

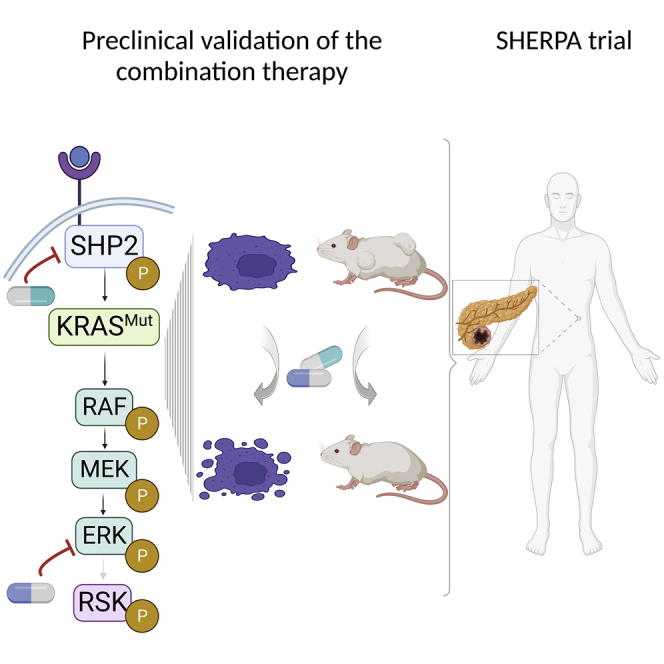
Summary
Over 90% of pancreatic cancers present mutations in KRAS, one of the most common oncogenic drivers overall. Currently, most KRAS mutant isoforms cannot be targeted directly. Moreover, targeting single RAS downstream effectors induces adaptive resistance mechanisms. We report here on the combined inhibition of SHP2, upstream of KRAS, using the allosteric inhibitor RMC-4550 and of ERK, downstream of KRAS, using LY3214996. This combination shows synergistic anti-cancer activity in vitro, superior disruption of the MAPK pathway, and increased apoptosis induction compared with single-agent treatments. In vivo, we demonstrate good tolerability and efficacy of the combination, with significant tumor regression in multiple pancreatic ductal adenocarcinoma (PDAC) mouse models. Finally, we show evidence that 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) can be used to assess early drug responses in animal models. Based on these results, we will investigate this drug combination in the SHP2 and ERK inhibition in pancreatic cancer (SHERPA; ClinicalTrials.gov: NCT04916236) clinical trial, enrolling patients with KRAS-mutant PDAC.
Keywords: pancreatic cancer, preclinical models, Kras, targeted therapies, SHP2, ERK, PDAC, combination therapy, MAPK, inhibitors
Graphical abstract
Highlights
-
•
SHP2 and ERK co-inhibition is synergistic and triggers apoptosis in PDAC in vitro
-
•
The combination is tolerated and promotes tumor regression in multiple in vivo models
-
•
Non-invasive PET-CT scans can monitor early response to the therapy
-
•
The SHERPA phase 1a/1b trial will investigate the combination clinically
KRAS mutant pancreatic tumors have poor prognosis and few therapeutic options. Here, Frank et al. show that the combination of RMC4550 (SHP2 inhibitor) and LY3214996 (ERK inhibitor) effectively impairs tumor growth and induces tumor regression in multiple in vivo models of PDAC.
Introduction
Pancreatic cancer is the third leading cause of cancer-related deaths in Western countries and the seventh worldwide and is predicted to become the second most common cause of cancer mortality in the US in the next 20 to 30 years.1,2,3 The 5-year survival rate of patients suffering from pancreatic ductal adenocarcinoma (PDAC) is only 10% as diagnosis is often made when disease is already advanced, and therapeutic options are limited. Over the last years, the genomic landscape of PDAC has emerged, and recurrent mutations have been identified.4,5 Recently, the discovery of a subset of patients with PDAC bearing germline alterations in BRCA1/2 and PALB2, which cause homologous repair deficiency (HRD), has led to the approval of PARP inhibitors as the first targeted therapy for HRD-pancreatic cancer.6 However, BRCA1/2 and PALB2 mutants are present in only 5%–9% of patients with PDAC7 compared with over 90% of patients that carry tumors bearing KRAS mutations, and, to date, no other non-cytotoxic, targeted therapies have been approved.
It has been widely demonstrated that KRAS mutations constitute an early initiating event in the pancreatic tumorigenic process8 and that pancreatic adenocarcinomas retain a high dependency on RAS signaling,9,10 thus making KRAS the ideal therapeutic target for pancreatic cancer. However, for more than three decades, research on the direct targeting of RAS has proven to be a very challenging task. Only recently have KRASG12C-specific inhibitors entered clinical development,11,12,13 and some, like sotorasib and adagrasib, have shown initial clinical responses,14,15,16 leading to sotorasib being the first drug approved by the US Food and Drug Administration (FDA) for the treatment of KRASG12C-driven non-small cell lung cancer.
Unfortunately, the G12C variant represents only 1% of KRAS mutations in pancreatic cancer, with the most frequent amino acid substitutions being G12D (41%), G12V (34%), and G12R (16%).17 For those most common mutations, targeted inhibitors have not yet been developed; therefore, most translational studies have been aimed at blocking downstream RAS effectors mainly in the MAPK or PI3K-AKT pathways.18,19,20,21 Unfortunately, attempts to target RAS downstream effectors have been hampered by compensatory feedback mechanisms, often involving reactivation of receptor tyrosine kinases.22 This notion, together with advances in KRAS biophysics and structural biology studies, undermined the old paradigm of mutant KRAS being constitutively active and made it clear that it is possible to reduce mutant RAS activation by combining inhibition of upstream and downstream nodes in the RAS-MAPK pathway. In particular, the ubiquitously expressed non-receptor protein tyrosine phosphatase SHP2, encoded by the PTPN11 gene, has been identified as a useful upstream target23,24,25 as it is involved in signal transduction downstream of multiple growth factor, cytokine, and integrin receptors.26 Potent and specific allosteric inhibitors of SHP2 have recently been developed and have entered clinical trials,27 holding great promise for receptor tyrosine kinase (RTK)-driven tumors. Nonetheless, so far, the question regarding the most beneficial drug combination for KRAS-driven pancreatic cancer remains.
MEK inhibitors have been tested extensively in KRAS-mutant PDAC, as well as other solid tumors, with poor results.28,29,30 This is primarily attributable to their highly toxic profile and adverse side effects as well as the above-mentioned feedback reactivation of the MAPK pathway.22,31 In contrast, inhibitors of ERK, directly downstream of MEK, have only recently been introduced into clinical studies32 and seem auspicious with regard to their toxicity profile. In the present study, we explore the tolerability and efficacy of combining the allosteric SHP2 inhibitor RMC-4550 with the ATP-competitive, selective ERK inhibitor LY3214996 in multiple in vitro and in vivo models of murine and human PDAC. Based on the data reported here, we developed the phase 1a/1b SHP2 and ERK inhibition in pancreatic cancer (SHERPA) clinical trial (ClinicalTrials.gov: NCT04916236).
Results
Combinatorial effect in vitro
In a previous study, we showed promising in vitro and in vivo results to support combined SHP2 and MEK inhibition for the treatment of pancreatic cancer.23,24 While the reported combinatorial strategy is sound, the frequently observed side effects and the common occurrence of resistance associated with MEK inhibitors like trametinib31,33,34 led us to search for possible alternatives. Since reactivation of ERK is a major mechanism hampering MEK inhibitor (MEKi) efficacy,22,33,35,36 we hypothesized that direct ERK inhibition might be a worthwhile strategy. The recently developed ERK inhibitor LY321499637 was shown to be a selective, potent, and reversible ATP-competitive inhibitor of ERK1/2 activity in KRAS- and BRAF-mutant cell lines. In parallel, RMC-4550 a selective allosteric SHP2 inhibitor was developed with a mode of action similar to the Novartis’ SHP099 but with slightly higher potency.38
Since LY3214996 and RMC-4550 have not yet been studied in combination, we first investigated the effects of combined treatment on MAPK activity, proliferation, and apoptosis in vitro. First, we analyzed the inhibitors’ capacity to block MAPK pathway activity. As SHP2 inhibitors alone have already proven not to hamper MAPK pathway activation in KRAS-mutant tumors under standard in vitro culture conditions,23 we decided to treat murine and human KRAS-mutant PDAC cell lines with either LY3214996 alone or LY3214996 + RMC-4550 for 6, 72, and 144 h. Western blot analysis was performed to examine protein expression levels of phosphorylated ribosomal S6 kinase 1 (pRSK-1), a direct downstream target of ERK.39 Compared with LY3214996 monotherapy, the combined RMC-4550 treatment inhibited MAPK pathway activity more effectively at all time points analyzed (Figure 1A), and, importantly, it was able to prevent the feedback reactivation of the route, which occurs at late time points when inhibiting ERK alone. After 6 days (144 h), a 100%, 80%, and 70% reduction in pRSK-1 levels compared with the control could still be observed for LY3214996 + RMC-4550-treated murine Kras;Trp53−/− (KCP) K2101 as well as human MiaPaCa-2 and Panc10.05 cells, respectively, compared with only 20%, 40%, and 30% reduction in the same cells with LY3214996 monotherapy. Of note, and as expected, the combined LY3214996 + RMC-4550 treatment also proved superior to the RMC-4550 monotherapy by preventing reactivation of the pathway in MiaPaCa-2 cells, as shown by pRSK-1 levels in Figure S1A.
Figure 1.

Evaluation of the combined effects of RMC-4550 (SHP2i) and LY3214996 (ERKi) administration in murine and human KRAS-mutant pancreatic cancer cell lines
(A) Western blot analysis with murine cancer cell line KCP_K2101 derived from KCP mouse model (KRASG12D) of spontaneous tumor formation and in human cancer cell lines: MiaPaCa-2 (KRASG12C) and Panc10.05 (KRASG12D). Cells were treated as depicted and collected for lysis at the indicated time points. Protein extracts were probed with specific antibodies against total RSK-1, phosphorylated RSK-1 (pRSK-1), and alpha-tubulin (as loading control). Numerical values indicate the pRSK-1/RSK-1 ratio quantified by densitometry. The blots are representative of at least three independent experiments. RSK-1, ribosomal S6 kinase 1.
(B) Synergistic effects of SHP2i and ERKi administration were evaluated by colony-formation assay in the KRAS-mutant cell lines used in (A). SHP2i and ERKi were combined at the indicated concentrations. Representative crystal violet staining of cells is shown (top panel). Box matrices below the plate scans depict quantification of growth inhibition in relation to control wells (middle panel). Bottom panel: calculation of the combination index (CI) scores from the growth inhibition values (shown above) via CompuSyn software demonstrating strong synergism between SHP2i and ERKi across a wide range of combinatorial concentrations. CI <0.75 (shades of green) indicates synergism, CI = 0.75–1.25 (shades of blue) indicates additive effects, and CI >1.25 (shades of red) indicates antagonism. Experiments were repeated independently at least three times each, with similar results.
(C) Apoptosis was analyzed in cell lines treated with either DMSO, SHP2i alone, ERKi alone, or a combination of SHP2i and ERKi at the indicated concentration in real time (top panel). GFP signal coupled to cleaved caspase 3 was quantified as readout. Bar plots for selected time points (48 h for KCP_K2101 and 76 h for MiaPaCa-2 and Panc10.05) show the fraction of GFP-positive cells (AU) (top panel) and the fold change GFP signal (bottom panel). AU, arbitrary units; GFP, green fluorescent protein. Experiments were repeated independently at least three times each. Results represent mean ± SD. ∗p < 0.05, ∗∗∗∗p < 0.0001, as determined by ordinary one-way ANOVA test.
See also Figure S1.
To better understand the potential clinical benefit of the combination therapy, we further tested the effect of LY3214996 + RMC-4550 treatment on cell proliferation (Figures 1B and S1B) and on the induction of cell death (Figure 1C). For the former, we performed 6-day colony-formation assays using two murine KCP and 5 different human PDAC cell lines, harboring KRASG12C, KRASG12V, or KRASG12D mutations (Figures 1B and S1B). The 96-well format allowed us to test a range of inhibitor concentrations both as monotherapy as well as in combination. Synergism (i.e., a combination index [CI] score below 0.75, indicated in shades of green in Figures 1B and S1B) was observed in at least 70% of all inhibitor combinations tested in all 7 cell lines, regardless of the type of KRAS mutation present. These data indicate that LY3214996 and RMC-4550 can synergistically inhibit PDAC cell growth in vitro at micromolar concentrations.
To study the effect of the drug combination on apoptosis, KCP_K2101, MiaPaCa-2, and Panc10.05 cells were treated with either LY3214996 or RMC-4550 alone at roughly half-maximal inhibitory concentration (IC50) concentrations or with the combination of the two inhibitors for up to 76 h, and the rate of apoptosis was measured by tracking GFP-labeled cleaved caspase 3 over time (Figure 1C, top panels). In murine KCP_K2101 cells, GFP-positive cleaved caspase 3 levels peaked after 48 h, and the fraction of caspase 3-positive cells was significantly higher in the combination-treated group compared with vehicle or LY3214996 and RMC-4550 monotherapy. Similarly, human cell lines MiaPaCa-2 and Panc10.05 showed a peak of GFP coupled to a cleaved caspase 3/7-specific recognition motif after 76 h, with a significant increase in combination-treated cells compared with either vehicle or monotherapies, indicating that apoptosis was triggered significantly with the combination. Based on the kinetics of the different cell lines, we identified the time point where apoptosis was maximally triggered to calculate the fold change in apoptosis for the monotherapies and combination treatment compared with vehicle (Figure 1C, bottom panels).
Taken together, our data show that LY3214996 and RMC-4550 act synergistically to both inhibit PDAC cell proliferation and MAPK signaling as well as induce significant levels of apoptosis in vitro, which prompted us to test the combination treatment in vivo.
In vivo tolerability
Due to the only recent development of both LY3214996 and RMC-4550, the scarcity of available in vivo data, as well as the non-existent data on combined toxicity, we performed a tolerability study in non-tumor-bearing wild-type (KrasLSLG12D;Trp53flox/flox - no Cre) and NOD-scid gamma (NSG) mice to determine the maximum tolerated dose (MTD) of combined LY3214996 and RMC-4550. The inhibitors were administered once per day via oral gavage for 14 consecutive days (Figure 2A). Following dosing recommendations by Eli Lilly and Revolution Medicines, we determined 9 different doses of inhibitor combinations labeled d1 (lowest) through d9 (highest), as illustrated in Figure 2B. We applied a modified “3 + 3” study design40,41 using cohorts of three animals per dose. As shown in Figure 2C, the first cohort was treated at a starting dose, and the subsequent cohorts were treated with ascending or descending doses according to the observed response. Dosing was increased until one or more mice per cohort experienced dose-limiting toxicities (DLTs). In case two or more mice experienced DLTs, the dose escalation was stopped, and the next lower dose, with no more than 1 in 6 mice showing signs of DLTs, was determined as the MTD. If only one in three mice experienced DLTs, the cohort was expanded to 6 mice, and the dose escalation continued if none of the additional three mice showed signs of DLTs; otherwise, the previous dose was determined as the MTD. Endpoints used as signs of DLT were weight loss of more than 20%, clinical score (abnormal behavior, signs of physical discomfort), and death. These parameters were evaluated daily, and animals were euthanized if either of these endpoints were met. Due to ethical and practical considerations, and to minimize the number of mice in the experiment, dose d5 was chosen as the starting dose. Figure 2 shows the body weight profile of both wild-type (Figure 2D) and NSG (Figure 2E) mice over the course of 14 days of treatment. All doses were well tolerated in wild-type mice (Figure 2D), while dose d9 (i.e., 100 mg/kg LY3214996 + 30 mg/kg RMC-4550) caused dose-limiting weight loss in NSG mice (Figure 2E).
Figure 2.

MTD study design
(A) Treatment schedule. Non-tumor-bearing wild-type and NOD scid gamma (NSG) mice were treated with the combination of RMC-4550 (SHP2i) and LY3214996 (ERKi) once per day via oral gavage for 14 consecutive days.
(B) Graphical representation of dose combinations. SHP2i and ERKi were combined in different concentrations to make up 9 combined doses.
(C) An illustration of the modified “3 + 3” study design. Each box represents a cohort comprising the indicated number of mice treated at a given dose level. DLT, dose-limiting toxicity; MTD, maximum tolerated dose.
(D) Individual body weight-time profile of the treatment groups in male wild-type (WT) mice: d5 (n = 3), d8 (n = 3), and d9 (n = 6).
(E) Individual body weight-time profile of the treatment groups in male NSG mice: d5 (n = 3), d8 (n = 3), and d9 (n = 3).
Thus, dose d8, i.e., 100 mg/kg LY3214996 + 10 mg/kg RMC-4550, was the highest dose that was well tolerated in both wild-type and NSG mice and was therefore used to assess anti-tumor efficacy.
In vivo efficacy
Once we found a well-tolerated dose for the combination therapy of LY3214996 + RMC-4550, we investigated its potential anti-tumor efficacy in vivo. First, we used a xenograft model of subcutaneously transplanted human PDAC cell lines (Figures 3A and 3B). Mice bearing tumors with a volume of approximately 200 mm3 were randomly assigned into either the vehicle, RMC-4550 (A), LY3214996 (B), or combination (C) cohort and were treated daily for 21 days via oral gavage (Figure 3A). While RMC-4550 alone was already partially effective at reducing MiaPaCa-2 xenograft tumor growth compared with vehicle, tumor volume reduction of more than 30% in 12 out of 16 mice was only achieved upon continuous combination treatment. We were able to confirm these results in a model of orthotopically transplanted KrasG12D/+; Trp53R172H/+; Pdx-1Cre tumors in immunocompetent C57BL/6J mice (KCPmut). Specifically, following post-surgical tumor expansion over 2 weeks, mice were randomized into either the baseline, vehicle, RMC-4550 (A), LY3214996 (B), or combination (C) cohort. Baseline mice were sacrificed to confirm the presence of well-integrated and uniform orthotopic tumors. All other mice were treated for 14 days via oral gavage as shown in Figure 3A. As Figures 3C and 3D show, significant inhibition in tumor growth, seen macroscopically and indicated by decreased tumor weight, was observed in both monotherapy groups as well as in the combination treatment group compared with the vehicle cohort. No difference in treatment efficacy was observed between weight-matched male and female mice (Figure 3D). Notably, and in agreement with the synergy described in vitro, the combination therapy (cohort C) was the most effective and induced a significantly stronger tumor volume reduction compared to LY3214996 or RMC-4550 monotherapies (Figures 3C and 3D). To confirm the on-target activity of the combination therapy, we show substantial reduction in transcriptional-based MAPK pathway activity score42 in vivo in both the orthotopic KCPmut tumors as well as the subcutaneous MiaPaCa-2 xenografts (Figure 3E) treated with continuous LY3214996 + RMC-4550 compared with vehicle-treated controls.
Figure 3.

In vivo assessment of treatment response in a xenograft and in an orthotopic PDAC model
(A) Schematic representation of the treatment schedule applied in a xenograft model of subcutaneously transplanted MiaPaCa-2 cell line and in a model of orthotopically transplanted KCPmut tumors. Cohort A: continuous treatment with RMC-4550 (SHP2i) alone daily (n = 15); cohort B: continuous treatment with LY3214996 (ERKi) alone daily (n = 15); and cohort C: continuous treatment with the combination of SHP2i and ERKi daily (n = 16). Control mice were continuously treated with vehicle (n = 15).
(B) Evaluation of ERKi and SHP2i monotherapy treatments and combined administration of SHP2i and ERKi. For all the xenograft experiments, 5 × 106 MiaPaCa-2 cells were subcutaneously injected into the right flank of NSG mice. When tumors reached 200–250 mm3, mice were randomly assigned into cohorts and treated by oral gavage with inhibitors or vehicle according to treatment schedule for 21 days, after which tumors were resected. The y axis shows tumor volume change in percentage from baseline. Each bar represents the difference in pancreatic volume in an individual animal. According to the RECIST criteria, black indicates progressive disease, dark gray indicates stable disease, and light gray indicates partial response. Significance was determined by one-way ANOVA with Bonferroni’s multiple comparison test.
(C and D) In vivo assessment of treatment response of orthotopically implanted tumors. ∼40 mm3 tumor pieces (KCPmut) were orthotopically implanted into the pancreata of 8-week-old male and female C57BL/6 mice. After 2 weeks, mice were either sacrificed as baseline (n = 12) or randomly assigned into cohorts and treated with inhibitors or vehicle according to the treatment schedule (A).
(C) Tumor weight (mean ± SD) was determined after 14 days of therapy as indicated: baseline (n = 12), vehicle (n = 17), cohort A (n = 7), cohort B (n = 8), cohort C (n = 12). ∗∗∗∗p < 0.0001, as determined by one-way ANOVA with Bonferroni’s multiple comparison test.
(D) Representative macroscopic photographs of tumors in (C).
(E) MAPK pathway activity scores in MiaPaCa-2 xenograft and in KCPmut orthotopic mouse models.
See also Figures S2 and S6.
While the lack of effectiveness of LY3214996 single treatment in vivo was congruent with the negligible effects we observed in vitro in terms of cell proliferation and pathway inhibition, the same was not true for RMC-4550, which also showed very little effect in vitro but induced a substantial impairment of tumor growth, in multiple in vivo models. Differences in response to SHP2 inhibitors in in vivo versus in vitro models have been previously attributed, in the context of KRAS mutant tumors, to either the serum concentration (accounting for growth factor availability)23 or to the tridimensional versus bidimensional growth.43 In order to better understand which factors played a role in our context, we grew MiaPaCa-2 and Panc10.05 cells in the presence of 10% or 3% serum, both in normal and ultra-low attachment 96-well plates. We then analyzed the response to RMC-4550 treatment using the CellTiter-Glo 3D assay as a readout for cell viability. As shown in Figure S2A, tridimensional growth seems to be the main factor influencing the response to the SHP2 inhibitor in the PDAC cell lines. In particular, MiaPaCa-2 and Panc10.05 were equally unresponsive to SHP2 inhibition in the presence of high or low serum concentrations in two-dimensional (2D) culture conditions. Nevertheless, cell viability was strongly impaired when both cell lines were treated with RMC-4550 in 3D conditions. Additionally, for Panc10.05, a significant difference in sensitivity to RMC-4550 was also observed in 3% versus 10% serum but only in the context of 3D growth.
Overall, despite RMC-4550 showing a stronger anti-tumor effect in vivo compared with in vitro, we could still observe a synergistic effect when used in combination with LY3214996 in multiple mouse models.
Optimal treatment regimen
Having shown a potent anti-tumor benefit of the LY3214996 + RMC-4550 combination treatment, compared with the monotherapies in MiaPaCa-2 xenografts and orthotopic KCPmut tumors, we decided to further expand our validation in additional PDAC models of murine and human origin and, at the same time, compare intermittent treatment schedules. With regards to future clinical application and the possibility of adverse effects in patients, we wanted to determine the optimal treatment regimen, defined as maximum anti-tumor effect with minimum toxicity. For the LY3214996 + RMC-4550 combination, these schedules included continuous administration (daily) of both drugs in combination (cohort C), as well as three different non-continuous (intermittent) schedules (cohorts D, E, and F). Control cohorts included daily vehicle treatment, as well as intermittent LY3214996 or RMC-4550 monotherapy (cohorts G, H, and I) (Figure 4A).
Figure 4.

In vivo assessment of optimal treatment regimen in a xenograft model
(A) Schematic representation of the treatment schedule applied in MiaPaCa-2 xenograft model. Cohort A: continuous treatment with SHP2i alone daily; cohort B: continuous treatment with ERKi alone daily; cohort C: continuous treatment with the combination of SHP2i and ERKi daily; cohort D: intermittent treatment with the combination of SHP2i and ERKi 5 days on/2 days off; cohort E: semi-continuous treatment schedule with daily dosing of SHP2i and intermittent dosing with ERKi 5 days on/2 days off; cohort F: continuous treatment with SHP2i and on alternate days with ERKi; cohort G: intermittent dosing with SHP2i alone 5 days on/2 days off; cohort H: intermittent dosing with ERKi alone 5 days on/2 days off; and cohort I: treatment with ERKi alone on alternate days. Control mice were continuously treated with vehicle. For all the xenograft experiments, 5 × 106 cells were subcutaneously injected into the right flank of NSG mice. When tumors reached 200–250 mm3, mice were randomly assigned into cohorts and treated by oral gavage with inhibitors or vehicle according to treatment schedule.
(B) Treatment response was assessed through tumor volume change using caliper measurements 3 times/week in MiaPaCa-2 (KRASG12C) xenograft model. Results represent mean ± SD.
(C) Tumor volume change at time point day 21(n = 15 for vehicle cohort, n = 16 for all other cohorts). The y axis shows tumor volume change in percentage from baseline. Each bar represents the difference in tumor volume in an individual animal. According to the RECIST criteria, black indicates progressive disease, dark gray indicates stable disease, and light gray indicates partial response. Vehicle and cohort C data from Figure 3 B are reported again for comparison. Significance was determined by one-way ANOVA with Bonferroni’s multiple comparison test.
See also Figure S2.
We tested these regimens in three different tumor models: the subcutaneous xenograft model with transplanted human PDAC cell lines (Figures 4 and S2B–S2E), the endogenous Kras; Trp53−/− (KCP) model of spontaneous PDAC formation (Figures 5, S3, and S4), and the subcutaneous model of transplanted patient-derived PDAC tissue xenografts (PDX) (Figures 6 and S5; Table S1) None of the tested schedules were associated with dose- or schedule-limiting toxicities (Figure S6) in any of the three tumor models.
Figure 5.

In vivo assessment of optimal treatment regimen in an endogenous murine PDAC model
(A) Schematic representation of the treatment schedule applied in the endogenous (KPC) murine model of spontaneous tumor formation as well as the magnetic resonance imaging (MRI) time points applied. Cohort C: continuous treatment with the combination of SHP2i and ERKi daily; cohort D: intermittent treatment with the combination of SHP2i and ERKi 5 days on/2 days off; and cohort E: semi-continuous treatment schedule with daily dosing of SHP2i and intermittent dosing with ERKi 5 days on/2 days off. Control mice were treated with vehicle for 14 consecutive days. All treated mice were sacrificed on day 15, and tumors were resected for histological analysis.
(B) Representative MRI scan slices depicting PDAC tumor sections of KCP mice treated with vehicle (n = 5), cohort C (n = 7), cohort D (n = 7), or cohort E (n = 6) at the indicated time points (days) following the start of therapy (pre), with similar results among the groups. Volumetric measurements indicate a decrease in pancreatic volume in mice treated with the combination of SHP2i and ERKi for 2 weeks compared with vehicle-treated mice. The y axis shows pancreatic volume change in percentage quantified by measurements of MRI scans. Each bar represents the difference in pancreatic volume in an individual animal from days 0 to 15. According to the RECIST criteria, black indicates progressive disease, dark gray indicates stable disease, and light gray indicates partial response. Significance was determined by one-way ANOVA with Bonferroni’s multiple comparison test. Volume-tracking curves for individual mice over the whole course of therapy are available in Figures S3B–S3E.
(C) Macroscopic images of pancreas and spleen (top row). Representative H&E-stained sections of pancreata from mice, treated as indicated. Scale bars represent 1,000 (middle) and 200 μm (bottom). Mice numbers are indicated below.
(D) Relative pancreatic weight was significantly lower in all groups treated with the combination of SHP2i and ERKi: cohort C (n = 7), cohort D (n = 6), and cohort E (n = 7) compared with vehicle-treated control mice (n = 9). Results represent mean ± SD. ∗p < 0.05, ∗∗∗p < 0.001, significance was determined by one-way ANOVA with Bonferroni’s multiple comparison test.
(E) Quantification of relative intact acinar area as ratio of whole pancreatic area. Analysis performed on n = 4 individual mice in vehicle group and on n = 6 individual mice in cohort C. Results represent mean ± SD.
(F) Ki67-positive cells in percentage quantified in pancreata of mice treated with vehicle (n = 6) or with the combination of SHP2i and ERKi daily (n = 7). Results represent mean ± SD. ∗∗p = 0.0044, significance was determined by unpaired t test.
See also Figures S3, S4, and S6.
Figure 6.

Evaluation of treatment response by the combined administration of RMC-4550 (SHP2i) and LY3214996 (ERKi) in patient-derived xenograft (PDX) models
(A) Treatment schedule. Mice were treated with the combination of RMC-4550 (SHP2i) and LY3214996 (ERKi) once per day via oral gavage for 14 consecutive days (cohort C) or 5 days on/2 days off (cohort D) or SHP2i continuously and ERKi5 days on/2 days off (cohort E). For PDX354 model, tumor pieces of 50 mm3 were subcutaneously implanted into both flanks of NSG mice (n = 7 mice per cohort). When tumors reached 200–250 mm3 (approximately 6–8 weeks after subcutaneous transplantation), mice were randomly assigned into cohorts and treated by oral gavage with inhibitors or vehicle according to treatment schedule for the indicated time, after which tumors were resected.
(B and C) Treatment response was assessed through tumor volume changes using daily caliper measurements (B) and tumor weight at endpoint (C) in PDX354 model. Results represent mean ± SD. ∗∗∗∗p < 0.0001, as determined by one-way ANOVA with Bonferroni’s multiple comparison test.
(D) Representative macroscopic images of resected tumors.
(E) Representative H&E-stained sections of vehicle- and combination-therapy-treated PDX354 tumors. Scale bars represent 1,000 (top) and 200 μm (bottom).
See also Figure S5.
The potent tumor-inhibitory effect of the continuous combination treatment (cohort C) shown in MiaPaCa-2 xenografts (Figure 4C) was also observed in Panc10.05, ASPC1, and YAPC xenografts (Figures S2B–S2E). While we observed varying degrees of sensitivity among the 4 PDAC cell lines, the common findings were that all combination schedules showed stronger anti-tumor efficacy compared with all monotherapies or vehicle controls and that the continuous schedule had the strongest inhibitory effect of all the combination regimens (Figures 4C and S2B–S2E). However, complete tumor elimination was not achieved, even with the continuous treatment schedule.
In agreement with these findings, our endogenous KCP model showed that the combination treatment was able to significantly inhibit tumor growth (Figures 5B–5D) or even induce pancreatic volume reduction. Of note, the continuous treatment was able to induce more than 30% pancreatic volume reduction (Figures 5B and S3C) and prevent tumor outgrowth from the microscopic to macroscopic scale if the mouse was treated early enough. This was indicated by morphological analysis, the relative pancreatic weight, and the considerable number of intact acini, as well as the significant reduction in proliferating, i.e., Ki67-positive, cells compared with the control (Figures 5C–5F and S4A). PDAC is associated with significant formation of desmoplastic stroma, which is recapitulated very well in the murine KCP model. This stroma compartment has been shown to be an obstacle for the tumor penetration of chemotherapeutic drugs.44,45 Here (Figures S4B and S4C), we show that RMC-4550 indeed takes longer to reach the fibrotic/cancerous pancreas tissue compared with healthy pancreas tissue. However, after 8 h, the levels of RMC-4550 recovered from both tissues are very similar. Interestingly, we see that the RMC-4550 levels decline more rapidly in cancerous pancreas tissue. LY3214996 penetration of the pancreas tissue seems less affected by the tumor stroma since the amount of recovered drug from cancerous and healthy pancreas tissue is very similar. We further found that LY3214996 does not persist in the pancreas like RMC-4550 does. Twenty h after treatment, we still recovered 1/6th of the maximal RMC-4550 amount, while LY3214996 levels were virtually undetectable.
Additionally, three different PDX models, each representing a different patient harboring KRASG12D mutations, corroborated the observation that all tested combination therapy regimens were able to significantly reduce tumor growth and even induce tumor volume reduction (Figures 6 and S5). While there was no statistically significant difference between the three treatment arms, the data seem to indicate a slight benefit of the continuous treatment compared with the intermittent schedules (Figures 6C, 6D, S5C, and S5G). Interestingly, all tumors from mice receiving the combination treatments contained lytic necrotic cores, suggesting tumor cell elimination in addition to the cytostatic affects observed (Figures 6E, S5D, and S5H).
Assessment of treatment response
Having shown efficacy data in multiple different KRAS-mutant PDAC mouse models, our aim is to bring this therapy to the clinic. While we have tried to diversify our models and thus account for inter-patient heterogeneity, it is still essential to distinguish responders from non-responders in a clinical setting, preferably using minimally invasive methods. Using a small cohort of tumor-bearing KCP mice (Figure S7), we were able to show that congruent with the in vitro setting (Figure 1), pRSK levels are also a good marker to show MAPK pathway inhibition in vivo. However, as the level of pRSK is highly dependent on the time elapsed between treatment and sample collection, pRSK is not a useful marker for the evaluation of treatment response in a clinical setting. Indeed, 1 or 2 h after the treatment, pRSK levels have not been decreased to the fullest extent, while this is only achieved after 4 h. However, 16 or 20 h after the treatment, pRSK levels are increasing again. We thus conclude that sample collection either too soon or too late after the last treatment could lead to detection of misleading pRSK levels.
Interestingly, Ying et al.10 described that oncogenic KRASG12D is required for PDAC tumor maintenance and reprograms PDAC metabolism by stimulating glucose uptake and glycolysis, while Bryant et al.46 found that both KRAS suppression and ERK inhibition decreased glucose uptake in PDAC. Based on those reports, we explored the possibility of using 18F-fluorodeoxyglucose (FDG) uptake, as measured by positron emission tomography (PET) scan, as an early response marker and a surrogate readout of MAPK activity.
To this aim, we treated mice bearing subcutaneous KCP tumors with either vehicle or the combination of RMC-4550 + LY3214996 daily for 7 days. PET scans were performed at days 0 (pretreatment), 3, and 7. Our results show that 18F-FDG uptake is readily detected by PET-CT scan of subcutaneously implanted KCP tumors and that a significant decrease in PET signal is already observable at a time when reduction in tumor volume is not yet significant (Figure 7). This finding raises the possibility of using FDG uptake in the clinic to monitor early response to this combinatorial therapy for patients with PDAC.
Figure 7.

Early non-invasive assessment of the treatment response in a subcutaneous tumor mouse model
(A) Schematic representation of the treatment schedule applied in the subcutaneous tumor mouse model as well as the [18F]-FDG-PET imaging time points applied. 2.5 × 106–3 × 106 cells were injected subcutaneously into the left and right flank of 10- to 15-week-old non-tumor-bearing littermates. Two to three weeks after subcutaneous injection, mice were randomly assigned into cohorts and treated by oral gavage with inhibitors or vehicle for 7 consecutive days. [18F]-FDG-PET scans were obtained at baseline before commencement of therapy (day 0) and at days 3 and 7 during treatment.
(B) Representative [18F]-FDG-PET images of tumor-bearing mice treated with vehicle or undergoing treatment with the combination of RMC-4550 (SHP2i) and LY3214996 (ERKi) once per day for 7 consecutive days (cohort C). Subcutaneous tumor areas are shown in dashed circles, and SUV of FDG uptake is indicated by color. White color indicates highest uptake, red color high uptake, yellow and green intermediate, and blue low uptake. [18F]-FDG-PET, 18-fluordesoxyglucose positron emission tomography; PET, positron emission tomography; SUV, standardized uptake value; FDG, fluorodeoxyglucose.
(C) Upper panel: relative total lesion glycolysis (TLG) on days 0, 3, and 7 in vehicle versus cohort C (n = 8–9 animals/group). Lower panel: relative tumor volume on days 0, 3, and 7 in vehicle versus cohort C. Results represent mean ± SD. ∗∗∗p < 0.001, significance was determined by unpaired, two-tailed t test.
See also Figure S7.
Discussion
Pancreatic cancer has one of the highest mortality rates among all tumor types, and innovative treatment options against this devastating cancer are urgently needed.2 Since targeted therapies are lacking and PDAC seems to be refractory to immunotherapy,47 classic chemotherapy is still the treatment of choice for the management of PDAC at all stages of the disease.48
We recently identified a strategy for targeting KRAS-mutant tumors, irrespective of the specific mutation, which consists of the concomitant blockade of the RAS downstream effector MEK and the upstream activator SHP2.23,24 Similarly, the combination of SOS1 and MEK inhibition has been proven effective in KRAS-mutant preclinical tumor models.49 In the present work, we validate this “up plus down” double blockade strategy in multiple in vivo models of PDAC by combining inhibition of the MEK downstream effector ERK (by using LY3214996) with upstream inhibition of SHP2 (with RMC-4550).
In agreement with our earlier findings, we show that ERK inhibitor monotherapy is insufficient to induce a durable suppression of the MAPK pathway, as demonstrated by a rebound in phosphorylated RSK1 levels and failure to induce apoptosis. We have previously demonstrated that the rebound in MAPK signaling following RAS downstream inhibition can be attributed to the feedback overexpression and activation of multiple RTKs. Therefore, the most effective way to short circuit this resistance-inducing loop is to disrupt the signal transmission from activated RTKs to RAS. The protein phosphatase SHP2 has been found to be recruited by virtually all phosphorylated growth factor receptors, as well as other cell-surface receptors like cytokine or hormone receptors, where it mediates the signal transmission to downstream protein effectors, making it the ideal target to prevent ERK inhibitor resistance.50 Indeed, we show that co-treatment with LY3214996 and RMC-4550 promptly induces apoptosis in cell cultures and tumor regression in several mouse models of PDAC, especially when administered continuously.
The previous failure of MEKis like selumetinib against KRAS-mutant tumors during clinical trials51 has been attributed not only to the above-mentioned resistance mechanisms22 but also to the highly toxic profile of such drugs. On the other hand, ERK inhibitors are compounds that only recently have entered the earliest phases of clinical testing, holding promise for improved tolerability and efficacy in the treatment of tumors with a MAPK pathway dependency. In the present study, we extensively evaluated the toxicity of the LY3214996 + RMC-4550 combination in multiple murine backgrounds and identified for each compound a dose that, in combination, is well tolerated as well as efficacious against PDAC tumor growth.
Apart from its role in the MAPK pathway, the SHP2 phosphatase is also recruited by the immune checkpoint receptor PD1, which is expressed on T and pro-B lymphocytes.52 PD1 activity is known to suppress T cell activation and therefore mediate cancer immune evasion.53,54 It has been reported that inhibition of SHP2 can stimulate an anti-tumor immune response by both promoting T cell function and depleting pro-tumorigenic M2 macrophages.55,56 Therefore, the use of SHP2 inhibitors could have a double beneficial effect: in the tumor cells, it would act synergistically with ERK inhibitors to suppress MAPK-induced proliferation, and in the tumor microenvironment, it could promote the anti-tumor immune response. Further investigation, for example using a pancreatic cancer model that can be transplanted both in immuno-competent and immuno-compromised hosts, will be needed in order to elucidate the non-tumor-intrinsic benefits of using SHP2 inhibitors.
As a translational approach to monitor the early response of patients, we searched for dynamic biomarkers that could be used in a clinical setting. So far, computed tomography (CT) is the method of choice to determine therapy response; however, this is usually done retrospectively. Thus, the idea of non-invasive but quick methods to evaluate an early drug response is gaining increasing attention in modern oncology as it allows monitoring of therapy failure, sparing the patient unnecessary toxicity. In 2006, Su et al.57 showed that inhibition of the MAPK pathway by treatment with anti-EGFR therapy in colorectal cancer models induced a rapid downregulation of glucose receptors, which was reflected by decreased FDG glucose uptake in PET scans. Recently, Bryant et al.46 reported that small interfering RNA (siRNA)-mediated silencing of KRAS or pharmacological ERK inhibition decreased glucose uptake and resulted in a clear reduction of key glycolytic intermediates in KRAS-driven PDAC. Furthermore, a pilot study by Wang et al.58 showed that 18F-FDG-PET and diffusion-weighted magnetic resonance imaging (DW-MRI) can be used as early treatment response assessment in patients with advanced PDAC. Based on these data, we suggest 18F-FDG imaging via PET scans as a tool to sensitively monitor tumor shrinkage shortly after therapy initiation to evaluate the early drug response. Indeed, we confirmed that co-inhibition of ERK and SHP2 in subcutaneous KRAS-mutant PDAC tumors induces a rapid and significant decrease in FDG uptake, which precedes the decrease in tumor volume.
In conclusion, LY3214996 + RMC-4550 combination has shown a positive tolerability profile as well as the capacity to synergistically induce a high percentage of partial response in several preclinical models of PDAC. In addition, the preliminary data of the PET scans to monitor early drug response warrants the ERK + SHP2 inhibitor combination to be explored at a clinical level. To this end, we started the phase 1a/1b SHERPA trial (ClinicalTrials.gov: NCT04916236) with the objective of testing the tolerability and early evidence of efficacy of the combination of RMC-4630 (the clinical equivalent of RMC-4550) and LY3214996, which may represent a promising targeted therapeutic option for the majority of patients with KRAS-mutant pancreatic cancer.
Limitations of the study
Although we have extensively validated the tolerability and efficacy of the RMC4550 plus LY3214996 combination in preclinical models, the results may vary when testing the combination in patients (where RMC4630 is used). In particular, pharmacokinetics and toxicity may be different in human subjects and may lead to a different dosing and schedule of the treatment.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ki67 antibody | Abcam | Cat# ab15580; RRID:AB_443209 |
| Alpha-Tubulin antibody | Sigma-Aldrich | T9026; RRID:AB_477593 |
| RSK-1 antibody | CST | #8408; RRID:AB_10828594 |
| P-RSK antibody | CST | #8753; RRID:AB_2783561 |
| P-RSK antibody | CST | #9344; RRID:AB_331650 |
| GAPDH antibody | CST | #5174; RRID:AB_10622025 |
| Vinculin antibody | Sigma-Aldrich | V9131; RRID:AB_477629 |
| Biological samples | ||
| Patient Derived Xenografts | Dr. Manuel Hidalgo, Spanish National Cancer Research Center (CNIO), Madrid, Spain | (Reference no. I409181220BSMH) |
| Chemicals, peptides, and recombinant proteins | ||
| LY3214996 (Temuterkib) | Lilly | Available at https://www.selleckchem.com/ Catalog No.S8534 |
| RMC-4550 | Revolution Medicines | Available at https://www.selleckchem.com/ Catalog No.S8718 |
| 37% Formaldehyde solution | Merck | 104,003 |
| Crystal Violet | Sigma-Aldrich | HT90132-1 L |
| Matrigel | Corning | REF 354230 LOT 9364004 |
| Critical commercial assays | ||
| CellTiter-Glo 3D | Promega | REF G9682 LOT 0000496328 |
| Caspase 3/7 reagent | Sartorius | #4440 |
| Deposited data | ||
| Uncropped Western Blots | Mendeley |
Mendeley Data: https://doi.org/10.17632/cz4vfsk28h.1 |
| RNAseq Data | GEO | GEO: GSE213706 |
| Experimental models: Cell lines | ||
| Murine KCP cell lines derived from KCP mice in house | This paper | N/A |
| MiaPaCa-2 | ATCC | CRL-1420; RRID: CVCL_0428 |
| ASPC1 | ATCC | CRL-1682; RRID: CVCL_0152 |
| Panc10.05 | ATCC | CRL-2547; RRID: CVCL_1639 |
| Panc-1 | ATCC | CRL-1469; RRID: CVCL_1794 |
| YAPC | DSMZ | REF 15070; RRID: CVCL_0480 |
| Experimental models: Organisms/strains | ||
| B6 (C57BL/6J) mice | Jackson Laboratory | RRID:IMSR_JAX:000,664 |
| NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice | Jackson Laboratory | RRID:IMSR_JAX:005,557 |
| KCP mice (Krastm4Tyj); (Ptf1atm1(Cre)Hnak); (Trp53tm1Brn) (KCP) | See References59,60,61 | N/A |
| KCPmut mice (Krastm4Tyj); (Tg(Pdx1-cre)6Tuv); Trp53tm1Tyj) |
See Reference62 | N/A |
| NU-Foxn1nu nude mice (Hsd:Athymic Nude-Foxn1nu/Foxn1+) | Envigo https://www.envigo.com/model/hsd-athymic-nude-foxn1nu-foxn1?selctry=France&ctry= |
N/A |
| Software and algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | RRID:SCR_003070 |
| CompuSyn |
https://www.combosyn.com/ See Reference63 |
N/A |
| Adobe Illustrator® | Adobe https://www.adobe.com/de/ |
RRID:SCR_010279 |
| GraphPad Prism 8 | GraphStats Technologies https://www.graphstats.net/ |
RRID:SCR_002798 |
| Nucline™ NanoScan Software | Mediso | N/A |
| Flywheel DICOM viewer | Flywheel https://docs.flywheel.io/hc/en-us |
N/A |
| Incucyte ZOOM | Essen Bioscience | N/A |
| Aperio ImageScope | Leica Biosystems https://www2.leicabiosystems.com/ |
|
| Axiovision | Zeiss https://www.micro-shop.zeiss.com/de/de/system/software-axiovision+software-produkte/1007/ |
|
| BioRender | https://biorender.com/ | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sara Mainardi (s.mainardi@nki.nl).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mouse strains
KrasG12D (Krastm4Tyj)59; p48-Cre (Ptf1atm1(Cre)Hnak)60; p53flox/flox(Trp53tm1Brn)61 (KCP) have been described previously and were bred in a mixed genetic background in our animal facility. Non-tumor-bearing littermates without mutational KrasG12D (Krastm4Tyj) and p48-Cre (Ptf1atm1(Cre)Hnak) were used in the dose finding study and for subcutaneous tumor transplantation. KrasG12D (Krastm4Tyj); Pdx1-Cre (Tg(Pdx1-cre)6Tuv); Trp53mut/+(Trp53tm1Tyj)62 (KCPmut) mouse strain with C57BL/6J background served as tumor donor for orthotopic transplantation experiments.
At the age of weaning and after death, genotypes were determined by PCR and gel electrophoresis. B6 (C57BL/6J) mice and NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were obtained from Jackson Laboratory. NU-Foxn1nu nude mice (Hsd:Athymic Nude-Foxn1nu/Foxn1+) were obtained from Envigo.
All mice were kept in an animal room (room temperature range between 20 and 22°C) with light-dark cycle of 12:12 h in groups of 2–4 animals in type III cages (Tecniplast) or in groups of 5 animals in IVC cages from (Innovive) with bedding and nesting material. All animals were provided with the standard maintenance food for mice (No. 1324–10 mm pellets, Altromin, or SDS diets Technilab BMI) and water ad libitum and housed under specific pathogen-free conditions in accordance with the European Directive 2012/63/EU.
All animal experiments and care were performed in accordance with the guidelines of institutional committees and European regulations (Directive 2012/63/UE) and approved by the local authorities, Regierung von Oberbayern (ROB-55.2-2532.Vet_02-15-143), the animal experiment committee at the Netherlands Cancer Institute (IVD 1.1.9082), the Dutch Central Authority for Scientific Procedures on Animals (AVD30100202010644), the Universidad Autónoma de Madrid Ethics Committee (CEI 60-1057-A068) and the Comunidad de Madrid (PROEX 335/14).
Cell culture and cell lines
Primary murine tumor cell lines were obtained from chopped pieces of explanted tumors without enzymatic digestion. All murine cell lines were routinely cultured in DMEM supplemented with 10% FBS and penicillin–streptomycin (100 U/mL, 100 μg/mL) (all Life Technologies). Human PDAC cell lines: YAPC (KRASp.G12V; p53p.H179R; SMAD4p.R515fs∗22) was purchased from DSMZ. ASPC1 (KRASp.G12D; p53p.C135fs∗35; SMAD4p.R100T; CDKN2Ap.L78fs∗41), Panc10.05 (KRASp.G12D; p53p.I255N), Panc1 (KRASp.G12D; p53p.R273H) and MiaPaCa-2 (KRASp.G12C; p53p.R248W; Homozygous for CDKN2A deletion) were purchased from the American Type Culture Collection (ATCC). Mutational status of the cell lines was compiled from the ATCC, Catalog of Somatic Mutations in Cancer (COSMIC; Wellcome Trust Sanger Institute) and Cancer Cell Line Encyclopedia (CCLE, Broad Institute) databases. Human cell lines were cultured in RPMI1640 (Life Technologies), supplemented with 10% FBS, penicillin/streptomycin (100 U/mL, 100 μg/mL, Life Technologies), and 2 mM L-glutamine (Thermo Fisher Scientific). All cells were kept at 37°C in a humidified incubator with 5% CO2.
Human pancreatic cancer cell line xenografts
MiaPaCa-2, Panc10.05, ASPC1, and YAPC cells were resuspended (5 × 106 cells per mouse) in a 1:1 mixture of RPMI and Matrigel (Corning) and injected subcutaneously into the right flanks of 8-week-old NSG mice. Tumor volume was monitored three times a week as described for the patient-derived tissue xenografts. Mice were randomized when the tumor reached a volume of approximately 200 mm3 and treated for a maximum period of 30 days (YAPC) or 42 days (MiaPaCa-2, Panc10.05, ASPC1). Mice were sacrificed after 1, 3 or 6 weeks of treatment (8 mice per time point and per cohort) or at humane end point. In this experimental set up, RMC-4550 (10 mg/kg) and LY3214996 (100 mg/kg) were dissolved in 2% HPMC E-50, 0.5% Tween-80 in 50 mM Sodium Citrate Buffer, pH 4.0, and administered according to the different schedules (cohorts A, B, C, D, E, F, G, H, I). Control groups were treated daily with the vehicle alone. End-of-treatment tumor material was partly snap frozen in liquid nitrogen and stored at −80°C.
Orthotopic PDAC mouse models
30 mm3 KCPmut tumor pieces were obtained from endogenous mouse models and subcutaneously transplanted into the flanks of female B6 host mice for expansion. Tumor growth was monitored as indicated for the patient-derived tissue xenografts by caliper analysis. After 4 weeks, subcutaneous KCPmut tumors from donor mice were harvested and chopped into ∼40 mm3 pieces and orthotopically transplanted into pancreata of 8-week-old female and weight matched 18–20 g male B6 mice, as previously described.63 Briefly: Mice were anesthetized via isoflurane inhalation, shaved and skin wiped with ethanol-containing skin antiseptic. A small cutaneous midline incision was performed (0.8 cm) and a small subcutaneous pocket was prepared. An equally small incision (0.8 cm) was made into the peritoneum, the pancreas was mobilized and exposed. Tumor pieces were orthotopically implanted into the pancreas. Then the pancreas and spleen were carefully repositioned in the abdomen and the peritoneum was closed with a single stitch suture using 6–0 Prolene sutures. The skin was then closed with surgical staples. Tumor growth was monitored by palpation. After 2 weeks, a representative number of mice were sacrificed to determine pre-treatment baseline pancreas/tumor weights, and the remaining mice were randomly grouped into cohort A, B, C, and vehicle and treated with inhibitors as described in the following. End-of-treatment tumor material was partly snap frozen in liquid nitrogen and stored at −80°C.
Subcutaneous cancer cell line mouse models
Pancreatic cancer cells from KCP endogenous donor mouse model were obtained and cultured as described above. 2.5 × 106–3 × 106 cells were suspended in 100 μL of a 1:1 mixture of DMEM and Matrigel (Corning) and injected subcutaneously into the left and right flank of 10–15-week-old non-tumor-bearing female and male littermates from mixed background mouse strain KrasG12D (Krastm4Tyj), p53flox/flox (Trp53tm1Brn). Tumor volume was monitored as indicated for the patient-derived tissue xenografts. Randomized therapy was initiated after tumors had reached a palpable volume of <300 mm3. Therefore, female, and male mice were treated continuously with inhibitors (cohort C) or vehicle alone. On day 0, 3 and 7 of treatment, mice were scanned by animal PET (Mediso), imaging radioactive labeled glucose (1⁸F-FDG) uptake. Mice were sacrificed after different time points, with a minimum treatment time of 3 days.
Method details
Drugs and inhibitors
SHP2 inhibitor RMC-4550 was kindly provided by Revolution Medicines, Redwood City, California U.S.A. RMC-4550 was diluted in 50 mM Sodium Citrate Buffer pH = 4 with 1% Hydroxyethylcellulose (Sigma-Aldrich), 0.25% Tween (Sigma-Aldrich) and 0.05% Antifoam A concentrate (Sigma-Aldrich). Erk1/2 inhibitor LY3214996 was kindly provided by Eli Lilly and Company, Indianapolis IN 46285 U.S.A. LY3214996 powder was dissolved in dH2O (Braun) with 1% Hydroxyethylcellulose (Sigma-Aldrich), 0.25% Tween (Sigma-Aldrich) and 0.05% Antifoam A concentrate (Sigma-Aldrich). Inhibitor combinations were used according to company’s recommendation.
In vitro drug synergy and quantitative analysis
Indicated cells were cultured and seeded into 96-well plates at a density of 300–2,000 cells per well, depending on growth rate. Twenty-four hours later, drugs were added at the indicated concentrations using the HP D300 Digital Dispenser (HP). After 72 h, medium and drugs were refreshed. The total duration of the experiment was 6 days (two treatments) for KCP_K2101, KCP_P0012, MiaPaCa-2 and Panc10.05, and 10 days (three treatments) for Panc1, ASPC1 and YAPC. Cells were fixed with 4% PFA diluted in PBS (37% Formaldehyde solution, Merck) and stained with 2% crystal violet solution (HT90132-1 L, Sigma-Aldrich). Drug synergy was calculated using CompuSyn software (version 1.0), which is based on the median-effect principle and the combination index–isobologram theorem.64 CompuSyn software generates combination index values, where combination index 0–0.75 indicates synergy, 0.75–1.25 indicates an additive effect and CI > 1.25 indicates antagonism.65 Following the instructions of the software, drug combinations at non-constant ratios were used to calculate the combination index in our study.
Cell viability assay
1000 cells were seeded in 96 Well-plates for adherent growth (Cellstar®, 655,180) and for growth in suspension 96 well-plate F-Bottom (Cellstar®, 655,970). After approximately 6 h 2 μM RMC-4550 was added using an HP D300 Digital Dispenser. PAO and DMSO were used as a positive and negative control, respectively. After 96 h measurement was conducted with EnVision plate reader (PerkinElmer) by adding CellTiter-Glo® 3D cell viability assay reagent. The data was corrected for PAO treated cells and normalized to DMSO treated cells. Statistical analyses compared the effect of SHP2 inhibition between 2D/3D growth both in 10% serum and 3% serum using ordinary one-way ANOVA test.
Incucyte cell-proliferation assay and apoptosis assay
Indicated cell lines were seeded into 96-well plates at a density of 200–2,000 cells per well, depending on growth rate and the design of the experiment. Approximately 24 h later, drugs were added at the indicated concentrations using the HP D300 Digital Dispenser (HP). Cells were imaged every 4 h using the Incucyte ZOOM (Essen Bioscience). Phase-contrast images were analyzed to detect cell proliferation on the basis of cell confluence. For cell apoptosis, caspase-3/caspase-7 green apoptosis-assay reagent (Essen Bioscience) was added to the culture medium (1:1000), and cell apoptosis was analyzed on the basis of green-fluorescent staining of apoptotic cells.
In vivo drug combination dose finding escalation
Dose finding was established according to modified “3 + 3” scheme. Non-tumor-bearing mice were put on continuous oral administration of both drugs over 14 days (NSG mice, KrasG12D; p53flox/flox, p53flox/flox, p53flox/wt or wildtype mice from KrasG12D; p48-Cre; p53flox/flox litter). Toxicity was evaluated daily by measuring mice body weight (endpoint at body weight loss >20%), general clinical signs (abnormal behavior, signs of physical discomfort). According to modified “3 + 3” design, mouse cohorts consisting of 3 animals were given an initial combination dose (d5), followed by increased dose d7 as no side effects were observed in all 3 mice. Up to six mice were assigned to one dose. If the combination dose showed side effects in 1/6 mice, the dose was designated as an admissible dose, opening next dose level for testing. If dose-limiting toxicity was observed in 2/6 mice, the combination was accepted as a maximum tolerated dose, closing higher doses for testing. If more than two of the six mice experienced dose-limiting toxicity, the dose was down staged. The following dose combinations were administered: dose d5 (10 mg/kg RMC-4550 + 75 mg/kg LY3214996), dose d7 (30 mg/kg RMC-4550 + 75 mg/kg LY3214996), dose d8 (10 mg/kg RMC-4550 + 100 mg/kg LY3214996) and dose d9 (30 mg/kg RMC-4550 + 100 mg/kg LY3214996). One cohort was administered vehicle (50 mM Sodium Citrate Buffer pH = 4 with 1% Hydroxyethylcellulose, 0.25% Tween and 0.05% Antifoam A concentrate) to monitor gavage-mediated side effects.
In vivo therapy treatment schedules
For in vivo application in KCP mice, human cell line xenografts, PDX xenografts, orthotopically transplanted KCPmut mice, and subcutaneously transplanted KCP mice, dose d8 (10 mg/kg RMC-4550 + 100 mg/kg LY3214996) was administered by oral gavage. Depending on the mouse model, the drug administration followed different schedules: continuous administration (daily) of both drugs (cohort C), administration of both drugs 5 days on/2 days off (cohort D), or continuous administration of RMC-4550 plus LY3214996 every other day (cohort E), or continuous administration of RMC-4550 plus LY3214996 5 days on/2 days off (cohort F). As controls, mice were treated daily with vehicle or with monotherapy. Monotherapy was scheduled either RMC-4550 continuous (cohort A) or 5 days on/2 days off (cohort G). Monotherapy with LY3214996 was accomplished continuously (cohort B), 5 days on/2 days off (cohort H) or every other day (cohort I).
Drug distribution of RMC-4550 and LY3214996 in tumor mice and non-tumor bearing controls
Tumors of female and male KCP mice were detected by palpation. Endogenous KCP tumor bearing mice and non-tumor-bearing control littermates without mutational KrasG12D (Krastm4Tyj) and p48-Cre (Ptf1atm1(Cre)Hnak) were administered a single dose d8 of the drug combination (10 mg/kg RMC-4550 + 100 mg/kg LY3214996) and sacrificed after 1, 2, 4, 8, 16, and 20 h, respectively. The control mice cohort includes two mice, the tumor mice cohort four mice for each time point. After sacrifice, half of the tumor was used for pharmacokinetics studies and the rest for protein analysis. After weighing, samples were homogenized using a FastPrep®-24 (MP-Biomedicals, NY) in 1% (w/v) bovine serum albumin in water. The biological samples were assayed by liquid chromatography triple quadrupole mass spectrometry (LC-MS/MS) using an API4000 detector (Sciex) for determination of LY3214996 and RMC-4550 using ion pairs 453.5/367.0 and 437.0/279.9, respectively. LC separation was achieved using a Zorbax Extend C18 column (100 × 2.0 mm: ID). Mobile phase A and B comprised 0.1% formic acid in water and methanol, respectively. The flow rate was 0.4 mL/min and a linear gradient from 20% B to 95% B in 2.5 min, followed by 95% B for 2 min, followed by re-equilibration at 20% B for 10 min was used for elution. Sample pre-treatment was accomplished by mixing 5 μL biological matrix with 30 μL of formic acid in acetonitrile (1 + 99). After centrifugation, the clear supernatant was diluted 1 + 8 with water and 2 μL was injected into the LC-MS/MS system.
PET imaging and 1⁸F-FDG in vivo
Two-to-six hours fasted female and male mice bearing subcutaneous KCP tumors were randomly divided into two groups (vehicle versus cohort C). For PET imaging, mice received 12–14 MBq of the radiotracer 18F-Fluordeoxyglucose (1⁸F-FDG) via injection through the lateral tail vein. PET images were acquired on a nanoScan PET system (Mediso, Budapest, Hungary) from 45–60 min post injection under isoflurane anesthesia (1–2% in medical air by precision vaporizer (Baxter Healthcare, Deerfield, IL, USA). During the imaging procedure, mice were placed on a heated bed and their heart rate was constantly monitored. For 3D whole body image reconstruction with a 0.4 mm3 voxel size, the Tera-Tomo 3D image reconstruction algorithm (integrated into Nucline NanoScan Software, Mediso) was applied (4 iterations, 6 subsets), without AC and scatter corrections. Image counts per voxel per second were converted into standardized uptake values (SUV) using the activity concentrations computed the Nucline NanoScan software normalized to the animal’s body weight. Quantification of tumor uptake was carried out using the Nucline NanoScan software, by drawing spherical regions of interest (ROIs), creating a volume that represented the entire tumor lesion. We recorded the volume, SUVmean, SUVmax and total lesion glycolysis (TLG) of each tumor.
Magnetic resonance imaging (MRI)
MR imaging for KCP male and female mice was started at an age of 24–37 days and repeated after 7 and 14 days of treatment (cohort C, D, E versus vehicle). Sedation was achieved via continuous inhalation of 2% isoflurane (Abbott) in 1.6% O2 using a veterinary anesthesia system (Vetland Medical). Body temperature was maintained and monitored, and eyes were protected by eye ointment. Image acquisition was achieved using a mouse 3T coil inside a preclinical 3T nano scan PET/MR (Mediso) and a T2 weighted fast spin echo sequence (resolution: 192 × 128–25 slices, echo time 55,52 ms; repetition time 3000 ms). Analysis, visualization, and calculation was done by Flywheel DICOM viewer. Solid tumor volumes were calculated by summating truncated pyramid volumes between tumor areas on vicinal slices. As drug treatment prevented tumor development in some of the endogenous KCP mice, pancreatic areas including tumor and non-neoplastic tissue were defined as regions of interest and summarized in the scanned slices to calculate pancreatic volume.
Histology
Tissue specimens were fixed in 4%-buffered paraformaldehyde for 48 h at 4°C, dehydrated and embedded in paraffin. H&E was performed as described previously on 2.5 μm cut sections.66 In brief, the paraffin-embedded tissue sections were deparaffinized in Roti®Histol (Roth) for 2 × 5 min followed by rehydration in ethanol of descending concentration (100%, 96%, 70%; 3–5 min each) and deionized water (3–5 min). Slides were then incubated in hematoxylin solution (Merck Millipore) for 5 min, washed with running tap water for 10 min and incubated in eosin solution (Merck) for 3.5 min. Subsequently, the stained tissue sections were dehydrated in 96% Ethanol and Isopropanol for 25 s each followed by Roti®Histol for 2 × 3 min. They were then covered with Pertex mounting medium (Medite GmbH) and coverslips (Merck). For immunohistochemistry, sections were probed with the antibody for Ki67 (ab15580, 1:2,500, abcam). Immunohistochemistry was performed using avidin-biotin-enhancement (Vector Laboratories). Slides were developed with DAB (3,3′ apos; -diaminobenzidine, Vector Laboratories) and counterstained with hematoxylin. Image acquisition was performed on a Zeiss AxioImager.A1 microscope. Quantitative interpretation of nuclear Ki67 staining was done with Aperio ImageScope (Leica). Quantitative analyses of tumor and acini areas were performed with Axiovision (Zeiss).
Sequencing and MAPK pathway activity score
For preparation of the MiaPaCa-2 and the Panc10.05 xenograft tumors, snap frozen material (3 mice from cohort C and vehicle, treated for 3 weeks), was cut at the cryostate (30 cryosections of 30 μm thickness per sample) and RNA was extracted using the RNeasy Mini Kit from Qiagen and analyzed using an Agilent 2100 Bioanalyzer system. For the orthotopically transplanted KCPmut tumors, snap frozen tissue (cohort C versus vehicle) was homogenized, and RNA was isolated with Maxwell® 16 LEV simplyRNA Purification Kit from Promega. Sample purity was evaluated by nanoDrop, and RNA quality validated by agarose gel electrophoresis. Transcript levels were quantified with Kallisto (v0.46), using the GENCODE reference transcriptome (mouse version m25 and human version h34). For the human cell line xenograft samples, the human and mouse reference transcriptome were combined, and only human transcripts were kept for downstream analysis. The transcript levels were summed to gene levels and the gene expression levels were normalized between samples with EdgeR (v3.26.8) using trimmed mean of M-values. MAPK activity scores were calculated as described,42 using the normalized log2 counts per million values for the following genes: SPRY2, SPRY4, ETV4, ETV5, DUSP4, DUSP6, CCND1, EPHA2, and EPHA4.
The AmpliSeq Cancer HotSpot Panel v2 for Illumina, which includes 50 oncogenes and tumor suppressor genes was used to prepare the sequencing library to analyze the Panc185 and Panc354 cells derived from PDX tumors. Sequencing was performed using the MiSeq instrument from Illumina with V2 MiSeq 2 × 150 (300-cycle) cartridges. Downstream analysis was performed using the Varsome Clinical software. The reads were aligned to the hg19 version of the genome and the Sentieon’s Tnhaplotyper2 algorithm was used to call the variants, which were filtered based on call quality using the standard parameters designed for the AmpliSeq Cancer HotSpot Panel.
Protein lysate preparation and immunoblotting
To prepare analysis of cell lysates, cells were plated in complete medium. The morning after, cells were refreshed with medium and drugs of interest. At the desired time points, the cells were washed with cold-PBS and lysed in RIPA buffer supplemented with Halt™ Protease & Phosphatase single-use inhibitors cocktail (100 X) (78,442) and Halt™ Protease single-use inhibitors cocktail (100 X) (78,430). For preparation of tissue lysates, PDAC tissue from mice was homogenized in MLB Buffer containing protease inhibitor (Serva) and phosphatase inhibitor cocktails (Serva). Protein quantification was performed with the BCA Protein Assay Kit (Pierce). The lysates were then resolved by electrophoresis in Bolt 4–12% Bis-Tris Plus Gels (Thermo Fisher Scientific) followed by western blotting as described previously.23 The following antibodies were used: Antibodies against RSK-1 (8408), phosphorylated RSK-1 (9344 and 8753) and GAPDH (5174) were purchased from Cell Signaling Technology (CST). Antibodies against alpha Tubulin (T9026) and Vinculin (V9131) were purchased from Sigma-Aldrich. Relative pRSK1 levels were quantified by densitometry using ImageJ.
Quantification and statistical analysis
All in vitro data are expressed as averages from at least two technical replicates ± SD, unless differently stated, and they have been independently reproduced at least twice with similar results. Significance was determined by ordinary one-way ANOVA test or by one-way ANOVA with Bonferroni’s multiple comparison test or by unpaired, two-tailed t test. Statistical analysis was performed with GraphPad PRISM 8.0 software.
Additional resources
Clinical trial
The clinical trial entitled Combination Therapy of RMC-4630 and LY3214996 in Metastatic KRAS Mutant Cancers (SHERPA), which arose from the results reported in this article, is registered under the ClinicalTrials.gov Identifier: NCT04916236. No data from this clinical trial are reported in this study.
Acknowledgments
We thank the Preclinical Imaging Core at TranslaTUM (PICTUM) at Klinikum rechts der Isar der Technischen Universität München for its support, specifically Markus Mittelhäuser and Hannes Rolbieski. We thank the NKI Intervention Unit for technical assistance with the human cell lines xenograft experiment, as well as Arthur Burylo from the NKI Pharmacology Department for helping with the pharmacokinetic studies. The graphical abstract was created with BioRender.com (agreement number TK24HPDSXK). This work was funded by the American Association for Cancer Research, Lustgarten Foundation, and Stand Up to Cancer as a Pancreatic Cancer Collective New Therapies Challenge grant (grant no. SU2C-AACR-PCC-01-18).
Author contributions
The authors confirm contribution to the paper as follows: R.B., H.A., S.M., M.L., B.S., K.J.F., A. Berninger, and A.M.-S. designed the study. In vitro cell culture and colony-formation assays were performed by A.M.-S., S.M., and A. Bosma. Apoptosis assay and immunoblotting were done by A.M.-S. In vitro data was analyzed by K.J.F. and A.M.-S. K.J.F., A. Berninger, M.L., K.G., N.W., K.N.D., E.K.-A., F.S., and L.K. conducted the KCP mouse model experiment. K.J.F. and A. Berninger performed the subcutaneous mouse model experiment. S.M., M.v.d.V., and N.P. designed and performed the human xenograft mouse model experiment. L.R.C. conducted the orthotopic mouse model experiment. The human PDX mouse model experiment was done by L.R.C. Imaging (MRI and PET) was performed by K.J.F., A. Berninger, K.N.D., N.W., and K.G. with the help of PICTUM. K.J.F. analyzed the MRI data. S.K. analyzed the PET data. RNA isolation and MAPK activity scoring were done by A Berninger, S.M., and B.T. O.v.T. performed the drug measurements for the pharmacokinetic studies. Histology was performed by A. Berninger. Image acquisition and quantification was performed by M.L. and A. Berninger. Maintenance of mouse strains and genotyping were done by A. Berninger, K.J.F., N.W., K.G., E.K.-A., D.K., K.N.D., F.S., L.K., M.v.d.V., and N.P. K.J.F., A. Berninger, A.M.-S., B.S., M.L., and S.M. selected and analyzed the data shown in this manuscript. M.L. generated the figures with the help of A. Berninger and K.J.F. K.J.F., A. Berninger, A.M.-S., M.L., and S.M. wrote the manuscript. The manuscript was edited by D.A.R., B.S., S.K., H.A., and R.B. Supervision was provided by R.B. and H.A. The shared first authors K.J.F., A.M.-S., and A. Berninger contributed equally to the experimental work, and their order was assigned considering that K.J.F. and A.M.-S. also produced preliminary data and K.J.F. coordinated inter-lab meetings.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100815.
Supplemental information
Data availability
-
•
Uncropped Western Blot images have been uploaded to Mendeley and can be accessed via Mendeley Data: https://doi.org/10.17632/cz4vfsk28h.1
-
•
RNAseq data was uploaded to GEO under the reference Series GSE213706 and can be accessed via GEO: GSE213706
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Carioli G., Bertuccio P., Boffetta P., Levi F., La Vecchia C., Negri E., Malvezzi M. European cancer mortality predictions for the year 2020 with a focus on prostate cancer. Ann. Oncol. 2020;31:650–658. doi: 10.1016/j.annonc.2020.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Mizrahi J.D., Surana R., Valle J.W., Shroff R.T. Pancreatic cancer. Lancet. 2020;395:2008–2020. doi: 10.1016/S0140-6736(20)30974-0. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. 2020. [DOI] [PubMed] [Google Scholar]
- 4.Pishvaian M.J., Bender R.J., Halverson D., Rahib L., Hendifar A.E., Mikhail S., Chung V., Picozzi V.J., Sohal D., Blais E.M., et al. Molecular profiling of patients with pancreatic cancer: initial results from the know your tumor initiative. Clin. Cancer Res. 2018;24:5018–5027. doi: 10.1158/1078-0432.CCR-18-0531. [DOI] [PubMed] [Google Scholar]
- 5.Pishvaian M.J., Blais E.M., Brody J.R., Lyons E., DeArbeloa P., Hendifar A., Mikhail S., Chung V., Sahai V., Sohal D.P.S., et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020;21:508–518. doi: 10.1016/S1470-2045(20)30074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Golan T., Hammel P., Reni M., Van Cutsem E., Macarulla T., Hall M.J., Park J.O., Hochhauser D., Arnold D., Oh D.Y., et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019;381:317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waddell N., Pajic M., Patch A.M., Chang D.K., Kassahn K.S., Bailey P., Johns A.L., Miller D., Nones K., Quek K., et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iacobuzio-Donahue C.A., Velculescu V.E., Wolfgang C.L., Hruban R.H. Genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. Clin. Cancer Res. 2012;18:4257–4265. doi: 10.1158/1078-0432.CCR-12-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins M.A., Bednar F., Zhang Y., Brisset J.C., Galbán S., Galbán C.J., Rakshit S., Flannagan K.S., Adsay N.V., Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ying H., Kimmelman A.C., Lyssiotis C.A., Hua S., Chu G.C., Fletcher-Sananikone E., Locasale J.W., Son J., Zhang H., Coloff J.L., et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar-Sagi D., Knelson E.H., Sequist L.V. A bright future for KRAS inhibitors. Nat. Can. 2020;1:25–27. doi: 10.1038/s43018-019-0016-8. [DOI] [PubMed] [Google Scholar]
- 12.Hallin J., Engstrom L.D., Hargis L., Calinisan A., Aranda R., Briere D.M., Sudhakar N., Bowcut V., Baer B.R., Ballard J.A., et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostrem J.M.L., Shokat K.M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016;15:771–785. doi: 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- 14.Hong D.S., Fakih M.G., Strickler J.H., Desai J., Durm G.A., Shapiro G.I., Falchook G.S., Price T.J., Sacher A., Denlinger C.S., et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020;383:1207–1217. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jänne P., Rybkin I., Spira A., Riely G., Papadopoulos K., Sabari J., Johnson M., Heist R., Bazhenova L., Barve M., et al. 3LBA late breaking - KRYSTAL-1: activity and safety of adagrasib (MRTX849) in advanced/metastatic non–small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur. J. Cancer. 2020;138:S1–S2. [Google Scholar]
- 16.Johnson M.L., Ou S., Barve M., Rybkin I., Papadopoulos K., Leal T., Velastegui K., Christensen J., Kheoh T., Chao R., Weiss J. 4LBA late breaking - KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with colorectal cancer (CRC) and other solid tumors harboring a KRAS G12C mutation. Eur. J. Cancer. 2020;138:S2. [Google Scholar]
- 17.Witkiewicz A.K., McMillan E.A., Balaji U., Baek G., Lin W.C., Mansour J., Mollaee M., Wagner K.U., Koduru P., Yopp A., et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellano E., Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011;2:261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engelman J.A., Chen L., Tan X., Crosby K., Guimaraes A.R., Upadhyay R., Maira M., McNamara K., Perera S.A., Song Y., et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatzivassiliou G., Haling J.R., Chen H., Song K., Price S., Heald R., Hewitt J.F.M., Zak M., Peck A., Orr C., et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 21.Waters A.M., Der C.J. The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018;8:a031435. doi: 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun C., Hobor S., Bertotti A., Zecchin D., Huang S., Galimi F., Cottino F., Prahallad A., Grernrum W., Tzani A., et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7:86–93. doi: 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- 23.Mainardi S., Mulero-Sánchez A., Prahallad A., Germano G., Bosma A., Krimpenfort P., Lieftink C., Steinberg J.D., de Wit N., Gonçalves-Ribeiro S., et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018;24:961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- 24.Ruess D.A., Heynen G.J., Ciecielski K.J., Ai J., Berninger A., Kabacaoglu D., Görgülü K., Dantes Z., Wörmann S.M., Diakopoulos K.N., et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018;24:954–960. doi: 10.1038/s41591-018-0024-8. [DOI] [PubMed] [Google Scholar]
- 25.Wong G.S., Zhou J., Liu J.B., Wu Z., Xu X., Li T., Xu D., Schumacher S.E., Puschhof J., McFarland J., et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018;24:968–977. doi: 10.1038/s41591-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neel B.G., Gu H., Pao L. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 27.Garcia Fortanet J., Chen C.H.T., Chen Y.N.P., Chen Z., Deng Z., Firestone B., Fekkes P., Fodor M., Fortin P.D., Fridrich C., et al. Allosteric inhibition of SHP2: identification of a potent, selective, and orally efficacious phosphatase inhibitor. J. Med. Chem. 2016;59:7773–7782. doi: 10.1021/acs.jmedchem.6b00680. [DOI] [PubMed] [Google Scholar]
- 28.Infante J.R., Somer B.G., Park J.O., Li C.P., Scheulen M.E., Kasubhai S.M., Oh D.Y., Liu Y., Redhu S., Steplewski K., Le N. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer. 2014;50:2072–2081. doi: 10.1016/j.ejca.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 29.Kasuga A., Nakagawa K., Nagashima F., Shimizu T., Naruge D., Nishina S., Kitamura H., Kurata T., Takasu A., Fujisaka Y., et al. A phase I/Ib study of trametinib (GSK1120212) alone and in combination with gemcitabine in Japanese patients with advanced solid tumors. Invest. N. Drugs. 2015;33:1058–1067. doi: 10.1007/s10637-015-0270-2. [DOI] [PubMed] [Google Scholar]
- 30.Tolcher A.W., Bendell J.C., Papadopoulos K.P., Burris H.A., 3rd, Patnaik A., Jones S.F., Rasco D., Cox D.S., Durante M., Bellew K.M., et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015;26:58–64. doi: 10.1093/annonc/mdu482. [DOI] [PubMed] [Google Scholar]
- 31.Welsh S.J., Corrie P.G. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther. Adv. Med. Oncol. 2015;7:122–136. doi: 10.1177/1758834014566428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pathania S., Rawal R.K. An update on chemical classes targeting ERK1/2 for the management of cancer. Future Med. Chem. 2020;12:593–611. doi: 10.4155/fmc-2019-0339. [DOI] [PubMed] [Google Scholar]
- 33.Hayes T.K., Neel N.F., Hu C., Gautam P., Chenard M., Long B., Aziz M., Kassner M., Bryant K.L., Pierobon M., et al. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell. 2016;29:75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J.C.-H., Lin C.-C., Chu C.-Y. In: IASLC Thoracic Oncology. Second Edition. Pass H.I., Ball D., Scagliotti G.V., editors. Elsevier, Philadelphia; 2018. 49 - management of toxicities of targeted therapies; pp. 490–500.e493. [Google Scholar]
- 35.Drosten M., Barbacid M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell. 2020;37:543–550. doi: 10.1016/j.ccell.2020.03.013. [DOI] [PubMed] [Google Scholar]
- 36.Ryan M.B., Der C.J., Wang-Gillam A., Cox A.D. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183–198. doi: 10.1016/j.trecan.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhagwat S.V., McMillen W.T., Cai S., Zhao B., Whitesell M., Shen W., Kindler L., Flack R.S., Wu W., Anderson B., et al. ERK inhibitor LY3214996 targets ERK pathway-driven cancers: a therapeutic approach toward precision medicine. Mol. Cancer Therapeut. 2020;19:325–336. doi: 10.1158/1535-7163.MCT-19-0183. [DOI] [PubMed] [Google Scholar]
- 38.Nichols R.J., Haderk F., Stahlhut C., Schulze C.J., Hemmati G., Wildes D., Tzitzilonis C., Mordec K., Marquez A., Romero J., et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-NF1- and RAS-driven cancers. Nat. Cell Biol. 2018;20:1064–1073. doi: 10.1038/s41556-018-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doehn U., Hauge C., Frank S.R., Jensen C.J., Duda K., Nielsen J.V., Cohen M.S., Johansen J.V., Winther B.R., Lund L.R., et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell. 2009;35:511–522. doi: 10.1016/j.molcel.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang X., Biswas S., Oki Y., Issa J.P., Berry D.A. A parallel phase I/II clinical trial design for combination therapies. Biometrics. 2007;63:429–436. doi: 10.1111/j.1541-0420.2006.00685.x. [DOI] [PubMed] [Google Scholar]
- 41.Le Tourneau C., Lee J.J., Siu L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 2009;101:708–720. doi: 10.1093/jnci/djp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagle M.C., Kirouac D., Klijn C., Liu B., Mahajan S., Junttila M., Moffat J., Merchant M., Huw L., Wongchenko M., et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis. Oncol. 2018;2:7. doi: 10.1038/s41698-018-0051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hao H.X., Wang H., Liu C., Kovats S., Velazquez R., Lu H., Pant B., Shirley M., Meyer M.J., Pu M., et al. Tumor intrinsic efficacy by SHP2 and RTK inhibitors in KRAS-mutant cancers. Mol. Cancer Therapeut. 2019;18:2368–2380. doi: 10.1158/1535-7163.MCT-19-0170. [DOI] [PubMed] [Google Scholar]
- 44.Jiang B., Zhou L., Lu J., Wang Y., Liu C., You L., Guo J. Stroma-targeting therapy in pancreatic cancer: one coin with two sides? Front. Oncol. 2020;10:576399. doi: 10.3389/fonc.2020.576399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neesse A., Algül H., Tuveson D.A., Gress T.M. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476–1484. doi: 10.1136/gutjnl-2015-309304. [DOI] [PubMed] [Google Scholar]
- 46.Bryant K.L., Stalnecker C.A., Zeitouni D., Klomp J.E., Peng S., Tikunov A.P., Gunda V., Pierobon M., Waters A.M., George S.D., et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019;25:628–640. doi: 10.1038/s41591-019-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balachandran V.P., Beatty G.L., Dougan S.K. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology. 2019;156:2056–2072. doi: 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nevala-Plagemann C., Hidalgo M., Garrido-Laguna I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020;17:108–123. doi: 10.1038/s41571-019-0281-6. [DOI] [PubMed] [Google Scholar]
- 49.Hofmann M.H., Gmachl M., Ramharter J., Savarese F., Gerlach D., Marszalek J.R., Sanderson M.P., Kessler D., Trapani F., Arnhof H., et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2021;11:142–157. doi: 10.1158/2159-8290.CD-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dance M., Montagner A., Salles J.P., Yart A., Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 2008;20:453–459. doi: 10.1016/j.cellsig.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Jänne P.A., van den Heuvel M.M., Barlesi F., Cobo M., Mazieres J., Crinò L., Orlov S., Blackhall F., Wolf J., Garrido P., et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. 2017;317:1844–1853. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z., Zhao Y., Fang J., Cui R., Xiao Y., Xu Q. SHP2 negatively regulates HLA-ABC and PD-L1 expression via STAT1 phosphorylation in prostate cancer cells. Oncotarget. 2017;8:53518–53530. doi: 10.18632/oncotarget.18591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Juneja V.R., McGuire K.A., Manguso R.T., LaFleur M.W., Collins N., Haining W.N., Freeman G.J., Sharpe A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017;214:895–904. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schoenfeld A.J., Hellmann M.D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443–455. doi: 10.1016/j.ccell.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quintana E., Schulze C.J., Myers D.R., Choy T.J., Mordec K., Wildes D., Shifrin N.T., Belwafa A., Koltun E.S., Gill A.L., et al. Allosteric inhibition of SHP2 stimulates antitumor immunity by transforming the immunosuppressive environment. Cancer Res. 2020;80:2889–2902. doi: 10.1158/0008-5472.CAN-19-3038. [DOI] [PubMed] [Google Scholar]
- 56.Zhao M., Guo W., Wu Y., Yang C., Zhong L., Deng G., Zhu Y., Liu W., Gu Y., Lu Y., et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm. Sin. B. 2019;9:304–315. doi: 10.1016/j.apsb.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su H., Bodenstein C., Dumont R.A., Seimbille Y., Dubinett S., Phelps M.E., Herschman H., Czernin J., Weber W. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin. Cancer Res. 2006;12:5659–5667. doi: 10.1158/1078-0432.CCR-06-0368. [DOI] [PubMed] [Google Scholar]
- 58.Wang Z.J., Behr S., Consunji M.V., Yeh B.M., Ohliger M.A., Gao K., Ko A.H., Cinar P., Tempero M.A., Collisson E.A. Early response assessment in pancreatic ductal adenocarcinoma through integrated PET/MRI. AJR Am. J. Roentgenol. 2018;211:1010–1019. doi: 10.2214/AJR.18.19602. [DOI] [PubMed] [Google Scholar]
- 59.Jackson E.L., Willis N., Mercer K., Bronson R.T., Crowley D., Montoya R., Jacks T., Tuveson D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakhai H., Sel S., Favor J., Mendoza-Torres L., Paulsen F., Duncker G.I.W., Schmid R.M. Ptf1a is essential for the differentiation of GABAergic and glycinergic amacrine cells and horizontal cells in the mouse retina. Development. 2007;134:1151–1160. doi: 10.1242/dev.02781. [DOI] [PubMed] [Google Scholar]
- 61.Marino S., Vooijs M., van Der Gulden H., Jonkers J., Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 62.Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 63.D'Errico G., Alonso-Nocelo M., Vallespinos M., Hermann P.C., Alcalá S., García C.P., Martin-Hijano L., Valle S., Earl J., Cassiano C., et al. Tumor-associated macrophage-secreted 14-3-3zeta signals via AXL to promote pancreatic cancer chemoresistance. Oncogene. 2019;38:5469–5485. doi: 10.1038/s41388-019-0803-9. [DOI] [PubMed] [Google Scholar]
- 64.Chou T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 65.Manchado E., Weissmueller S., Morris J.P., 4th, Chen C.C., Wullenkord R., Lujambio A., de Stanchina E., Poirier J.T., Gainor J.F., Corcoran R.B., et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534:647–651. doi: 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lesina M., Kurkowski M.U., Ludes K., Rose-John S., Treiber M., Klöppel G., Yoshimura A., Reindl W., Sipos B., Akira S., et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Uncropped Western Blot images have been uploaded to Mendeley and can be accessed via Mendeley Data: https://doi.org/10.17632/cz4vfsk28h.1
-
•
RNAseq data was uploaded to GEO under the reference Series GSE213706 and can be accessed via GEO: GSE213706
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.