

Abstract
T cells are essential for mounting defense against various pathogens and malignantly transformed cells. Thymic development and peripheral T cell differentiation are highly orchestrated biological processes that require precise gene regulation. Higher-order genome organization on multiple scales, in the form of chromatin loops, topologically associating domains and compartments, provides pivotal control of T cell gene expression. CTCF and the cohesin machinery are ubiquitously expressed architectural proteins responsible for establishing chromatin structures. Recent studies indicate that transcription factors, such as T lineage-defining Tcf1 and TCR-induced Batf, may have intrinsic ability and/or engage CTCF to shape chromatin architecture. Here we summarize current knowledge on the dynamic changes in genome topology that underlie normal or leukemic T cell development, CD4+ helper T cell differentiation, and CD8+ cytotoxic T cell functions. The knowledge lays a solid foundation for elucidating the causative link of spatial chromatin configuration to transcriptional and functional output in T cells.
The mammalian genome contains billions of base pairs that are packed into the limited space of a cell nucleus through highly organized folding. Classical microscopy-based and modern next-generation sequencing studies demonstrate hierarchical organization of the genome. Each chromosome constitutes a condensed unit and occupies a physically distinguishable ‘chromosome territory’ in the nucleus (1). Within a ‘chromosome territory’, positioned towards the nuclear interior are A compartments that are gene-rich and transcriptionally active, while B compartments, which are gene-poor and transcriptionally inactive, are in the nuclear periphery and associated with the nuclear lamina (2). In the compartments, genomic regions in the megabase size show strong self-association, as revealed by high-throughput chromosome conformation capture (Hi-C) assay, and are termed topologically associating domains (TADs) (3, 4). Based on higher resolution Hi-C, TADs harbor subTADs or insulated neighborhoods on a smaller, sub-megabase scale in a nested configuration (5, 6) (Fig. 1A). Regardless of the scale difference, TADs and subTADs share two fundamental features, strong chromatin interactions within the TAD/subTAD and sparse chromatin interactions between neighboring TADs/subTADs, with the latter manifested as an “insulation” effect (7, 8) (Fig. 1A). Demarcating the boundaries of TADs/subTADs are long-range chromatin loops formed between two distal anchor regions, which are enriched with CTCF and cohesin binding (9, 10) (Fig. 1B). CTCF was originally identified as an insulator protein that blocks enhancer activities, and is now recognized as an essential genome organizer that establishes insulated neighborhoods and mediates enhancer-promoter interactions (11, 12). The cohesin complex consists of three core proteins, SMC1, SMC3, and RAD21, which form a ring-shaped structure (13) (Fig. 1B). According to the prevailing loop extrusion model, the cohesin complex loads onto the genome and “extrudes” the intervening DNA using its intrinsic ATPase activity (14, 15); this process is then stalled at a pair of CTCF binding sites in a convergent orientation (i.e., the asymmetric CTCF motifs facing each other) (Fig. 1B), forming the boundaries that separate a TAD/subTAD from its neighbors (16–18). As such, CTCF and cohesin colocalize on the genome, and acute deletion of either protein disrupts chromatin loops and TAD integrity (10, 19).
Figure 1. Organization of the genome into topologically associating domains (TADs) is orchestrated by architectural proteins.
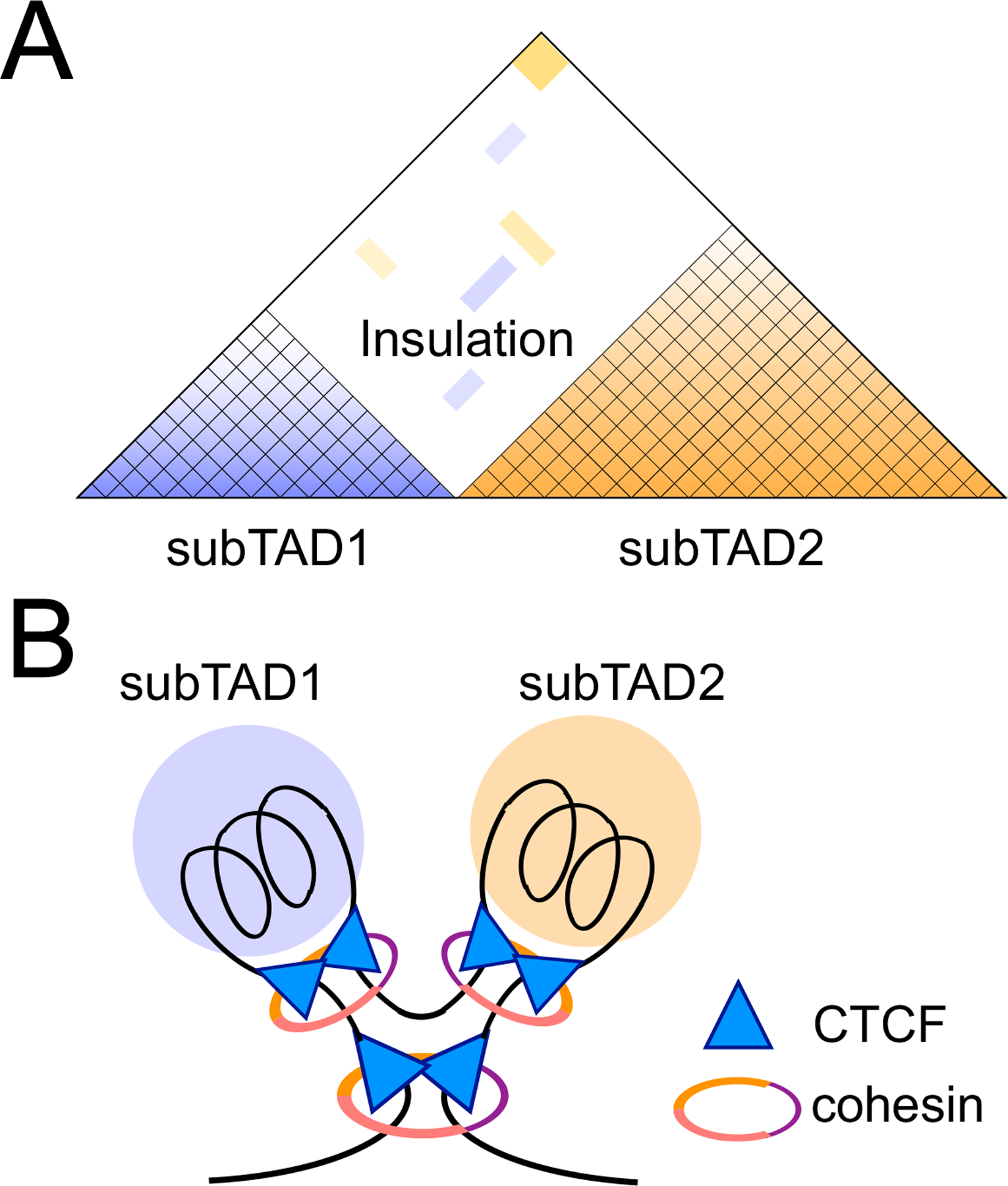
A. Diagram representation of a TAD that harbors subTADs in a nested configuration. Color gradient in each subTAD denotes chromatin interaction frequency as measured with Hi-C, and scarcity of chromatin contacts between subTADs represents an insulation effect.
B. Illustration showing positioning of CTCF and cohesin architectural proteins in organizing a TAD and subTADs. Different colors in the cohesin ring denote its major components, SMC1, SMC3, and RAD21. The top vertices of CTCF triangles are positioned to represent the most frequently observed convergent orientation of CTCF motifs at the TAD/subTAD boundaries. It should be noted that not all boundary CTCF motifs are in the convergent orientation on the linear genome sequence.
The genome organization on all these scales is not static. During cell differentiation, chromosome territories associated with the nuclear lamina can move into the nuclear interior (20), and compartments can switch between A and B states (21). While TADs appear to be relatively invariant across cell types, the boundary strength of some existing subTADs can be weakened, and new subTADs can form in more differentiated cells (7). These genome topological changes actively participate in gene regulation and cellular functional output (6, 7). T lymphocytes provide versatile protection against various pathogens and transformed cells, and their higher-order genome organization has gained more attention in recent years. Key advances shed new light on the contribution of sequence-specific transcription factors (TFs) to shaping chromatin architecture in T cells. For the unique aspects of genome reorganization that facilitate the recombination events at the antigen receptor gene loci, please refer to recent excellent reviews (22, 23). Here we focus on 3D chromatin architectural changes and their biological impacts during T cell development and differentiation under physiological and pathological conditions.
Reconfiguration of genome topology during T cell development.
Hematopoietic stem cells (HSCs) and their downstream progenitors such as common lymphoid progenitors (CLPs) seed the thymus, and give rise to early thymic progenitors (ETPs) (24). Activation of Notch signaling pathway in ETPs is essential for initiating a cascade of transcriptional activity, propelling T-lineage specification and commitment. Among the induced TFs, Tcf1 is rapidly induced by Notch signaling in ETPs, and Bcl11b and Gata3 are induced in the ensuing CD4−CD8− double negative 2 (DN2) stage (25–27). These TFs lay the groundwork for DN3 thymocytes to fully commit to the T-lineage with rearranged Tcrb locus and pre-TCR expression, and exert continued regulatory functions in maturation of DN3 to the CD4+CD8+ double positive (DP) cells, and in selection and lineage choice processes for DP thymocytes to become CD4+ or CD8+ single positive, mature T cells (28–31).
A comprehensive mapping of 3D genome organization using Hi-C from HSCs to DP thymocytes reveals systematic changes on the compartment and TAD levels, with the most drastic changes occurring at the transition of DN2 to DN3 cells (21). The compartments containing the stemness genes (e.g., Hmga2 and Meis1) switch from active A compartments in HSCs/CLPs to repressive B compartments in DN3 cells, while those containing T-lineage genes (e.g., Bcl11b and Ets1) switch from B compartments in HSCs/CLPs to A compartments in DN3 cells. On these large scales, the changes in compartment states and intra-TAD activity (i.e., chromatin interaction strength among anchor points within the TADs) are highly concordant with alterations in chromatin accessibility (ChrAcc) and gene expression accompanying differentiation of HSCs to T-lineage cells (21). The full commitment to T-cell lineage at the DN3 stage is thus associated with extensive reconfiguration of chromatin architecture, which facilitates continued activation of T-lineage transcriptional program while eliminating the potential to access non-T cell fate. One remarkable observation is on the timing of chromatin reorganization and gene expression changes. The stemness-associated genes are silenced before their resident compartments enter a repressive state (21), suggesting that the lagging architectural changes serve as a reinforcement to prevent fate reversion and ensure the progression of T-lineage maturation. On the other hand, intra-TAD activity is increased before the expression of enclosed T-lineage genes (21), demonstrating that a more permissive architectural framework is preconfigured to facilitate the induction of T-lineage transcriptional program. Such asynchronous timing arrangement appears to be an efficient strategy that locks in the T cell fate and prioritizes the forward T-lineage progression.
The force driving the extensive chromatin reorganization in developing thymocytes is of keen interest. An elegant case study of initiating Bcl11b expression reveals a requirement for the transcription of non-coding RNA ThymoD (thymocyte differentiation factor) in DN2 thymocytes (32). ThymoD is located ~850kb downstream of Bcl11b and spans an ~55kb region, with its transcription start site (TSS) overlapping with a known Bcl11b enhancer (27, 33) and its gene body containing several ChrAcc sites as potential regulatory elements. ThymoD transcription is essential for releasing the Bcl11b enhancer from the nuclear lamina to interior during differentiation of HSCs to DN2 cells (32). ThymoD transcription erases DNA methylation at the CpG islands within the locus, which in turn allows binding by CTCF and SMC3. Cohesin-mediated chromatin extrusion then brings the distal enhancers to the proximity of Bcl11b promoter to drive its timely expression at the DN2 stage (32). Upon induced expression, Bcl11b appears to directly contribute to genome reconfiguration, as Bcl11b binding sites are associated with elevated intra-TAD chromatin interactions during DN to DP maturation (21). Ablating Bcl11b in mature CD4+ T cells indeed causes reduction in global chromatin interactions, which is particularly profound at anchors enriched with strong Bcl11b binding (21).
Tcf1 is expressed in ETPs and further elevated in DN2 cells. Tcf1 has a high-mobility group (HMG) DNA binding domain, and the HMG family TFs share a characteristic feature of bending DNA upon binding the minor grooves of DNA double helix (34). Tcf1 has instructive roles at different stages of T cell development (35–37), and its impact on genome topology is starting to be elucidated. Ablating Tcf1 and its homologue Lef1 in mature CD8+ T cells affects genome organization on multiple scales, from compartments, TADs to chromatin loops (detailed below) (38). The density of Tcf1 binding sites in DP thymocytes is highly correlated with increased intra-TAD activity in DP over DN cells (39), based on Tcf1-centered analysis of Hi-C data in developing thymocytes at these early stages (21). Hi-C analysis of Tcf1-deficient DN3-like cell line and primary DP thymocytes consistently demonstrates a dependence of chromatin interactions on Tcf1 on the compartment and TAD scales (39). Based on ChIP-seq data in DP thymocytes, Tcf1 binding sites overlap with a substantial portion of CTCF-occupied genomic locations. CTCF solo sites (not co-occupied by Tcf1) consistently exhibit an insulation effect throughout CLP to DP stages as determined with Hi-C analysis (21, 39). On the other hand, the insulation by Tcf1+CTCF+ cobound sites is observed at CLP to DN2 stages but is lost from DN3 to DP thymocytes, coinciding with T-lineage specification and commitment (39). Although one has to assume that CTCF binding sites remain invariant throughout CLP to DP stages, it is clear that the “future Tcf1-bound” elements are at the boundaries of insulated regions in CLP-to-DN2 cells, and Tcf1 binding to these elements at DN3-to-DP cells diminishes the boundary strength, facilitating interactions between neighboring TADs/subTADs (39). These findings establish a critical contribution by T lineage-defining TFs to genome topological changes during T cell development. Tcf1 and Bcl11b TFs are induced earlier than full T-lineage commitment at the DN3 stage, and they have essential roles in launching the T-lineage transcriptional programs at the ETP/DN2 stages (25–27). It is also demonstrated that ectopically expressed Tcf1 gains access to regions marked with repressive histone modifications in fibroblasts and creates ChrAcc sites therein (40). It is therefore plausible to speculate that after their early induction, Tcf1 and Bcl11b establish de novo ChrAcc sites in ETP/DN2 cells, likely by acting in concert with other constitutively expressed TFs. The new ChrAcc sites can serve at least two purposes: one is to function as enhancer elements to promote transcription of proximal T-lineage genes, and the other is to facilitate chromatin interactions between their flanking regions and bring distal enhancers to the physical proximity of T-lineage genes. The latter may occur in existing TADs/subTADs, manifested as increased intra-TAD activities at Tcf1 and Bcl11b binding sites (21), as well as in previously insulated regions, manifested as “intermingling” of neighboring TADs/subTADs (40). These architectural changes could be incremental in magnitude in ETP/DN2 cells, and the gradual accumulation of the early events amounts to stabilized chromatin architectural reorganization that becomes more readily detectable in T-lineage committed DN3 cells. Although asynchronous in timing, the positive regulatory roles by Tcf1 and Bcl11b in inducing ChrAcc, priming gene transcription, and promoting chromatin interactions act synergistically to achieve complete and sustained activation of T-lineage program genes.
Chromatin interaction alterations during T helper cell differentiation
Thymus-derived mature CD4+ T cells that populate secondary lymphoid organs consist of two major subsets, Foxp3+CD25+CD4+ regulatory T (TREG) and Foxp3−CD25−conventional CD4+ T (TCONV) cells. TCONV cells have versatile helper functions by differentiating into various subtypes with distinctive cytokine production profiles, tailored to the pathogen types (41, 42). T helper 1 (TH1) cells produce IFN-γ in response to infection by intracellular bacteria and viruses, while TH2 cells produce IL-4, IL-5, and IL-13 cytokines to mount defense against parasites, in particular helminths, but elicit pathological responses to various allergens. Early studies of chromatin interactions in TH1 and TH2 cells were performed with chromosome conformation capture (3C) analysis. 3C-based scanning of the Ifng gene locus shows direct interactions of −70 kb upstream and +66 kb downstream CTCF-bound elements with the first intron of Ifng in naïve CD4+ T cells (43). These interactions are enhanced in TH1 cells in a CTCF- and Tbet-dependent manner (where Tbet is a TH1 lineage-defining TF), but weakened in TH2 cells (43). The TH2 cytokine-encoding genes (Il4, Il5, and Il13), along with Rad50, are distributed in tandem on the same chromosome. 3C analysis shows extensive interactions between TH2 cytokine gene promoters and ChrAcc sites within this region in naïve CD4+ T cells, and the interaction strength is weakened in the absence of Stat6 in TH2 cells and appears to require Gata3 (a TH2 lineage-defining TF) for maintenance (44). In addition, CTCF binds to the TH2 cytokine gene loci, and loss of CTCF impairs TH2 cytokine production (45). Besides the intra-chromosome interactions, the TH2 cytokine gene loci interact with Ifng and Il17a genes on separate chromosomes in naïve CD4+ T cells (46, 47), and the inter-chromosomal association between Ifng and the TH2 cytokine gene loci is nonetheless lost after CD4+ T cell activation (46).
TH1 cells cultured in a condition with higher IL-2 concentration show increased CTCF binding strength at key gene loci such as Slc2a1 and Hk2 (encoding Glucose transporter 1 and hexokinase 2, respectively) than those cultured with lower dose of IL-2, and the increased CTCF binding is associated with higher chromatin interaction probability, as determined with Hi-C (48). Addition of α-ketoglutarate, a glycolytic metabolite, to low IL-2 TH1 cells can partly enhance CTCF binding and associated chromatin interactions (48). These observations suggest that chromatin interactions in TH1 cells show dynamic changes in response to the metabolic state and may underlie the regulation of IL-2-dependent effector program. Unlike strong dependence on CTCF in TH2 cells, genetic ablation of CTCF only modestly reduces Tbet induction and IFN-γ production (45), and CTCF knockdown also moderately diminishes expression of IL-2-dependent genes (48). These observations suggest that other factors besides CTCF are necessary for bridging enhancer-promoter interactions during TH1 cell differentiation. In line with this notion, stimulation of CD4+ T cells primed under non-polarizing (TH0) condition with IL-2 potently induces chromatin interaction loops engaging RNA polymerase II-associated promoters such as Il2ra and Socs1, as determined with Chromatin Interaction Analysis by Paired-End Tag (ChIA-PET) (49). These IL-2-induced loop anchors are enriched in Stat5 binding sites, which rarely overlap with CTCF binding detected in naïve CD4+ T cells (49).
TH17 cells are important for defense against extracellular bacteria and fungi, and produce characteristic IL-17A and IL-17F cytokines, as well as IL-21 and IL-23. Batf, an AP-1 family TF, is considered as a pioneer factor that initiates chromatin remodeling during TH17 cell differentiation (50). Under TH17 polarizing or TH0+IL-6 condition, Batf directly recruits CTCF, as exemplified at the Il21 gene locus (51). Hi-C analysis shows that loss of Batf causes globally diminished chromatin interactions in TH0+IL-6-activated CD4+ T cells; at the Il21 locus specifically, 3C analysis validates that Batf is critical for establishing chromatin loops that link a +28 kb downstream region to its −41 to −80 kb upstream regions (51). In addition, loss of Ets1 diminishes CTCF binding to Batf-bound locations, and Batf deficiency impairs Ets1 expression and its binding to key TH17 gene loci (51). The analyses suggest that TFs act cooperatively to stabilize CTCF binding and chromatin architectural changes to induce the TH17 program.
Unlike TCONV cells that confer immune protection against various pathogens, TREG cells are immunosuppressive and essential for self-tolerance, with Foxp3 as the TREG lineage-defining TF (52). H3K27ac-based HiChIP (a combination of Hi-C and chromatin immuno-precipitation targeting a specific protein) analysis reveals extensively different configuration of enhancer-promoter loops between TREG and TCONV cells, and the subset-specific loop strength is positively correlated with corresponding gene expression levels, as observed at TREG-specific Il2ra and TCONV-specific Lef1 gene loci (53). By comparing H3K27ac HiChIP between WT TREG and Foxp3-null TREG-like cells, Foxp3 deficiency diminishes enhancer-promoter loop strength, which is more evident at Foxp3-dependent genes such as Ikzf2 (53). Furthermore, Foxp3-based HiChIP in TREG cells shows that Foxp3 is directly involved in chromatin interactions connecting distal regulatory regions to ~5,000 promoters, beyond the known ~500 TREG signature genes; nonetheless, high-density Foxp3-anchored chromatin interactions are more strongly associated with Foxp3-dependent TREG signature genes and Foxp3-dependent enhancer-promoter loops (53). While Foxp3-bound promoter anchors are enriched in TATA-binding motifs, Foxp3-bound enhancer anchors have CTCF as one of the over-represented motifs (53). These data highlight that a major mechanistic action taken by Foxp3 is to directly mediate enhancer-promoter interactions for transactivation of a subset of Treg signature genes, in addition to its previously recognized repressive role in gene regulation.
Based on the knowledge accumulated over the past two decades, a likely scenario emerges for the interplay between TFs and genomic organization that directs helper subtype differentiation. The helper subtype-associated cytokine gene loci have pre-existing chromatin configuration that positions potential enhancers in architectural proximity of promoters in naïve CD4+ TCONV cells, where the genes remain transcriptionally silent due to DNA hypermethylation and/or repressive histone modifications. Upon activation by their cognate antigens, the cytokine gene loci are demethylated, as demonstrated for Il4 and Ifng genes (54, 55), repressive histone marks erased and/or replaced with active ones, resulting in activation of existing or de novo enhancers, leading to cytokine gene transcription. During this process, the TCR-induced early response TFs such as Batf, cytokine-mobilized Stat TFs, and/or subtype/subset lineage-defining TFs not only have direct impact on ChrAcc and enhancer activation, but also engage architectural proteins including CTCF to establish de novo chromatin interactions and/or enforce pre-configured enhancer-promoter contacts that are appropriate for the helper subtypes. It is conceivable that beyond helper subtype-characteristic cytokine gene loci, there is extensive genome reorganization across different subtypes, and applying modern technology for a systematic comparison of chromatin architecture among the helper subtypes will advance our understanding of their lineage stability and plasticity in the 3D genomic space.
Fluidity of chromatin architecture in CD8+ T cell identity, homeostasis, and memory formation.
CD8+ T cells have remarkable cytotoxicity that destructs cells infected with intracellular pathogens or transformed cells expressing neoantigens (56, 57). After populating the peripheral lymphoid organs, CD8+ T cells need to maintain 1) their identity as dedicated cytotoxic cells, 2) a naïve state to prevent premature activation before encountering their cognate antigens, and 3) a stable pool size to sustain immunocompetence. Tcf1 and Lef1 TFs are essential for all these aspects, through positively regulating T cell identity genes, repressing non-T lineage or effector-associated genes, and supporting a transcriptional program required for homeostatic proliferation (38, 58). Significantly, these regulatory functions by Tcf1/Lef1 are intimately linked to their ability to modulate 3D genome organization. Hi-C analysis of naïve CD8+ T cells shows strong correlation between the density of Tcf1 binding sites and chromatin interaction scores in compartments, TADs, and chromatin loops; importantly, genetic ablation of Tcf1/Lef1 in mature CD8+ T cells diminishes interaction scores on all these genomic scales (38). Tcf1/Lef1 also sustain ChrAcc state and super enhancer activities in naïve CD8+ T cells, and Tcf1/Lef1-dependent ChrAcc sites and super enhancers are highly enriched in anchors of Tcf1/Lef1-dependent chromatin loops (38). For example, Tcf1/Lef1 positively regulate Myb expression in naïve CD8+ T cells. Upon loss of Tcf1/Lef1, a TAD of ~1.2 Mbp harboring the Myb gene locus maintains the overall structural integrity but shows potent reduction in chromatin interactions within the Myb-containing sub-TAD and those between the Myb-containing subTAD and its neighboring subTAD (Fig. 2A). Within the Myb-containing subTAD, a ~250kb chromatin interaction hub covering the Myb gene locus and its upstream regions is most profoundly affected by Tcf1/Lef1 deficiency (Fig. 2B,C). At the hub center is a cluster of Tcf1/Lef1-dependent ChrAcc sites and enhancers, which shows diminished interaction with the Myb promoter region in Tcf1/Lef1-deficient CD8+ T cells (Fig. 2C,D). These findings underscore the highly coordinated nature of TF binding, ChrAcc, super enhancer activity, and chromatin interactions, which exerts integrative control of transcriptional and functional output by naïve CD8+ T cells.
Figure 2. Tcf1 and Lef1 transcription factors shape genome organization on multiple scales.
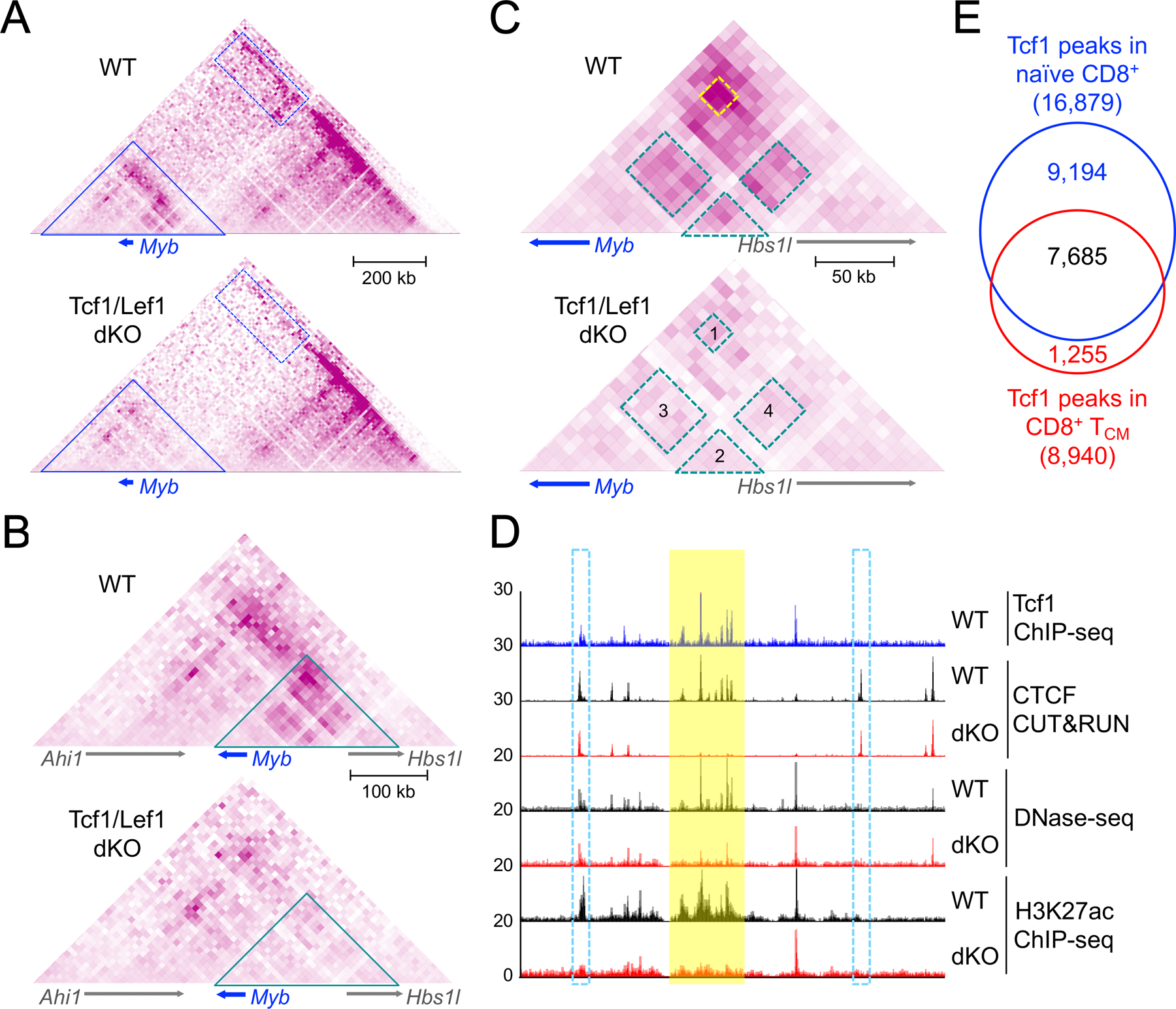
A–C. Hi-C heatmaps showing the impact of Tcf1/Lef1 deficiency on genome organization surrounding the Myb gene locus on the TAD (A), subTAD (B), and hub (C) scales. A illustrates a 1.2 Mbp TAD in WT and Tcf1/Lef1 double knockout (dKO) naïve CD8+ T cells, where rectangles with dotted blue lines denote Tcf1/Lef1-dependent interactions between subTADs. The Myb-containing subTAD (denoted with triangles with blue lines) is enlarged in B, where triangles with cyan lines denote chromatin interaction hubs showing strong dependence on Tcf1 and Lef1. Enlarged hubs are displayed in C, where rectangles with dotted lines highlight four Tcf1/Lef1-dependent chromatin interaction patches. Hi-C data are from Refs. 38 and 58, and displayed in 10kb resolution, with arrows marking transcription orientation of key genes.
D. Molecular characteristics at the genomic element level, aligned with chromatin interaction anchors in C. Shown are sequencing tracks of Tcf1 and CTCF binding, ChrAcc (determined with DNase-seq), and H3K27ac marking active promoters/enhancers. Yellow bar highlights a cluster of Tcf1+CTCF+ cobound sites (also part of a super enhancer spanning Myb) showing Tcf1/Lef1-dependent CTCF recruitment, ChrAcc, and enhancer activity, and the intra-cluster chromatin interaction (patch #2 in C) shows Tcf1/Lef1 dependence. The ChrAcc/enhancer cluster shows Tcf1/Lef1-dependent interactions with Myb and Hbs1l promoter regions (patches #3 and #4 in C, respectively). Open bars with dotted blue lines denote a Tcf1+CTCF+ cobound site at the Myb promoter and a CTCF solo site in Hbs1l intron, which form a strong, Tcf1/Lef1-dependent interaction loop (patch #1 in C), likely marking the boundaries of a ‘semi-insulated’ hub.
E. Venn diagram showing Tcf1 peaks in naive CD8+ and TCM cells. Tcf1 ChIP-seq data in naive CD8+ and TCM cells are from Ref. 38 (GSE164713) and Ref. 62 (GSE177064), respectively, and after mapping to the mm9 mouse genome, high-confidence Tcf1 binding sites were called with MACS2 by the requiring ≥ 4-fold enrichment over a negative control (i.e., Tcf1-deficient naive CD8+ T cells) and false discovery rate < 0.05, with genomic regions on the blacklist removed.
Global mapping of CTCF binding in naïve CD8+ T cells shows remarkable colocalization of CTCF and Tcf1, with >50% Tcf1 binding sites also occupied by CTCF. Both proteins physically interact, and at ~1/3 of Tcf1+CTCF+ cobound sites (detected in non-promoter regions), CTCF binding depends on Tcf1/Lef1. Importantly, the prevalent Tcf1-CTCF colocalization and Tcf1-depending CTCF recruitment in CD8+ T cells are independently validated by both CUT&RUN and ChIP-seq methods (58). CTCF solo sites have weak to negligible signals of ChrAcc and H3K27Ac, and are more frequently found at TAD boundaries with strong insulation effect; in contrast, Tcf1+CTCF+ cobound sites are mostly ChrAcc sites with strong H3K27Ac signals, and are more frequently found within the TAD with minimal insulation, if any (58). Furthermore, Tcf1/Lef1-dependent CTCF binding sites show diminished chromatin interaction scores, reduced ChrAcc and H3K27ac in Tcf1/Lef1-deficient CD8+ T cells (58). Taking the Myb gene locus as an example, the cluster of Tcf1/Lef1-dependent ChrAcc sites and enhancers shows Tcf1/Lef1-dependent CTCF recruitment (Fig. 2D), which directly accounts for reduced chromatin interactions within the ChrAcc/enhancer cluster and reduced interactions of the ChrAcc/enhancer cluster with its upstream Myb and downstream Hbs1l promoters (Fig. 2C). These data indicate that Tcf1/Lef1 directly engage and cooperate with CTCF, and this cooperativity can serve at least two purposes: Tcf1/Lef1 recruit CTCF as a transcriptional coactivator to increase ChrAcc and endow enhancer activity, and Tcf1/Lef1 integrate the genome-organizing capacity of CTCF with their own to further promote chromatin interactions. These mechanisms act additively or in synergy to achieve precise regulation of CD8+ T cell functionality.
CD8+ T cells undergo homeostatic proliferation in response to cytokines including IL-7 and IL-15 (59). Ex vivo stimulation of CD8+ T cells with IL-7/IL-15 mobilizes not only the well-established Stat5 pathway but also unexpectedly CTCF, that is, CTCF binding sites show increased binding strength and are created de novo after stimulation with these homeostatic cytokines (58). Hi-C analysis of the IL-7/15-stimulated CD8+ T cells demonstrates that the cytokine-mobilized CTCF sites can forge stronger chromatin interactions among each other and with invariant CTCF binding sites (i.e., sites not affected by cytokines). Approximately half of the cytokine-induced CTCF binding sites depends on intact Tcf1/Lef1 expression, and the cytokine-induced chromatin interactions also show Tcf1/Lef1 dependence, especially those anchored at the Tcf1/Lef1-dependent CTCF binding sites, as observed at the Tnfsf8 (encoding CD30L) and Eomes gene loci (58). Whereas it remains to be elucidated how CTCF is mobilized, these data suggest that chromatin interactions are fluidic and responsive to environmental stimuli, and can actively participate in gene regulation to meet biological needs. In this process, both lineage-specific Tcf1/Lef1 and the ubiquitously expressed CTCF act together to integrate local chromatin changes and higher-order genome organization to induce the transcriptional program underlying homeostatic proliferation of CD8+ T cells. In support of this notion, Tcf1/Lef1 and CTCF do regulate a similar set of genes that promote cell cycle progression, DNA replication, and protein synthesis, as determined with transcriptomic analysis of CD8+ T cells exposed to lymphogenic environment (58).
Upon encountering their cognate antigens, CD8+ T cells are activated and differentiate into effector cells equipped with cytotoxic molecules. Hi-C analysis of human CD8+ T cells that are activated in vitro for 72 hrs show extensive changes in chromatin interactions compared with naive cells, and these changes are concordantly associated with changes in ChrAcc and gene expression on a global scale (60). TAD boundary strength is also altered, and remarkably, the in vitro activated CD8+ T cells have stronger intra-TAD interactions in shorter range, resulting in an increased number of identifiable subTADs (60). A fraction of antigen-experienced CD8+ T cells persists as memory cells for a long term to provide enhanced protection against the same pathogens. Compared with naive CD8+ T cells, CD62L+ central memory CD8+ T (TCM) cells expressed Tcf1 at a decreased level (61) and have about half as many high-confidence Tcf1 binding sites as determined with ChIP-seq (38, 62), while Tcf1 peaks detected in TCM cells mostly overlap with those in naive CD8+ T cells (Fig. 2E). Systematic comparative analysis of genome organization between naive and memory CD8+ T cell has not been performed to date; however, Hi-C analysis of wild-type and Tcf1-deficient TCM cells reveal a critical requirement for Tcf1 to mediate chromatin interactions and preprogram responsiveness of TCM cells to recall stimulation (62). Unlike naïve CD8+ T cells undergoing homeostatic proliferation where Tcf1 expression is preserved, TCM cells rapidly downregulate Tcf1 upon recall stimulation. Among recall-induced ChrAcc sites in wild-type TCM cells, approximately half of the sites show insufficient ChrAcc induction in the absence of Tcf1, as observed at the Id3 and many glycolytic gene loci; however, only <3% of those Tcf1-dependent, recall-induced ChrAcc sites are preoccupied by Tcf1 in TCM cells (62), indicating that Tcf1 rarely functions as a placeholder for TCR-induced enhancer elements. Therefore, Tcf1-mediated ‘preprogramming’ is at least partially ascribed to its ability to mediate chromatin interactions in resting TCM cells. The Tcf1-dependent chromatin interactions in TCM cells act in at least two formats: one is to bridge direct interaction of the “would-be” enhancers to target gene promoters, and the other is to connect two distal boundary-like elements which span the “would-be” enhancers and target gene promoters in an architecturally poised state (62). These architectural configurations in TCM cells can thus allow recall-induced enhancers to reach their target promoters more rapidly or in higher frequency, leading to more efficient induction of Id3, mobilization of glycolysis, and cytotoxic machineries in the secondary effector cells. Collectively, these detailed analyses in CD8+ T cells reveal that genome reorganization on multiple scales constitutes an important regulatory mechanism for target gene expression at homeostatic state and in response to external stimuli, and during this process, TFs such as Tcf1 have pivotal contributions to shaping the chromatin architecture, alone or in cooperativity with CTCF.
Aberrant chromatin interactions in pathological T cells
Besides physiological development and differentiation processes, T cells exhibit alterations in genome organization in pathological conditions. Hyperactive T cells are a major cause of autoimmune diseases, and genome-wide association studies have identified large genomic regions that contain sequence variants associated with higher risks of autoimmunity (63). Non-obese diabetic (NOD) mouse strain is a useful genetic model of Type 1 diabetes (TID), and comparisons between NOD and diabetes-resistant C57BL/6 mice identify dozens of “insulin-dependent diabetes” (Idd) regions that are linked to diabetes susceptibility (64). A recent study employs SMC1-based HiChIP to compare genome organization in DP thymocytes from disease-free young NOD and C57BL/6 mice (65). This analysis identifies densely inter-connected enhancer-enhancer and enhancer-promoter interaction regions, defined as ‘3D cliques’, and the top ‘cliques’ harboring T cell identity genes such as Bcl11b and Ets1 show similar chromatin configuration between both strains. In contrast, the diabetes-prone NOD DP thymocytes have unique ‘cliques’ harboring Idd9.2 and Idd6.AM loci, and the ‘clique’ boundaries are marked with NOD strain-specific CTCF binding sites (65). Fluorescent in situ hybridization (FISH) assay confirms that compared with wild-type cells, NOD DP thymocytes show closer physical proximity among the inter-connected regions in the Idd ‘cliques’ in a single cell. KRAB-ZFP family genes in the Idd9.2 locus show elevated expression in pancreas-infiltrating immune cells from the T1D patients, especially among the fast progressor group (65). These findings suggest that genetic variants are associated with differential CTCF distribution and chromatin configuration changes, and the resulting chromatin hyper-connectivity predisposes T cells for aberrant gene expression linked to increased risk of autoimmunity.
T cell acute lymphoblastic leukemia (T-ALL) derives from malignant transformation of thymocytes at early developmental stages (66). T-ALL is genetically heterogeneous, resulting from activation of various oncogenes due to point mutations, gene amplifications, and chromosomal translocations. SMC1-based ChIA-PET analysis of Jurkat T-ALL cells shows that TAL1 and LMO1 oncogenes reside in subTADs/insulated neighborhoods, and CRISPR/Cas9-mediated excision of CTCF/SMC1-occupied boundary sites results in increased expression of both oncogenes in a non-T cell, fibroblast cell line (67). In various types of cancers, recurrent mutations are more frequently observed in boundary than non-boundary CTCF sites (67). This pioneering work demonstrates that genetic changes disrupting normal genome organization are a novel causative event leading to oncogene activation and malignant transformation of T cells.
Systematic Hi-C analyses comparing T-ALL (primary or cell lines with activating mutations in NOTCH1) with normal T cells demonstrate prevalent changes in compartment activity and intra-TAD chromatin interactions upon T cell transformation, and these genome topological changes are concordant with those in gene expression, super enhancer activity, and CTCF binding strength at the TAD boundaries (68). Gain-of-function NOTCH1 mutations are a leading cause of T cell progenitor transformation (69), and the activating mutations result in release of intracellular domain of NOTCH1 (ICN). ICN then translocates into the nuclei and forms a complex with RBPJ transcription factor, leading to activation of a wide array of target genes (70). NOTCH1 target genes such as APCDD1 and IKZF2 show greatly elevated chromatin interactions in their resident TADs in T-ALL over normal T cells. The MYC oncogene is regulated by a super-enhancer that is more than 1 Mbp downstream (71), and ICN acts on select component enhancer(s) to promote normal thymic development and T-ALL leukemogenesis (72, 73). In normal T cells, the MYC gene and its super-enhancer are segregated into two neighboring TADs as observed in Hi-C, and there are few interactions between the MYC promoter and enhancers as determined with a promoter-centered, circular 3C (a.k.a. 4C) assay (68). In contrast, in NOTCH1-mutated primary T-ALL and CUTLL T-ALL cell line, the interactions between the two TADs are increased so intensely that the MYC promoter and super-enhancer are in a reorganized larger TAD, where the MYC promoter forms strong interaction loops with multiple component enhancers (68). Of note, the CTCF binding that demarcates the boundary between MYC promoter and super-enhancer TADs in normal T cells is lost in T-ALL cells, with the boundary virtually erased after malignant transformation (68). Significantly, the elevated chromatin interactions, in particular the promoter-enhancer loops, in T-ALL cells show distinct sensitivity to small molecule inhibitors, as determined with the 4C assay in CUTLL cells. For example, the interaction strength of a loop linking APCDD1 promoter with its enhancer is diminished by a γ-secretase inhibitor (GSI) which blocks NOTCH1 activation, while that of loops connecting MYC and IKZF2 promoters with their respective enhancers is not sensitive to GSI, but is diminished by an inhibitor of cyclin-dependent kinase 7 (68). These findings underscore that chromatin architecture in transformed cells can be a druggable target to inhibit oncogenic events, representing a new frontier for exploring novel therapeutic options.
The multi-scale genome topological aberrations in T-ALLs require input from lineage-defining TFs. In a murine T-ALL model where ICN is ectopically expressed in bone marrow HSCs and progenitor cells, ICN-induced T-ALL is abrogated in the absence of Tcf1 (74). While loss of Tcf1 does not profoundly impact compartment and TAD activities in ICN-expressing HSCs as determined with Hi-C, a substantial portion of ICN-induced chromatin interaction loops depends on Tcf1, including that linking Bcl11b promoter and its distal enhancer (74). In addition, two ICN-induced distal enhancers for Myc depend on Tcf1 to remain in ChrAcc state, and germline deletion of either enhancer abrogates T-ALL development (72, 74). These data suggest that Tcf1-mediated chromatin interactions contribute to oncogenic events induced by aberrant NOTCH1 signaling. It should be noted that Tcf1 could function as a tumor suppressor in early thymocytes (28, 75), and further studies are needed to reconcile these findings and the underlying chromatin architectural changes.
NOTCH1-mutated T-ALL cells are initially sensitive to GSI but grow resistant after repeated exposure, undermining NOTCH1-targeted cancer therapy. Analysis of GSI-resistant, NOTCH1-mutated DND41 T-ALL cell line with Hi-C and SMC1-based HiChIP reveals extensive genome topological alterations compared with the parental, GSI-sensitive cells, including compartment activity, TAD boundary strength, and enhancer-promoter interaction, and these changes are concordant with those in enhancer activity, ChrAcc, and gene expression (76). GSI-resistant cells greatly diminish Tcf1 and Lef1 expression, but acquire the expression of Ebf1, a B cell lineage-defining TF. Notably, SMC1, CTCF, and YY1 (a ubiquitously expressed TF with an architectural role) (77) show colocalization with Tcf1/Lef1 in parental T-ALL cells, but are redistributed to Ebf1 binding sites in GSI-resistant T-ALL cells; the redistribution of architectural proteins is concordant with loss of Tcf1/Lef1-anchored and gain of Ebf1-anchored chromatin loops in GSI-resistant cells (76). These findings highlight a critical contribution by TFs to dynamic genome reorganization under pathological conditions. In fact, ectopic expression of Ebf1 or Tcf1 in NOTCH1-mutated breast cell line establishes de novo chromatin loops (76).
Conclusions.
Technical advances in the past two decades have greatly improved our understanding of genome organization in T cells. It becomes clear that during thymic development and mature T cell responses to various stimuli, chromatin interactions undergo dynamic changes on different scales including compartment switching, subTAD reorganization, and chromatin loop rewiring. These genome topological changes are critical for establishing and sustaining gene expression programs appropriate for specific cell fates. In this process, lineage-specific TFs such as Tcf1 and Batf have pivotal roles, especially those possessing the ability to initiate nucleosome displacement in a sequence-specific manner, establish chromatin accessibility and facilitate deposit of active histone marks. These factors can directly establish novel chromatin interactions or indirectly recruit CTCF to facilitate new loop formation. In this context, either the TF or TF-CTCF complex can function as one anchor point to direct loading of the cohesin complex, which then drives DNA extrusion till encountering the other anchor, demarcated by dynamic or constitutive CTCF binding sites (Fig. 3A,B). As exemplified at the Myb gene locus, the Myb promoter is co-occupied by Tcf1 and CTCF and forms strong chromatin interaction loop with a CTCF solo site located in the Hbs1l intron; and this loop is destabilized upon Tcf1/Lef1 ablation in naïve CD8+ T cells (Fig. 2C,D). NIPBL and MAU2 are responsible for loading of the cohesin complex onto chromatin (13, 78). At >2,000 Tcf1 binding sites, H3K27ac, SMC1, and NIPBL binding show concordant reduction in Tcf1-deficient DN3 cells (39). Coupled with the unique ability of HMG TFs to bend DNA at the local element level (34), it is tempting to speculate that Tcf1-bound, Tcf1-dependent enhancers could serve as cohesin loading sites (Fig. 3C). The TF-initiated chromatin interactions can have at least two major impacts: one is to promote juxtaposition of distal enhancer(s) and promoter for target gene regulation, and the other is to create a ‘semi-insulated’ interaction hub where enhancer(s) and promoter have increased contact frequencies. In the latter case, the size of the hub does not need to be on par with a subTAD or insulated neighborhood, and the boundaries of the hub do not need to be strong enough to achieve complete insulation, so that the hub has ‘autonomy’ in regulating its encompassing genes and yet retains the ability of communicating with its neighboring hubs. The combinatorial use of these mechanisms may thus establish and sustain lineage/subset-specific transcriptional programs.
Figure 3. Projected working models of transcription factors (TFs) acting cooperatively with architectural proteins in T cell genome organization.
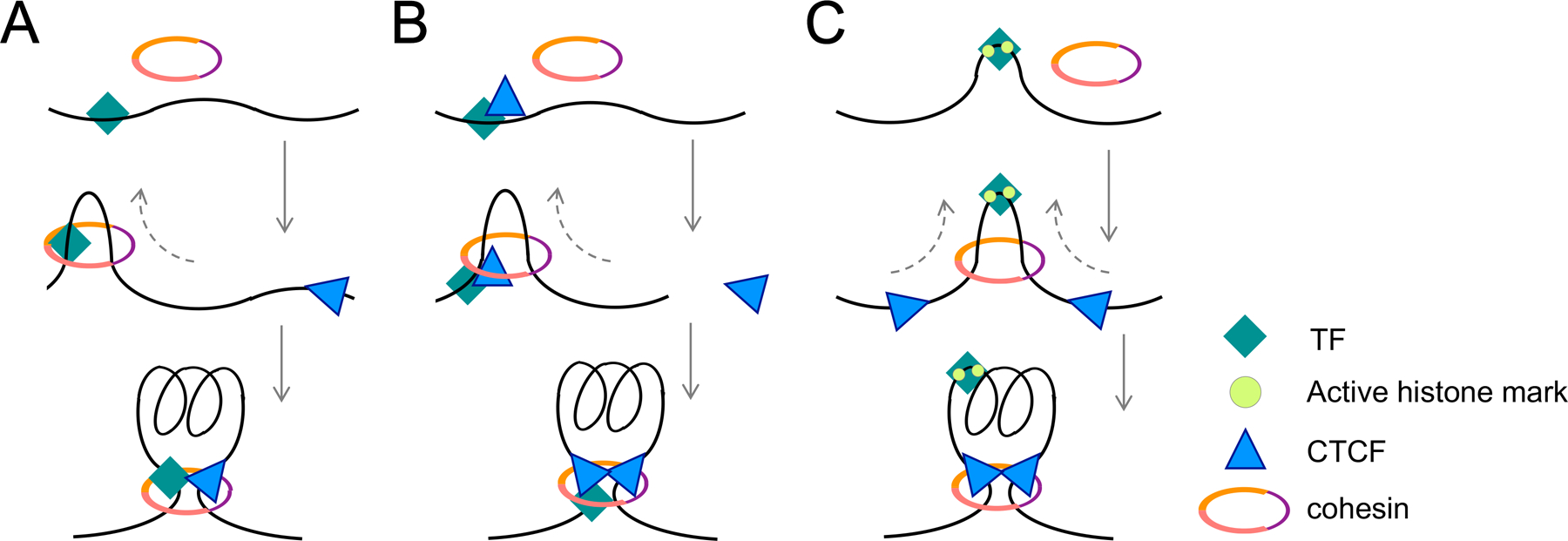
T lineage-specific TFs bind to specific DNA sequences and create nucleosome-free elements. Taking Tcf1 as an example, Tcf1 may act alone based on its intrinsic architectural function (A) or recruit CTCF (B) to assist loading of cohesin complex, which then extrudes DNA till encountering the other boundary-demarcating CTCF on the genome. TFs including Tcf1 facilitate chromatin opening and deposit of active histone marks; coupled with the DNA-bending effect by the HMG domain, Tcf1-bound enhancer elements may contribute to cohesin loading, and the ensuing symmetrical extrusion forms chromatin loops (C). Dotted arrows denote extrusion direction.
These simplistic models clearly require refinements in many aspects. It is frequently demonstrated that changes in genomic architecture and gene expression are concordant on large scales, but more often than not, there is a disconnect when a specific gene is examined, that is, a gene expression change is not linearly linked to a specific chromatin loop or an interaction hub. One underlying issue is the resolution of Hi-C data, which is usually at 5–10 kb at the best, partly owing to the scarcity of DN thymocytes and antigen-specific memory T cells. With ultra-deep sequencing, base-pair resolution is achievable (79), which is nonetheless associated with substantial cost for sequencing and collecting sufficient rare cells. Single-cell Hi-C starts to gain traction (80), yet the resolution challenge remains. Another significant challenge is the productive processing of Hi-C data, which is often hampered by the enormous data size and high levels of variability among batches and laboratories. Integrative use of multiple algorithms is necessary to establish replicate reproducibility, calibrate proximal interactions, and define chromatin structure on multiple scales. Outstanding issues include Hi-C normalization across conditions to facilitate comparative analysis, and identification of function-linked, structural units that go beyond loops between two focal anchor points without reaching TADs/subTADs in the size of megabases. The recently developed HiCHub algorithm (81) employs a network approach to define hyper-connected ‘hubs’ on an intermediate scale between loops and TADs, and such ‘hubs’ show strong association with concordant changes in chromatin state and gene expression. HiCHub also works well with data of limited sequencing coverage and facilitates the integration of one-dimensional epigenetic features onto the chromatin architecture. Improvements in Hi-C data processing and analysis, with the perspective of integrating machine learning (82, 83), require multi-disciplinary collaborations to ensure that the data output makes mathematical and biological senses. Lastly, an important goal is to establish causative effect of chromatin interactions on functional alterations. CRISPR-based genome editing will help discern the impact through disrupting an enhancer-promoter interaction or boundaries of TADs, subTADs, and ‘semi-insulated’ interaction hubs. Dissecting functionally consequential chromatin interactions in high resolution may identify novel therapeutic targets, from the unique angle of genome topology, which might achieve more specific effects on enhancing anti-pathogen T cell immunity, blocking malignant T cell proliferation, and dampening self-reactive T cells.
Acknowledgements
We thank Ms Blanca Teran for careful proofreading.
H.-H.X and W. P. are supported by the NIH (AI121080 and AI139874). H.-H.X. is also supported by AI112579 and Veteran Affairs (BX005771).
References
- 1.Misteli T 2007. Beyond the sequence: cellular organization of genome function. Cell 128: 787–800. [DOI] [PubMed] [Google Scholar]
- 2.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, and Dekker J. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, and Heard E. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hnisz D, Day DS, and Young RA. 2016. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 167: 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beagan JA, and Phillips-Cremins JE. 2020. On the existence and functionality of topologically associating domains. Nat Genet 52: 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu M, and Ren B. 2017. The Three-Dimensional Organization of Mammalian Genomes. Annu Rev Cell Dev Biol 33: 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon JR, Gorkin DU, and Ren B. 2016. Chromatin Domains: The Unit of Chromosome Organization. Mol Cell 62: 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, Michalski P, Piecuch E, Wang P, Wang D, Tian SZ, Penrad-Mobayed M, Sachs LM, Ruan X, Wei CL, Liu ET, Wilczynski GM, Plewczynski D, Li G, and Ruan Y. 2015. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163: 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG. 2017. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169: 930–944 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braccioli L, and de Wit E. 2019. CTCF: a Swiss-army knife for genome organization and transcription regulation. Essays Biochem 63: 157–165. [DOI] [PubMed] [Google Scholar]
- 12.Ren G, Jin W, Cui K, Rodrigez J, Hu G, Zhang Z, Larson DR, and Zhao K. 2017. CTCF-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol Cell 67: 1049–1058 e1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill VK, Kim JS, and Waldman T. 2016. Cohesin mutations in human cancer. Biochim Biophys Acta 1866: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA. 2016. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep 15: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, Sanborn AL, Canela A, Maman Y, Oksanen A, Resch W, Li X, Lee B, Kovalchuk AL, Tang Z, Nelson S, Di Pierro M, Cheng RR, Machol I, St Hilaire BG, Durand NC, Shamim MS, Stamenova EK, Onuchic JN, Ruan Y, Nussenzweig A, Levens D, Aiden EL, and Casellas R. 2018. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 175: 292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merkenschlager M, and Nora EP. 2016. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu Rev Genomics Hum Genet 17: 17–43. [DOI] [PubMed] [Google Scholar]
- 17.Phillips JE, and Corces VG. 2009. CTCF: master weaver of the genome. Cell 137: 1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ong CT, and Corces VG. 2014. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15: 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wutz G, Varnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, Walther N, Koch B, Kueblbeck M, Ellenberg J, Zuber J, Fraser P, and Peters JM. 2017. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J 36: 3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng X, Hu J, Yue S, Kristiani L, Kim M, Sauria M, Taylor J, Kim Y, and Zheng Y. 2018. Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol Cell 71: 802–815 e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, Jin W, Ried T, Liu P, Zhu J, Rothenberg EV, and Zhao K. 2018. Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity 48: 227–242 e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pongubala JMR, and Murre C. 2021. Spatial Organization of Chromatin: Transcriptional Control of Adaptive Immune Cell Development. Front Immunol 12: 633825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johanson TM, Chan WF, Keenan CR, and Allan RS. 2019. Genome organization in immune cells: unique challenges. Nat Rev Immunol 19: 448–456. [DOI] [PubMed] [Google Scholar]
- 24.Hosokawa H, and Rothenberg EV. 2021. How transcription factors drive choice of the T cell fate. Nat Rev Immunol 21: 162–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, Arnett KL, Blacklow SC, Aifantis I, Aster JC, and Gounari F. 2011. T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc Natl Acad Sci U S A 108: 20060–20065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, and Bhandoola A. 2011. A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476: 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kueh HY, Yui MA, Ng KK, Pease SS, Zhang JA, Damle SS, Freedman G, Siu S, Bernstein ID, Elowitz MB, and Rothenberg EV. 2016. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol 17: 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu S, Zhou X, Steinke FC, Liu C, Chen SC, Zagorodna O, Jing X, Yokota Y, Meyerholz DK, Mullighan CG, Knudson CM, Zhao DM, and Xue HH. 2012. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 37: 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, Kawamoto H, Zhu J, Tan K, and Xue HH. 2014. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat Immunol 15: 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kojo S, Tanaka H, Endo TA, Muroi S, Liu Y, Seo W, Tenno M, Kakugawa K, Naoe Y, Nair K, Moro K, Katsuragi Y, Kanai A, Inaba T, Egawa T, Venkatesh B, Minoda A, Kominami R, and Taniuchi I. 2017. Priming of lineage-specifying genes by Bcl11b is required for lineage choice in post-selection thymocytes. Nat Commun 8: 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Wildt KF, Zhu J, Zhang X, Feigenbaum L, Tessarollo L, Paul WE, Fowlkes BJ, and Bosselut R. 2008. Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4(+) T cells. Nat Immunol 9: 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP, Wiest DL, and Murre C. 2017. Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 171: 103–119 e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Zhang JA, Dose M, Kueh HY, Mosadeghi R, Gounari F, and Rothenberg EV. 2013. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood 122: 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grosschedl R, Giese K, and Pagel J. 1994. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet 10: 94–100. [DOI] [PubMed] [Google Scholar]
- 35.Gounari F, and Khazaie K. 2022. TCF-1: a maverick in T cell development and function. Nat Immunol 23: 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinke FC, and Xue HH. 2014. From inception to output, Tcf1 and Lef1 safeguard development of T cells and innate immune cells. Immunol Res 59: 45–55. [DOI] [PubMed] [Google Scholar]
- 37.Zhao X, Shan Q, and Xue HH. 2022. TCF1 in T cell immunity: a broadened frontier. Nat Rev Immunol 22: 147–157. [DOI] [PubMed] [Google Scholar]
- 38.Shan Q, Li X, Chen X, Zeng Z, Zhu S, Gai K, Peng W, and Xue HH. 2021. Tcf1 and Lef1 provide constant supervision to mature CD8(+) T cell identity and function by organizing genomic architecture. Nat Commun 12: 5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, Chandra A, Goldman N, Yoon S, Ferrari EK, Nguyen SC, Joyce EF, and Vahedi G. 2022. TCF-1 promotes chromatin interactions across topologically associating domains in T cell progenitors. Nat Immunol 23: 1052–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, Pear WS, Bhandoola A, Wherry EJ, and Vahedi G. 2018. Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cells. Immunity 48: 243–257 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butcher MJ, and Zhu J. 2021. Recent advances in understanding the Th1/Th2 effector choice. Fac Rev 10: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonelli M, Shih HY, Hirahara K, Singelton K, Laurence A, Poholek A, Hand T, Mikami Y, Vahedi G, Kanno Y, and O’Shea JJ. 2014. Helper T cell plasticity: impact of extrinsic and intrinsic signals on transcriptomes and epigenomes. Curr Top Microbiol Immunol 381: 279–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sekimata M, Perez-Melgosa M, Miller SA, Weinmann AS, Sabo PJ, Sandstrom R, Dorschner MO, Stamatoyannopoulos JA, and Wilson CB. 2009. CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-gamma locus. Immunity 31: 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spilianakis CG, and Flavell RA. 2004. Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat Immunol 5: 1017–1027. [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro de Almeida C, Heath H, Krpic S, Dingjan GM, van Hamburg JP, Bergen I, van de Nobelen S, Sleutels F, Grosveld F, Galjart N, and Hendriks RW. 2009. Critical role for the transcription regulator CCCTC-binding factor in the control of Th2 cytokine expression. J Immunol 182: 999–1010. [DOI] [PubMed] [Google Scholar]
- 46.Spilianakis CG, Lalioti MD, Town T, Lee GR, and Flavell RA. 2005. Interchromosomal associations between alternatively expressed loci. Nature 435: 637–645. [DOI] [PubMed] [Google Scholar]
- 47.Kim LK, Esplugues E, Zorca CE, Parisi F, Kluger Y, Kim TH, Galjart NJ, and Flavell RA. 2014. Oct-1 regulates IL-17 expression by directing interchromosomal associations in conjunction with CTCF in T cells. Mol Cell 54: 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, Leon B, Crossman DK, Murre C, Myers RM, and Weinmann AS. 2017. CCCTC-Binding Factor Translates Interleukin 2- and alpha-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity 47: 251–267 e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li P, Mitra S, Spolski R, Oh J, Liao W, Tang Z, Mo F, Li X, West EE, Gromer D, Lin JX, Liu C, Ruan Y, and Leonard WJ. 2017. STAT5-mediated chromatin interactions in superenhancers activate IL-2 highly inducible genes: Functional dissection of the Il2ra gene locus. Proc Natl Acad Sci U S A 114: 12111–12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R, and Littman DR. 2012. A validated regulatory network for Th17 cell specification. Cell 151: 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pham D, Moseley CE, Gao M, Savic D, Winstead CJ, Sun M, Kee BL, Myers RM, Weaver CT, and Hatton RD. 2019. Batf Pioneers the Reorganization of Chromatin in Developing Effector T Cells via Ets1-Dependent Recruitment of Ctcf. Cell Rep 29: 1203–1220 e1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wing JB, Tanaka A, and Sakaguchi S. 2019. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 50: 302–316. [DOI] [PubMed] [Google Scholar]
- 53.Ramirez RN, Chowdhary K, Leon J, Mathis D, and Benoist C. 2022. FoxP3 associates with enhancer-promoter loops to regulate Treg-specific gene expression. Sci Immunol 7: eabj9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yano S, Ghosh P, Kusaba H, Buchholz M, and Longo DL. 2003. Effect of promoter methylation on the regulation of IFN-gamma gene during in vitro differentiation of human peripheral blood T cells into a Th2 population. J Immunol 171: 2510–2516. [DOI] [PubMed] [Google Scholar]
- 55.Lee DU, Agarwal S, and Rao A. 2002. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity 16: 649–660. [DOI] [PubMed] [Google Scholar]
- 56.Chung HK, McDonald B, and Kaech SM. 2021. The architectural design of CD8+ T cell responses in acute and chronic infection: Parallel structures with divergent fates. J Exp Med 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henning AN, Roychoudhuri R, and Restifo NP. 2018. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol 18: 340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shan Q, Zhu S, Chen X, Liu J, Yuan S, Li X, Peng W, and Xue HH. 2022. Tcf1-CTCF cooperativity shapes genomic architecture to promote CD8(+) T cell homeostasis. Nat Immunol 23: 1222–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawabe T, Yi J, and Sprent J. 2021. Homeostasis of Naive and Memory T Lymphocytes. Cold Spring Harb Perspect Biol 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bediaga NG, Coughlan HD, Johanson TM, Garnham AL, Naselli G, Schroder J, Fearnley LG, Bandala-Sanchez E, Allan RS, Smyth GK, and Harrison LC. 2021. Multi-level remodelling of chromatin underlying activation of human T cells. Sci Rep 11: 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao DM, Yu S, Zhou X, Haring JS, Held W, Badovinac VP, Harty JT, and Xue HH. 2010. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J Immunol 184: 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shan Q, Hu SS, Zhu S, Chen X, Badovinac VP, Peng W, Zang C, and Xue HH. 2022. Tcf1 preprograms the mobilization of glycolysis in central memory CD8(+) T cells during recall responses. Nat Immunol 23: 386–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kochi Y 2016. Genetics of autoimmune diseases: perspectives from genome-wide association studies. Int Immunol 28: 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steward CA, Gonzalez JM, Trevanion S, Sheppard D, Kerry G, Gilbert JG, Wicker LS, Rogers J, and Harrow JL. 2013. The non-obese diabetic mouse sequence, annotation and variation resource: an aid for investigating type 1 diabetes. Database (Oxford) 2013: bat032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fasolino M, Goldman N, Wang W, Cattau B, Zhou Y, Petrovic J, Link VM, Cote A, Chandra A, Silverman M, Joyce EF, Little SC, Consortium H, Kaestner KH, Naji A, Raj A, Henao-Mejia J, Faryabi RB, and Vahedi G. 2020. Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity 52: 257–274 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belver L, and Ferrando A. 2016. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 16: 494–507. [DOI] [PubMed] [Google Scholar]
- 67.Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, Reddy J, Borges-Rivera D, Lee TI, Jaenisch R, Porteus MH, Dekker J, and Young RA. 2016. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351: 1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, Chen X, Hu H, Bakogianni S, Wang J, Fu Y, Boccalatte F, Zhong H, Paietta E, Trimarchi T, Zhu Y, Van Vlierberghe P, Inghirami GG, Lionnet T, Aifantis I, and Tsirigos A. 2020. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet 52: 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aster JC, Blacklow SC, and Pear WS. 2011. Notch signalling in T-cell lymphoblastic leukaemia/lymphoma and other haematological malignancies. J Pathol 223: 262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castel D, Mourikis P, Bartels SJ, Brinkman AB, Tajbakhsh S, and Stunnenberg HG. 2013. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev 27: 1059–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, Zeisberger P, Benk AS, Amit I, Zandstra PW, Lupien M, Dick JE, Trumpp A, and Spitz F. 2018. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553: 515–520. [DOI] [PubMed] [Google Scholar]
- 72.Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, Clappier E, Soulier J, and Ferrando AA. 2014. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 20: 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, Knoechel B, Lanauze C, Louis L, Forsyth KS, Chen S, Chung Y, Schug J, Blobel GA, Liebhaber SA, Bernstein BE, Blacklow SC, Liu XS, Aster JC, and Pear WS. 2014. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci U S A 111: E4946–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Antoszewski M, Fournier N, Ruiz Buendia GA, Lourenco J, Liu Y, Sugrue T, Dubey C, Nkosi M, Pritchard CEJ, Huijbers IJ, Segat GC, Alonso-Moreno S, Serracanta E, Belver L, Ferrando AA, Ciriello G, Weng AP, Koch U, and Radtke F. 2022. Tcf1 is essential for initiation of oncogenic Notch1-driven chromatin topology in T-ALL. Blood 139: 2483–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tiemessen MM, Baert MR, Schonewille T, Brugman MH, Famili F, Salvatori DC, Meijerink JP, Ozbek U, Clevers H, van Dongen JJ, and Staal FJ. 2012. The nuclear effector of Wnt-signaling, Tcf1, functions as a T-cell-specific tumor suppressor for development of lymphomas. PLoS Biol 10: e1001430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou Y, Petrovic J, Zhao J, Zhang W, Bigdeli A, Zhang Z, Berger SL, Pear WS, and Faryabi RB. 2022. EBF1 nuclear repositioning instructs chromatin refolding to promote therapy resistance in T leukemic cells. Mol Cell 82: 1003–1020 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, Guo YE, Hnisz D, Jaenisch R, Bradner JE, Gray NS, and Young RA. 2017. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171: 1573–1588 e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murayama Y, and Uhlmann F. 2014. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 505: 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hua P, Badat M, Hanssen LLP, Hentges LD, Crump N, Downes DJ, Jeziorska DM, Oudelaar AM, Schwessinger R, Taylor S, Milne TA, Hughes JR, Higgs DR, and Davies JOJ. 2021. Defining genome architecture at base-pair resolution. Nature 595: 125–129. [DOI] [PubMed] [Google Scholar]
- 80.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, and Fraser P. 2013. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li X, Yuan S, Zhu S, Xue H-H, and Peng W. 2022. HiCHub: A Network-Based Approach to Identify Domains of Differential Interactions from 3D Genome Data. bioRxiv: 2022.2004.2016.488566. [Google Scholar]
- 82.Kai Y, Andricovich J, Zeng Z, Zhu J, Tzatsos A, and Peng W. 2018. Predicting CTCF-mediated chromatin interactions by integrating genomic and epigenomic features. Nat Commun 9: 4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwessinger R, Gosden M, Downes D, Brown RC, Oudelaar AM, Telenius J, Teh YW, Lunter G, and Hughes JR. 2020. DeepC: predicting 3D genome folding using megabase-scale transfer learning. Nat Methods 17: 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]