

Summary
Combinatorial mutagenesis is a method where multiple user-defined mutations are encoded at defined positions in a sequence. Combinatorial mutagenic libraries can be used in a variety of applications including evaluating fundamental questions about molecular evolution, directed evolution workflows for enzyme engineering, and in better understanding of biological processes like antibody affinity maturation. Here we show a method of combinatorial mutagenesis utilizing the template-based nicking mutagenesis with several modifications. We show an example for generating a combinatorial library with 14 mutated positions, a total of 16,384 library variants, and a protocol for the generation of large, user-defined combinatorial libraries. The reader can use this protocol to create such libraries in two days.
Keywords: Combinatorial Mutagenesis, Protein Engineering, Epistasis, Molecular Evolution, Fitness Landscapes, Directed Evolution, Enzyme Engineering, Antibody Engineering, Nicking Mutagenesis
1. Introduction
Combinatorial mutagenesis is a method where multiple user-defined mutations are encoded at defined positions in a sequence (Fig. 1a). There are several different applications for combinatorial mutagenic libraries. First, combinatorial mutagenic libraries can be applied to directed evolution workflows, where multiple beneficial mutations can be combined. This end-use is especially complementary with deep mutational scanning approaches that allow for comprehensive screening of all single point mutations in a protein sequence (1). A second end-use enables quantitative evaluation of epistasis (2, 3) and addresses other fundamental questions in molecular evolution. Our lab is particularly interested in using combinatorial mutagenic libraries to recapitulate in vitro the process of somatic hypermutation in antibody maturation and development. To facilitate such studies we have developed a combinatorial mutagenesis protocol based on nicking mutagenesis (4). While there are other established protocols better suited for construction of multi-site saturation combinatorial mutagenesis libraries (5–8), the protocol described in this chapter has a niche in circumstances where the desired combinatorial library contains one or two user-defined mutations per codon (2). For example, consider a directed evolution campaign identifying twelve beneficial mutations at twelve different codons. The method described here could be used to construct the full set of 212 (4,096) possible combinations of the beneficial mutations in two days.
Fig. 1.
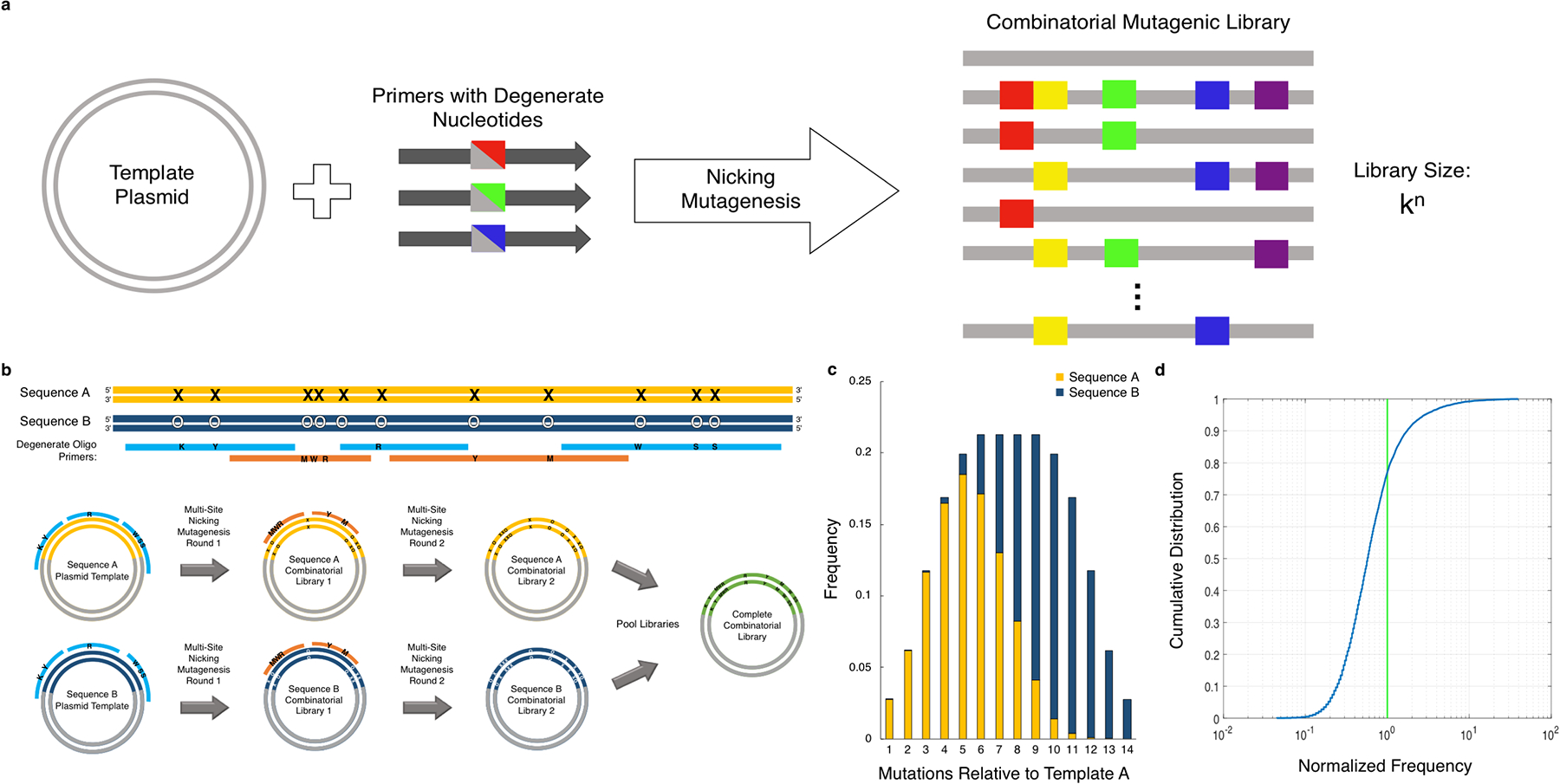
(a) Method overview. The combinatorial mutagenesis method is a variation on nicking mutagenesis where a reaction involving plasmid DNA with degenerate oligonucleotides produces large combinatorial libraries with kn number of variants (k: amino acid changes per residue, n: total number of mutated residues) (b) A generalized schematic of two rounds of nicking mutagenesis with two parental sequences is shown, yielding a plasmid-encoded library of the complete set of 214 differences between the two sequences. (c) The expected distribution of variants as a function of number of codon changes relative to a starting sequence using two parental plasmids is shown. (d) The combinatorial library has an expected cumulative distribution function shown (blue). A uniformly distributed library (green) is shown for comparison.
The following protocol is an extension of multi-site nicking mutagenesis (4). Nicking mutagenesis is a template-based mutagenic method, conceptually similar to Kunkel mutagenesis (9), wherein mutations are encoded using oligonucleotides containing mismatches with the parental DNA sequence. Oligonucleotides anneal to an ssDNA plasmid template prepared from enzymatic degradation of dsDNA. After generation of the complementary strand containing the mutation, the ssDNA template is then selectively nicked and degraded. The complement of the mutagenic strand is then regenerated, leaving mutagenic plasmid dsDNA. A molar ratio of 5:1 mutagenic oligonucleotides to template allows multiple primers to anneal at the same time, affording a high number of mutations encoded per gene. The mutagenic oligonucleotides contain degenerate codons that allow for either the parental sequence residue(s) or the user-defined mutation(s) to be encoded at that specific position. We determined empirically that primer annealing efficiency, and thus mutational incorporation, is strongly dependent on the number of mismatches with the template (M.B.K. and T.A.W. unpublished data). Though the protocol described here has relatively even mutation incorporation (Fig. 1c–d), the current protocol is limited to mutations at eight different positions using a single plasmid as the parental DNA. For combinatorial libraries with a greater number of mutated positions, two different plasmid sequences should be used as the parental DNA – sequence A would contain the starting sequence, while sequence B would contain the complete set of mutations relative to sequence A. Here, we show successful creation of a combinatorial library with 14 mutated positions using two parental plasmids encoding a mature broadly neutralizing antibody fragment CR6261 to Influenza A Hemagglutinin (10) (sequence A) and the unmutated common ancestor (UCA) for this antibody (11) (sequence B) (Fig. 1b). This straightforward and cost-effective method can be performed in as little as two days, and up to three with the larger libraries if two sequential rounds of nicking mutagenesis are required.
With this technique, the user can expect to create large combinatorial libraries with near-complete (>99%) coverage of the set of combinatorial mutations at up to 14 different positions (a library size of 214 or 16384 variants) over several hundred nucleotides in length, and with low carry-over of the wild-type parental DNA. The expected frequency per number of mutations relative to the parental sequence and cumulative distribution are shown in Fig. 1c and Fig. 1d, respectively. Theoretically 16 positions or more could be mutated, though we have not demonstrated this in our lab. For libraries covering longer genes, the presented method is compatible with different barcoding protocols haplotyping a unique molecular identifier with a given set of combinatorial mutations (12, 13).
2. Materials
2.1. Strains:
Bacterial strain: Escherichia coli strain XL1-Blue high-efficiency electrocompetent cells (Agilent Cat # 200228).
2.2. Reagents, Media, & Plates:
Parental DNA plasmids containing a single BbvCI restriction enzyme sites, or multiple sites all in the same orientation
Nuclease-free water (NF H2O) (e.g. New England Biolabs, Cat # B1500S/L).
Custom mutagenic primers with degenerate nucleotides (Integrated DNA Technologies).
Single primer that anneals to the template strand at a non-mutagenized location with an opposite orientation compared with mutagenic oligos
10X T4 Polynucleotide Kinase Buffer (New England Biolabs).
10X CutSmart Buffer (New England Biolabs Cat # B7204S).
5X Phusion HF Buffer (New England Biolabs Cat # B0518S).
10 mM ATP (e.g. New England Biolabs Cat # P0756S/L).
50 mM DTT (e.g. GoldBio DTT10).
50 mM NAD+ (e.g. New England Biolabs Cat # B9007S).
10 mM dNTPs (e.g. New England Biolabs Cat # N0447S).
SOC Media (e.g. BD Cat # 244310 supplemented with 20mM Glucose)
LB agar plates: 2.5% w/v of LB powder (e.g. BD Cat # 244620) and 1.5% w/v of agar. Sterilize by autoclaving. When pouring agar plates, supplement with desired antibiotic for selection.
PCR tubes (e.g. VWR Cat # 52509-304)
Glass Pasteur Pipette
AMPure XP (Beckman Coulter Cat # A63881)
2.3. Enzymes:
10 U/μL T4 Polynucleotide Kinase (New England Biolabs Cat # M0201S/L).
10 U/μL Nt.BbvCI (New England Biolabs Cat # R0632S/L).
10 U/μL Nb.BbvCI (New England Biolabs Cat # R0631S/L).
100 U/μL exonuclease III (New England Biolabs Cat # M0206S/L).
20 U/μL exonuclease I (New England Biolabs Cat # M0293S/L).
2 U/μL Phusion High-Fidelity DNA Polymerase (New England Biolabs Cat # M0530S/L).
40 U/μL Taq DNA ligase (New England Biolabs Cat # M0208S/L).
20 U/μL DpnI (New England Biolabs Cat # R0176S/L).
Shrimp Alkaline Phosphatase rSAP (New England Biolabs Cat #M0371S/L).
2.4. Equipment & Materials:
Monarch® PCR & DNA Cleanup Kit (New England Biolabs Cat # T1030S/L).
Corning square bioassay dishes, 245 mm × 245 mm × 25 mm (Cat # 431111).
Thermal Cycler (e.g. Eppendorf™ Mastercycler™ Pro Cat #950040025)
Microcentrifuge (e.g. any microcentrifuge capable of spinning 16,000 × g, ~13,000 RPM, Cat #13-100-675; does not need to be refrigerated)
Electroporator (e.g. Eppendorf Eporator® Cat # 4309000027).
Monarch® Plasmid Miniprep Kit (New England Biolabs Cat # T1010S/L)
3. Methods
3.1. Preparation of Parental DNA Plasmid(s)
Plasmids should be prepared with gene of interest already inserted and sequence verified, and isolated with any standard plasmid miniprep kit. Sequences of plasmids used in the example are included with the gene of interest, BbvCI site, and residues to be mutated highlighted (See Note 1 for the example antibody parental plasmid sequences, and Note 2 for all primers used in the protocol for these specific sequences).
The parental plasmid(s) must contain a BbvCI site (Nt.BbvCI – CCTCAGC; Nb.BbvCI – GCTGAGG). It is acceptable for the plasmid to contain multiple BbvCI sites only if all are in the same orientation. If not already present, the site can be added using any standard site-directed mutagenesis procedure (e.g. New England Biolabs Cat #E0554S). If using the nicking enzymes in the order presented here (Nb.BbvCI first, Nt.BbvCI second), the orientation of the nicking site will determine if the mutagenic primers should match the sense or antisense strand of the plasmid (Fig. 2) (See Notes 3–4).
Nicked plasmid DNA will be degraded during nicking mutagenesis, so plasmid preparations should be freshly prepared from a dam+ bacterial strain using a commercially available plasmid miniprep kit (e.g. New England Biolabs T1010S/L). 0.76 pmol (typically 2–3 μg) of dsDNA plasmid must be prepared for each parental sequence. (See Note 5).
Fig. 2.
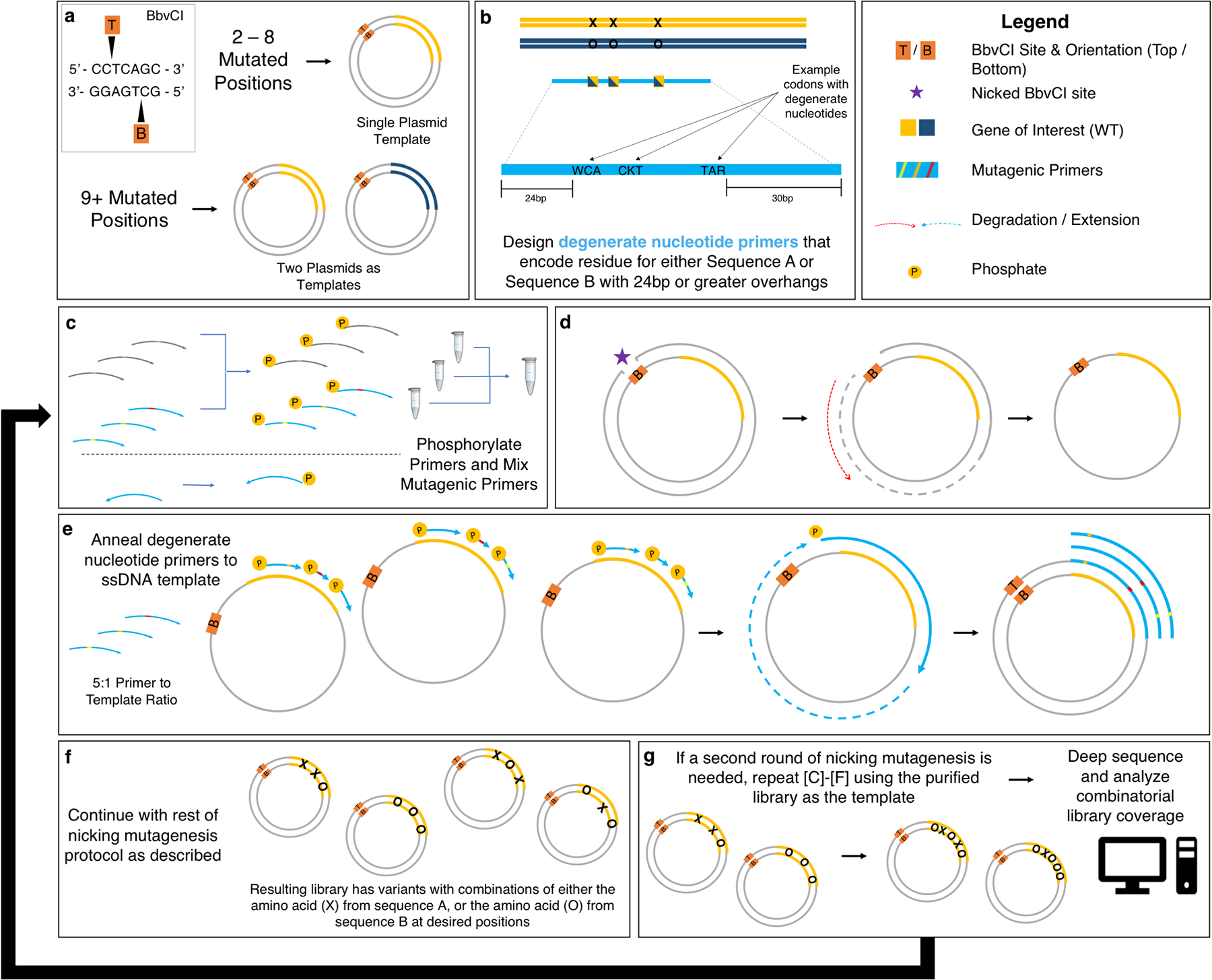
Flowchart for combinatorial mutagenesis. (a) Gene(s) of interest inserted into plasmid. One or two parental plasmids are needed depending on number of mutated positions desired. (b) Design of degenerate nucleotide primers to encode mutations in library. (c) Phosphorylate primers individually and mix together prior to mutagenesis. (d) Selectively nick and degrade one strand of plasmid DNA leaving circular ssDNA template. (e) Anneal mutagenic primers to template, extend around template and ligate to form heteroduplex DNA. (f) The remaining steps follow nicking mutagenesis protocol as previously described. (g) If a second round of nicking mutagenesis is required, repeat steps (c)-(f) using the purified library from the first round as the parental plasmid for the second round. The library can then be prepared for sequencing, sequenced, and analyzed.
3.2. Design of custom and mutagenic oligonucleotides
In this step we design the mutagenic oligonucleotides necessary for creating the combinatorial library (See example primers in Note 2 corresponding to sequence A and B in Note 1).
Identify the sets of mutations to be included in the combinatorial library. For the example described in this protocol CR6261 and UCA_CR6261 gene sequences were aligned in order to determine the positions with mutations.
Determine the distance in basepairs (bp) between each position to mutate. Residues that are close together should be incorporated into one oligonucleotide, while residues that are 30bp or greater apart should be incorporated in different oligonucleotides. The primers should be designed to have 30bp homology arms where possible (See Note 6). The total oligo length should not exceed 100bp.
The specific degenerate codons to use in the primer are determined by the sets of mutations the user wishes to encode by choosing compatible codons that differ by 1 or 2 bps for the given residues at each position.
All set of mutations will be incorporated only if all oligonucleotides can anneal at the same time to one ssDNA template. Where simultaneous annealing of all oligos is impossible, two rounds of nicking mutagenesis will need to be performed. If two rounds are necessary, the oligonucleotides used in the second round of nicking mutagenesis must not overwrite any encoded positions from the first round. For the example provided, two rounds of nicking mutagenesis were performed. The first required three oligos encoding mutations at eight positions and the second round required two oligos encoding the mutations at the remaining six positions.
A secondary primer with a melting temperature (Tm) above 55 °C should be designed that anneals to a non-mutagenized region of the plasmid in the opposite orientation compared to the mutagenic primers.
Order oligonucleotides from Integrated DNA Technologies or preferred vendor and resuspend the lyophilized primers to 100 μM in NF H2O.
3.3. Preparation of Combinatorial Mutagenic Libraries by Nicking Mutagenesis
This portion of the protocol introduces the designed library mutations in the parental plasmid(s). Many of the steps have been previously described (4, 14, 15) (See Note 7).
Phosphorylate each mutagenic oligo and the secondary primer on ice in individual PCR tubes. Into each tube add 18 μL of NF H2O, 3 μL of T4 Polynucleotide Kinase Buffer, 7 μL of 100 μM oligo, 1 μL of 10 mM ATP, 1 μL of T4 Polynucleotide Kinase. Incubate each reaction at 37 °C for 1 hour. Phosphorylated oligos can be stored at −20 °C up to a week but are best used freshly phosphorylated. The reaction volume is 30 μL. Mix well (See Notes 8–9).
Phosphorylated oligos should be diluted and mixed the morning of mutagenesis. 2 μL of each mutagenic oligo should be mixed together in a PCR tube with NF H2O up to a total volume of 40 μL. 2 μL of the secondary primer should be mixed with 38 μL of NF H2O. Mix well. The mixture of phosphorylated oligos should contain primers that do not overlap when annealing to the parental sequence. For the first round of mutagenesis in the example provided, oligonucleotides 1, 2, and 3 are mixed and anneal to sequence A and oligonucleotides 1, 3, and 6 are mixed and anneal to sequence B. Oligonucleotide 7 is the secondary primer for both parental DNA sequences.
To prepare the ssDNA, add the following to a PCR tube on ice: 0.76 pmol of dsDNA plasmid, 2 μL 10X CutSmart Buffer, 1 μL of 1:10 diluted exonuclease III (final concentration of 10 U/μL), 1 μL of Nb.BbvCI, 1 μL of exonuclease I, and NF H2O up to 20 μL final volume. Mix well. There should be one reaction per parental sequence so for the example there is one ssDNA template prepared from parental sequence A, and a separate template prepared from sequence B in parallel. The diluent for the enzymes is 1X CutSmart Buffer (See Note 4).
Preheat the thermal cycler to 37 °C and place the PCR tube(s) in it with the following program: 60 minutes at 37 °C, 20 minutes at 80 °C, hold at 4 °C.
To anneal the mutagenic oligos to the ssDNA template, take the PCR tube(s) off thermal cycler and onto ice and add 16.7 μL NF H2O, 10 μL 5X Phusion HF Buffer, and 3.3 μL of the 1:20 diluted mutagenic oligo mixture. The total reaction volume is 50 μL. Mix well (See Note 10).
Place the PCR tube(s) on a preheated thermal cycler block at 98 °C and run the following program: 2 minutes at 98 °C, 5 minutes at 55 °C, hold at 55 °C.
While keeping the PCR tube(s) on the thermal cycler block, add the following to a separate PCR tube on ice: 11 μL NF H2O, 10 μL 5X Phusion HF Buffer, 20 μL 50 mM DTT, 1 μL 50 mM NAD+, 2 μL 10 mM dNTPs, 5 uL Taq DNA Ligase, 1 μL Phusion HF Polymerase. Transfer the PCR tube(s) to the thermal cycler block to briefly warm to 55 °C.
Add the mixture from the PCR tube(s) used in step 7 to the reaction PCR tube(s) for a total reaction volume of 100 μL, mix well, and run the following program: 72 °C for 10 minutes, 45 °C for 20 minutes, hold at 4 °C.
Purify each reaction separately using the Monarch® PCR & DNA Cleanup Kit to a final volume of 15 μL using NF H2O according the manufacturer’s instructions with the following exception: wait five minutes after applying the eluant to the column before centrifugation.
To degrade the complementary template strand(s), transfer 14 μL of each of the purified DNA product(s) to new PCR tube(s) and add the following: 2 μL 10X CutSmart Buffer, 2 μL 1:50 diluted exonuclease III (final concentration 2 U/μL), 1 μL of 1:10 diluted Nt.BbvCI (final concentration 1 U/μL), and 1 μL exonuclease I. The total reaction volume is 20 μL. Mix well. The diluent for the enzymes is 1X CutSmart Buffer (See Note 4).
Put the PCR tube(s) on the thermal cycler block preheated to 37 °C with the following program: 60 minutes at 37 °C, 20 minutes at 80 °C, hold at 4 °C.
To synthesize the complementary mutagenic strand, remove the PCR tube(s) from the thermal cycler block and add the following to the reaction tube(s) on ice: 27.7 μL NF H2O, 20 μL Phusion HF Buffer, 3.3 μL of 1:20 diluted and phosphorylated secondary oligo, 20 μL of 50 mM DTT, 1 μL of 50 mM NAD+, 2 μL 10mM dNTPs, 5 μL Taq DNA Ligase, and 1 μL Phusion HF Polymerase. The total reaction volume is 100 μL. Mix well. In the example, the secondary primer used for both sequence A and sequence B is #7.
Preheat the thermal cycler to 98 °C, place the reaction tube(s) on the cycler block and run the following program: 30 seconds at 98 °C, 45 seconds at 55 °C, 10 minutes at 72 °C, 20 minutes at 45 °C, and hold at 4 °C.
Add 2 μL of DpnI into each reaction tube, mix well, and incubate at 37 °C for 1 hour to degrade the methylated and hemimethylated wild-type DNA (See Note 11).
Purify the reaction using the Monarch® PCR & DNA Cleanup Kit to a final volume of 6 μL using NF H2O according the manufacturer’s instructions with the exception of waiting five minutes after applying the eluant to the column before centrifugation.
Transform the entire 6 μL of the purified reaction product into E. coli strain XL1 Blue high efficiency cells following standard electrocompetent transformation protocols (See Note 12).
After recovery of the transformed cells in SOC media, bring the final volume of the transformation to 2 mL with additional sterile media.
Prepare six tenfold serial dilutions and plate 10 μL of each on standard agar plates supplemented with antibiotic in order to determine the transformation efficiency (See Note 12).
Spread the remaining transformation mixture onto the prepared large bioassay dish supplemented with antibiotic and wait for plate to dry before incubating at 37 °C overnight.
The next day if a sufficient number of transformants were produced according to the dilution plate (See NOTE 12), scrape the large bioassay dish to remove and resuspend the grown colonies in sterile media. Add sufficient media to remove all of the colonies from the agar using a plate scraper, either bought or prepared by bending a heated glass Pasteur pipette.
Extract the mutagenic plasmid DNA from an aliquot of the collected cells using the Monarch® Plasmid Miniprep Kit according to the manufacturer’s instructions. If only one round of nicking mutagenesis was necessary, the library is ready for deep sequencing.
If an additional round is necessary, as is in the example (Fig. 1b), the library created from the first round will serve as the parental plasmid DNA for the second round.
Follow steps 1–21 for the second round of nicking mutagenesis with the library generated in round one as the parental plasmid and the remaining oligonucleotides are used in the primer mixture. The oligonucleotides 4 and 5 are mixed together and used for both the sequence A library and sequence B library in the example.
After isolating the plasmid library generated in the second round of nicking mutagenesis, the libraries are ready to be prepared for sequencing.
In cases where two parental DNA plasmids were used as templates the user will have two sub-libraries that can be pooled together to create the complete combinatorial library (See Note 14).
3.4. Sequence Verification of Combinatorial Library
Prepare the linear amplicon for deep sequencing by adding the Illumina adapters and barcodes to the mutagenic region in the plasmid library via PCR. Preparing the libraries for sequencing can be done following established protocol (16).
In a PCR tube add the following: 10 μL of 5X Phusion HF Buffer, 1 μL of 10 mM dNTPs, 2.5 μL of 10 μM forward inner primer, 2.5 μL of 10 μM reverse inner primer, 100 ng of dsDNA library plasmid, 0.5 μL of Phusion HF Polymerase, and NF H2O up to a total reaction volume of 50 μL (See Note 2 for primer sequences). The inner primers are gene specific and must be designed for each combinatorial library. In the example, primers 7 and 8 are the inner primers for both the library generated from sequence A and the library generated from sequence B.
Place the PCR tube on a preheated thermal cycler block preheated to 98 °C and run the following program: 30 seconds at 98 °C, 16x cycles of 5 seconds at 98 °C, 15 seconds at 53 °C, and 15 seconds 72 °C, and then hold at 4 °C.
To the same PCR tube, add 5 μL of exonuclease I and 10 μL of shrimp alkaline phosphatase and run the following thermal cycler program: 37 °C for 15 minutes, 80 °C for 15 minutes, and hold at 4 °C.
To add the Illumina adapters and barcodes add the following to a new PCR tube on ice: 18.5 μL of NF H2O, 1 μL of DNA (from the cleaning step after the 1st PCR reaction), 10 μL of 5X Phusion HF Buffer, 2.5 μL of 10 μM forward outer primer, 2.5 μL of 10 μM reverse outer primer, 1 μL of 10 mM dNTPs, and 0.5 μL of Phusion HF Polymerase (See Note 2 for primer sequences).
Place the PCR tube on a preheated thermal cycler block preheated to 98 °C and run the following program: 30 seconds at 98 °C, 16x cycles of 5 seconds at 98 °C, 15 seconds at 53 °C, and 15 seconds 72 °C, and then hold at 4 °C.
Purify and clean the PCR product using Agencourt AMPure XP following manufacturer instructions. It was not necessary to clean the PCR product prior to purification with AMPure XP.
Follow the instructions from your sequencing facility for quantifying and submitting purified PCR product for NGS (See Note 15).
To confirm the library coverage of the combinatorial library produced, a variety of deep sequencing analysis programs are freely available to use, including PACT: Protein Analysis and Classifier Toolkit (17). This program can be modified to filter and merge sequencing reads in the combinatorial library by following the published notes and specifically can incorporate the degenerate codons used in the mutagenic oligos in the program input file (See Note 16 for complete commandline information for the examples shown in this protocol).
4. Notes
-
1
Plasmid Sequences used for examples:
scFv sequence, BbvCI site, mutated residues, location of secondary primer
>CR6261_pETcon (Sequence A)
ATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCATGATTACGCCAAGCTCGAAATTAACCCTCACTAAAGGGAACAAAAGCTGGTACCAATTCCTTGAATTTTCAAAAATTCTTACTTTTTTTTTGGATGGACGCAAAGAAGTTTAATAATCATATTACATGGCATTACCACCATATACATATCCATATCTAATCTTACTTATATGTTGTGGAAATGTAAAGAGCCCCATTATCTTAGCCTAAAAAAACCTTCTCTTTGGAACTTTCAGTAATACGCTTAACTGCTCATTGCTATATTGAAGTACGGATTAGAAGCCGCCGAGCGGGTGACAGCCCTCCGAAGGAAGACTCTCCTCCGTGCGTCCTCGTCTTCACCGGTCGCGTTCCTGAAACGCAGATGTGCCTCGCGCCGCACTGCTCCGAACAATAAAGATTCTACAATACTAGCTTTTATGGTTATGAAGAGGAAAAATTGGCAGTAACCTGGCCCCACAAACCTTCAAATGAACGAATCAAATTAACAACCATAGGATGATAATGCGATTAGTTTTTTAGCCTTATTTCTGGGGTAATTAATCAGCGAAGCGATGATTTTTGATCTATTAACAGATATATAAATGCAAAAACTGCATAACCACTTTAACTAATACTTTCAACATTTTCGGTTTGTATTACTTCTTATTCAAATGTAATAAAAGTATCAACAAAAAATTGTTAATATACCTCTATACTTTAACGTCAAGGAGAAAAAACCCCGGATCGAATTCCCTACTTCATACATTTTCAATTAAGATGCAGTTACTTCGCTGTTTTTCAATATTTTCTGTTATTGCTTCAGTTTTAGCACAGGAACTGACAACTATATGCGAGCAAATCCCCTCACCAACTTTAGAATCGACGCCGTACTCTTTGTCAACGACTACTATTTTGGCCAACGGGAAGGCAATGCAAGGAGTTTTTGAATATTACAAATCAGTAACGTTTGTCAGTAATTGCGGTTCTCACCCCTCAACAACTAGCAAAGGCAGCCCCATAAACACACAGTATGTTTTTAAGGACAATAGCTCGACGATTGAAGGTAGATACCCATACGACGTTCCAGACTACGCTCTGCAGGCTAGTGGTGGAGGAGGCTCTGGTGGAGGCGGTAGCGGAGGCGGAGGGTCGGCTAGCCATATGGAAGTTCAATTGGTTGAGTCCGGTGCTGAGGTCAAAAAACCTGGTTCCTCCGTCAAAGTTTCCTGTAAGGCTTCTGGTGGTCCATTCAGATCCTACGCCATCTCCTGGGTTAGACAAGCTCCTGGTCAAGGACCTGAGTGGATGGGAGGAATCATTCCAATCTTCGGTACTACCAAATACGCTCCAAAATTCCAGGGTAGAGTCACCATTACTGCCGATGATTTCGCTGGAACCGTTTACATGGAGTTGAGTTCATTGAGATCCGAGGACACTGCCATGTACTACTGTGCCAAACACATGGGATACCAAGTCAGAGAAACCATGGATGTCTGGGGTAAAGGAACAACCGTCACTGTCTCTAGTGGCGGTGGAGGTAGTGGAGGAGGTGGCTCAGGTGGCGGTGGATCTGTCTTGACCCAACCACCATCTGTCTCTGCTGCCCCTGGTCAAAAAGTCACCATCTCATGCTCTGGTTCTTCATCAAACATCGGAAACGACTACGTTTCATGGTACCAACAATTGCCTGGTACTGCCCCAAAATTGTTGATCTACGACAACAACAAAAGACCATCCGGTATTCCAGACAGATTCTCCGGTTCCAAATCCGGAACTTCCGCCACTTTGGGAATCACTGGATTGCAAACCGGAGATGAGGCCAACTACTACTGTGCCACCTGGGATAGAAGACCAACTGCCTACGTTGTTTTCGGTGGTGGAACCAAATTGACCGTCTTGGGACAACCACTCGAGGGGGGCGGATCCGAACAAAAGCTTATTTCTGAAGAGGACTTGTAATAGAGATCTGATAACAACAGTGTAGATGTAACAAAATCGACTTTGTTCCCACTGTACTTTTAGCTCGTACAAAATACAATATACTTTTCATTTCTCCGTAAACAACATGTTTTCCCATGTAATATCCTTTTCTATTTTTCGTTCCGTTACCAACTTTACACATACTTTATATAGCTATTCACTTCTATACACTAAAAAACTAAGACAATTTTAATTTTGCTGCCTGCCATATTTCAATTTGTTATAAATTCCTATAATTTATCCTATTAGTAGCTAAAAAAAGATGAATGTGAATCGAATCCTAAGAGAATTGAGCTCCAATTCGCCCTATAGTGAGTCGTATTACAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCTTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGACGCGCCCTGTAGCGGCGCATTAAGCGCGGCGGGTGTGGTGGTTACGCGCAGCGTGACCGCTACACTTGCCAGCGCCCTAGCGCCCGCTCCTTTCGCTTTCTTCCCTTCCTTTCTCGCCACGTTCGCCGGCTTTCCCCGTCAAGCTCTAAATCGGGGGCTCCCTTTAGGGTTCCGATTTAGTGCTTTACGGCACCTCGACCCCAAAAAACTTGATTAGGGTGATGGTTCACGTAGTGGGCCATCGCCCTGATAGACGGTTTTTCGCCCTTTGACGTTGGAGTCCACGTTCTTTAATAGTGGACTCTTGTTCCAAACTGGAACAACACTCAACCCTATCTCGGTCTATTCTTTTGATTTATAAGGGATTTTGCCGATTTCGGCCTATTGGTTAAAAAATGAGCTGATTTAACAAAAATTTAACGCGAATTTTAACAAAATATTAACGCTTACAATTTCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATAGATCGGCAAGTGCACAAACAATACTTAAATAAATACTACTCAGTAATAACCTATTTCTTAGCATTTTTGACGAAATTTGCTATTTTGTTAGAGTCTTTTACACCATTTGTCTCCACACCTCCGCTTACATCAACACCAATAACGCCATTTAATCTAAGCGCATCACCAACATTTTCTGGCGTCAGTCCACCAGCTAACATAAAATGTAAGCTTTCGGGGCTCTCTTGCCTTCCAACCCAGTCAGAAATCGAGTTCCAATCCAAAAGTTCACCTGTCCCACCTGCTTCTGAATCAAACAAGGGAATAAACGAATGAGGTTTCTGTGAAGCTGCACTGAGTAGTATGTTGCAGTCTTTTGGAAATACGAGTCTTTTAATAACTGGCAAACCGAGGAACTCTTGGTATTCTTGCCACGACTCATCTCCATGCAGTTGGACGATATCAATGCCGTAATCATTGACCAGAGCCAAAACATCCTCCTTAGGTTGATTACGAAACACGCCAACCAAGTATTTCGGAGTGCCTGAACTATTTTTATATGCTTTTACAAGACTTGAAATTTTCCTTGCAATAACCGGGTCAATTGTTCTCTTTCTATTGGGCACACATATAATACCCAGCAAGTCAGCATCGGAATCTAGAGCACATTCTGCGGCCTCTGTGCTCTGCAAGCCGCAAACTTTCACCAATGGACCAGAACTACCTGTGAAATTAATAACAGACATACTCCAAGCTGCCTTTGTGTGCTTAATCACGTATACTCACGTGCTCAATAGTCACCAATGCCCTCCCTCTTGGCCCTCTCCTTTTCTTTTTTCGACCGAATTAATTCTTAATCGGCAAAAAAAGAAAAGCTCCGGATCAAGATTGTACGTAAGGTGACAAGCTATTTTTCAATAAAGAATATCTTCCACTACTGCCATCTGGCGTCATAACTGCAAAGTACACATATATTACGATGCTGTTCTATTAAATGCTTCCTATATTATATATATAGTAATGTCGTGATCTATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAGTTAAGCCAGCCCCGACACCCGCCAACACCCGCTGACGCGCCCTGACGGGCTTGTCTGCTCCCGGCATCCGCTTACAGACAAGCTGTGACCGTCTCCGGGAGCTGCATGTGTCAGAGGTTTTCACCGTCATCACCGAAACGCGCGAGACGAAAGGGCCTCGTGATACGCCTATTTTTATAGGTTAATGTCATGATAATAATGGTTTCTTAGACGGATCGCTTGCCTGTAACTTACACGCGCCTCGTATCTTTTAATGATGGAATAATTTGGGAATTTACTCTGTGTTTATTTATTTTTATGTTTTGTATTTGGATTTTAGAAAGTAAATAAAGAAGGTAGAAGAGTTACGGAATGAAGAAAAAAAAATAAACAAAGGTTTAAAAAATTTCAACAAAAAGCGTACTTTACATATATATTTATTAGACAAGAAAAGCAGATTAAATAGATATACATTCGATTAACGATAAGTAAAATGTAAAATCACAGGATTTTCGTGTGTGGTCTTCTACACAGACAAGATGAAACAATTCGGCATTAATACCTGAGAGCAGGAAGAGCAAGATAAAAGGTAGTATTTGTTGGCGATCCCCCTAGAGTCTTTTACATCTTCGGAAAACAAAAACTATTTTTTCTTTAATTTCTTTTTTTACTTTCTATTTTTAATTTATATATTTATATTAAAAAATTTAAATTATAATTATTTTTATAGCACGTGATGAAAAGGACCCAGGTGGCACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTATGAGTATTCAACATTTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAACGCTGGTGAAAGTAAAAGATGCTGAAGATCAGTTGGGTGCACGAGTGGGTTACATCGAACTGGATCTCAACAGCGGTAAGATCCTTGAGAGTTTTCGCCCCGAAGAACGTTTTCCAATGATGAGCACTTTTAAAGTTCTGCTATGTGGCGCGGTATTATCCCGTATTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATACACTATTCTCAGAATGACTTGGTTGAGTACTCACCAGTCACAGAAAAGCATCTTACGGATGGCATGACAGTAAGAGAATTATGCAGTGCTGCCATAACCATGAGTGATAACACTGCGGCCAACTTACTTCTGACAACGATCGGAGGACCGAAGGAGCTAACCGCTTTTTTGCACAACATGGGGGATCATGTAACTCGCCTTGATCGTTGGGAACCGGAGCTGAATGAAGCCATACCAAACGACGAGCGTGACACCACGATGCCTGTAGCAATGGCAACAACGTTGCGCAAACTATTAACTGGCGAACTACTTACTCTAGCTTCCCGGCAACAATTAATAGACTGGATGGAGGCGGATAAAGTTGCAGGACCACTTCTGCGCTCGGCCCTTCCGGCTGGCTGGTTTATTGCTGATAAATCTGGAGCCGGTGAGCGTGGGTCTCGCGGTATCATTGCAGCACTGGGGCCAGATGGTAAGCCCTCCCGTATCGTAGTTATCTACACGACGGGGAGTCAGGCAACTATGGATGAACGAAATAGACAGATCGCTGAGATAGGTGCCTCACTGATTAAGCATTGGTAACTGTCAGACCAAGTTTACTCATATATACTTTAGATTGATTTAAAACTTCATTTTTAATTTAAAAGGATCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTCCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCTGCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGCCGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATACCAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCACCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAAGTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCGGGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAACTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGGCGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTCCAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAGCGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACGCGGCCTTTTTACGGTTCCTGGCCTTTTGCTGGCCTTTTGCTCACATGTTCTTTCCTGCGTTATCCCCTGATTCTGTGGATAACCGTATTACCGCCTTTGAGTGAGCTGATACCGCTCGCCGCAGCCGAACGACCGAGCGCAGCGAGTCAGTGAGCGAGGAAGCGGAAGAGCGCCCAATACGCAAACCGCCTCTCCCCGCGCGTTGGCCGATTCATTAATGCAGCTGGCACGACAGGTTTCCCGACTGGAAAGCGGGCAGTGAGCGCAACGCAATTAATGTGAGTTAGCTCACTCATTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGA
>CR6261_UCA_pETcon (Sequence B)
ATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCATGATTACGCCAAGCTCGAAATTAACCCTCACTAAAGGGAACAAAAGCTGGTACCAATTCCTTGAATTTTCAAAAATTCTTACTTTTTTTTTGGATGGACGCAAAGAAGTTTAATAATCATATTACATGGCATTACCACCATATACATATCCATATCTAATCTTACTTATATGTTGTGGAAATGTAAAGAGCCCCATTATCTTAGCCTAAAAAAACCTTCTCTTTGGAACTTTCAGTAATACGCTTAACTGCTCATTGCTATATTGAAGTACGGATTAGAAGCCGCCGAGCGGGTGACAGCCCTCCGAAGGAAGACTCTCCTCCGTGCGTCCTCGTCTTCACCGGTCGCGTTCCTGAAACGCAGATGTGCCTCGCGCCGCACTGCTCCGAACAATAAAGATTCTACAATACTAGCTTTTATGGTTATGAAGAGGAAAAATTGGCAGTAACCTGGCCCCACAAACCTTCAAATGAACGAATCAAATTAACAACCATAGGATGATAATGCGATTAGTTTTTTAGCCTTATTTCTGGGGTAATTAATCAGCGAAGCGATGATTTTTGATCTATTAACAGATATATAAATGCAAAAACTGCATAACCACTTTAACTAATACTTTCAACATTTTCGGTTTGTATTACTTCTTATTCAAATGTAATAAAAGTATCAACAAAAAATTGTTAATATACCTCTATACTTTAACGTCAAGGAGAAAAAACCCCGGATCGAATTCCCTACTTCATACATTTTCAATTAAGATGCAGTTACTTCGCTGTTTTTCAATATTTTCTGTTATTGCTTCAGTTTTAGCACAGGAACTGACAACTATATGCGAGCAAATCCCCTCACCAACTTTAGAATCGACGCCGTACTCTTTGTCAACGACTACTATTTTGGCCAACGGGAAGGCAATGCAAGGAGTTTTTGAATATTACAAATCAGTAACGTTTGTCAGTAATTGCGGTTCTCACCCCTCAACAACTAGCAAAGGCAGCCCCATAAACACACAGTATGTTTTTAAGGACAATAGCTCGACGATTGAAGGTAGATACCCATACGACGTTCCAGACTACGCTCTGCAGGCTAGTGGTGGAGGAGGCTCTGGTGGAGGCGGTAGCGGAGGCGGAGGGTCGGCTAGCCATATGGAAGTTCAATTGGTTGAGTCCGGTGCTGAGGTCAAAAAACCTGGTTCCTCCGTCAAAGTTTCCTGTAAGGCTTCTGGTGGTACATTCAGCTCCTACGCCATCTCCTGGGTTAGACAAGCTCCTGGTCAAGGACTTGAGTGGATGGGAGGAATCATTCCAATCTTCGGTACTGCCAACTACGCTCAAAAATTCCAGGGTAGAGTCACCATTACTGCCGATGAATCCACTAGCACCGCTTACATGGAGTTGAGTTCATTGAGATCCGAGGACACTGCCGTGTACTACTGTGCCAGACACATGGGATACCAACTCAGAGAAACCATGGATGTCTGGGGTAAAGGAACAACCGTCACTGTCTCTAGTGGCGGTGGAGGTAGTGGAGGAGGTGGCTCAGGTGGCGGTGGATCTGTCTTGACCCAACCACCATCTGTCTCTGCTGCCCCTGGTCAAAAAGTCACCATCTCATGCTCTGGTTCTTCATCAAACATCGGAAACGACTACGTTTCATGGTACCAACAATTGCCTGGTACTGCCCCAAAATTGTTGATCTACGACAACAACAAAAGACCATCCGGTATTCCAGACAGATTCTCCGGTTCCAAATCCGGAACTTCCGCCACTTTGGGAATCACTGGATTGCAAACCGGAGATGAGGCCAACTACTACTGTGCCACCTGGGATAGAAGACCAACTGCCTACGTTGTTTTCGGTGGTGGAACCAAATTGACCGTCTTGGGCGGATCCGAACAAAAGCTTATTTCTGAAGAGGACTTGTAATAGAGATCTGATAACAACAGTGTAGATGTAACAAAATCGACTTTGTTCCCACTGTACTTTTAGCTCGTACAAAATACAATATACTTTTCATTTCTCCGTAAACAACATGTTTTCCCATGTAATATCCTTTTCTATTTTTCGTTCCGTTACCAACTTTACACATACTTTATATAGCTATTCACTTCTATACACTAAAAAACTAAGACAATTTTAATTTTGCTGCCTGCCATATTTCAATTTGTTATAAATTCCTATAATTTATCCTATTAGTAGCTAAAAAAAGATGAATGTGAATCGAATCCTAAGAGAATTGAGCTCCAATTCGCCCTATAGTGAGTCGTATTACAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCTTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGACGCGCCCTGTAGCGGCGCATTAAGCGCGGCGGGTGTGGTGGTTACGCGCAGCGTGACCGCTACACTTGCCAGCGCCCTAGCGCCCGCTCCTTTCGCTTTCTTCCCTTCCTTTCTCGCCACGTTCGCCGGCTTTCCCCGTCAAGCTCTAAATCGGGGGCTCCCTTTAGGGTTCCGATTTAGTGCTTTACGGCACCTCGACCCCAAAAAACTTGATTAGGGTGATGGTTCACGTAGTGGGCCATCGCCCTGATAGACGGTTTTTCGCCCTTTGACGTTGGAGTCCACGTTCTTTAATAGTGGACTCTTGTTCCAAACTGGAACAACACTCAACCCTATCTCGGTCTATTCTTTTGATTTATAAGGGATTTTGCCGATTTCGGCCTATTGGTTAAAAAATGAGCTGATTTAACAAAAATTTAACGCGAATTTTAACAAAATATTAACGCTTACAATTTCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATAGATCGGCAAGTGCACAAACAATACTTAAATAAATACTACTCAGTAATAACCTATTTCTTAGCATTTTTGACGAAATTTGCTATTTTGTTAGAGTCTTTTACACCATTTGTCTCCACACCTCCGCTTACATCAACACCAATAACGCCATTTAATCTAAGCGCATCACCAACATTTTCTGGCGTCAGTCCACCAGCTAACATAAAATGTAAGCTTTCGGGGCTCTCTTGCCTTCCAACCCAGTCAGAAATCGAGTTCCAATCCAAAAGTTCACCTGTCCCACCTGCTTCTGAATCAAACAAGGGAATAAACGAATGAGGTTTCTGTGAAGCTGCACTGAGTAGTATGTTGCAGTCTTTTGGAAATACGAGTCTTTTAATAACTGGCAAACCGAGGAACTCTTGGTATTCTTGCCACGACTCATCTCCATGCAGTTGGACGATATCAATGCCGTAATCATTGACCAGAGCCAAAACATCCTCCTTAGGTTGATTACGAAACACGCCAACCAAGTATTTCGGAGTGCCTGAACTATTTTTATATGCTTTTACAAGACTTGAAATTTTCCTTGCAATAACCGGGTCAATTGTTCTCTTTCTATTGGGCACACATATAATACCCAGCAAGTCAGCATCGGAATCTAGAGCACATTCTGCGGCCTCTGTGCTCTGCAAGCCGCAAACTTTCACCAATGGACCAGAACTACCTGTGAAATTAATAACAGACATACTCCAAGCTGCCTTTGTGTGCTTAATCACGTATACTCACGTGCTCAATAGTCACCAATGCCCTCCCTCTTGGCCCTCTCCTTTTCTTTTTTCGACCGAATTAATTCTTAATCGGCAAAAAAAGAAAAGCTCCGGATCAAGATTGTACGTAAGGTGACAAGCTATTTTTCAATAAAGAATATCTTCCACTACTGCCATCTGGCGTCATAACTGCAAAGTACACATATATTACGATGCTGTTCTATTAAATGCTTCCTATATTATATATATAGTAATGTCGTGATCTATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAGTTAAGCCAGCCCCGACACCCGCCAACACCCGCTGACGCGCCCTGACGGGCTTGTCTGCTCCCGGCATCCGCTTACAGACAAGCTGTGACCGTCTCCGGGAGCTGCATGTGTCAGAGGTTTTCACCGTCATCACCGAAACGCGCGAGACGAAAGGGCCTCGTGATACGCCTATTTTTATAGGTTAATGTCATGATAATAATGGTTTCTTAGACGGATCGCTTGCCTGTAACTTACACGCGCCTCGTATCTTTTAATGATGGAATAATTTGGGAATTTACTCTGTGTTTATTTATTTTTATGTTTTGTATTTGGATTTTAGAAAGTAAATAAAGAAGGTAGAAGAGTTACGGAATGAAGAAAAAAAAATAAACAAAGGTTTAAAAAATTTCAACAAAAAGCGTACTTTACATATATATTTATTAGACAAGAAAAGCAGATTAAATAGATATACATTCGATTAACGATAAGTAAAATGTAAAATCACAGGATTTTCGTGTGTGGTCTTCTACACAGACAAGATGAAACAATTCGGCATTAATACCTGAGAGCAGGAAGAGCAAGATAAAAGGTAGTATTTGTTGGCGATCCCCCTAGAGTCTTTTACATCTTCGGAAAACAAAAACTATTTTTTCTTTAATTTCTTTTTTTACTTTCTATTTTTAATTTATATATTTATATTAAAAAATTTAAATTATAATTATTTTTATAGCACGTGATGAAAAGGACCCAGGTGGCACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTATGAGTATTCAACATTTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAACGCTGGTGAAAGTAAAAGATGCTGAAGATCAGTTGGGTGCACGAGTGGGTTACATCGAACTGGATCTCAACAGCGGTAAGATCCTTGAGAGTTTTCGCCCCGAAGAACGTTTTCCAATGATGAGCACTTTTAAAGTTCTGCTATGTGGCGCGGTATTATCCCGTATTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATACACTATTCTCAGAATGACTTGGTTGAGTACTCACCAGTCACAGAAAAGCATCTTACGGATGGCATGACAGTAAGAGAATTATGCAGTGCTGCCATAACCATGAGTGATAACACTGCGGCCAACTTACTTCTGACAACGATCGGAGGACCGAAGGAGCTAACCGCTTTTTTGCACAACATGGGGGATCATGTAACTCGCCTTGATCGTTGGGAACCGGAGCTGAATGAAGCCATACCAAACGACGAGCGTGACACCACGATGCCTGTAGCAATGGCAACAACGTTGCGCAAACTATTAACTGGCGAACTACTTACTCTAGCTTCCCGGCAACAATTAATAGACTGGATGGAGGCGGATAAAGTTGCAGGACCACTTCTGCGCTCGGCCCTTCCGGCTGGCTGGTTTATTGCTGATAAATCTGGAGCCGGTGAGCGTGGGTCTCGCGGTATCATTGCAGCACTGGGGCCAGATGGTAAGCCCTCCCGTATCGTAGTTATCTACACGACGGGGAGTCAGGCAACTATGGATGAACGAAATAGACAGATCGCTGAGATAGGTGCCTCACTGATTAAGCATTGGTAACTGTCAGACCAAGTTTACTCATATATACTTTAGATTGATTTAAAACTTCATTTTTAATTTAAAAGGATCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTCCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCTGCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGCCGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATACCAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCACCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAAGTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCGGGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAACTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGGCGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTCCAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAGCGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACGCGGCCTTTTTACGGTTCCTGGCCTTTTGCTGGCCTTTTGCTCACATGTTCTTTCCTGCGTTATCCCCTGATTCTGTGGATAACCGTATTACCGCCTTTGAGTGAGCTGATACCGCTCGCCGCAGCCGAACGACCGAGCGCAGCGAGTCAGTGAGCGAGGAAGCGGAAGAGCGCCCAATACGCAAACCGCCTCTCCCCGCGCGTTGGCCGATTCATTAATGCAGCTGGCACGACAGGTTTCCCGACTGGAAAGCGGGCAGTGAGCGCAACGCAATTAATGTGAGTTAGCTCACTCATTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGA
-
2Primer SequencesOligonucleotide Sequences:
Primer ID Primer Name Sequence 1 CR6261_28&30 Primer TGGTTCCTCCGTCAAAGTTTCCTGTAAGGCTTCTGGTGGTMCATTCAGMTCCTACGCCATCTCCTGGGTTAGACAAGCTCCTGGTCAAG 2 CR6261_58,59&62 Primer ACCTGAGTGGATGGGAGGAATCATTCCAATCTTCGGTACTRCCAAMTACGCTCMAAAATTCCAGGGTAGAGTCACCATTACTGCCGATGATTTCG 3 CR6261_93,98&104 Primer TTACATGGAGTTGAGTTCATTGAGATCCGAGGACACTGCCRTGTACTACTGTGCCARACACATGGGATACCAASTCAGAGAAACCATGGATGTCTGGGGTAAAGGAACAACCGTCA 4 CR6261_45 Primer CTACGCCATCTCCTGGGTTAGACAAGCTCCTGGTCAAGGACYTGAGTGGATGGGAGGAATCATTCCAATCTTCGGTACT 5 CR6261_74,75,76,77,79 Primer AAATTCCAGGGTAGAGTCACCATTACTGCCGATGAWTYCRCTRGMACCGYTTACATGGAGTTGAGTTCATTGAGATCCGAGGACACTGCC 6 CR6261_UCA_58,59,62 Primer AAATTCCAGGGTAGAGTCACCATTACTGCCGATGAWTYCRCTRGMACCGYTTACATGGAGTTGAGTTCATTGAGATCCGAGGACACTGCC 7 Secondary Primer CAAGTCCTCTTCAGAAATAAGCTTTTG The red colored nucleotides correspond to the codons with degenerate nucleotides that will encode mutations.Deep Sequencing Primer Sequences:Primer ID Primer Name Sequence 8 Forward Inner Primer GTTCAGAGTTCTACAGTCCGACGATCGCTTCTGGTGGT 9 Reverse Inner Primer CCTTGGCACCCGAGAATTCCACATGGTTTCTCT 10 Forward Outer Primer AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGATC 11 Reverse Outer Primer CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA
Where NNNNNN is the indexing barcode, for full sequences refer to (16)The Illumina universal sequences are colored blue for the forward primer set and green for the reverse primer set. The gene specific portion of the primer is colored orange. -
3
If the number of mutated residues in the desired library is between 2–8, a single plasmid can be used. If the desired library has greater than 8 mutated positions using two plasmid sequences, one with the starting sequence at each position and one with a mutated residue at each position, should be used in order to obtain complete combinatorial coverage (Fig. 1b). Use of two plasmid templates is recommended since the frequency of observed variants is log-normally distributed where variants with many mutations occurring less frequently. 8 positions is the recommended limit to obtain the distributions observed in Fig. 1c, d.
-
4
Based on the orientation of the BbvCI site in the sequence A and sequence B parental DNA plasmids, Nb.BbvCI is used first to generate the ssDNA template to which the mutagenic oligos anneal to. The orientation of the BbvCI site and design of mutagenic oligos determines which enzyme to use first in the protocol.
-
5
Plasmids prepared for nicking mutagenesis should not be subjected to multiple freeze-thaw cycles to preserve covalently closed molecules. In our hands, plasmid can be prepared in single aliquot use and frozen for up to a month in a non-defrosting −20 °C freezer.
-
6
Method optimization revealed that mutational efficiency improves with increasing primer length. A reasonable starting point for primer length would including 30bp homology arms upstream and downstream of any mutations, although depending on the location of the mutated residues may not be possible. In such a case, using primers with 24bp overhangs is sufficient.
-
7
Helpful troubleshooting with the nicking mutagenesis protocol is located in the published bio-protocol (15). Common issues include low numbers of transformants, difficulties in preparing the ssDNA template, or regenerating the complementary strand. Additionally, a positive control plasmid pEDA5-GFPmut3-Y66H is described in the bio-protocol that can help identify issues with any enzymes, buffers, or reagents used for generation of the combinatorial library.
-
8
T4 Ligase Buffer can be used with T4 Polynucleotide Kinase and does not require the addition of 1 μL of 10 mM ATP to be added to the reaction.
-
9
Sufficient mixing is crucial for even mutational incorporation, where each variant in the desired combinatorial library is expressed in relatively even frequencies. What we have found to be most reproducible is after each step to briefly spin down the PCR reaction and pipette up and down in the reaction tube with a pipette set to half of the total reaction volume.
-
10
The primer annealing step is critical to the success of the method. Sufficient mixing at each step is important, especially at this point. The optimization of this step revealed the importance of a quick anneal step, where the reaction drops in temperature from 98 °C to 55 °C rapidly instead of over 15 minutes as its written in the nicking mutagenesis original protocol. We found during optimization that slow annealing leads to preferential annealing of oligonucleotides with no or few mismatches. By quenching the temperature rapidly as opposed to over a 15 minute period, we were able to incorporate a more even distribution of the desired mutations. We also found that primers with a silent mutation to the template-matched oligonucleotide lead to more even distributions between template and desired mutant.
-
11
Intact wild-type parental DNA will be preferentially transformed over the ligated library. The DpnI step removes template to background levels. We find that additional mixing by pipetting up and down can help remove excess wild-type plasmid during the DpnI incubation.
-
12
The exact cell type for transformation is not important as long as the cells are high-efficiency with >109 CFU/μg plasmid DNA. A good target for the number of transformants desired is 100 transformants per library member to ensure >99% coverage of the desired library. This protocol typically yields on the order of 10 million transformants.
-
13
Sequencing the libraries prior to pooling is recommended in case errors arise in either library.
-
14
It is important to verify that the sequencing primers are amplifying a single clear region of your plasmid prior to NGS. This can be done by running 5 μL of the product from the second PCR reaction on a 2% agarose gel with proper gel stain to visualize band size. We use SYBR™ Safe DNA Gel Stain (ThermoFisher Cat # S33102).
-
15
Detailed description of fitness file inputs found in supplementary materials of Klesmith et al. 2018 (17). For the example CR6261 and CR6261_UCA, the following is an example input file with CR6261 (sequence A) as the reference sequence.
[pact]
pact_config_version: 2018.6
pact_protocol: fitness
[workflow]
fastq_merge_sel: True
fastq_merge_ref: False
fastq_filter_translate_ref: False
fastq_filter_translate_sel: True
filter_counter_sel: True
filter_counter_ref: False
enrichment: True
fitness: False
multiple_freq_mi: False
library_stats: True
[global]
wtdna:GAAGTTCAATTGGTTGAGTCCGGTGCTGAGGTCAAAAAACCTGGTTCCTCCGTCAAAGTTTCCTGTAAGGCTTCTGGTGGTCCATTCAGATCCTACGCCATCTCCTGGGTTAGACAAGCTCCTGGTCAAGGACCTGAGTGGATGGGAGGAATCATTCCAATCTTCGGTACTACCAAATACGCTCCAAAATTCCAGGGTAGAGTCACCATTACTGCCGATGATTTCGCTGGAACCGTTTACATGGAGTTGAGTTCATTGAGATCCGAGGACACTGCCATGTACTACTGTGCCAAACACATGGGATACCAAGTCAGAGAAACCATGGATGTCTGGGGTAAAGGAACAACCGTCACTGTCTCTAGT
wtaa: EVQLVESGAEVKKPGSSVKVSCKASGGPFRSYAISWVRQAPGQGPEWMGGIIPIFGTTKYAPKFQGRVTITADDFAGTVYMELSSLRSEDTAMYYCAKHMGYQVRETMDVWGKGTTVTVSS
processes: 4
directory: ./
output_prefix: CR6261_A
firstaamutated: 28
lastaamutated: 104
mutationtype: multiple
mutthreshold: 14
mutcodons: [[28,30,45,58,59,62,74,75,76,77,79,93,98,104]]
[fastq_merge_sel]
forward_fastq: CR6162-L2-MK_S2_L001_R1_001.fastq
reverse_fastq: CR6162-L2-MK_S2_L001_R2_001.fastq
directory:
min_coverage: 0.2
[fastq_merge_ref]
forward_fastq:
reverse_fastq:
directory:
min_coverage: 0.2
[fastq_filter_translate_sel]
fiveprimeanchor: TCTGGTGGT
enable_anchors: True
qaverage: 20
qlimit: 0
fastq_file:
[fastq_filter_translate_ref]
fiveprimeanchor:
enable_anchors:
qaverage:
qlimit:
fastq_file:
[filter_counter_sel]
read_file:
[filter_counter_ref]
read_file:
[enrichment]
ref_count_wildtype:
sel_count_wildtype:
ref_count:
sel_count:
ref_count_rejected:
sel_count_rejected:
ref_count_threshold:
sel_count_threshold: 10
strict_count_threshold: False
[fitness]
pact_enrichment_summary:
pact_enrichment_accept_nonsynon:
pact_enrichment_wtsynon:
manual_log2:
metric: e-wt
evalue_type: facs
evalue_facs_cellcount:
growth_gp:
facs_sd: 0.0
facs_pc: 0.0
[multiple_freq_mi]
pact_fitness_nonsynon:
frequency_log2_filter: False
[library_stats]
pact_enrichment_summary:
pact_fitness_nonsynon:
pact_fitness_wtsynon:
codon_type: {‘MCA’:[[28]],’AGM’:[[30]],’CYT’:[[45]],’RCC’:[[58]],’AAM’:[[59]],’CMA’:[[62]],’GAW’:[[74]],’TYC’:[[75]],’RCT’:[[76]],’RGM’:[[77]],’GYT’:[[79]],’RTG’:[[93]],’ARA’:[[98]],’STC’:[[104]]}References
- 1.Starr TN and Thornton JW (2016) Epistasis in protein evolution. Protein Sci 1204–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi GCG, Zhou P, Yuen CTL, et al. (2019) Combinatorial mutagenesis en masse optimizes the genome editing activities of SpCas9. Nat Methods 16:722–730 [DOI] [PubMed] [Google Scholar]
- 3.Poelwijk FJ, Socolich M, and Ranganathan R (2019) Learning the pattern of epistasis linking genotype and phenotype in a protein. Nat Commun 10:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wrenbeck EE, Klesmith JR, Stapleton JA, et al. (2016) Plasmid-based one-pot saturation mutagenesis. Nat Methods 13:928–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng L, Baumann U, and Reymond JL (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Püllmann P, Ulpinnis C, Marillonnet S, et al. (2019) Golden Mutagenesis: An efficient multi-site-saturation mutagenesis approach by Golden Gate cloning with automated primer design. Sci Rep 9:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams E, Copp JN, and Ackerley DF (2014) Site-Saturation Mutagenesis by Overlap Extension PCR. Methods Mol Biol 1179:315–33 [DOI] [PubMed] [Google Scholar]
- 8.Belsare KD, Andorfer MC, Cardenas FS, et al. (2017) A Simple Combinatorial Codon Mutagenesis Method for Targeted Protein Engineering. ACS Synth Biol 6:416–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunkel TA (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A 82:488–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekiert DC, Bhabha G, Elsliger M, et al. (2009) Antibody recognition of a highly conserved influenza virus epitope : implications for universal prevention and therapy. Science (80- ) 324:246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pappas L, Foglierini M, Piccoli L, et al. (2014) Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 516:418–422 [DOI] [PubMed] [Google Scholar]
- 12.Blundell JR and Levy SF (2014) Beyond genome sequencing: Lineage tracking with barcodes to study the dynamics of evolution, infection, and cancer. Genomics 104:417–430 [DOI] [PubMed] [Google Scholar]
- 13.Davidsson M, Diaz-Fernandez P, Schwich OD, et al. (2016) A novel process of viral vector barcoding and library preparation enables high-diversity library generation and recombination-free paired-end sequencing. Sci Rep 6:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina-Cucurella AV and Whitehead TA (2018) Characterizing Protein-Protein Interactions Using Deep Sequencing Coupled to Yeast Surface Display. Methods Mol Biol 1764:101–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steiner PJ, Baumer ZT, and Whitehead TA (2020) A Method for User-defined Mutagenesis by Integrating Oligo Pool Synthesis Technology with Nicking Mutagenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kowalsky CA, Klesmith JR, Stapleton JA, et al. (2015) High-resolution sequence-function mapping of full-length proteins. PLoS One 10:1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klesmith JR and Hackel BJ (2018) Improved mutant function prediction via PACT: Protein Analysis and Classifier Toolkit. 35:2707–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]