

Abstract
During protein synthesis, aminoacyl-tRNA synthetases covalently link amino acids with their cognate tRNAs. Amino acid mutations in glycyl-tRNA synthetase can disrupt protein synthesis and lead to a neurological disorder known as Charcot-Marie-Tooth disease type 2D (CMT-2D). Several studies employing diverse techniques have identified potential disease mechanisms at the molecular level. The majority of CMT-2D mutations in glycyl-tRNA are found within its dimer interface. However, no atomic structures bearing these mutations have been solved. Consequently, the specific disease-causing structural changes that occur in glycyl-tRNA synthetase have not been definitively established. Here we use molecular dynamics simulations to probe conformational changes in glycyl-tRNA synthetase caused by one mutation within the dimer interface: G240R. Our results show that the mutation alters the number of native interactions at the dimer interface and also leads to altered dynamics of two regions of glycyl-tRNA synthetase associated with tRNA binding. Additionally, we use our simulations to make predictions about the effects of other clinically reported CMT-2D mutations. Our results identify a region of the glycyl-tRNA synthetase structure that may be disrupted in a large number of CMT-2D mutations. Structural changes in this region may be a common molecular mechanism in glycyl-tRNA synthetase CMT-2D pathologies. Statement of significance: In this study, we use molecular dynamics simulations to elucidate structural conformations accessible to glycyl-tRNA synthetase (GlyRS), an enzyme that ligates cytosolic glycine with tRNA-Gly. This protein contains multiple flexible regions with dynamics that elude in vitro structural characterization. Our computational approach provides unparalleled atomistic details of structural changes in GlyRS that contribute to its role in protein synthesis. A number of mutations in GlyRS are associated with a peripheral nerve disorder, Charcot-Marie-Tooth disease type 2D (CMT-2D). Mutation-induced structural and dynamic changes in GlyRS have similarity that elude in vitro structural characterization. Our simulations provide insights into disease mechanisms for one such mutation: G240R. Additionally, we leverage our computational data to identify regions of GlyRS critical to its function and to predict the effects of other disease-associated mutations. These results open up new directions for research into the molecular characterization of GlyRS and into hypothesis-driven studies of CMT-2D disease mechanisms.
Keywords: GlyRS, Charcot-Marie-Tooth, Molecular dynamics simulations
1. Introduction
Aminoacyl-tRNA synthetases (ARSs) are an ancient and phylogenetically ubiquitous group of enzymes responsible for attaching amino acids to their corresponding tRNAs. Eukaryotic genomes contain about 30 ARS genes. Most of the 20 amino acids translationally incorporate into cytoplasmic proteins via interaction with a unique ARS gene. However, for some amino acids, additional ARS genes support mitochondrial protein synthesis. ARS genes are subdivided into 2 classes based on primary sequence similarity[1], [2]. Class I and Class II ARS genes evolved independently and no similarity in folded structure or active site configuration exists between proteins of the two classes, despite sharing the common function of loading amino acids onto cognate tRNAs to facilitate mRNA translation[1].
Mutations in genes encoding several different ARSs cause peripheral motor and sensory neuropathies that form part of the Charcot-Marie-Tooth (CMT) family of inheritable disorders[3]. Within this group, the GARS1 gene, encoding glycyl-tRNA synthetase (GlyRS), was the first member identified whose mutations were implicated in a human genetic disease and, specifically, in causing a genetically dominant axonal form of CMT, type 2D (CMT-2D)[4]. Genetic evidence excludes haploinsufficiency as a cause for dominant inheritance, leaving dominant-negative effects or neomorphic toxic effects as possible mechanisms for GlyRS mutant CMT. A current theory for a mechanism linking mutant GlyRS to CMT pathology is toxic neomorphic gain-of-function resulting from mutationally-induced conformational changes that trigger association with particular neuronal proteins. Specifically, mutant GlyRS proteins are reported to bind and modulate activity of histone deacetylase 6 (HDAC6)[5], TrkA/B/C neurotrophin receptors[6], and neuropilin1 VEGF receptors[7]. However, while some disease-causing mutations in the GARS1 gene enhance GlyRS-HDAC6 interactions, other mutations do not[5]. Furthermore, differences in the capacity for specific mutations to affect the aminoacylation function of GlyRS have also been reported[8–10]. These inconsistencies raise questions about how mutations in the GlyRS gene cause disease and how specific sequence changes alter the structure and functions of the resulting GlyRS protein.
Understanding how mutations in the GlyRS gene cause neuropathic disease will help facilitate the development of novel therapies against these severely debilitating conditions. Furthermore, such information will increase our understanding of how protein synthesis occurs in axons and supports the stability and functionality of synaptic terminals that exist at great distances from their respective cell soma. As such, research on this subject has the potential to make important contributions to our understanding of neuron survival and will serve to increase our knowledge of the basic mechanisms that underpin central and peripheral nervous system function.
X-ray crystallographic studies have provided significant insight into the structure of the GlyRS protein (Figure 1a,b). GlyRS exists as a homodimer (in higher eukaryotes, Fig 1b) and possesses a modular architecture, comprising a catalytic core consisting of seven antiparallel β-strands, a C-terminal anticodon-binding domain, an N-terminal WHEP-TRS domain, and several appended motifs (Figure 1a)[11]. Motif 1 is believed to be involved in active site formation and enzyme dimerization while Motifs 2 and 3 contain ATP-binding residues. An insertion domain (Insertion 1) separates Motifs 1 and 2 and is predicted to interact with tRNA[12]. Insertion 2 comprises two short helices and β-strands and is appended after Motif 2. Its function is difficult to determine since it lies away from the dimer interface and the tRNA-binding site. Insertion 3 is located after Motif 3 and is predicted to interact with tRNA. The anticodon-binding domain consists of three α-helices surrounding a five-stranded mixed parallel and anti-parallel β-sheet.
Figure 1. GlyRS Structure and Domain Organization.
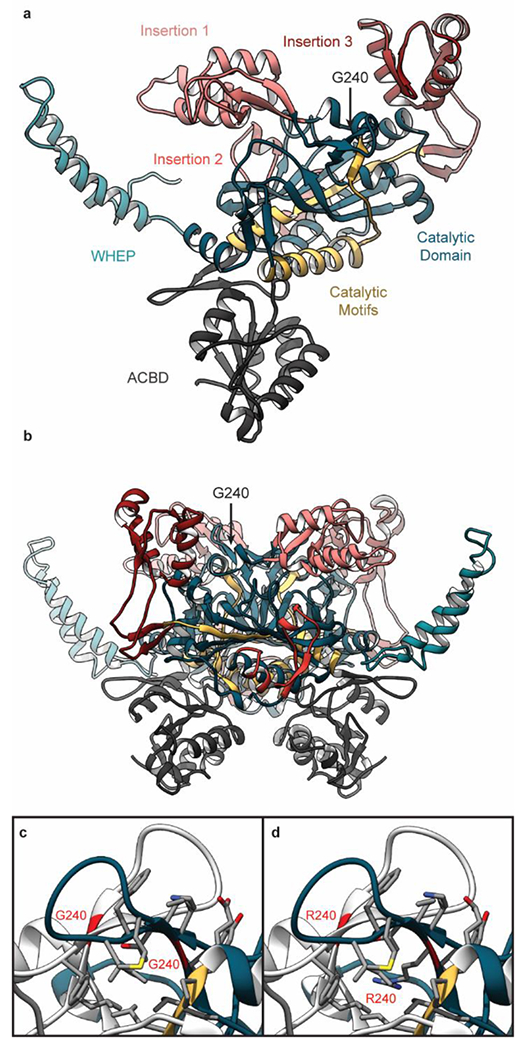
Each GlyRS monomer (a) contains a central catalytic domain (blue) connected to an N-terminal WHEP domain (cyan) and an anticodon binding domain (ACBD, grey). 3 motifs (yellow) within the catalytic domain contribute to GlyRS enzymatic activity and three sequence insertions (#1-3, pink, red, crimson) coordinate GlyRS-tRNA interactions. In this image, the GlyRS dimer-dimer interface is facing toward the page (b) GlyRS exists functionally as a homodimer. (c) G240 is located in a β-hairpin at the GlyRS dimer interface. The ribbon structure of chain A is colored as in panel a and the ribbon of chain B is colored grey. The ribbons of the two G240 residues are colored red. Atoms for side chains within 5 Å of the mutation side are shown.(d) The structure of G240R GlyRS is shown after the mutation was introduced as described in Methods.
Several lines of structural and biochemical evidence have demonstrated that CMT-2D mutations cause conformational changes in GlyRS that perturb its ability to form homodimers, alter the overall dimeric structure, or perturb interactions with tRNA[13], [14]. The molecular details of these pathological structural changes have not been definitively established and may be ‘hidden’ to X-ray crystallography. Currently, there are 14 crystal structures of GlyRS from H. sapiens in the Protein Data Bank (PDB). These structures include both wild-type (WT) and mutant GlyRS (E71G, S581L, and G526R); free GlyRS and GlyRS complexed with tRNA and/or ATP. However, this set of structures does not sufficiently probe conformational states accessible to GlyRS, particularly those populated because of CMT-2D mutations. Primarily because none of the crystallized GlyRS bear mutations within the dimer interface. Further details of the structural consequences of these mutations are needed to refine and test potential hypotheses. Computational models of protein dynamics therefore offer attractive opportunities to understand how the normal dynamics and conformation(s) of GlyRS become disrupted in pathological conditions. Here we use molecular dynamics (MD) simulations of WT and mutant (G240R) GlyRS dimers to explore mutation-induced structural changes. The G240R GlyRS mutation was first reported in 2003 by Antonellis et al.[4]. The mutation was discovered in a genetic study of five families with CMT-2D and/or distal spinal muscular atrophy type V[4]. This mutation is located within a β-hairpin of the catalytic domain that is close to the dimer interface (Figure 1c). In the dimer structure, the two β-hairpins associate with one another at the dimer interface to form an extended 4-stranded β-sheet. We find that G240R alters the dimer interface of GlyRS and also changes the dynamics of insertion 1 and 3 that are likely related to tRNA binding. Using these results, we make predictions – at the protein structural level – about the molecular mechanisms connecting GlyRS mutations to CMT-2D pathologies. To our knowledge, these are the longest (3 μs net sampling) all-atom, explicit solvent, continuous MD simulations of GlyRS ever reported.
2. Methods
Scheme 1, Adapted from Xie et al 2007
Scheme 2, Adapted from Xie et al 2007
2.1. General Model Building
To survey conformations accessible to GlyRS in crystallographic states, 14 GlyRS coordinate sets were obtained from the PDB. All 14 structures were solved by X-ray crystallography in 6 separate studies and capture various states of GlyRS including both apo- and holo- forms, wild type and mutant forms, and conformations in various stages of a two-step enzymatic aminoacylation reaction (Scheme 1 & 2). Coordinates for insertion 3 (residues 412-518) were missing in most structures, but present in the 4KQE structure that harbors the E71G mutation.
2.2. MD Model Building
Starting coordinates for the MD simulations were obtained from an X-ray structure of E71G GlyRS in the PDB (www.rcsb.org)[15]. This structure (PDB ID: 4KQE, 2.6 Å, residues 64-674) was solved in the absence of its enzymatic cofactors, substates, and products[16]. This structure was chosen because it contains coordinates for most residues in insertion 3 that are missing in most other structures and the E71G mutation did not otherwise alter the structure in comparison with WT GlyRS structures (e.g., 2PME[17]). Coordinates for the N-terminal WHEP domain were modeled using a tRNAGly-bound WT GlyRS structure as a template (PDB ID: 5E6M)[18]. G71 was replaced by E71 in the rotameric state matching the WT 2PME structure. Missing heavy atoms were built using Modeller[19]. Starting coordinates for the G240R simulations were obtained via in silico mutation of the WT structure using UCSF Chimera[20]. The Rotamers tool in UCSF Chimera was used to mutate the Gly side chain to Arg[21]’ [22]. Introduction of the larger Arg side chain led to several atomic clashes between the mutation and residues L129, T230, and I232. Residues within 5 Å of the mutation site were minimized. During minimization, a ~0.7 Å displacement of the T230 side chain accommodated the Arg side chain within a cavity (Figure 1d). Next, hydrogen atoms were modeled onto the initial structure using the tleap module of AMBER and each protein was solvated with explicit water molecules in a periodic, truncated octahedral box that extended at least 10 Å beyond any protein atom. Na+ and Cl− counterions were added to neutralize the solvated protein systems.
2.3. Force Field and Molecular Mechanics
Simulations were performed with the AMBER20 package[23] and the ff14SB force field[24]. Water molecules were treated with the TIP3P force field[25]. Metal ions were modeled using the Li and Merz parameter set[26–28]. The SHAKE algorithm was used to constrain the motion of hydrogen-containing bonds. Long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) method.
2.4. Pre-production protocols
Each system was minimized in three stages. First, hydrogen atoms were minimized for 1000 steps in the presence of 100 kcal mol−1 restraints on all heavy atoms. Second, all solvent atoms were minimized for 1000 steps in the presence of 25 kcal mol−1 restraints on all protein atoms. Third, all atoms were minimized for 8000 steps in the presence of 25 kcal mol−1 restraints on all backbone heavy atoms (N, O, Cα, and C atoms) After minimization, systems were heated to 310 K during 3 successive stages. In each stage, the system temperature was increased by ~100 K over 100 ps (50,000 steps) using the canonical NVT (constant number of particles, volume, and temperature) ensemble. During all heating stages, 25 kcal mol−1 restraints were present on the backbone heavy atoms (N, O, Ca, and C atoms). After the system temperatures reached 310 K, the systems were equilibrated over 5 successive stages using the isobaric-isothermal NPT (constant number of particles, pressure, and temperature) ensemble. During each stage, the systems were equilibrated for 5.4 ns in the presence of restraints on backbone atoms. The strength of the restraints was decreased from 25 kcal mol−1 during the first stage to 1 kcal mol−1 during the fourth stage. During the final equilibration stage, the systems were equilibrated in the absence of restraints.
2.5. MD protocol
Production dynamics for conventional molecular dynamics simulations were then performed using the canonical NVT ensemble using an 8 Å nonbonded cutoff and a 2 fs time step. WT and G240R GlyRS dimers were simulated for 500 ns in triplicate (1,500 ns net sampling per simulated system). Unless specified otherwise, frames were selected for analysis every 10 picoseconds and the results of replicate simulations were averaged together. The minimization, heating, and equilibration portions of simulation times are not counted in the reported timepoints. The 0.0 ns timepoint corresponds to the start of production dynamics.
2.6. MD analysis
The Cα root-mean-squared deviation (RMSD) and interatomic contacts were calculated with cpptraj[29]. The Cα RMSD was calculated after alignment of all Ca atoms to the minimized structure. Two residues were considered in contact with one another if at least one pair of heavy atoms were within 5 Å of one another. All protein images were prepared using UCSF Chimera.
3. Results
3.1. Large-amplitude motions in GlyRS
We detected large scale structural fluctuations within GlyRS by measuring the Cα RMSD of MD-derived snapshots relative to the initial, equilibrated starting structure. The GlyRS dimer is a large, dynamic enzyme and conformational variability has been previously reported [16]’ [17] in three regions of the structure: the N-terminal WHEP domain (residues 1-67), insertion 1 (residues 117-247), and insertion 3 (residues 412-518). In MD simulations, the ensemble average Cα RMSD for all residues in the dimer was 11.6 ± 1.6 Å for the WT system and 13.9 ± 2.5 Å for the G240R system. However, the RMSD values were much higher than is typical for compact and well-folded proteins. To quantify the relative conformational variability present in various regions of GlyRS, we calculated a set of Cα RMSD values after aligning to a set of ‘core’ residues that had low structural variability. The core residues included the ACBD domain and the catalytic domain (residues 68-116, 248-411, and 519-674). The MD simulations were aligned to these core residues and then Cα RMSD values were calculated for the core residues, N-terminal WHEP domain, insertion 1, and insertion 3 (Table 1).
Table 1.
Cα RMSD values in MD simulations of WT and G240R GlyRS Dimers
| Region | RMSD WT (Å) | G240R (Å) |
|---|---|---|
| All Residues 1 | 11.6 ± 1.62 | 13.9 ± 2.5 |
| Core Residues 3,4 | 1.7 ± 0.24 | 1.8 ± 0.25 |
| WHEP Domain 3,5 | 32.2 ± 7.5 | 41.0 ± 8.9 |
| Insertion 1 3,6 | 6.4 ± 2.1 | 6.1 ± 3.1 |
| Insertion 3 3,7 | 13.4 ± 6.0 | 12.1 ± 9.2 |
Trajectories were aligned on all residues and the RMSD was reported for all Cα atoms.
The reported standard deviations were calculated from the ensemble averaging over replicate simulations and both chains.
Trajectories were aligned on the core residues (68-116, 248-411, and 519-674).
The RMSD was reported for (Cα atoms of the ‘core’ residues (68-116, 248-411, and 519-674)
The RMSD was reported for Cα atoms of WHEP domain residues (1-67)
The RMSD was reported for Cα atoms of insertion 1 residues (117-247)
The RMSD was reported for (Cα atoms of insertion 3 residues (412-518)
The RMSD data indicate that GlyRS is comprised of a stable and well-folded set of core residues joined with three dynamic regions. The magnitudes of structural changes were similar in the WT and G240R systems, through there was more variation in the WHEP domain, insertion 1, and insertion 3 in the presence of the mutation. Because the RMSD values report deviations from a single structure, it is difficult to assess relative differences in conformational heterogeneity between the WT and mutant or to proscribe any meaningful description of conformational changes induced by the mutation from these data alone.
3.2. Conformational heterogeneity in GlyRS sampled during MD
We used the program ttclust[30] to cluster the ensemble of GlyRS conformations sampled during MD. Residues in the WHEP domain were excluded from all clustering. In an initial calculation, we clustered on Cα atoms within non-WHEP residues in both chains of the dimer. The clustering segregated the ensembles on the substantial conformational changes in insertions 1 and 3. There were no appreciable correlated motions between insertions 1 and 3 within a single monomer nor between insertions of both dimers. This led to a large number of lowly-populated clusters describing the various dimer conformations observed, but there were few clusters that were observed multiple times within a single simulation or across multiple simulations. Therefore, we re-clustered the simulations, but both monomer chains were clustered together. Again, the clustering segregated the simulations based on conformational changes within insertions 1 and 3, but due to the increased effective ensemble size and diminished variability (because we clustered on the monomers not dimers), we observed a smaller number of larger-sized clusters that were populated across multiple simulations. The clustering also indicated that G240R conferred greater conformational variation within the GlyRS structure (Figure 2). The x-ray crystal-like cluster (C1) was populated by 43% of the WT ensemble, but only 26% of the G240R ensemble. The remaining clusters were differentiated by the conformations of insertion 1 and 3.
Figure 2. Conformations sampled by GlyRS during MD.
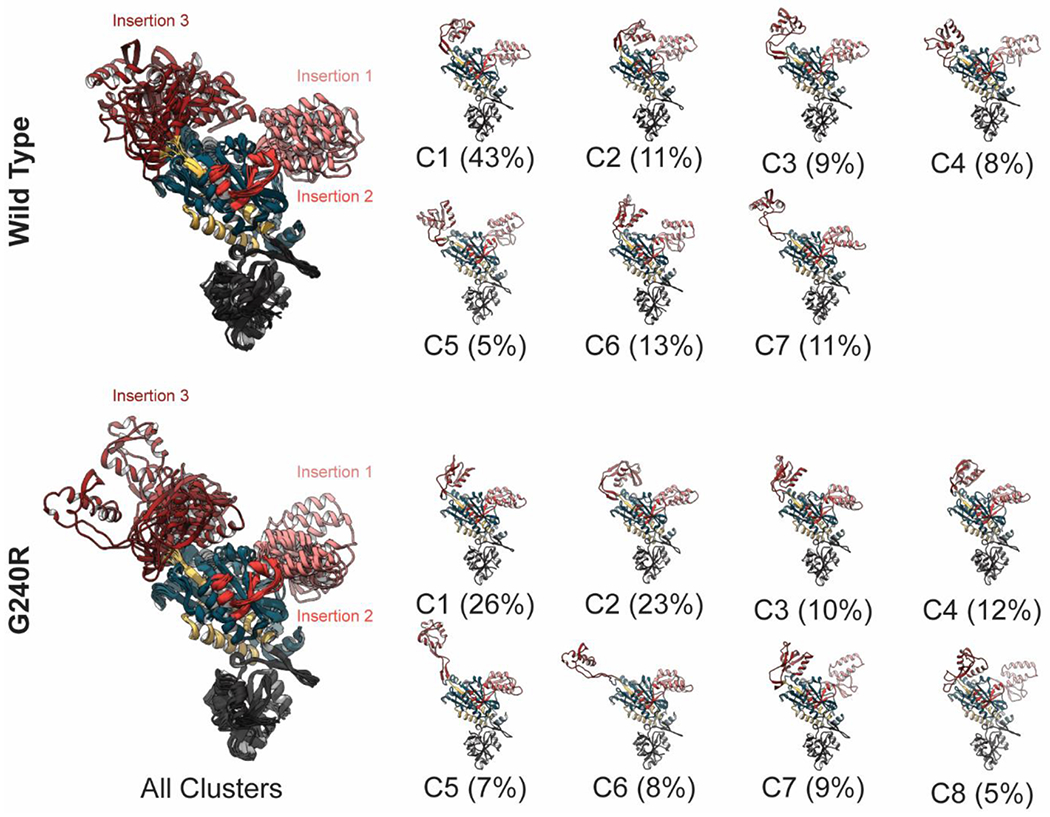
The individual chains of WT (top row) and G240R (bottom row) GlyRS sampled multiple conformations during MD. The clustering yielded 7 clusters for the WT ensemble and 8 for G240R. The large images on the left contain all cluster representatives, which have been aligned on the ‘core’ residues. Since the individual chains of the GlyRS dimers were clustered together, the representative structures depict conformations of individual GlyRS chains that were sampled during MD simulations of GlyRS dimers. To highlight the variation associated with insertions 1 and 3, residues in the WHEP domain are hidden and the chains are oriented such that the dimer interface is directed away from the page. Coloring is as in Figure 1. Snapshots of the individual cluster representatives are shown in the smaller images, and the fraction of the ensemble allocated to each cluster is reported below its image.
Insertion 3 experienced the greatest structural changes (Table 1). In the crystal-like structure, insertion 1 is compact and folded over like a hook and the region is stabilized by interactions with the ‘top’ of the catalytic domain of GlyRS. In most of the other clusters, insertion 3 maintained a bent and compact conformation, but formed interactions with different regions of the catalytic domain (see for example WT clusters 2 and 4; G240R clusters 2, 4, 7, and 8). Insertion 3 also sampled alternate conformations in which the distal portion of the fishhook interacted with the ‘side’ of GlyRS or projected into solvent (Figure 2, WT clusters 3 and 7; G240R clusters 3, 4, 5, and 6).
Some of these alternate conformations were similar to the conformation observed in x-ray crystal structures of GlyRS:tRNA complexes (e.g. PDB 4QEI[16]). In the G240R simulations, insertion 3 spent a greater amount of time in the alternate conformations and also experienced greater deviation from the 4KQE x-ray structure (Figure 2 C5).
Insertion 1 also had distinct conformational sampling in the WT and G240R simulations. In the crystal-like conformation, insertion 1 rests against the catalytic domain evenly situated between the insertion 3s of the two monomers. Insertion 1 also sampled conformations in which it moved away from the catalytic core and towards insertion 3 of the other monomer. Such conformations were captured in WT clusters 4, 5, and 6; G240R clusters 2, 4, 7, 8 (Figure 2). As with insertion 3, the ‘upward’ conformation of insertion 1 is associated with tRNA binding (see PDB: 4QEI[16]) and occurred in both the WT and mutant simulations.
3.3. The breathing motion of insertions 1 and 3 was disrupted by G240R
In all three WT simulations and all three G240R simulations, insertion 1 experienced conformational changes as described by the clustering to some degree. In the WT simulations, insertion 3 detached from the catalytic domain, projected into solvent, and then returned to a bent, x-ray-like conformation multiple times in replicate simulations (Figure 3). In runs 1 and 3 of the G240R simulations, insertion 3 did not return to an x-ray-like conformation after detaching from the catalytic domain, though projection of insertion 3 into solvent occurred later in those simulations. (Figure 3). In the G240R run 2 simulation, the insertion 3 domains did not detach. These results indicate that G240R disrupted the conformational distribution of insertion 3 relative to the WT simulations.
Figure 3. G240R led to increased extension of insertion 3.
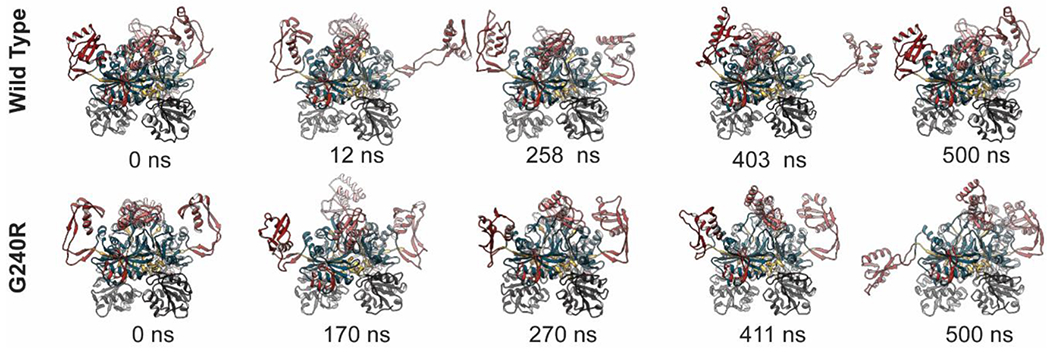
Snapshots extracted from MD simulations of WT and G240R GlyRS are shown. The 0 ns snapshots correspond to the beginning of production dynamics, differences in the WT and G240R conformations occurred during minimization, heating, and equilibration. The structures are colored as in Figure 1 and the WHEP domain is excluded for clarity. Snapshots from WT run 3 are shown in the top row and snapshots from G240R run 3 are shown in the bottom row. The timestamps for each snapshot are annotated below.
We analyzed residue-residue contacts to identify regions of GlyRS that had altered structure due to the G240R mutation. We calculated the percent simulation time for which each pair of residues was in contact (defined as having at least one heavy atom in each of two residues within 5 Å of one another). Next, we applied a Student’s t-test to identify contacts that were present for statistically distinct % simulation times in the WT and G240R simulations (p < 0.05). Finally, we excluded those contacts that had % simulation time differences < 35%. Of ~13,000 observed residue-residue contacts, 138 remained after our selection criteria. We have high confidence that these remaining contacts reflect significant structural changes at the residue level between the WT and G240R simulations. These contact differences are mapped onto the GlyRS structure in Figure 4. The contact differences were mostly restricted to the vicinity of the mutation site (which occurs in a β-hairpin at the ‘top’ of the catalytic domain. The changes in contacts included: altered interactions between catalytic domain residues across the dimer-dimer interface, between insertion 1 and the β-hairpin of the opposite monomer, internal insertion 1 interactions, internal insertion 3 interactions, and regions that structurally connect the mutation site and insertion 3 (residues 80-96, 129-144, and 413-422. These altered interactions were generally symmetric for both dimers. We also assessed interactions at the dimer:dimer interface by tracking the number of atom:atom contacts at the interface and divided them into ‘native’ (observed in the x-ray structure and/or MD) and ‘nonnative’ contacts (observed during MD only). The WT simulations preserved a greater number of native dimer:dimer contacts versus the G240R simulations (831 vs. 597, p = 0.02). There were not statistically significant differences in the number of nonnative contacts formed (718 vs 807) or the total number of contacts (1550 vs 1405).
Figure 4. Statistically significant contact differences in WT and G240R GlyRS.
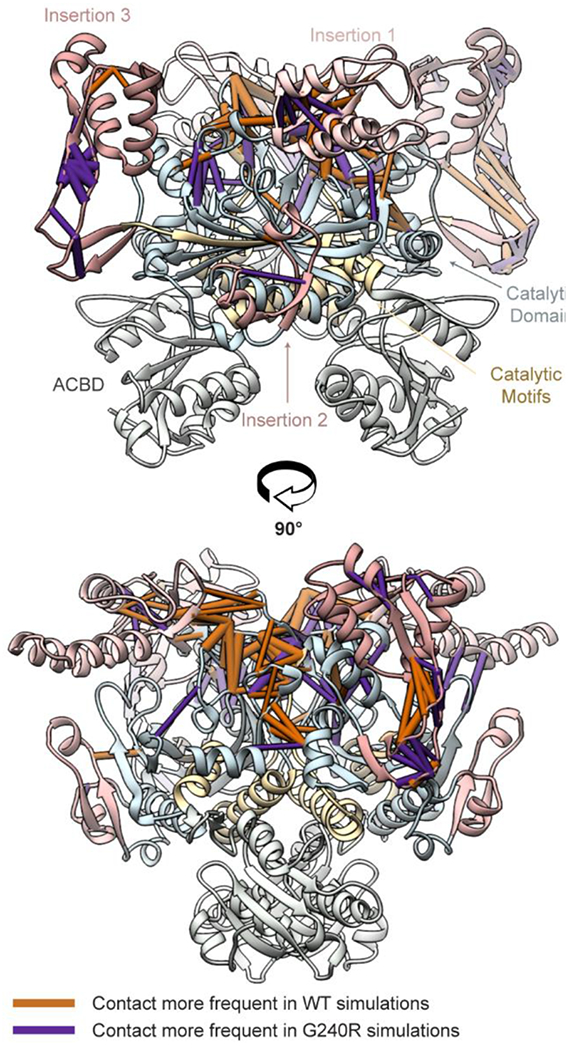
We identified statistically significant (p < 0.05) contacts where the % contact simulation time between the G240R and WT simulations was > 35%. Pipes within this snapshot represent these residue-residue contacts. The pipe cylinder radii are proportional to the % difference in simulation time that the contacts differed in the WT and G240R simulations. Orange pipes were observed more frequently in the WT simulations and purple pipes were observed more frequently in the G240R simulations. The contact differences have been mapped onto a reference WT structure. For clarity, the protein ribbon colors have been muted.
The structural origin of altered interactions in the immediate vicinity of the mutation site was clear: G240R replaced the smallest, flexible residue with a large, charged one. To fit within the native conformation, Arg was buried in a hydrophobic region of the protein interior. As this was energetically unfavorable, in MD simulations we observed that the hairpins opened to allow the Arg side chains to become hydrated by solvent (Figure 5). This adjustment of the hairpins led to a series of structural changes. The conformations of residues 80-96 of the opposite dimer and 129-144 adjusted to accommodate the new structure of the mutation-containing hairpins. This led to a loss of stabilizing interactions between residues 129-144 and 413-422, the latter of which are included in insertion 3 (Figure 6). We propose that these lost stabilizing interactions gave rise to the altered dynamics of insertion 3 observed in the G240R simulations.
Figure 5. Local effects of G240R.
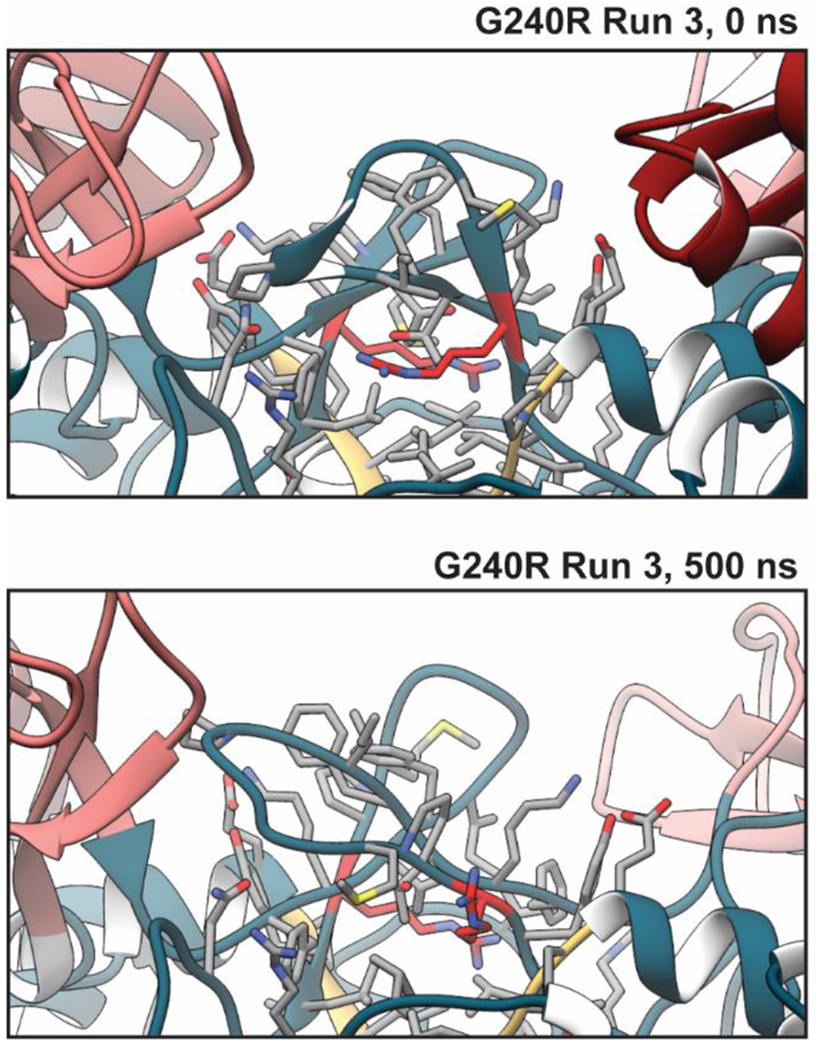
Replacement of the small, flexible Gly with large and charged Arg led to disruption of local structure. The effects of these structural changes began during equilibration, as there were differences in the WT and G240R starting structures. The structural environment of residue position 240 required that the side chain be inserted into an interior, hydrophobic region of the protein (at least in the reference structure). This was unfavorable for the electrically charged Arg. Over the course of the simulations, the β-hairpins containing R240 (colored red) furled upward and allowed the R240 sidechain to become solvated by water.
Figure 6. Molecular mechanism underlying increased insertion 3 flexibility with G240R.
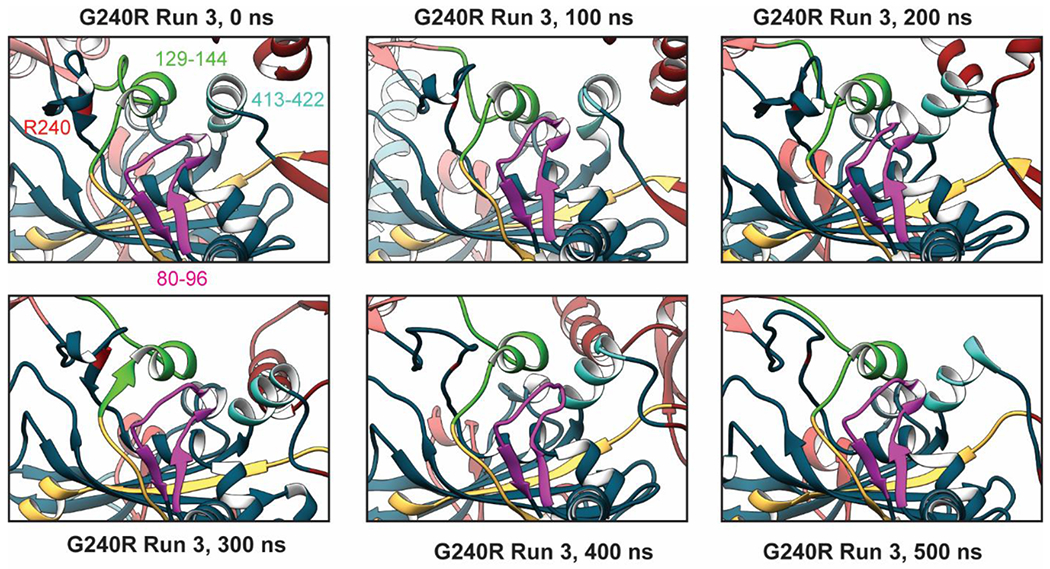
Disruption of local structure around residue 240 propagated to nearby regions that effect the stability/structure of insertion 3 an N-terminal region of the opposite monomer (residues 80-96, magenta), an α-helix connected to insertion 1 and a catalytic motif (residues 129-144, green), and an a-helix at the N-terminal end of insertion 3 (residues 413-422, cyan). Unfurling of the b-hairpin that contains R240 unfurled and projection of the R240 sidechain into solvent was associated with a reorganization of these three regions. This ultimately led to a change in contacts that affected interactions between insertion 3 and the rest of GlyRS. We propose that such altered interactions give rise to altered dynamics of insertion 3.
4. Discussion
The introduction of the G240R mutation in silico is a limitation of our study as it assumes that the mutant protein can access the crystallographically observed conformation (which was the case for the E71G, S581L, and G526R GlyRS structures). However, this approach provides rapid access to predictions about the dynamics of wild type and mutant GlyRS that cannot be made by static structures alone. Our simulations generate and refine hypotheses that describe the pathological mechanisms by which G240R and other GlyRS mutations lead to CMT-2D. These predictions should inform future computational and experimental studies of CMT-2D mutations in ARS proteins.
4.1. Large-amplitude structural changes may be essential for enzymatic function
The largest structural changes were observed during MD related to motions of the WHEP domain, insertion 1, and insertion 3. Similar conformations were observed in the WT and G240R simulations, though the relative distribution of conformations was distinct. This suggests that tRNA binding occurs via a conformational-selection mechanism made possible when insertion 3 becomes detached from the catalytic domain. Recent in vitro studies have reported that certain GlyRS mutations – including G240R – prolong GlyRS:tRNA interactions despite similar aminoacylation rates to WT GlyRS[31]. Our simulations suggest that G240R may disrupt the equilibrium between x-ray-like and detached conformations of insertion 3 as well as the rate of insertion 3 detachment. However, we did not obtain sufficient sampling to make statistically meaningful claims about the effect of G240R on insertion 3 detachment kinetics. Our simulations suggest that prolonged GlyRS: tRNA interactions may occur because mutations alter the dynamics of insertion 3. Altered insertion 3 dynamics may either favor conformations of insertion 3 than can bind tRNA – or other proteins - more easily or disfavor insertion 3 conformations that are bent and attached to the catalytic domain. Additional structural studies that investigate CMT-2D mutations in GlyRS:tRNA complexes are required to test these hypotheses. It is noteworthy that the only x-ray structure able to resolve insertion1/3 coordinates in the absence of tRNA (PDB: 4KQE which is the base model in our study) included a mutation (E71G) that is close to the structural pathway by which we predict G240R led to increased insertion 3 flexibility[16]. It is possible that the E71G mutation affects the conformation of residues 80-96, 129-144, 413-422 in a way that stabilizes insertion 3. This anecdotal evidence supports a hypothesis that GlyRS mutations alter protein synthesis by stabilizing or generating GlyRS conformations that variously stabilize GlyRS:tRNA interactions or lead to neomorphic interactions with other proteins.
WHEP domains, and the remarkable conformational flexibility of the GlyRS WHEP domain in particular, is of interest because WHEP domains are present in 5 ARS proteins, and in no other protein, yet little is known about the functions of these domains. WHEP domains were appended to these ARSs in three separate events during metazoan evolution, implying a strong selective driving force that is specific to ARS function [32]. The only specific function ascribed to a WHEP domain is the WHEP domain of EPARS, which stabilizes its association with a large multienzyme complex of 8 ARS proteins[33]. If the WHEP domain of GlyRS mediates interaction with protein partners that remain to be identified, the increased mobility of this domain associated with the G240R mutation may influence the state of such protein-protein interactions.
4.2. Putative effects of the other CMT-2D GlyRS mutations
At least 21 mutations in GlyRS are clinically associated with CMT-2D phenotypes. A majority of these mutations localize nearby insertion 1 and the dimer:dimer interface (Figure 7a). The P234KY insertion is associated with a CMT-2D phenotype in mice. In prior studies, CMT-2D mutations have been variously associated with a disruption of GlyRS dimer stability[13], opening of a neomorphic surface[34], and altered GlyRS-tRNA interactions[35]. In the simulations reported here, we found that the G240R mutation decreased the number of native (i.e. crystallographic) contacts at the dimer;dimer interface and also caused conformational changes in a region that appears to regulate insertion 3 dynamics (residues 80-96, 129-144, and 413-422). To inform future computational and experimental studies, we utilized our MD data to examine whether other mutations may similarly modulate insertion 3 dynamics. To do so, we compared the list of reported GlyRS mutations (Figure 7) with the contacts that were altered in MD simulations of WT and G240R GlyRS (Figure 4)[36]. Of the 21 mutations reported in Figure 7, 12 mutation sites had altered residue-residue interactions in the G240R simulations. These 12 mutation sites (L129P, D146N, D146Y, C157R, D161H, P234KY, M238R, G240R, P244L, E279D, I280F, and H418R) participate in a network of contacts that connect the β-hairpins that contain G240, the dimer interface, and residues hypothesized to regulate insertion 3 dynamics (residues 80-96, 129-144, or 413-422). The 9 mutation sites that did not have altered residue-residue in the G240R simulations (A57V, E71G, H162R, S211F, L218Q, D500N, S581L, G526R, G598A) are distributed throughout the GlyRS structure. This categorization predicts one set of mutations that may impair insertion 3 dynamics. Altered insertion 3 conformations may either disrupt native GlyRS-tRNA interactions or provide a surface through which GlyRS forms neomorphic interactions with other molecules. The remaining mutation sites are predicted to operate through other molecular mechanisms. However, this prediction is naïve as it is based on simulations of a single mutation. Some mutations, such as E71G and D500N, may alter insertion 3 dynamics in other ways. A unifying framework by which GlyRS mutations lead to CMT-2D remains elusive. However, the simulations reported here should guide the design of future studies and assist in identifying the pathological origins of GlyRS-associated CMT. Of particular interest are those mutations that did not modify contact networks involved in insertion 3 dynamics. Our data suggest that studies on these mutations may provide additional insights into the precise molecular mechanisms linking GlyRS mutations and CMT-2D. A57 is located in the N-terminal WHEP domain, and the structural significance of this residue is uncertain. H162, S211 and L218 are within insertion 1 and likely form interactions with tRNA. D500 is within insertion 3 and forms salt bridges with neighboring residues that may stabilize certain insertion 3 conformations. S581, G598, and G598 are in regions associated with tRNA binding. G526 is in a catalytic motif of the catalytic domain.
Figure 7. Structural locations of CMT-2D GlyRS mutations.
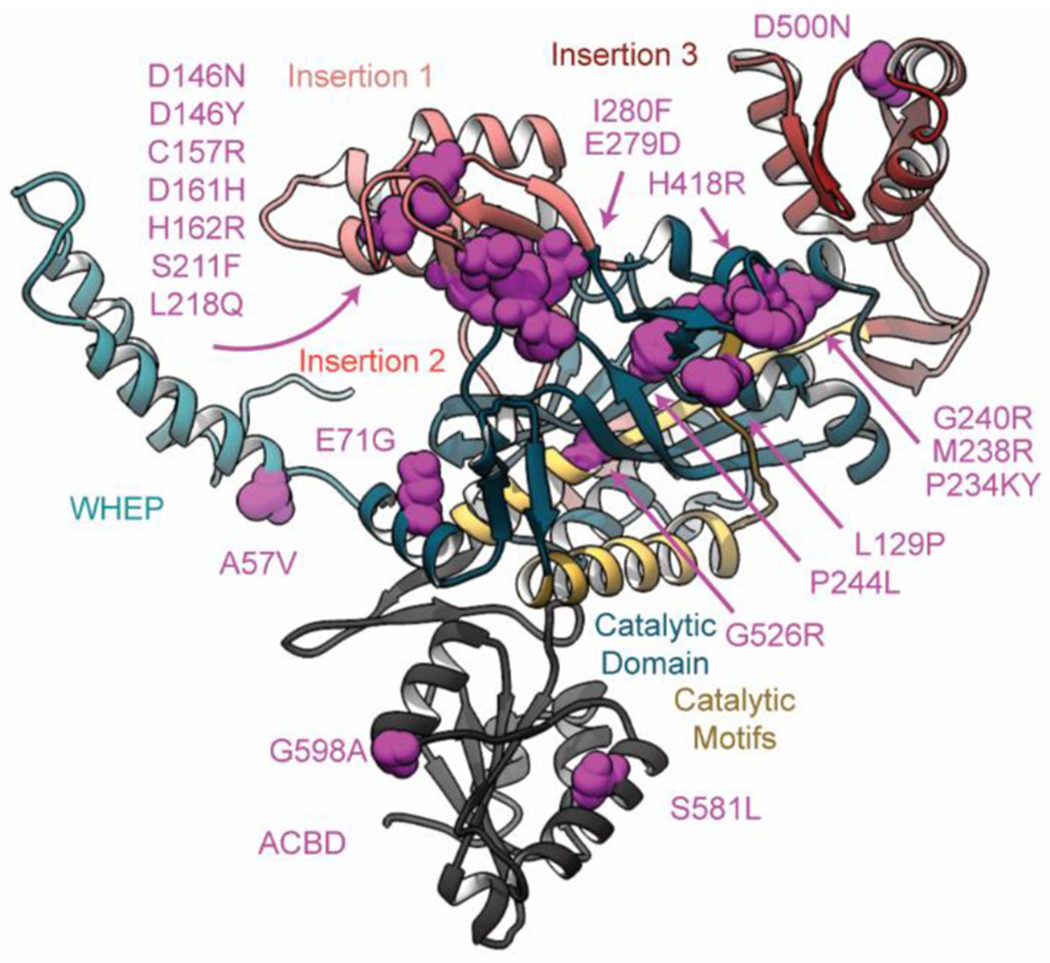
GlyRS mutations that are associated with CMT-2D pathologies are distributed throughout the structure, but primarily localize to the dimer interface. Here, the atoms of mutation sites are represented as magenta spheres. For clarity only a GlyRS monomer is shown.
5. Conclusion
Our MD simulations revealed large-scale amplitude motions. The alternate conformations observed in MD are also captured in X-ray crystal structures of GlyRS in complex with its tRNA. Therefore, we conclude that these alternate conformations are natively sampled by GlyRS. We also saw that large-amplitude motions in insertion 1 and 3, which are likely related to tRNA binding, became disrupted in the presence of G240R. Much emphasis has been placed on the capacity of CMT-2D mutations to destabilize the dimer:dimer interface of GlyRS. However, our MD data along with in vitro reports of altered GlyRS:tRNA interactions indicate that altered insertion 1/3 dynamics may also be relevant to the structural origins of CMT-2D pathologies. Altered conformations of insertions 1 and 3 may affect tRNA binding or the formation of neomorphic interactions with other biomolecules. Other ARS proteins associated with CMT pathologies may have similar dynamics tied to interactions or other molecules that may be disrupted by mutations. The results described here provide insight into how mutations in GlyRS proteins lead to Charcot-Marie-Tooth pathology in human patients. Compounds capable of inhibiting these structural changes may have potential to ameliorate symptoms in these patients and are a promising direction for future study.
Acknowledgements
The project described was supported by a Cardiovascular Research Training Grant to MCC via Award Number T32HL007828 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) resource COMET through allocation TG-MCB200100 to M.C.C. and M.R. XSEDE is supported by National Science Foundation grant number ACI-1548562. Partial funding for M.C.C and M.R. comes from the UW Center for Translational Muscle Research, sponsored by NIH NIAMS P30AR074990
Footnotes
Ethical Statement
- This material is the authors’ own original work, which has not been previously published elsewhere.
- The paper is not currently being considered for publication elsewhere.
- The paper reflects the authors’ own research and analysis in a truthful and complete manner.
- The paper properly credits the meaningful contributions of coauthors and co-researchers.
- The results are appropriately placed in the context of prior and existing research.
- All sources used are properly disclosed (correct citation). Literally copying of text must be indicated as such by using quotation marks and giving proper reference.
- All authors have been personally and actively involved in substantial work leading to the paper, and will take public responsibility for its content.
The violation of the Ethical Statement rules may result in severe consequences.
To verify originality, your article may be checked by the originality detection software iThenticate. See also http://www.elsevier.com/editors/plagdetect.
I agree with the above statements and declare that this submission follows the policies of Brain Multiphysics as outlined in the Guide for Authors and in the Ethical Statement.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].de Pouplana L Ribas, Schimmel P, Aminoacyl-tRNA Synthetases as Clues to Establishment of the Genetic Code, editor, in: de Pouplana L Ribas (Ed.), The Genetic Code and the Origin of Life, Springer US, Boston, MA, 2004, pp. 119–133. [Google Scholar]
- [2].Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs, Nature 347 (6289) (1990) 203–206. [DOI] [PubMed] [Google Scholar]
- [3].Boczonadi V, Jennings MJ, Horvath R, The role of tRNA synthetases in neurological and neuromuscular disorders, FEBS Lett. 592 (5) (2018) 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, et al. , Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V, Am. J. Hum. Genet 72 (5) (2003) 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mo Z, Zhao X, Liu H, Hu Q, Chen XQ, Pham J, et al. , Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy, Nat Commun 9 (1) (2018) 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sleigh JN, Dawes JM, West SJ, Wei N, Spaulding EL, Gómez-Martín A, et al. , Trk receptor signaling and sensory neuron fate are perturbed in human neuropathy caused by Gars mutations, Proc. Natl. Acad. Sci 114 (16) (2017) E3324. -E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sleigh JN, Gómez-Martín A, Wei N, Bai G, Yang XL, Schiavo G, Neuropilin 1 sequestration by neuropathogenic mutant glycyl-tRNA synthetase is permissive to vascular homeostasis, Sci Rep 7 (1) (2017) 9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Oprescu SN, Chepa-Lotrea X, Takase R, Golas G, Markello TC, Adams DR, et al. , Compound heterozygosity for loss-of-function GARS variants results in a multisystem developmental syndrome that includes severe growth retardation, Hum Mutat 38 (10) (2017) 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Achilli F, Bros-Facer V, Williams HP, Banks GT, AlQatari M, Chia R, et al. , An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy, Dis Model Mech 2 (7–8) (2009) 359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Griffin LB, Sakaguchi R, McGuigan D, Gonzalez MA, Searby C, Züchner S, et al. , Impaired Function is a Common Feature of Neuropathy-Associated Glycyl-tRNA Synthetase Mutations, Hum Mutat 35 (11) (2014) 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cader MZ, Ren J, James PA, Bird LE, Talbot K, Stammers DK, Crystal structure of human wildtype and S581L-mutant glycyl-tRNA synthetase, an enzyme underlying distal spinal muscular atrophy, FEBS Lett. 581 (16) (2007) 2959–2964. [DOI] [PubMed] [Google Scholar]
- [12].Logan DT, Mazauric MH, Kern D, Moras D, Crystal structure of glycyl-tRNA synthetase from Thermus thermophilus, EMBO J. 14 (17) (1995) 4156–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nangle LA, Zhang W, Xie W, Yang X-L, Schimmel P, Charcot–Marie–Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect, Proc. Natl. Acad. Sci 104 (27) (2007) 11239–11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].He W, Bai G, Zhou H, Wei N, White NM, Lauer J, et al. , CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase, Nature 526 (7575) (2015) 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. , The protein data bank, Nucleic Acids Res. 28 (1) (2000) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Deng X, Qin X, Chen L, Jia Q, Zhang Y, Zhang Z, et al. , Large Conformational Changes of Insertion 3 in Human Glycyl-tRNA Synthetase (hGlyRS) during Catalysis*, J. Biol. Chem 291 (11) (2016) 5740–5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xie W, Nangle LA, Zhang W, Schimmel P, Yang X-L, Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase, Proc. Natl. Acad. Sci 104 (24) (2007) 9976–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Qin X, Deng X, Chen L, Xie W, Crystal Structure of the Wild-Type Human GlyRS Bound with tRNAGly in a Productive Conformation, J. Mol. Biol 428 (18) (2016) 3603–3614. [DOI] [PubMed] [Google Scholar]
- [19].Webb B, Sali A, Comparative protein structure modeling using MODELLER, Curr Protoc Bioinformatics 54 (1) (2016) 1–5, 56637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pettersen EF, Goddard TD, Huang CC, Couch GS, DM Greenblatt, Meng EC, et al. , UCSF Chimera—a visualization system for exploratory research and analysis, J. Comput. Chem 25 (13) (2004) 1605–1612. [DOI] [PubMed] [Google Scholar]
- [21].Scouras AD, Daggett V, The Dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein science : a publication of the, Protein Society 20 (2) (2011) 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Towse C-L, J Rysavy Steven, M Vulovic Ivan, Daggett V, New Dynamic Rotamer Libraries: Data-Driven Analysis of Side-Chain Conformational Propensities, Structure 24 (1) (2016) 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Case DA, Belfon K, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, et al. , AMBER 2020, University of California, San Francisco, 2020. [Google Scholar]
- [24].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput 11 (8) (2015) 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys 79 (2) (1983) 926–935. [Google Scholar]
- [26].Li P, Merz KM Jr., Taking into account the ion-induced dipole interaction in the nonbonded model of ions, J. Chem. Theory Comput 10 (1) (2014) 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li P, Song LF, Merz KM Jr., Parameterization of highly charged metal ions using the 12-6-4 LJ-type nonbonded model in explicit water, J. Phys. Chem. B 119 (3) (2015) 883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li P, Song LF, Merz KM Jr., Systematic parameterization of monovalent ions employing the nonbonded model, J. Chem. Theory Comput 11 (4) (2015) 1645–1657. [DOI] [PubMed] [Google Scholar]
- [29].Roe DR, Cheatham TE III, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput 9 (7) (2013) 3084–3095. [DOI] [PubMed] [Google Scholar]
- [30].Tubiana T, Carvaillo J-C, Boulard Y, Bressanelli S, TTClust: a versatile molecular simulation trajectory clustering program with graphical summaries, J. Chem. Inf. Model 58 (11) (2018) 2178–2182. [DOI] [PubMed] [Google Scholar]
- [31].Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW, An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model, Neuron 51 (6) (2006) 715–726. [DOI] [PubMed] [Google Scholar]
- [32].Guo M, Schimmel P, Yang X-L, Functional expansion of human tRNA synthetases achieved by structural inventions, FEBS Lett. 584 (2) (2010) 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rho SB, Lee KH, Kim JW, Shiba K, Jo YJ, Kim S, Interaction between human tRNA synthetases involves repeated sequence elements, Proc. Natl. Acad. Sci 93 (19) (1996) 10128–10133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].He W, Zhang H-M, Chong YE, Guo M, Marshall AG, Yang X-L, Dispersed disease-causing neomorphic mutations on a single protein promote the same localized conformational opening, Proc. Natl. Acad. Sci 108 (30) (2011) 12307–12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mendonsa S, von Kuegelgen N, Bujanic L, Chekulaeva M, Charcot–Marie–Tooth mutation in glycyl-tRNA synthetase stalls ribosomes in a pre-accommodation state and activates integrated stress response, Nucleic Acids Res. 49 (17) (2021) 10007–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Benson NC, Daggett V A chemical group graph representation for efficient high-throughput analysis of atomistic protein simulations, J. Bioinform. Comput. Biol 10 (4) (2012), 1250008. [DOI] [PMC free article] [PubMed] [Google Scholar]