from the singularity image, assuming all files have the standard names described in the protocol
singularity exec ibpcnv.simg python3 /opt/eyeCNV/visualizer.py \
$workingdir putative_cnvs.txt loci.txt samples_list.txt GC_YES- Name the project (box a), the name will dictate the name of the output file, namely visual_output_projectname.txt. The project name should be meaningful, e.g., when user “abc” is evaluating deletions in the TAR locus, a good project name could be “TAR_del_abc”.
- Select the loci to inspect. Left clicking on the button b will open a secondary window (i) where it is possible to select the loci to inspect in the current run.
- Select the condition. Left clicking on button c will open a menu with the following options: true, false, unknown, all. In a new project “all” should be selected, while re-evaluating a previous project the user can select only a portion of the calls (e.g., unknown).
- Select the type. Left clicking on button d will open a menu with the following options: duplications, deletions, any. This lets the user select a specific type of CNV to inspect.
- Once this is set, the user can click button e, “start”.
- If at least one CNV call was selected, the right panel will appear. The panel consists of two plots (h), the top is for LRR and the bottom is for BAF. Each dot represents a marker. The red dots are within the locus of interest, the blue dots are outside.
- For each plot the user can evaluate if a CNV is present within the red region and record the decision using the dedicated buttons (f):
- True: there is a CNV in the red region (with the correct CN, as shown in g).
- False: there is not a CNV in the red region (or the CN is not correct).
- Unknown: it is not possible to tell whether a CNV is present or not, likely due to excessive noise in the region.
- Error: useful to mark samples with problematic intensity data.
- The progress bar and text in g mark the session’s progress. Once all selected calls have been inspected the result file will be written in $workingdir. The last column, “visual_output”, contains the record of the visual inspection as integers: 1 (true), 2 (false), 3 (unknown), −7 (error). This file can be used again as putative CNV file, for example to re-evaluate unknown calls only.
Visual evaluation concludes basic protocol 2. The file visual_output_projectname.txt will contain the results. The visual output codes (step 5e) can be used to filter the relevant CNVs. See also the final section “Understanding Results”.
Figure 3:
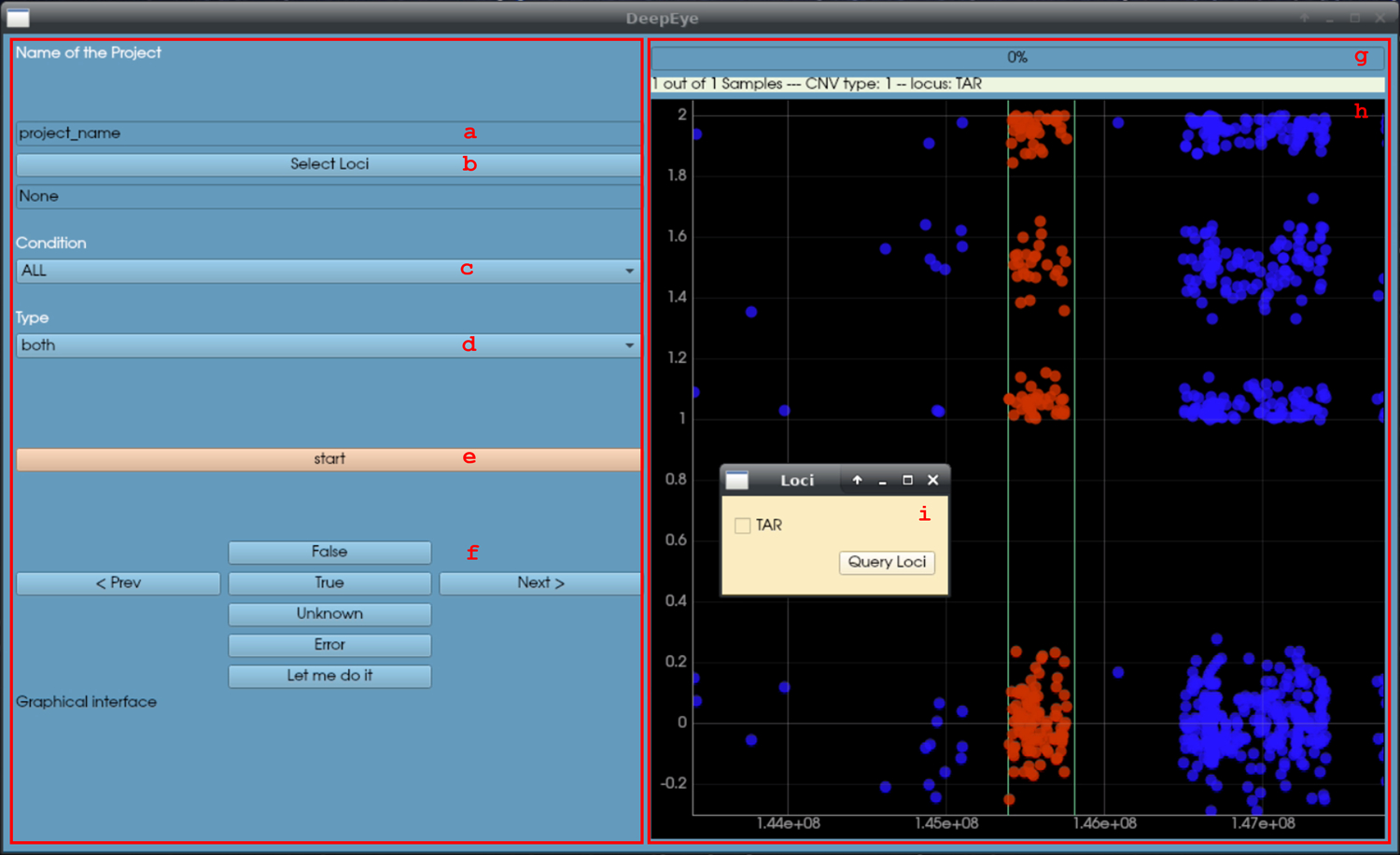
DeepEYE graphical interface (no CNV is present in the region, simulated data). Refer to step 5c of basic protocol 2 for the usage instruction.
SUPPORT PROTOCOL 1
Quality Control & Quality Assessment
This protocol, similar to any other CNV calling pipeline, implements a certain set of filters. We propose valid default values based on the scientific literature and our research experience (Stefansson et al., 2014; Calle Sánchez et al., 2021). However, since this protocol is mostly aimed at CNV calling in recurrent loci where the actual CNVs are quite rare, we need to be extremely careful that our processing does not introduce any false negatives that may severely bias our estimates (false positives are controlled via visual inspection). In large cohorts, this ultimately means balancing the strength of the filters and the amount of manual validation required. Finally, some filters are applied sample-wise on values that directly reflect the noise in the data, for the specific sample (LRRSD and BAF drift in particular). We found that it is often quite possible to use relaxed filters, thus eliminating fewer samples. However, in doing so, one must be able to assess that the ability to detect CNVs is not significantly different in “noisier” samples, otherwise biases may be introduced in the results. Here we show how to produce a series of plots that help identify such potential issues in the results of the protocol, and briefly discuss the interpretation of each one. Specific problems and solutions are then highlighted in the troubleshooting section.
Protocol steps with step annotations
- Setup. This support protocol is meant to help evaluate the performance of the CNV calling protocol. First, start an R session. From $workingdir,
singularity exec ibpcnv.simg R library(data.table); vi_res <- fread(“visual_output_ALL.txt”). - Create the plots. The function qc_plots_cnvs() will create three plots for the main filtering arguments and more supporting ones. Here we will discuss briefly only the main one, for the complete discussion refer to the commentary. As an example, to create the QC plots for the CNVs in all loci, simply run
library(QCtreeCNV); qc_plots_cnvs(vi_res, “all_loci”). - Interpret the results. Figure 4 shows a good and a bad example of the two plot types. See also the final section “Understanding Results” for more discussion.
- Plot 1 (example in figure 4A) shows the CNV prevalence in different LRRSD chunks (low, medium, high), separated for deletions and duplications. It illustrates the ability to detect true CNVs in different groups of samples from the noise perspective (high LRRSD can be considered the main indication of a noisy sample). Ideally, the prevalence should not differ significantly across the different groups, in particular the one with the highest LRRSD. A significant change means that the ability to detect CNVs is significantly affected in noisy samples, usually lowering it. This means the LRRSD filter threshold may need to be increased. On the other hand, if a strong LRRSD filter was used and plot 1 shows very high consistency, the user may want to explore lowering it to possibly exclude less samples from the analysis.
- Plots 2 and 3 (example in figure 4B) show the distribution of the number of SNPs and overlap proportion with the locus per call for true CNVs, numsnp and overlap respectively. Both these measures are used as filters when selecting putative CNV calls. Ideally the distribution should not seem to be “cut” at the threshold values for numsnp and overlap. If that is the case, it might indicate that some potential true CNVs are being filtered out and it may be worth trying to relax the filters. This is especially important for very rare CNV loci, where a small increase in carriers can have an impact on power. Note that plot 2 may be more meaningful when used on individual loci or a group of loci with very similar marker coverage, since it is the absolute number of markers of each call. If different thresholds were used for different loci, they must be treated separately to obtain meaningful results.
- Plot 4 and 5 can be interpreted in the same way as plot 1 (figure 4A) but regarding two other noise measures, BAF drift and GCWF, respectively. They can be considered secondary since these dimensions are less prone to affect the CNV detection accuracy.
- Deal with possible detection bias. If the dataset includes sample groups selected differently (e.g., cases and controls) it may be a good idea to analyse them separately. The CNV prevalence is often expected to differ in different groups (e.g., some recurrent CNVs are more frequent in neuropsychiatric case groups than in population controls), thus combining them may lead to confusion in the interpretation of the QC plots, especially if LRRSD distribution also differs across those groups. Possible problems in these plots are described in the last two points of the troubleshooting section. Assuming the sample_IDs for one group are in vector groupA QC analysis can be run separately with
qc_plots_cnvs(vi_res[sample_ID %in% groupA,], “groupA”) - Explore an individual locus or groups of loci. The process described in previous steps 2 and 3 can be applied to groups of loci as well as to individual loci. This is useful especially when there seems to be some problems but only in a fraction of the loci or in a particular one. For example, to look only at the results for loci “A” and “B” run,
qc_plots_cnvs(vi_res[locus %in% c(“A”, “B”),], “loci_A_B”)
Figure 4:
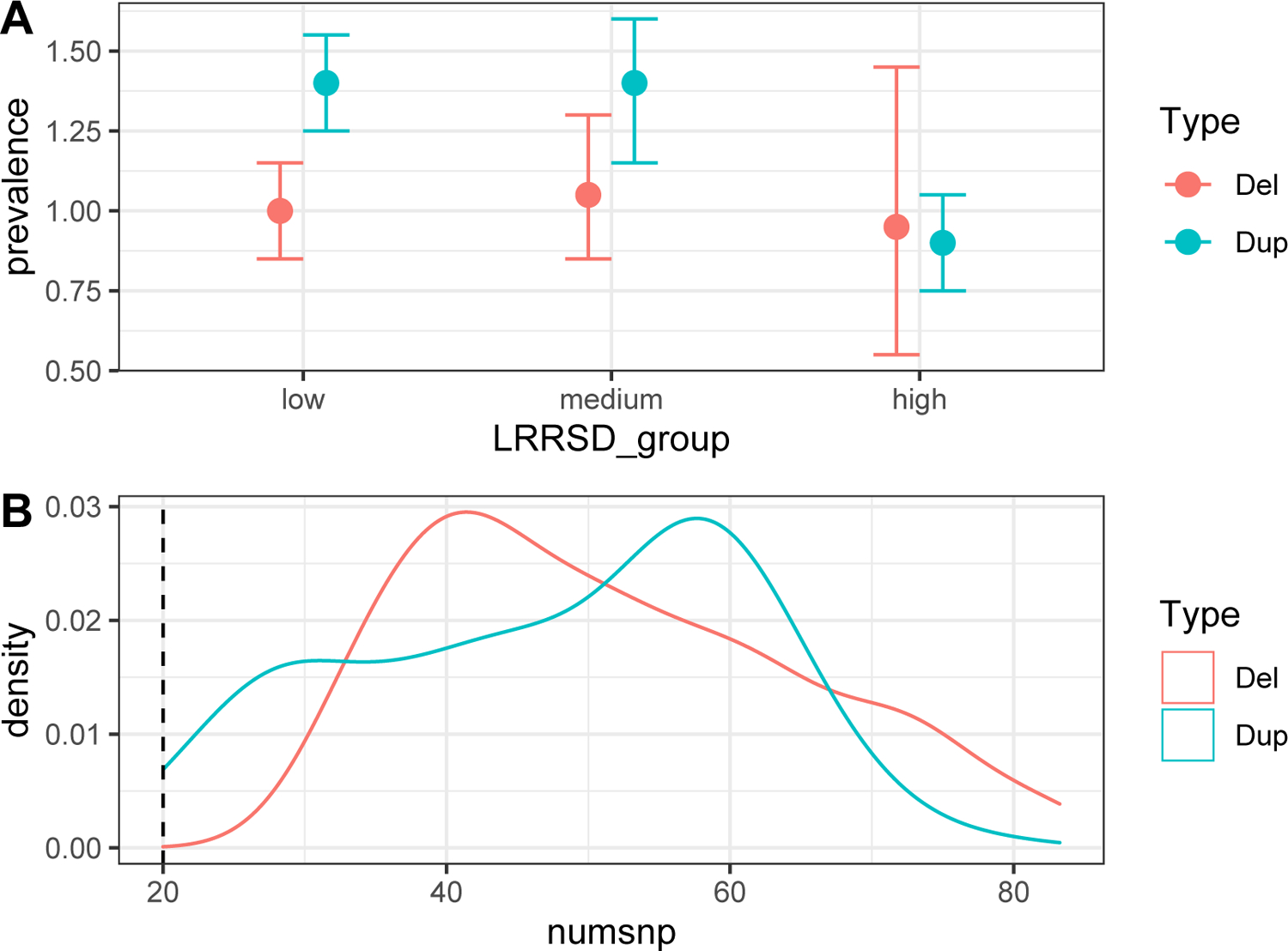
A good and a problematic example of QC plots 1, 4 and 5 (panel A), and 2 and 3 (panel B). In both plots the deletion in light red represent the ideal situation and the duplication in light blue represent the problematic situation. A, the group of samples with high LRRSD have less true duplications that the other two groups, and that the confidence interval is relatively small. B, the threshold value we selected for numsnp appears to be cutting the left tail of the distribution for the true duplications.
SUPPORT PROTOCOL 2
Install the necessary software
We provide a docker image containing all software required to run the protocol on Docker Hub at https://hub.docker.com/r/sinomem/docker_cnv_protocol. This container has the following software installed: htslib (Bonfield et al., 2021; Danecek et al., 2021) (1.14), PennCNV (Wang et al., 2007) (1.0.5), R (R Core Team, 2018) (4.1.2), DeepEYE2 as well as several R packages, including data.table (Dowle et al., 2020), fpc (Hennig, 2020) and VariantAnnotation (Obenchain et al., 2014).
Regarding the in-house software used in this protocol, QCtreeCNV R package is available on GitHub at https://github.com/SinomeM/QCtreeCNV. Instructions to install the package are given in the README. DeepEYE2 is available at https://github.com/XabierCS/eyeCNV. All other scripts are collected in a GitHub repository at https://github.com/SinomeM/IBPcnv.
The statistical programming language R is available at https://www.r-project.org/. Tabix is part of the HTSlib suite, together with SAMtools and BCFtools. It can be obtained at https://www.htslib.org/download/. PennCNV is the de facto standard in CNV calling from array data, in particular Illumina. It is available at http://penncnv.openbioinformatics.org/en/latest/user-guide/download/. In the following section we detail how to install the docker/singularity image and use it to run the protocol.
Protocol steps with step annotations
- Setup. Throughout the protocol $workingdir is used to indicate the main project folder. This folder will contain all the input and output files. For simplicity the user can define an environmental variable,
export workingdir=/path/to/workingdir Install Singularity. The software should be already installed on most modern HPC. If not, the user should ask the system administrator to install it for them. To install it on a Linux workstation, one should follow the official instructions available here, https://sylabs.io/guides/3.0/user-guide/installation.html. A precompiled RPM package is available at https://dl.fedoraproject.org/pub/epel/8/Everything/x86_64/Packages/s/singularity-3.8.0-1.el8.x86_64.rpm and the program alien can be used to convert it to DEB (https://github.com/apptainer/singularity/issues/5390). Finally, in systems where the use of conda environments is encouraged or enforced, it should be possible to use this conda package https://anaconda.org/conda-forge/singularity.
- Download the provided container image. We provide the container on DockerHub and it can be pulled directly by singularity. To do so, first move in the desired folder (cd $workingdir) and then type
singularity pull ibpcnv.simg docker://sinomem/docker_cnv_protocol:latest - Download the IBPcnv repository. We provide two versions, one that uses SLURM (srun/sbatch) and one that uses PBS (qsub). They can be obtained running
wget https://github.com/SinomeM/IBPcnv/archive/refs/heads/master.zip && \
unzip master.zip && mv IBPcnv-master IBPcnv && rm master.zip
# or
wget https://github.com/SinomeM/IBPcnv/archive/refs/heads/pbs.zip && \
unzip pbs.zip && mv IBPcnv-pbs IBPcnv && rm pbs.zipexport ibpcnvdir=${workingdir}/IBPcnv Add the SLURM/PBS account if needed. The four scripts that use the job scheduler (03, 03.1, 03.2, and 04 in $ibpcnvdir/penncnv_pipeline/) are designed to be easily edited if the system requires the use of a specific account name. Throughout the text we provide both versions of the commands when run interactively.
Run the protocol. All scripts assume that the singularity image is used. The pipeline in basic protocol 1 is designed to take advantage of the SLURM or PBS job scheduler, depending on which branch of the IBPcnv repository was chosen.
- Docker vs singularity. To download the container using docker we run
docker pull sinomem/docker_cnv_protocol:latest singularity exec /path/to/ibpcnv.simg tabix -help
# or
docker run sinomem/docker_cnv_protocol:latest tabix --help.
COMMENTARY
Background information
The main scope of this protocol is to provide a framework to enable the creation of high-quality datasets of recurrent CNVs from large scale SNP-genotyped collections. We condense years of experience in the field into easy-to-use pipelines and an extensive set of best practices, providing strong default values and great customizability for all major parameters. The framework is composed of four main elements: a docker/singularity container, a standardised PennCNV pipeline, an R package for data handling and cleaning, and a graphical interface to perform visual validation of the CNVs. The singularity image (support protocol 1) contains all necessary software in the correct version, and the whole protocol is designed to use it. Using a container, software installation is not a variable in the process, and instructions are always ensured to work as intended. Distributing it in both docker and singularity formats, we made it accessible to most systems. The CNV calling pipeline (basic protocol 1) has multiple qualities. It is meant to work on most HPC systems, freeing researchers from the task of designing ad-hoc pipelines thus providing standardisation and user friendliness. Moreover, it integrates several, seemingly simple, steps that required years of expertise to be refined, and can have a huge impact on the final results. The R package (basic protocol 2) serves multiple functions. It standardises putative CNV selection and filtering. It also implements a novel, more advanced, filtering algorithm designed to reduce the number of putative CNV to inspect, but with a strong focus on minimising false negatives. Quality control after visual validation (support protocol 2) is also handled by this package. Finally, the graphical interface is a very powerful program that can be used not only to validate CNVs in fixed loci, but to manually refine the boundaries of a CNV call (feature not showcased in this manuscript). Together with the possibility of using GCWF-adjusted values for LRR, in our opinion this makes it superior to any similar solution.
While CNVs can be detected and studied with different approaches, we designed the protocol with a clear focus, namely effective and precise detection of rare recurrent CNVs in specific and characterised genomic loci using large collections of SNP-array data. Nonetheless, we believe the graphical interface, as well as the PennCNV pipeline, provide a high value also when used on their own. As an example, when analysing small collections or for studies not focused on recurrent CNVs. To our knowledge, no comparable software packages are available. In contrast, the novel filtering algorithm we developed (step 4 of protocol 2) is narrower in its approach. While the use of the R package to manage and select the CNV call from PennCNV is advised to all users, qctree() has been specifically constructed in order to deal with a high number of calls, where the user knows (or suspects from exploratory analysis) that a good portion of the putative calls are false. This can be due to noise, if the collection has for example a high LRRSD or GCWF, or due to the presence of smaller irrelevant CNVs within the locus of interest. The algorithm can deal with both these problems and by default will behave quite conservatively, meaning that for a call to be filtered out two or more measures need to decisively point towards the exclusion. In other words, in a dubious situation, the decision should always be made by the analyst. In accord with this model, visual validation always needs to be performed. If the number of putative calls to inspect is manageable, and/or if it is suspected that no large groups of false positives are present, then it is advised to skip the step and perform visual validation directly.
Design choices
We made several design choices during the creation of the protocol; we discuss the major ones in this section. In basic protocol 1, the pipeline is structured to be very easy to use also for inexperienced analysts while including several advanced steps that would require some level of expertise. However, all design choices we made can be considered fairly standard and coherent with current standard practice. In contrast, basic protocol 2 makes use of tabix indexed intensity files (introduced in protocol 1), which is a novel idea in CNV studies. Tabix indexed files provide an enormous speed advantage when accessing specific sections of the file (such as a genomic region in a BED file), by avoiding the need to load the entire file into the computer RAM or to screen the file from the beginning. This advantage is used throughout all functions that need access to raw data in QCtreeCNV, as well as to create the CNV plots on-the-fly in DeepEYE. Of note, when creating the indexed file, we also add an additional column containing the GCWF-adjusted values for LRR. The GCWF-adjusted LRR value can be used in DeepEYE, meaning that, in contrast to any other methods to our knowledge, the user can actually “see” the raw data trends in the same way as PennCNV did. This is because GCWF correction is performed by PennCNV when calling the CNVs, however the adjusted values are usually not stored and thus not accessible by the user. This whole process comes with a cost as well, as basically a full copy of each intensity file needs to be generated (CPU time and high I/O on the system) and stored (disk space). We strongly believe the advantages exceed the costs in disk space and propose this format as the new standard for all future programs that deals with SNP-array data in the form of intensity files. Another important choice was to use the R data.table structures and grammar in QCtreeCNV internal functions as well as in basic protocol 2. This package provides several advantages, including implicit parallelization of each operation if multiple cores are available and a simple and extremely powerful grammar. We believe that in small and very specific R packages like ours there is no need to avoid relying on external dependencies. Moreover, the aforementioned advantages (implicit parallelization in particular) vastly outdo the small added complexity in the protocol commands. We also state clearly that all objects can be reverted back to simple data.frame after completion if the user wishes to continue with downstream analysis. Moreover, we believe that exploratory analysis is a necessary part in this kind of studies, and that no protocol can substitute them. For this reason and considering the small number of commands and the large number of tuneable parameters, we show how to run the QCtreeCNV pipeline in an interactive R session and we do not provide an R script. Testing multiple settings and exploring the results is strongly encouraged. An RStudio session may be ideal for some users but we choose to not include RStudio in the container as it would make it more complex and heavier. We note that all R packages required to run the basic protocol 2 are listed in support protocol 2. Finally, we show how to run the protocol as a whole in all loci of interest but this may be limiting in some cases. One example is when the user is using a list of loci where one or few are very different from the others (e.g., very small or covered by very few markers). In this case it may be beneficial to treat a particular locus or group of loci differently from the other with regards to some critical parameters such as minimum number of SNPs, length or overlap. This applies to basic protocol 2 and support protocol 1, as all samples must be treated the same in basic protocol 1.
Technical details
In this section we provide more technical details, in particular about CNVRs and the advanced filtering function qctree(). CNV regions can be defined in different ways depending on the scope, but in general they are regions of the genome where very similar CNVs are present, across samples. Recurrent CNVs loci can be considered CNVRs, but they also can be smaller. In this protocol we use CNVRs only to extract false positives from the putative CNVs. In practice, the function cnvr_fast() performs 2D clustering on the normalised centre position and length of all putative CNVs in each locus of interest (separately). If multiple subgroups are found, they are then categorised and potentially treated differently in the qctree() function, meaning that CNV belonging to large CNVRs will be harder to mark as false positives, CNVs belonging to small CNVRs will be easier to exclude, and those from medium CNVRs will receive an in-between treatment. Supplementary figure 1 provides a schematic representation of the filtering pipeline. Step 1 is applied sample-wise and will exclude any QC outlier, based on LRR standard deviation, BAF drift and GC waviness factor. It is the more canonical step and the more accessible to customization. Step 2 separate CNVs calls based on the CNVR they belong to. The reasoning is that CNVs calls that are very similar to the locus of interest should be almost always passed to visual inspection, while CNVs from smaller CNVRs may be filtered more aggressively. Step 3 further separates CNVs based on CNVRs frequency, sending the putative calls either directly to step 5 or first to step 4. All the dimensions on which step 4 and 5 are applied are derived directly from the intensity files, thus do not depend on PennCNV processing. Broadly speaking, they measure how LRR and BAF trends (see supplementary figure 2 for a visual interpretation on how BAF is used in this context) behave and are compared to threshold values we derived from a broad collection of human-validated true and false CNVs calls (~15,000) in thirty different recurrent loci. In short, from step 3, if the CNVR is small as well as frequent, it may indicate the presence of a smaller true recurrent CNVs locus within the main one. Thus, CNVs with these characteristics proceed to step 4 where an aggressive filter is applied to test whether the raw data is consistent with the assumption. In contrast, if the CNVR is small but infrequent, it is likely that the CNVs are either noise or true CNVs called only partially by PennCNV. Thus, a lighter filter should be sufficient to separate the two groups.
Critical parameters
In this section we briefly discuss the most important parameters of the entire protocol. In basic protocol 1 the user can mainly change the parameters of step 3b and 4d, namely the SNPs filtering and the first round of CNV calls stitching. In 3b it is suggested to keep the default value of minMAF (the minimum value of Minor Allele Frequency), however the user can set the last parameter to FALSE if they feel too many SNPs are excluded because of duplicated SNP IDs or positions. In basic protocol 2 the critical parameters are in steps 3c and 4a. Step 3c parameters control how the putative CNVs are selected; minsnp, maxgap, and minoverlap control the required minimum number of markers per call (applied after stitching), the maximum gap allowed before stitching two calls, and the minimum amount of overlap between CNV and locus of interest. Step 4a consists of the advanced filtering function, as already stated the most accessible parameters are the sample-wise filters maxLRRSD, maxBAFdrift, maxGCWF, minGCWF. These control respectively the maximum LRR standard deviation and B allele frequency drift, and the GCWF range allowed. The first two can be considered the major knobs the user can turn when managing the noise-to-signal ratio, other than the qctree() function itself.
Troubleshooting
| Problem | (Possible) Cause | Suggested Solution |
|---|---|---|
| Errors in support protocol 2 | No formal software installation is required thus the most likely problems are: required programs (singularity, wget, unzip) are missing and no internet connection. | Refer to the official website for singularity installation. If wget is not installed on the system one may use curl. Another alternative is to use git clone. When using git, remember to switch to the PBS branch if necessary! |
| The computing system does not provide a job scheduler (PBS or SLURM) | While common practice in large HPCs, smaller systems may not use a job scheduler to manage the computing load. | Only a section of basic protocol 1 requires a job scheduler (step 4), the actual CNV calling pipeline. In a small dataset, a quick solution is to manually run each batch in a separate session ($ibpcnvdir/penncnv_pipeline/03_1_per_wave.sh), avoiding intra-batch parallelization (with small modifications of $ibpcnvdir/penncnv_pipeline/03_2_cnv_calling.sh). |
| Bash and PBS/SLURM errors in basic protocol 1 | The main cause of errors are erroneous paths or typos in naming the required files. | Check multiple times that all required files are in the expected format, in particular the file name, field separator (TAB) and columns header. Check also that all paths are complete (starting from `/`) and do not include any links (as an example going through the user home directory). |
| PBS/SLURM job scheduler throws errors regarding incorrect “account” or “partition” | To interact with the job scheduler, one may be required to use a specific account or queue. | As described in step 5 of support protocol 2, all scripts that use PBS/SLURM are designed to be easily modified in this case. Also, all commands in the protocol that used srun or qsub must be modified accordingly. For srun, adding the flag --account=account_name. |
| Step 4c of basic protocol 1 shows some samples were not processed by PennCNV | Samples are called in chunks (within each batch) of ~200 per job. Some jobs may fail for multiple reasons. | A simple solution is to rerun the chunk or the whole batch manually, as shown in step 4c. |
| Step 4c of basic protocol 1 shows some samples were not processed by PennCNV | A second reason some samples may fail processing is that the intensity file is in the wrong format or corrupted. For convenience reasons, step 2b will check that around 25% of all intensity files exist, but only 100 will be opened to check for the correct format. It is possible that some files have errors or are missing. | Check the format of all intensity files. Lines 19–37 of $ibpcnvdir/penncnv_pipeline/01_preprocess.R can be used as template. |
| Errors in basic protocol 2 R session, such as “file not found” | The commands shown in the protocol require precise file naming and formats. | Check multiple times the paths and file names used correspond to the commands run (e.g., some output files may not have standard names), and change the suggested commands when necessary. |
| QC plot 1, 4 or 5 show problems in support protocol 1 | As described in the protocol, these plots are meant to show if the ability to detect CNVs is affected by the noise. The high noise category is the most likely to show problems, i.e., a different prevalence than the other groups. The relevant filtering happens in step 4b of basic protocol 2. | This must be interpreted with some care, as often the high noise group is also the smaller one. If the numbers are too low and the estimate is unstable (especially for a single locus) it is quite hard to recommend a specific action. In general, here it is a matter of balancing losing samples with a stronger filter and getting potentially biased results from too-noisy data. Key parameters to remove the noisy samples are explained in step 4b of basic protocol 2. |
| QC plots 2 or 3 show problems in support protocol 1 | As described in the protocol, these plots are meant to show if the minsnp and overlap filters in step. The relevant filtering happens in 3c of basic protocol 2. | The distribution can take various shapes but, with high numbers, it should be somewhat gaussian. The main problem is if the distribution appears to be “cut” at the threshold value for the measure of interest. This is better observed with a single locus or a group of similar loci. If such a problem presents, it means that some true CNVs were “tagged” by very small calls or calls only very partially overlapping the locus of interest. The solution is to try reducing the relevant threshold value. However this could lead to a higher number of false positives, even though qctree() in the following step should remove most of them. |
Understanding Results
The ultimate results of this protocol are a file containing visually inspected CNVs (basic protocol 2, step 5e and 6) and a collection of QC plots describing the prevalence of those CNVs that were deemed true across bins of increasing values of sample-quality relevant metrics (LRRSD, BAFDRIFT, GCWF) as well as the distribution of those true calls with respect to measures relevant to CNV detection sensitivity (number of SNPs per call, and the level of CNV call overlap with the test locus, see Support Protocol 1). In this section we will briefly describe both. The output of basic protocol 2 can be considered the result of the entire methodology, it will contain all visually inspected CNVs. Depending on the specific application the user is working on, it may be more or less important to obtain the highest possible precision. In situations where maximum precision is required, it can be helpful for more than one analysist to inspect the same set of CNVs (basic protocol 2, steps 5–6) and then to merge the results, re-analysing together any CNVs where there was no consensus among the analysts. In any case, at the end of the process, the most users will want to have a final table containing only the true validated CNVs. This can be achieved, in R, as shown in support protocol 2 step 1 to load the table, followed by vi_res_true <- vi_res[visual_output == 1, ] to select only the true ones. The object vi_res_true can then be saved in the preferred disk location. Regarding the QC plots, we will describe the provided example, figure 4. The last two points of the troubleshooting section provide some guidance on how to interpret such plots. Following them we can see in both figure 4A and 4B that the prevalence and call size distribution of the verified deletions look unproblematic, while those of the verified duplications do not. In figure 4A the deletion prevalence is consistent with respect to LRR SD. The wider prevalence error bars observed for the group with the highest LRR SD reflects the fact that relatively few samples have LRR SD values in this range. In contrast, in figure 4A we observe a lower duplication prevalence in the high LRR SD group than in the other two LRR SD groups. This indicates that the duplication detection sensitivity in this LRR-SD group may be reduced, as prevalence should not vary depending on LRR SD. In this example the duplication prevalence error bars are narrow, indicating that the lower prevalence in this group may not be a chance finding (i.e. that we are not detecting all true duplications in samples within this LRR SD range). The solution depends on the application, in a discovery study no adjustment may be needed, however, in a prevalence study the filtering cut-off for LRR SD should be lowered, to ensure that we are able to detect true deletions and true duplications with the same efficiency. In figure 4B, the call size distribution of the deletions is unproblematic; with a sample large enough the density curve should approach a gaussian distribution. In contrast, the density curve for duplications appears to be cut at the threshold value of numsnp, indicating that some true duplications may have been filtered out in step 3c of basic protocol 2. The filtering value should in this case be lowered to ensure optimal capture of true CNVs at the locus in question.
Time Considerations
The time required to run the protocol depends on the amount of sample the user needs to process, as well as the coverage of the specific SNP array used. A collection of approximately 500,000 samples should take less than 72 hours to complete basic protocol 1. However, given the fact that most steps make strong use of parallel computation this estimate is strongly dependent on the actual computing capabilities available (i.e. the number of nodes and amount of CPU cores and RAM per node) and thus the amount of concurrent jobs possible. Basic protocol 2 (except step 5) and support protocol 1 are exploratory in nature, however, they should not require more than one day of analyst work each. Step 5 of basic protocol 2 (i.e. the visual confirmation) is the most human-labour intensive step of the entire protocol. Based on our experience, an expert analyst needs between 2 and 10 seconds on average to evaluate each putative call.
Supplementary Material
ACKNOWLEDGEMENTS:
Our research was supported by the NIH (R01 MH124789–01) but the funding source had no involvement in the design of the protocol, nor in collection, analysis or interpretation of data. While protocol is designed as a general software, it was initially conceptualised in conjunction with CNV analysis of data from The Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH).
Footnotes
CONFLICT OF INTEREST STATEMENT:
The authors declare no conflict of interests.
DATA AVAILABILITY STATEMENT:
We provide a docker image containing all software required to run the protocol on Docker Hub at https://hub.docker.com/r/sinomem/docker_cnv_protocol. The protocol is designed to handle large collections of human genetic data. For privacy reasons, all plots shown were created using simulated data. QCtreeCNV R package is available on GitHub at https://github.com/SinomeM/QCtreeCNV, the code to generate figure 4 is located in the code to generate tmp/dev.R. DeepEYE is available at https://github.com/XabierCS/eyeCNV, toy data (used in figure 3) is available in the code to generate toydata/.
LITERATURE CITED
- Bonfield JK, Marshall J, Danecek P, Li H, Ohan V, Whitwham A, … Davies RM (2021). HTSlib: C library for reading/writing high-throughput sequencing data. GigaScience, 10(2), giab007. 10.1093/gigascience/giab007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle Sánchez X, Helenius D, Bybjerg-Grauholm J, Pedersen C, Hougaard DM, Børglum AD, Nordentoft M, Mors O, Mortensen PB, Geschwind DH, Montalbano S, Raznahan A, Thompson WK, Ingason A, & Werge T (2021). Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders. JAMA Psychiatry. 10.1001/jamapsychiatry.2021.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, & Ragoussis J (2007). QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Research, 35(6), 2013–2025. 10.1093/nar/gkm076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K, Bracher-Smith M, Owen D, Kendall KM, Rees E, Pardiñas AF, … Kirov G (2019). Medical consequences of pathogenic CNVs in adults: Analysis of the UK Biobank. Journal of Medical Genetics, 56(3), 131–138. 10.1136/jmedgenet-2018-105477 [DOI] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, … Li H (2021). Twelve years of SAMtools and BCFtools. GigaScience, 10(2), giab008. 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowle M, Srinivasan A, Gorecki J, Chirico M, Stetsenko P, Short T, … Eddelbuettel D (2020). data.table: Extension of ‘data.frame’ (Version 1.13.2). Retrieved from https://CRAN.R-project.org/package=data.table
- Driscoll DA, Spinner NB, Budarf ML, McDonald-McGinn DM, Zackai EH, Goldberg RB, … Jones MC (1992). Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. American Journal of Medical Genetics, 44(2), 261–268. 10.1002/ajmg.1320440237 [DOI] [PubMed] [Google Scholar]
- Hennig C (2020). fpc: Flexible Procedures for Clustering (Version 2.2–9). Retrieved from https://CRAN.R-project.org/package=fpc [Google Scholar]
- Malhotra D, & Sebat J (2012). CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell, 148(6), 1223–1241. 10.1016/j.cell.2012.02.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, & Morgan M (2014). VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics, 30(14), 2076–2078. 10.1093/bioinformatics/btu168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2018). R: A Language and Environment for Statistical Computing.
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, … Eichler EE (2005). Segmental duplications and copy-number variation in the human genome. American Journal of Human Genetics, 77(1), 78–88. 10.1086/431652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, & Lupski JR (2002). Genome architecture, rearrangements and genomic disorders. Trends in Genetics: TIG, 18(2), 74–82. 10.1016/s0168-9525(02)02592-1 [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietiläinen OPH, Ingason A, Steinberg S, … Stefansson K (2008). Large recurrent microdeletions associated with schizophrenia. Nature, 455(7210), 232–236. 10.1038/nature07229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Bjornsdottir G, Walters GB, Jonsdottir GA, Doyle OM, Tost H, Grimm O, Kristjansdottir S, Snorrason H, Davidsdottir SR, Gudmundsson LJ, Jonsson GF, Stefansdottir B, Helgadottir I, … Stefansson K (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature, 505(7483), 361–366. 10.1038/nature12818 [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, … Korbel JO (2015). An integrated map of structural variation in 2,504 human genomes. Nature, 526(7571), 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, … Hurles ME (2008). Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nature Genetics, 40(1), 90–95. 10.1038/ng.2007.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, … Bucan M (2007). PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Research, 17(11), 1665–1674. 10.1101/gr.6861907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Symmons O, Spitz F, & Korbel JO (2013). Phenotypic impact of genomic structural variation: Insights from and for human disease. Nature Reviews Genetics, 14(2), 125–138. 10.1038/nrg3373 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We provide a docker image containing all software required to run the protocol on Docker Hub at https://hub.docker.com/r/sinomem/docker_cnv_protocol. The protocol is designed to handle large collections of human genetic data. For privacy reasons, all plots shown were created using simulated data. QCtreeCNV R package is available on GitHub at https://github.com/SinomeM/QCtreeCNV, the code to generate figure 4 is located in the code to generate tmp/dev.R. DeepEYE is available at https://github.com/XabierCS/eyeCNV, toy data (used in figure 3) is available in the code to generate toydata/.