

SUMMARY
N-glycans are processed mainly in the Golgi, and a well-organized Golgi structure is required for accurate glycosylation. However, during mitosis the Golgi undergoes severe fragmentation. The resulting trafficking block leads to an extended exposure of cargo molecules to Golgi enzymes. It is unclear how cells avoid glycosylation defects during mitosis. In this study, we report that Golgi α-1,2-mannosidase IA (MAN1A1), the first enzyme that cargo proteins encounter once arriving the Golgi, is phosphorylated at serine 12 by CDK1 in mitosis, which attenuates its activity, affects the production of glycan isomers, and reduces its interaction with the subsequent glycosyltransferase, MGAT1. Expression of wild-type MAN1A1, but not its phosphomimetic mutant, rescues the glycosylation defects in mannosidase I-deficient cells, whereas expression of its phosphorylation-deficient mutant in mitosis increases the formation of complex glycans. Our study reveals that glycosylation is regulated by cytosolic signaling during the cell cycle.
Graphical abstract
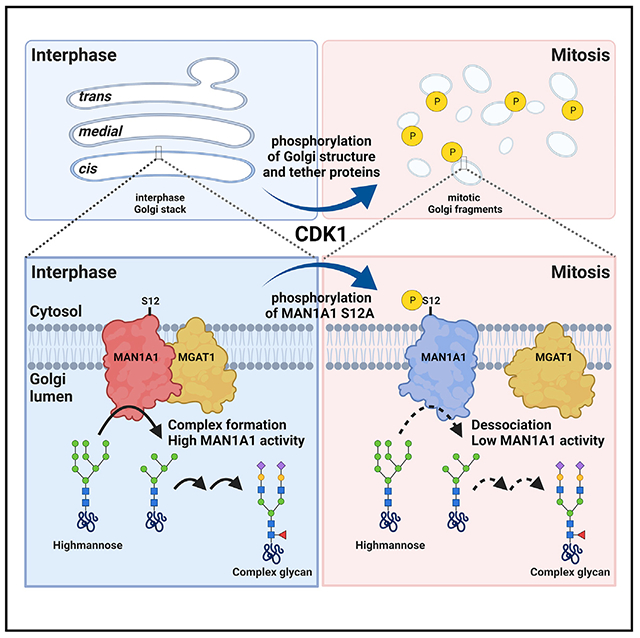
In brief
In this study, Huang et al. discover that MAN1A1, the first enzyme that cargo proteins encounter once arriving the Golgi, is phosphorylated in mitosis. This attenuates its activity likely by reducing its interaction with the subsequent glycosyltransferase, MGAT1, suggesting that glycosylation is regulated by cytosolic signaling during the cell cycle.
INTRODUCTION
Asparagine-linked (N-linked) protein glycosylation is a common form of co-translational and post-translational modification of membrane and secreted proteins. The N-glycosylation pathway initiates in the endoplasmic reticulum (ER) with the synthesis and transfer of a pre-formed, high-mannose oligosaccharide precursor to nascent polypeptide chains (Kornfeld and Kornfeld, 1985). After the removal of three glucose residues by the ER glucosidases and one mannose by the ER mannosidase MAN1B1, the oligosaccharide is further processed in the Golgi, first by α-mannosidase I (MAN1A1, MAN1A2, MAN1C1), which removes three mannose residues; followed by α-1,3-mannosyl-glycoprotein 2-β-N-acetylglucosaminyltransferase (MGAT1; also called GnT1), which adds a GlcNAc residue; Golgi α-mannosidases II (MAN2A1, MAN2A2), which cleaves two terminal mannose residues; and GnT2, which adds another GlcNAc residue. The produced GlcNAcMan3GlcNAc2-Asn core structure is then further processed by branching and capping reactions mediated by other enzymes to form complex type oligosaccharides (Moremen and Nairn, 2014).
The Golgi stack consists of an ordered series of compartments that are biochemically and functionally distinct from each other. The cis-Golgi, being closest to the ER from which it receives cargo molecules, contains Golgi MAN1 that removes three mannose residues from the oligosaccharides. The medial-Golgi includes the central layers in the stack where MAN2 removes two mannose residues and several glycosyltransferases add sugars (e.g., GlcNAc) to the glycan chain. The trans-Golgi, which is farthest from the ER, hosts additional glycosyltransferases that add more sugars (e.g., galactose and sialic acid) to the glycoproteins. The trans-Golgi network (TGN), a tubular network extended from the trans-Golgi, sorts cargo molecules for delivery to different destinations. An ordered organization of glycosidases and glycosyltransferases in the subcompartments of the Golgi stack is required for sequential and accurate processing of N-glycans (Zhang and Wang, 2016). Disruption of the Golgi structure impairs accurate glycosylation (Puthenveedu et al., 2006; Xiang et al., 2013), and glycosylation defects are often linked to Golgi structural disorganization in diseases (Condon et al., 2013; Kornak et al., 2008; Percival and Froehner, 2007; D’Souza et al., 2020).
Golgi structure disorganization also occurs under physiological conditions. During mitosis, the Golgi undergoes a series of stepwise disassembly processes controlled by cytoplasmic factors (Huang and Wang, 2017; Tang and Wang, 2013; Wang and Seemann, 2011). A key player is cyclin-dependent kinase 1 (CDK1), which phosphorylates and inactivates multiple Golgi structural proteins including GRASP65 and GM130, leading to mitotic Golgi disassembly (Lowe et al., 1998; Wang et al., 2003). Previous research suggested that intra-Golgi transport is stopped during mitotic Golgi fragmentation (Collins and Warren, 1992). This raises a question: how does the cell avoid inaccurate glycosylation when the cargo molecules and enzymes are trapped together for an extended exposure during mitosis?
One possibility is that the activities of glycosylation enzymes are regulated in mitosis. Given that phosphorylation is the driver of cell cycle progression as well as the major cause of Golgi fragmentation, it is possible that Golgi enzymes are also regulated by mitotic kinases. Several Golgi glycosylation enzymes, such as MAN1A1 and MAN1C1 (Bongini et al., 2014), MAN2A1 (Villen et al., 2007), and MGAT4A (Tagliabracci et al., 2015), have been identified in phosphoproteomic studies of interphase cells. However, there is so far no report on cell cycle-dependent phosphorylation or regulation of Golgi glycosylation enzymes.
Here, we performed phosphoproteomic studies of mitotic Golgi membranes and discovered that MAN1A1 is highly phosphorylated at serine 12 (S12) at its cytoplasmic domain by CDK1 in mitosis, which inhibits MAN1A1 activity through reducing its interaction with the subsequent glycosylation enzyme MGAT1.Our study reveals that glycosylation is regulated by phosphorylation in the cell cycle.
RESULTS
Phosphoproteomic analysis reveals that MAN1A1 is highly phosphorylated in mitosis
To identify candidate proteins regulated by phosphorylation during the cell cycle, we purified interphase Golgi membranes from rat liver (RLG), prepared mitotic Golgi fragments (MGFs) by incubating RLG with mitotic cytosol (Tang and Wang, 2015; Wang et al., 2006; Tang et al., 2010), and performed mass spectrometry (MS) analysis to identify phosphorylated proteins (Kweon and Andrews, 2013). The results revealed previously reported mitotic phosphorylation of Golgi structure proteins, including GM130 (Lowe et al., 2000), GRASP65 (Tang et al., 2012), Giantin (Dephoure et al., 2008; Olsen et al., 2010), and Golgin 84 (Diao et al., 2003; Dephoure et al., 2008) (Table S1), as well as a number of Golgi enzymes, including MAN1A1, MAN1A2, ST6GAL1, and GALNT11 (Figure 1A; Table S1). Among these, serine 12 on MAN1A1 was most highly phosphorylated in mitosis than interphase, while S11 on MAN1A1 was phosphorylated at a very low abundance in only one of the two MS results and was not specific to mitosis (Table S1) and thus was not a focus in this study. In addition, threonine 2 (T2), threonine 3 (T3), and serine 10 (S10) on MAN1A2 were also phosphorylated (Figure 1A).
Figure 1. MAN1A1 is highly phosphorylated at S12 in mitosis.
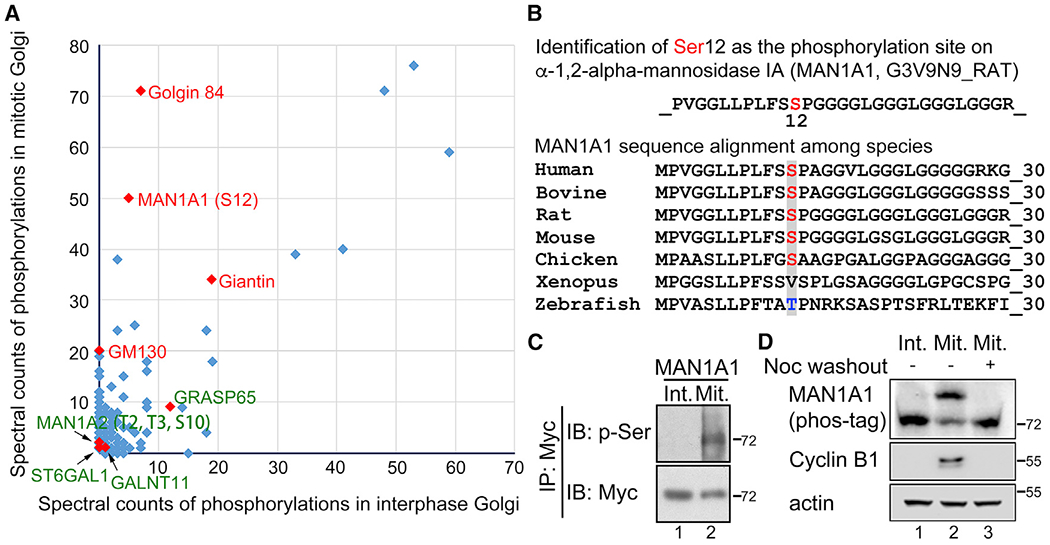
(A) Mass spectrometry analysis of interphase and mitotic Golgi phosphoproteomes identified that MAN1A1 is highly phosphorylated at S12 in mitotic but not interphase Golgi membranes. Proteins highly phosphorylated only in mitotic Golgi membranes are indicated in red. Proteins relatively weakly phosphorylated in both interphase and mitosis are labeled in green.
(B) Alignment of MAN1A1 sequence across different species demonstrates that S12 is conserved.
(C) MAN1A1 is phosphorylated in mitotic cells. Int, interphase; Mit, mitosis; p-Ser, phosphor-serine.
(D) Endogenous MAN1A1 is phosphorylated in mitotic cells. Nocodazole (Noc) washout, 4 h.
Alignment of MAN1A1 amino acid sequences showed that S12 is highly conserved across mammalian species where mitotic Golgi fragmentation has been documented (Figure 1B). To confirm MAN1A1 phosphorylation in mitotic cells, we enriched mitotic cells by nocodazole synchronization (Tang et al., 2012), immunoprecipitated MAN1A1 from mitotic and interphase cells, and determined its phosphorylation by western blot using a phospho-serine (p-Ser) antibody. The result showed that MAN1A1 was phosphorylated in mitotic but not interphase cells (Figure 1C). Similarly, T2, T3, and S10 on MAN1A2 are also conserved among species (Figure S1A), and mitotic phosphorylation of MAN1A2 was confirmed using a phospho-threonine (p-Thr) antibody (Figure S1B). As a negative control, MAN2A1 was not phosphorylated in our phosphoproteomic and biochemical analyses (Figure S1C). Moreover, phosphorylation of both endogenous and exogenous MAN1A1 in mitosis was confirmed by phos-tag gel analysis (Figures 1D and S1D). The same experiment also revealed that phosphorylation of MAN1A1 is specific to mitosis (indicated by cyclin B1 expression) and is reversible; the upshifted MAN1A1 band in mitosis was downshifted when the cells were released into interphase by nocodazole washout (Figures 1D and S1D). Taken together, MAN1A1 is highly phosphorylated in mitosis but not interphase.
MAN1A1 is phosphorylated at S12 by CDK1
Given that S12 in MAN1A1 fits the S/T-P consensus sequence for CDK1 phosphorylation, we tested the possibility that MAN1A1 might be phosphorylated by CDK1. We treated mitotic HeLa cells with a highly selective CDK1 inhibitor, RO-3306, a more general CDK inhibitor, roscovitine, or a general kinase inhibitor, staurosporine, and analyzed MAN1A1 phosphorylation by phos-tag gel electrophoresis and western blot. As shown in Figures 2A and 2B, inhibition of CDK1 effectively abolished mitotic phosphorylation of MAN1A1 as indicated by the band-shift on the phos-tag gel. In these experiments, cyclin B protein level remained high after CDK1 inhibition (Figure 2A, lanes 3–7; Figure 2B, lane 3), indicating that such a short-term CDK1 inhibition did not lead to mitotic exit.
Figure 2. MAN1A1 is phosphorylated at S12 by CDK1.
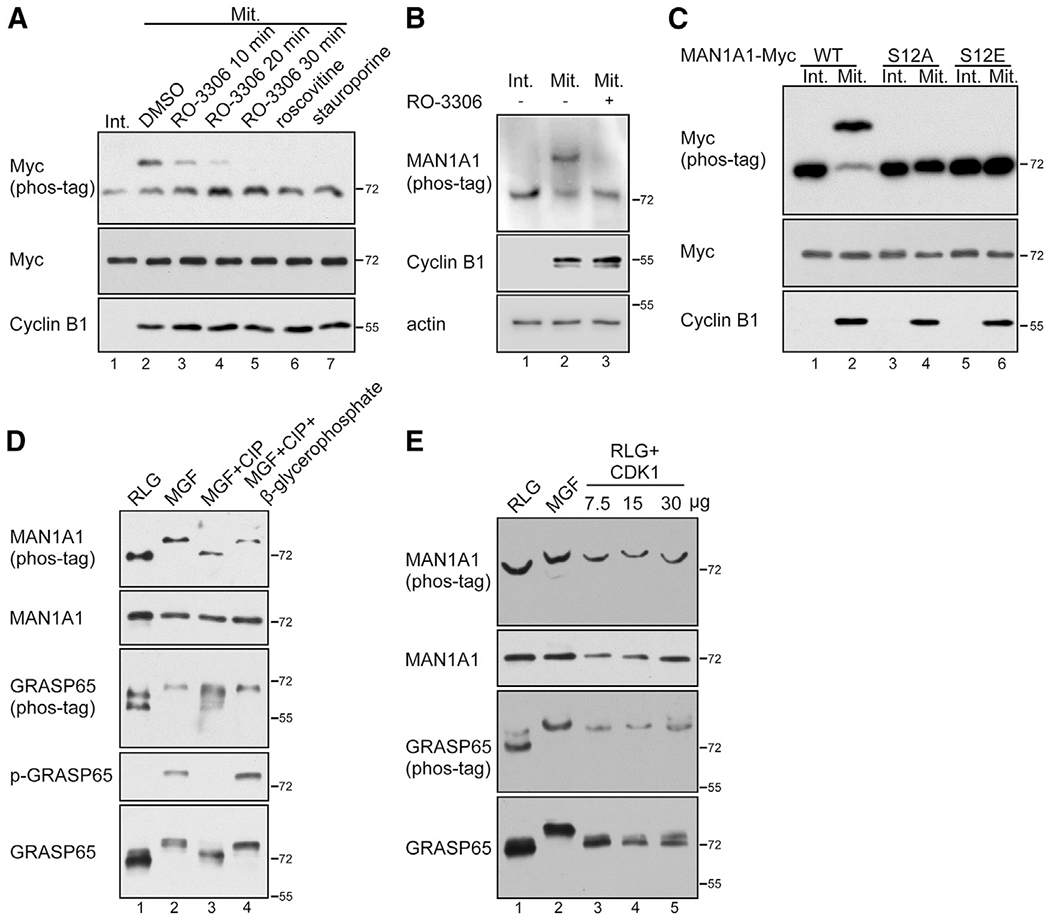
(A) MAN1A1 is phosphorylated by CDK1 in mitotic (Mit) but not interphase (Int) cells.
(B) Endogenous MAN1A1 is phosphorylated by CDK1 in mitotic cells. RO-3306, 10 μM for 30 min.
(C) Mutation of S12 abolishes MAN1A1 phosphorylation in mitotic cells.
(D) MAN1A1 is phosphorylated in mitotic Golgi fragments (MGF) but not interphase Golgi membranes (RLG). p-GRASP65, GRASP65 was blotted with a phospho-specific antibody.
(E) In vitro phosphorylation of MAN1A1 by CDK1 in purified Golgi membranes. Purified RLG membranes were incubated with mitotic cytosol (MGF) or with purified cyclin B1/CDK1 (7.5, 15 or 30 μg in lanes 3–5) and analyzed using western blot.
To further specify that S12 of MAN1A1 is phosphorylated in mitosis, we generated the phospho-deficient S12A and phosphomimetic S12E mutants. When expressed in cells, only wild-type (WT) MAN1A1, but not its mutants, was phosphorylated in mitotic cells (Figure 2C). Phosphorylation of WT MAN1A1 was inhibited by RO-3306, whereas S12A and S12E were not affected (Figure S2). These results demonstrate that MAN1A1 is phosphorylated at S12 by CDK1 in mitosis.
We then confirmed that MAN1A1 phosphorylation occurs on purified mitotic but not interphase Golgi membranes (Figure 2D). Further treatment of MGFs with calf intestinal alkaline phosphatase (CIP) reversed MAN1A1 phosphorylation, and the effect of CIP was inhibited by β-glycerophosphate, a general phosphatase inhibitor (Figure 2D). To verify that MAN1A1 is directly phosphorylated by CDK1 on Golgi membranes, we treated purified Golgi membranes with purified CDK1 (cyclin B1/CDK1 protein complex), which effectively phosphorylated MAN1A1, as indicated by a similar shift of the MAN1A1 band by CDK1 and by mitotic cytosol (Figure 2E, lanes 3–5 versus 2). In comparison with MAN1A1, CDK1 was less effective in phosphorylating GRASP65, which is consistent with previous reports that GRASP65 phosphorylation requires both CDK1 and Plk1 (Preisinger et al., 2005; Wang et al., 2003, 2005). In conclusion, MAN1A1 is phosphorylated at S12 by CDK1 on mitotic Golgi membranes.
MAN1A1 phosphorylation inhibits its activity
In the Golgi, mannose residues on the Man8 isomer (Man8B) are cleaved by a combination of MAN1A1, MAN1A2, and MAN1C1 to trim high-mannose oligosaccharide chains to the Man5 isomer (Figure 3A; also see Figure 4A). To determine whether MAN1A1 phosphorylation affects its enzymatic activity, we performed a Golgi MAN1 assay using RLG and MGFs. In brief, purified RLG or MGF membranes were lysed and incubated with a mannosidase glycan substrate, pyridylaminated Man9GlcNAc2 (Man9-PA). In this reaction, the long mannose chain of the Man9-PA substrate was trimmed to shorter chains (Man8, Man7, Man6, and Man5) which were distinguished and quantified on the basis of the retention time on high-performance liquid chromatography (HPLC) (Figure 3B). The result indicated that MGFs exhibited lower MAN1 activity than RLG, as indicated by the reduction of the trimmed mannose index (Figure 3C). A time course incubation showed that MGFs exhibited consistently lower MAN1 activity than RLG under different incubation times (Figures S3A–S3E). Meanwhile, Golgi MAN2 activity showed no difference between interphase and mitosis (Figures 3D and S3F–S3H).
Figure 3. Mitotic phosphorylation reduces Golgi MAN1 activity.
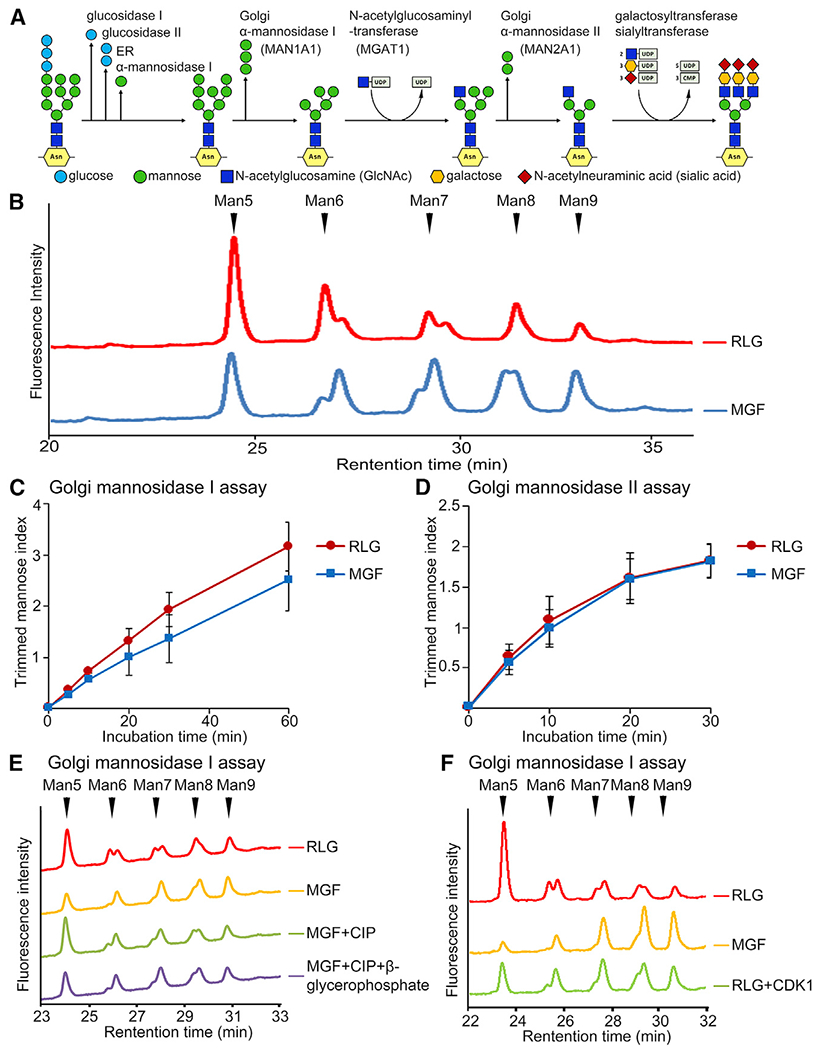
(A) A schematic of the N-glycosylation pathway.
(B) Mitotic Golgi fragments (MGF) exhibit reduced MAN1 activity than interphase Golgi membranes (RLG). Shown are representative HPLC profiles from 3 independent experiments after 1 h incubation. Note the reduced MAN1 activity that trims long mannose chains (peaks on the right) to shorter chains (peaks on the left) in MGF (blue line).
(C) Trimmed mannose index shows reduced MAN1 activity in MGF. Results are expressed as mean ± SD from three independent experiments.
(D) MAN2 activity does not change in mitosis.
(E) MAN1 activity in Golgi membranes is regulated by phosphorylation and dephosphorylation.
(F) MAN1A1 activity is regulated by CDK1-dependent phosphorylation.
Figure 4. Mitotic phosphorylation of Golgi membranes alters the production of different isomers of glycans.
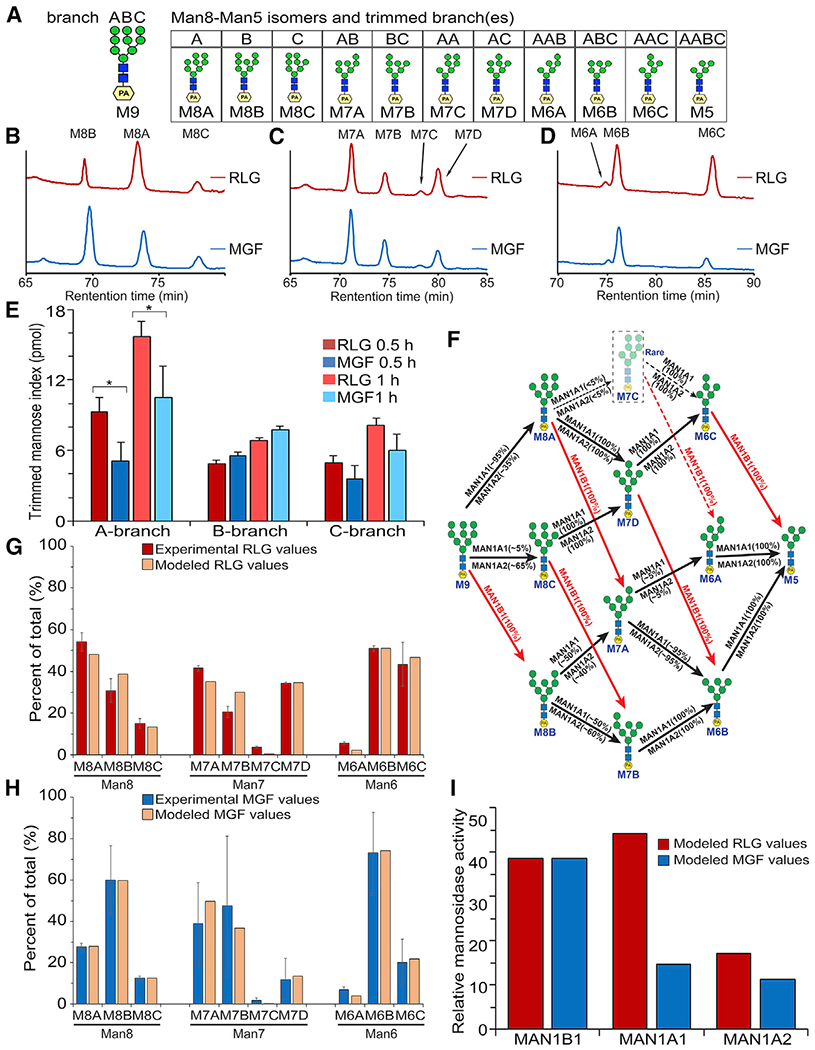
(A) A schematic diagram of the branched Man9-PA substrate including the residue nomenclature and glycosidic linkages for mannose and N-acetylglucosamine residues, and the produced Man8-Man5 isomers.
(B–D) Mitotic phosphorylation of MAN1A1 alters the production of Man8, Man7, and Man6 isomers. Shown are the dual-gradient, reversed-phase HPLC profiles.
(E) Analyses of the trimming activity of each branch on the basis of the production of all isomers from (B)–(D). Results are expressed as mean ± SD from three independent experiments. The p value was determined using Student’s t test. *p < 0.05.
(F) Established substrate specificities for MAN1B1, MAN1A1, and MAN1A2 for the digestion series of Man9 to Man5. All predicted isomer intermediates for Man8, Man7, and Man6 are displayed, and MAN1B1 actions are shown in red.
(G) The abundances of the Man8, Man7, and Man6 isomer intermediates were determined for the 30 min digestion of Man9-PA by the RLG membrane preparation on the basis of dual-gradient, reversed-phase HPLC (Figure S4A). Values for each isomer abundance in its respective size fraction were normalized as a percentage of total for that size fraction. Results represent mean ± SD from triplicate analyses (red bars). The data were then modeled on the basis of the established substrate specificities of MAN1B1, MAN1A1, and MAN1A2 (Figure 4F), and the abundances of the three enzymes were adjusted to result in a modeled dataset (tan bars) that best matched the profiles of the respective experimental isomer intermediates.
(H) The abundances of the Man8, Man7, and Man6 isomer intermediates were determined for the 30 min digestion of Man9-PA by the MGF membrane preparation as in (G). Results represent mean ± SD from triplicate analyses (blue bars). The data were then modeled as in (F) and the abundances of the three enzymes were adjusted to result in a modeled dataset (tan bars) that best matched the profiles of the respective experimental isomer intermediates.
(I) The modeled activities of MAN1B1, MAN1A1, and MAN1A2 for 30 min digestion of Man9-PA by RLG (G) and MGF (H) fractions were used to display changes in relative enzyme activities in the two membrane fractions. It was assumed that MAN1B1 activity did not change between the two membrane fractions, and the activities of MAN1A1 and MAN1A2 were then plotted relative to MAN1B1 to determine their respective changes between RLG and MGF.
We then confirmed that the reduced MAN1 activity in mitotic Golgi membranes was due to phosphorylation, as dephosphorylation of MAN1A1 with CIP (Figure 2D) recovered MAN1 activity, whereas inhibition of CIP by β-glycerophosphate failed to do so (Figure 3E). Reversely, phosphorylation of MAN1A1 by purified CDK1 reduced MAN1 activity (Figure 3F). In addition, MAN1 activity was not affected by swainsonine, a MAN2 inhibitor (Figure S3I), indicating high specificity of the assay. Taken together, mitotic phosphorylation inhibits MAN1A1 activity.
MAN1A1 phosphorylation affects the production of different isomers of glycans
When examining the peaks of the different oligosaccharide products in our MAN1 activity assay, we noticed that mitotic phosphorylation affected not only the levels of the peaks but also the sub-peaks that represent different isomers within the same length oligosaccharides, such as Man8, Man7, and Man6 (Figure 3B). There are three Man8 isomers that can be generated following mannosidase digestion, Man8A, Man8B, and Man8C, depending on which branch is trimmed on Man9 (Xiang et al., 2016). Similarly, four Man7 isomers and three Man6 isomers can be produced during trimming to the Man5 processing intermediate (Figure 4A) (Lal et al., 1998). These isomers were detected as sub-peaks on the HPLC profiles (Figure 3B), and the changes in the sub-peaks indicate that mitotic phosphorylation may affect MAN1A1 activity differently on different branches. Therefore, we further analyzed the glycan isomers by dual-gradient, reversed-phase HPLC (Figures 4B–4D). At 30 min, while interphase Golgi produced mainly Man8A and a lesser amount of Man8B (54.2% and 30.8% of total Man8, respectively, calculated on the basis of the results shown in Figure S4A), as previously reported (Lal et al., 1998), mitotic Golgi produced Man8A and Man8B in the opposite ratio (27.7% Man8A and 59.9% Man8B) (Figure S4A). Similar results were obtained at 60 min incubation (Figures S4A and S4B). When considering all products, interphase Golgi exhibited more activity to trim the A- and C-branches, which were inhibited by mitotic phosphorylation (Figure 4E). These results demonstrate that MAN1A1 phosphorylation affects the production of different isomers of glycans.
Our previous quantitative proteomic studies demonstrated that within the 4 forms of MAN1 (MAN1A1, MAN1A2, MAN1B1, and MAN1C1), MAN1A1 is the most abundant in purified Golgi membranes, with the ratio of MAN1A1, MAN1A2 and MAN1B1 approximately 2:1:1 (to be more accurate, 27:14:12) according to the peptides detected, whereas MAN1C1 was not detected and so might have a low abundance in Golgi membranes (Chen et al., 2010). Of note, MAN1B1, which exclusively produces the Man8B isomer in the ER and Golgi, is also found in the isolated Golgi membrane fractions. Although the abundance of the MAN1A1, MAN1A2, and MAN1B1 proteins does not change during mitosis (Figures 1C and 12; also see Figure 7) (Chen et al., 2010), the differences in glycan isomer abundance during in vitro digestions of Man9 between RLG and MGF membranes indicate that the respective enzyme activities are altered during mitosis.
Figure 7. S12 phosphorylation impairs MAN1A1 interaction with MGAT1.
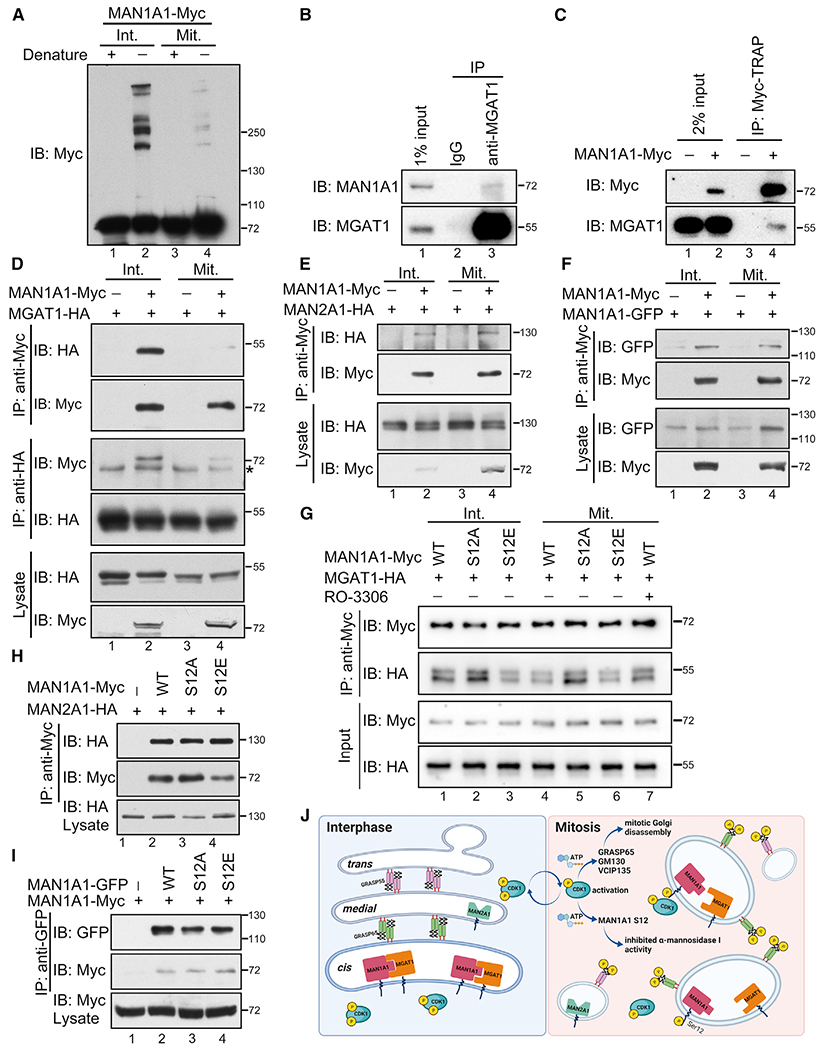
(A) MAN1A1 forms large protein complexes in interphase but not mitotic cells. Interphase (Int.) and mitotic (Mit.) cells expressing MAN1A1-myc were analyzed using western blot with or without denaturing. Note the high molecular weight bands of MAN1A1 in interphase cells without denaturing (lane 2).
(B) Endogenous MAN1A1 interacts with MGAT1. HeLa cells were lysed and immunoprecipitated with rabbit IgG or a MGAT1 antibody as indicated followed by western blot of both proteins.
(C) Endogenous MGAT1 interacts with MAN1A1-myc.
(D) MAN1A1 interacts with MGAT1 in interphase but not mitotic cells. *IgG heavy chain.
(E) MAN1A1 interacts with MAN2A1 in both interphase and mitotic cells.
(F) MAN1A1 self-interacts in both interphase and mitotic cells.
(G) CDK1-mediated mitotic S12 phosphorylation attenuates MAN1A1-MGAT1 interaction. RO-3306, 10 μM for 30 min.
(H) S12 mutation of MAN1A1 does not affect MAN1A1-MAN2A1 interaction in interphase.
(I) S12 mutation of MAN1A1 does not affect MAN1A1 self-interaction in interphase.
(J) A schematic model showing the mitotic regulation of Golgi morphology and MAN1A1 activity by CDK1. Upon mitotic activation, CDK1 phosphorylates Golgi structural and fusion proteins, such as GRASP65, GM130, and VCIP135, leading to mitotic Golgi disassembly. Meanwhile, CDK1 phosphorylates MAN1A1 at S12, which inhibits MAN1A1 interaction with MGAT1 and reduces its α-mannosidase activity.
Previous studies have determined the respective substrate specificities and isomer profiles for MAN1B1, MAN1A1, and MAN1A2 in their cleavage of Man9 to Man5 (Karaveg and Moremen, 2005; Tempel et al., 2004; Vallee et al., 2000). Thus, the relative ratios of isomer intermediates generated during glycan digestion could be predicted and modeled (Figure 4F) on the basis of the known specificities of the enzymes. Time course studies on digestion of the Man9 substrate were performed in triplicate for both RLG and MGF membrane preparations and two time points (30 and 60 min) were analyzed (Figures 4G and 4H). A simple modeling of the Man8, Man7, and Man6 isomer profiles was then performed using the known specificities of MAN1B1, MAN1A1, and MAN1A2, and their relative contributions to the overall glycan digestion and isomer ratios were varied to best replicate the experimental data.
Modeling of the 30 min time point for RLG suggests a ratio of MAN1B1 (38.6%), MAN1A1 (44.3%), and MAN1A2 (17%) activities would produce an isomer profile that most closely reflects the isomer profile of the experimental data (Figure 4G). By comparison, modeling of the enzyme activities for the MGF sample would suggest that a ratio of MAN1B1 (59.6%), MAN1A1 (22.8%), and MAN1A2 (17.5%) would produce an isomer ratio pattern that most effectively fit the experimental profile data (Figure 4H). If one assumes that MAN1B1 activity remains unchanged between the two samples, comparison of the RLG and MGF modeled enzyme ratios would suggest that there was an overall reduction in MAN1A1 activity by ~3-fold in the MGF fraction (Figure 4I). A similar analysis of the 60 min glycan digestion time point suggests an enzyme ratio of MAN1B1 (32.5%), MAN1A1 (62.5%), and MAN1A2 (5.0%) for the RLG sample and a ~5.6-fold decrease in MAN1A1 activity for the MGF sample (Figures S4C–S4E). Thus, our modeling data suggests that mitotic phosphorylation significantly attenuates MAN1A1 enzymatic activity to trim A- and C-branches of the oligosaccharide.
MAN1A1 phosphorylation alters protein glycosylation in cells
To determine the functional consequence of S12 phosphorylation on MAN1A1 activity in cells, we took advantage of a previously established MAN1-deficient Chinese hamster ovary (CHO) cell line in which all four forms of MAN1 (MAN1A1/1A2/1B1/1C1, referred to as MAN1 QKO in this study) were knocked out by CRISPR-Cas9-mediated genome editing (Lamriben et al., 2018). Following a previously established protocol (Jin et al., 2018), we stained non-permeabilized cells with Phaseolus vulgaris leucoagglutinin (PHA-L), a lectin that recognizes complex-type glycans. The QKO cell line exhibited defective N-glycosylation as indicated by the lower PHA-L signal compared with WT cells (Figure 5A). Expression of WT MAN1A1, or its phosphorylation-deficient mutant S12A, effectively rescued the glycosylation defect as indicated by the increased PHA-L signals (Figure 5B). In contrast, expression of the phosphomimetic mutant S12E failed to fully rescue the glycosylation defect (Figure 5B). Quantitation of the PHA-L intensity confirmed that S12E had significantly lower activity than WT and S12A in recovering PHA-L signals in interphase QKO cells (Figure 5C), although the three forms were expressed at a comparable level (Figure 5D). These results were confirmed by flow cytometry (Figures 5E–5G). In conclusion, S12E had less activity than WT MAN1A1 and S12A in rescuing the glycosylation defects in MAN1 QKO cells.
Figure 5. MAN1A1 phosphorylation reduces complex glycosylation in interphase cells.
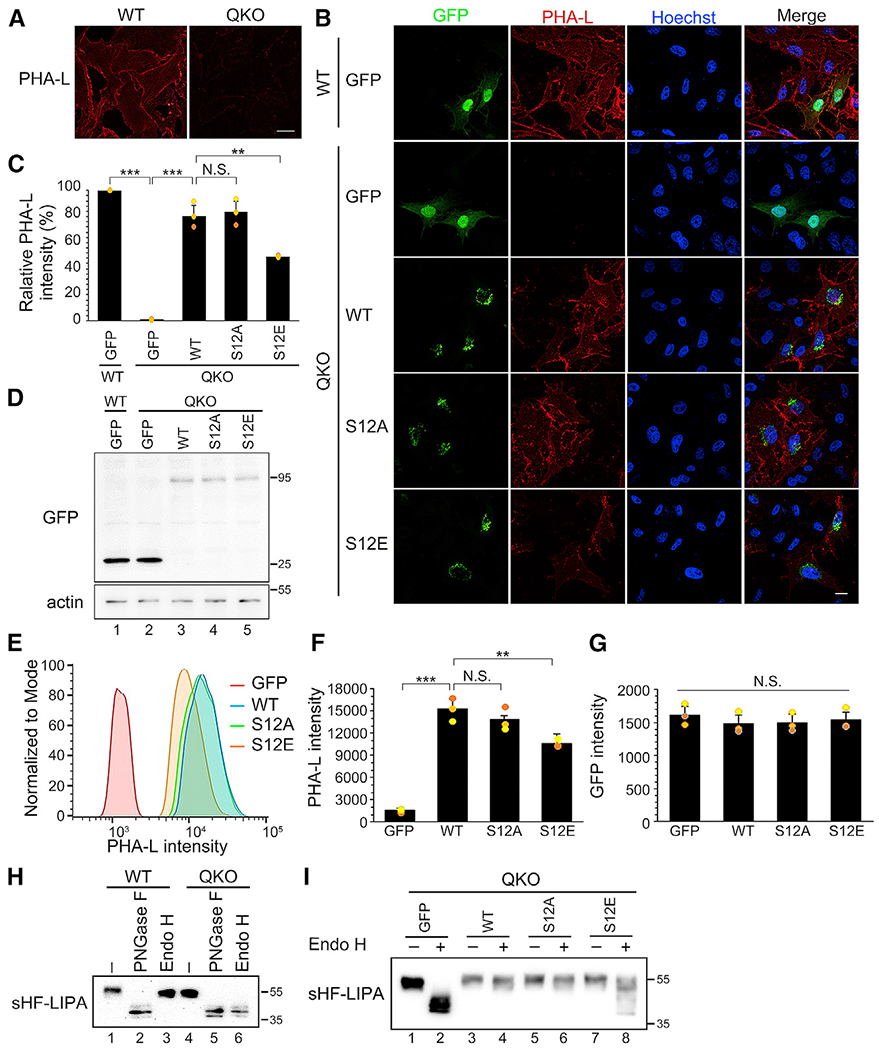
(A) Protein glycosylation is defective in MAN1 QKO cells. PHA-L was used to detect complex glycans. Scale bar, 20 μm.
(B) Expression of the phosphomimetic mutant of MAN1A1 (S12E) fails to fully rescue the glycosylation defects in MAN1 QKO cells. Scale bar, 20 μm.
(C) Quantification of the cell surface PHA-L intensity in (B). Results are expressed as mean ± SD from three independent experiments. The p value was determined using one-way ANOVA with post hoc Tukey honestly significant difference (HSD) test. N.S., not significant. **p < 0.01 and ***p < 0.001.
(D) MAN1A1 and its S12 mutants are expressed at a comparable level analyzed using western blot.
(E) Flow cytometry analysis of cell surface PHA-L intensity in MAN1 QKO cells expressing MAN1A1 WT or mutants.
(F) Quantification of the PHA-L intensity in (E). Results are expressed as mean ± SD from three independent experiments. The p value was determined using one-way ANOVA with post hoc Tukey HSD test. N.S., not significant. **p < 0.01 and ***p < 0.001.
(G) Quantification of the GFP intensity in (E).
(H) MAN1 QKO cells are defective in producing complex-type sugars assessed using sHF-LIPA as a reporter protein.
(I) Expression of WT MAN1A1, but not its phosphomimetic mutant S12E, rescues sHF-LIPA glycosylation defects in MAN1 QKO cells.
To further validate the results observed above, we expressed a reporter glycoprotein His6-FLAG tagged lysosomal acid lipase (sHF-LIPA, where sHF refers to an ER signal sequence with a His6-FLAG-tag) in WT and QKO cells, collected secreted sHF-LIPA in conditioned media with a nickel column, treated it with PNGase F, which removes all N-linked polysaccharides, or with endoglycosidase H (Endo H), which cleaves only high-mannose N-linked glycans, and analyzed its glycosylation by band-shift on western blot. As shown in Figure 5H, sHF-LIPA secreted by WT but not MAN1 QKO cells was resistant to Endo H treatment (lane 6 versus 3). Expression of WT MAN1A1, or its S12A mutant, rescued its glycosylation defect (Figure 5I, lanes 4 and 6). In contrast, the S12E mutant failed to fully rescue sHF-LIPA glycosylation, indicated by a mixture of Endo H-sensitive and Endo H-resistant forms of sHF-LIPA (Figure 5I, lane 8).
The phosphorylation-deficient mutant of MAN1A1 remains active in mitosis
On the basis of the results supporting the inhibitory effect of MAN1A1 mitotic phosphorylation, one hypothesis can be raised that expression of the phospho-deficient mutant S12A would lead to aberrant accumulation of complex glycans in mitosis. However, it is impossible to show that MAN1A1 S12A is active in mitosis by cell surface lectin staining of mitotic cells because MAN1A1 substrates in the Golgi cannot be delivered to the cell surface because of the mitotic trafficking block. To overcome this difficulty, we performed live cell imaging to capture glycosylation changes at the cell surface upon the arrival of cargo proteins at the onset of mitotic exit when membrane trafficking resumes. Briefly, we transfected MAN1 QKO cells with GFP-tagged MAN1A1 WT, S12A, or S12E for 6 h to allow expression of MAN1A1 and then blocked cells to prometaphase by nocodazole treatment. During this time, MAN1A1 and its mutants were expressed. If active, they would modify the accessible substrates in the Golgi. Upon nocodazole washout, cells exit mitosis and membrane trafficking resumes, allowing cargo molecules in the Golgi to be transported to the cell surface. To monitor the glycosylation status of this population of cargo molecules, we added fluorescently labeled PHA-L into the cell culture medium and performed live cell imaging. It would be readily recruited to the cell surface upon the arrival of cargo proteins with complex glycans and thereby can use be used to indicate the glycosylation states of these proteins. Post-mitotic Golgi reassembly was monitored by the GFP signal tagged to MAN1A1. The results showed that cells expressing S12A exhibited more rapid accumulation of complex glycosylated proteins at the cell surface than WT MAN1A1, whereas S12E-expressing cells displayed significantly reduced PHA-L signal at the cell surface throughout the entire recording time (Figure 6; Videos S1, S2, S3, and S4). These results demonstrate that the S12A mutant remains highly active in mitosis, supporting the conclusion that mitotic phosphorylation inhibits MAN1A1 activity.
Figure 6. The S12A phospho-deficient mutant of MAN1A1 remains highly active in mitosis.
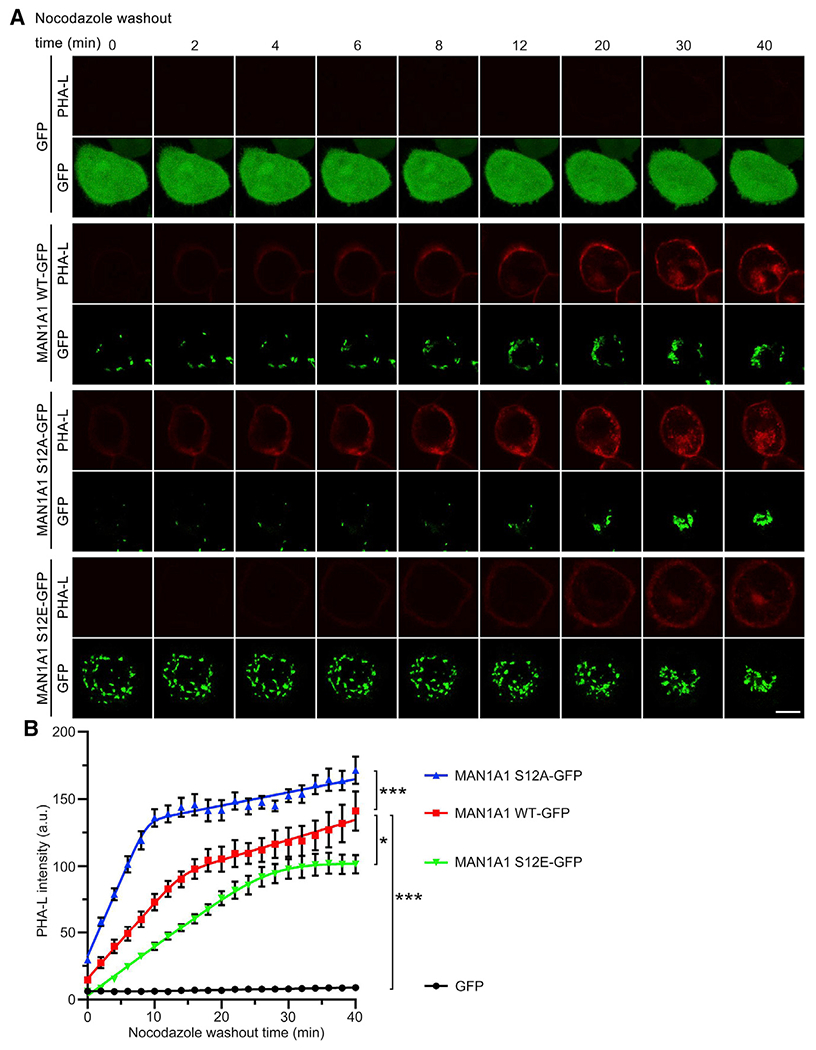
(A) Expression of the S12A mutant of MAN1A1 in mitosis increases the formation of complex glycans. Shown are still images of chosen time points from live cell imaging (Videos S1, S2, S3, and S4). Note that the red signal (PHA-L) in cells expressing S12A appears earlier than that in WT MAN1A1-expressing cells, whereas this signal in S12E expressing cells was comparatively lower. Scale bar, 10 μm.
(B) Quantification of cell surface PHA-L intensity in (A). Results are expressed as mean ± SEM from five cells. The p value was determined using one-way ANOVA with post hoc Tukey HSD test. *p < 0.05 and ***p < 0.001.
S12 phosphorylation impairs MAN1A1 interaction with MGAT1
One critical property of Golgi glycosylation enzymes is to form large protein complexes with themselves or with subsequent enzymes, which has been known to enhance their enzymatic activity (Seko and Yamashita, 2008; Hassinen et al., 2011). Indeed, when cell lysates were analyzed without denaturation, MAN1A1 exhibited high molecular bands in interphase but not mitotic cells (Figure 7A). To understand the nature of these large protein complexes, we determined MAN1A1 interaction with its immediate downstream enzymes MGAT1 and MAN2A1, as well as MAN1A1 itself, in interphase and mitotic cells by co-immunoprecipitation. The results showed that MAN1A1 and MGAT1 co-immunoprecipitated with each other in interphase cells regardless of which protein was used as the bait (Figures 7B–7D), while the interaction was dramatically reduced in mitosis (Figure 7D). Moreover, MAN1A1 also weakly interacted with MAN2A1 and MAN1A1 itself, but these interactions were not regulated in the cell cycle (Figures 7E and 7F). These results demonstrate that MAN1A1 interacts with MGAT1 in interphase cells, and the interaction is inhibited in mitosis, likely because of phosphorylation of MAN1A1.
We then tested whether S12 mutation of MAN1A1 affects its interaction with MGAT1. WT MAN1A1 and S12A strongly interacted with MGAT1 in interphase cells, while S12E mutation reduced this interaction (Figure 7G, lane 3 versus 1 and 2). Consistently, S12A and MGAT1 interaction remained strong in mitosis (Figure 7G, lane 5), while inhibition of CDK1 with RO-3306 in mitotic cells significantly increased the interaction between WT MAN1A1 and MGAT1 (Figure 7G, lane 7 versus 4). On the other hand, MAN1A1 interaction with MAN2A1 or MAN1A1 itself was not affected by S12 mutation (Figures 7H and 7I). Moreover, all proteins used in this study were correctly targeted to the Golgi and their expression did not affect the Golgi structure (Figure S5). S12 mutation also did not affect the stability of MAN1A1 protein (Figure S6). In conclusion, MAN1A1 phosphorylation at S12 by CDK1 in mitosis suppresses its mannosidase activity likely by disrupting its interaction with MGAT1.
DISCUSSION
In this study, we show that glycosylation is regulated by cytosolic signaling during the cell cycle. Given that CDK1 also regulates Golgi membrane dynamics by phosphorylating Golgi structure proteins, our results indicate that both Golgi structure and function (i.e., glycosylation) are coordinately regulated by the same cytosolic signaling pathway (e.g., CDK1), at least during mitosis (Figure 7J). As MAN1 is the first glycan-processing enzyme that cargo molecules encounter when reaching the Golgi, this provides a possible mechanism by which cells avoid glycosylation defects during mitosis when cargo molecules and glycosylation enzymes are trapped together in the mitotic Golgi fragments. Interestingly, MAN1A1 phosphorylation not only controls MAN1A1 activity in mitosis but also affects the production of oligosaccharide isomers (Figure 4), which could be explained by the composition and specificity of different Golgi enzymes toward different branches on the oligosaccharide. So far, it is unclear whether Golgi structural defects observed in human diseases affect MAN1 activity as well.
It is not surprising that MAN1A1 is phosphorylated by CDK1 at an evolutionally conserved site. CDK1 is a master kinase that drives cell cycle progression. It triggers mitotic Golgi fragmentation by phosphorylating and inhibiting several Golgi structure proteins, membrane tethers, and membrane fusion proteins such as GRASP65, GM130, and VCIP135 (Wang and Seemann, 2011; Zhang and Wang, 2015; Zhang et al., 2014). Similarly, CDK1 phosphorylates nuclear lamin to induce mitotic disassembly of the nuclear envelope (Guttinger et al., 2009). These results together reveal an essential role for CDK1 in regulating both the organization and function of membrane organelles in addition to cell cycle control.
MAN1A1 is a type II transmembrane protein; the phosphorylation site S12 at the cytosolic domain is rather far from the catalytic domain in the Golgi lumen. Thus, the finding that S12 phosphorylation inhibits MAN1A1 enzymatic activity is unexpected. Microscopy analysis excluded the possibility that S12 phosphorylation affects MAN1A1 localization or turnover. How S12 phosphorylation reduces MAN1A1 activity at the molecular level remains unknown, but likely through the disruption of complex formation. First, MAN1A1 forms large protein complexes in interphase but not mitosis (Figure 7A). Second, MAN1A1 interacts with MGAT1 in interphase when MAN1A1 is dephosphorylated (Figure 7), consistent with previous studies that homo- and hetero-oligomerization of Golgi enzymes enhances their activities (Seko and Yamashita, 2008; Hassinen et al., 2011). Third, it is possible that the large protein complexes seen in interphase are consisted of multiple proteins including MAN1A1 and MGAT1. If so, more proteins may be affected in mitosis. Notably, treatment of MGF with CIP in Figure 3E or RLG with purified CDK1 in Figure 3F did not fully reverse MAN1 activity to the level in RLG or MGF, respectively. One likely explanation is that CIP or CDK1 treatment could only reverse the phosphorylation state of MAN1A1 but not the organization of the Golgi membranes because of the lack of membrane budding and fusion machineries in these in vitro reactions and thus MAN1A1-MGAT1 interaction. Future studies are needed to confirm that the reduced MAN1A1-MGAT1 interaction is the cause of the reduced MAN1A1 activity in mitosis.
The concept that Golgi glycosylation enzymes form complexes emerged years ago (Nilsson et al., 1993, 2009). Several enzyme complexes, such as the β1,3-N-acetylglucosaminyltransferase-2 (B3GNT2)-B3GNT8 complex (Seko and Yamashita, 2008) and the β-1,4-galactosyltransferase (B4GALT1)-β-galactoside α-2,6-sialyltransferase 1 (ST6GALI) complex (Hassinen et al., 2011), have been revealed to have higher activity than either of the enzymes alone. In our study, MGAT1 is an immediate downstream enzyme of MAN1 in N-glycan processing and is also the first enzyme in the Golgi that extends sugar residues to N-glycans. It is therefore reasonable to speculate that MAN1A1-MGAT1 interaction may facilitate the next step of N-glycan processing by MGAT1 following the trimming of high-mannose substrates by MAN1.
Limitations of the study
One major limitation of our study is that it does not provide a molecular mechanism of how mitotic phosphorylation at a cytosolic serine residue regulates the enzymatic activity conferred by the luminal catalytic domain. A structural analysis of the MAN1A1 may provide mechanistic insights into the conformational changes for substrates affinity in the catalytic pocket. N-glycan processing is complex and sequential, and the four MAN1 isoforms (A1, A2, B1, and C1) play similar roles in removing mannose residues from core oligosaccharides. This increases the difficulty of pinpointing the phenotype when only MAN1A1 activity is suppressed. Although we took advantage of our knowledge of the abundance of each isoform in the Golgi to calculate the activities, the results were estimates rather than actual measurements of the enzymatic activity of a single protein. In the future it may be helpful to conduct high throughput glycomic analysis in a series of cell lines in which different glycosylation enzymes are sequentially knocked out. Last, some protein-protein interactions were tested using exogenously expressed proteins because of the lack of proper antibodies, and our results do not establish a cause-and-effect relationship between MAN1A1-MGAT1 interaction and MAN1A1 activity. The best way to address this question is to map the interaction sites between MAN1A1 and MAGT1, interrupt their interactions by point mutations, and determine the effects on MAN1A1 activity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yanzhuang Wang (yzwang@umich.edu).
Materials availability
Plasmids and other reagents generated in the study will be available from the lead contact upon request with a completed Material Transfer Agreement (MTA).
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Wild type (WT) parental and MAN1 quadrant knockout CHO (QKO MAN1A1/1A2/1B1/1C1) cells were kindly gifted by Drs. Daniel N. Hebert (University of Massachusetts) and Henrik Clausen (University of Copenhagen, Denmark) (Lamriben et al., 2018). CHO cells were maintained in Gibco Minimum Essential Medium Alpha medium (ThermoFisher) supplemented with 10% Iron-supplemented bovine calf serum (Cytiva HyClone). HeLa cells (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (ThermoFisher) with 10% Iron-supplemented bovine calf serum. Cells were incubated at 37°C in 5% CO2. All cells used in this study were tested negative for mycoplasma.
METHOD DETAILS
Reagents, antibodies, and plasmids
All reagents were purchased from Sigma-Aldrich (St. Louis, MO), ThermoFisher Scientific (Waltham, MA), and Roche (Mannheim, Germany) unless otherwise specified. Roscovitine was purchased from Selleck (S1153), Staurosporine (81590) and RO-3306 (15149) were purchased from Cayman Chemical (Ann Arbor, MI). Myc-Trap Magnetic Agarose was purchased from ChromoTek (Islandia, NY; ytma-20).
The following antibodies were used: monoclonal antibodies against cyclin B1 (BD Biosciences, 554176), Flag (Sigma-Aldrich, M1804), GFP (Proteintech, 66002-1-Ig), HA (Covance, 16B12), and Myc (The University of Michigan Hybridoma Core Facility, 9E10); polyclonal antibodies against human GRASP65 (Joachim Seemann, UT Southwestern, Dallas, TX), human MAN1A1 (Sigma, M3694; Abcam, ab140613), human MGAT1 (Abcam, ab180578), phosphoserine (Zymed Technologies, 61–8100) and phospho-threonine (Zymed Technologies, 71–8200).
MAN1A1-myc mutants were generated by site-directed mutagenesis based on the template of full-length WT MAN1A1 construct (Vallee et al., 2000). MAN1A1-GFP constructs are cloned by restriction enzyme digestion of MAN1A1 from pcDNA3.1-myc-MAN1A1 and inserted into pEGFP-N1 vector. pcDNA3.1-MGAT1-HA coding full-length MGAT1 and pcDNA3.1-MAN2A1-HA coding full-length MAN2A1 were kindly gifted by Dr. Pamela Stanley (Albert Einstein College of Medicine) (Huang et al., 2015). pHEK293Ultra-sHF-LIPA coding mature LIPA was described previously (Jin et al., 2018). All new constructs generated in this study were confirmed by DNA sequencing. Protein sequence alignment was performed using the Clustal Omega Multiple Sequence Alignment program.
Cell transfection, treatment, and synchronization
For transfection, HeLa cells were transfected using Polyethylenimine (25 kDa linear PEI, Polysciences, Inc.), and CHO cells were transfected with Lipofectamine 2000 or 3000 (ThermoFisher) following manufacturer’s instructions.
To enrich mitotic cells, HeLa cells were synchronized to mitosis by 100 ng/mL nocodazole treatment for 18 h (Tang et al., 2010; Jackman and O’Connor, 1998). For kinase inhibitor treatment, nocodazole-arrested mitotic cells were treated with 10 μM RO-3306 for 10, 20, or 30 min, 100 μM roscovitine for 1 h, or 50 nM staurosporine for 1 h. Cells were harvested and subjected to Western blot. For cycloheximide (CHX) treatment, 100 ng/mL CHX was added into growth medium for indicated times (0–4 h) before sample collection.
Preparation of interphase and mitotic golgi membranes
Rat liver Golgi membranes and HeLa cell cytosol were prepared as previously reported (Tang et al., 2010). To prepare mitotic Golgi fragments (MGF), 3 μg purified rat liver Golgi (RLG) membranes were incubated with 300 μg mitotic HeLa cell cytosol and an ATP-regenerating system (10 mM creatine phosphate, 0.1 mM ATP, 20 μg/mL creatine kinase, 20 μg/mL cytochalasin B) in MEB buffer (50 mM Tris-HCl, pH 7.4, 0.2 M sucrose, 50 mM KCl, 20 mM β-glycerophosphate, 15 mM EGTA, 10 mM MgCl2, 2 mM ATP, 1 mM GTP, 1 mM glutathione, and protease inhibitors) at 37°C for 1 h. Mitotic Golgi fragments in each reaction were isolated and soluble proteins were removed by centrifugation (136,000 g for 60 min in a TLA55 rotor) through a 0.4 M sucrose cushion in MEB buffer onto a 6 μL 2 M sucrose cushion (Tang et al., 2010). To prepare interphase Golgi membrane, 3 μg RLG membranes were incubated with 300 μg interphase HeLa cell cytosol and an ATP-regenerating system in KHM buffer (20 mM HEPES-KOH, pH 7.0, 0.2 M sucrose, 60 mM KCl, 5 mM Mg(OAc)2, 2 mM ATP, 1 mM GTP, 1 mM glutathione, and protease inhibitors) and then pelleted by ultra-centrifugation as described above. Treatment of Golgi membranes with CDK1, CIP, or β-glycerophosphate were performed as previously reported (Wang et al., 2003).
For CDK1 treatment of Golgi membranes, a 5 μg aliquot of RLG was mixed with indicated amount of purified cyclin B1/CDK1 protein complex in the presence of 5 μM ATP at 37°C for 60 min. For CIP treatment of Golgi membranes, a 5 μg aliquot of MGFs was treated with 20 U of CIP at 37°C for 60 min with or without 50 mM β-glycerophosphate. The membranes were pelleted through a 0.4 M sucrose cushion by centrifugation at 55,000 r.p.m. (186,000 x g) for 30 min in a TLA55 rotor (Beckman) (Wang et al., 2003; Tang et al., 2008, 2010).
Mass spectrometry and phosphoproteomic analysis
Mass spectrometry and phosphoproteomic analysis were done as previously described (Kweon and Andrews, 2013). Briefly, interphase and mitotic Golgi membranes prepared above were lysed in lysis buffer in the presence of protease and phosphatase inhibitors. Equal amounts of interphase and mitotic Golgi proteins were digested with trypsin after dithiothreitol (DTT) reduction and IAA alkylation. Phosphopeptides were enriched from interphase and mitotic Golgi peptides using two sequential IMAC enrichments and subsequent ZrO2 enrichment. Enriched fractions derived from three sequential enrichment steps were analyzed by reverse-phase LC-MS/MS using an Orbitrap Fusion Tribrid mass spectrometer (ThermoFisher).
For peptide and protein identification, MS/MS data were analyzed by MaxQuant (version 1.5) matching them against protein database (UniProt rat proteome). Mass tolerance of 2.5 ppm was allowed for the precursor ion search. Up to two miscleavages were allowed for trypsin digestion. Variable modifications of phosphorylation (STY), oxidation (M), and acetylation (N-term), and static modification of carbamidomethyl (C) were allowed. Peptides and proteins were identified by applying 1% FDR filter.
Preparation of oligosaccharide substrates
The pyridylaminated Man9GlcNAc2 (Man9-PA) was prepared as follows: mns1 Δ mnn1 Δ och1 Δ strain [MATa mnn1 Δ:kanMX4 mns1-Δ:hphNT2 och1 Δ:His3MX6 leu2 Δ0 met15 Δ0 ura3 Δ0 (BY4741 background)] was constructed by one-step PCR method for gene disruption (Longtine et al., 1998). Yeast cells were grown in YPD medium (1% yeast extract, 2% peptone, 2% glucose) with osmotic stabilizer (250 mM KCl). All incubations were performed at 25°C unless noted otherwise. Yeast cells (1000 mL culture) were collected and suspended in 50 mL of 10 mM citrate buffer (pH 7.0) and lysed by autoclaving at 121 °C for 120 min. Three volumes of cold ethanol were added to the supernatant to precipitate the proteins. The precipitates thus obtained were subjected to trypsin digestion (Thermo), followed by incubation with PNGase F (Roche) to release N-linked glycans. For desalting, released N-glycans were applied onto AG1-X2 (200–400 mesh, acetate form) and AG50-X8 (200–400 mesh, H+ form) (Bio-Rad) columns, and the flow-through fraction was loaded onto a graphitized carbon column (InertSep GC; GL Science, Tokyo, Japan). After washing the column twice with 3 mL of water, N-glycans were eluted with 25% acetonitrile (v/v) and subjected to 2-aminopyridylation (PA-labeling) as described previously (Hirayama et al., 2010) with some modifications. Desalted Man9 glycans were reacted with 20 μL PA reagent solution (552 mg of 2-aminopyridine dissolved in 200 μL of acetic acid) at 80°C for 1 h, and then reduced with 20 μL reducing reagent solution (20 mg dimethylamine-borane in 40 μL acetic acid) at 80°C for 1 h. After adding 10 μL water and 450 μL acetonitrile to the reaction mixture, excess free PA was removed using a monolithic silica spin column (MonoFas® spin column, GL-Science, 501021451). The spin column was first washed once with 200 μL water and then three times with 200 μL acetonitrile. After loading and passing the sample solution through the spin column, the column was washed twice with 400 μL acetonitrile and twice with 650 μL 95% acetonitrile (v/v). The PA-oligosaccharides were then eluted from the column with 200 μL water.
PA-labeled GlcNAc-Man5-GlcNAc2 (GN1M5-PA) was prepared from Gal-GlcNAc-Man5-GlcNAc2-PA (Masuda Chemical Industries Co., Ltd., Kagawa, Japan) by incubation with β-galactosidase in 50 mM sodium citrate buffer (pH 5.5) for 16 h at 37°C. The reaction was terminated by heat denaturation at 100°C for 5 min. Three volumes of cold ethanol were added to the reaction mixtures, followed by centrifugation at 15,000 g for 15 min at 4°C to remove proteins. The supernatant was dried in vacuo and dissolved in water. PA-labeled glycans were analyzed by HPLC as described below.
Measurement of mannosidase I and mannosidase II activities in golgi membranes
10 μg of Golgi membranes were resuspended in 20 μL of reaction buffer (50 mM MES-NaOH, pH 6, 0.1% Triton X-100, 1 mM CaCl2, 10 μM swainsonine, 5 pmol of Man9-PAasthe substrate) and incubated for the indicated times at 37°C. Reactions were incubated for 1 h, unless otherwise indicated. The reaction was terminated by heating at 100°C for 5 min, and proteins were removed by 75% (v/v) ethanol precipitation. The supernatants were analyzed by size fractionation HPLC. In detail, PA-glycans were separated by size fractionation HPLC using a Shodex NH2P-40 3E column (3.0 × 250 mm; Shodex, Tokyo, Japan) (Hirayama et al., 2010), with some modifications. The elution was performed using two solvent gradients: solvent A, 93% acetonitrile in 0.3% acetate (pH adjusted to 7.0 with ammonia), and solvent B, 20% acetonitrile in 0.3% acetate (pH adjusted to 7.0 with ammonia). The flow rate was set to 450 μL/min, and the column temperature was 25°C. The gradient program was as follows: 0–0.5 min, 1–10% solvent B; 0.5–3 min, 10–24.5% solvent B; 3–33 min, 24.5–55% solvent B; 33–35 min, isocratic 70% solvent B. PA-oligosaccharides were detected by measuring fluorescence (excitation wavelength, 310 nm; emission wavelength, 380 nm).
For isomer structure analysis, double volume of Golgi membrane and reaction mixture was used, and the reaction products were fractionated by size fractionation HPLC. The isomer structures were analyzed using dual-gradient, reversed-phase HPLC with an Inertsil ODS-3 column (2.1 × 150 mm; GL Sciences) (Suzuki et al., 2008). The elution was achieved by two solvent gradients: solvent A (0.1 M ammonium acetate buffer, pH 6.4) and solvent B (0.1 M ammonium acetate buffer, pH 4.0, and 0.5% 1-butanol). The flow rate was 200 μL/min, and the column temperature was 25°C. The gradient program was as follows: 0–10 min, isocratic 99% solvent A; 10–110 min, 99–30% solvent A; 110–150 min, isocratic 99% solvent A. Fluorescence of the labeled glycans was detected at the excitation wavelength (320 nm) and the emission wavelength (400 nm).
For enzyme assay of α-mannosidase II in Golgi membranes, 10 μg of Golgi membranes were resuspended in 20 μL of reaction buffer (50 mM MES-NaOH, pH 6, 0.1% Triton X-100, 1 mM CaCl2, 5 pmol of GN1M5-PA as a substrate), and incubated for the indicated times at 37°C. Trimmed mannose index was calculated by the sum of GN1M4-PA percentage × 1 and GN1M3-PA percentage × 2 to reflect the activity toward the final product.
Modeling of mannosidase I activities in golgi membrane preparations
The proteomics studies from the rat liver Golgi membrane fractions indicated that three of the four MAN1 isoforms, MAN1B1, MAN1A1 and MAN1A2, were present in the RLG and MGF membrane preparations, but the fourth isoform, MAN1C1, was absent (Chen et al., 2010). Since the substrate specificities of these three enzyme isoforms have been previously studied in detail (Lal et al., 1998; Xiang et al., 2016; Karaveg and Moremen, 2005; Tempel et al., 2004; Vallee et al., 2000), it is possible to create a matrix of enzyme isoform contributions to the time course digestion of the Man9-PA substrate to Man5-PA in the Golgi membrane preparations, including the formation of all isomer intermediates (Figure 4F). This matrix of possible isomer intermediates was used to estimate the contributions of each of the three MAN1 isoforms to generate a modeled dataset that most closely matched the isomer ratios obtained from the respective membrane preparations. An Excel spreadsheet was employed to create a modeling template and a simple manual adjustment of relative enzyme activities was performed to minimize the differences between the modeled data for isomer abundance and the experimental data in the RLG and MGF samples at both 30 min and 60 min time points. In order to assess the magnitude of the change in MAN1A1 activity, it was assumed that MAN1B1 activity was unchanged in the experiment and the data was subsequently scaled based on normalization of the modeled MAN1B1 abundance. The resulting normalized abundance for the MAN1 isoforms was then plotted to reveal the apparent reduction in MAN1A1 activity.
Western blot and phos-tag gels
For immunoblot, cells were scraped, pelleted, lysed in lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, protease and phosphatase inhibitors), and cleared by centrifugation at 20,000 g and 4°C for 10 min in a benchtop microcentrifuge. Cell lysates were boiled in SDS loading buffer (6×, 300 mM Tris-HCl, pH 6.8, 6% SDS, 0.06% Bromophenol Blue, 36% (v/v) glycerol, 12 mM DTT) and analyzed by SDS-PAGE and Western blot. For non-denaturing samples, cells were lysed with loading buffer without SDS (6×, 300 mM Tris-HCl 6.8, 0.06% Bromophenol Blue, 36% (v/v) glycerol, 12 mM DTT) and analyzed by SDS-PAGE without boiling. Phos-tag conjugated acrylamide was purchased from Wako Chemicals (304–93521), and gels containing phos-tag were prepared according to the manufacturer’s instructions.
Co-immunoprecipitation
HeLa cells were scraped and lysed in immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100 (0.5% for co-immunoprecipitation of endogenous proteins), protease and phosphatase inhibitors) for 20 min at 4°C and cleared by centrifugation at 20,000 g and 4°C for 20 min in a benchtop centrifuge. For co-immunoprecipitation of endogenous proteins, cell lysate was pre-cleaned by incubation with protein A beads for 1 h at 4°C with gentle agitation. Myc or HA antibody pre-loaded to protein G beads, MGAT1 antibody pre-loaded to protein A beads, or Myc-TRAP magnetic agarose prewashed with immunoprecipitation buffer, were added to the lysate and incubated overnight at 4°C with gentle rotation. After extensive washing with the immunoprecipitation buffer, proteins were eluted by heating at 95°C in 2× loading buffer and analyzed by SDS-PAGE and Western blot.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as previously described (Huang et al., 2016). Briefly, cells were rinsed with phosphate buffered saline (PBS), fixed with 4% (w/v) paraformaldehyde (PFA) for 15 min, and quenched with 50 mM NH4Cl for 10 min, followed by permeabilization with 0.2% (v/v) Triton X-100 in PBS for 10 min. Cells were then blocked with 1% (w/v) BSA Fraction V (Dot Scientific, DSA30075-100) for 1 h and incubated sequentially with a primary antibody and FITC- or TRITC-labeled secondary antibody diluted in 1% BSA in PBS (PBSB). DNA was stained with 1 μg/mL Hoechst 33342 (ThermoFisher). Coverslips were mounted on glass slides with Moviol and images were taken with a Zeiss Observer Z1 epifluorescence microscope with a 63× oil lens. For super-resolution microscopy, samples were prepared as previously described (Ireland et al., 2020). Briefly, Alexa Fluor 488-labeled secondary antibodies (ThermoFisher) were used. After washing, coverslips were mounted using ProLong Diamond antifade super-resolution imaging mountant (ThermoFisher). Super-resolution images were taken with a 100× oil lens on a Leica (Wetzlar, Germany) TCS SP8 STimulated Emission Depletion (STED) super-resolution microscope. Images were processed using the NIH ImageJ software. Brightness and contrast were adjusted linearly across all samples to clearly show the Golgi structure.
Lectin staining and microscopy
Rhodamine labeled Phaseolus Vulgaris Leucoagglutinin (PHA-L) was purchased from Vector Labs (RL-1112). Lectin staining was performed as previously described (Blackburn and Lupashin, 2016) with minor modifications. Briefly, cells were cultured as indicated to 70–80% confluency on collagen-coated coverslips. Cells were washed with PBS and fixed in 1% PFA for 15 min, quenched with 50 mM NH4Cl for 10 min, and washed with PBS, followed by a 30 min block with PBSB. The coverslips were incubated in PBSB containing 2 μg/mL PHA-L in a 4°C cold-room (kept in the dark during incubation) with gentle rocking for 30 min. Then, coverslips were stained with Hoechst 33342, washed with PBS three times, and mounted for imaging. The images were obtained using a Nikon NIS-Elements C confocal microscope with a 60× oil lens and Z-stacks at 0.3 μm intervals. Maximum intensity projections were processed and quantified using Nikon NIS-Elements analysis software. All images in the same experiment were captured and processed with the same setting.
Live cell imaging
MAN1 QKO CHO cells were seeded onto poly-lysine coated glass-bottom dishes at the density of 1.5 × 105 cells/mL on day 1. Cells were transfected with pEGFP-N1, pEGFP-N1-MAN1A1 WT, pEGFP-N1-MAN1A1 S12A, and pEGFP-N1-MAN1A1 S12E plasmids using Lipofectamine™ 3000 on day 2. Six hours after transfection, cells were incubated with fresh medium containing 150 ng/mL nocodazole for 18 h to block cells to prometaphase. Cells were then changed to imaging medium (ThermoFisher, 21063029) containing 150 ng/mL nocodazole, transferred to a live-cell imaging chamber at 37°C with 5% CO2 coupled to a Nikon ECLIPSE Ti2 Confocal microscope, and observed under a 60× oil objective to identify mitotic cells whose locations under the microscope were saved. Cells were then extensively washed and incubated with imaging medium containing 100 μg/mL CHX and 2 μg/mL Rhodamine-PHA-L (without nocodazole). Overall, nocodazole washout took about 3 min and a live cell imaging program was run immediately after it. Images were taken every 2 min with 5 stacks (0.8 μm/stack). Videos were processed with maximum intensity projection; time 0 is 5 min after nocodazole washout due to the time needed for the procedure and equipment setting. Quantifications were performed using the Nikon NIS-Elements AR analysis software to calculate the PHA-L intensity of 5 cells from one representative replicate of 3 independent experiments.
Flow cytometry
Cells were cultured as indicated to 70–80% confluency. Cells were detached with 20 mM EDTA, resuspended in PBS by gentle pipetting, and placed in an Eppendorf tube. Cells were then pelleted at 800 g for 3 min, fixed with 1% PFA for 15 min, quenched with 50 mM NH4Cl for 10 min, and washed with PBS, followed by a 30 min block with PBSB. Cells were pelleted, resuspended in PBSB containing the lectin of choice, and incubated at 4°C with gentle rotation for 30 min. Then, cells were washed and resuspended in PBS for flow cytometry analysis. The flow cytometry data were gained using the BD Fortessa cytometer at the University of Michigan flow cytometry core and analyzed using FlowJo software.
sHF-LIPA glycosylation assay
sHF-LIPA glycosylation assay was performed as previously described (Jin et al., 2018) with minor modifications. Briefly, WT and QKO CHO cells were co-transfected with sHF-LIPA and control pEGFP-N1 vector or pEGFP-N1-MAN1A1 constructs as indicated and cultured for 72 h. 10 mL of the conditioned medium was collected and spun at 1000 g for 3 min at 4°C to remove dead cells, followed by incubation with 100 μL of prewashed HisPurTM Ni-NTA Resin (Thermo, 88222) with gentle rotation at 4°C for 2 h. Ni-NTA Resin were washed with His-buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 2 mM DTT, 40 mM imidazole) and sHF-LIPA were eluted from the resin using 200 mM imidazole in pH 7.4 PBS. Eluted proteins were denatured by a 10× Endo H denaturing buffer (New England Biolabs) at 95°C for 5 min. Each denatured sample was divided equally to two parts, untreated or treated with 250 U Endo H or PNGase F (New England Biolabs) following the manufacturer’s instructions. Both parts were incubated on a 37°C shaker for 1 h. Reactions were terminated by adding a 6× SDS loading buffer and heating at 95°C for 3 min. Samples were then analyzed by SDS-PAGE and Western blot. sHF-LIPA was detected using a mouse monoclonal anti-FLAG antibody (Sigma-Aldrich, M1804, 1:2,000).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data represent the mean ± SD (standard deviation) of at least three independent experiments unless stated. A statistical analysis was conducted with one-way ANOVA with post-hoc Tukey HSD test or two-tailed Student t test. Quantitation results of live cell imaging in Figure 6B are expressed as mean ± SEM and the p value was determined by one-way ANOVA with post-hoc Tukey HSD test. Differences in means were considered statistically significant at p < 0.05. Significance levels are: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Flow cytometry data were analyzed using FlowJo software. Immunoblot and immunofluorescence analyses were performed using ImageJ or Nikon NIS-Elements analysis software. Figures were assembled with Adobe Photoshop CS6.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Cyclin B1 | BD Biosciences | Cat# 554176; RRID:AB_395287 |
| Mouse monoclonal anti-Flag (clone M2) | Sigma-Aldrich | Cat# M1804 |
| Mouse monoclonal anti-GFP (clone 1E10H7) | Proteintech | Cat# 66002-1-Ig; RRID:AB_11182611 |
| Mouse monoclonal anti-HA (clone 16B12) | Covance | Cat# MMS-101R; RRID:AB_291262 |
| Rabbit polyclonal anti-human GRASP65 | Joachim Seemann, UT Southwestern, Dallas, TX | N/A |
| Rabbit polyclonal anti-α1,2-Mannosidase 1A1 | Sigma-Aldrich | Cat# M3694; RRID:AB_477183 |
| Rabbit polyclonal anti-α1,2-Mannosidase 1A1 | Abcam | Cat#ab140613 |
| Mouse monoclonal anti-Myc (clone 9E10) | The University of Michigan Hybridoma Core Facility | N/A |
| Rabbit polyclonal anti-MGAT1 | Abcam | Cat# ab180578; RRID:AB_2800510 |
| Rabbit polyclonal anti-phospho-serine | Zymed Technologies | Cat# 61–8100 |
| Rabbit polyclonal anti-phospho-threonine | Zymed Technologies | Cat# 71–8200 |
| Chemicals, peptides, and recombinant proteins | ||
| Roscovitine | Selleck Chemicals | S1153; CAS: 186692-46-6 |
| Staurosporine | Cayman Chemical | 81590; CAS: 62996-74-1 |
| RO-3306 | Cayman Chemical | 15149; CAS: 872573-93-8 |
| Nocodazole | ThermoFisher | AC35824 |
| Gal-GlcNAc-Man5-GlcNAc2-PA | Masuda Chemical Industries Co., Ltd., Kagawa, Japan | PA-066 |
| Dimethylamine-Borane | Wako | 026–08402 |
| 2-Aminopyridine | Wako | 011–14181 |
| Cycloheximide | AG Scientific | C1189 |
| Rhodamine labeled Phaseolus Vulgaris Leucoagglutinin (PHA-L) | Vector Labs | RL-1112 |
| Phos-tag (TM) Acrylamide | Wako Chemicals | 304–93521 |
| Myc-Trap Magnetic Agarose | ChromoTek | ytma-20 |
| Lipofectamine 2000 | ThermoFisher | 11668027 |
| Lipofectamine 3000 | ThermoFisher | L3000015 |
| Polyethylenimine | Polysciences, Inc. | 23966-1; CAS: 9002-98-6, 26913-06-4 |
| DMEM high glucose, pyruvate | ThermoFisher | 11995073 |
| MEM α no nucleosides | ThermoFisher | 12561056 |
| DMEM, high glucose, HEPES, no phenol red | ThermoFisher | 21063029 |
| Penicillin-Streptomycin | ThermoFisher | 15140122 |
| Bovine calf serum | Hyclone | SH3007203 |
| Bacterial and virus strains | ||
| E. coli DH5α | ThermoFisher | 18265017 |
| Experimental models: Cell Lines | ||
| MAN1 quadrant knockout CHO (QKO MAN1A1/1A2/1B1/1C1) cells | (Lamriben et al., 2018) | N/A |
| Wild type (WT) parental CHO cells | (Lamriben et al., 2018) | N/A |
| HeLa | AATC | CRM-CCL-2™ |
| Recombinant DNA | ||
| pcDNA3.1-myc-MAN1A1 | Kelley W. Moremen, The University of Georgia | N/A |
| pcDNA3.1-myc-MAN1A1 S12A | This paper | N/A |
| pcDNA3.1-myc-MAN1A1 S12E | This paper | N/A |
| pEGFP-N1-MAN1A1 | This paper | N/A |
| pEGFP-N1-MAN1A1 S12A | This paper | N/A |
| pEGFP-N1-MAN1A1 S12E | This paper | N/A |
| pcDNA3.1-MGAT1-HA | (Huang et al., 2015) | N/A |
| pcDNA3.1-MAN2A1-HA | (Huang et al., 2015) | N/A |
| pHEK293Ultra-sHF-LIPA | (Jin et al., 2018) | N/A |
| Software and algorithms | ||
| Nikon NIS-Elements analysis software | Nikon | N/A |
| Prism 9.0 | GraphPad | https://www.graphpad.com |
Highlights.
The Golgi mannosidase MAN1A1 is phosphorylated at S12 by CDK1 in mitosis
Phosphorylation reduces MAN1A1 activity and affects glycan isomer production
Expression of MAN1A1 phosphorylation mutants alters glycosylation in cells
MAN1A1 mitotic phosphorylation reduces its interaction with glycosyltransferase MGAT1
ACKNOWLEDGMENTS
We are grateful to Dr. Daniel N. Hebert and Dr. Henrik Clausen for providing the MAN1 QKO and parental WT CHO cell lines. We thank Dr. Pamela Stanley for providing cDNA constructs and intellectual input. We thank Dr. Vladimir Lupashin for antibodies and other reagents. We thank Dr. Gregg Sobocinski (University of Michigan) for technique support with microscopy. We thank members of the Wang lab for suggestions and reagents. This work was supported by the National Institutes of Health (grant R35GM130331), the Mizutani Foundation for Glycoscience, and the Fast Forward Protein Folding Disease Initiative of the University of Michigan to Y.W. and the National Institutes of Health (grant R01GM130915) to K.W.M. Research in the Suzuki laboratory is supported by Pioneering Research Project “glyco-lipidologue Initiative” (RIKEN), AMED-CREST, under grant JP21gm14100003, and MEXT/JSPS KAKENHI grant JP18H03990 (to T.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111679.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Blackburn JB, and Lupashin VV (2016). Creating knockouts of conserved oligomeric Golgi complex subunits using CRISPR-mediated gene editing paired with a selection strategy based on glycosylation defects associated with impaired COG complex function. Methods Mol. Biol 1496, 145–161. 10.1007/978-1-4939-6463-5_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongini L, Melli L, Lombardi V, and Bianco P (2014). Transient kinetics measured with force steps discriminate between double-stranded DNA elongation and melting and define the reaction energetics. Nucleic Acids Res. 42, 3436–3449. 10.1093/nar/gkt1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Simon ES, Xiang Y, Kachman M, Andrews PC, and Wang Y (2010). Quantitative proteomics analysis of cell cycle-regulated Golgi disassembly and reassembly. J. Biol. Chem 285, 7197–7207. 10.1074/jbc.M109.047084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RN, and Warren G (1992). Sphingolipid transport in mitotic HeLa cells. J. Biol. Chem 267, 24906–24911. [PubMed] [Google Scholar]
- Condon KH, Ho J, Robinson CG, Hanus C, and Ehlers MD (2013). The Angelman syndrome protein Ube3a/E6AP is required for Golgi acidification and surface protein sialylation. J. Neurosci 33, 3799–3814. 10.1523/JNEUROSCI.1930-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’souza Z, Taher FS, and Lupashin VV (2020). Golgi inCOGnito: from vesicle tethering to human disease. Biochim. Biophys. Acta. Gen. Subj 1864, 129694. 10.1016/j.bbagen.2020.129694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, and Gygi SP (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 105, 10762–10767. 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao A, Rahman D, Pappin DJC, Lucocq J, and Lowe M (2003). The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J. Cell Biol 160, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güttinger S, Laurell E, and Kutay U (2009). Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol 10, 178–191. 10.1038/nrm2641. [DOI] [PubMed] [Google Scholar]
- Hassinen A, Pujol FM, Kokkonen N, Pieters C, Kihlström M, Korhonen K, and Kellokumpu S (2011). Functional organization of Golgi N- and O-glycosylation pathways involves pH-dependent complex formation that is impaired in cancer cells. J. Biol. Chem 286, 38329–38340. 10.1074/jbc.M111.277681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama H, Seino J, Kitajima T, Jigami Y, and Suzuki T (2010). Free oligosaccharides to monitor glycoprotein endoplasmic reticulum-associated degradation in Saccharomyces cerevisiae. J. Biol. Chem 285, 12390–12404. 10.1074/jbc.M109.082081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HH, Hassinen A, Sundaram S, Spiess AN, Kellokumpu S, and Stanley P (2015). GnT1IP-L specifically inhibits MGAT1 in the Golgi via its luminal domain. Elife 4. 10.7554/eLife.08916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Tang D, and Wang Y (2016). Monoubiquitination of syntaxin 5 regulates Golgi membrane dynamics during the cell cycle. Dev. Cell 38, 73–85. 10.1016/j.devcel.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, and Wang Y (2017). Golgi structure formation, function, and post-translational modifications in mammalian cells. F1000Res. 6, 2050. 10.12688/f1000research.11900.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland S, Ramnarayanan S, Fu M, Zhang X, Zhang J, Li J, Emebo D, and Wang Y (2020). Cytosolic Ca(2+) modulates Golgi structure through PKCalpha-mediated GRASP55 phosphorylation. iScience 28, 100952. 10.1016/j.isci.2020.100952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman J, and O’connor PM (1998). Methods for synchronizing cells at specific stages of the cell cycle. Curr. Protoc. Cell Biol Chapter 8, Unit 8.3. 3.1-8.3.20. 10.1002/0471143030.cb0803s00. [DOI] [PubMed] [Google Scholar]
- Jin ZC, Kitajima T, Dong W, Huang YF, Ren WW, Guan F, Chiba Y, Gao XD, and Fujita M (2018). Genetic disruption of multiple alpha1, 2-mannosidases generates mammalian cells producing recombinant proteins with high-mannose-type N-glycans. J. Biol. Chem 293, 5572–5584. 10.1074/jbc.M117.813030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaveg K, and Moremen KW (2005). Energetics of substrate binding and catalysis by class 1 (glycosylhydrolase family 47) alpha-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J. Biol. Chem 280, 29837–29848. 10.1074/jbc.M505130200. [DOI] [PubMed] [Google Scholar]
- Kornak U, Reynders E, Dimopoulou A, Van Reeuwijk J, Fischer B, Rajab A, Budde B, Nürnberg P, Foulquier F, Lefeber D, et al. ; ARCL Debré-type Study Group (2008). Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet 40, 32–34. 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, and Kornfeld S (1985). Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem 54, 631–664. 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Kweon HK, and Andrews PC (2013). Quantitative analysis of global phosphorylation changes with high-resolution tandem mass spectrometry and stable isotopic labeling. Methods 61, 251–259. 10.1016/j.ymeth.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Pang P, Kalelkar S, Romero PA, Herscovics A, and Moremen KW (1998). Substrate specificities of recombinant murine Golgi alpha1, 2-mannosidases IA and IB and comparison with endoplasmic reticulum and Golgi processing alpha1, 2-mannosidases. Glycobiology 8, 981–995. 10.1093/glycob/8.10.981. [DOI] [PubMed] [Google Scholar]
- Lamriben L, Oster ME, Tamura T, Tian W, Yang Z, Clausen H, and Hebert DN (2018). EDEM1’s mannosidase-like domain binds ERAD client proteins in a redox-sensitive manner and possesses catalytic activity. J. Biol. Chem 293, 13932–13945. 10.1074/jbc.RA118.004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, Mckenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lowe M, Gonatas NK, and Warren G (2000). The mitotic phosphorylation cycle of the cis-Golgi matrix protein GM130. J. Cell Biol 149, 341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Rabouille C, Nakamura N, Watson R, Jackman M, Jämsä E, Rahman D, Pappin DJ, and Warren G (1998). Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 94, 783–793. [DOI] [PubMed] [Google Scholar]
- Moremen KW, and Nairn AV (2014). Mannosidase, alpha, class 1 (MAN1A1 (Golgi alpha-mannnosidase IA), Man1A2 (Golgi alpha-mannosidase IB), MAN1B1(ER alpha-mannosidase I), MAN1C1 (Golgi alpha-mannosidase IC)). In Handbook of Glycosyltransferases and Related Genes, Taniguchi N, Honke K, Fukuda M, Narimatsu H, Yamaguchi Y, and Angata T, eds. (Springer; ), pp. 1297–1312. [Google Scholar]
- Nilsson T, Au CE, and Bergeron JJM (2009). Sorting out glycosylation enzymes in the Golgi apparatus. FEBS Lett. 583, 3764–3769. 10.1016/j.febslet.2009.10.064. [DOI] [PubMed] [Google Scholar]
- Nilsson T, Slusarewicz P, Hoe MH, and Warren G (1993). Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 330, 1–4. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, et al. (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal 3, ra3. 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- Percival JM, and Froehner SC (2007). Golgi complex organization in skeletal muscle: a role for Golgi-mediated glycosylation in muscular dystrophies? Traffic 8, 184–194. 10.1111/j.1600-0854.2006.00523.x. [DOI] [PubMed] [Google Scholar]
- Preisinger C, Körner R, Wind M, Lehmann WD, Kopajtich R, and Barr FA (2005). Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J. 24, 753–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu MA, Bachert C, Puri S, Lanni F, and Linstedt AD (2006). GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol 8, 238–248. [DOI] [PubMed] [Google Scholar]
- Seko A, and Yamashita K (2008). Activation of beta1, 3-N-acetylglucosaminyltransferase-2 (beta3Gn-T2) by beta3Gn-T8. Possible involvement of beta3Gn-T8 in increasing poly-N-acetyllactosamine chains in differentiated HL-60 cells. J. Biol. Chem 283, 33094–33100. 10.1074/jbc.M806933200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Matsuo I, Totani K, Funayama S, Seino J, Taniguchi N, Ito Y, and Hase S (2008). Dual-gradient high-performance liquid chromatography for identification of cytosolic high-mannose-type free glycans. Anal. Biochem 381, 224–232. 10.1016/j.ab.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Tagliabracci VS, Wiley SE, Guo X, Kinch LN, Durrant E, Wen J, Xiao J, Cui J, Nguyen KB, Engel JL, et al. (2015).A single kinase generates the majority of the secreted phosphoproteome. Cell 161, 1619–1632. 10.1016/j.cell.2015.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Mar K, Warren G, and Wang Y (2008). Molecular mechanism of mitotic Golgi disassembly and reassembly revealed by a defined reconstitution assay. J. Biol. Chem 283, 6085–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, and Wang Y (2013). Cell cycle regulation of Golgi membrane dynamics. Trends Cell Biol. 23, 296–304. 10.1016/j.tcb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, and Wang Y (2015). Golgi isolation. Cold Spring Harb. Protoc 2015, 562–567. 10.1101/pdb.prot075911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Xiang Y, and Wang Y (2010). Reconstitution of the cell cycle-regulated Golgi disassembly and reassembly in a cell-free system. Nat. Protoc 5, 758–772. 10.1038/nprot.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Yuan H, Vielemeyer O, Perez F, and Wang Y (2012). Sequential phosphorylation of GRASP65 during mitotic Golgi disassembly. Biol. Open 1, 1204–1214. 10.1242/bio.20122659BIO20122659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel W, Karaveg K, Liu ZJ, Rose J, Wang BC, and Moremen KW (2004). Structure of mouse Golgi alpha-mannosidase IA reveals the molecular basis for substrate specificity among class 1 (family 47 glycosylhydrolase) alpha1, 2-mannosidases. J. Biol. Chem 279, 29774–29786. 10.1074/jbc.M403065200. [DOI] [PubMed] [Google Scholar]
- Vallee F, Karaveg K, Herscovics A, Moremen KW, and Howell PL (2000). Structural basis for catalysis and inhibition of N-glycan processing class I alpha 1, 2-mannosidases. J. Biol. Chem 275, 41287–41298. 10.1074/jbc.M006927200. [DOI] [PubMed] [Google Scholar]
- Villésurnamen J, Beausoleil SA, Gerber SA, and Gygi SP (2007). Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. USA 104, 1488–1493. 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Satoh A, and Warren G (2005). Mapping the functional domains of the Golgi stacking factor GRASP65. J. Biol. Chem 280, 4921–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Seemann J (2011). Golgi biogenesis. Cold Spring Harb. Perspect. Biol 3, a005330. 10.1101/cshperspect.a005330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Seemann J, Pypaert M, Shorter J, and Warren G (2003). A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. EMBO J. 22, 3279–3290. 10.1093/emboj/cdg317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Taguchi T, and Warren G (2006). Purification of rat liver Golgi stacks. In Cell Biology: A Laboratory Handbook, Third Edition, Celis J, ed. (Elsevier Science; ), pp. 33–39. [Google Scholar]
- Xiang Y, Karaveg K, and Moremen KW (2016). Substrate recognition and catalysis by GH47 alpha-mannosidases involved in Asn-linked glycan maturation in the mammalian secretory pathway. Proc. Natl. Acad. Sci. USA 113, E7890–E7899. 10.1073/pnas.1611213113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Zhang X, Nix DB, Katoh T, Aoki K, Tiemeyer M, and Wang Y (2013). Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun 4, 1659. 10.1038/ncomms2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Wang Y (2015). Cell cycle regulation of VCIP135 deubiquitinase activity and function in p97/p47-mediated Golgi reassembly. Mol. Biol. Cell 26, 2242–2251. 10.1091/mbc.E15-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Wang Y (2016). Glycosylation quality control by the Golgi structure. J. Mol. Biol 428, 3183–3193. 10.1016/j.jmb.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang H, and Wang Y (2014). Phosphorylation regulates VCIP135 function in Golgi membrane fusion during the cell cycle. J. Cell Sci 127, 172–181. 10.1242/jcs.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.