

Summary
Natural killer (NK) cells and type 1 innate lymphoid cells (ILC1s) are populations of non-T, non-B lymphocytes in peripheral tissues. Although NK and ILC1 subsets have been described, their identification and characteristics remain unclear. We performed single-cell RNA sequencing and CITE-seq to explore NK and ILC1 heterogeneity between tissues. We observed that although NK1 and NK2 subsets are conserved in spleen and liver, ILC1s are heterogeneous across tissues. We identified sets of genes expressed by related subsets or characterizing unique ILC1 populations in each organ. The syndecan-4 appeared as a marker discriminating murine ILC1 from NK cells across organs. Finally, we revealed that the expressions of EOMES, GZMA, IRF8, JAK1, NKG7, PLEK, PRF1, and ZEB2 define NK cells and that IL7R, LTB, and RGS1 differentiate ILC1s from NK cells in mice and humans. Our data constitute an important resource to improve our understanding of NK-ILC1 origin, phenotype, and biology.
Keywords: immunology, innate immunity, innate lymphoid cells, NK, ILC1, single-cell RNA-seq, CITE-seq
Graphical abstract
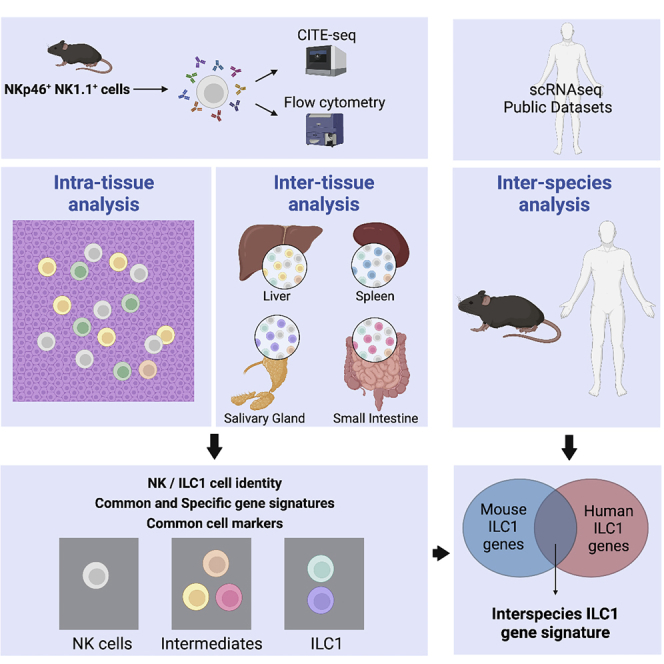
Highlights
-
•
scRNA-seq reveals inter- and intra-tissue heterogeneity of NKp46+NK1.1+ cells
-
•
ILC1 heterogeneity stems from distinct differentiation pathways and maturation state
-
•
Syndecan-4 appears as a useful marker discriminating murine ILC1 from NK cells
-
•
Common gene-specific signature of NK cells and ILC1s across organs and species
Lopes et al. use CITE-seq to dissect NK and ILC1 heterogeneity. They identify multiple NK-ILC1 subsets with tissue-specific transcriptional profiles. Syndecan-4 constitutes a novel marker for discriminating murine ILC1 from NK cells. They also reveal genes shared by mouse and human NK cells or ILC1s across organs.
Introduction
Innate lymphoid cells (ILCs) are heterogenous innate non-T, non-B lymphocytes involved in immunity, inflammation, and tissue homeostasis.1 ILCs can be classified into five main subsets: natural killer (NK) cells, lymphoid-tissue inducer cells (LTis), type 1 ILCs (ILC1s), type 2 ILCs (ILC2s), and type 3 ILCs (ILC3s). ILC1s express T-Bet and produce interferon-γ (IFN-γ), ILC2s are dependent on GATA-3 and secrete interleukin (IL)-12 and -5, and ILC3s express RORγt and produce IL-17 and IL-22.1
While NK cells recirculate in the blood, ILC1s are tissue resident.2 Both cell types have several features in common, including expression of the cell-surface markers NK1.1 and NKp46 in mice, the transcription factor T-Bet, and IFN-γ secretion. Few markers have been proposed to discriminate ILC1s from NK cells, such as integrin α1 (CD49a), CD200R, IL-7 receptor (CD127), CD69, and C-X-C chemokine receptor type 6 (CXCR6).3 ILCs are highly plastic, with a phenotype shaped by their microenvironment. For example, transforming growth factor β (TGF-β) has been shown to guide the transdifferentiation of NK cells into ILC1s.4,5
Recent advances in single-cell RNA sequencing (scRNA-seq) improved the characterization of NK cells and ILC1s across tissues in mice and humans.6,7,8,9,10,11 Our previous data on blood and splenic NK cells from mice and humans identified three subsets, NK0, NK1, and NK2, ranging from immature to mature cells.8 ScRNA-seq on murine NKp46+NK1.1+ cells from several tissues has revealed differences in cell identity and gene expression programs between tissue-specific and non-tissue-specific NKp46+NK1.1+ subsets.11 However, the relationships between these various populations at both the intra- and inter-tissues levels remain to be understood. Here, we used CITE-seq (cellular indexing of transcriptomes and epitopes by sequencing) technology, which combines scRNA-seq with the use of oligonucleotide-labeled antibodies carrying unique barcodes, to investigate NK and ILC1 heterogeneity between tissues. Based on both phenotypic and transcriptomic profiling, we identified several NK and ILC1 populations with tissue-specific imprints. These data reveal a much wider heterogeneity than previously anticipated and prompt us to refine the definition of NK and ILC1 identity to take into account their heterogeneity between and within tissues. Taken together, our data represent a resource to improve our understanding of the role of NK cells and ILC1s in physiology.
Results
Phenotypic diversity of NKp46+NK1.1+ cells between tissues
We first investigated the phenotypic heterogeneity of NKp46+NK1.1+ cells from liver, spleen, salivary glands (SGs), and lamina propria (LP) in mice by flow cytometry. As previously observed, NK cell markers (CD49b, Ly49H, Ly49D, and CD11b) were expressed by only Eomes+ cells, whereas all the ILC1 markers (CD49a, CD200R, LAG-3, TRAIL, and CXCR6) were expressed by Eomes− cells. However, we found Eomes− and Eomes+ cells in every population defined by the expression of the classical markers CD49a and CD49b (Figure S1A). Furthermore, Eomes+ and Eomes− NKp46+NK1.1+ cells had highly variable expression profiles depending on the tissue of origin, both in terms of frequency and intensity (Figure S1B). Some markers such as CD90.2 and CD122 were expressed by both Eomes− and Eomes+ populations.
Using UMAP and FlowSOM clustering, we identified nine distinct cell clusters with tissue-specific distribution patterns (Figures S1C–S1F). Clusters 1 and 2, corresponding to classical NK cells, were present in all tissues, while cluster 8 was shared between the liver, SG, and LP. Other clusters had a marked tissue-specific distribution: cluster 9 in the liver, clusters 3 and 4 in the SG, and clusters 6 and 7 in the LP. The detailed analysis of each cluster revealed that the expression of the NK cell activation receptors Ly49H and Ly49D segregate two major groups of populations, indicating the contribution of cells with two distinct origins, NK and non-NK, to the NKp46+NK1.1+ cell pool between tissues (Figure S1E). Further, with the exception of clusters 1 and 2, each cluster was defined by a graded and specific combinatorial expression of ILC1 markers, including CD49a, CD200R, CD127, and LAG-3. Finally, clusters 3 and 4, expressing both CD49a and CD49b but differing in terms of CD90.2 expression, fitted the definition of intermediate (INT) ILC1s (Figures S1D–S1F). Overall, these data highlighted a broad heterogeneity of NK and ILC1 subsets between tissues and unexpected variability in the expression of markers restricted to ILC1s depending on each subset.
Tissue-specific transcriptional profiles and diversity in NKp46+NK1.1+ cells
For a more in-depth analysis of this heterogeneity, we sorted Lin−NKp46+NK1.1+ cells from the liver, spleen, SG, and LP and subjected them to CITE-seq12 using antibodies against CD49a, CD49b, CD200R, CD90.2, CD122, LAG-3, and CD127 (Figure S2).
In agreement with our flow cytometry data, unbiased phenotypically based clustering separated the cells into multiple distinct clusters, with marked tissue-specific patterns of distribution (Figures 1A–1C) and marker expression levels (Figure 1D). Clusters 1 and 5 exhibited high levels of CD49a, exhibited low levels of CD49b and CD122, and segregated according to the expression of CD90.2. These clusters displayed inversely correlated levels of CD200R and CD127 (Figures 1D and S1D–S1F), suggesting that the expression of these two genes might be controlled by common regulatory mechanisms. At the transcriptomic level, these clusters were characterized by the common expression of ILC1-related genes, such as Cxcr6, Tmem176a, Tmem176b, Cd160, and Tnfsf10 (encoding TRAIL), whereas several other genes, such as Nr4a1, Ikzf2, Litaf, and Icam1, displayed tissue-restricted expression patterns (Figure 1D). The SG-associated clusters 4 and 7 also had closely related phenotypes. Consistent with the description of an “INT”-ILC1 population,4,5 these cells express CD49a and CD90.2, as well as intermediate levels of CD49b, and contain transcripts for both NK cell markers (Klra family genes and Eomes) and ILC1 markers (Il21r and Cd69) (Figure 1D). Finally, clusters 6, 3, and 2, in the liver and spleen, displayed high levels of CD49b and CD122, but not CD49a, CD127, CD200R, or LAG-3. At the transcriptomic level, these cells expressed NK cell marker genes Prf1, Zeb2, Gzma, Lgals1, Klf2, and Irf8 (Figure 1D). This classical NK cell phenotype, together with their conserved transcriptomic profiles between liver and spleen, suggested that these clusters represent non-tissue-specific circulating NK cells. UMAP dimensionality reduction based on RNA levels of NKp46+NK1.1+ single cells from all organs showed a clear segregation between cells according to their tissue of origin, particularly for LP (Figure 1E). Furthermore, computation of cell signature scores indicated that cells from clusters 6, 3, and 2 expressed high levels of genes associated to circulating cells, whereas other clusters were associated with a tissue-resident cell transcriptional signature13,14 (Figure 1F).
Figure 1.

Tissue-specific transcriptional profile of ILC1s
(A) UMAP showing the clustering of 17,820 NKp46+NK1.1+ cells from the liver, spleen, SG, and lamina propria (LP) on the basis of phenotypic markers from CITE-seq data, annotated by organ (left) and cluster label (right).
(B and C) Tissue distribution frequency analysis across phenotypic clusters (B) and representation of clusters by organ (C).
(D) Heatmap displaying the top 30 DEGs (bottom) by antibody-based clusters of NKp46+NK1.1+ cells (top) (fold change [FC] > 0.35 and p < 0.05).
(E) UMAP of NKp46+NK1.1+ single cells from all organs based on RNA levels.
(F) Module score analysis for tissue-resident and circulating lymphocytes; cell transcriptomic signature by cell surface phenotype-based cluster identified in (B) (mean ± SD).
(G) Cell surface phenotype-based clusters projected onto an RNA-level-based UMAP.
Interestingly, cells from SG and LP exhibited an NK cell phenotype but marked differences in their gene-expression program, indicating the presence of tissue-restricted NK cell subsets (Figure 1D). Along this line, cell surface phenotype-based clusters projected onto an RNA-level-based UMAP indicated that cell populations with identical phenotypes could still display markedly distinct gene expression programs between tissues (Figure 1G). Furthermore, with the exception of hepatic and splenic NK cells, UMAP and clustering based on transcriptomic data indicated that the other NK1.1+NKp46+ cell subsets clearly segregated according to tissue of origin. Overall, these results show that besides non-tissue-specific circulating NK cell populations, each organ includes tissue-resident cells derived from either NK cells or classical ILC1. They also highlight that the classical definition of NK cell and ILC1 phenotypes does not adequately take into account the heterogeneity of cell identity across tissues.
Liver NK and ILC1 subsets can be defined on the basis of maturation state
Given the heterogeneity of NKp46+NK1.1+ cells between tissues, we then investigated whether a spectrum of populations could also be found within each tissue. Principal-component analysis (PCA) on hepatic NKp46+NK1.1+ cells showed that principal component 1 (PC1) scores segregated two groups of cells, characterized by the expression of NK cell-associated genes (Klf2, Zeb2, Prf1, Itga4, Cma1, Sell, Klra8, Irf8, Klra4, Gzma, Eomes, Lgals1, and S1pr5) and expression of ILC1-related genes (Tnsf10, Cxcr3, Cd160, S100a4, Ly6e, Itga1, Cd3g, Ltb, Cxcr6, Xcl1, and Cd7) (Figures 2A and 2B). Cell clustering analysis further identified five subsets (Figure 2C). Based on the identity of the top 10 differentially expressed genes (DEGs), and the expression of NK and ILC1 signature genes between clusters, we annotated three subsets of NK cells (NK_Liv1–3) and two subsets of ILC1 (ILC1_Liv1 and ILC1_Liv2) (Figures 2C and 2D).
Figure 2.

Different transcriptional features between hepatic NK cell and ILC1 populations
(A) Principal-component analysis (PCA) of 16,262 hepatic NKp46+NK1.1+ cells.
(B) Heatmap showing the top 15 genes with the lowest or highest PC1 scores, ranked according to their score values. Cells and genes are sorted by principal-component scores.
(C) UMAP projection of hepatic NKp46+NK1.1+ cells clustered according to RNA levels. The donut graph shows the percentage of each liver NKp46+NK1.1+ subset identified.
(D) Heatmap of the top 10 upregulated DEGs of the identified NKp46+NK1.1+ clusters, ranked by adjusted p value (FC > 0.35 and p < 0.05).
(E) UMAP plot overlaid with tissue-resident and circulating lymphocyte signatures.
(F) Module score analysis for tissue-resident and circulating lymphocyte signatures for clusters identified in (C) (mean ± SD).
(G) Module score analysis of “early” and "effector" ILC1 transcriptomic signatures for the hepatic ILC1 subsets identified in (C) (mean ± SD).
(H) Violin plots showing mRNA expression profiles of selected genes across Seurat clusters.
(I and J) Left panel: pseudotime analysis of cells included in NK (I) and ILC1 (J) clusters. UMAPs are colored according to pseudotime scores (left) and cell clusters (right).
(K and L) Normalized expression of genes along the pseudotime axis calculated for the cells included in NK (K) and ILC1 (L) clusters. Color bars indicate cluster identity.
NK_Liv1–3 accounted for 36%, 22%, and 8% of the NKp46+NK1.1+ cells, respectively (Figure 2C). ILC1_Liv1 was the major ILC1 subset (27%), whereas ILC1_Liv2 represented only 7% of total hepatic NKp46+NK1.1+ cells. Consistent with our previous findings (Figure 1E), NK cell subsets preferentially expressed genes associated to circulating lymphocyte signature, whereas ILC1s were associated with a tissue-resident cell signature (Figures 2E and 2F).
NK_Liv1 expressed high levels of the transcription factors Zeb2 and Klf2 known to be required for NK maturation,15,16 as well as Prf1, Klrg1, and Ly6c2, indicative of a mature phenotype (Figure 2D). By contrast, NK_Liv2 expressed genes associated with an immature cell profile, such as the hematopoietic stem/progenitor cell regulators embigin (Emb) and Tcf7, the homing receptor CD62L (Sell), and the chemokine receptor CCR2, involved in NK cell migration. Consistent with a recent study,17 the NK_Liv3 subset was associated with expression of the stress-response genes, indicating that they likely represent a population of “stressed” cells. Gene Ontology (GO) enrichment analysis further indicated that, consistent with their mature phenotype, NK_Liv1 cells expressed genes associated with IFN-γ production, whereas NK_Liv2 cells express genes associated with T cell differentiation and activation (Figure S3A). By contrast, NK_Liv3 cells expressed genes involved in many biological processes, reinforcing the idea of a stressed cellular state.
Liver ILC1s express high levels of Itga1, Cxcr6, Cd200r2, Tnfsf10, and Cd7. Interestingly, they segregated into two subsets owing to a graded expression of the genes constituting this signature. ILC1_Liv1 cells preferentially expressed Il7r, Tmem176a and Tmem176b, Ikzf2, and Cd160, whereas ILC1_Liv2 had high levels of Itga1, Cd3g, Cd7, and Gzmc (Figures 2D and 2H). A cKit+CD127hiTCF-1hi “early” ILC1 subset with the potential to mature into CD127−TCF-1− effector ILC1s was recently identified in the liver.18 By comparing the gene expression profiles of the two hepatic ILC1 subsets with these previously described populations,18 we found that ILC1_Liv1 and ILC1_Liv2 matched to the early and effector ILC1 populations, respectively (Figure 2G). Furthermore, ILC1_Liv1 cells preferentially express genes associated with cell adhesion, IFN-γ production, and defense response to bacteria, reminiscent of an effector phenotype. ILC1_Liv2 cells expressed genes strongly associated with cytokine production and the Ag-receptor-mediated signaling pathway (Figure S3A).
To investigate whether these hepatic NK cells or ILC1s have a distinct origin or might represent cellular intermediates on a continuous differentiation pathway, we performed a trajectory reconstruction analysis using the Monocle algorithm (Figures 2I and 2J). In line with our previous study,8 the progressive downregulation of genes associated with an immature differentiation stage (Cd160, Emb, Tcf7, and Xcl1), paralleled by the upregulation of genes associated with effector functions and terminal maturation (Gzmb, Klrg1, or Zeb2), was associated with a transition between NK_Liv2 and NK_Liv1 (Figure 2K). Also, consistent with previous studies,17,18,19 pseudotime ordering of ILC1 pictures a transition from ILC1_Liv1 to ILC1_Liv2, with the downregulation of genes associated with stem-cell-like and immature state (Emb, Tcf7, Il7r, and Il18r1), balanced by an upregulation of effector genes (Gzma, Gzmb, Gzmc, and Tnfsf10) (Figure 2L).
Taken together, these data reveal that similar to NK cells, the liver ILC1 population is heterogeneous and includes two distinct subsets with marked differences in their differentiation and effector status.
Subsets of NK cells and ILC1s in the spleen
In contrast with the liver, PCA on splenic NKp46+NK1.1+ cells did not separate clear populations (Figure 3A). Still, it indicated that high PC1 scores were associated with the expression of the mature NK cell-associated genes (Figure 3B). We identified two major NK cell clusters, NK_Sp1 and NK_Sp2, accounting for 52% and 43% of total splenic NKp46+NK1.1+ cells, respectively, and two additional minor NK (NK_Sp3) and ILC1 (ILC1_Sp1) subsets (Figure 3C). As observed in the liver, splenic NK cells and ILC1s were respectively associated with a circulating and a resident cell transcriptional signature (Figures 3D and 3E). Interestingly, 6 of the top 10 DEGs were shared between the NK_Sp1 and NK_Liv1 populations (Figure 3F). We also identified similarities between NK_Sp2 and NK_Liv2 cells and between NK_Sp3 and NK_Liv3 cells. In agreement, with their highly related transcriptional programs and our previous study,8 trajectory reconstruction ordered the three NK cell clusters along a maturation process associated with regulation of gene expression. Of note, the set of genes modulated during splenic NK cell differentiation is the same than for hepatic NK cells (Figures 2K and 3H). Consistent with the functional features previously reported for these cells,8 NK_Sp1 and NK_Sp2, respectively, expressed genes associated with apoptosis/leukocyte migration and lymphocyte activation and differentiation (Figure S3B). NK_Sp3 cells expressed genes involved in the response to cytokines.
Figure 3.

scRNA-seq identifies liver-like splenic NK cells and one splenic ILC1 subset
(A) PCA of 8,062 splenic NKp46+NK1.1+ cells.
(B) Heatmap showing the top 15 genes with the lowest or highest PC1 score, ranked according to their score value.
(C) UMAP projection of splenic NKp46+NK1.1+ cells clustered on the basis of RNA levels. The donut graph shows the percentage of each spleen NKp46+NK1.1+ subset identified.
(D) UMAP plot overlaid with tissue-resident and circulating lymphocyte signatures.
(E) Module score analysis of tissue-resident and circulating lymphocyte signature for clusters identified in (C) (mean ± SD).
(F) Heatmap of the top 10 upregulated DEGs of identified NKp46+NK1.1+ clusters, ranked by adjusted p value (FC > 0.35 and p < 0.05).
(G) Pseudotime analysis of cells included in NK clusters. UMAPs are colored according to pseudotime scores (left) and cell cluster (right).
(H) Normalized expression of genes along the pseudotime axis calculated for the cells included in NK cell clusters. Color bars indicate cluster identity.
(I) Module score analysis of splenic NK1, NK2, and NK3 transcriptomic signatures for the splenic and hepatic ILC1 subsets identified in (C) (mean ± SD).
We recently identified mature (NK1) and immature (NK2) NK cell populations in mouse spleen and blood and in human blood.8 The NK_Liv1/NK_Sp1 and NK_Liv2/NK_Sp2 populations were respectively characterized by high similarity scores for the NK1 and NK2 gene signatures, indicating that these populations are closely related to blood NK cells (Figure 3I) and likely represent non-tissue-specific circulating NK cells.
Finally, we found a small but homogenous subset of NKp46+NK1.1+ cells distinct from classical NK cells, expressing the early ILC1-associated genes Il7r, Cd160, Tmem176a, Tmem176b, and Cxcr6 (Figures 3A, 3C, and S4).
Overall, these results confirm that the NK cells found in both the liver and the spleen correspond to non-tissue-specific circulating NK cells and also reveal a small population of splenic cells displaying “early ILC1” features.
Two distinct origins for ILC1 in SG
We then focused our transcriptional analysis on NKp46+NK1.1+ cells from the SG, where ILC1s have unique phenotypic features because of TGF-β imprinting.4 PCA on NKp46+NK1.1+ cells did not reveal any clear segregation between populations but a continuum of cells along PC1, driven by ILC1- and NK-related genes (Figures 4A and 4B). We identified five cell subsets, including a large population of unconventional NK cells (NK_SG1) displaying a mixed expression profile with genes associated to effector and immature NK cells, two populations of ILC1s expressing high levels of Gzmb and Gzmc (ILC1_SG1) similar to the liver effector ILC1s, and the ILC1_SG2 expressing Il7r, Tmem176a, Tmem176b, Il18r1, and Emb, similar to the liver early ILC1s (Figures 4C–4E). Two additional subsets (INT_SG1 and INT_SG2) expressed the NK cell receptor genes Klra4, Klra7, and Klra8, indicating that they belong to the NK cell lineage (Figure 4E). They also displayed intermediate features between NK and ILC1, such as lower levels of Eomes and Zeb2 but increased levels of Itga1 and Tnfsf10 (Figure 4E). The INT_SG1 subset shares a significant part of its transcriptional signature with NK_SG1 cells but differed by a lower expression of Sell, Klf2, Il18r1, Emb, and Gzma. The INT_SG2, closely related to ILC1_SG1 cells, did not express these genes but expressed Cxcr6, Tnfsf10, and Gzmc. Consistent with a differentiation of NK cells into ILC1s upon TGF-β stimulation,4 these two intermediate populations expressed the TGF-β target genes, Smad7 and Itgae. Also, pseudotime analysis ordered these populations from NK_SG1 to ILC1_SG1, via INT_SG1 and INT_SG2, because of the progressive downregulation of NK and immature cell-associated genes (Zeb2, Klf2, Eomes, Cxcr3, Tcf7, Emb, Il18r1, and Cd160), balanced by an upregulation of Itgae and the effector ILC1-related genes Itga1, Cxcr6, Gzmb, and Gzmc (Figures 4F and 4G). In contrast with the liver and spleen, we found a progressive transition from a circulating to a resident lymphocyte signature, which parallels the transition of NK cells to ILC1s along the pseudotime axis (Figures 4H and 4I).
Figure 4.

Transcriptional heterogeneity among NKp46+NK1.1+ subsets in the salivary glands
(A) PCA of 4,455 NKp46+NK1.1+ cells from the SG.
(B) Heatmap showing the top 15 genes with the lowest or highest PC1 score, ranked according to their score value.
(C) UMAP projection of SG NKp46+NK1.1+ cells clustered on the basis of RNA levels. The donut graph shows the percentage of each of the NKp46+NK1.1+ subsets identified.
(D) Heatmap of the top 10 upregulated DEGs of the identified NKp46+NK1.1+ clusters, ranked by adjusted p value (FC > 0.35 and p < 0.05).
(E) Violin plots showing mRNA expression profiles of selected genes across Seurat clusters.
(F) Pseudotime analysis of cells included in NK clusters. UMAPs are colored according to pseudotime scores (left) and cell cluster (right).
(G) Normalized expression of genes along the pseudotime calculated for the cells included in NK and ILC1 clusters. Color bars indicate cluster identity.
(H) UMAP plot overlaid with tissue-resident and circulating lymphocyte signatures.
(I) Module score analysis for tissue-resident and circulating lymphocyte signature for clusters identified in (C) (mean ± SD).
GO analysis revealed that NK_SG1 cells were enriched in the expression of genes associated with IFN-γ and TNF-α production and defense responses, consistent with their activated phenotype (Figure S3C). INT_SG1 cells expressed genes associated with cell adhesion and T cell activation, and INT_SG2 cells expressed genes associated with cytolysis. Finally, although ILC1_SG1s are associated with a cytotoxicity-related gene signature, ILC1_SG2 cells are enriched in genes involved in activation, proliferation, and differentiation, reminiscent of effector and early differentiating gene expression programs.
Highlighting the heterogeneity of the SG NKp46+NK1.1+ cell population, these results indicate that the ILC1_SG1 subset derives from tissue-resident NK cells, whereas the ILC1_SG2 arises from a distinct origin. They also reveal a striking parallel with the liver ILC1s, as these two ILC1 populations also exhibit an immature and effector status.
Transcriptomic heterogeneity and complexity among LP NKp46+NK1.1+ cells
In agreement with our data and previous reports,11,20 scRNA-seq analysis indicated that the NKp46+NK1.1+ cell population from the LP is highly heterogeneous and differs from the ones infiltrating other tissues (Figures 1E and S1). Similar to the SG, PCA indicated that cells from LP constitute a continuum driven on PC1 by ILC1-related genes and NK cell signature genes (Figures 5A and 5B).
Figure 5.

Transcriptomic analysis reveals that NKp46+NK1.1+ subsets from the LP have unique features
(A) PCA of 15,658 NKp46+NK1.1+ cells from the LP.
(B) Heatmap showing the top 15 genes with the lowest or highest PC1 score, ranked according to their score value.
(C) UMAP projection of NKp46+NK1.1+ cells from the LP clustered on the basis of RNA levels. The donut graph shows the percentage of each LP NKp46+NK1.1+ subset identified.
(D) Heatmap of the top 10 upregulated DEGs of the identified NKp46+NK1.1+ clusters, ranked by adjusted p value (FC > 0.35 and p < 0.05).
(E) Violin plots showing mRNA expression profiles of selected genes across Seurat clusters.
(F and G) Pseudotime analysis of cells included in NK (F) and ILC1 (G) clusters. UMAPs are colored according to pseudotime scores (left) and cell cluster (right).
(H and I) Normalized expression of genes along the pseudotime axis calculated for the cells included in NK (H) and ILC1 (I) clusters. Color bars indicate cluster identity.
(J) UMAP plot overlaid with tissue-resident and circulating lymphocyte signatures.
(K) Module score analysis for tissue-resident and circulating lymphocyte signatures for clusters identified in (C) (mean ± SD).
We next identified one NK cell subset (NK_LP1) accounting for 10% of total NKp46+NK1.1+ cells, two intermediate clusters (INT_LP1/INT_LP2) accounting for 20% and 12% of the cells, and two ILC1 subsets (ILC1_LP1/ILC1_LP2) accounting for 43% and 15% of the whole-cell population (Figure 5C).
Both NK_LP1 and INT_LP1 expressed the NK cell receptor genes Klra4, 7, 8, 9, and Eomes but could be distinguished by the expression of GzmA, Sell, and Klf2 (Figures 5D and 5E). A close analysis of the INT_LP2 subset revealed that it included both a small fraction of cells expressing Klra genes and Eomes and cells expressing Itga1, Tnfsf10, and Cxcr6. Yet, they homogenously express high levels of stress-response-related genes (Hspa1a, Hspa1b, Jun, Hspe1, and Hsph1) and the IFN-γ-inducible gene Ifi47 (Figure 5D). This suggested that they could represent a mixed population of closely related stressed or activated cells but with distinct NK or non-NK origins. Consistently, the INT-LP2 subset seemed to segregate into two subpopulations, one in close contact with the INT_LP1 and one close to the ILC1_LP2 (Figure 5C). In agreement, ILC1_LP1 and ILC1_LP2 did not express Klra genes and Eomes but the core ILC1 signature genes Itga1, Tnfsf10, and Cxcr6 (Figure 5E).
In line with the continuum of LP NKp46+NK1.1+ populations, trajectory reconstruction analyses revealed a proper transition of Klra-expressing cells from mature NK cells to cells with an ILC1 identity, with the downregulation of Eomes, Zeb2, Klrg1, and Gzma, balanced by an upregulation of Cxcr6, Rora, Il7r, Tmem176a, and Tmem176b (Figures 5E, 5F, and 5H). Further, among non-NK ILC1, a transition from ILC1_LP1 to ILC1_LP2 was associated with the downregulation of the early ILC1-associated genes, Il7r and Tmem176a and Tmem176b, and the upregulations of genes encoding effector molecules (Figures 5E, 5G, and 5I). Similar to SG, we observed a progressive transition from a dominant circulating to a dominant resident cell signature (Figures 5J and 5K). Importantly, ILC1_LP1 included cells expressing the Il22 and Rorc genes, suggesting that at least part of this population represented ILC3-derived ILC1 (Figure 5D). Hence, as for the liver and the SG, the NKp46+NK1.1+ cell population from the LP displaying a core ILC1 gene expression program includes two populations with distinct maturation and effector status. Further, they include cells from multiple origins, such as classical and ILC3-derived ILC1.
Common and specific signatures of NKp46+NK1.1+ populations
TGF-β signaling is required for the formation of resident memory T cells in multiple organs, including the LP and the SG, but not the liver. Consistently, we found that a TGF-β-induced gene signature is highly enriched in cells from the LP and SG and mildly in the liver (Figure S6). Also, we recently showed that a tissue-specific differentiation pathway controls the differentiation of liver ILC1.21 These data supported the hypothesis that ILC1s are under the influence of tissue-specific niches, whether it would stem from local environmental signals or developmental pathways, leading to significant differences in their transcriptional programs (Figure 1E). Yet, our results indicated that ILC1 populations from each peripheral tissue segregate into two subsets with an early and effector state, highlighting a significant conservation of cell identities between tissues.
To evaluate the extent of these similarities, we performed a multidataset integration using the Harmony algorithm.22 The projection of every subset from all organs on a UMAP representation provided an overview of their relationships (Figures 6A and S5). Three well-defined compartments corresponding to NK, intermediate cells, and ILC1 subsets segregated together between tissues, showing that despite their transcriptional profile heterogeneity (Figure 1E), each tissue-resident NKp46+NK1.1+ cell pool includes highly related cell subsets.
Figure 6.

Inter-tissue-specific signatures of NK and ILC1 subpopulations
(A) Transcriptome-based clusters computed for each organ separately, projected onto the multi-organ integrated UMAP obtained with Harmony.
(B) Heatmap representing overlap coefficient dissimilarity between transcriptome-based clusters calculated separately for each organ.
(C) Dot plot of NK cell-specific versus ILC1-specific markers. Intermediate clusters were not taken into account.
(D) Dot plot of specific markers for each cluster.
(E) Flow cytometry profiles of CD49a, DNAM-1, PD-1H, and SDC4 expression, analyzed in Lin−NKp46+NK1.1+ cells. Histograms represent the frequency of CD49a−CD49b+ NK cells and CD49a+CD49b− ILC1s for each indicated marker across organs. Data are shown as mean ± SEM and are pooled from two independent experiments. Each point represents a pool of two mice.
We then computed the Szymkiewicz-Simpson coefficient for each pair of cell subsets and show that all the tissue-resident ILC1 populations clustered together regardless of their origin (Figure 6B). Also, ILC1_Sp1, ILC1_SG2, and ILC1_LP1 were closely related to the liver early ILC1 population (ILC1_Liv1). In parallel, INT_SG2, ILC1_SG1, and ILC1_LP2 were the closest populations to the liver ILC1_Liv2 effector population within each tissue. In agreement with our tissue-specific analyses, these results further supported the hypothesis that comparable populations of immature and effector ILC1 populations could be found within each tissue.
We also identified a common gene signature for all NK cells versus ILC1 subsets regardless of the tissue of their origin (Figure 6C). Compared with ILC1s, NK cells homogenously express genes from the Klra family encoding NK cell receptors of the Ly49 family and high levels of Klf2, Gzma, Zeb2, Cma1, Sell, Eomes, Itgam, Itga2, and Prf1. They also express Scimp, encoding a protein acting as a scaffold for Src-family kinases,23,24 integrin β4 (Itga4), involved in cytolytic cell-cell interactions with target cells, pleckstrin (Plek) and the CD3ζ subunit (Cd247). Conversely, the ILC1s differ from NK cells by the expression of DNAX accessory molecule DNAM-1 (Cd226), the integral membrane protein (Itm2c), the transcriptional regulator Rbpj playing a central role in Notch signaling,25 and Ltb. In addition, the differential expression of Tnfsf10, Tmem176a and Tmem176b, Il7r, and Cxcr6, as well as Itga1, segregate ILC1 subpopulations corresponding to early and effector transcriptional profiles within each tissue. Finally, we identified sets of genes characterizing unique ILC1 populations in each organ (Figure 6D).
We next tested several markers from our NK-ILC1 gene signature by flow cytometry (Figures 6C and 6D). Although DNAM-1 has been shown to be expressed by NK cells in spleen, liver, and tumors,5,26 it is also expressed by more than 70% of ILC1s in all organs (Figure 6E). Interestingly, DNAM-1 is more highly expressed by hepatic ILC1s as compared with NK cells. Furthermore, PD-1H, also known as VISTA, is also expressed by a majority of ILC1s across organs, as well as NK cells from the liver and the spleen (Figure 6E). Importantly, we identified that syndecan-4 (SDC4), a transmembrane heparan sulfate proteoglycan, is specifically expressed by almost all ILC1s and not by NK cells, suggesting that this marker will be useful to discriminate NK cells from ILC1s.
Identification of cross-organ NK and ILC1-specific markers between mouse and human
A recent study analyzed the heterogeneity of human ILCs and NK cells by scRNA-seq across four tissues: blood, tonsil, lung, and colon.9 We sought to determine whether our murine NK cell versus ILC1 signatures could also identify these populations in human. We thus performed PCA, clustering, and UMAP on this human ILC scRNA-seq dataset with the human orthologous genes. We found that ourmurine liver and SG signatures allow us to accurately discriminate human NK cells from ILC1s (Figure 7A). When we used signatures from murine spleen or LP, we were also able to separate human NK cells from ILC1s, except for the tonsil where these subsets clustered (Figure 7A).
Figure 7.

Transfer murine transcriptomic signatures for human dataset
(A) Clustering and UMAP calculated with organ-specific murine orthologous signature. Cells are colored by cluster (upper panel) or by organ (lower panel).
(B) Venn diagram displaying cross-species and cross-organ NK or ILC1 signature intersections: HS.LUNG (human signature from lung9), HS.TONSIL (human signature from tonsil9), MM.LIVER (murine signature from liver), MM.SPLEEN (murine signature from spleen), MM.GUT (murine signature from gut), and MM.SG (murine signature from salivary glands).
(C) Dot plot of cross-species and cross-organ NK cell-specific and ILC1-specific markers. Dot size encodes the percentage of positive cells and dot intensity the average expression.
To identify common NK and ILC1 markers in mouse and human, we performed differential expression analysis between NK cells and ILC1 markers from both species in six different organs: human lung, human tonsil, murine liver, murine spleen, murine SG, and murine LP. We found eight cross-species and cross-organ NK-specific markers, EOMES, GZMA, IRF8, JAK1, NKG7, PLEK, PRF1, and ZEB2 (Figure 7B). Importantly, these eight markers also discriminated human NK cells from the other ILC subtypes and CD4 T cells (Figure 7C). We identified three cross-species and cross-organ ILC1-specific markers: IL7R, LTB, and RGS1 (Figure 7B). However, these three markers were also expressed by other ILC populations and CD4 T cells (Figure 7C).
Discussion
Distinguishing between NK cells and ILC1s is a major challenge to study the individual functions of these cells. However, it remains difficult to identify these two cell types unambiguously because of their high degree of similarity and their plasticity in a particular environment. We used CITE-seq and high-throughput scRNA-seq to investigate the similarities and differences between NK cells and ILC1s in multiple organs and showed that their current phenotypic definition does not adequately take into account the heterogeneity of cell identity across tissues.
We previously identified two major NK subsets from spleen and blood in mouse and human: NK1 defined as human CD56dim and mouse CD27−CD11b+ mature NK cells and NK2 as human CD56bright and mouse CD27+CD11b− immature NK cells.8 Here, we showed that NK1 and NK2 are found in spleen and liver, revealing that these populations are closely related to blood NK cells and represent non-tissue-specific circulating NK cells. Consistently, the transcriptional profile of these cells was found to be homogeneous, probably because these cells circulate in the bloodstream and are not specific to a tissue.
NK cells from SG have a singular transcriptomic profile and constitute an activated and mature subset. This cell type also expressed a set of genes implicated in defense responses, consistent with a study showing that SG NK cells play a critical role during chronic viral infection.27 NK cells from the LP are also unique. In particular, they express the gene encoding vascular endothelial growth factor A (Vegfa) involved in angiogenesis. Interestingly, uterine NK cells also produce this angiogenic factor.28 Whether NK cells in the gut could have protumorigenic effects because of their support for tumor angiogenesis remains to be investigated.29
In line with their tissue-resident status,2,11 the transcriptomic profile of ILC1s was found to be specific to individual tissues. By using CITE-seq, scRNA-seq, and Monocle analysis, we provide data supporting that hepatic ILC1s could differentiate from an immature to an effector subset, which is consistent with recent findings.18 It has been reported that this conversion is mediated by Hobit because germline deletion of Zfp683 (encoding Hobit) leads to an increase in immature cells and a decrease of cytotoxic ILC1s. However, other data reported that the conditional deletion of Zfp683 in NKp46+ cells reduced both subsets, suggesting that cytotoxic ILC1s develop from another progenitor in a Hobit-dependent manner.17 Furthermore, recent results indicate that these two liver ILC1 populations derive from distinct ontogenic pathways.30 Thus, further investigations are required to better characterize the origin and differentiation of hepatic ILC1s.
We also found two distinct populations in the SG and the LP closely related to these liver ILC1 subsets, which display an early and effector phenotype. Consistently, a recent study identified a mature and cytotoxic ILC1 subset from mouse liver and SG that expressed granzyme C and contributed to antitumor immunity and neonatal autoimmunity.31 ILC1s have been described as poor cytotoxic cells. We and others17,18 found that ILC1s expressed granzymes A, B, and C, whereas NK cells expressed granzymes A and B. Altogether, these data demonstrate that a lytic granule-mediated cytotoxicity program is not restricted to NK cells and could also occur in subsets of ILC1s.
We also noticed that despite these similarities in their gene expression programs, ILC1s arise from distinct developmental pathways. In contrast to ILC1_Liv2 and ILC1_LP2, the ILC1_SG1s derive from NK cells. Also, ILC1_LP1s express Il22 and Rorc, indicating they might derive from ILC3, as described in both mice and humans.
The presence of ILC1s in the spleen remains a matter of debate. Here, we identified a discrete population of cells with a gene expression program reminiscent of immature ILC1s identified in liver. Interestingly, it has been recently identified that splenic ILC1s exhibit an immature phenotype and a broader cytokine profile than conventional NK cells.32 These ILC1-like NK cells directly recognize MHC class I-deficient or virus-infected cells and mounted cytotoxic, adaptive-NK live responses as efficiently as conventional NK cells. Furthermore, our data showed that splenic ILC1s highly expressed genes enriched in lymphocyte differentiation and activation processes. Accordingly, these cells play a role in inducing conventional type 1 dendritic cell clustering and consequently enhancing CD8+ T cell priming during murine cytomegalovirus infection,32 indicating that this ILC1 subset constituted an early sentinel of viral infection and was able to contribute to adaptive-like immune responses.
We identified two intermediate subsets in the SG (INT_SG1 and INT_SG2) with transcriptomic profiles resembling those of NK cells (NK_SG1) and ILC1s (ILC1_SG1 and ILC1_SG2). In parallel, we found an NK cell-like intermediate subset in the LP with cell-killing functions (INT_LP1), while a second population (INT_LP2) had a profile consistent with a role in stress responses. Although these cells intermediate between the NK cell and ILC1 compartments appeared to be unique, they do share similarities with NK cells or ILC1s, possibly reflecting the differentiation of NK cells into ILC1s as a result of environmental cues such as TGF-β.4,5 Consistently, a Monocle analysis ranked these cell populations between NK cells and ILC1s along a pseudotime axis.
Finally, we performed a transcriptomic comparison of NK cells, ILC1s, and intermediate cells, across tissues, and identified sets of genes expressed by related subsets or characterizing unique ILC1 populations in each organ. We used this specific NK-ILC1 gene signature to identify additional universal cell-surface markers to better distinguish these two cell types by flow cytometry. Although NK cells express both DNAM-1 and PD-1H, ILC1s specifically expressed high levels of these two receptors. Interestingly, a recent study showed that ILC1s target leukemia stem cells through DNAM-1 to suppress leukemogenesis, revealing an important role of DNAM-1 in their anti-tumor function.33 We also identified SDC4 (ryudocan) as a possible marker for ILC1s because it was expressed by almost all ILC1s and not by NK cells. SDC4 is a transmembrane heparan sulfate, which is involved in signal transduction processes, cell proliferation, migration, and adhesion. Its extracellular domain binds several growth factors such as fibroblast growth factor 2 (FGF2),34 hepatocyte growth factor (HGF),35 platelet-derived growth factor (PDGF),36 and also different cytokines, including monocyte chemoattractant protein-1 (MCP-1)37 or stromal cell-derived factor-1 (SDF-1), also referred to as CXCL12.38 It can also directly bind fibronectin.39 It has been shown that SDC4 is a peculiar T cell coinhibitory receptor that does not include any immunoreceptor-based tyrosine inhibition motif. Instead, SDC4 employs the protein tyrosine phosphatase CD148 to inhibit T cell activation.40 Furthermore, SDC4 is also crucial for dendritic cell adhesion and motility,41 as well as their migration during allergic inflammation.42 However, the role of SDC4 in ILC1s remains to be elucidated.
Recent studies aimed to characterize the transcriptional programs of ILC1s and NK cells in humans, and as in the mouse ILC1s, identification in humans is not straightforward. Consistent with our analysis, they revealed a considerable heterogeneity and a substantial impact of the microenvironment on the transcriptomic profiles of these cells.6,7,8,9,10 We took advantage of our data to analyze the similarities and the differences between the NK cell and ILC1 subsets identified in humans. We found that our murine NK-ILC1 signatures can also discriminate these populations in humans, and we identified metagenes consisting of eight cross-species and cross-organ genes for NK cells (EOMES, GZMA, IRF8, JAK1, NKG7, PLEK, PRF1, and ZEB2) and three genes for ILC1 (IL7R, LTB, and RGS1). Taken together with the multidimensional comparison of NK and ILC1 subsets across four tissues in the mouse, these resource data pave the way for improving the characterization and understanding of NK cell and ILC1 biology.
Limitations of the study
Our results indicate that the ILC1 compartment is highly heterogeneous, owing to the coexistence of distinct tissue-resident NKp46+NK1.1+ populations, cell states, and maturation stages. This will have to be carefully taken into account in future studies. Nevertheless, our results indicate that related populations are found between tissues, suggesting developmental relationships between them that remain to be fully investigated. Also, we provide an in-depth snapshot analysis of ILC1 populations at steady state, but our study does not take into account parameters such as their evolution across time or pathological conditions. Whether and how these variables alter ILC1 heterogeneity will have to be addressed in follow-up studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PeCy5 anti-mouse CD3 (145-2C11) | BD Biosciences | Cat# 553065; RRID:AB_394598 |
| PeCy5 anti-mouse TCRβ (H57-597) | BD Biosciences | Cat# 553173; RRID:AB_394685 |
| BV650 anti-mouse CD226/DNAM-1 (10E5) | BD Biosciences | Cat# 750916; RRID:AB_2875003 |
| BUV805 anti-mouse CD45 (104) | BD Biosciences | Cat# 741957; RRID:AB_2871265 |
| BV785 anti-mouse CD49a (Hα31/8) | BD Biosciences | Cat# 740919; RRID:AB_2740560 |
| BUV395 anti-mouse CD49b (HMα2) | BD Biosciences | Cat# 740250; RRID:AB_2739996 |
| A700 anti-mouse NKp46 (29A1.4) | BD Biosciences | Cat# 561169; RRID:AB_10561840 |
| BV510 anti-mouse NK1.1 (PK136) | BD Biosciences | Cat# 563096; RRID:AB_2738002 |
| APC.Cy7 anti-mouse CD90.2 (53-2.1) | BD Biosciences | Cat# 561641; RRID:AB_10898013 |
| BUV661 anti-mouse CD122 (5H4) | BD Biosciences | Cat# 741537; RRID:AB_2870975 |
| BV421 anti-mouse CD223/LAG-3 (C9B7W) | BD Biosciences | Cat# 740072; RRID:AB_2739836 |
| BV650 anti-mouse Ly49D (4E5) | BD Biosciences | Cat# 742558; RRID:AB_2740868 |
| BV605 anti-mouse VISTA/PD-1H (MIH64) | BD Biosciences | Cat# 742722; RRID:AB_2740998 |
| PeCy5 anti-mouse Ter-119 (Ter-119) | eBiosciences | Cat# 15-5921-83; RRID:AB_468811 |
| PerCP-eFluor 710 anti-mouse CD200R (OX110) | eBiosciences | Cat# 46-5201-82; RRID:AB_10804765 |
| PeCy7 anti-mouse CD127 (A7R34) | eBiosciences | Cat# 25-1271-82; RRID:AB_469649 |
| eFluor660 anti-mouse Eomes (Dan11mag) | eBiosciences | Cat# 50-4875-82; RRID:AB_2574227 |
| APC anti-mouse CD253/TRAIL (N2B2) | eBiosciences | Cat# 17-5951-82; RRID:AB_2573248 |
| PeCy5 anti-mouse TCRγδ (GL3) | eBiosciences | Cat# 15-5711-82; RRID:AB_468804 |
| FITC anti-mouse Ly49H (3D10) | eBiosciences | Cat# 11-5886-82; RRID:AB_1257159 |
| PeCy5 anti-mouse CD19 (1D3) | eBiosciences | Cat# 15-0193-82; RRID:AB_657672 |
| APC anti-mouse SDC4 (REA640) | Miltenyi Biotec | Cat# 130-109-831; RRID:AB_2653641 |
| PE/Dazzle 594 CXCR6 (SA051D1) | BioLegend | Cat# 151117; RRID:AB_2721700 |
| A700 anti-mouse CD11b (M1/70) | BioLegend | Cat# 101222; RRID:AB_493705 |
| Biological samples | ||
| Healthy liver, spleen, salivary glands and lamina propria from adult mice | N/A | |
| Human ILC single-cell dataset | Mazzurana et al., 20219 | Downloaded from the NCBI GEO depository (GSE150050) |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase IV | Sigma Aldrich | C5138 |
| Collagenase VIII | Sigma Aldrich | C2139 |
| DNase | Sigma Aldrich | DN25 |
| Fetal bovine serum | Thermo Fisher scientific | 10437028 |
| RPMI1640 | Corning | 10-041-CV |
| Phosphate Buffered Saline | Corning | 21-040-CMR |
| EDTA | Invitrogen | AM9260G |
| Foxp3 transcription factor staining buffer kit | Invitrogen | 00-5523-00 |
| LIVE/DEAD Fixable Near-IR dead cell stain kit | Life technologies | L10119 |
| Critical commercial assays | ||
| Easysep Mouse NK Cell Isolation Kit | Stemcell technologies | 19855 |
| 10× Genomics Chromium Next GEM Single Cell 3′ v3.1 kit | Fisher scientific | NC1690752 |
| Qubit High Sensitivity assay kit | Invitrogen | Q32854 |
| Deposited data | ||
| Raw and analyzed data (scRNA-seq) | This paper | GEO: GSE189807 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6JR | Janvier Labs | https://janvier-labs.com/fiche_produit/2-c57bl-6jrj/ |
| Software and algorithms | ||
| Flowjo software v10 | Tree Star | https://www.flowjo.com/RRID: SCR_008520 |
| Graph Pad Prism Version 9 | Graphpad | https://www.graphpad.com/RRID: SCR_002798 |
| CellRanger software v4.0.0 | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| CITE-seq-Count 1.4.3 | https://hoohm.github.io/CITE-seq-Count/ | |
| Seurat v4.0.0 | Hao et al., 202143 | https://satijalab.org/seurat/ |
| Monocle 0.3.1 | N/A | https://cole-trapnell-lab.github.io/monocle3/ |
| ClusterProfiler 3.18.1 | N/A | https://guangchuangyu.github.io/software/clusterProfiler/ |
| Harmony 0.1.0 | Korsunsky, I et al. Nat Methods, 201922 | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
Resource availability
Lead contact
Further information and resource requests should be directed to and will be fulfilled by the lead contact, Eric Vivier (vivier@ciml.univ-mrs.fr).
Materials availability
This study did not generate new materials.
Experimental model and subject details
C57BL/6J WT mice were purchased from Janvier Laboratories. All mice were maintained under specific pathogen-free conditions at the Centre d’Immunologie de Marseille-Luminy, France. Standard food and water were provided ad libitum. Eight-week-old females were used for this study. All experiments were performed in accordance with national and European regulations and were approved by the Ethics Committee for Animal Experimentation of Marseille (Comité National de Réflexion Ethique sur l’Expérimentation Animale no.014).
Method details
Mouse tissue collection
Splenocytes and liver cells were isolated by forcing the tissues through 70 μm-mesh and 100 μm-mesh cell strainers. SG were cut into small pieces and digested by incubation with 500 IU/mL collagenase IV and 5 IU/mL DNase I for 45 min at 37°C. Cell suspensions from SG were then passed through a 18G needle. The gut was resected and cleaned of residual fat tissue. Peyer’s patches were removed and the intestine was cut open longitudinally and washed with phosphate-buffered saline (PBS) on ice. The gut tissue was then incubated in 10% fetal bovine serum, 5 mM EDTA and 15 mM HEPES in PBS for 30 min, at 37°C, with shaking. The supernatant was discarded and the remaining tissue was incubated with 10% FCS, 15 mM HEPES and 300 IU/mL collagenase VIII in PBS for 30 min at 37°C, with shaking. The digested material was then ground and passed through a 18G needle. Red blood cells were lysed in RBC lysis buffer (BioLegend). Cells isolated from the liver, SG and LP were separated by gradient centrifugation with 37.5 and 67.5% (for liver and SG) and 40 and 60% (for LP) Percoll.
Flow cytometry and cell sorting
Cells were analyzed by flow cytometry with standard procedures. For surface staining, cells were incubated for 20 min at 37°C with antibodies and LIVE/DEAD Fixable Near-IR in FACs buffer (1xPBS, 2% FCS, 1 mM EDTA). Cells were stained with antibodies against CD3, TCRβ, CD45, CD49a, CD49b, NKp46, NK1.1, CD90.2, CD122, LAG-3, Ly49D, CD226, PD-1H, F4/80, CD19, Ter-119, CD200R, CD127, TCRγδ, Eomes, Ly49H, TRAIL CXCR6, CD11b and SDC4. Flow cytometry analysis was performed with a FACS Symphony (BD Biosciences) cell sorter and data were analyzed with FlowJo software (BD Biosciences). The cells for cell sorting were isolated from the spleen, liver, SG and LP and were enriched by centrifugation on Percoll. For the LP, after purification on a Percoll gradient, the cells were stained with a biotin-conjugated EpCAM antibody for 15 min at 4°C, and enriched with a Stemcell EasySep™ Mouse NK Cell Isolation Kit. Lin (CD3, CD19, Ter-119, F4/80, TCRβ, TCRγδ)−CD45+NKp46+Nk1.1+ cells were purified on a FACSAriaIII Cell Sorter (BD Biosciences).
Library preparation and sequencing
According to the instructions of 10× Genomics, cells were resuspended in 50 μL staining buffer and incubated with Fc receptor blocker for 10 min. They were stained simultaneously with sorting antibodies (Live/Dead, CD45.2, NK1.1, NKp46, Lin: CD3, CD19, F4/80, TCRb, TCRgd, Ter119) and TotalSeq-A antibodies from Biolegend (Table S1), for 45 min at 4°C. Cells were then washed three times in staining buffer, and stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain for 10 min at 4°C.
Cells were sorted and resuspended at a density of 1000 cells/μL in 1× PBS + 0.04% bovine serum albumin (BSA). Libraries were then prepared with the Chromium Next GEM Single Cell 3′ v3.1 kit from 10× Genomics. RNA and protein libraries were pooled in a 9:1 ratio and sequenced in 2 × 100 bp paired-end mode on a NextSeq 550 at the GenomEast platform of the Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) of Strasbourg. All libraries were quantified and qualified in Qubit High Sensitivity assays and with the Agilent Bioanalyzer High Sensitivity kit.
Preprocessing of samples
Raw FASTQ files were processed with CellRanger software (v4.0.0), which performs alignment, filtering, barcode counting and UMI counting. CellRanger software was used to align reads with the mm10 genome. Antibodies were counted with Cite-seq-Count v1.4.3. Subsequent analyses were performed with Seurat v3.1.5. We first analyzed each sample independently as follows. Low-quality cells were excluded in an initial quality-control step, which removed genes expressed in fewer than three cells and cells expressing fewer than 200 genes. Cells with less than 7% ribosomal genes or more than 10% of mitochondrial were excluded. Library size normalization was performed on the gene expression values for each cell barcode after UMI collapsing, with scaling by the total number of transcripts and multiplication by 10,000. The data were then log-transformed before further downstream analysis with Seurat. We selected genes with a high variance, using the FindVariableGenes function with default parameters. We then reduced the dimensionality of our data by PCA and cells were clustered with Seurat’s FindClusters function with the Louvain algorithm. We applied the CellCycleScoring function to identify cells associated with the S or G2 phases of the cell cycle for removal (we set the threshold at 0.15 rather than 0).
Pooled sample analysis by organ
We then merged the samples by tissue and reperformed the selection of variable genes, normalization and clustering. Small clusters found in less than 2.5% of cells were removed. In total, we obtained 16262, 8062, 4455, 15658 cells for the liver, spleen, SG and gut, respectively. The two SG samples differed in depth. We therefore applied a regression to nCount_RNA during the ScaleData step. For visualization, we applied RunUmap to the cell, using the previously selected PCs with default parameters to view the cells in two dimensions. We identified cluster markers with FindAllMarkers, with the parameter only.pos set to TRUE and logFC = 0.35 to obtain exclusively upregulated genes as markers of a cluster relative to all other cells.
Pooled multi-organ sample analysis
We then pooled all the samples together. We ensured that the mixture contained equal numbers of cells per organ, by randomly downsampling 4455 cells from the liver, spleen and LP, to match cell numbers for these organs with the number of cells for the SG, the organ with the fewest cells. For mRNA clustering, we selected variable genes, reduced dimensionality and performed clustering as described above. ADT counts were normalized by the CLR method, with the margin parameter set to 2. Given the small number of ADTs, they were clustered on a standard Euclidean distance matrix.
Heatmap ADT/mRNA
This heatmap was generated with the ComplexHeatmap package. The ADT compartment shows normalized ADT expression and the mRNA compartment shows scaled normalized expression. Genes were selected from the markers of the different clusters.
Overlap coefficient-based matrix
Cluster markers were calculated for the pooled multi-organ dataset by applying the FindAllMarkers function to the clusters identified in the pooled analysis by organ. Organ-specific markers common to clusters from the same organ were removed by organ-based differential expression analysis with FindAllMarkers using the following parameters: min.pct = 0.2 and logfc.treshold = 0.35. The organ-specific markers obtained were removed from the list of cluster markers. The overlap coefficient between two lists of markers was obtained with the following formula: overlapcoefficient (X, Y) = |X∩Y| ⁄ min (|X|,|Y|). We calculated a distance based on the overlap coefficient as follows: d(X,Y) = 1-overlap_coefficient(X,Y).
Harmony
For multi-organ dataset integration, we used the RunHarmony function from Harmony package 0.1.0.
Comparison with known gene signatures
Scoring analysis was performed with the Seurat AddModuleScore function, which computes a score at single-cell level. The mean expression level for each gene in the defined expression profiles was calculated for each cell, and the aggregated expression for the control gene set was then subtracted. All analyzed genes were binned on the basis of mean expression level, and the control genes were randomly selected from each bin. Violin plots were used to assess the distribution of module scores for each NK cell grouped by subset.
Pseudotime analysis
Pseudotime analysis was performed with monocle 3 v0.1.3. We set the starting point of the trajectory to the cell with the higher immature NK (mNK_Sp2) scoring after removing cells associated with “real” ILC1 clusters in spleen and SG. We obtained two trajectories in liver cells, first one associated with NK cells and the second one for ILC1. We also computed two trajectories in the LP, one for NK and intermediate cells and the other one for ILC1.
Human analysis
Human ILC single-cell RNAseq dataset9 is downloaded from the NCBI GEO depository, accession number GSE150050. Cells are qualified with the following criteria: total number of reads ≥ 105, number of expressed genes ≥1500, percentage of mitochondrial reads < 20% and percentage of ribosomal reads ≤ 10%. For each common organ between NK cells and ILC1 (Lung & tonsil), we performed differential analysis with the FindAllMarkers function (R package Seurat version 4.0.4) to define an organ-specific NK vs ILC1 signature.
Human orthologous for murine signatures described in this paper were defined with Ensembl Biomart using the R package biomaRt (version 2.48.3). Missing and multiple orthologous are completed and validated manually.
To validate the transfer of murine signature to predict human cells, we selected NK cells and ILC1 sorted by the authors to define a filtered gene-barcode matrix with a subset of cells. We normalized this matrix with the ‘LogNormalize’ method in Seurat v4, with default parameters. We performed the next analyses step (scaling, PCA, clustering and UMAP) on a sub-matrix with NK/ILC1 murine differentially expressed genes, organ by organ. Venn diagrams are used to show the NK-specific or ILC1-specific markers shared across organs and species. We used the R package ggVennDiagram version 1.2.1.
Quantification and statistical analysis
Data are presented as the mean ± standard error of the means (SEM), or standard deviations (SD). GraphPad Prism 6 was used for statistical analysis. Marker genes were defined as genes with an adjusted p value of <0.05 in nonparametric Wilcoxon rank-sum tests after Bonferroni correction. Enrichment scores (FDR) were calculated with a hypergeometric test followed by Benjamini-Hochberg correction. The significance threshold was set at FDR = 0.05.
Acknowledgments
We thank the animal and flow cytometry facilities at the Centre d’Immunologie de Marseille-Luminy for technical support. The E.V. laboratory at CIML and Assistance-Publique des Hôpitaux de Marseille was supported by funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (TILC grant 694502; MInfla-TILC grant 875102), the Agence Nationale de la Recherche including the PIONEER Project (ANR-17-RHUS-0007), MSDAVENIR, Innate Pharma, and institutional grants awarded to the CIML (INSERM, CNRS, and Aix-Marseille University) and Marseille Immunopole.
Author contributions
Y.M.K. and E.V. conceived the project. J.G., N.L., and Y.M.K. performed the experiments. N.L., J.G., and Y.M.K. analyzed the data. B.E. performed all bioinformatic analyses. S.C. analyzed human data and provided insightful comments. N.L., J.G., E.V., and Y.M.K. wrote the manuscript with the help of the other co-authors.
Declaration of interests
E.V. is a cofounder and employee of Innate Pharma. S.C. is an employee of Innate Pharma.
Published: November 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100812.
Contributor Information
Yann M. Kerdiles, Email: kerdiles@ciml.univ-mrs.fr.
Eric Vivier, Email: vivier@ciml.univ-mrs.fr.
Supplemental information
Data and code availability
-
•
All the sequencing data have been deposited in the NCBI GEO depository and are accessible under accession number GEO: GSE189807.
-
•
No new code was generated in this study.
-
•
Any additional information required to reanalyze the data reported in the paper is available from the lead contact upon request.
References
- 1.Vivier E., Artis D., Colonna M., Diefenbach A., Di Santo J.P., Eberl G., Koyasu S., Locksley R.M., McKenzie A.N.J., Mebius R.E., et al. Innate lymphoid cells: 10 Years on. Cell. 2018;174:1054–1066. doi: 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Gasteiger G., Fan X., Dikiy S., Lee S.Y., Rudensky A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seillet C., Brossay L., Vivier E. Natural killers or ILC1s? That is the question. Curr. Opin. Immunol. 2021;68:48–53. doi: 10.1016/j.coi.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cortez V.S., Cervantes-Barragan L., Robinette M.L., Bando J.K., Wang Y., Geiger T.L., Gilfillan S., Fuchs A., Vivier E., Sun J.C., et al. Transforming growth factor-beta signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity. 2016;44:1127–1139. doi: 10.1016/j.immuni.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao Y., Souza-Fonseca-Guimaraes F., Bald T., Ng S.S., Young A., Ngiow S.F., Rautela J., Straube J., Waddell N., Blake S.J., et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017;18:1004–1015. doi: 10.1038/ni.3800. [DOI] [PubMed] [Google Scholar]
- 6.Cella M., Gamini R., Sécca C., Collins P.L., Zhao S., Peng V., Robinette M.L., Schettini J., Zaitsev K., Gordon W., et al. Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat. Immunol. 2019;20:980–991. doi: 10.1038/s41590-019-0425-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins P.L., Cella M., Porter S.I., Li S., Gurewitz G.L., Hong H.S., Johnson R.P., Oltz E.M., Colonna M. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell. 2019;176:348–360.e12. doi: 10.1016/j.cell.2018.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crinier A., Milpied P., Escalière B., Piperoglou C., Galluso J., Balsamo A., Spinelli L., Cervera-Marzal I., Ebbo M., Girard-Madoux M., et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. 2018;49:971–986.e5. doi: 10.1016/j.immuni.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazzurana L., Czarnewski P., Jonsson V., Wigge L., Ringnér M., Williams T.C., Ravindran A., Björklund Å.K., Säfholm J., Nilsson G., et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. 2021;31:554–568. doi: 10.1038/s41422-020-00445-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yudanin N.A., Schmitz F., Flamar A.L., Thome J.J.C., Tait Wojno E., Moeller J.B., Schirmer M., Latorre I.J., Xavier R.J., Farber D.L., et al. Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity. 2019;50:505–519.e4. doi: 10.1016/j.immuni.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McFarland A.P., Yalin A., Wang S.Y., Cortez V.S., Landsberger T., Sudan R., Peng V., Miller H.L., Ricci B., David E., et al. Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity. 2021;54:1320–1337.e4. doi: 10.1016/j.immuni.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackay L.K., Minnich M., Kragten N.A.M., Liao Y., Nota B., Seillet C., Zaid A., Man K., Preston S., Freestone D., et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 14.Mackay L.K., Rahimpour A., Ma J.Z., Collins N., Stock A.T., Hafon M.L., Vega-Ramos J., Lauzurica P., Mueller S.N., Stefanovic T., et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 15.van Helden M.J., Goossens S., Daussy C., Mathieu A.L., Faure F., Marçais A., Vandamme N., Farla N., Mayol K., Viel S., et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J. Exp. Med. 2015;212:2015–2025. doi: 10.1084/jem.20150809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rabacal W., Pabbisetty S.K., Hoek K.L., Cendron D., Guo Y., Maseda D., Sebzda E. Transcription factor KLF2 regulates homeostatic NK cell proliferation and survival. Proc. Natl. Acad. Sci. USA. 2016;113:5370–5375. doi: 10.1073/pnas.1521491113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yomogida K., Bigley T.M., Trsan T., Gilfillan S., Cella M., Yokoyama W.M., Egawa T., Colonna M. Hobit confers tissue-dependent programs to type 1 innate lymphoid cells. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2117965118. e2117965118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedrich C., Taggenbrock R.L.R.E., Doucet-Ladevèze R., Golda G., Moenius R., Arampatzi P., Kragten N.A.M., Kreymborg K., Gomez de Agüero M., Kastenmüller W., et al. Effector differentiation downstream of lineage commitment in ILC1s is driven by Hobit across tissues. Nat. Immunol. 2021;22:1256–1267. doi: 10.1038/s41590-021-01013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Censo C., Marotel M., Mattiola I., Müller L., Scarno G., Pietropaolo G., Peruzzi G., Laffranchi M., Mazej J., Hasim M.S., et al. Granzyme A and CD160 expression delineates ILC1 with graded functions in the mouse liver. Eur. J. Immunol. 2021;51:2568–2575. doi: 10.1002/eji.202149209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klose C.S.N., Flach M., Möhle L., Rogell L., Hoyler T., Ebert K., Fabiunke C., Pfeifer D., Sexl V., Fonseca-Pereira D., et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157:340–356. doi: 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 21.Bai L., Vienne M., Tang L., Kerdiles Y., Etiennot M., Escalière B., Galluso J., Wei H., Sun R., Vivier E., et al. Liver type 1 innate lymphoid cells develop locally via an interferon-gamma-dependent loop. Science. 2021;371:eaba4177. doi: 10.1126/science.aba4177. [DOI] [PubMed] [Google Scholar]
- 22.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kralova J., Fabisik M., Pokorna J., Skopcova T., Malissen B., Brdicka T. The transmembrane adaptor protein SCIMP facilitates sustained dectin-1 signaling in dendritic cells. J. Biol. Chem. 2016;291:16530–16540. doi: 10.1074/jbc.M116.717157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo L., Bokil N.J., Wall A.A., Kapetanovic R., Lansdaal N.M., Marceline F., Burgess B.J., Tong S.J., Guo Z., Alexandrov K., et al. SCIMP is a transmembrane non-TIR TLR adaptor that promotes proinflammatory cytokine production from macrophages. Nat. Commun. 2017;8:14133. doi: 10.1038/ncomms14133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarriault S., Brou C., Logeat F., Schroeter E.H., Kopan R., Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 26.Chan C.J., Martinet L., Gilfillan S., Souza-Fonseca-Guimaraes F., Chow M.T., Town L., Ritchie D.S., Colonna M., Andrews D.M., Smyth M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014;15:431–438. doi: 10.1038/ni.2850. [DOI] [PubMed] [Google Scholar]
- 27.Schuster I.S., Wikstrom M.E., Brizard G., Coudert J.D., Estcourt M.J., Manzur M., O'Reilly L.A., Smyth M.J., Trapani J.A., Hill G.R., et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity. 2014;41:646–656. doi: 10.1016/j.immuni.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z., Zhang J., Hatta K., Lima P.D.A., Yadi H., Colucci F., Yamada A.T., Croy B.A. DBA-lectin reactivity defines mouse uterine natural killer cell subsets with biased gene expression. Biol. Reprod. 2012;87:81. doi: 10.1095/biolreprod.112.102293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bassani B., Baci D., Gallazzi M., Poggi A., Bruno A., Mortara L. Natural killer cells as key players of tumor progression and angiogenesis: old and novel tools to divert their pro-tumor activities into potent anti-tumor effects. Cancers. 2019;11:E461. doi: 10.3390/cancers11040461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y., Wang X., Hao X., Li B., Tao W., Zhu S., Qu K., Wei H., Sun R., Peng H., Tian Z. Ly49E separates liver ILC1s into embryo-derived and postnatal subsets with different functions. J. Exp. Med. 2022;219:e20211805. doi: 10.1084/jem.20211805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nixon B.G., Chou C., Krishna C., Dadi S., Michel A.O., Cornish A.E., Kansler E.R., Do M.H., Wang X., Capistrano K.J., et al. Cytotoxic granzyme C-expressing ILC1s contribute to antitumor immunity and neonatal autoimmunity. Sci. Immunol. 2022;7:eabi8642. doi: 10.1126/sciimmunol.abi8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flommersfeld S., Böttcher J.P., Ersching J., Flossdorf M., Meiser P., Pachmayr L.O., Leube J., Hensel I., Jarosch S., Zhang Q., et al. Fate mapping of single NK cells identifies a type 1 innate lymphoid-like lineage that bridges innate and adaptive recognition of viral infection. Immunity. 2021;54:2288–2304.e7. doi: 10.1016/j.immuni.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z., Ma R., Ma S., Tian L., Lu T., Zhang J., Mundy-Bosse B.L., Zhang B., Marcucci G., Caligiuri M.A., Yu J. ILC1s control leukemia stem cell fate and limit development of AML. Nat. Immunol. 2022;23:718–730. doi: 10.1038/s41590-022-01198-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y., Li J., Partovian C., Sellke F.W., Simons M. Syndecan-4 modulates basic fibroblast growth factor 2 signaling in vivo. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H2078–H2082. doi: 10.1152/ajpheart.00942.2001. [DOI] [PubMed] [Google Scholar]
- 35.Cornelison D.D.W., Wilcox-Adelman S.A., Goetinck P.F., Rauvala H., Rapraeger A.C., Olwin B.B. Essential and separable roles for Syndecan-3 and Syndecan-4 in skeletal muscle development and regeneration. Genes Dev. 2004;18:2231–2236. doi: 10.1101/gad.1214204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambert J., Makin K., Akbareian S., Johnson R., Alghamdi A.A.A., Robinson S.D., Edwards D.R. ADAMTS-1 and syndecan-4 intersect in the regulation of cell migration and angiogenesis. J. Cell Sci. 2020;133:jcs235762. doi: 10.1242/jcs.235762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slimani H., Charnaux N., Mbemba E., Saffar L., Vassy R., Vita C., Gattegno L. Interaction of RANTES with syndecan-1 and syndecan-4 expressed by human primary macrophages. Biochim. Biophys. Acta. 2003;1617:80–88. doi: 10.1016/j.bbamem.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Charnaux N., Brule S., Hamon M., Chaigneau T., Saffar L., Prost C., Lievre N., Gattegno L. Syndecan-4 is a signaling molecule for stromal cell-derived factor-1 (SDF-1)/CXCL12. FEBS J. 2005;272:1937–1951. doi: 10.1111/j.1742-4658.2005.04624.x. [DOI] [PubMed] [Google Scholar]
- 39.Tumova S., Woods A., Couchman J.R. Heparan sulfate chains from glypican and syndecans bind the Hep II domain of fibronectin similarly despite minor structural differences. J. Biol. Chem. 2000;275:9410–9417. doi: 10.1074/jbc.275.13.9410. [DOI] [PubMed] [Google Scholar]
- 40.Chung J.S., Cruz P.D., Jr., Ariizumi K. Inhibition of T-cell activation by syndecan-4 is mediated by CD148 through protein tyrosine phosphatase activity. Eur. J. Immunol. 2011;41:1794–1799. doi: 10.1002/eji.201041233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bühligen J., Himmel M., Gebhardt C., Simon J.C., Ziegler W., Averbeck M. Lysophosphatidylcholine-mediated functional inactivation of syndecan-4 results in decreased adhesion and motility of dendritic cells. J. Cell. Physiol. 2010;225:905–914. doi: 10.1002/jcp.22301. [DOI] [PubMed] [Google Scholar]
- 42.Polte T., Petzold S., Bertrand J., Schütze N., Hinz D., Simon J.C., Lehmann I., Echtermeyer F., Pap T., Averbeck M. Critical role for syndecan-4 in dendritic cell migration during development of allergic airway inflammation. Nat. Commun. 2015;6:7554. doi: 10.1038/ncomms8554. [DOI] [PubMed] [Google Scholar]
- 43.Hao Y., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All the sequencing data have been deposited in the NCBI GEO depository and are accessible under accession number GEO: GSE189807.
-
•
No new code was generated in this study.
-
•
Any additional information required to reanalyze the data reported in the paper is available from the lead contact upon request.