

Graphical abstract

Keywords: Betulonic acid derivatives, Omicron, Membrane fusion, Structure–activity relationships
Abstract
The recent global Omicron epidemics underscore the great need for the development of small molecule therapeutics with appropriate mechanisms. The trimeric spike protein (S) of SARS-CoV-2 plays a pivotal role in mediating viral entry into host cells. We continued our efforts to develop small-molecule SARS-CoV-2 entry inhibitors. In this work, two sets of BA derivatives were designed and synthesized based on the hit BA-1 that was identified as a novel SARS-CoV-2 entry inhibitor. Compound BA-4, the most potent one, showed broad inhibitory activities against pOmicron and other pseudotyped variants with EC50 values ranging 2.73 to 5.19 μM. Moreover, pSARS-CoV-2 assay, SPR analysis, Co-IP assay and the cell–cell fusion assay coupled with docking and mutagenesis studies revealed that BA-4 could stabilize S in the pre-fusion step to interfere with the membrane fusion, thereby displaying promising inhibition against Omicron entry.
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that leads to the coronavirus disease 2019 (COVID-19), has rapidly spread around the world, devastating consequences for the health sector and the economy since the late December of 2019 [1], [2]. Currently, the public become more aware of the devastation caused by the emerging variants and mutations of SARS-CoV-2, as exampled by Omicron variant, which is posing a great challenge to public health and safety globally [3], [4], [5]. As of September 2022, there have been>610 million confirmed cases and 6.5 million deaths worldwide despite a ring vaccination program with the FDA-approved BNT162b2 and others, underlining the urgency for developing effective antiviral agents to prevent these lethal infections. Remdesivir [6], a RdRp inhibitor, was approved by the United States Food and Drug Administration (FDA) in May 2020 for the treatment of severe COVID-19 patients. In December 2021, a nucleoside analog molnupiravir that was originally used for influenza, was also approved by FDA [7]. The third drug approved by FDA is paxlovid, which was found to reduce the risk of hospitalization and death by 89 % in the Phase 2/3 EPIC-HR study [8]. In addition, other small-molecule drugs targeting the replication cycle of SARS-CoV-2 are currently being developed in clinic [9], [10]. For example, small-molecule inhibitors GC373 and GC376 can effectively inhibit the enzymatic activity of 3CLpro by covalent modification with the amino acid residue Cys145 of the catalytic site to display potent inhibitory potency coupled with low toxicity, which represent potential candidate drugs for the treatment of COVID-19 [11].
SARS-CoV-2 is a new member of single-stranded RNA and enveloped β-coronaviruses family, of which infection process starts from the viral entry into host cells. Evidence has shown that the spike protein (S) of SARS-CoV-2 is a “Type I” viral transmembrane glycoprotein, which plays a vital role in viral entry [12], [13]. The S protein consists of two subunits, namely S1 and S2, of which the former can recognize and bind to human angiotensin-converting enzyme 2 (ACE2) receptor of host cells through RBD domain and the latter is responsible for regulating S-mediated viral/cell membrane fusion [14], [15]. Structurally, S2 subunit is composed of fusion peptide (FP), heptapeptide repeat 1 (HR1), heptapeptide repeat 2 (HR2), transmembrane domain (TM), and cytoplasmic domain (CP) [16]. After SARS-CoV-2 S1 binds to ACE2, the S protein needs to be activated by cellular proteases to permit insertion of FP into the host membrane, the anchoring process. Subsequently, the HR1 and HR2 regions of the trimeric viral transmembrane protein interact to form a six-helix bundle, which pulls the viral and cellular membranes together and mediates fusion, thereby leading to the release of the viral genome into the cytoplasm [17]. All these steps, attachment to the cellular receptors, conformational changes of S1/S2, FP insertion, rearrangement, and 6HB formation, are critical for SARS-CoV-2 infection and more importantly, the S2 subunit sequence is more conservative than the S1 subunit sequence [18]. Collectively, the SARS-CoV-2 S2 subunit mediates viral fusion and entry, which represents the main target for the development of neutralizing antibodies, and small-molecule fusion inhibitors.
So far, multiple potential SARS-CoV-2 fusion inhibitors have been identified, which showed good antiviral activities in vivo/vitro, as exampled by HR-derived peptides EK1 and its analogs [19], niclosamide [20], bergamottin (1, Fig. 1 A) [3], clofazimine [21], and other natural products such as angeloylgomisin O (2, Fig. 1A), schisandrin B (3, Fig. 1A) [22]. For example, EK1 and its analogs have been shown to target the HR1 or HR2 domain to interact with virus-cell fusion, thereby exhibiting potent inhibition against SARS-CoV-2 and its variants in vivo [19]. Zhou and co-workers reported that bergamottin could act at multiple stages of the SARS-CoV-2 life cycle to reduce viral entry into cells by both blocking the S-mediated membrane fusion stage and inhibiting the expression of ACE2 [3]. Angeloylgomisin O and schisandrin B that were extracted from Schisandra chinensis, a plant used to treat hepatitis, were found to exhibit strong inhibitory effects on membrane fusion and show more potent antiviral activity against SARS-CoV-2 than remdesivir [22]. In addition, our group has previously conducted multiple high-throughput screens of various small-molecule libraries to identify salvianolic acid C (Sal-C, 4, Fig. 1A) [23] and estradiol (5, Fig. 1A) [24] as potential anti-SARS-CoV-2 agents, which could inhibit SARS-CoV-2 infection in vitro by blocking the formation of six-helix bundle core of S to block S-mediated membrane fusion. However, only a few SARS-CoV-2 fusion inhibitors have advanced to clinical trials up to now.
Fig. 1.


A. Chemical structures of representative small-molecule SARS-CoV-2 fusion inhibitors 1–5Fig. 1B. Chemical structures of betulinic acid 6, betulonic acid 7, the hit compound BA-1 and the lead compound BA-4.
Betulinic acid (BA, 6, Fig. 1B), a naturally occurring pentacyclic triterpene, represents a promising structure type for a wide variety of agents with good antiviral use against HIV, influenza virus, HSV and others [25], [26], [27]. For example, the BA core is present in bevirimat, an HIV maturation inhibitor, which has undergone phase 2 clinical evaluation. Interestingly, BA was found to possess anti-SARS-CoV activity in the μM range in vitro and in particular, betulonic acid (7, Fig. 1B), an oxidized analog at C-3 position of BA, exhibited improved anti-SARS-CoV potency with an EC50 of 0.63 μM [28]. Recently, a class of BA derivatives with a 1, 2, 3-triazolo-fused BA structure have been shown to be potent inhibitors of HCoV-229E nsp15 replication by Naesens and co-workers [29]. Encouraged by these results, we decided to investigate if BA and its derivatives will also have anti-SARS-CoV-2 activity in vitro.
Here, we report identification of a class of SARS-CoV-2 fusion inhibitors with a 3-O-β-chacotriosyl BA structure based on the hit BA-1. We describe their hit-to-lead modification, structure–activity relationship (SAR), and the mechanistic findings, giving rise to the lead compound BA-4 that can directly target S protein as a novel Omicron fusion inhibitor. These biological data consisted well with the binding model that we obtained by the lead compound BA-4 docking in the Omicron S protein structure, which was supported by site-specific mutation. We demonstrate that the interface in Omicron S where the lead BA-4 binds, can be as a potential target for developing Omicron and other SARS-CoV-2 fusion inhibitors.
2. Results and discussion
2.1. Chemical synthesis
Compound BA-1 was prepared according to our previous procedure [34]. The synthetic routes for title compounds BA-3 – BA-16 and amide analogs BA-N-1 as well as BA-N-2 were illustrated in Scheme 1 . Esterification of BA with benzyl bromide in the presence of potassium carbonate afforded the known intermediate 8 [35]. 3β-acetoxylup-20 (29)-ene-3, 28-diol 9 [36] was obtained from the commercially available betulin following literature procedures. The TfOH catalyzed coupling reaction between 9 and benzyl 2, 2, 2-trichloroacetimidate furnished benzyl-substituted ether 10, which then went through the hydrolysis reaction under basic conditions (LiOH) to yield the intermediate 11.
Scheme 1.

Reagents and conditions: (a) BnBr, K2CO3, DMF; (b) benzyl 2, 2, 2-trichloroacetimidate, TfOH, CH2Cl2; (c) LiOH, THF-MeOH-H2O; (d) TMSOTf, 4 Å Ms, CH2Cl2; (e) CH3ONa, MeOH; (f) PivCl, CH2Cl2; (g) (i) TMSOTf, 4 Å Ms, CH2Cl2; (ii) NaOH, MeOH-THF-H2O; (h) 10 % Pd/C, H2, MeOH-THF; (i) (i) Ac2O, DMAP, pyridine; (j) (i) various bromide alkanes, K2CO3, DMF; (ii) CH3ONa, MeOH; (k) (i) (COCl)2, CH2Cl2 (ii) R1R2N•HCl, Et3N, CH2Cl2; (iii) CH3ONa, CH3OH.
With glycosyl acceptor 8 or 11 as well as the known the glycosyl donor 2, 3, 4, 6-tetra-O-benzoyl-d-glucopyranosyl trichloroacetimidate 12 [30] in hand, TMSOTf-catalyzed glycosylations were performed to provide compound 13 or 14, followed by the hydrolysis reaction under basic conditions (CH3ONa in MeOH) to yield 3-O-β-glucopyranoside 15 or 16, respectively. Subsequently, the pivaloyl (Piv) group could be selectively installed at the 3, 6-OHs of the β-glucopyranosyl residues in 15 or 16 at a controlled low temperature to afford 17 or 18, respectively. With the glycosyl donor 2, 3, 4-tri-O-acetyl-l-rhamnopyranosyl trichloroacetimidate 19 [30] and acceptor 17 or 18, the glycosylation reaction was then performed under TMSOTf activation to provide crude trisaccharides, followed by sodium hydroxide (NaOH)-mediated deprotection to give the title compound BA-16 or BA-20, respectively. Using 10 % Pd/C as a catalyst, hydrogenolysis of the benzyl group in BA-16 or BA-20 was carried out smoothly to provide the title saponin BA-2 or BA-17, respectively. Then the intermediate 20 was obtained from BA-2 through a direct acetylation reaction with acetic anhydride, which served as the coupling partner for subsequent diversifications, respectively. Under the basic conditions, the corresponding alkyl residues were incorporated at the C-28 position of BA in 20, followed by removing all the acetyl groups using the similar method as 15 and 16 to afford the subseries BA-3 -- BA-15 (Table 2), with different hydrophobic substituents at the C-28 position of BA core. On the other hand, 20 was treated with oxalyl chloride to furnish 28‑acyl chloride, which was then condensed with appropriate amines, followed by removal of all the Ac groups with MeONa to give the corresponding target saponins BA-N-1 and BA-N-2, respectively.
Table 2.
Inhibitory activities of saponins BA-1-BA-16 against infection of 293 T-ACE2 cells by pSARS-CoV-2.
| Compound | R | EC50a (μM) | CC50b(μM) | SIc |
|---|---|---|---|---|
| BA-1 | CH3 | 4.64 ± 0.52 | 40.88 ± 0.25 | 8.81 |
| BA-2 | OH | >20.00 | NT | NT |
| BA-3 | Et | 3.70 ± 0.72 | 36.12 ± 1.05 | 9.76 |
| BA-4 | n-propyl | 3.12 ± 0.40 | 39.13 ± 0.73 | 12.54 |
| BA-5 | 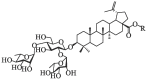 |
6.42 ± 0.20 | 76.49 ± 1.23 | 11.91 |
| BA-6 |  |
5.37 ± 0.37 | 24.36 ± 0.33 | 4.54 |
| BA-7 | 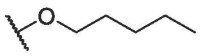 |
5.54 ± 0.81 | 16.39 ± 0.19 | 2.96 |
| BA-8 | 6.05 ± 0.38 | 47.25 ± 0.63 | 7.81 | |
| BA-9 | 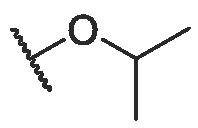 |
>20.00 | NT | NT |
| BA-10 | 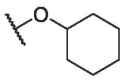 |
7.67 ± 0.31 | 51.81 ± 1.35 | 6.75 |
| BA-11 |  |
9.03 ± 0.56 | 46.12 ± 0.31 | 5.11 |
| BA-12 | 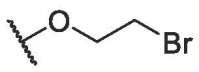 |
>20.00 | NT | NT |
| BA-13 |  |
8.61 ± 0.47 | 36.21 ± 0.75 | 4.21 |
| BA-14 | 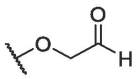 |
8.23 ± 0.55 | 84.62 ± 0.76 | 10.28 |
| BA-15 | 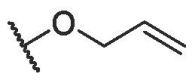 |
15.90 ± 0.82 | 75.52 ± 1.63 | 4.75 |
| BA-16 | 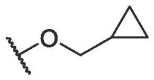 |
3.13 ± 0.42 | 12.79 ± 0.25 | 4.09 |
| Sal-C | / | 4.06 ± 0.51 | >100.00 | >24.63 |
The samples were examined in 293 T-ACE2 cells in triplicate. 293 T-ACE2 cells were incubated with test compounds and pSARS-CoV-2, and the concentration of test compound resulting in 50 % cell protection was reported as the EC50. Values are the mean of three experiments, presented as the mean ± standard deviation (SD). b50% cellular cytotoxicity concentration (CC50). cSI: selectivity index as CC50/EC50.
The following attempts were made to decorate the hydroxymethylene moiety at the C-17 position of BA-17 to expand our chemical diversity. As depicted in Scheme 2 , treatment of BA-20 with acetic anhydride as did 20 gave rise to 21, followed by hydrogenolysis of the benzyl group in 21 over palladium/carbon to yield the important intermediate 22, which served as the starting partner for subsequent diversifications, respectively. On the one hand, BA-17 was converted to the corresponding aldehyde BA-18 by reaction of 22 with the freshly prepared PCC reagent, which then undergo hydrolysis reaction with CH3ONa similarly as compounds 15 and 16. On the other hand, nucleophilic substitution of 22 with methyl iodide, followed by CH3ONa-mediated deprotection of all Ac groups gave rise to BA-19. In addition, by treating with thionyl chloride, compound 22 was readily converted into chlorides, of which all the Ac groups were then hydrolyzed using CH3ONa to afford the title saponin BA-21.
Scheme 2.

Reagents and conditions: (a) Ac2O, DMAP, pyridine; (b) 10 % Pd/C, H2, MeOH-THF; (c) (i) PCC, CH2Cl2; (ii) CH3ONa, MeOH; (d) (i) CH3I, Ag2O, ACN; (ii) CH3ONa, MeOH; (e) (i) SOCl2, CH2Cl2; (ii) CH3ONa, MeOH.
2.2. Hit discovery
Previous screening efforts focused on human CoVs (SARS-CoV, HCoV-229E) and consequently revealed these BA-based molecules with potential could inhibit SARS-CoV-2 or other variants in the SARS-CoV-2 family. Since the chacotrioside moiety, a 2, 4-branched trisaccharide residue, has been characterized as an antiviral-privileged fragment [30], [31], we supposed that introducing this moiety into BA might enhance the potency of pharmacologically active molecules. Thus, we decided to fuse this unique fragment to BA at the C-3 position, giving rise to the saponin BA-1 (Fig. 1B). Initial attempts to confirm the inhibitory effects of BA and BA-1 on infectious SARS-CoV-2 virus (wuhan-HU-1 variant) were made in a BSL-3 facility, wherein we determined their EC50 values against authentic SARS-CoV-2 in Vero-E6 cells using a full-time treatment model. Encouragingly, BA-1 proved to be a highly effective SARS-CoV-2 inhibitor with an EC50 value of 0.51 μM, which did not exhibit cytotoxicity against Vero E6, even at a concentration of 50 μM (Fig. 2 A). This data demonstrated that BA-1 might interfere only slightly with the growth of Vero E6 cells and could inhibit specifically SARS-CoV-2 in cell cultures. In contrast to BA-1, the starting compound BA was virtually inactive (Table 1 ), implying that the privileged β-chacotriosyl moiety is critical for the anti-SARS-CoV-2 activity. Briefly, these results suggested that the 3-O-β-chacotriosyl betulonic acid methyl ester BA-1 possessed excellent efficiency against SARS-CoV-2 and promising safety, which should be identified as a hit for further development.
Fig. 2.

(A) Evaluation on cytotoxicity of BA-1 and inhibitory activity against authentic SARS-CoV-2 virus (wuhan-HU-1 variant) infection in Vero-E6 cells. (B) SPR analysis of the interaction between BA-1 with SARS-CoV-2 3CL.
Table 1.
anti-SARS-CoV-2 and inhibitory activities against 3CL of BA and BA-1.
| Compound | Anti-SARS-CoV-2 EC50a (µM) |
inhibition rate against 3CL (%)b |
||
|---|---|---|---|---|
| 100 | 50 | 25 | ||
| BA | >20 | 72.2 | 51.4 | 30.2 |
| BA-1 | 0.51 ± 0.19 | 31.3 | 22.6 | 18.5 |
| Ebselen | 0.08 ± 0.01 | 98.5 | 97.6 | 96.2 |
The samples were examined in Vero-E6 cells in triplicate. Vero-E6 cells were incubated with test compounds and SARS-CoV-2 (wuhan-HU-1 variant), and the concentration of test compound resulting in 50 % cell protection was reported as the EC50. Values are the mean of three experiments, presented as the mean ± standard deviation (SD). bInhibitory rate against 3CL based on the FRET assay. Data are expressed as the mean ± SD of three experiments.
Due to its pivotal role in the SARS-CoV-2 life cycle that is involved in the viral maturation process to cleave the virus-encoded polyproteins, the 3CL protease has become a key target for discovery of anti-SARS-CoV-2 agents. It has been confirmed that BA possessed moderate inhibitory effects on SARS 3CL protease activity with an IC50 value of 10 μM [28]. SARS 3CL and SARS-CoV-2 3CL are structurally similar members of the human CoV family, sharing high homology and similarity in sequences, structures, and functions [32]. Thus, in parallel, these two compounds were also evaluated for inhibition of SARS-CoV-2 3CL protease activity based on a quenched fluorescence energy transfer (FRET) method where Ebselen was used as a positive control. As expected, BA displayed an acceptable enzyme inhibitory effect, especially at a high concentration 100 μM (Table 1). In contrast, BA-1 only exhibited weak inhibitory ability with a 31.3 % inhibition rate at 100 μM, consistent with a low equilibrium dissociation constant (K D) value of 46.3 μM (Fig. 2B) on the basis of a surface plasmon resonance (SPR) analysis, suggesting that BA-1 inhibited replication of SARS-CoV-2 through a different mechanism or pathway from BA and the positive control Ebselen. Collectively, these results demonstrate that BA-1 has a potent anti-SARS-CoV-2 activity with a high selectivity index in cell culture models but its antiviral potency is independent of inhibition toward 3CL protease.
2.3. Target identification
Evidence from several reports has illustrated that BA derivatives could effectively interfere with the fusion of the incoming virus to the host cell membrane to block HIV/H5N1/SARS and other viral entry into test cells in the low micromolar range [28], [33]. Considering the similarity between the viral fusion proteins such as gp41/HIV-1, HA2/H5N1, GP/EBOV and S2 of SARS-CoV-2, all of which play key roles in virus-induced membrane fusion, we speculated that a further anti-SARS-CoV-2 mechanism of BA-1 might be the blocking of SARS-CoV-2 entry by inhibiting the membrane fusion, thereby disrupting viral entry into the host cells. To confirm our hypothesis, BA-1 was firstly evaluated in a luciferase-expressing pseudovirus encoding SARS-CoV-2 S protein (pSARS-CoV-2) inhibition assay, which allowed for direct comparison of S protein function with a common lentiviral core and reporter [23]. Notably, we found that BA-1 exhibited the similar capability in effectively inhibiting pSARS-CoV-2 as the positive control Sal-C, a small-molecule SARS-CoV-2 entry inhibitor previously shown to bind S directly [23], and the inhibitory effect was concentration-dependent with an EC50 value of 4.64 μM (Fig. 3 A). It was interesting that BA-1 displayed not only negligible inhibition toward VSV-G pseudovirus (Fig. 3A) but also marginal cytotoxicity against 293 T-ACE2 cells (HEK293T cells overexpressing human angiotensin-converting enzyme 2) within the effective concentration range (Fig. 3B). These results highlighted that BA-1 could exert inhibitory activity against SARS-CoV-2 entry by targeting the S protein and the similarity in the potency of BA-1 between the pseudovirus and infectious virus assays supported the validity of the S/HIV-based anti-SARS-CoV-2 assay used in 293 T-ACE2 cells.
Fig. 3.

(A) Dose-response curves and EC50 of BA-1 on inhibiting the entry of SARS-CoV-2 PsV and VSV-G in 293 T-ACE2 cells. (B) Evaluation on cytotoxicity of BA-1 in 293 T. (C) BA-1 inhibited pSARS-CoV-2 infection by dose-dependent blocking of S-mediated membrane fusion.
Having identified S as the potential target, we then utilized the cell–cell fusion assay mediated by SARS-CoV-2 S protein to explore whether BA-1 had any effect on the viral membrane fusion, the critical step for entry of SARS-CoV-2 viruses into host cells for initiation of virus infection. As shown in Fig. 3C, BA-1 was identified to potently interfere with the membrane fusion of S-overexpressed-HEK293T and Vero-E6 cells in a marked dose-dependent manner at 24 h, demonstrating that its antiviral potency apparently involved action on SARS-CoV-2 S-mediated membrane fusion. Taken together, BA-1 represents a novel SARS-CoV-2 fusion inhibitor, which was selected as a promising structure for further research and optimization.
2.4. Proposed binding mode of hit to the spike protein
In view of SARS-CoV-2 S as the important target and the membrane fusion interaction between virus and host cells as the critical interruption event, we performed blind docking calculations based on the X-ray crystal structures of SARS-CoV-2 S (PDB code: 6VXX) to investigate the potential binding site. A proposed binding mode of BA-1 was established (Fig. 4 ), which indicated that BA-1 could occupy well a cavity between the S1 and S2 subunits at the entrance to a large tunnel that links with equivalent tunnels from the other monomers of the trimer at the threefold axis. As shown in Fig. 4, at the upper region of the cavity, the hydrophilic chacotriosyl residue of BA-1 made multiple stable hydrogen bonds with the backbone of residues Thr961, Leu303, Arg765, and Lys964 to result in increased interaction with S, further supporting the proposition that that the trisaccharide moiety was vital to improve antiviral activity. Hydrophobic pentacyclic triterpenoid skeleton of BA-1 occupied a large lipophilic region located in the middle of the cavity, creating a tight hydrophobic interaction with the side chain of Val772 to maintain the active conformation of BA-1.
Fig. 4.

Molecular docking of BA-1 to spike protein (PDB: 6VXX). S1 subunit, S2 subunit, and BA-1 were shown as orange ribbon, blue ribbon, magenta sticks, respectively. Green dashes in the interaction plot indicating hydrogen bond. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To further confirm the above binding mode, we conducted the single amino acid mutagenesis of pSARS-CoV-2 S to understand the molecular basis of fusion inhibition by the hit BA-1. As indicated by the preliminary mutagenesis studies (Fig. 5 ), the pSARS-CoV-2 N764A/R765A/Q957A/K964 A mutations resulted in a significant loss of potency toward BA-1 in dose-dependent fashion relative to WT S. In short, the docking result, supported by the mutagenesis studies, led us to propose a potential SARS-CoV-2-S binding pose of BA-1, which reflected some characteristics that could guide subsequent structural modification and optimization.
Fig. 5.

(A) Inhibitions of BA-1 against SARS-CoV-2 PsV and its mutants infections in 293 T-ACE2 cells, including N764A/R765A/Q957A/K964A. (B) Antiviral efficacy of BA-1 against SARS-CoV-2 PsV mutants caused by site-directed mutation, including N764A/R765A/Q957A/K964A.
2.5. Design of novel SARS-CoV-2 fusion inhibitors
The proposed mode analysis indicated that the hit BA-1 fitted well in the binding region, where important hydrogen-bond networks were observed between the chacotriosyl moiety and the cavity. However, there was still some space for further modification to fit better in the binding site. On the basis of the proposed mode, we identified a hydrophobic region under the aglycone core that is formed by D663, P665, V772, D775 and K776 residues. As depicted in Fig. 4, the C-17 position of BA-1 seemed well positioned to extend into this pocket but there is no chemical structure that can form stable interactions with this region. We reasoned that this cavity could be presumably occupied by an alternative bulky group like an ethyl ester substituent or another type of linear/ring structure to yield better intermolecular interactions to improve potency. Here, our strategy was to enhance antiviral activity by extending from the 17-position of the aglycone BA to fill the bottom area of the binding cavity. At the other side of the molecule, the β-chacotriosyl fragment moiety probably needs to be kept since it forms multiple hydrogen bonds with S protein. It is noteworthy that most of the pocket residues are conserved, which highlights the relevance of this S interface pocket for new SARS-CoV-2 fusion inhibitors design. Briefly, we attempted to improve potency further by increasing steric bulk to more completely occupy this area of the binding pocket and derive additional hydrophobic contacts, which resulted in a set of 3-O-β-chacotriosyl BA derivatives.
2.6. Sars of the BA saponins modified at 28-position
We focused our SAR campaign on investigating the alkyl groups at the C-28 position of BA, to a lesser extent, the linker at the C-17 position. Initially, we changed the ester moiety at the C-17 position of BA-1 into amide moiety as its bioisosteric surrogates (compound BA-N-1 and BA-N-2) to investigate their influence on biological activity. Unexpectedly, BA-N-1 and BA-N-2 showed significantly reduced inhibition against pSARS-CoV-2 entry relative to BA-1 (Figure S1), presumably due to unfavorable conformation. For example, BA-N-1 and BA-N-2 only exhibited about 50 % inhibition rate against pSARS-CoV-2 at high concentration 20 μM. Thus, further amide modifications incorporated at the 17-position of BA were not pursued and we turned attention to the ester linker in this study. To identify novel and potent SARS-CoV-2 entry inhibitors, we performed a screen of the above two series of 3-O-β-chacotriosyl BA derivatives BA-2--BA-16 based on the established pseudovirus model of SARS-CoV-2 (S/HIV) under low level containment (biosafety level 2) while VSVG/HIV pseudoviral transduction was used as a specificity control to exclude inhibitory effect on post-entry for HIV infection. As shown in Table 2, the variety of structural modifications described herein, especially the incorporation of bulky alkyl or aryl substituents, furnished compounds either almost equipotent or more potent against pSARS-CoV-2 virus while no effects on VSVG (Figure S2). Among these analogs, the 28-n-propyl-substituted analogue BA-4 showed the strongest inhibition toward pSARS-CoV-2 virus coupled with the highest selectivity index (SI = 12.54).
To continue probing interactions with the potential binding pocket, our initial efforts were made by changing the length, size or type of the fragments linked by ester group at the C-28 position of BA to fine-tune the hydrophobicity effects on the antiviral activities, exampled by the analogs BA-2--BA-16. As illustrated in Table 2, the free carboxylic acid BA-2 exhibited a significant decrease in potency against pSARS-CoV-2 relative to BA-1 (EC50 > 20.00 μM), probably due to limited membrane permeability. Careful examination on the chemical structure of BA-2 revealed that the carboxylic acid moiety may be responsible for its inferior cellular activities, which is known to negatively impact cell membrane permeability. However, ethyl ester BA-3 displayed a slight increased potency (EC50 = 3.63 μM) compared to BA-1. These findings reveal that the substitutions at the C-28 position of BA may play an important role in the drug-target interactions and appropriate C-28 substitutions are helpful in improving the antiviral potency, especially against SARS-CoV-2 virus. Based on the docking analysis, we inferred that this cavity could accomodate an alternative larger substituent than ethyl group and further chemical optimization at the side chain of BA-1 probably led to more potent entry inhibitors. This hypothesis gave rise to analogs BA-4--BA-8 (Table 2) with improved (BA-4 vs BA-1) or maintained inhibitory activities. Notably, augmenting the length and hydrophobicity of R substituent with n-propyl group (BA-4) led to 1.5-fold enhanced inhibitory activity (EC50 = 3.12 μM) BA-1, rendering compound BA-4 as the most potent candidate against pSARS-CoV-2 entry identified in the preliminary SAR optimization attempt. The improvement in potency may be attributed to the fact that the n-propyl group (BA-4) can occupy the binding pocket more because of its bigger bulk than methyl group to enhance the intermolecular hydrogen and hydrophobic interactions with SARS-CoV-2 S (see Fig. 9B). However, an increase in the length (BA-5, BA-6 and BA-7) or volume (BA-8) of the hydrophobic side chain via the inclusion of a n-butyl, n-pentyl, n-hexyl or isopropyl moiety at the 28-position of BA did not lead to more active compounds but coupled with different effects on cytotoxicity against 293 T-ACE2 cells. For example, the replacement of methyl moiety with n-butyl group (BA-5) or isopropyl residue (BA-8) resulted in a slight drop in antiviral potency while there was concomitant decrease in cytotoxicity against 293 T-ACE2 cells. In contrast to compound BA-5, the replacement by longer n-pentyl (BA-6) or n-hexyl (BA-7) presented a 1.7- to 2.5-fold increased toxic while keeping similar anti-SARS-CoV-2 activities. These data suggest that the side chain length at the C-28 position of BA is a critical component of both antiviral activity and selectivity index for this chemotype. In additional to these linear alkyl residues, the substitution of the methyl group in BA-1 with ring structures such as a cyclohexyl moiety generated BA-9, and surprisingly, the activity observed in pseudoviruses was entirely lost against pSARS-CoV-2 (Table 2). This data revealed that the volume size of R subsite was limited and this moiety was intolerant to cyclic alkyl chains. Collectively, these results demonstrated that a hydrophobic alkyl side chain with a length between 1 and 4 atoms at the position 28 of BA is optimal for inhibition against SARS-CoV-2 entry, which seems to accept a short and linear structure.
Fig. 9.

(A) Molecular docking of BA-4 to Omicron spike protein (PDB: 7TF8). S1 subunit, S2 subunit, and BA-4 were shown as magenta ribbon, purple ribbon, orange sticks, respectively. Yellow dashes in the interaction plot indicating hydrogen bond. Red dashes in the interaction plot indicating hydrophobic interaction. (B) Molecular docking of BA-4 to spike protein (PDB: 6VXX). S1 subunit, S2 subunit, and BA-4 were shown as orange ribbon, blue ribbon, green sticks, respectively. Yellow dashes in the interaction plot indicating hydrogen bond. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The encouraging antiviral profiles of compounds with small linear alkyl substituents, exampled by BA-3 and BA-4 prompted an examination of introduction of functional groups into the preferred ethyl or n-propyl substituent to form additional potential interactions, exampled by the analogs BA-10--BA-16 (Table 2). With the exception of 2′- hydroxyl derivative BA-12, these analogs exhibited comparable or slightly reduced antiviral activity compared to BA-4, as a result, their potency was still potent enough to emphasize the significance of the modification of side chains attached to C-28 position of BA. Among this set of derivatives, chlorine derivative BA-10 was more active than the corresponding bromine derivative BA-11 though 2.4-fold decreased potency relative to BA-4. However, insertion of a hydroxyl group into ethyl moiety (BA-12) led to a total loss of potency, of while oxidic product BA-13 could maintain comparable potency to BA-1, supporting the need for the general high hydrophobicity required for the side scaffold at the C-28 position of BA. To weaken cytotoxic activity, we incorporated in our chemical optimization campaign modifications to the preferred BA-4 based on the conformational constraints strategy, anticipated to enhance selectivity index. As shown in Table 2, the incorporation of a rigid carbon–carbon double bond (BA-14) led to a 2.6-fold reduced potency coupled with remarkably decreased cytotoxicity compared to BA-4, thereby displaying a similar SI as BA-4 but superior to BA-1. This result implied that the unsaturated fragment was tolerated on the alkyl side chain region in this set of SARS-CoV-2 entry inhibitors. In contrast to compound BA-14, the cyclized derivative BA-15 suffered a significant 4.9-fold loss of inhibition against pSARS-CoV-2 probably due to steric clashes with S protein, though it displayed reduced cytotoxicity as did BA-14. To address this gap, we hypothesized that it was better to incorporate the cyclized aromatic group through a flexible linker at the C-28 position of BA to form additional interaction with S protein. As expected, benzyl ester (BA-16) demonstrated comparable potency to BA-4 against pSARS-CoV-2 (EC50 = 3.13 μM) while showing increased cytotoxicity. As seen in Fig. S3A, the docking model demonstrated that the introduced benzyl moiety was extended to the inside of hydrophobic pockets and formed tight Van der Waals interactions with Pro665 and Val772 residues, and thus made a good functional ligand–protein interaction. In short, these results again emphasize the importance of the property, type, and size of the R substituent at the C-28 position of BA for exhibiting inhibition against pSARS-CoV-2.
2.7. Sars of the betulin derivatives
For better orientation of the tail region of BA-1 into the new hydrophobic pocket, we then shifted our focus onto the more flexible ether linker moiety at the 17-position of of BA. Firstly, we proceed to investigate the effect of the 17-COOH of BA on the anti-SARS-CoV-2 activity through reduction of the carboxyl group. As depicted in Table 3 , the betulin saponin BA-17 displayed no potencies in cellular assays as did the unsubstituted acid analogue BA-2, supporting the highly hydrophobic nature of the potential new SARS-CoV-2-S binding site. Interestingly, further optimization for potencies through oxidation of hydroxyl group at the 28 position of BA-17 was achieved in betulinicaldehyde saponin BA-18 with an EC50 value of 5.12 μM, which indicated the requirement of the hydrophobic properties at the 17 position of the aglycone skeleton to maintain highly potent inhibitory activity against pSARS-CoV-2. The observation prompted us to examine the potential of more hydrophobic modification around the hydroxyl group in betulin at the 28 positon. Therefore, a small set of 3-O-β-chacotriosyl betulin derivatives BA-19 -- BA-21 differing only in the substituent at the 17 position were picked for the preliminary SARs study. Although a bit less potent than the ester analogue BA-1, methyl ether of OH (BA-19) induced a markedly increase in inhibitory activity relative to BA-17, again highlighting that the enhancement of potency appeared to be correlated to the lipophilicity of the substituents at the 17 position. Interestingly, the introduction of benzyl group at the 28-position of BA-17 yielded compound BA-20 with moderate potency, which was 4.81-fold less active than its benzyl ester analogue BA-16. As seen in Fig. S3B, reduction of carbonyl group to methylene resulted in more flexible conformation of side chain linked at the 28 position, which would not stabilize the BA skeleton orientation and make the head chacotriosyl moiety shift toward the inside of the binding cavity, thus failing to form hydrogen bond with the critical residue Lys964. Since the hydrophobic interaction between the side chain and the new hydrophobic pocket was critical for increased potency of these SARS-CoV-2 entry inhibitors, we attempted to enhance the hydrophobic interaction by replacing hydroxyl group at the 28 position of BA-17 with one chlorine atom to produce BA-21. Surprisingly, BA-21 presented a substantial increase in SARS-CoV-2 entry inhibitory potency though it exhibited poor SI because of high toxic. One possible reason for the increased antiviral activity of BA-21 was that the incorporation of chlorine atom into the end of the side chain at the 28 position led to a greater binding interaction energy with the active pocket in the S protein, as illustrated in Fig. S3C. This result reinforced the importance of the chlorine atom as a versatile design element for lead optimization while needing to balance between the potency and cytotoxicity.
Table 3.
Inhibitory activities of saponins BA-17-BA-21 against infection of 293 T-ACE2 cells by pSARS-CoV-2.
| Compound | R | EC50a (μM) | CC50b(μM) | SIc |
|---|---|---|---|---|
| BA-17 | 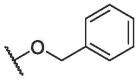 |
>20.00 | NT | NT |
| BA-18 | 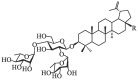 |
5.12 ± 0.31 | 29.86 ± 0.22 | 5.83 |
| BA-19 | 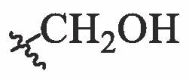 |
11.38 ± 1.41 | 34.18 ± 0.93 | 3.01 |
| BA-20 | 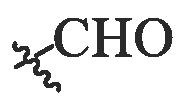 |
15.07 ± 1.05 | 96.20 ± 1.88 | 6.38 |
| BA-21 | 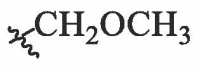 |
3.53 ± 0.18 | 15.01 ± 0.24 | 4.25 |
| Sal-C | / | 4.06 ± 0.51 | >100.00 | >24.63 |
The samples were examined in 293 T-ACE2 cells in triplicate. 293 T-ACE2 cells were incubated with test compounds and pSARS-CoV-2, and the concentration of test compound resulting in 50 % cell protection was reported as the EC50. Values are the mean of three experiments, presented as the mean ± standard deviation (SD). b50% cellular cytotoxicity concentration (CC50). cSI: selectivity index as CC50/EC50.
Taken together, through our SARs effort, we discovered that the introduction of hydrophobic side chain at the 17 position of the aglycone BA was favorable to enhance anti-SARS-CoV-2 activities as a result of increased interaction with S. In the present SARs study, the type of linker at the 17 position of the aglycone may affect the preferential binding conformation between saponins and the S protein, which in turn affects inhibitory potency toward pSARS-CoV-2. Similarly, the intensity of the hydrogen bond between the chacotriosyl residue and the binding pocket as well as hydrophobic interaction formed by the aglycone with S may also change due to the introduction of the different substituent group at the 28 position, which will lead to changes in compound activity. In addition, the potency change was probably attributed to various factors including the length, volume and type of the substituent group at the 28 position, not just the hydrophobic properties. In general, the substitution of short and small-volume hydrophobic groups did improve the inhibitory effects of these saponins. Among them, BA-4 stood out with the most potent antiviral activity in vitro and best selectivity index, rendering compound BA-4 as the lead compound against SARS-CoV-2 entry identified in the SARs optimization attempt.
2.8. Broad inhibitory activities against Omicron and other variants
More recently, the emerged Omicron and Delta variants that bear multiple mutations in their S proteins have exhibited increased adaptability and transmissibility. The good potencies of representative compounds BA-1 and BA-4 against pSARS-CoV-2 prompted us to examine the inhibitory activity of these two saponins against emerging variants such as Omicron, Delta, and other variants with N501Y, D614G, E484K, or P681H single mutation in their S proteins, respectively. As shown in Table 4 , the broad antiviral effects of BA-1 and BA-4 against these SARS-CoV-2 pseudoviruses containing multiple mutations in S protein were observed in micromole levels, implying that these newly developed BA saponins are broad-spectrum anti-SARS-CoV-2 agents that can block the S-mediated SARS-CoV-2 entry process. Notably, saponins BA-1 and BA-4 demonstrated comparable potency against Omicron pseudovirus to pSARS-CoV-2 with EC50s of 7.04 μM and 4.66 μM, which was in good agreement with the SARs. Briefly, broad and appreciable inhibition of viral entry for all pSARS-CoV-2 variants tested, along with good selectivity index, highlights the lead compound BA-4 as a potential antiviral candidate for the treatment of Omicron infections.
Table 4.
Inhibitory activities of BA-1 and BA-4 against Omicron pseudovirus and other variants.
| varians compds |
EC50a(μM) |
|||||
|---|---|---|---|---|---|---|
| Omicron | Delta | N501Y | D614G | E484K | P681H | |
| BA-1 | 7.04 ± 0.35 | 8.79 ± 0.22 | 5.84 ± 0.60 | 7.70 ± 0.41 | 8.41 ± 0.63 | 9.62 ± 0.50 |
| BA-4 | 4.66 ± 0.52 | 4.25 ± 0.37 | 2.73 ± 0.31 | 3.01 ± 0.25 | 4.75 ± 0.58 | 5.19 ± 0.86 |
The samples were examined in 293 T-ACE2 cells in triplicate. 293 T-ACE2 cells were incubated with test compounds and pSARS-CoV-2, and the concentration of test compound resulting in 50 % cell protection was reported as the EC50.
2.9. Validation of Omicron s binding
Given the robust activity of the lead compound BA-4 against Omicron, we used this virus to reveal its mechanism of action and appreciate how its anti-SARS-CoV-2 was. To explore whether the findings in the present SARS-CoV-2 study could be extended to Omicron, a similar VSV-based Omicron S protein-bearing pseudovirus (pv) was firstly used to assess the efficacy of BA-4 on virus entry. As shown in Fig. 6 A, BA-4 exhibited a dose-dependent inhibition of Omicron pv infection while no inhibition was observed on VSV-G pseudoviral transduction. Moreover, it was found that BA-4 showed strong binding affinity to S of Omicron variant with a K D value of 0.36 μM based on a SPR assay (Fig. 6B), demonstrating that BA-4 could directly target the Omicron S protein to block virus entry into hose cells.
Fig. 6.

(A) Dose-response curves and EC50 of BA-4 on inhibiting the entry of Omicron and VSV-G in 293 T-ACE2 cells. (B) SPR analysis of the interaction between BA-4 with Omicron S-trimer.
2.10. BA-4 could mediate membrane fusion of viral entry
Omicron entry into host cells can divided into two major steps: virus attachment to host cell receptor and virus-cell membrane fusion. As the lead compound BA-4 could inhibit Omicron entry into hose cells by targeting S, we further dissected which steps were blocked by BA-4. As shown in Fig. 7 A, BA-4 displayed little effect on the interaction of Omicron S1 subunit with its ACE2 receptor based on a Co-Immunoprecipitation (Co-IP) assay, the critical step for recognition and attachment of Omicron to host cells for initiation of virus infection, suggesting that BA-4 may be acting during Omicron S2 mediated fusion stage. Notably, BA-4 was able to interfere with the membrane fusion of A549 cells mediated by Omicron S in a concentration-dependent fashion (as seen in Fig. 7B), supporting our hypothesis. Interestingly, we found that BA-4 bound strongly to the Omicron S2 subunit, displaying a potent dose-dependent response, with a much higher K D value of 85.2 pM (Fig. 7C) relative to S; no specific binding to Omicron S1 subunit was found for BA-4 in the parallel experiment (Fig. 7D). Taken together, these results revealed that the lead compound BA-4 had a specific affinity to S2, and thus interfered with the viral and cell membrane fusion, by which Omicron entry into host cells could be blocked.
Fig. 7.

(A) With addition of BA-4 (20 uM), Co-IP assays showed no affection of the binding of SARS-CoV-2 S protein and ACE2 (anti-Flag). (B) BA-4 inhibited SARS-CoV-2 Omicron mutant infection via blocking Omicron protein-mediated membrane fusion. (C) SPR analysis of the interaction between BA-4 with Omicron S2. (D) SPR analysis of the interaction between BA-4 with Omicron S1.
2.11. BA-4 could target the prefusion state during viral-host fusion
Receptor engagement by RBD will induce conformation change of Omicron S2 subunit from the pre-fusion state to a post-fusion trimer-of-hairpins conformation to result in viral membrane fusion, where the 6-HB structure formed by HR1 and HR2 regions in the S2 subunit has been identified as a critical element of the trimer-of-hairpins [18]. For a better understanding of possible mechanism during the fusion of Omicron with cellular membranes treated by BA-4, we determined the biophysical change of 6-HB by using circular-dichroism (CD) spectroscopy as described before [23]. Unlike Sal-C that can target the 6-HB of SARS-CoV-2 (Figure S4), BA-4 had negligible effect on inhibiting viral 6-HB formation (Fig. 8 ), demonstrating that BA-4 exerted potent inhibitory effect on Omicron-cell membrane fusion by the different action mechanism from Sal-C. Based on these data, we speculated that a further anti-Omicron mechanism of BA-4 might be the maintaining S protein in the pre-fusion step during the fusion of virus particle into host cells to inhibit Omicron entry.
Fig. 8.

The CD curve of the SARS-CoV Omicron HR1P/HR2P complex (purple) shows a characteristic α-helix spectrum with a minimum at 208 or 222 nm. The secondary structure of 6-HB in the HR1P/HR2P mixture was unaffected by the addition of BA-4 (20 µM), as shown by the purple and red models. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To investigate the potential binding mode of our new compounds, selected BA-4 was docked into Omicron S (PDB code: 7TF8) that is physically blocked in the pre-fusion state. As depicted in Fig. 9A, at the head region, stable hydrogen bonds between the β-chacotriosyl moiety and Lys964, Thr761, Arg765, Thr302, Glu309 as well as Leu303 are formed to create a critical interaction with the Omicron S protein, which is conducive to stabilizing the pre-fusion state of S to prevent its conformational rearrangements. In the center of the binding pocket, BA aglycone makes multiple Van der Waals interactions with Arg765, Val772, Pro665 and Ile312 residues, which is helpful in maintaining the active conformation of BA-4. In the underpart region, the n-propyl side chain at the 28 position of BA forms additional hydrophobic contacts with Lys310, validating that extension of methyl side chain in BA-1 is contributing positively to binding with S. Consistent well with this result, BA-4 adopts the similar binding mode with SARS-CoV-2 S (Fig. 9B), suggesting that there is a general similarity between the structure of the SARS-CoV-2 S binding pocket and that of other variants bearing S mutants. While a hydrophobic interaction between Lys310 in the binding cacity and the n-propyl residue appears to be weakened, both chacotriosyl moiety and BA skeleton of BA-4 are able to be involved in multiple similar interactions with Lys964, Leu303, and other residues in this corresponding hydrophobic pocket as that with Omicron S. Thus, we hypothesize that the observed broad inhibition against pSARS-CoV-2 Omicron and other variants maybe results from the similar binding mode in this region with corresponding S protein. Taken together, the high conservation of residues located in this binding cavity among different SARS-CoV-2 strains, makes this cavity an ideal target for designing novel SARS-CoV-2 fusion inhibitors that can disrupt the the viral and cell membrane fusion to display broad antiviral activities.
2.12. Site-specific mutation of Omicron-S supports s binding as blocking inhibition of viral entry
Based on the CD spectroscopy and docking analysis, the potential mechanism of antiviral activity of the lead compound BA-4 against SARS-CoV-2 is more intriguing as BA-4-binding site is physically blocked in the Omicron-S prefusion state. We next used the single amino acid mutagenesis of pOmicron S to confirm our hypothesis, where the representative residues K964 and R765 in the fusion loop of conserved S2 subunit were evaluated. When compared to WT Omicron, pOmicron S mutant K964A caused a right shift and an over 2.5-fold loss of potency in response to BA-4 (Fig. 10 A). Similarly, the pOmicron S mutant R765A showed only a modest right shift of the dose–response curve, possibly due to weaker hydrogen bond between R765 and S relative to K964 as seen in Fig. 9A. Furthermore, the similar trend of the response to BA-1 in the pOmicron S mutant K964A or R765A was observed (Fig. 10B). The SARs and docking, supported by the mutagenesis studies, confirmed the binding of BA-4 to the site near the key residue K964 in the binding cavity between the attachment (S1) and fusion (S2) subunits.
Fig. 10.

(A) Inhibitions of BA-4 against SARS-CoV-2 PsV mutants caused by site-directed mutation, including R765A, K964A. (B) Inhibitions of BA-1 against SARS-CoV-2 PsV mutants caused by site-directed mutation, including R765A, K964A.
2.13. Compound BA-4 exhibited promising liver microsomes, intestinal S9-UDPGA and stability in mouse plasma
Given that the lead compound BA-4 displayed braod and promising anti-SARS-CoV-2 activities in vitro, we further evaluated the stability of BA-4 in vitro metabolic stability in mouse liver, mouse intestinal S9-UDPGA and in mouse plasma, respectively. First, we evaluated the metabolic stability of BA-4 in a mouse liver microsomes assay while propafenone with moderate metabolic stability was used as the control compound. As depicted in Table 5 , compound BA-4 displayed acceptable metabolic stability with a half-life value of 16.1 min in mouse liver microsomes, which was superior to 6.8 min of propafenone. Meanwhile, BA-4 also exhibited reasonable clearance rates with the intrinsic clearance (CL) value of 59.3 μL/min/mg, which was 4-fold lowe than that of propafenone in the same assay (CL = 201.6 μL/min/mg). In addition, the stability of BA-4 in mouse intestinal S9-UDPGA was also evaluated where clozapine was tested for comparison. Notably, 35 showed promising stability in mouse intestinal S9-UDPGA with higher half-life values of 77.8 min and lower CL value of 14.1 μL/min/mg compared to that of microsomal stability, though was inferior to those of clozapine.
Table 5.
Metabolic stability in the presence of mouse liver microsomes and stability in the intestinal S9-UDPGA of BA-4.
| compd | mouse liver microsomes |
mouse S9-UDPGA |
||
|---|---|---|---|---|
| T1/2a (min) |
CLint(mic)b (μL/min/mg) |
T1/2a (min) |
CLint(in vitro)b (μL/min/mg) |
|
| BA-4 | 16.1 | 59.3 | 77.8 | 14.1 |
| propafenone | 6.8 | 201.6 | / | / |
| clozapine | / | / | >145 | <6.8 |
T1/2 is the half-life and CLint (mic) is the intrinsic clearance. bCLint (mic) = 0.693/half-life/mg microsome protein per milliliter.
Then we examined the stability of BA-4 in mouse plasma using propantheline bromide as a reference. As shown in Table 6 , BA-4 exhibited moderate stability in mouse plasma, displaying an approximately 50 % compound retention after 120 min incubation, which was superior to that of the reference propantheline bromide. Collectively, these results reveal that the lead compound BA-4 possesses acceptable metabolic stability in mouse liver microsomes and stability in mouse plasma as well as reasonable S9-UDPGA, which meets the basic requirements of ADMET.
Table 6.
Stability of compound BA-4 in mouse plasma.
| incubation time (min) |
BA-4 remained | Propantheline bromide remained |
|---|---|---|
| mouse plasma (%) | mouse plasma (%) | |
| 0 | 100 | 100 |
| 30 | 73.6 | 45.5 |
| 60 | 61.3 | 18.2 |
| 90 | 53.4 | 10.3 |
| 120 | 47.5 | 2.0 |
3. Conclusions
This study presented here discovered a hit compound BA-1 that showed good inhibition against infectious and pseudotyped SARS-CoV-2 virus by directly targeting the S protein. Based on the structure BA-1, rational drug design and subsequent chemical optimization resulted in the development of the lead compound BA-4, as a novel Omicron fusion inhibitor. Utilizing the SPR assay, CD spectroscopy, docking and mutagenesis studies, we confirmed that the anti-Omicron mechanism of BA-4 was through directly binding to the S protein, which was capable of stabilizing S in the pre-fusion step to block Omicron entry into host cells. Moreover, the lead compound BA-4 was found to have a broad-spectrum entry inhibition against all SARS-CoV-2 variants tested and display favorable SI values. Overall, BA-4 represents a novel and potent Omicron fusion inhibitor and justifies further development as a potential candidate for treatment of SARS-CoV-2 infections.
4. Experimental
4.1. Chemistry
Solvents were purified in a conventional manner. Thin layer chromatography (TLC) was performed on precoated E. Merck silica gel 60 F254 plates. Flash column chromatography was performed on silica gel (200–300 mesh, Qingdao, China). 1H NMR and 13C NMR spectra were taken on a JEOL JNM-ECP 600 spectrometer with tetramethylsilane as an internal standard, and chemical shifts are recorded in ppm values. Mass spectra were recorded on a Q-TOF Global mass spectrometer.
4.1.1. 28-(Benzyloxy)-3β-acetoxy-lup-20 (29)-ene-3-ol (10)
To a solution of 9 (3.00 g, 6.19 mmol), benzyl 2, 2, 2-trichloroacetimidate (3.42 g, 13.62 mmol) and 4 Å molecular sieves in dry CH2Cl2 (50 mL) was added TfOH (0.14 g, 0.93 mmol) at −10 ℃ under N2 atmosphere. The reaction mixture was kept at −10 °C for 2 h and warmed to room temperature for 1 h. After the reaction was complete detected by TLC, triethylamine was added to quench the reaction. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleumether-EtOAc-CH2Cl2, 30:1:1) to yield 10 (3.16 g, 89 %) as a white solid. 1H NMR (600 MHz, CDCl3): δ 7.45–7.20 (m, 5H, Ar-H), 4.64 (s, 1H, C = CH2-1), 4.56 (s, 2H, Ar-CH2), 4.48 (s, 1H, C = CH2-2), 3.51 (d, 1H, J = 8.9 Hz), 3.09 (d, 1H, J = 8.9 Hz), 2.35 (td, 1H, J = 10.6, 5.6 Hz), 2.03 (s, 3H, COCH3), 1.66 (s, 3H, CH3), 0.94 (s, 6H, 2 × CH3), 0.84 (s, 9H, 3 × CH3), 0.79 (d, 1H, J = 9.5 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 171.14, 150.85 (C-20), 139.13, 128.43 (two), 127.67 (two), 127.56, 109.64 (C-29), 81.07, 73.50, 68.15, 55.47, 50.38, 48.94, 48.10, 47.38, 42.71, 40.96, 38.47, 37.91, 37.54, 37.16, 34.98, 34.21, 30.10, 30.05, 28.07, 27.22, 25.25, 23.81, 21.44, 20.94, 19.18, 18.31, 16.62, 16.27, 15.89, 14.86. HRMS (ESI) m/z: calcd for C39H59O3 [M + H]+, 575.4464; found, 575.4478.
4.1.2. 28-(Benzyloxy)-3β-hydroxy-lup-20 (29)-ene-3-ol (11)
To a solution of 10 (3.16 g, 5.50 mmol) and LiOH (2.87 g, 0.12 mmol) in THF-MeOH-H2O (90 mL) and then the reaction mixture was stirred at 50 ℃ for 12 h. After the reaction was complete detected by TLC, 1 M HCl was added to adjust pH = 7. The mixture was concentrated in vacuo. The residue was dissolved in EtOAc (150 mL), then extracted with water (3 × 50 mL) and brine (3 × 50 mL). The combined organic layer was concentrated under vacuum after drying over Na2SO4. The resulting crude was then purified by column chromatography (CH2Cl2-MeOH, 30:1) to give 11 (2.1 g, 72 %) as a white solid. 1H NMR (600 MHz, CDCl3): δ 7.37–7.31 (m, 5H, Ar-H), 4.64 (s, 1H, C = CH2-1), 4.55 (s, 2H, Ar-CH2), 4.48 (s, 1H, C = CH2-2), 3.51 (d, 1H, J = 8.9 Hz), 3.09 (d, 1H, J = 8.9 Hz), 2.35 (td, 1H, J = 10.7, 5.6 Hz), 1.66 (s, 3H, CH3), 0.96, 0.94, 0.84, 0.79, 0.75 (each s, each 3H, CH3), 0.66 (d, 1H, J = 9.4 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 150.85 (C-20), 139.11, 128.43 (two), 127.67 (two), 127.56, 109.61 (C-29), 79.07, 73.50, 68.14, 55.38, 50.47, 48.96, 48.07, 47.38, 42.71, 40.94, 38.97, 38.79, 37.55, 37.24, 34.98, 34.28, 30.11, 30.06, 28.11, 27.50, 27.23, 25.29, 20.92, 19.20, 18.43, 16.20, 15.89, 15.49, 14.89. HRMS (MALDI) m/z: calcd for C37H56O2Na [M + Na]+, 555.4178; found, 555.4192.
4.1.3. General procedure for 13 and 14
To a solution of 8 or 11 (1 eq), 2, 3, 4, 6-tetra-O-benzoyl-d-glucopyranosyl trichloroacetimidate 12 (1.5 eq) and 4 Å molecular sieves in dry CH2Cl2 (30 mL) was added TMSOTf (0.15 eq) at −5 ℃ under N2 atmosphere. The reaction mixture was kept at −5 °C for 0.5 h and then warmed to room temperature for 1 h. After the reaction was complete detected by TLC, the reaction was quenched with trimethylamine. The mixture was filtered and the filtrate was concentrated under vacuum. Then the residue was purified by silica gel column chromatography (petroleumether-EtOAc-CH2Cl2, 8:1:1) to produce 13 or 14 as a white solid, respectively.
4.1.3.1. Benzyl-3β-O-(2, 3, 4, 6-tetra-O-benzoyl-β-d-glucopyranosyl)-lup-20 (29)-ene-28-oic acid (13)
Compound 13 was obtained as a white solid. 1H NMR (600 MHz, CDCl3): δ 7.95–7.65 (m, 15H, Ar-H), 7.46–7.09 (m, 10H, Ar-H), 5.77 (t, 1H, J = 9.7 Hz, H-3′), 5.45 (t, 1H, J = 9.5 Hz, H-4′), 5.00 (d, 1H, J = 12.2 Hz, Ar-CH2-1), 4.95 (d, 1H, J = 12.3 Hz, Ar-CH2-2), 4.72 (s, 1H, C = CH2-1), 4.70 (s, 1H, C = CH2-2), 4.64–4.59 (m, 1H, H-1′), 4.51–4.48 (m, 1H, H-2′), 4.46 (dd, 1H, J = 11.9, 3.4 Hz, H-6′-1), 4.40 (dd, 1H, J = 11.9, 6.6 Hz, H-6′-2), 4.06–3.96 (m, 1H, H-5′), 2.92 (dd, 1H, J = 11.7, 4.5 Hz, H-3), 2.13 (d, 1H, J = 12.4 Hz), 1.57, 0.77, 0.59, 0.56, 0.52, 0.47 (each s, each 3H, CH3), 0.40 (d, 1H, J = 9.7 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 175.85 (C-28), 166.12, 165.96, 165.40, 165.11, 150.69 (C-20), 136.53, 133.56, 133.32, 133.18, 130.16, 129.93 (two), 129.84 (four), 129.82 (three), 129.74, 129.66, 129.47, 128.91, 128.85, 128.58 (three), 128.52 (two), 128.47 (three), 128.37 (two), 128.34 (three), 128.16 (two), 109.64 (C-29), 103.31 (C-1′), 90.81, 73.05, 72.22, 72.05, 70.37, 65.83, 63.52, 56.62, 55.65, 50.61, 49.52, 46.97, 42.41, 40.68, 38.93, 38.67, 38.23, 37.03, 36.86, 34.30, 32.19, 30.71, 29.60, 27.58, 26.06, 25.63, 20.92, 19.60, 18.12, 16.11, 16.06, 15.87, 14.69. HRMS (ESI) m/z: calcd for C71H81O12 [M + H]+, 1125.5728; found, 1125.5750.
4.1.3.2. 28-(Benzyloxy)-3β-O-(2, 3, 4, 6-tetra-O-benzoyl-β-d-glucopyranosyl)-lup-20 (29)-ene-3-ol (14)
Compound 14 was obtained as a white solid. 1H NMR (600 MHz, CDCl3): δ 8.05–7.80 (m, 10H, Ar-H), 7.56–7.22 (m, 15H, Ar-H), 5.91 (t, 1H, J = 9.9, H-3′), 5.56 (t, 1H, J = 10.1 Hz, H-4′), 4.86 (s, 1H, C = CH2-1), 4.84 (s, 1H, C = CH2-2), 4.68–4.63 (m, 1H, H-1′), 4.62–4.56 (m, 3H, H-2′, Ar-CH2), 4.58–4.49 (m, 1H, H-6′-1), 4.50–4.42 (m, 1H, H-6′-2), 4.19–4.10 (m, 1H, H-5′), 3.49 (d, 2H, J = 8.9 Hz, C-CH2), 3.07 (dd, 1H, J = 11.2, 6.6 Hz, H-3), 2.40–2.31 (m, 1H), 1.69, 0.90, 0.78, 0.72, 0.67, 0.61 (each s, each 3H, CH3), 0.54 (d, 1H, J = 10.1 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 166.05, 165.90, 165.34, 165.06, 150.76 (C-20), 138.98, 133.48, 133.24, 133.10, 133.08, 129.87 (two), 129.78 (four), 129.75 (three), 129.71, 129.44, 128.88, 128.82, 128.45 (two), 128.38 (three), 128.32 (four), 127.56 (three), 127.47, 109.49 (C-29), 103.26 (C-1′), 90.70, 73.40, 73.01, 72.19, 72.01, 70.33, 68.02, 63.46, 55.54, 50.36, 48.86, 47.92, 47.28, 42.56, 40.80, 38.87, 38.59, 37.41, 36.76, 34.89, 34.12, 30.01, 27.52, 27.09, 25.99, 25.21, 20.79, 19.20, 18.06, 15.98 (two), 15.74, 14.72. HRMS (ESI) m/z: calcd for C71H81O11 [M + H]+, 1111.5935; found, 1111.5947.
4.1.4. Benzyl 3β-O-(d-glucopyranosyl)-lup-20 (29)-ene-28-oic acid (15)
To a stirred solution of compound 13 (12.11 g, 10.7 mmol) in CH2Cl2 (60 mL) and MeOH (60 mL), CH3ONa was added until pH = 10. Stirring was continued overnight at room temperature. Then, the mixture was neutralized with Dowex 50 × 8 (H+) resin until pH = 7, filtered and then evaporated to remove excess solvent under vacuum. The residue was purified by silica gel column chromatography (CH2Cl2-MeOH, 10:1) to give 15 (7.05 g, 92 %) as a white solid. 1H NMR (600 MHz, CD3OD): δ 7.42–7.29 (m, 5H, Ar-H), 5.16 (d, 1H, J = 12.1 Hz, Ar-CH2-1), 5.08 (d, 1H, J = 12.1 Hz, Ar-CH2-2), 4.70 (s, 1H, C = CH2-1), 4.59 (s, 1H, C = CH2-2), 4.30 (d, 1H, J = 7.8 Hz, H-1′), 3.90 (s, 1H), 3.83 (dd, 1H, J = 11.9, 2.3 Hz, H-6′-1), 3.67 (dd, 1H, J = 11.9, 5.2 Hz, H-6′-2), 3.38–3.27 (m, 1H, H-5), 3.19–3.09 (m, 1H), 3.07–2.96 (m, 1H, H-3), 2.28–2.21 (m, 2H), 1.68, 1.02, 0.97, 0.83, 0.82, 0.74 (each s, each 3H, CH3), 0.71 (d, 1H, J = 9.7 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.06 (C-28), 151.67 (C-20), 137.90, 129.61 (two), 129.54 (two), 129.23, 110.40 (C-29), 106.73 (C-1′), 90.73, 78.18, 77.58, 75.57, 71.50, 66.80, 62.71, 57.77, 57.09, 51.90, 50.60, 48.44, 43.49, 41.88, 40.25, 39.55, 38.00, 37.88, 35.53, 33.13, 31.62, 30.69, 28.43, 27.64, 27.61, 27.16, 26.82, 22.06, 19.65, 19.26, 16.86, 16.62, 15.22. HRMS (ESI) m/z: calcd for C43H65O8 [M + H]+, 709.4679; found, 709.4691.
4.1.5. 28-(Benzyloxy)-3β-O-(d-glucopyranosyl)-lup-20 (29)-ene-3-ol (16)
Compound 16 was obtained from 14 as a white solid using the similar method as 15. 1H NMR (600 MHz, CD3OD): δ 7.32–7.00 (m, 5H, Ar-H), 4.60 (s, 1H, C = CH2-1), 4.50 (s, 1H, C = CH2-2), 4.34 (dd, 1H, J = 12.3, 6.8 Hz, H-6′-1), 4.24 (d, 1H, J = 7.7 Hz, H-1′), 3.90–3.72 (m, 1H, H-6′-2), 3.67–3.40 (m, 2H), 3.21–3.04 (m, 6H), 1.92 (s, 2H), 1.60 (s, 3H, CH3), 0.97 (s, 6H, 2 × CH3), 0.76 (s, 9H, 3 × CH3), 0.65 (d, 1H, J = 10.0 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 150.38 (C-20), 138.82, 128.04 (two), 127.60 (two), 127.57, 108.98 (C-29), 105.36 (C-1′), 89.38, 76.86, 76.23, 74.25, 72.97, 70.19, 67.69, 67.53, 61.40, 55.68, 50.38, 48.74, 42.33, 40.69, 38.90, 38.62, 37.43, 36.61, 34.07, 33.92, 29.83, 29.66, 27.10, 26.91, 25.82, 25.17, 20.62, 17.93, 15.53, 15.48, 15.22, 14.02. HRMS (MALDI) m/z: calcd for C43H66O7Na [M + Na]+, 717.4706; found, 717.4732.
4.1.6. General procedure for 17 and 18
To a stirred solution of 15 or 16 (1.00 mmol) in 30 mL of dry pyridine and CH2Cl2 (V: V = 1: 1), PivCl (5.00 mmol) was added slowly to the mixture at −15 ℃ under argon atmosphere. Stirring was continued for 12 h at that temperature and then the reaction was quenched with CH3OH. Excess solvent was removed in vacuo. The residue was extracted with dichloromethane and washed with saturated NaHCO3 solution and brine. The organic layer was dried over Na2SO4 and concentrated under vacuum to furnish a crude product that was further purified by column chromatography (petroleumether-EtOAc-CH2Cl2, 8:1:1) to produce 17 or 18, respectively.
4.1.6.1. Benzyl-3β-O-(3, 6-di-O-pivaloyl-β-d-glucopyranosyl)-lup-20 (29)-ene-28-oic acid (17)
Compound 17 was obtained in 84 % yield as white solid. 1H NMR (600 MHz, CDCl3): δ 7.39–7.28 (m, 5H, Ar-H), 5.14 (d, 1H, J = 12.3 Hz, Ar-CH2-1), 5.08 (d, 1H, J = 12.3 Hz, Ar-CH2-2), 4.91–4.81 (m, 1H), 4.72 (s, 1H, C = CH2-1), 4.60 (s, 1H, C = CH2-2), 4.45 (dd, 1H, J = 11.8, 2.2 Hz, H-6′-1), 4.39 (d, 1H, J = 7.8 Hz, H-1′), 4.18 (dd, 1H, J = 12.0, 7.2 Hz, H-6′-2), 3.62–3.50 (m, 1H, H-5′), 3.45 (t, 1H, J = 9.4 Hz), 3.12 (dd, 1H, J = 11.9, 4.5 Hz), 3.03 (dd, 1H, J = 10.9, 4.4 Hz, H-3), 2.27 (d, 2H, J = 12.3 Hz), 1.68 (s, 3H, CH3), 1.24 (s, 9H, C(CH3)3), 1.20 (s, 9H, C(CH3)3), 0.96, 0.93, 0.79, 0.78, 0.74 (each s, each 3H, CH3), 0.66 (d, 1H, J = 9.2 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 180.42, 178.72 (C-28), 175.92, 150.78 (C-20), 136.57, 128.61 (two), 128.36 (two), 128.18, 109.67 (C-29), 104.84 (C-1′), 90.54, 78.07, 74.21, 72.76, 70.26, 65.86, 63.93, 56.67, 55.72, 50.61, 49.55, 47.04, 42.47, 40.76, 39.17, 38.70, 38.26, 36.99, 34.34, 32.21, 30.72, 29.66, 28.29, 27.20 (two), 26.06, 25.65, 20.98, 19.54, 18.27, 16.59, 16.21, 15.94, 14.74. HRMS (ESI) m/z: calcd for C53H81O10 [M + H] +, 877.5830; found, 877.5862.
4.1.6.2. 28-(Benzyloxy)-3β-O-(3, 6-di-O-pivaloyl-β-d-glucopyranosyl)-lup-20 (29)-ene-3-ol (18)
Compound 18 was obtained as a white solid. 1H NMR (600 MHz, CDCl3): δ 7.41–7.18 (m, 5H, Ar-H), 4.89–4.87 (m, 1H), 4.65 (d, 1H, J = 2.5 Hz), 4.61–4.52 (m, 2H, C = CH2), 4.47 (d, 1H, J = 6.1 Hz), 4.47–4.41 (m, 1H), 4.40 (d, 1H, J = 7.8 Hz, H-1′), 4.18 (dd, 1H, J = 11.8, 7.2 Hz, H-6′-1), 3.62–3.40 (m, 4H), 3.17–3.06 (m, 1H, H-3), 2.39 (d, 1H, J = 12.9 Hz), 1.67 (s, 3H, CH3), 1.24 (s, 9H, C(CH3)3), 1.20 (s, 9H, C(CH3)3), 0.97, 0.93, 0.83 (each s, each 3H, CH3), 0.80 (s, 6H, 2 × CH3), 0.67 (d, 1H, J = 10.0 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 180.29, 178.62, 150.83 (C-20), 138.99, 128.33 (two), 127.56 (two), 127.47, 109.47 (C-29), 104.73 (C-1′), 90.38, 77.95, 74.12, 73.39, 72.70, 70.14, 68.01, 63.83, 55.56, 50.32, 48.86, 47.96, 47.29, 42.58, 40.84, 39.07 (two), 38.84, 38.57, 37.42, 36.85, 34.12, 29.99, 28.18, 27.17 (four), 27.10 (five), 25.95, 25.19, 20.81, 19.14, 18.18, 16.48, 16.04, 15.78, 14.73. HRMS (ESI) m/z: calcd for C53H83O9 [M + H] +, 863.6037; found, 863.6051.
4.1.7. General procedure for BA-16 and BA-20
To a mixture of 17 or 18 (1.0 mmol) and 4 Å molecular sieves in dried CH2Cl2 (20 mL) at −40 °C under argon was added TMSOTf (0.20 mmol), followed by a solution of the 2, 3, 4-tri-O-acetyl-l-rhamnopyranosyl trichloroacetimidate 19 (5.00 mmol) in dry CH2Cl2 (5 mL). After stirring at − 40 °C for 3 h, the reaction mixture was warmed to 0 °C and stirred for 5 h under argon. After the reaction was complete detected by TLC, the reaction was quenched with Et3N. The solid was filtered, and the filtrate was concentrated in reduced pressure and then purified by column chromatography (petroleum ether-EtOAc, 1:1) to afford the crude trisaccharide product. Subsequently, to a stirred solution of this crude product in 20 mL THF and CH3OH (V:V = 1:1), 4 M NaOH (10 mL) was added. After stirred at 45 °C for 10 h, 1 M HCl was added to adjust pH = 7. The resulting precipitate was filtered off and washed with CH3OH, and then concentrated under vacuum. The obtained crude product was further purified by column chromatography, eluting with CH2Cl2/CH3OH mixtures, with gradient from 8:1 to 4:1, to furnish the target compound BA-16 or BA-20, respectively.
4.1.7.1. Benzyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-16)
Compound BA-16 was obtained as a white powder. 1H NMR (600 MHz, CD3OD): δ 7.42–7.30 (m, 5H, Ar-H), 5.35 (d, 1H, J = 1.7 Hz, Rha-H-1), 5.17 (d, 1H, J = 12.1 Hz, Ar-CH2-1), 5.09 (d, 1H, J = 12.1 Hz, Ar-CH2-2), 4.82 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.70 (s, 1H, C = CH2-1), 4.60 (s, 1H, C = CH2-2), 4.41 (d, 1H, J = 7.7 Hz, H-1′), 4.11 (s, 1H), 4.03–3.93 (m, 2H), 3.92–3.90 (m, 2H), 3.90–3.79 (m, 1H, H-5′), 3.75 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.68 (d, 1H, J = 3.7 Hz), 3.63 (dd, 1H, J = 9.6, 3.2 Hz, Rha-H-3), 3.57–3.53 (m, 2H), 3.48–3.33 (m, 2H), 3.34–3.25 (m, 2H), 3.12 (dd, 1H, J = 11.7, 4.4 Hz, H-3), 3.05–2.96 (m, 1H), 2.29–2.35 (m, 1H), 1.68 (s, 3H, CH3), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.02 (s, 3H, CH3), 0.97 (s, 3H, CH3), 0.83 (s, 6H, 2 × CH3), 0.74 (s, 3H, CH3), 0.70 (d, 1H, J = 9.8 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.16 (C-28), 151.71 (C-20), 137.88, 130.21, 129.93, 129.61 (two), 129.55 (two), 129.25, 110.37 (C-29), 105.48 (C-1′), 103.02 (Rha-C-1), 101.98 (Rha-C-1), 90.40, 80.29, 79.19, 78.10, 76.40, 73.91, 73.66, 72.41, 72.10, 71.97, 70.72, 69.99, 66.83, 61.94, 57.80, 57.39, 51.96, 50.62, 47.81, 43.49, 41.89, 40.32, 39.58, 38.01, 37.87, 35.53, 33.12, 31.62, 30.68, 28.39, 27.35, 26.85, 22.05, 19.61, 19.24, 18.03, 17.90, 16.99, 16.92, 16.59, 15.18. HRMS (ESI) m/z: calcd for C55H84O16Na [M + Na]+, 1023.5657; found, 1023.5623.
4.1.7.2. 28-(Benzyloxy)-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-3-ol (BA-20)
Compound BA-20 was obtained as a white powder. 1H NMR (600 MHz, CD3OD): δ 7.39–7.21 (m, 5H, Ar-H), 5.37 (d, J = 1.7 Hz, 1H, Rha-H-1), 4.86 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.72 (s, 1H, C = CH2-1), 4.66 (s, 1H, C = CH2-2), 4.58 (d, 2H, J = 12.5 Hz, Ar-CH2), 4.43 (d, 1H, J = 7.9 Hz, H-1′), 4.40 (d, 1H, J = 2.3 Hz), 4.01–3.93 (m, 2H), 3.95–3.87 (m, 1H, H-5′), 3.84 (t, 1H, J = 9.4 Hz, Rha-H-4), 3.76 (dd, 1H, J = 9.7, 3.5 Hz, Rha-H-3), 3.66 (dd, 1H, J = 9.7, 3.6 Hz, Rha-H-3), 3.66–3.58 (m, 3H), 3.60–3.48 (m, 3H), 3.48–3.35 (m, 3H), 3.15–3.09 (m, 1H, H-3), 2.45–2.33 (m, 2H), 1.98 (s, 1H), 1.67 (s, 3H, CH3), 1.26 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.02 (s, 3H, CH3), 0.96 (s, 3H, CH3), 0.83 (s, 6H, 2 × CH3), 0.82 (s, 3H, CH3), 0.70 (d, 1H, J = 9.5 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 150.40 (C-20), 138.78, 128.03 (two), 127.66 (two), 127.31, 108.92 (C-29), 104.10 (C-1′), 101.60 (Rha-C-1), 100.55 (Rha-C-1), 94.36, 89.03, 78.91, 77.83, 76.72, 75.03, 72.92, 72.83, 72.53, 72.29, 71.05, 70.74, 70.62, 69.33, 68.61, 67.82, 67.43, 60.56, 55.95, 50.43, 42.29, 40.67, 38.96, 38.89, 37.42, 36.59, 34.51, 34.02, 29.75, 29.60, 27.02, 26.87, 25.98, 25.16, 20.57, 18.07, 17.88, 16.75, 16.65, 16.53, 15.63, 15.50, 15.11, 13.93. HRMS (MALDI) m/z: calcd for C55H86O15Na [M + Na]+, 1009.5864; found, 1009.5882.
4.1.8. General procedure for BA-2 and BA-17
To a stirred solution of BA-16 or BA-20 (1.00 mmol) in 20 mL of dry methanol and tetrahydrofuran (V: V = 1:1), 10 % Pd/C (100 mg) was added at r.t. under argon atmosphere. Then the solution was stirred at room temperature for 12 h under hydrogen atmosphere. The mixture was filtered and concentrated under reduced pressure. The resultant crude material was purified by column chromatography (CH2Cl2-CH3OH, 5:1) to afford the title compound BA-2 or BA-17 as a white solid, respectively.
4.1.8.1. 3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-2)
Saponin BA-2 was obtained as a white powder. 1H NMR (600 MHz, CD3OD): δ 5.38 (s, 1H, Rha-H-1), 4.86 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.70 (s, 1H, C = CH2-1), 4.59 (s, 1H, C = CH2-2), 4.42 (d, 1H, J = 7.6 Hz, H-1′), 4.03–3.96 (m, 2H), 3.98–3.85 (m, 1H, H-5′), 3.79 (t, 1H, J = 10.0 Hz, Rha-H-4), 3.67–3.63 (m, 2H), 3.57 (t, 1H, J = 9.8 Hz, Rha-H-4), 3.48–3.37 (m, 1H), 3.35–3.32 (m, 2H), 3.23–3.19 (m, 4H), 3.14 (dd, 1H, J = 11.5, 4.2 Hz, H-3), 3.05–3.03 (m, 1H), 2.23 (d, 1H, J = 12.0 Hz), 1.69 (s, 3H, CH3), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.0 Hz, Rha-H-6), 1.03, 1.00, 0.96, 0.86, 0.83 (each s, each 3H, CH3), 0.76–0.70 (m, 1H, H-5); 13C NMR (151 MHz, CD3OD): δ 172.73 (C-28), 151.99 (C-20), 110.19 (C-29), 105.48 (C-1′), 102.81 (Rha-C-1), 101.77 (Rha-C-1), 90.38, 80.07, 79.13, 78.02, 76.38, 74.16, 73.84, 73.63, 73.02, 72.40 (two), 72.07, 71.97, 70.58, 69.89, 69.09, 61.83, 57.53, 57.38, 51.98, 50.37, 43.53, 41.90, 40.32 (two), 39.55, 38.01 (two), 35.57, 31.68, 30.83, 28.37, 27.36, 26.86, 22.07, 19.57, 19.26, 18.01, 17.90, 16.98, 16.93, 16.68, 15.15. HRMS (ESI) m/z: calcd for C48H77O16 [M + H]+, 909.5290; found, 909.5277.
4.1.8.2. 3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-3, 28-diol (BA-17)
Compound BA-17 was obtained as a white powder. 1H NMR (600 MHz, CD3OD): δ 5.16 (s, 1H, Rha-H-1), 4.81 (s, 1H, Rha-H-1), 4.47 (s, 1H, C = CH2-1), 4.35 (s, 1H, C = CH2-2), 4.20 (d, 1H, J = 7.7 Hz, H-1′), 3.83–3.64 (m, 5H), 3.56 (t, 1H, J = 8.8 Hz), 3.47–3.40 (m, 4H), 3.36 (t, 1H, J = 8.5 Hz), 3.23–3.20 (m, 2H), 3.09 (s, 2H, C-CH2), 2.89–2.83 (m, 1H, H-3), 2.20 (d, 1H, J = 11.5 Hz), 1.46 (s, 3H, CH3), 1.05 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.00 (d, 3H, J = 6.0 Hz, Rha-H-6), 0.85, 0.81, 0.78, 0.65, 0.62 (each s, each 3H, CH3), 0.59–0.48 (m, 1H, H-5); 13C NMR (151 MHz, CD3OD): δ 151.81 (C-20), 110.27 (C-29), 105.45 (C-1′), 102.77 (Rha-C-1), 101.74 (Rha-C-1), 90.39, 80.16, 79.24, 77.98, 76.39, 73.65, 72.41, 72.12, 71.99, 70.57, 69.90, 61.89, 60.32, 57.33, 51.83, 50.00, 43.94, 43.76, 42.16, 42.12, 40.32 (two), 38.64, 37.97, 35.45, 34.98, 30.78, 30.33, 28.38, 27.35, 26.58, 23.45, 22.63, 21.99, 19.39, 19.26, 17.99, 17.88, 16.98, 16.85, 16.53, 15.25, 9.09, 7.84. HRMS (MALDI) m/z: calcd for C48H79O15 [M + H]+, 895.5497; found, 895.5473.
4.1.9. General procedure for 20 and 21
Compound BA-2 or BA-20 (1.0 mmol) was dissolved in 20 mL of dry pyridine, Ac2O (16.0 mmol) and DMAP (0.8 mmol) were added at 0 ℃. The reaction mixture was warmed to 60 ℃ and stirred for 24 h under N2 atmosphere. After excess solvent was removed in vacuo, the crude product was extracted with ethyl acetate, which was then washed with 1 M HCl, saturated NaHCO3 solution and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo to provide a crude residue. The residue was further purified by column chromatography (petroleumether-EtOAc-CH2Cl2, 3:1:1) to yield 20 or 21, respectively.
4.1.9.1. 3β-O-[2, 4-Di-O-(2, 3, 4-tri-O-Acetyl-α-l-rhamnopyranosyl)-β-(3, 6-di-O-acetyl)-d-glucopyranosyl]-lup-20(29)-ene-28-oic acid (20)
Compound 20 was obtained as a white powder. 1H NMR (600 MHz, CDCl3): δ 5.28–5.20 (m, 3H), 5.17 (dd, 1H, J = 10.2, 3.2 Hz, Rha-H-3), 5.10 (dd, 1H, J = 3.5, 1.8 Hz, Rha-H-2), 5.05–5.01 (m, 4H), 4.80 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.74 (s, 1H, C = CH2-1), 4.62 (s, 1H, C = CH2-2), 4.53 (d, 1H, J = 7.7 Hz, H-1′), 4.46 (dd, 1H, J = 12.4, 2.1 Hz, H-6′-1), 4.27 (dd, 1H, J = 13.4, 5.3 Hz, H-6′-2), 4.25–4.17 (m, 1H, H-5′), 3.91–3.81 (m, 2H), 3.76 (t, 1H, J = 9.3 Hz, Rha-H-4), 3.71–3.58 (m, 3H), 3.14–3.12 (m, 1H), 3.05–2.94 (m, 1H, H-3), 2.27 (d, 1H, J = 12.5 Hz), 2.14, 2.13, 2.11, 2.10, 2.04, 2.01, 1.99, 1.97 (each s, each 3H, each CH3CO), 1.70 (s, 3H, CH3), 1.17 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.15 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.01, 0.98, 0.92, 0.82, 0.78 (each s, each 3H, CH3), 0.71 (d, 1H, J = 10.3 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 181.96, 170.76, 170.37, 170.27, 170.22 (C-28), 170.14 (two), 169.81, 150.48 (C-20), 109.85 (C-29), 103.83 (C-1′), 99.58 (Rha-C-1), 97.03 (Rha-C-1), 90.15, 78.05, 77.48, 77.16, 75.59, 75.43, 72.17, 71.20, 70.59, 70.00, 69.80, 68.69, 68.58, 68.00, 66.75, 62.31, 56.44, 56.07, 50.57, 49.27, 46.98, 42.51, 40.77, 39.24, 39.20, 38.45, 36.99, 34.35, 32.23, 30.65, 29.74, 27.80, 26.23, 25.54, 21.60, 21.07, 21.01, 20.97, 20.94 (three), 20.86, 20.81, 19.45, 18.25, 17.34, 17.24, 16.34, 16.13, 16.02, 14.75. HRMS (ESI) m/z: calcd for C64H95O24 [M + H]+, 1247.6213; found, 1247.6235.
4.1.9.2. 28-(Benzyloxy)-3β-O-[2, 4-Di-O-(2, 3, 4-tri-O-Acetyl-α-l-rhamnopyranosyl)-β-(3, 6-di-O-acetyl)-d-glucopyranosyl]-lup-20 (29)-ene-3-ol (21)
Compound 21 was obtained as a white powder. 1H NMR (600 MHz, CDCl3): δ 7.37–7.23 (m, 5H, Ar-H), 5.49–5.30 (m, 3H), 5.26 (dd, 1H, J = 9.7, 3.4 Hz, Rha-H-3), 5.15–5.01 (m, 2H), 4.84 (s, 1H, Rha-H-1), 4.65 (s, 1H, Rha-H-1), 4.56 (s, 1H, C = CH2-1), 4.48 (s, 1H, C = CH2-2), 4.42 (d, 1H, J = 8.1 Hz, H-1′), 4.34–4.17 (m, 3H), 4.08 (dd, 1H, J = 9.8, 7.0 Hz, H-2′), 3.99 (d, 1H, J = 2.5 Hz), 3.73 (t, 1H, J = 9.5 Hz, Rha-H-4), 3.61 (t, 1H, J = 9.1 Hz, Rha-H-4), 3.54–3.40 (m, 3H), 3.09 (d, 2H, J = 9.1 Hz), 2.40–2.32 (m, 1H), 2.18–1.94 (m, 24H, 8 × CH3CO), 1.67 (s, 3H, CH3), 1.27 (d, 3H, J = 6.4 Hz, Rha-H-6), 1.19 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.05 (s, 3H, CH3), 0.94 (s, 3H, CH3), 0.83 (s, 3H, CH3), 0.81 (s, 6H, 2 × CH3), 0.70 (d, 1H, J = 9.5 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 171.93 (C-28), 170.20, 170.12 (two), 170.10 (two), 170.00 (two), 169.78, 149.64 (C-20), 140.06, 128.45, 127.69, 127.58, 118.61 (C-29), 104.07 (C-1′), 99.31 (Rha-C-1), 97.63 (Rha-C-1), 85.23, 83.18, 73.99, 71.64, 70.09, 69.45, 69.23, 68.12, 67.33, 65.05, 57.77, 56.60, 54.34, 53.95 (three), 51.52, 50.15, 48.71, 46.99, 45.85, 42.58, 42.15, 42.00, 41.39, 40.82, 39.10, 38.70, 38.22, 36.30, 33.53, 33.14, 30.14, 27.94, 27.52, 25.75, 25.25, 21.62, 21.14 (two), 20.99 (two), 20.91 (three), 17.86, 16.11, 14.78. HRMS (ESI) m/z: calcd for C71H103O23 [M + H]+, 1323.6890; found, 1323.6898.
4.1.10. General procedure for BA-3 -- BA-15
To a solution of BA-2 (1.00 mmol) in DMF (20 mL) was added K2CO3 (5 mmol) at 30 °C under N2 atmosphere. After stirring at 30 °C for 2 h, the corresponding halogenated hydrocarbon (3.00 mmol) was added. Stirring was continued overnight at that temperature. After the mixture was evaporated to remove excess solvent under reduced pressure, the residue was dissolved in EtOAc (100 mL), then extracted with water (3 × 50 mL) and brine (3 × 50 mL). The combined organic layer was concentrated in vacuo after drying over Na2SO4. Then, the residue was re-dissolved in MeOH (10 mL) and CH2Cl2 (10 mL), CH3ONa was added until pH = 10. After the reaction mixture was stirred at r.t. for 5 h, Dowex 50 × 8 (H+) resin was added until pH = 7. The reaction mixture was filtered and concentrated under vacuum. The residue was purified by silica gel column chromatography (CH2Cl2-MeOH, 6:1) to give title saponins BA-3--BA-15.
4.1.10.1. Ethyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-3)
Similarly, BA-3 was prepared as a white solid in 82 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.37 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.82 (s, 1H, Rha-H-1), 4.74 (s, 1H, C = CH2-1), 4.62 (s, 1H, C = CH2-2), 4.43 (d, 1H, J = 7.7 Hz, H-1′), 4.24–4.07 (m, 2H), 4.03–3.96 (m, 2H), 3.92 (dd, 1H, J = 9.5, 3.2 Hz, Rha-H-3), 3.86 (dd, 1H, J = 3.3, 1.8 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.1, 2.0 Hz, H-6′-1), 3.76 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.70–3.68 (m, 1H), 3.66 (dd, 1H, J = 3.4, 1.7 Hz), 3.66–3.60 (m, 1H), 3.58–3.56 (m, 1H), 3.52–3.34 (m, 2H), 3.34–3.29 (m, 2H), 3.15 (dd, 1H, J = 11.7, 4.4 Hz, H-3), 2.26 (d, 1H, J = 8.2 Hz), 1.71 (s, 3H, CH3), 1.28 (s, 3H, CH3), 1.27 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.05, 1.02, 0.96, 0.88, 0.85 (each s, each 3H, CH3), 0.75 (d, 1H, J = 9.8 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.58 (C-28), 151.78 (C-20), 110.34 (C-29), 105.49 (C-1′), 103.03 (Rha-C-1), 101.99 (Rha-C-1), 90.40, 80.30, 79.20, 78.11, 76.42, 73.91, 73.67, 72.41, 72.11, 71.97, 70.73, 69.99, 61.95, 61.02, 57.72, 57.43, 52.00, 50.57, 43.52, 41.97, 40.34 (two), 39.64, 38.05, 37.95, 36.97, 35.57, 33.14, 31.66, 30.75, 28.40, 27.36, 26.88, 22.09, 19.62, 19.27, 18.02, 17.90, 16.99, 16.93, 16.64, 15.21, 14.71 (two). HRMS (ESI) m/z: calcd for C50H82O16Na [M + Na]+, 961.5501; found, 961.5423.
4.1.10.2. n-Propyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-4)
Similarly, BA-4 was prepared as a white solid in 80 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.38 (s, 1H, Rha-H-1), 4.85 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.73 (s, 1H, C = CH2-1), 4.63 (s, 1H, C = CH2-2), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 4.14–4.00 (m, 2H), 4.03–3.95 (m, 2H), 3.92 (dd, 1H, J = 9.6, 3.3 Hz, Rha-H-3), 3.88–3.78 (m, 1H), 3.76 (dd, 1H, J = 9.5, 3.3 Hz, Rha-H-3), 3.66 (t, 1H, J = 10.6 Hz, Rha-H-4), 3.58 (t, 1H, J = 8.1 Hz), 3.50–3.38 (m, 3H), 3.38–3.29 (m, 3H), 3.15 (dd, 1H, J = 11.7, 4.4 Hz, H-3), 3.10–2.98 (m, 1H), 2.28 (d, 1H, J = 10.3 Hz), 1.72 (s, 3H, CH3), 1.50–1.35 (m, 2H, CH2), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.23 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.05, 1.03, 1.01, 0.96, 0.89, 0.86 (each s, each 3H, CH3), 0.75 (d, 1H, J = 9.9 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.69 (C-28), 151.75 (C-20), 110.36 (C-29), 105.50 (C-1′), 103.02 (Rha-C-1), 102.00 (Rha-C-1), 90.40, 80.29, 79.21, 78.11, 76.42, 73.91, 73.67, 72.41, 72.10, 71.96, 70.73, 69.99, 66.78, 61.95, 57.89, 57.43, 51.99, 50.59, 43.54, 41.97, 40.34 (two), 39.70, 38.05, 36.97, 35.58, 33.21, 31.68, 30.79, 28.41, 27.36, 26.88, 23.20 (two), 22.10, 19.63, 19.28, 18.03 (two), 17.90 (two), 16.99, 16.94, 16.67, 15.24. HRMS (ESI) m/z: calcd for C51H84O16Na [M + Na]+, 975.5557; found, 975.5582.
4.1.10.3. n-Butyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-5)
Similarly, BA-5 was prepared as a white solid in 78 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.36 (s, 1H, Rha-H-1), 4.86 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.72 (s, 1H, C = CH2-1), 4.61 (s, 1H, C = CH2-2), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.17–4.02 (m, 2H), 4.01–3.93 (m, 2H), 3.91 (dd, 1H, J = 9.5, 7.7 Hz, H-2′), 3.87–3.77 (m, 1H), 3.75 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.71–3.59 (m, 2H), 3.57 (t, 1H, J = 9.0 Hz, Rha-H-4), 3.48–3.34 (m, 3H), 3.33–3.30 (m, 2H), 3.14 (dd, 1H, J = 11.6, 4.3 Hz, H-3), 3.10–2.98 (m, 1H), 1.95 (d, 1H, J = 9.7 Hz), 1.70 (s, 3H, CH3), 1.46–1.43 (m, 4H), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.03, 1.01 (each s, each 3H, CH3), 0.97 (t, 3H, J = 7.4 Hz, CH3), 0.94, 0.87, 0.84 (each s, each 3H, CH3), 0.74 (d, 1H, J = 9.3 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.72 (C-28), 151.76 (C-20), 110.34 (C-29), 105.48 (C-1′), 103.04 (Rha-C-1), 101.99 (Rha-C-1), 90.39, 80.34, 79.21, 78.12, 76.41, 73.91, 73.67, 72.41, 72.11, 71.98, 70.73, 69.99, 64.84, 61.95, 57.89, 57.41, 51.99, 50.59, 43.54, 41.97, 40.34 (two), 39.73, 38.04, 38.00, 35.58, 33.20, 31.96 (two), 31.68, 30.79, 28.39, 27.36, 26.88, 22.09, 20.43 (two), 19.59, 19.26, 18.02, 17.90, 16.99, 16.92, 16.68, 15.20, 14.04. HRMS (ESI) m/z: calcd for C52H87O16 [M + H]+, 967.5994; found, 967.6018.
4.1.10.4. n-Pentyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-6)
Similarly, BA-6 was prepared as a white solid in 76 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.38 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.83 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.73 (s, 1H, C = CH2-1), 4.63 (s, 1H, C = CH2-2), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 4.19–4.03 (m, 2H), 4.03–3.95 (m, 2H), 3.94–3.92 (m, 1H), 3.86 (dd, 1H, J = 3.2, 1.8 Hz, Rha-H-2), 3.82 (dd, 1H, J = 12.0, 2.0 Hz, H-6′-1), 3.76 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.72–3.63 (m, 2H), 3.64 (dd, 1H, J = 3.4, 1.1 Hz, Rha-H-2), 3.58 (t, 1H, J = 9.0 Hz, Rha-H-4), 3.50–3.38 (m, 2H), 3.35–3.30 (m, 2H), 3.16 (dd, 1H, J = 11.7, 4.2 Hz, H-3), 3.10–2.98 (m, 2H), 2.27 (d, 1H, J = 9.4 Hz), 1.72 (s, 3H, CH3), 1.47–1.38 (m, 6H), 1.29 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.23 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.05, 1.03, 0.96, 0.96, 0.88, 0.86 (each s, each 3H, CH3), 0.76 (d, 1H, J = 9.9 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.77 (C-28), 151.78 (C-20), 110.35 (C-29), 105.49 (C-1′), 103.05 (Rha-C-1), 102.00 (Rha-C-1), 90.38, 80.34, 79.21, 78.13, 76.42, 73.92, 73.68, 72.43, 72.11, 71.99, 70.74, 69.99, 65.13, 61.96, 57.91, 57.41, 51.98, 50.58, 49.85, 43.55, 41.98, 40.34, 39.76, 38.05, 35.59, 33.23, 31.70, 30.79, 29.59 (three), 28.39, 27.36, 26.88, 23.36 (two), 22.09, 19.60, 19.27, 18.02, 17.90, 16.99, 16.92, 16.72, 15.19, 14.43 (two). HRMS (ESI) m/z: calcd for C53H89O16 [M + H]+, 981.6151; found, 981.6183.
4.1.10.5. n-Hexyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-7)
Similarly, BA-7 was prepared as a white solid in 75 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.38 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.85 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.73 (s, 1H, C = CH2-1), 4.63 (s, 1H, C = CH2-2), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 4.19–4.03 (m, 2H), 4.03–3.95 (m, 2H), 3.93 (dd, 1H, J = 9.5, 7.3 Hz, H-2′), 3.86 (dd, 1H, J = 3.2, 1.8 Hz, Rha-H-2), 3.82 (dd, 1H, J = 12.0, 2.0 Hz, H-6′-1), 3.76 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.72–3.63 (m, 2H), 3.64 (d, 1H, J = 3.4 Hz, Rha-H-2), 3.58 (t, 1H, J = 9.0 Hz, Rha-H-4), 3.50–3.38 (m, 2H), 3.33 (brs, 1H), 3.16 (dd, 1H, J = 11.7, 4.2 Hz, H-3), 3.10–2.98 (m, 1H), 2.27 (d, 1H, J = 9.4 Hz), 1.72 (s, 3H, CH3), 1.47–1.38 (m, 8H), 1.29 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.23 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.05, 1.03, 0.96, 0.96, 0.88, 0.86 (each s, each 3H, CH3), 0.76 (d, 1H, J = 9.9 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.80 (C-28), 151.79 (C-20), 110.36 (C-29), 105.50 (C-1′), 103.06 (Rha-C-1), 102.01 (Rha-C-1), 90.39, 80.36, 79.22, 78.14, 76.44, 73.93, 73.68, 72.43, 72.12, 71.99, 70.75, 70.00, 65.15, 61.97, 57.92, 57.42, 51.98, 50.59, 43.55, 41.99, 40.35, 40.31, 39.78, 38.05, 35.59, 33.25, 32.57, 31.71, 30.82, 29.87, 28.40, 27.37, 27.07, 26.88, 23.73 (two), 22.10, 19.60, 19.27, 18.02, 17.90, 16.99, 16.92, 16.73, 15.20, 14.40. HRMS (ESI) m/z: calcd for C54H91O16 [M + H]+, 995.6307; found, 995.6335.
4.1.10.6. Isopropyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-8)
Similarly, BA-8 was prepared as a white solid in 78 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.36 (s, 1H, Rha-H-1), 4.82 (d, 1H, J = 1.1 Hz, Rha-H-1), 5.06–4.94 (m, 1H), 4.72 (s, 1H, C = CH2-1), 4.61 (s, 1H, C = CH2-2), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.03–3.95 (m, 2H), 3.91 (dd, 1H, J = 9.5, 7.2 Hz, H-2′), 3.84 (m, 1H), 3.80 (d, 1H, J = 10.9 Hz), 3.75 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.71–3.59 (m, 2H), 3.57 (t, 1H, J = 9.7 Hz, Rha-H-4), 3.48–3.34 (m, 2H), 3.32–3.30 (m, 2H), 3.14 (dd, 1H, J = 11.5, 4.2 Hz, H-3), 3.07–2.97 (m, 1H), 2.24 (d, 1H, J = 8.5 Hz), 1.70 (s, 3H, CH3), 1.28–1.25 (m, 6H), 1.25 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.03, 1.01, 0.95, 0.87, 0.84 (each s, each 3H, CH3), 0.74 (d, 1H, J = 9.6 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.14 (C-28), 151.82 (C-20), 110.31 (C-29), 105.48 (C-1′), 103.02 (Rha-C-1), 101.99 (Rha-C-1), 90.39, 80.31, 79.21, 78.11, 76.41, 73.90, 73.66, 72.41, 72.10, 71.97, 70.72, 69.98, 68.36, 61.95, 57.67, 57.42, 51.99, 50.51, 43.53, 42.01, 40.34 (two), 39.69, 38.04 (two), 35.56, 33.14, 31.70, 30.72, 28.40, 27.36, 26.90, 22.11 (three), 22.05 (two), 19.62, 19.26, 18.02, 17.90, 16.98, 16.93, 16.70, 15.20. HRMS (ESI) m/z: calcd for C51H85O16 [M + H]+, 953.5838; found, 953.5852.
4.1.10.7. Cyclohexyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-9)
Similarly, BA-9 was prepared as a white solid in 73 % yield for two steps; 1H NMR (400 MHz, CD3OD): δ 5.34 (s, 1H, Rha-H-1), 4.85 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.68 (s, 1H, C = CH2-1), 4.56 (s, 1H, C = CH2-2), 4.40 (d, 1H, J = 7.7 Hz, H-1′), 3.98–3.95 (m, 2H), 3.92–3.69 (m, 3H), 3.68–3.46 (m, 3H), 3.46–3.32 (m, 2H), 3.12 (d, 1H, J = 9.6 Hz, H-3), 2.70–2.66 (m, 10H, cyclohexanol-H), 2.22 (d, 1H, J = 12.8 Hz), 1.67 (s, 3H, CH3), 1.24 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.18 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.00, 0.98, 0.94, 0.83, 0.81 (each s, each 3H, CH3), 0.71 (d, 1H, J = 9.9 Hz, H-5); 13C NMR (101 MHz, CD3OD): δ 170.73 (C-28), 152.26 (C-20), 110.12 (C-29), 105.46 (C-1′), 102.97 (Rha-C-1), 101.96 (Rha-C-1), 90.40, 80.22, 79.19, 78.08, 76.40, 73.90, 73.67, 72.41, 72.08 (two), 71.98, 70.70, 69.98, 61.91, 57.40, 52.01, 50.42, 43.56, 41.93, 40.32 (two), 39.57, 38.03, 35.60, 35.38 (two), 31.73, 30.86, 28.38, 27.35, 26.90, 22.09, 19.57, 19.26, 18.01 (two), 17.91 (two), 16.97 (two), 16.90 (two), 16.73, 15.13. HRMS (ESI) m/z: calcd for C54H89O16 [M + H]+, 993.6151; found, 993.6175.
4.1.10.8. 2′-Chloroethyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-10)
Similarly, BA-10 was prepared as a white solid in 74 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.38 (s, 1H, Rha-H-1), 4.82 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.75 (s, 1H, C = CH2-1), 4.63 (s, 1H, C = CH2-2), 4.44 (d, 1H, J = 7.9 Hz, H-1′), 4.40–4.32 (m, 2H), 3.97–3.95 (m, 2H), 3.82–3.80 (m, 3H), 3.72–3.51 (m, 2H), 3.44–3.41 (m, 2H), 3.16 (d, 1H, J = 10.7 Hz, H-3), 2.29 (d, 1H, J = 10.1 Hz, H-13), 1.73 (s, 3H, CH3), 1.29 (d, 3H, J = 6.0 Hz, Rha-H-6), 1.23 (d, 3H, J = 6.0 Hz, Rha-H-6), 1.05, 1.04, 0.97, 0.89, 0.86 (each s, each 3H, CH3), 0.76 (d, 1H, J = 9.3 Hz, H-5; 13C NMR (151 MHz, CD3OD): δ 177.21 (C-28), 151.74 (C-20), 110.38 (C-29), 105.50 (C-1′), 103.06 (Rha-C-1), 102.01 (Rha-C-1), 90.41, 80.35, 79.21, 78.14, 76.44, 73.93, 73.68, 72.43, 72.12 (two), 71.99, 70.75, 70.00, 65.09, 61.97, 57.98, 57.43, 52.00, 50.63, 43.54, 43.26, 42.00, 40.35 (two), 39.72, 38.05, 35.58, 33.08, 31.63, 30.82, 28.40, 27.38, 26.88, 19.60, 19.27, 18.02 (two), 17.91, 16.99 (two), 16.94 (two), 16.65, 15.21. HRMS (ESI) m/z: calcd for C50H82O16Cl [M + H]+, 973.5291; found, 973.5317.
4.1.10.9. 2′-Bromoethyl 3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-11)
Similarly, BA-11 was prepared as a white solid in 70 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.34 (d, 1H, J = 1.8 Hz, Rha-H-1), 4.83 (s, 1H, Rha-H-1), 4.70 (s, 1H, C = CH2-1), 4.58 (s, 1H, C = CH2-2), 4.40 (d, 1H, J = 7.7 Hz, H-1′), 4.16–4.08 (m, 2H), 3.99–3.91 (m, 2H), 3.89 (dd, 1H, J = 9.6, 7.4 Hz, H-2′), 3.83–3.81 (m, 1H), 3.78 (dd, 1H, J = 12.0, 2.0 Hz, H-6′-1), 3.72 (dd, 1H, J = 9.7, 3.1 Hz, Rha-H-3), 3.64 (dd, 1H, J = 13.0, 5.6 Hz, H-6′-2), 3.63–3.56 (m, 2H), 3.59–3.49 (m, 1H), 3.46–3.34 (m, 2H), 3.32–3.27 (m, 2H), 3.16–3.07 (m, 1H, H-3), 3.03–2.93 (m, 1H), 2.27 (d, 1H, J = 12.8 Hz), 1.67 (s, 3H, CH3), 1.24 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.19 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.01, 0.98, 0.92, 0.84, 0.81 (each s, each 3H, CH3), 0.71 (d, 1H, J = 9.6 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.62 (C-28), 151.83 (C-20), 110.42 (C-29), 105.46 (C-1′), 103.02 (Rha-C-1), 101.97 (Rha-C-1), 90.37, 80.32, 79.20, 78.11, 76.40, 73.90, 73.67, 72.41, 72.10 (two), 71.98, 70.71, 69.97, 66.33, 61.12, 57.88, 57.40, 51.99, 49.85, 43.52, 41.95 (two), 40.32 (two), 39.57, 38.03 (two), 33.04, 30.72, 28.37, 27.35, 26.86, 22.05, 19.57, 19.25, 18.00 (two), 17.90 (two), 16.97 (two), 16.89 (two), 16.59, 15.16. HRMS (ESI) m/z: calcd for C50H82O16Br [M + H]+, 1017.4786; found, 1017.4803.
4.1.10.10. 2′-Hydroxylethyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-12)
Similarly, BA-12 was prepared as a white solid in 70 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.37 (s, 1H, Rha-H-1), 4.82 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.73 (s, 1H, C = CH2-1), 4.61 (s, 1H, C = CH2-2), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.16–4.14 (m, 2H), 3.97–3.95 (m, 2H), 3.87–3.71 (m, 3H), 3.66–3.64 (m, 1H), 3.57 (t, 1H, J = 8.7 Hz), 3.47–3.35 (m, 2H), 3.34–3.32 (m, 3H), 3.19–3.11 (m, 1H, H-3), 3.02–3.00 (m, 2H), 2.31 (d, 1H, J = 11.9 Hz), 1.71 (s, 3H, CH3), 1.27 (d, 3H, J = 6.0 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.04, 1.01, 0.95, 0.87, 0.84 (each s, each 3H, CH3), 0.73 (d, 1H, J = 9.7 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 178.06 (C-28), 151.73 (C-20), 110.36 (C-29), 105.48, 105.30 (C-1′), 103.02 (Rha-C-1), 101.97 (Rha-C-1), 90.37, 80.30, 79.17, 78.12, 76.41, 73.91, 73.67, 72.41, 72.11 (two), 71.98, 70.72, 69.97, 61.94, 57.86, 57.41, 56.45, 51.97, 51.84, 50.62, 43.50, 41.90, 40.33 (two), 39.63, 38.04, 37.86, 35.55, 33.13, 31.62, 30.80, 28.40, 27.36, 26.84, 22.06, 19.60, 19.27, 18.03, 17.91, 16.99, 16.92, 16.59, 15.21. HRMS (ESI) m/z: calcd for C50H83O17 [M + H]+, 955.5630; found, 955.5658.
4.1.10.11. 2′-Oxopethyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-13)
Similarly, BA-13 was prepared as a white solid in 65 % yield for two steps; 1H NMR (500 MHz, CD3OD): δ 9.60 (s, 1H, CHO), 5.33 (s, 1H, Rha-H-1), 4.87 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.82 (brs, 2H), 4.68 (s, 1H, C = CH2-1), 4.57 (s, 1H, C = CH2-2), 4.38 (d, 1H, J = 7.8 Hz, H-1′), 3.92–3.87 (m, 3H), 3.74 (dd, 1H, J = 9.6, 7.5 Hz, H-2′), 3.63–3.61 (m, 4H), 3.53 (t, 1H, J = 9.2 Hz, Rha-H-4), 3.42–3.33 (m, 1H), 3.28–3.26 (m, 2H), 3.20–3.14 (m, 3H), 3.12–3.10 (m, 1H, H-3), 3.01–2.91 (m, 2H), 2.19 (d, 1H, J = 11.3 Hz), 1.66 (s, 3H, CH3), 1.23 (d, 3H, J = 6.0 Hz, Rha-H-6), 1.17 (d, 3H, J = 6.0 Hz, Rha-H-6), 0.99, 0.97, 0.90, 0.83, 0.80 (each s, each 3H, CH3), 0.70 (d, 1H, J = 9.4 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 207.02, 178.08 (C-28), 151.72 (C-20), 110.38 (C-129), 105.49 (C-1′), 103.00 (Rha-C-1), 101.98 (Rha-C-1), 90.39, 80.23, 79.17, 78.10, 76.41, 73.89, 73.65, 72.40, 72.08 (two), 71.96, 70.70, 69.98, 61.91, 57.85, 57.40, 51.96, 51.86, 50.61, 43.49, 41.89, 40.33 (two), 39.62, 38.03 (two), 37.86, 35.54, 33.12, 31.60, 30.79, 28.39, 27.35, 26.82, 22.06, 19.59, 19.26, 18.03 (two), 17.91 (two), 16.99, 16.93, 16.58, 15.21. HRMS (ESI) m/z: calcd for C50H81O17 [M + H]+, 953.5474; found, 953.5496.
4.1.10.12. Allyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-14)
Similarly, BA-14 was prepared as a white solid in 68 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 6.04–5.90 (m, 1H), 5.38 (s, 1H, Rha-H-1), 5.35 (d, 1H, J = 11.7 Hz), 5.25 (d, 1H, J = 10.4 Hz), 4.85 (s, 1H, Rha-H-1), 4.73 (s, 1H, C = CH2-1), 4.61 (s, 1H, C = CH2-2), 4.58 (d, 2H, J = 8.2 Hz), 4.43 (d, 1H, J = 7.7 Hz, H-1′), 4.03–3.94 (m, 2H), 3.92 (dd, 1H, J = 9.5, 7.2 Hz, H-2′), 3.88–3.77 (m, 2H), 3.75 (dd, 1H, J = 9.6, 3.3 Hz, Rha-H-3), 3.71–3.60 (m, 2H), 3.62–3.53 (m, 1H), 3.49–3.35 (m, 3H), 3.33–3.31 (m, 1H), 3.15 (dd, 1H, J = 11.3, 4.1 Hz, H-3), 3.02–3.00 (m, 1H), 2.89–2.87 (m, 1H), 2.27 (d, 1H, J = 9.4 Hz), 1.71 (s, 3H, CH3), 1.28 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.04, 1.02, 0.94, 0.87, 0.85 (each s, each 3H, CH3), 0.74 (d, 1H, J = 9.5 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 177.18 (C-28), 151.78 (C-20), 61.97, 57.91, 57.42, 52.01, 50.64, 43.53, 42.00, 40.35, 39.66, 38.05, 37.92, 36.97, 35.56, 35.38 (two), 33.13, 31.62, 30.77, 28.38, 27.37, 26.86, 22.07, 19.55, 19.26, 18.01, 17.90, 16.98, 16.89, 16.64, 15.14. HRMS (ESI) m/z: calcd for C51H83O16 [M + H]+, 951.5681; found, 951.5703.
4.1.10.13. Cyclopropylmethyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-28-oic acid (BA-15)
Similarly, BA-15 was prepared as a white solid in 70 % yield for two steps; 1H NMR (600 MHz, CD3OD): δ 5.37 (s, 1H, Rha-H-1), 4.82 (s, 1H, Rha-H-1), 4.74 (s, 1H, C = CH2-1), 4.62 (s, 1H, C = CH2-2), 4.43 (d, 1H, J = 7.7 Hz, H-1′), 4.04–3.94 (m, 4H), 3.91 (dd, 1H, J = 9.6, 7.2 Hz, H-2′), 3.90–3.78 (m, 1H), 3.76 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.72–3.60 (m, 3H), 3.58 (t, 1H, J = 8.4 Hz), 3.48–3.35 (m, 3H), 3.33–3.31 (m, 2H), 3.15 (dd, 1H, J = 11.4, 4.3 Hz, H-3), 3.10–2.99 (m, 2H), 2.29 (d, 1H, J = 10.1 Hz), 1.71 (s, 3H, CH3), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.04, 1.02, 0.96, 0.88, 0.85 (each s, each 3H, CH3), 0.75 (d, 1H, J = 9.7 Hz, H-5), 0.58 (q, 2H, J = 7.7 Hz), 0.32 (q, 2H, J = 7.5 Hz); 13C NMR (151 MHz, CD3OD): δ 177.72 (C-28), 151.81 (C-20), 110.34 (C-29), 105.50 (C-1′), 103.04 (Rha-C-1), 102.00 (Rha-C-1), 90.40, 80.31, 79.21, 78.12, 76.42, 73.91, 73.67, 72.42, 72.11 (two), 71.98, 70.73, 69.99, 69.68, 61.95, 57.87, 57.43, 52.00, 50.60, 43.56, 41.97, 40.35 (two), 39.71, 38.05, 35.60, 33.21, 31.70, 30.78, 28.41, 27.37, 26.90, 22.12, 19.63, 19.28, 18.03 (two), 17.90 (two), 16.99 (two), 16.94 (two), 16.73, 15.24, 10.99. HRMS (ESI) m/z: calcd for C52H85O16 [M + H]+, 965.5838; found, 965.5856.
4.1.11 General procedure forBA-N-1andBA-N-2. To a solution of compound 20 (1.0 mmol) in 10 mL dried CH2Cl2 was added oxalyl chloride (1 mL) under argon. Then the mixture was stirred at room temperature for 12 h and concentrated to dryness in vacuo. To a dried CH2Cl2 (10 mL) solution of methylamine hydrochloride or dimethylamine hydrochloride (2.0 mmol) was added to the crude acid chloride. The reaction mixturewas was stirred at r.t. for 3 h under argon and then concentratedunder reduced pressure. The obtained residue was re-dissolved in 2:1 MeOH/CH2Cl2 (15 mL) and then NaOMe was added until pH = 10. After stirred at r.t. for 3 h, the solution was neutralized with Dowex 50 × 8 (H+) resin until pH = 7, filtered and concentrated in vacuo. Then the residue was purified by silica gel column chromatography (CH2Cl2-MeOH, 6:1) to yield compounds BA-N-1 and BA-N-2, respectively.
4.1.11. N-{β-O-[2, 4-Di-O-(α-l-rhamnopyranosyl)-β-Dglucopyranosyl]-lup-20 (29)-ene-28-oyl}-methylamine (BA-N-1)
BA-N-1 was prepared as a white solid in 85 % yield for three steps. 1H NMR (600 MHz, CD3OD): δ 5.37 (s, 1H, Rha-H-1), 4.83 (s, 1H, Rha-H-1), 4.71 (s, 1H, C = CH2-1), 4.59 (s, 1H, C = CH2-2), 4.43 (d, 1H, J = 7.4 Hz, H-1′), 3.94–3.90 (m, 3H), 3.83 (d, 1H, J = 12.0 Hz), 3.76 (t, 1H, J = 11.2 Hz), 3.71–3.52 (m, 3H), 3.45–3.36 (m, 3H), 2.70 (s, 3H, NH-CH3), 2.57–2.55 (m, 1H), 2.11 (d, 1H, J = 12.2 Hz), 1.69 (s, 3H, CH3), 1.25–1.19 (m, 6H, 2 × Rha-H-6), 1.03, 1.00, 0.96, 0.87, 0.84 (each s, each 3H, CH3), 0.78–0.70 (m, 1H, H-5); 13C NMR (151 MHz, CD3OD): δ 179.75 (C-28), 152.37 (C-20), 109.93 (C-29), 105.45 (C-1′), 103.04 (Rha-C-1), 101.94 (Rha-C-1), 90.34, 80.34, 79.13, 78.17, 76.42, 73.93, 73.70, 72.44, 72.11 (two), 72.03, 70.74, 69.96, 61.92, 57.43, 56.94, 52.09, 51.43, 48.17, 43.48, 41.97, 40.33 (two), 39.34, 38.97, 38.05, 35.59, 34.13, 31.93, 30.54, 28.39, 27.38, 26.98, 26.42, 22.14, 19.63, 19.27, 18.04, 17.96, 16.99, 16.91, 16.72, 15.09. HRMS (ESI) m/z: calcd for C49H81O15NNa [M + Na]+, 946.5498; found, 946.5447.
4.1.12. N-{β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-Dglucopyranosyl]-lup-20(29)-ene-28-oyl}-dimethylamine (BA-N-2)
BA-N-2 was prepared as a white solid in 82 % yield for three steps; 1H NMR (600 MHz, CD3OD): δ 5.37 (s, 1H, Rha-H-1), 4.81 (s, 1H, Rha-H-1), 4.70 (s, 1H, C = CH2-1), 4.58 (s, 1H, C = CH2-2), 4.43 (d, 1H, J = 7.7 Hz, H-1′), 4.00–3.95 (m, 3H), 3.92 (dd, 1H, J = 9.5, 6.8 Hz, H-2′), 3.87–3.78 (m, 3H), 3.75 (dd, 1H, J = 9.5, 3.4 Hz, Rha-H-3), 3.70–3.62 (m, 2H), 3.57–3.55 (m, 1H), 3.47–3.35 (m, 2H), 3.2 (s, 6H, 2 × NCH3), 3.07–2.95 (m, 1H, H-3), 2.34 (d, 1H, J = 13.4 Hz), 1.70 (s, 3H, CH3), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.03, 1.00, 0.95, 0.87, 0.84 (each s, each 3H, CH3), 0.74 (s, 1H, H-5); 13C NMR (151 MHz, CD3OD): δ 177.13 (C-28), 152.63 (C-20), 109.83 (C-29), 105.47 (C-1′), 103.09 (Rha-C-1), 101.98 (Rha-C-1), 90.40, 80.44, 79.19, 78.19, 76.44, 73.94, 73.70, 72.45, 72.13, 72.03, 70.76, 70.00, 61.96, 57.51, 56.09, 53.92, 52.28, 47.49, 43.03, 41.94, 40.36, 38.49, 38.08, 36.83, 35.62, 32.92, 32.44, 31.02, 28.37, 27.38, 27.03, 22.34, 19.75, 19.29, 18.01, 17.91, 17.48, 17.29, 17.10, 16.98, 16.68, 15.10. HRMS (ESI) m/z: calcd for C50H83O15NNa [M + Na]+, 960.5660; found, 960.5691.
4.1.13. 3β-O-[2, 4-Di-O-(2, 3, 4-tri-O-Acetyl-α-l-rhamnopyranosyl)-β-(3, 6-di-O-acetyl)-d-glucopyranosyl]-lup-20 (29)-ene-3, 28-diol (22)
Compound 21 (360 mg, 0.27 mmol) dissolved in THF (5 mL) and MeOH (5 mL), and 10 % Pd/C (40 mg) was added to the solution. The reaction mixture was stirred under atmospheric pressure hydrogen at r.t. for 1 h. After Pd/C was filtered off, the mixture was concentrated under vacuum to give a crude residue, which was purified by column chromatography (petroleumether-EtOAc-CH2Cl2, 2:1:1) to obtain 22 (302 mg, 90 %) as a white powder. 1H NMR (600 MHz, CDCl3): δ 5.52–5.21 (m, 2H), 5.11–5.07 (m, 3H), 4.86 (s, 1H, C = CH2-1), 4.82 (s, 1H, C = CH2-2), 4.69–4.67 (m, 1H), 4.58 (d, 1H, J = 7.9 Hz, H-1′), 4.42 (t, 1H, J = 9.7 Hz, Rha-H-4), 4.36–4.16 (m, 3H), 4.16–3.88 (m, 4H), 3.73 (t, 1H, J = 9.2 Hz, H-4′), 3.61 (t, 1H, J = 8.8 Hz, Rha-H-4), 3.55–3.43 (m, 2H, C-CH2), 3.31 (d, 1H, J = 10.6 Hz), 3.18–3.05 (m, 1H, H-3), 2.15, 2.12, 2.12, 2.06, 2.04, 2.00, 1.99, 1.95 (each s, each 3H, each CH3CO), 1.67 (s, 3H, CH3), 1.24 (d, 3H, J = 6.3 Hz, CH3), 1.17 (d, 3H, J = 6.3 Hz, CH3), 1.04 (s, 6H, 2 × CH3), 1.00 (s, 3H, CH3), 0.81 (s, 6H, 2 × CH3), 0.75 (d, 1H, J = 7.0 Hz, H-5); 13C NMR (151 MHz, CDCl3): δ 170.27, 170.13 (three), 170.09 (three), 169.82, 150.01 (C-20), 111.18 (C-29), 104.02 (C-1′), 99.14 (Rha-C-1), 97.36 (Rha-C-1), 90.28, 82.28, 76.33, 75.63, 75.56, 75.42, 71.67, 71.22, 70.70, 70.02, 69.73, 69.62, 69.28, 69.11, 68.71, 68.21, 66.66, 62.66, 60.42, 56.05, 50.44, 48.92, 47.88, 42.81, 41.08, 39.26, 36.94, 34.26, 34.08, 29.35, 28.28, 27.84, 26.99, 26.29, 25.88, 23.02, 22.58, 21.06, 20.92 (three), 20.88 (three), 20.84 (three), 20.76 (two), 18.29, 17.49 (two), 17.42 (two), 16.24 (two), 16.23, 14.89, 14.80. HRMS (ESI) m/z: calcd for C64H97O23 [M + H]+, 1233.6421; found, 1233.6443.
4.1.14. 3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (30)-en-28-al (BA-18)
To a solution of 21 (0.20 g, 0.16 mmol, 1.0 eq) in CH2Cl2-MeOH (20 mL, V: V = 1: 1) and was added PCC (0.10 g, 0.41 mmol) at 0 ℃ under argon. After the mixture was stirred at r.t. for 4 h, the reaction mixture was filtered and the filtrate was concentrated under vacuum. The residue was dissolved in EtOAc (60 mL), then extracted with water (3 × 30 mL) and brine (3 × 30 mL). The organic phase was concentrated under reduced pressure. The residue was re-dissolved in MeOH (10 mL) and CH2Cl2 (10 mL), CH3ONa was added until pH = 10. The reaction mixture was stirred at r.t. until the reaction was complete detected by TLC. Then, the mixture was neutralized with Dowex 50 × 8 (H+) resin until pH = 7. The reaction mixture was filtered and evaporated to remove excess solvent under vacuum. The residue was purified by silica gel column chromatography (CH2Cl2-MeOH, 5:1) to produce BA-18 (0.13 g, 84 % for two steps). 1H NMR (600 MHz, CD3OD): δ 9.58 (s, 1H, CHO), 5.29 (s, 1H, Rha-H-1), 4.89 (s, 1H, Rha-H-1), 4.56 (s, 2H, C = CH2), 4.35 (d, 1H, J = 7.7 Hz, H-1′), 3.87 (m, 2H), 3.79–3.63 (m, 1H), 3.62–3.44 (m, 3H), 3.42–3.29 (m, 3H), 3.25–3.23 (m, 3H), 3.12–3.00 (m, 1H, H-3), 2.86–2.73 (m, 2H), 2.03–1.80 (m, 1H), 1.64 (s, 3H, CH3), 1.17 (m, 6H, 2 × Rha-H-6), 1.00–0.67 (m, 15H, 5 × CH3); 13C NMR (151 MHz, CD3OD): δ 207.06 (CHO), 149.80 (C-20), 110.21 (C-29),104.09 (C-1′), 101.65 (Rha-C-1), 100.62 (Rha-C-1), 89.04, 78.95, 77.84, 76.74, 75.05, 72.54, 72.30, 71.04, 70.73, 70.61, 69.35, 68.63, 60.58, 59.13, 56.53, 56.00, 50.64, 42.50, 42.28, 42.17, 40.66, 40.55, 38.95, 38.69, 36.66, 34.20, 29.43, 26.99, 25.98, 20.68, 19.76, 18.19, 17.86, 16.63, 16.52, 15.61, 15.51, 15.26, 15.09, 13.75, 13.68, 13.30. HRMS (ESI) m/z: calcd for C49H81O15 [M + H]+, 909.5575; found, 909.5593.
4.1.15. 28-(Methoxy)-3β-O-[2, 4-di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-3-al (BA-19)
To a solution of 21 (0.20 g, 0.16 mmol) in ACN (10 mL) was added Ag2O (0.19 g, 0.81 mmol). After stirring for 20 min, CH3I (0.12 g, 0.81 mmol) was added quickly while the mixture was at 60 ℃ under argon atmosphere. Stirring was continued overnight at that temperature. Then the mixture was cooled to room temperature, filtered and the filtrate was concentrated under vacuum. The residue was dissolved in EtOAc (100 mL), then extracted with water (3 × 50 mL) and brine (3 × 50 mL). The organic phase was concentrated under vacuum. Then, the residue was re-dissolved in MeOH (10 mL) and CH2Cl2 (10 mL), CH3ONa was added until pH = 10. After the reaction mixture was stirred at r.t. for 5 h, Dowex 50 × 8 (H+) resin was added until pH = 7. The reaction mixture was filtered and concentrated under vacuum. The residue was purified by silica gel column chromatography (CH2Cl2-MeOH, 6:1) to afford BA-19 (0.12 g, 83 % for two steps). 1H NMR (600 MHz, CD3OD): δ 5.39 (s, 1H, Rha-H-1), 4.85 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.72 (s, 1H, C = CH2-1), 4.60 (s, 1H, C = CH2-2), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 3.99 (dd, 1H, J = 3.5, 1.7 Hz, Rha-H-2), 3.93 (dd, 1H, J = 9.5, 7.4 Hz, H-2′), 3.86 (dd, 1H, J = 3.3, 1.9 Hz, Rha-H-2), 3.86–3.78 (m, 1H), 3.77 (dd, 1H, J = 9.7, 3.5 Hz, Rha-H-3), 3.67 (dd, 1H, J = 9.7, 3.8 Hz, Rha-H-3), 3.58 (t, 1H, J = 9.3 Hz, Rha-H-3), 3.50–3.38 (m, 3H), 3.36–3.30 (m, 4H), 3.20–3.11 (m, 2H), 2.49–2.45 (m, 1H), 1.71 (s, 3H, CH3), 1.29 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.23 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.10, 1.06, 1.03, 0.90, 0.87 (each s, each 3H, CH3), 0.76 (d, 1H, J = 9.8 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 151.77 (C-20), 110.33 (C-29), 105.49 (C-1′), 103.05 (Rha-C-1), 102.00 (Rha-C-1), 90.39, 80.36, 79.21, 78.14, 76.43, 73.93, 73.68, 72.43, 72.12, 72.00, 70.75, 70.00, 61.96, 59.88, 57.38, 51.87, 50.13, 48.41, 43.78, 42.15, 40.35, 40.29, 38.88, 38.01, 35.77, 35.47, 31.07, 30.99, 30.77, 28.40, 28.35, 27.37, 26.59, 22.00, 19.42, 19.28, 18.02, 17.90, 17.00, 16.90, 16.58, 15.33. HRMS (MALDI) m/z: calcd for C49H82O15Na [M + Na]+, 933.5551; found, 933.5573.
4.1.16. 28-Chloro-3β-O-[2, 4-di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-lup-20 (29)-ene-3-al (BA-21)
To a solution of 21 (0.20 g, 0.16 mmol, 1.0 eq) in dry CH2Cl2 (10 mL) was added SOCl2 (18 µL, 0.21 mmol) at 0 ℃ under N2 atmosphere. After the mixture was stirred at r.t. for 12 h, the reaction was quenched with NaHCO3 at 0 ℃. Then, the reaction mixture was concentrated under vacuum. The residue was dissolved in EtOAc (60 mL), then extracted with water and brine, dried over Na2SO4, and concentrated in reduced pressure. The obtained crude product was re-dissolved in MeOH (10 mL) and CH2Cl2 (10 mL), CH3ONa was added until pH = 10. The reactant was then held at 30 °C until TLC indicated the reaction was complete. Dowex 50 × 8 (H + ) resin was added until pH = 7. Removal of the precipitate by filtration provided a yellowish solution, which was concentrated under vacuum. The residue was purified by silica gel column chromatography (CH2Cl2-MeOH, 6:1) to yield BA-21 (0.11 g, 76 % for two steps). 1H NMR (600 MHz, CD3OD): δ 5.38 (s, 1H, Rha-H-1), 4.85 (s, 1H, Rha-H-1), 4.70 (s, 1H, C = CH2-1), 4.63 (s, 1H, C = CH2-2), 4.45 (d, 1H, J = 7.7 Hz, H-1′), 4.04–3.95 (m, 2H), 3.93 (dd, 1H, J = 9.5, 7.4 Hz, H-2′), 3.89–3.78 (m, 2H), 3.77 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.73–3.59 (m, 2H), 3.55–3.56 (m, 1H), 3.53–3.43 (m, 2H), 3.47–3.36 (m, 2H), 3.34–3.31 (m, 2H), 3.18 (dd, 1H, J = 11.7, 4.6 Hz, H-3), 3.03–3.01 (m, 1H), 2.90–2.88 (m, 1H), 2.04–1.94 (m, 1H), 1.32–1.30 (m, 2H), 1.23 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.05 (d, 3H, J = 6.2 Hz, Rha-H-6), 0.98, 0.94, 0.92, 0.87, 0.85 (each s, each 3H, CH3), 0.75 (d, 1H, J = 9.7 Hz, H-5); 13C NMR (151 MHz, CD3OD): δ 150.19 (C-20), 108.89 (C-29), 104.25 (C-1′), 101.01 (Rha-C-1), 100.45 (Rha-C-1), 88.45, 87.20, 77.39, 76.90, 75.68, 72.39, 71.19, 71.09, 70.74, 70.62, 69.20, 69.17, 68.55, 60.59, 55.97, 50.93, 46.63, 41.37, 40.72, 39.16, 38.97, 36.94, 36.53, 36.37, 36.27, 34.72, 34.21, 33.90, 33.16, 29.27, 27.68, 26.46, 26.43, 26.36, 26.31, 24.71, 21.25, 18.32 (two), 18.10, 16.89, 16.44, 15.86, 13.75, 11.84. HRMS (MALDI) m/z: calcd for C48H79O14ClNa M + Na]+, 937.5056; found, 937.5072.
4.2. Biology assay
4.2.1. Cell lines and plasmids
HEK-293 T (Human, embryonic kidney) and Vero-E6 (African green monkey, kidney) cells were cultured in Dulbecco's Modified Eagle Medium (Gibco) supplemented with 10 % fetal bovine serum (Capricorn Scientific) and 1 % penicillin (100 units/mL) /streptomycin (100 μg/mL) (Gibco, USA). 293 T-ACE2 (293 T cells stably expressing human ACE2) were constructed by our laboratory and cultivated under the same conditions as above.
Plasmid pcDNA3.1-SARS-CoV-2-Sipke and pAAV-IRES-GFP-SARS-CoV-2-Sipke were given by the laboratory of Professor Shibo Jiang. Plasmid pAAV-IRES-EGFP was purchased from Hedgehogbio Science and Technology ltd. Expression plasmids for full-length vesicular stomatitis virus (VSV) glycoprotein (VSV-G) and pNL4-3.Luc.R-E- plasmids were obtained from Addgene (Cambridge, MA). Based on Plasmid pcDNA3.1-SARS-CoV-2 Spike, its mutants N501Y, D614G and Delta were all retained in our laboratory. Plasmid pcDNA3.1-SARS-CoV-2-Omicron and its 2 mutant plasmids were constructed by our laboratory. In brief, primers containing mutant sites were designed to amplify the specified DNA fragments with pcDNA3.1-SARS-CoV-2-Omicron as a template. The product obtained by PCR in the previous step was subjected to homologous recombination according to the manufacturer's instructions (Vazyme, China). The recombinant plasmids were used to transform stabl3 and inoculated in culture plates containing the corresponding resistance. After incubation in 37 ℃ for 12–16 h, single colonies on plates were selected and sequenced. Mutation sites and corresponding primers were shown in followed table [30]:
| 765-Foward | GCTTCTGCACCCAGCTGAAGGCAGCCCTGACCGGCATCGCCGTGG |
|---|---|
| 765-Reverse | CTTCAGCTGGGTGCAGAAGCTGCC |
| 964-Foward | CCAGGCCCTGAACACCCTGGTGGCGCAGCTGTCCAGCAAGTTCGG |
| 964-Reverse | CACCAGGGTGTTCAGGGCCTGG |
| Circ-Foward | GCGTGAAGCTGCACTACACCGGCGGCACCGAGACATCTCAGG |
| Circ-Reverse | GGCTAGCACGGAAGCGACCAGCATC |
4.2.2. Pseudotyped SARS-CoV-2 infection assay
HEK293T cells were seeded in 6-well plates and cultured overnight at 37 °C. 1 µg pNL4-3.Luc.R-E-plasmid and 0.5 µg pcDNA3.1-SARS-CoV-2-S plasmid were transfected into 293 T cells, and the supernatant virus liquid was collected after culturing at 37 °C for 48 h. 293 T-ACE2 cells were seeded in 96-well cell plates one day before infection. The concentration gradient drug and SARS-CoV-2 pseudovirus were mixed for 30 min at room temperature, and then added to the cells for 48 h of infection. Cells were lysed and luciferase activities were quantified by Luciferase assay system (Promega, USA) [30].
4.2.3. Cytotoxicity assay
Cells were seeded in a 96-well plate at a density of 1*104 cells/well, and cultured at 37 °C overnight. After 48 h of concentration gradient administration, 20 µL of MTT working solution (5 mg/mL) was added to each well, and cultured at 37 °C for 4 h. After discarding the culture supernatant, 150 µL of DMSO was added to each well, and the absorbance at 570 nm was measured by a microplate reader after sufficient shaking to dissolve. According to the measured OD value, the survival rate of cells under the action of the corresponding concentration of drugs compared with the control group was calculated, respectively [31].
4.2.4. Authentic SARS-CoV-2 inhibition assay
Authentic SARS-CoV-2 inhibition assay was performed by Wuhan institute of virology, Chinese academy of sciences. Vero-E6 cells were seeded in a 48-well plate at a cell density of 3*105 cells/well and cultured overnight at 37 °C, 5 % CO2. SARS-CoV-2 virus dilution (MOI = 0.05) and serially diluted drugs were pre-incubated at 37 °C for 1 h to infect cells. After that, the supernatant of the infectious material was fully removed and 200 µL of complete medium was added to each well to continue the culture. After 24 h, 150 µL of cell culture supernatant was collected and viral RNA was extracted with an RNA extraction kit (Takara, Japan). The reverse transcribed product was determined by qRT-PCR for viral copy number in the supernatant (Takara TB Green® Premix Ex Taq™ II, Japan) [31].
4.2.5. Co-immunoprecipitation and western blotting
HEK-293 T cells were seeded in six-well plates at a density of 4*105 cells/well one day in advance. 2 ug plasmids pcDNA3.1-ACE2-Flag and 2 ug pcDNA3.1-SARS-Omicron were co-transfected into each well and drugs were added in at the same time. After 48 h, total cell protein was extracted and incubated with protein A Sepharose bound by anti-labeled antibody or mouse IgG. The protein samples were separated by polyacrylamide gel electrophoresis after 12–16 h incubation with the antibody and were transferred to nitrocellulose membranes (Roche, Germany). SARS-Omicron and ACE2 were detected by anti-SARS-CoV-S (Sinol biological Inc., China) and anti-Flag (Sigma, USA) with mouse anti-goat-horseradish peroxidase (HRP) (Fude biological Technology Co., ltd., China) as the secondary antibody [31].
4.2.6. Cell-cell fusion assays
HEK-293 T cells were seeded in 6-well plates at a density of 4*105/well and cultured overnight. After transfection with pAAV-IRES-GFP-SARS-CoV-2-S plasmid expressing both SARS-CoV-2-S protein and green fluorescent protein GFP or pAAV-IRES-GFP vector plasmid, the cells were cultured at 37 °C for 48 h. Target cells Vero-E6 were seeded in 96-well plates at a density of 1*104 cells/well 6 h before cell fusion experiments. 293 T/SARS-CoV-2-S/EGFP or 293 T/EGFP effector cells were incubated with the concentration gradient drug for 30 min and then added to the target cells. After 24 h, three random fields were imaged by inverted fluorescence microscope (Zeiss, Germany) [19].
4.2.7. Surface plasmon resonance (SPR) measurement
Compound BEA-1, or BEA-4 was fixed on the chip by photo-crosslinking, then recombinant SARS-CoV-2 S-trimer protein (DRA 47, Novoprotein Inc. Shanghai) at indicated concentrations was injected sequentially into the chamber in buffer PBST (0.1 % Tween 20, pH 7.4). The interaction of S-trimer with BEA-1, or BEA-4 fixed was detected by PlexArrayTM HT SPRi (Seattle, US). The reaction temperature was controlled at 4 ℃, binding time was 600 s, disassociation time was 360 s, flow rate was 0.5 μL/s. The chip was regenerated with Glycine Hydrochloride (pH 2.0). The data of interaction signals was retrieved and analyzed with PlexeraDE software [30].
4.2.8. Circular dichroism (CD) spectroscopy
CD spectra were recorded on a Chirascan plus ACD (Applied Photophysics ltd, England). HR1P and HR2P were dissolved in buffer (0.1 M KCl, 0.05 M KH2PO4, pH 7.2) at a final concentration of 10 μM. Briefly, HR1P was incubated with PBS or BA-4 (20 μM) at 25 ℃ for 30 min, followed by addition of HR2P (10 μM). After further incubation at 25 ℃ for 30 min, the CD wave scans were measured from 190 to 260 nm at 4 ℃ with the bandwidth of 2 nm and the step size of 1 nm [18], [19].
4.2.9. Molecular docking
A molecular docking study was performed using Discovery Studio 3.0. The 3D crystal structure of SARS-CoV-2 spike glycoprotein was downloaded from RCSB Protein Date Bank (https://www.rcsb.org) using PDB IDs of 6VXX or 7TF8, water and glycosyl molecules removed by manual. The protein and the ligand were prepared by minimization with CHARMM force field. Then the binding site of the protein was defined and prepared for docking by using Define Site (From Receptor Cavities) protocol. Molecular docking results were carried out using CDOCKER protocol without constraint and ranked by -CDOCKER_ENERGY [37].
4.2.10. Plasma stability, microsomal stability and intestinal S9-UDPGA stability
Plasma stability was determined as following steps [38]: (1) prepare a 10 mM DMSO stock of BA-4. (2) Dilute the 10 mM stock to 1 μM with mouse plasma. (3) Transfer 50 μL plasma into a new tube and stop reaction using 250 μL acetonitrile. (4) Incubate the plasma sample in a water bath at 37 °C. (5) Stop reaction at 10, 30, 60, and 90 min, respectively. (6) Measure the compound concentration by LC-MS/MS; microsomal stability was determined using 10 μM BA-4 to incubate with mouse microsomes (0.5 mg/mL) for 5 min at 37 °C in phosphate buffer (100 mM, pH = 7.4) before 1 mM NADPH was added to start the reaction. Then, the cold acetonitrile was utilized to precipitate the protein. Lastly, the samples were centrifuged for further analysis by LC-MS/MS; the experimental procedures of mouse intestinal S9-UDPGA were similar as previously reported [39].
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82073722 to G.S. and 82130101 to S.L.), Guangdong Basic and Applied Basic Research Foundation (2022A1515010016) to G.S., the Major scientific and technological projects of Guangdong Province (2019B020202002) and Chinese Academy of Traditional Chinese Medicine (ZZ13-035-02, 2019XZZX-LG04) to S. L; Youth Innovative Talents Project from the Department of Education of Guangdong Province (2022KQNCX245) to X. W; and Science and Technology Program of Huizhou (2022CZ010192) to X. W.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2022.106316.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
No data was used for the research described in the article.
References
- 1.Bedford J., Enria D., Giesecke J., Heymann D.L., Ihekweazu C., Kobinger G., Lane H.C., Memish Z., Oh M.D., Sall A.A., Schuchat A., Ungchusak K., Wieler L.H. COVID-19: towards controlling of a pandemic. Lancet. 2020;395:1015–1018. doi: 10.1016/S0140-6736(20)30673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARSCoV-2 and COVID-19. Nat. Rev. Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou M.M., Liu Y., Cao J.Y., Dong S.Q., Hou Y.X., Yu Y., Zhang Q.Y., Zhang Y.L., Jia X.Y., Zhang B., Xiao G.F., Li G., Wang W. Bergamottin, a bioactive component of bergamot, inhibits SARS-CoV-2 infection in golden Syrian hamsters. Antivir. Res. 2022;204 doi: 10.1016/j.antiviral.2022.105365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann M., Krüger N., Schulz S., Cossmann A., Rocha C., Kempf A., Nehlmeier I., Graichen L., Moldenhauer A.S., Winkler M.S., Lier M., Dopfer-Jablonka A., Jäck H.M., Behrens G.M.N., Pöhlmann S. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell. 2022;185:447–456. doi: 10.1016/j.cell.2021.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan Y., Li X., Zhang L., Wan S., Zhang L., Zhou F. SARS-CoV-2 Omicron variant: recent progress and future perspectives. Signal. Transduct. Target. Ther. 2022;7:141. doi: 10.1038/s41392-022-00997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kundu S., Sarkar D. Synthetic attempts towards eminent anti-viral candidates of SARS-CoV. Mini. Rev. Med. Chem. 2022;22:232–247. doi: 10.2174/1389557521666210712205655. [DOI] [PubMed] [Google Scholar]
- 7.Jayk Bernal A., Gomes da Silva M.M., Musungaie D.B., Kovalchuk E., Gonzalez A., Delos Reyes V., Martín-Quirós A., Caraco Y., Williams-Diaz A., Brown M.L., Du J., Pedley A., Assaid C., Strizki J., Grobler J.A., Shamsuddin H.H., Tipping R., Wan H., Paschke A., Butterton J.R., Johnson M.G., De Anda C. MOVe-OUT study group. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 2022;386:509–520. doi: 10.1056/NEJMoa2116044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Najjar-Debbiny R., Gronich N., Weber G., Khoury J., Amar M., Stein N., Goldstein L.H., Saliba W. Effectiveness of paxlovid in reducing severe COVID-19 and mortality in high risk patients. Clin. Infect. Dis. 2022:ciac443. doi: 10.1093/cid/ciac443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drayman N., DeMarco J.K., Jones K.A., Azizi S.A., Froggatt H.M., Tan K., Maltseva N.I., Chen S., Nicolaescu V., Dvorkin S., Furlong K., Kathayat R.S., Firpo M.R., Mastrodomenico V., Bruce E.A., Schmidt M.M., Jedrzejczak R., Muñoz-Alía M.Á., Schuster B., Nair V., Han K.Y., O'Brien A., Tomatsidou A., Meyer B., Vignuzzi M., Missiakas D., Botten J.W., Brooke C.B., Lee H., Baker S.C., Mounce B.C., Heaton N.S., Severson W.E., Palmer K.E., Dickinson B.C., Joachimiak A., Randall G., Tay S. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science. 2021;373:931–936. doi: 10.1126/science.abg5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan H.Z., Hu Y.M., Jadhav P., Tan B., Wang J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J. Med. Chem. 2022;65:7561–7580. doi: 10.1021/acs.jmedchem.2c00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M.A., Young H.S., Tyrrell D.L., Vederas J.C., Lemieux M.J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.L.Y ang, R.J. Pei, H. Li, X.N. Ma, Y. Zhou, F.H. Zhu, P.L. He, W. Tang, Y.C. Zhang, J. Xiong, S.Q. Xiao, X.K. Tong, B. Zhang, J.P. Zuo, Identification of SARS-CoV-2 entry inhibitors among already approved drugs. Acta Pharmacol. Sin. 2021;42:1347–1353. doi: 10.1038/s41401-020-00556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.S. Yu, X. Zheng, B. Zhou, J. Li, M. Chen, R. Deng, G. Wong, D. Lavillette, G. Meng, SARS-CoV-2 spike engagement of ACE2 primes S2' site cleavage and fusion initiation, Proc. Natl. Acad. Sci. U. S. A. 119 (2022) e2111199119. [DOI] [PMC free article] [PubMed]
- 14.Gur M., Taka E., Yilmaz S.Z., Kilinc C., Aktas U., Golcuk M. Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states. J. Chem. Phys. 2020;153:75101. doi: 10.1063/5.0011141. [DOI] [PubMed] [Google Scholar]
- 15.Braga L., Ali H., Secco I., Chiavacci E., Neves G., Goldhill D., Penn R., Jimenez-Guardeño J.M., Ortega-Prieto A.M., Bussani R., Cannatà A., Rizzari G., Collesi C., Schneider E., Arosio D., Shah A.M., Barclay W.S., Malim M.H., Burrone J., Giacca M. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature. 2021;594:88–93. doi: 10.1038/s41586-021-03491-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schütz D., Ruiz-Blanco Y.B., Münch J., Kirchhoff F., Sanchez-Garcia E., Müller J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug. Deliv. Rev. 2020;167:47–65. doi: 10.1016/j.addr.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia S., Liu M.Q., Wang C., Xu W., Lan Q.S., Feng S.L., Qi F.F., Bao L.L., Du L.Y., Liu S.W., Qin C., Sun F., Shi Z.L., Zhu Y., Jiang S.B., Lu L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia S., Lan Q.S., Zhu Y., Wang C., Xu W., Li Y.T., Wang L.J., Jiao F., Zhou J., Hua C., Wang Q., Cai X., Wu Y., Gao J., Liu H., Sun G., Münch J., Kirchhoff F., Yuan Z.H., Yo H., Xie F., Sun S.B., Jiang L.L. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal. Transduct. Target. Ther. 2021;6:288. doi: 10.1038/s41392-021-00712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drożdżal S., Rosik J., Lechowicz K., Machaj F., Szostak B., Przybyciński J., Lorzadeh S., Kotfis K., Ghavami S., Łos M.J. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updat. 2021;59 doi: 10.1016/j.drup.2021.100794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan S.F., Yin X., Meng X.Z., Chan J.F., Ye Z.W., Riva L., Pache L., Chan C.C., Lai P.M., Chan C.C., Poon V.K., Lee A.C., Matsunaga N., Pu Y., Yuen C.K., Cao J., Liang R., Tang K., Sheng L., Du Y.S., Xu W., Lau C.Y., Sit K.Y., Au W.K., Wang R.M., Zhang Y.Y., Tang Y.D., Clausen T.M., Pihl J., Oh J., Sze K.H., Zhang A.J., Chu H., Kok K.H., Wang D., Cai X.H., Esko J.D., Hung I.F., Li R.A., Chen H., Sun H., Jin D.Y., Sun R., Chanda S.K., Yuen K.Y. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature. 2021;593:418–423. doi: 10.1038/s41586-021-03431-4. [DOI] [PubMed] [Google Scholar]
- 22.Cao J.Y., Liu Y., Zhou M.M., Dong S.Q., Hou Y.X., Jia X.Y., Lan X.H., Zhang Y.L., Guo J., Xiao G.F., Wang W. Screening of botanical drugs against SARS-CoV-2 entry reveals novel therapeutic agents to treat COVID-19. Viruses. 2022;14:353. doi: 10.3390/v14020353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C., Pan X.Y., Xu X.F., Cheng C., Huang Y., Li L., Jiang S.B., Xu W., Xiao G.F., Liu S.W. Salvianolic acid C potently inhibits SARS-CoV-2 infection by blocking the formation of six-helix bundle core of spike protein. Signal. Transduct. Target. Ther. 2020;5:220. doi: 10.1038/s41392-020-00325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang C., Pan X.Y., Huang Y., Cheng C., Xu X.F., Wu Y., Xu Y.X., Shang W.J., Niu X.G., Wan Y.H., Li Z.F., Zhang R., Liu S.W., Xiao G.F., Xu W. Drug repurposing of itraconazole and estradiol benzoate against COVID-19 by blocking SARS-CoV-2 spike protein-mediated membrane fusion. Adv Ther (Weinh). 2021;4:2000224. doi: 10.1002/adtp.202000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J., Wei W.H., Zhang X.F., Cao S.Q., Hu B.T., Ye Y., Jiang M., Wang T.Q., Zuo J.P., He S.J., Yang C.H. Synthesis and biological evaluation of C-17-amino-substituted pyrazole-fused betulinic acid derivatives as novel agents for osteoarthritis treatment. J. Med. Chem. 2021;64:13676–13692. doi: 10.1021/acs.jmedchem.1c01019. [DOI] [PubMed] [Google Scholar]
- 26.Wang H., Xu R.Y., Shi Y.Y., Si L.L., Jiao P.X., Fan Z.B., Han X., Wu X.Y., Zhou X.S., Yu F., Zhang Y.M., Zhang L., Zhang L.R., Zhou D.M., Xiao S.L. Design, synthesis and biological evaluation of novel L-ascorbic acid-conjugated pentacyclic triterpene derivatives as potential influenza virus entry inhibitors. Eur. J. Med. Chem. 2016;110:376–388. doi: 10.1016/j.ejmech.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Visalli R.J., Ziobrowski H., Badri K.R., He J.J., Zhang X.G., Arumugam S.R., Zhao H. Ionic derivatives of betulinic acid exhibit antiviral activity against herpes simplex virus type-2 (HSV-2), but not HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2015;25:3168–3171. doi: 10.1016/j.bmcl.2015.05.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen C.C., Kuo Y.H., Jan J.T., Liang P.H., Wang S.Y., Liu H.G., Lee C.K., Chang S.T., Kuo C.J., Lee S.S., Hou C.C., Hsiao P.W., Chien S.C., Shyur L.F., Yang N.S. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007;50:4087–4095. doi: 10.1021/jm070295s. [DOI] [PubMed] [Google Scholar]
- 29.Stevaert A., Krasniqi B., Van Loy B., Nguyen T., Thomas J., Vandeput J., Jochmans D., Thiel V., Dijkman R., Dehaen W., Voet A., Naesens L. Betulonic acid derivatives interfering with human coronavirus 229E replication via the nsp15 endoribonuclease. J. Med. Chem. 2021;64:5632–5644. doi: 10.1021/acs.jmedchem.0c02124. [DOI] [PubMed] [Google Scholar]
- 30.Li H., Cheng C., Shi S.S., Wu Y., Gao Y.F., Liu Z.H., Liu M.J., Li Z.D., Huo L.J., Pan X.Y., Liu S.W., Song G.P. Identification, optimization, and biological evaluation of 3-O-β-chacotriosyl ursolic acid derivatives as novel SARS-CoV-2 entry inhibitors by targeting the prefusion state of spike protein. Eur. J. Med. Chem. 2022;238 doi: 10.1016/j.ejmech.2022.114426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H., Cheng C., Li S.M., Wu Y., Liu Z.H., Liu M.J., Chen J.X., Zhong Q.Y., Zhang X.S., Liu S.W., Song G.P. Discovery and structural optimization of 3-O-β-chacotriosyl oleanane-type triterpenoids as potent entry inhibitors of SARS-CoV-2 virus infections. Eur. J. Med. Chem. 2021;215 doi: 10.1016/j.ejmech.2021.113242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai W.H., Zhang B., Jiang X.M., Su H.X., Li J., Zhao Y., Xie X., Jin Z.M., Peng J.J., Liu F.J., Li C.P., Li Y., Bai F., Wang H.F., Cheng X., Cen X.B., Hu S.L., Yang X.N., Wang J., Liu X., Xiao G.F., Jiang H.L., Rao Z.H., Zhang L.K., Xu Y.C., Yang H.T., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Si L.L., Meng K., Tian Z.Y., Sun J.Q., Li H.Q., Zhang Z.W., Soloveva V., Li H.W., Fu G., Xia Q., Xiao S.L., Zhang L.H., Zhou D.M. Triterpenoids manipulate a broad range of virus-host fusion via wrapping the HR2 domain prevalent in viral envelopes. Sci Adv. 2018;4:eaau8408. doi: 10.1126/sciadv.aau8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song G.P., Shen X.T., Li S.M., Li Y.B., Liu Y.P., Zheng Y.S., Lin R.H., Fan J.H., H.M. Ye,W. Liu, Structure-activity relationships of 3-O-β-chacotriosyl ursolic acid derivatives as novel H5N1 entry inhibitors. Eur. J. Med. Chem. 2015;93:431–442. doi: 10.1016/j.ejmech.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 35.Wolfram R.K., Heller L., Csuk R. Targeting mitochondria: esters of rhodamine B with triterpenoids are mitocanic triggers of apoptosis. Eur. J. Med. Chem. 2018;152:21–30. doi: 10.1016/j.ejmech.2018.04.031. [DOI] [PubMed] [Google Scholar]
- 36.Brandes B., Hoenke S., Fischer L., Csuk R. Design, synthesis and cytotoxicity of BODIPY FL labelled triterpenoids. Eur. J. Med. Chem. 2020;185 doi: 10.1016/j.ejmech.2019.111858. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z.H., Gu S.Y., Zhu X., Lu M.J., Cao Z.Q., Qiu P.S., Li S.M., Liu S.W., Song G.P. Discovery and optimization of new 6, 7-dihydroxy-1, 2, 3, 4-tetrahydroisoquinoline derivatives as potent influenza virus PAN inhibitors. Eur. J. Med. Chem. 2022;227 doi: 10.1016/j.ejmech.2021.113929. [DOI] [PubMed] [Google Scholar]
- 38.Wang L., Jiang J.S., Zhang L.X., Zhang Q.Y., Zhou J.R., Li L., Xu X.L., You Q.D. Discovery and optimization of small molecules targeting the protein-protein interaction of heat shock protein 90 (Hsp90) and cell division cycle 37 as orally active inhibitors for the treatment of colorectal cancer. J. Med. Chem. 2020;63:1281–1297. doi: 10.1021/acs.jmedchem.9b01659. [DOI] [PubMed] [Google Scholar]
- 39.Kazmierski W.M., Xia B., Miller J., De la Rosa M., Favre D., Dunham R.M., Washio Y., Zhu Z., Wang F., Mebrahtu M., Deng H., Basilla J., Wang L., Evindar G., Fan L., Olszewski A., Prabhu N., Davie C., Messer J.A., Samano V. DNA-Encoded library technology-based discovery, lead optimization, and prodrug strategy toward structurally unique indoleamine 2,3-dioxygenase-1 (IDO1) inhibitors. J. Med. Chem. 2020;63:3552–3562. doi: 10.1021/acs.jmedchem.9b01799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.