

Abstract
Enhanced aerobic glycolysis contributes to the metastasis of pancreatic cancer metastasis, but the mechanism underlying the abnormal activation of glycolysis has not been fully elucidated. The E3 ligase tripartite motif 16 (TRIM16) is involved in the progression of many cancers. However, the role of and molecular mechanism by which TRIM16 acts in pancreatic cancer are unclear. In this study, we report that TRIM16 was significantly upregulated in pancreatic cancer tissues, and high expression of TRIM16 was associated with poor prognosis in patients with pancreatic cancer. Multivariate analyses showed that TRIM16 was an independent predictor of poor outcomes among patients with pancreatic cancer. In addition, in vitro and in vivo evidence showed that TRIM16 promoted pancreatic cancer cell metastasis by enhancing glycolysis. Furthermore, we revealed that TRIM16 controlled glycolysis and pancreatic cancer cell’s metastasis by regulating sine oculis homeobox 1 (SIX1), an important transcription factor that promotes glycolysis. TRIM16 upregulated SIX1 by inhibiting its ubiquitination and degradation, which was mediated by NF-κB-inducing kinase (NIK), an upstream regulator of SIX1. Hence, NIK inhibitor can suppress SIX1 expression, glycolysis and metastasis in TRIM16-overexpressing pancreatic cancer cells. Mechanistic investigations demonstrated that TRIM16 competed with NIK’s E3 ligase, TNF receptor-associated factor 3 (TRAF3), at the ISIIAQA sequence motif of NIK, and then stabilized NIK protein. Our study identified the TRIM16-NIK-SIX1 axis as a critical regulatory pathway in aerobic glycolysis and pancreatic cancer metastasis, indicating that this axis can be an excellent therapeutic target for curing pancreatic cancer.
Keywords: Pancreatic cancer, TRIM16, aerobic glycolysis, metastasis, SIX1, NIK
Introduction
Pancreatic cancer is one of the most lethal malignancies worldwide [1]. Although the incidence of pancreatic cancer is not high, it is associated with extremely high mortality [2]. Despite developments in the detection and management of pancreatic cancer, the 5-year survival rate of patients is dismal (less than 9%) [3]. Most pancreatic cancer patients are diagnosed at an advanced stage, when most treatment regimens are ineffective [3]. Tumor metastasis is the leading cause of therapy failure in patients with pancreatic cancer. According to clinical experience and a review of the literature, distant metastasis occurs in 90% of deaths [4]. One obstacle underlying the treatment of pancreatic cancer is our limited understanding of metastasis. Therefore, novel approaches or targets are urgently needed to improve treatments that will prevent the distal metastasis of pancreatic cancer.
Aerobic glycolysis, also known as the Warburg effect, is a hallmark of cancer [5]. Enhanced glycolysis is found in many human cancers, and tumor tissues exhibit increased glucose uptake and the overexpression of many glycolytic enzymes, such as glucose transporter 1 (GLUT1), hexokinase 2 (HK2), phosphoglycerate kinase 1 (PGK1) and lactate dehydrogenase A (LDHA) [6]. Studies have confirmed that enhanced glycolysis promotes proliferation, EMT, invasion, metastasis and chemotherapy resistance in many cancers [7]. Abnormal glycolytic metabolism also plays an important role in the invasion and metastasis of pancreatic cancer [8]. Due to the distinctive glucose metabolism associated with carcinogenesis and cancer progression, glycolysis can be regarded as an attractive target that shows high selectivity for cancer cells. For example, scientists have developed a small molecule inhibitor, WZB117, that targets GLUT1, and its feasibility and effect in repressing glycolysis and tumors have been extensively tested in vitro and in vivo [9]. In addition, the PGK1 inhibitor CBR-470-1 and the LDHA inhibitor FX11 significantly inhibit glycolysis and tumor progression [10,11]. Moreover, these inhibitors also show a synergistic antitumor effect when administered in combination with other agents. However, glycolysis is under sophisticated control, and most therapeutic strategies being developed to target glycolysis in cancer are generally for a single rate-limiting enzyme, indicating that more-elaborate studies are needed to determine the mechanism underlying the abnormal activation of glycolysis and to identify agents that extensively inhibit glycolysis.
Increasing evidence shows that oncogenes and tumor suppressors regulate altered glucose metabolism [12]. The oncoprotein sine oculis homeobox 1 (SIX1) is an important transcription factor that regulates the process of glycolysis, which directly promotes the expression of several glycolytic enzymes, such as GLUT1, HK2, PGK1 and LDHA [13]. SIX1 belongs to the PAX-SIX-EYA-DACH Network (PSEDN), which orchestrates the development of multiple organs in mammals [14]. Recently, increasing evidence has shown that SIX1 is dysregulated in cancer and plays a key role in the tumorigenesis of various human cancers, including breast cancer, hepatocellular carcinoma and colorectal cancer [15]. In pancreatic cancer, SIX1 drives tumor growth and metastasis [16,17]. Various factors, especially noncoding RNAs, can regulate SIX1 expression via posttranscriptional mechanisms and are involved in the regulation of cancer progression [18,19]. NF-κB-inducing kinase (NIK, also known as MAP3K14) is also a regulator of SIX1, and SIX1 is upregulated in differentiated macrophages by the NIK-mediated suppression of the ubiquitin proteasome pathway [20]. Although increased SIX1 levels contribute to the aerobic glycolysis and metastasis of pancreatic cancer, the upstream regulatory mechanisms of SIX1 remain unclear.
It is well known that the dysregulation of oncogene proteins is closely related to ubiquitin proteasome-mediated degradation [21]. The substrate to be degraded is ubiquitinated, and then the proteasome cleaves it. Ubiquitination requires the sequential action of three enzymes: a ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzymes (E2) and a ubiquitin ligase (E3) [22]. As a substrate interaction module, E3 ubiquitin ligases are the most heterogeneous and have attracted great attention. Tripartite motif 16 (TRIM16), an E3 ubiquitin ligase belonging to the tripartite motif (TRIM) protein family (a RING-type E3 ubiquitin ligase subfamily), mediates the ubiquitination of its target proteins and tags them for degradation [23]. TRIM16 harbors two B-box domains, a coiled-coil domain and a C-terminal domain called SPRY. TRIM16 is devoid of a typical RING domain, and the B box domains are important for its E3 ligase activity [24]. Previous studies have defined the functions of TRIM16 in various biological processes, such as cell proliferation, cell cycle regulation, differentiation, apoptosis and autophagy [25]. Nevertheless, its effect in cancer is controversial. TRIM16 has been ascribed both tumor-suppressive and oncogenic roles in different types of tumors. In neuroblastoma, TRIM16 is identified as a DNA-binding protein with histone acetylase activity that suppresses neuroblastoma cell growth and reduces tumorigenicity in vivo [26]. TRIM16 also inhibits cell growth and migration in ovarian cancer and breast cancer [27,28]. On the other hand, a study demonstrated that TRIM16 was upregulated in colon cancer tissues and cell lines and that silencing TRIM16 could inhibit the viability of colon cancer cells [29]. In gastric cancer, the expression of TRIM16 was increased in distant metastases, and the upregulation of TRIM16 could promote cell invasion and migration [30]. The expression and role of TRIM16 in pancreatic cancer have not yet been reported. Importantly, we observed that the expression of TRIM16 mRNA was elevated in pancreatic cancer tissues included in The Cancer Genome Atlas (TCGA) dataset (Supplementary Figure 1A). These data suggested that TRIM16 may play a role in pancreatic cancer progression. However, the molecular function of TRIM16, its target protein substrates, and its clinical significance in pancreatic cancer are unclear.
In this study, we aimed to determine the role of TRIM16 in the progression of pancreatic cancer. We further explored the mechanism of abnormally high expression of SIX1 in pancreatic cancer and found that TRIM16 positively regulated SIX1 expression by upregulating NIK. Furthermore, we elucidated that the molecular mechanism by which TRIM16 upregulates NIK was independent of its ubiquitin ligase activity. Our work identified an interplay among these proteins that promotes pancreatic cancer metastasis by enhancing aerobic glycolysis.
Materials and methods
Detailed methods and materials of this manuscription are provided in the Supplementary Information.
Animal studies
Animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85-23, revised 1996) and approved by the Animal Ethics Committee of Nanchang University.
Statistical analysis
All results are shown as the mean ± SD and were analyzed using GraphPad Prism 8 (GraphPad Software, USA) from at least three independent experiments. Two-tailed unpaired Student’s t test was used for analyzing the comparison between the two groups and one-way ANOVA was used for analyzing the comparison among multiple groups. Chi-square tests were used for analyzing the counting data. P values were two-sided, and differences were considered statistically significant at P<0.05. *P<0.05, **P<0.01, ***P<0.001.
Results
TRIM16 is highly expressed in pancreatic cancer tissues and is closely associated with a poor prognosis in patients with pancreatic cancer
To reveal the role of TRIM16 in the progression of pancreatic cancer, we initially explored the expression patterns of TRIM16 in 42 pancreatic cancer tissues and compared them with those in adjacent normal tissues. RT-qPCR results showed that the mRNA expression of TRIM16 in fresh pancreatic cancer tissues was higher than that in corresponding normal tissues (Figure 1A and 1B). We further examined the protein expression of TRIM16 in pancreatic cancer tissues by using western blotting. Consistent with the RT-qPCR results, the protein level of TRIM16 was also higher in pancreatic cancer tissues than in normal tissues (Figure 1C and 1D). In addition, we used immunohistochemistry (IHC) to detect the protein expression of TRIM16 in 96 pancreatic cancer samples. As shown in Figure 1E and 1F, aberrant TRIM16 expression occurred in pancreatic cancer tissues, whereas a weak positive signal was detected in corresponding normal tissues. These results revealed that TRIM16 is highly expressed in pancreatic cancer tissues.
Figure 1.
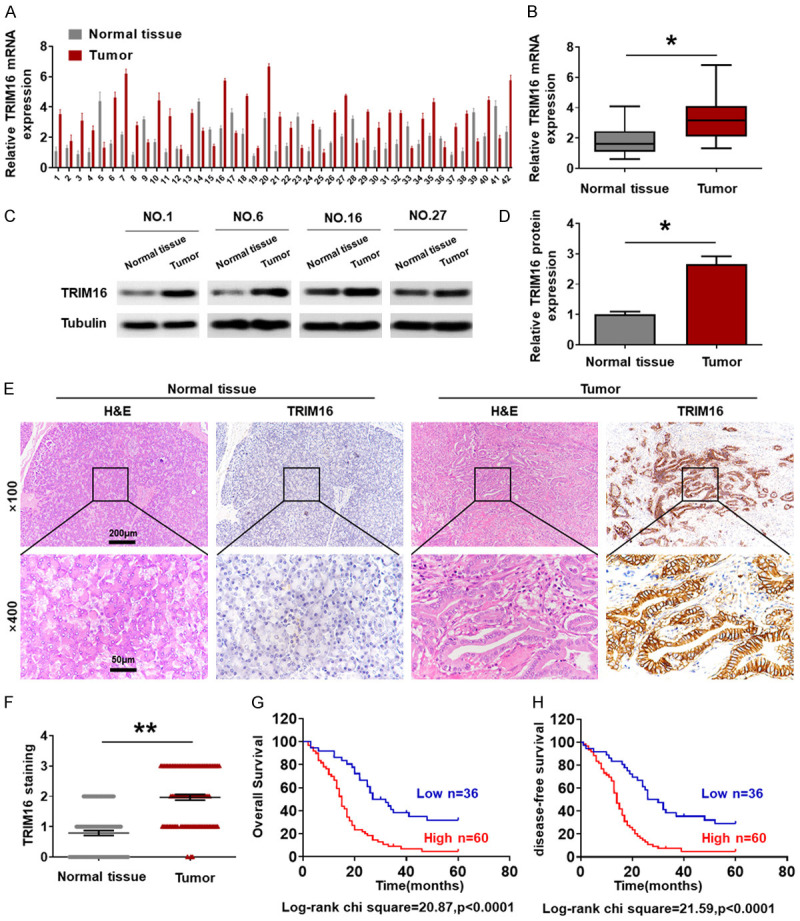
High TRIM16 expression correlates with poor prognosis in pancreatic cancer patients. (A, B) Determination and quantification of TRIM16 mRNA levels in pancreatic cancer tissues and paired normal tissues by qRT-PCR (*P<0.05). (C, D) Determination and quantification of TRIM16 protein levels in pancreatic cancer tissues and paired normal tissues by western blotting (*P<0.05). (E, F) Representative image (E) and quantification (F) of IHC staining of TRIM16 in pancreatic cancer tissues and paired normal tissues. Magnification, 100 ×, scale bar, 200 μm; magnification, 400 ×, scale bar, 50 μm. **P<0.01. (G, H) Kaplan-Meier plots representing probabilities of overall survival and disease-free survival in 96 pancreatic cancer patients according to expression level of TRIM16. Statistical analysis was conducted using Student’s t-test and Log Rank test.
Next, to determine whether TRIM16 might be an effective target for predicting pancreatic cancer patient survival, we analyzed the relationship between the expression of TRIM16 and prognostic factors in 96 pancreatic cancer patients (Supplementary Table 1). The data showed no significant association between TRIM16 expression and age, sex, tumor size or CEA level in pancreatic cancer patients, but high expression of TRIM16 was significantly correlated with vessel invasion (P<0.001), lymph node metastasis (P<0.001) and TNM stage (P = 0.001). Furthermore, we found that the overall survival (OS) and disease-free survival (RFS) of pancreatic cancer patients with high TRIM16 protein expression were significantly poorer than in those with low TRIM16 protein expression (Figure 1G and 1H). We additionally analyzed the relationship between TRIM16 mRNA expression and pancreatic cancer patient survival time included in The Cancer Genome Atlas (TCGA) database. The results showed that the cumulative survival rate was significantly lower in pancreatic cancer patients with high TRIM16 mRNA expression than in those with low TRIM16 mRNA expression (Supplementary Figure 1B and 1C). Multivariate Cox regression analysis further revealed that high TRIM16 expression was an independent predictor of poor survival in patients with pancreatic cancer (Supplementary Table 2). Taken together, our data suggest that TRIM16 may be involved in the progression of pancreatic cancer.
TRIM16 promotes the metastasis of pancreatic cancer cells in vitro and in vivo
As shown in Supplementary Table 1, the high expression of TRIM16 was related to TNM stage, lymph node metastasis and vessel invasion, and we speculated that TRIM16 might be involved in regulating pancreatic cancer cell metastasis. To prove our conjecture, we were going to knock down the expression of TRIM16 in pancreatic cancer cells and detect biological changes in the cells. Firstly, we screened several shRNAs targeting TRIM16 (Supplementary Figure 2A and 2B) and used the shRNA (shTRIM16-c) with the best effect of knockdown to conduct experiments. Then, we transfected two pancreatic cancer cell lines exhibiting high endogenous TRIM16 expression (AsPC-1 and BxPC-3) with shTRIM16 plasmids to knock down TRIM16 expression (Figure 2A-C). We found that TRIM16-silenced cells showed a significantly lower in vitro invasive capacity than shNC-transfected cells (Figure 2D and 2E). Consistently, RTCA assay data also showed that downregulation of TRIM16 suppressed AsPC-1 and BxPC-3 cell migration (Figure 2F and 2G). Besides, we used another shRNA targeting TRIM16 (shTRIM16-b) to carry out key silencing experiments. The results showed that shTRIM16-b can significantly reduce the expression of TRIM16 and inhibit the invasion and migration of pancreatic cancer cells (Supplementary Figure 2C-H). To exclude the off-target effect, we conduct rescue experiments, we generated a TRIM16 cDNA harboring silent mutations in the shRNA-targeting sequence that made the mRNA insensitive to this shRNA. Rescue experiments showed that shRNA-resistant-TRIM16 significantly restored and stabilized the expression of TRIM16 in BxPC-3 cells with TRIM16 interference, and cell’s migration was also restored (Supplementary Figure 2I-L). In contrast, we increased the TRIM16 level by transfecting a TRIM16 overexpression plasmid into PANC-1 and CaPan-1 cells (Supplementary Figure 3A and 3B). Compared with the control cells transfected with vector plasmid, the invasion ability of TRIM16-overexpressing cells was enhanced (Supplementary Figure 3C and 3D). RTCA assays also demonstrated that the upregulation of TRIM16 enhanced the migratory ability of both pancreatic cancer cell lines (Supplementary Figure 3E and 3F). Our data confirmed that TRIM16 promoted the migration and invasion of pancreatic cancer cells in vitro.
Figure 2.

TRIM16 promotes the metastasis of pancreatic cancer cells in vitro and in vivo. A. The protein level of TRIM16 was detected by using western blotting assay in fiver pancreatic cancer cells and the normal pancreatic duct epithelial cell line HPDE6-C7. Tubulin was used as a loading control. B, C. Western blotting analyses were used to detect the expression level of TRIM16 in AsPC-1 and BxPC-3 cells stably transfected with the TRIM16-silenced vector. Tubulin was used as a loading control. D, E. Transwell migration and transwell invasion assays of AsPC-1 and BxPC-3 cells transfected with TRIM16-silenced vector. The image was captured at 400 × magnification. Scale bar, 50 μm. *P<0.05. F, G. RTCA assays were performed to detect the metastasis ability of AsPC-1 and BxPC-3 cells transfected with TRIM16-silenced vector. H. AsPC-1 cells transfected with TRIM16-silenced vector were injected into the tail vein of nude mice, and the in vivo liver metastatic signal detection were imaged by a Lumina Series III IVIS instrument. I. The incidence of liver metastasis were measured after 6-8 weeks. n = 6, **P<0.01. J. Representative image (left; magnification: × 100) and quantification (right) of H&E staining of liver metastatic nodules. n = 6. Scale bar, 50 μm. *P<0.05.
Next, we further examined the effect of altered TRIM16 expression on pancreatic cancer metastasis in vivo. Control cells and TRIM16-silenced cells were injected into the tail veins of BALB/c nude mice, and we found that knocking down TRIM16 significantly reduced the incidence of liver metastasis (Figure 2H and 2I). Additionally, H&E-stained serial liver sections revealed that decreasing TRIM16 markedly suppressed experimental liver metastasis (Figure 2J). However, the overexpression of TRIM16 increased the liver metastasis of pancreatic cancer (Supplementary Figure 3G-I). These data collectively indicate that TRIM16 promotes the metastasis of pancreatic cancer cells in vivo.
Glycolysis is crucial for the pro-metastatic function of TRIM16
To determine the mechanism by which TRIM16 influences the metastasis of pancreatic cancer cells, we used iTRAQ to find global changes in the proteome when TRIM16 was knocked down in BxPC-3 cells. KEGG analysis revealed that the top downregulated gene set in TRIM16-silenced cells was associated with glycolysis (Figure 3A). Many studies and our previous study have shown that glycolysis can promote cancer cell metastasis [8,9]. Therefore, we further evaluated whether TRIM16 could modulate the glycolytic phenotype in pancreatic cancer cells. As expected, the overexpression of TRIM16 increased glucose uptake, lactate production and ATP generation in PANC-1 and CaPan-1 cells (Figure 3B-D). Furthermore, the extracellular acidification rate (ECAR), which represents overall glycolytic flux, was also increased in TRIM16-overexpressing pancreatic cancer cells (Figure 3E and 3F). Conversely, the downregulation of TRIM16 inhibited the glycolytic phenotype in AsPC-1 and BxPC-3 cells (Supplementary Figure 4A-E). Collectively, our results showed that TRIM16 can promote glycolysis in pancreatic cancer cells.
Figure 3.

TRIM16 promotes migration and invasion of pancreatic cancer cells by modulating aerobic glycolysis. A. KEGG analysis of iTRAQ data revealed that the top down-regulated gene set in TRIM16-silenced cells was associated with glycolysis. B-D. Glucose consumption, lactate production, and ATP levels in PANC-1 and CaPan-1 cells stably transfected with the TRIM16-overexpressing vector (His-TRIM16). Three independent experiments were performed. *P<0.05, **P<0.01. E, F. ECAR data showing the glycolytic rate and capacity in PANC-1 and CaPan-1 cells stably transfected with the TRIM16-overexpressing vector. Glucose (10 mM), oligomycin (1.0 μM) and 2-deoxyglucose (2-DG, 50 mM) were sequentially injected into each well at the indicated time points. *P<0.05. G. Transwell migration and transwell invasion assays of PANC-1 cells transfected with TRIM16-overexpressing vector with or without the glycolytic inhibitor 2-DG. The image was captured at 400 × magnification. Scale bar, 50 μm. *P<0.05. H. PANC-1 cells transfected with TRIM16-overexpressing vector were injected into the tail vein of nude mice, and oral administration of 2-DG from the date of injection. The incidence of liver metastasis was measured after 6-8 weeks. n = 6, **P<0.01. I. Representative image (left; magnification: × 100) and quantification (right) of H&E staining of liver metastatic nodules. n = 6. Scale bar, 50 μm. *P<0.05.
To verify whether glycolysis mediates the prometastatic effect of TRIM16 in pancreatic cancer cells, the glycolytic inhibitor 2-deoxy-D-glucose (2-DG) was used to block glycolysis in PANC-1 and CaPan-1 cells transfected with TRIM16-overexpressing plasmids (Supplementary Figure 4F and 4G). The results of in vitro invasion assays showed that the increase in pancreatic cancer cell metastatic ability induced by TRIM16 overexpression was attenuated by 2-DG (Figure 3G-I, Supplementary Figure 4H). These findings suggest that TRIM16 exerts its metastasis-promoting function by enhancing glycolysis in pancreatic cancer cells.
TRIM16 enhances glycolysis by increasing SIX1 expression in pancreatic cancer
Previous studies have demonstrated that SIX1 is a key transcription factor that regulates aerobic glycolysis [13], and the iTRAQ data indicated that the protein expression of SIX1 was reduced in TRIM16-silenced cells (Figure 4A). Thus, we speculated that TRIM16-mediated glycolysis enhancement might result from elevated levels of SIX1 in pancreatic cancer cells. As expected, western blotting data indicated that downregulating TRIM16 significantly reduced SIX1 protein expression, whereas overexpressing TRIM16 increased the protein level of SIX1 in pancreatic cancer cells (Figure 4B and 4C, Supplementary Figure 5A and 5B). However, TRIM16 alteration had no effect on the expression of SIX1 mRNA (Supplementary Figure 5C and 5D). We then investigated the expression of SIX1 target genes related to glycolysis, including GLUT1, HK2, PGK1 and LDHA, in TRIM16-overexpressing or TRIM16-silenced pancreatic cancer cells. Our data showed that the mRNA and protein levels of these genes were increased in TRIM16-overexpressing pancreatic cancer cells, whereas the expression of these genes was decreased in TRIM16-silenced pancreatic cancer cells (Supplementary Figure 5E-H).
Figure 4.

TRIM16 enhances glycolysis by increasing SIX1 expression in pancreatic cancer. A. Heat map of top 10 differentially regulated proteins in the glycolysis set in shNC and shTRIM16 cells. B. Protein levels of SIX1 in PANC-1 cells stably transfected with the TRIM16-overexpressing vector. C. Protein levels of SIX1 in AsPC-1 cells transfected with TRIM16-silenced vector. D. After knocking down SIX1 in TRIM16-overexpressing PANC-1 cells, the protein levels of TRIM16 and SIX1 were detected. Tubulin was used as a loading control. E-H. Glucose consumption, lactate production, ATP levels and glycolytic rate and capacity were measured. Three independent experiments were performed. *P<0.05. I. protein levels of GLUT1, HK2, PGK1 and LDHA were detected. Tubulin was used as a loading control. J, K. cell migration and invasive capacity were measured using the transwell assay. *P<0.05. L. After overexpression of SIX1 in TRIM16-silenced AsPC-1 cells, the protein levels of TRIM16 and SIX1 were detected. Tubulin was used as a loading control. M-P. Glucose consumption, lactate production, ATP levels and glycolytic rate and capacity were measured. Three independent experiments were performed. *P<0.05. Q. protein levels of GLUT1, HK2, PGK1 and LDHA were detected. Tubulin was used as a loading control. R, S. cell migration and invasive capacity were measured using the transwell assay. *P<0.05.
To further determine the correlation between TRIM16 and SIX1, we measured the protein level of SIX1 in 32 pancreatic cancer tissues that had upregulated levels of TRIM16. Our western blotting results showed that the level of SIX1 protein was significantly higher in pancreatic cancer tissues than in adjacent nontumor tissues (Supplementary Figure 5I and 5J). In addition, correlation analysis indicated that the protein level of SIX1 was positively correlated with the protein level of TRIM16 in pancreatic cancer tissues (Supplementary Figure 5K). Moreover, immunohistochemistry (IHC) staining of pancreatic cancer specimens showed that the expression of SIX1 was high in the tumor tissue compared with the adjacent nontumor tissue, which was consistent with TRIM16 expression (Supplementary Figure 5L).
Next, we sought to explore whether SIX1 was responsible for the oncogenic functions of TRIM16 in pancreatic cancer cells. We transfected an shRNA plasmid targeting SIX1 into pancreatic cancer cells overexpressing TRIM16 to inhibit SIX1 and examined the change in cell biological function. As shown in Figure 4D-H, we found that knocking down SIX1 attenuated the increase in glucose uptake, lactate production, ATP generation and ECAR caused by TRIM16 overexpression. In addition, inhibiting SIX1 decreased the expression of GLUT1, HK2, PGK1 and LDHA in TRIM16-overexpressing cells and counteracted the oncogenic effect of TRIM16 (Figure 4I-K). On the other hand, we transfected a SIX1 overexpression plasmid into TRIM16-silenced pancreatic cancer cells and examined the change in cell biological function. The results showed that restoring the protein level of SIX1 rescued the decrease in glucose uptake, lactate production, ATP generation and ECAR induced by TRIM16 inhibition (Figure 4L-P). Additionally, the downregulated expression of GLUT1, HK2, PGK1 and LDHA in TRIM16-silenced cells was also reversed by the overexpression of SIX1 (Figure 4Q). Moreover, the inhibitory effects of silencing TRIM16 on invasion and metastasis were also reversed by SIX1 restoration (Figure 4R and 4S). Our data confirmed that SIX1 mediates the oncogenic effect of TRIM16 in pancreatic cancer cells.
TRIM16 stabilizes SIX1 protein by inhibiting its ubiquitination in pancreatic cancer cells
We further probed the mechanisms by which TRIM16 regulates SIX1. Our RT-qPCR results showed that the expression of SIX1 mRNA was not regulated by TRIM16 (Supplementary Figure 5C and 5D), suggesting that TRIM16 may regulate the degradation of the SIX1 protein. A study reported that SIX1 was degraded by the ubiquitin proteasome system (UPS). To determine whether SIX1 was also degraded by the UPS in pancreatic cancer cells, we first investigated the interaction between SIX1 and ubiquitin in pancreatic cancer cells. Coimmunoprecipitation (co-IP) results showed that endogenous SIX1 and ubiquitin bound directly in pancreatic cancer cells (Figure 5A). The conjugated ubiquitin chains on SIX1 were primarily Lys 48 (K48) Ub linkages (Figure 5B). Moreover, treatment with the proteasome inhibitor MG132 for the indicated time caused a significant accumulation of endogenous SIX1 protein in pancreatic cancer cells (Figure 5C). These results demonstrated that the SIX1 protein is also degraded by the UPS in pancreatic cancer cells.
Figure 5.

TRIM16 stabilizes SIX1 protein by inhibiting its ubiquitination. A. Representative western blot showing that Ubiquitin interacts with SIX1 in pancreatic cancer cells. Pancreatic cancer cells were lysed, and an immunoprecipitation assay was performed with anti-IgG or anti-ub antibodies, followed by western blot with the indicated antibodies. B. Ubiquitinated SIX1 in the lysates of AsPC-1 and PANC-1 cells was precipitated with anti-IgG or anti-SIX1 after the cells were treated with MG132 (15 mmol/l) for 12 h, followed by western blot to detect ubiquitin (linkage-specific K48). C. AsPC-1 and PANC-1 cells were treated with MG132 for the indicated times, the levels of SIX1 protein were detected by western blot. D. Transfecting TRIM16-silenced vector or TRIM16-overexpressing vector into AsPC-1 and PANC-1 cells, and using western blot to detect the protein levels of TRIM16 and SIX1 with or without MG132 treatment. E, F. AsPC-1 and PANC-1 cells were stably transfected with the TRIM16-silenced or TRIM16-overexpressing vector. The cells were subjected to cycloheximide (CHX, 20 mmol/L) exposure for the indicated times and the protein level of SIX1 was detected. The degradation rate was converted into a statistical linear graph (*P<0.05, **P<0.01, nonlinear regression, exponential one-phase decay). G. AsPC-1 and PANC-1 cells were transfected with increasing amounts of His-trim16 plasmid. The protein levels of SIX1 were detected with anti-SIX1 antibody. H. AsPC-1 and PANC-1 cells were stably transfected with the TRIM16-silenced or TRIM16-overexpressing vector. The level of ubiquitin-attached SIX1 was precipitated with anti-SIX1 after the cells were treated with MG132 for 12 h, followed by western blot to detect ub.
Next, to assess the role of TRIM16 in the process of SIX1 degradation, we sought to determine whether TRIM16 was involved in regulating the degradation of the SIX1 protein. We silenced or overexpressed TRIM16 and detected the effects of altering TRIM16 on SIX1 expression with or without MG132. We found that MG132 abolished the regulatory effect of TRIM16 on SIX1 (Figure 5D). Second, we tested the effect of changing TRIM16 on the degradation rate of the SIX1 protein. A degradation dynamics assay showed that the half-life of endogenous SIX1 was significantly shortened in TRIM16-silenced pancreatic cancer cells but was prolonged in TRIM16-overexpressing cells (Figure 5E and 5F). Third, we found that the ectopic dose-dependent effect of TRIM16 overexpression caused an accumulation of endogenous SIX1 proteins in pancreatic cancer cells (Figure 5G). Finally, we further explored the effect of TRIM16 on the ubiquitination level of SIX1. The results showed that knocking down TRIM16 increased the ubiquitination level of SIX1, whereas overexpressing TRIM16 inhibited the ubiquitination level of SIX1 (Figure 5H). These data demonstrate that TRIM16 inhibits the UPS-mediated degradation of SIX1, thereby stabilizing the SIX1 protein.
TRIM16 affects the ubiquitination of SIX1 by regulating the expression of NIK
Studies have shown that TRIM16 generally binds to various substrates to exert its effects. Therefore, we investigated the interaction between TRIM16 and SIX1. However, coimmunoprecipitation (co-IP) results showed that TRIM16 did not interact with SIX1 (Figure 6A). A GST pulldown experiment also confirmed that there was no direct interaction between the TRIM16 and SIX1 proteins (Figure 6B), indicating that TRIM16 did not directly affect the ubiquitination of SIX1. Study has confirmed that SIX1 was degraded via the E3 anaphase-promoting complex (APC) with its activating subunit Cdh1 [31], thus we discuss whether TRIM16 can directly target Cdh1 to regulate the ubiquitination of SIX1. As shown in Supplementary Figure 6A, TRIM16 does not regulate the expression of Cdh1. What’s more, TRIM16 can’t directly target Cdh1 (Supplementary Figure 6B). It has been reported that NIK can inhibit the ubiquitination of SIX1, thereby stabilizing the SIX1 protein in mouse primary bone marrow-derived macrophages (BMDMs) and human fibroblasts [21]. We also confirmed that NIK regulated the ubiquitination and protein levels of SIX1 in pancreatic cancer cells, as upregulating NIK inhibited the ubiquitination of SIX1, thereby increasing the protein level of SIX1, while knocking down NIK enhanced the ubiquitination of SIX1 and then decreasing the protein level of SIX1 (Supplementary Figure 6C-E). Therefore, we speculated that TRIM16 regulates the ubiquitination of SIX1 via NIK. To test this hypothesis, we first explored whether TRIM16 regulates NIK expression. Western blotting data indicated that overexpressing TRIM16 upregulated NIK protein expression, whereas downregulating TRIM16 reduced the protein level of NIK in pancreatic cancer cells (Figure 6C and 6D, Supplementary Figure 6F and 6G). However, the expression of NIK mRNA was not regulated by TRIM16 (Supplementary Figure 6H and 6I). In addition, we transfected a shRNA plasmid targeting NIK into TRIM16-overexpressing cells to inhibit NIK and detected changes in the protein level and ubiquitination level of SIX1. As shown in Figure 6E and 6F, the upregulation of TRIM16 led to a decrease in the ubiquitination level and an increase in the SIX1 protein level, but this phenomenon was reversed when the expression of NIK was knocked down. Moreover, increasing NIK also reversed the increase in the ubiquitination level and the decrease in the SIX1 protein level caused by silencing TRIM16 (Figure 6G and 6H). In addition, we evaluated the effect of NIK SMI1 (a selective inhibitor of NIK) on TRIM16-mediated glycolysis and metastasis of pancreatic cancer in vitro and in vivo. As shown in Figure 6I-L, NIK SMI1 suppressed the expression of SIX1 and glycolysis-related genes, the glycolytic phenotype and pancreatic cancer cell metastasis. Moreover, NIK SMI1 blocked the increase in glycolysis and tumor metastasis mediated by TRIM16 overexpression (Figure 6I-L). These findings were similar in AsPC-1 cells (Supplementary Figure 6J-M). Taken together, our data demonstrate that the TRIM16-mediated regulation of SIX1 ubiquitination and protein expression is dependent on NIK.
Figure 6.
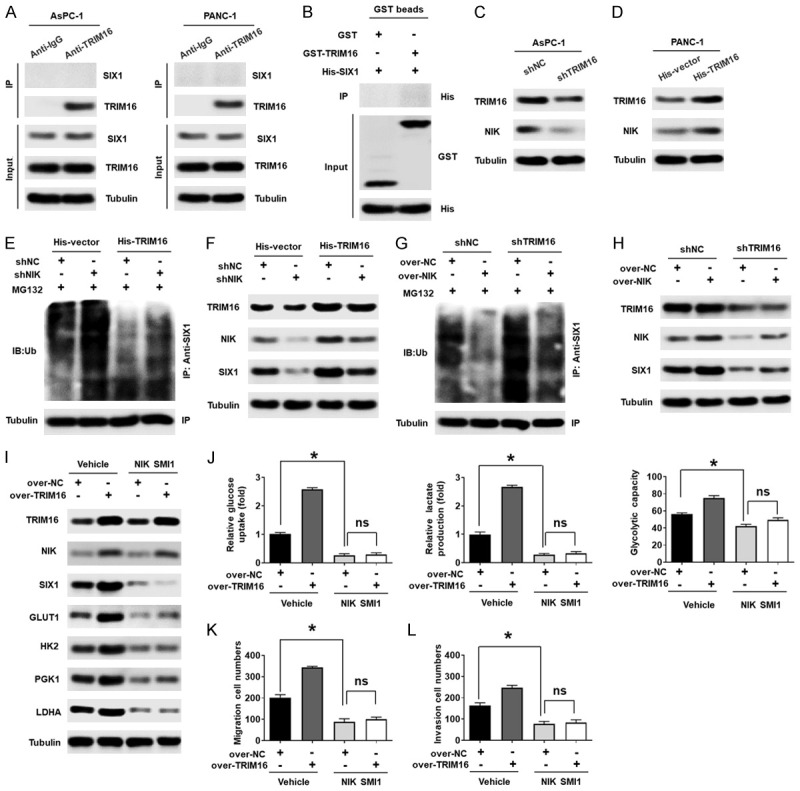
TRIM16 affects the ubiquitination of SIX1 by regulating the expression of NIK. A. Representative western blot showing that TRIM16 don’t interact with SIX1 in pancreatic cancer cells. Pancreatic cancer cells were lysed, and an immunoprecipitation assay was performed with anti-IgG or anti-TRIM16 antibodies, followed by western blot with the indicated antibodies. B. GST pull-down assay performed to detect the direct interaction between TRIM16 and SIX1. C. Protein levels of NIK in AsPC-1 cells transfected with TRIM16-silenced vector. D. Protein levels of NIK in PANC-1 cells stably transfected with the TRIM16-overexpressing vector. E, F. After silencing NIK in TRIM16-overexpressing PANC-1 cells, the ubiquitination level of SIX1 was precipitated with anti-SIX1 after the cells were treated with MG132 for 12 h, followed by western blot to detect ub. The protein level of TRIM16, NIK and SIX1 were detected by western blot. G, H. After upregulating NIK in TRIM16-silenced AsPC-1 cells, the ubiquitination level of SIX1 was precipitated with anti-SIX1, followed by western blot to detect ub. The protein level of TRIM16, NIK and SIX1 were detected by western blot. I. The protein level of TRIM16, NIK, SIX1, GLUT1, HK2, PGK1 and LDHA were detected by western blot in TRIM16-overexpressing PANC-1 cells with or without NIK SMI1. J. Glucose consumption, lactate production and glycolytic capacity were measured in TRIM16-overexpressing PANC-1 cells with or without NIK SMI1. *P<0.05, ns, no significant. K, L. The cells’ migration and invasion abilities were measured by using transwell assays in TRIM16-overexpressing PANC-1 cells with or without NIK SMI1. *P<0.05, ns, no significant.
TRIM16 competed with TRAF3 at the ISIIAQA sequence motif of NIK and stabilized NIK protein
Finally, we further explored the molecular mechanism by which TRIM16 regulates NIK. As shown in Figure 7A, endogenous NIK in pancreatic cancer cells interacted with TRIM16, as assessed by coimmunoprecipitation (co-IP) using anti-NIK antibodies. The results of GST pulldown assays confirmed that TRIM16 directly binds to NIK (Figure 7B). Next, we determined the specificity of the TRIM16-NIK interaction by mapping the domains of TRIM16 required for the interaction with NIK. We found that the SPRY-domain-deleted TRIM16 protein was unable to interact with NIK, suggesting that SPRY domains are required for the TRIM16-NIK interaction (Figure 7C). A reciprocal pull-down assay also mapped the N-terminus (aa 1-331) of NIK as the major TRIM16-interaction region (Figure 7D). To examine whether TRIM16 catalyzed the ubiquitination of NIK, we assembled an in vitro ubiquitylation system that contained ATP, an E1 Ub-activating enzyme (Uba1), an E2 Ub conjugating enzyme (UbcH7), the substrate (NIK) and the E3 Ub ligase (TRIM16). As shown in Figure 7E, TRIM16 did not ubiquitylate NIK in vitro. However, knocking down endogenous TRIM16 in pancreatic cancer cells efficiently increased the formation of K48-linked polyubiquitin chains on NIK while having no effects on K63-linked polyubiquitin chains on NIK (Figure 7F, Supplementary Figure 7A). In contrast, upregulating TRIM16 reduced the formation of K48-linked polyubiquitin chains on NIK (Figure 7G, Supplementary Figure 7B). Our data suggested that TRIM16 stabilized NIK protein in a ligase-independent manner.
Figure 7.

TRIM16 dissociates NIK from TRAF3 and upregulates NIK protein. A. Representative western blot showing that TRIM16 interact with NIK in pancreatic cancer cells. Pancreatic cancer cells were lysed, and an immunoprecipitation assay was performed with anti-IgG or anti-TRIM16 antibodies, followed by western blot with the indicated antibodies. B. GST pull-down assay performed to detect the direct interaction between TRIM16 and NIK protein. C. Mapping of TRIM16 regions interacting with NIK. Lysates of HEK293T cells co-expressing Flag-NIK and GST-TRIM16 variants (see scheme in left panel) were subjected to immunoprecipitation with anti-GST and blots were probed as indicated antibodies. D. Mapping of NIK regions interacting with TRIM16. Lysates of HEK293T cells co-expressing GST-TRIM16 and Flag-NIK variants (see scheme in left panel) were subjected to immunoprecipitation with anti-Flag and blots were probed as indicated antibodies. E. TRIM16 doesn’t ubiquitylate NIK in vitro. In vitro ubiquitylation assay was carried out with the recombinant proteins: NIK, Uba1 as E1, UbcH7 as E2, and TRIM16 as E3 together with indicated components. F, G. AsPC-1 cells were stably transfected with the TRIM16-silenced vector, PANC-1 cells were stably transfected with the TRIM16-overexpressing vector. The level of ubiquitin-attached NIK was precipitated with anti-NIK after the cells were treated with MG132 for 12 h, followed by western blot to detect ub (linkage-specific K48). H. The protein level of TRIM16, c-IAP1, c-IAP2, TRAF2 and TRAF3 were detected by using western blot in TRIM16-silenced AsPC-1 cells. I. The binding amount of NIK with TRAF3 or TRIM16 were detected by immunoprecipitation with anti-NIK and blots were probed as indicated antibodies in TRIM16-silenced AsPC-1 cells. J. The protein level of TRIM16, c-IAP1, c-IAP2, TRAF2 and TRAF3 were detected by using western blot in TRIM16-overexpressing PANC-1 cells. K. The binding amount of NIK with TRAF3 or TRIM16 were detected in TRIM16-overexpressing PANC-1 cells. L. TRIM16 was combined with a specific sequence motif (ISIIAQA) of NIK. GST pulldown assays were used to detect the interaction between TRIM16 and NIK variants. M. BIAcore-binding properties of NIK to TRAF3 with (KD = 4.38±0.83 µM) or without TRIM16 (KD = 12.76±1.28 µM).
It has been reported that NIK is ubiquitylated by a TRAF-cIAP E3 complex consisting of cellular inhibitor of apoptosis 1 and 2 (c-IAP1/2) and tumor necrosis factor receptor-associated factors 2 and 3 (TRAF2/3), in which TRAF3 functions as the substrate-binding component [32]. NIK is directly bound by TRAF3 and targeted for ubiquitin-proteasome-mediated degradation. Based on this information, we explored whether TRIM16 regulates the c-IAP-TRAF-NIK complex. Firstly, we tested whether TRIM16 can directly target TRAF3, and the GST pull down ST pulldown experiment showed that TRIM16 didn’t interact with TRAF3 proteins (Supplementary Figure 7C). Secondly, although we found that knocking down TRIM16 did not change the protein levels of c-IAP1, c-IAP2, TRAF2 or TRAF3, it enhanced the binding of NIK and TRAF3, accompanied by a decrease in the binding of TRIM16 to NIK in pancreatic cancer cells (Figure 7H and 7I, Supplementary Figure 7D and 7E). The upregulation of TRIM16 lessened the binding of NIK and TRAF3 and increased the binding of TRIM16 to NIK (Figure 7J and 7K, Supplementary Figure 7F and 7G). These results indicated that TRIM16 may compete with TRAF3 in combination with NIK. To further confirm this idea, we examined whether TRIM16 was also combined with a specific sequence motif (ISIIAQA) of NIK, which is required for the interaction of NIK with TRAF3 [33]. We deleted this sequence motif to generate a NIK mutant, NIKΔ78-84. GST pulldown assays confirmed that NIKΔ78-84 was largely defective in TRIM16 binding (Figure 7L), indicating that TRIM16 was also combined with this specific sequence motif of NIK. Furthermore, surface plasmon resonance (SPR) experiments demonstrated that the purified recombinant TRAF3 protein could bind to NIK, whereas adding TRIM16 proteins led to reduced binding between TRAF3 and NIK (Figure 7M). In summary, our results confirm that TRIM16 competes with TRAF3 at the ISIIAQA sequence motif of NIK, which in turn leads to an increase in NIK protein levels.
Discussion
Cancer cells in solid tumors generally display high rates of aerobic glycolysis [5]. This metabolic reprogramming gives cancer cells an advantage for metastasis [34]. Pancreatic cancer has a high metastasis rate, which is closely related to the enhancement of glycolysis [8]. However, the mechanism of increased glycolysis in pancreatic cancer has not been fully elucidated. In our research, we demonstrated that TRIM16 was highly expressed in pancreatic cancer tissues and promoted the metastasis of pancreatic cancer. Using different cancer cell lines, we showed that TRIM16 upregulated the expression of SIX1, a key transcription factor for the regulation of glycolysis, and increased glucose uptake, lactate production, ATP generation, and the expression of many glycolytic genes, facilitating tumor metastasis. The regulation of SIX1 expression by TRIM16 is mediated by NIK, which can inhibit the ubiquitination and degradation of SIX1. TRIM16 bound to NIK directly and competed with TRAF3 for NIK, resulting in the dissociation of NIK from TRAF3 and an increase in NIK protein expression (Figure 8). Our study established the TRIM16-NIK-SIX1 axis as a critical regulatory pathway in the Warburg effect and in pancreatic cancer metastasis; these findings will provide a new theoretical basis and therapeutic targets for the treatment of pancreatic cancer.
Figure 8.
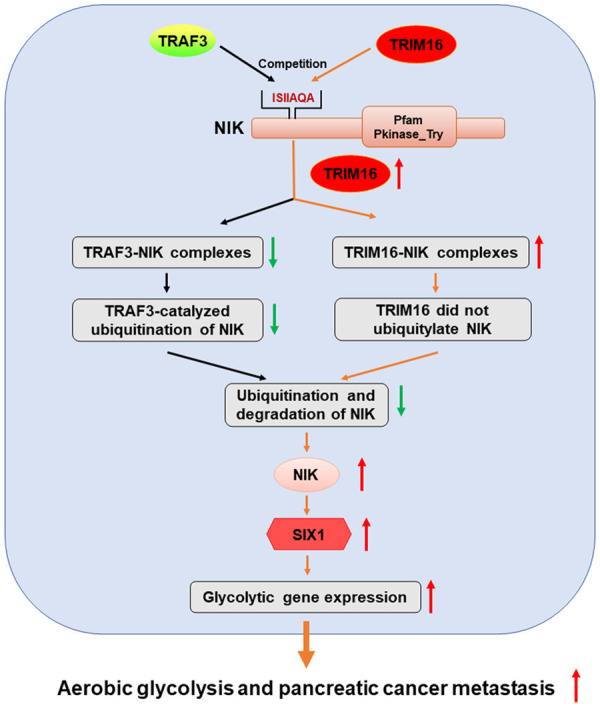
Proposed model of this paper. TRIM16 promotes SIX1-mediated aerobic glycolysis and pancreatic cancer metastasis by competing with TRAF3 at the ISIIAQA sequence motif of NIK, thereby stabilizing NIK protein and upregulating SIX1.
TRIM16 is associated with diverse cellular processes, such as apoptosis and differentiation [25]. However, the effect of TRIM16 on apoptosis is paradoxical. In human breast cancer and neuroblastoma cells, TRIM16 induces apoptosis [35]. However, under stressful conditions, TRIM16 can promote the process of stress-induced aggregate clearance and protect cells against oxidative/proteotoxic-stress-induced apoptosis [24]. The role of TRIM16 in cancer is also controversial. In neuroblastoma, ovarian cancer and breast cancer, TRIM16 acts as a tumor suppressor [26-28]. In colon cancer and gastric cancer, TRIM16 acts as an oncoprotein [29,30]. Therefore, TRIM16 should not be generalized as a tumor suppressor or oncoprotein, as its impact on cancer varies from cancer to cancer. Currently, there is no information on the role of or mechanism by which TRIM16 acts in pancreatic cancer. In this study, we found that TRIM16 was highly expressed in pancreatic cancer tissues, and an increase in TRIM16 expression was associated with the malignant phenotype of patients with pancreatic cancer. Our results, and data included in TCGA, showed that patients with high TRIM16 expression had significantly shorter overall survival. Univariate and multivariate analyses showed that high TRIM16 expression is an independent predictor of poor prognosis in patients with pancreatic cancer. In addition, we examined the role of TRIM16 in pancreatic cancer metastasis using cell culture approaches and animal-based tumor models. We found that TRIM16 promoted pancreatic cancer cell migration and invasion in vitro and metastasis in vivo. Our data implied that TRIM16 serves as an oncogene and promotes tumor metastasis in pancreatic cancer.
The Warburg effect is a hallmark of cancer cells. Enhanced glycolysis plays a crucial role during tumor metastasis [8]. Therefore, understanding the potential regulatory mechanisms of glycolysis in pancreatic cancer will be beneficial for the prevention and treatment of this malignant cancer. Transcription factors, such as HIF1α, p53, c-Myc and SIX1, play a critical role in glycolysis by directly regulating the expression of several glycolytic enzyme-related genes [12,13]. Glycolysis mediated by these transcription factors is regulated by a variety of signals, thus forming a complex network to contribute to the Warburg effect. Here, our iTRAQ data showed that SIX1 and glycolytic genes were significantly reduced when TRIM16 was downregulated in pancreatic cancer cells. However, TRIM16 did not affect the expression of HIF1α, p53 or c-Myc. In addition, silencing TRIM16 inhibited the expression of SIX1 and several glycolytic genes that were target genes of SIX1, as well as the metabolic phenotype of glycolysis. In contrast, increasing TRIM16 enhanced the expression of SIX1 and glycolytic activity in pancreatic cancer cells. More importantly, restoring SIX1 rescued the decrease in glycolytic activity and metastasis induced by TRIM16 inhibition, but inhibiting SIX1 decreased the expression of glycolytic genes in TRIM16-overexpressing cells and counteracted the oncogenic effect of TRIM16. Hence, our data indicated that the prometastatic role of TRIM16 was dependent on SIX1-mediated glycolysis. Since the TRIM16/SIX1 axis is deregulated in cancer, correlates with prognosis and controls glycolysis, this axis is expected to be an excellent therapeutic target for curing pancreatic cancer.
SIX1, a homeobox transcription factor, is a key regulator during the embryonic development of numerous organs [36]. SIX1 is largely downregulated after embryogenesis but is overexpressed in many cancers, such as pancreatic cancer, breast cancer, ovarian carcinoma, colorectal cancer and hepatocellular carcinoma [17,37-39]. SIX1 can promote tumor growth, the epithelial-mesenchymal transition, tumor metastasis and cancer stem cell properties. Recently, SIX1 was identified as a key regulator of glycolytic gene expression, and it promotes the Warburg effect, which is closely associated with cancer progression [13]. Various factors, including noncoding RNA and O-GlcNAcylation modification [18,40], affect cancer progression by regulating the expression of SIX1. However, studies on the ubiquitination and degradation of SIX1 are rare. In the present study, we demonstrated that TRIM16 inhibited the ubiquitination of SIX1, thus blocking its protein degradation. In addition, we clarified the mechanisms by which TRIM16 mediates the ubiquitination modification of SIX1 in pancreatic cancer. We found that TRIM16 did not interact with SIX1 but inhibited SIX1 ubiquitination through NIK. Upregulating NIK could reverse the increase in the ubiquitination level and the decrease in the protein level of SIX1 caused by silencing TRIM16. Importantly, NIK SMI1, a selective inhibitor of NIK, suppressed SIX1 expression, glycolysis and cell invasion in TRIM16-overexpressing pancreatic cancer cells. TRIM16 and SIX1 are difficult to target at present, so NIK inhibitors have prospects for broad application because they can decrease SIX1 expression and extend the inhibition of glycolysis. Notably, NIK inhibitors are currently for research use only. In the future, understanding in vivo target engagement, time of residency and their effects in other cancer models are necessary to fully assess the therapeutic value of NIK inhibitors.
Studies have shown that NIK is a key regulator of SIX1. NIK inhibits the ubiquitination level of SIX1 and proteasome-mediated degradation in mouse primary bone marrow-derived macrophages (BMDMs) and human fibroblasts [20]. We confirmed that this regulatory mechanism is also present in pancreatic cancer cells. NIK is constitutively active in pancreatic cancer cells and promotes pancreatic cancer cell proliferation and tumorigenicity [41,42]. The activation of NIK is primarily controlled through its protein accumulation [32]. The regulatory mechanism of NIK protein stability is dependent on TRAF-cIAP E3 complex-mediated ubiquitination and proteasome degradation events [33]. In this study, we found that TRIM16 was involved in the NIK degradation process but did not serve as an E3 ubiquitin ligase for NIK in pancreatic cancer cells. This notion is supported by three lines of experimental evidence. First, TRIM16 bound to NIK directly but did not ubiquitylate NIK in vitro. Second, inhibiting TRIM16 efficiently increased the formation of K48-linked polyubiquitin chains on NIK, while upregulating TRIM16 reduced the ubiquitination level of NIK. Third, TRIM16 competed with TRAF3 for NIK, resulting in the dissociation of the TRAF-cIAP E3 complex from NIK, thereby inhibiting the ubiquitination and degradation of NIK. Although TRIM16 can act as an E3 ligase, our study reveals that TRIM16 can also compete with other E3 ligases to stabilize substrates. This novel mechanism is consistent with that reported in other studies [24,25].
Our study confirmed that TRIM16 is upregulated and the TRAF-cIAP E3 complex is downregulated in pancreatic cancer cells [42], indicating that the imbalance between them might be a major cause of NIK’s constitutive activation in pancreatic cancer. Thus, targeting NIK may make cancer therapy more effective than targeting TRIM16. However, further investigation of the potential side effects of NIK inhibition is required, as NIK is a central kinase of the noncanonical NF-κB pathway and is also involved in the canonical NF-κB pathway [43].
Acknowledgements
This research was supported by grants from the Science and Technology Project of Jiangxi Provincial Department of Education (GJJ210180).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crippa S, Angelini C, Mussi C, Bonardi C, Romano F, Sartori P, Uggeri F, Bovo G. Surgical treatment of metastatic tumors to the pancreas: a single center experience and review of the literature. World J Surg. 2006;30:1536–1542. doi: 10.1007/s00268-005-0464-4. [DOI] [PubMed] [Google Scholar]
- 5.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 6.Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152. doi: 10.1186/1476-4598-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang M, Xiong H, Luo D, Xu B, Liu H. CSN5 upregulates glycolysis to promote hepatocellular carcinoma metastasis via stabilizing the HK2 protein. Exp Cell Res. 2020;388:111876. doi: 10.1016/j.yexcr.2020.111876. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Ren B, Yang G, Wang H, Chen G, You L, Zhang T, Zhao Y. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305–321. doi: 10.1007/s00018-019-03278-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, Hines J, Chen X. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012;11:1672–1682. doi: 10.1158/1535-7163.MCT-12-0131. [DOI] [PubMed] [Google Scholar]
- 10.Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, Chin EN, Ahmad I, Chatterjee AK, Lairson LL, Schultz PG, Moellering RE. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature. 2018;562:600–604. doi: 10.1038/s41586-018-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Liang Y, Kang L, Liu Y, Gao S, Chen S, Li Y, You W, Dong Q, Hong T, Yan Z, Jin S, Wang T, Zhao W, Mai H, Huang J, Han X, Ji Q, Song Q, Yang C, Zhao S, Xu X, Ye Q. Transcriptional regulation of the warburg effect in cancer by SIX1. Cancer Cell. 2018;33:368–385. e7. doi: 10.1016/j.ccell.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Kingsbury TJ, Kim M, Civin CI. Regulation of cancer stem cell properties by SIX1, a member of the PAX-SIX-EYA-DACH network. Adv Cancer Res. 2019;141:1–42. doi: 10.1016/bs.acr.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Wu W, Ren Z, Li P, Yu D, Chen J, Huang R, Liu H. Six1: a critical transcription factor in tumorigenesis. Int J Cancer. 2015;136:1245–1253. doi: 10.1002/ijc.28755. [DOI] [PubMed] [Google Scholar]
- 16.Camolotto SA, Belova VK, Torre-Healy L, Vahrenkamp JM, Berrett KC, Conway H, Shea J, Stubben C, Moffitt R, Gertz J, Snyder EL. Reciprocal regulation of pancreatic ductal adenocarcinoma growth and molecular subtype by HNF4alpha and SIX1/4. Gut. 2021;70:900–914. doi: 10.1136/gutjnl-2020-321316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lerbs T, Bisht S, Scholch S, Pecqueux M, Kristiansen G, Schneider M, Hofmann BT, Welsch T, Reissfelder C, Rahbari NN, Fritzmann J, Brossart P, Weitz J, Feldmann G, Kahlert C. Inhibition of Six1 affects tumour invasion and the expression of cancer stem cell markers in pancreatic cancer. BMC Cancer. 2017;17:249. doi: 10.1186/s12885-017-3225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Z, Rong Z, Luo Z, Yu Z, Zhang J, Qiu Z, Huang C. Circular RNA circNHSL1 promotes gastric cancer progression through the miR-1306-3p/SIX1/vimentin axis. Mol Cancer. 2019;18:126. doi: 10.1186/s12943-019-1054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X, Zhu X, Yan Z, Li C, Zhao H, Ma L, Zhang D, Liu J, Liu Z, Du N, Ye Q, Xu X. MiR-489-3p/SIX1 axis regulates melanoma proliferation and glycolytic potential. Mol Ther Oncolytics. 2019;16:30–40. doi: 10.1016/j.omto.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Mar KB, Hanners NW, Perelman SS, Kanchwala M, Xing C, Schoggins JW, Alto NM. A NIK-SIX signalling axis controls inflammation by targeted silencing of non-canonical NF-kappaB. Nature. 2019;568:249–253. doi: 10.1038/s41586-019-1041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 22.Morreale FE, Walden H. Types of ubiquitin ligases. Cell. 2016;165:248–248. e1. doi: 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Bell JL, Malyukova A, Holien JK, Koach J, Parker MW, Kavallaris M, Marshall GM, Cheung BB. TRIM16 acts as an E3 ubiquitin ligase and can heterodimerize with other TRIM family members. PLoS One. 2012;7:e37470. doi: 10.1371/journal.pone.0037470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jena KK, Kolapalli SP, Mehto S, Nath P, Das B, Sahoo PK, Ahad A, Syed GH, Raghav SK, Senapati S, Chauhan S, Chauhan S. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018;37:e98358. doi: 10.15252/embj.201798358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Zhai Q, Liu H, Xi X, Chen S, Liu D. TRIM16 promotes osteogenic differentiation of human periodontal ligament stem cells by modulating CHIP-mediated degradation of RUNX2. Front Cell Dev Biol. 2021;8:625105. doi: 10.3389/fcell.2020.625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall GM, Bell JL, Koach J, Tan O, Kim P, Malyukova A, Thomas W, Sekyere EO, Liu T, Cunningham AM, Tobias V, Norris MD, Haber M, Kavallaris M, Cheung BB. TRIM16 acts as a tumour suppressor by inhibitory effects on cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells. Oncogene. 2010;29:6172–6183. doi: 10.1038/onc.2010.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan H, Qi J, Chu G, Liu Z. Tripartite motif 16 inhibits the migration and invasion in ovarian cancer cells. Oncol Res. 2017;25:551–558. doi: 10.3727/096504016X14758370595285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim PY, Tan O, Liu B, Trahair T, Liu T, Haber M, Norris MD, Marshall GM, Cheung BB. High TDP43 expression is required for TRIM16-induced inhibition of cancer cell growth and correlated with good prognosis of neuroblastoma and breast cancer patients. Cancer Lett. 2016;374:315–323. doi: 10.1016/j.canlet.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 29.Ju B, Liu Z, Nai C, Zhu X. Long non-coding RNA CASC2 induces apoptosis and autophagy in human colon cancer cells via modulation of TRIM16 expression. Am J Transl Res. 2020;12:2695–2702. [PMC free article] [PubMed] [Google Scholar]
- 30.Yan Y, Shen Z, Gao Z, Cao J, Yang Y, Wang B, Shen C, Mao S, Jiang K, Ye Y, Wang S. Long noncoding ribonucleic acid specific for distant metastasis of gastric cancer is associated with TRIM16 expression and facilitates tumor cell invasion in vitro. J Gastroenterol Hepatol. 2015;30:1367–1375. doi: 10.1111/jgh.12976. [DOI] [PubMed] [Google Scholar]
- 31.Christensen KL, Brennan JD, Aldridge CS, Ford HL. Cell cycle regulation of the human Six1 homeoprotein is mediated by APC(Cdh1) Oncogene. 2007;26:3406–3414. doi: 10.1038/sj.onc.1210122. [DOI] [PubMed] [Google Scholar]
- 32.Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545–558. doi: 10.1038/nri.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 34.Ghanavat M, Shahrouzian M, Deris Zayeri Z, Banihashemi S, Kazemi SM, Saki N. Digging deeper through glucose metabolism and its regulators in cancer and metastasis. Life Sci. 2021;264:118603. doi: 10.1016/j.lfs.2020.118603. [DOI] [PubMed] [Google Scholar]
- 35.Kim PY, Rahmanto AS, Tan O, Norris MD, Haber M, Marshall GM, Cheung BB. TRIM16 overexpression induces apoptosis through activation of caspase-2 in cancer cells. Apoptosis. 2013;18:639–651. doi: 10.1007/s10495-013-0813-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blevins MA, Towers CG, Patrick AN, Zhao R, Ford HL. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert Opin Ther Targets. 2015;19:213–225. doi: 10.1517/14728222.2014.978860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou H, Blevins MA, Hsu JY, Kong D, Galbraith MD, Goodspeed A, Culp-Hill R, Oliphant MUJ, Ramirez D, Zhang L, Trinidad-Pineiro J, Mathews Griner L, King R, Barnaeva E, Hu X, Southall NT, Ferrer M, Gustafson DL, Regan DP, D’Alessandro A, Costello JC, Patnaik S, Marugan J, Zhao R, Ford HL. Identification of a small-molecule inhibitor that disrupts the SIX1/EYA2 complex, EMT, and metastasis. Cancer Res. 2020;80:2689–2702. doi: 10.1158/0008-5472.CAN-20-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A, Ford HL. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007;67:3036–3042. doi: 10.1158/0008-5472.CAN-06-3755. [DOI] [PubMed] [Google Scholar]
- 39.Ng KT, Man K, Sun CK, Lee TK, Poon RT, Lo CM, Fan ST. Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br J Cancer. 2006;95:1050–1055. doi: 10.1038/sj.bjc.6603399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu Y, Jiang M, Wu N, Xu B, Li W, Liu H, Su S, Shi Y, Liu H, Gao X, Fu X, Chen D, Li X, Wang W, Liang J, Nie Y, Fan D. O-GlcNAcylation of SIX1 enhances its stability and promotes hepatocellular carcinoma proliferation. Theranostics. 2020;10:9830–9842. doi: 10.7150/thno.45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishina T, Yamaguchi N, Gohda J, Semba K, Inoue J. NIK is involved in constitutive activation of the alternative NF-kappaB pathway and proliferation of pancreatic cancer cells. Biochem Biophys Res Commun. 2009;388:96–101. doi: 10.1016/j.bbrc.2009.07.125. [DOI] [PubMed] [Google Scholar]
- 42.Doppler H, Liou GY, Storz P. Downregulation of TRAF2 mediates NIK-induced pancreatic cancer cell proliferation and tumorigenicity. PLoS One. 2013;8:e53676. doi: 10.1371/journal.pone.0053676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pflug KM, Sitcheran R. Targeting NF-kappaB-inducing kinase (NIK) in immunity, inflammation, and cancer. Int J Mol Sci. 2020;21:8470. doi: 10.3390/ijms21228470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.