

Abstract
Respiratory failure is a common characteristic of systemic inflammatory response syndrome (SIRS) and sepsis. Trauma and severe blood loss cause the release of endogenous molecules known as damage-associated molecular patterns (DAMPs). Mitochondrial N-formyl peptides (F-MITs) are DAMPs that share similarities with bacterial N-formylated peptides, and are potent immune system activators. Recently, we observed that hemorrhagic shock-induced increases in plasma levels of F-MITs associated with lung damage, and that antagonism of formyl peptide receptors (FPR) ameliorated hemorrhagic shock-induced lung injury in rats. Corroborating these data, in the present study, it was observed that F-MITs expression is higher in plasma samples from trauma patients with SIRS or sepsis when compared to control trauma group. Therefore, to better understand the role of F-MITs in the regulation of lung and airway function, we studied the hypothesis that F-MITs lead to airway contraction and lung inflammation. We observed that F-MITs induced concentration-dependent contraction in trachea, bronchi and bronchioles. However, pre-treatment with mast cells degranulator or FPR antagonist decreased this response. Finally, intratracheal challenge with F-MITs increased neutrophil elastase expression in lung and inducible nitric oxide synthase and cell division control protein 42 expression in all airway segments. These data suggest that F-MITs could be a putative target to treat respiratory failure in trauma patients.
Keywords: Trauma, Mitochondrial N-formyl peptides, Airway and lung inflammation
1. Introduction
Systemic inflammatory response syndrome (SIRS) is a serious condition associated with multiple organ dysfunction and failure. In 1992, the American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference concluded that the diagnosis of SIRS requires the same criteria as sepsis, minus the presence of infection [1]. Therefore, the diagnostic criteria for SIRS include the presence of two or more of the following: hypothermia or hyperthermia, tachycardia, tachypnea, and abnormal white blood cell count. If unresolved, SIRS may lead to hypotension, vascular leakage, disseminated coagulation, organ failure and death [1]. SIRS can be seen following a variety of insults, such as trauma, burns or acute myocardial infarction.
A frequent complication of SIRS and sepsis is the development of increased airway resistance and lung inflammation [2,3]. For example, Uhlig et al. [3] demonstrated that rat lungs exposed to endotoxin present increased airway resistance due to constriction of terminal bronchioles [3] Moreover, it has been demonstrated that N-formyl-methionyl-leucyl-phenylalanine (FMLP), a bacterial chemotactic peptide, induces contraction of human airway [4] and lung inflammation in mice [5]. Therefore, during the onset of infection or sepsis it is clear that the presence of the pathogen is the primary cause of increased airway resistance and lung injury. However, the reason why patients that have sepsis-like symptoms or SIRS (sterile condition) also develop airway constriction and lung inflammation is still unknown.
It has been proposed that cell components from traumatized tissue are the primary instigators of sterile inflammation or SIRS [6–8]. These cell components are called damage-associated molecular patterns (DAMPs). DAMPs are endogenous molecules released from cells or tissues following injury, which activate the innate immune system in a similar manner to pathogens [7]. Mitochondrial N-formyl peptides are DAMPs that share similarities with bacterial N-formyl peptides. Although these peptides play a crucial role in the protein synthesis of bacteria and mitochondria [9], they are not used in cytosolic protein synthesis of eukaryotes. Therefore, mitochondrial and bacterial N-formyl peptides are recognized by the innate immune system as pathogens and are known to play a role in the initiation of inflammation by activating the formyl peptide receptor (FPR) [10,11].
The FPR has been identified as a subfamily of G protein-coupled receptors [10]. It is well known that FPR-1 and FPR-2 are expressed at high levels on leukocytes, and that they mediate cell chemotaxis [10,12]. More recently, it has been demonstrated that lung and tracheobronchial epithelial cells also express FPR and its expression is higher in response to scratch injury [13]. Interestingly, we were the first to observe that mitochondrial N-formyl peptides (F-MITs) are able to activate FPR in lung and cardiovascular system [6,11]. This activation led to sepsis-like symptoms, including cardiovascular collapse and lung damage [11]. Based on these data, we hypothesized in the current investigation that F-MITs, which are released during traumatic injury, are able to induce airway contraction and lung inflammation. Therefore, if this hypothesis is correct, F-MITs may be an important instigator of airway dysfunction and respiratory failure observed in trauma-induced SIRS.
2. Methods
2.1. Human plasma analyses
Blood samples from patients (18–44 years old) with moderate to severe trauma as defined by the Injury Severity Score Index (score ≥ 15) were collected [14,15]. The patients were divided into three groups: 1) control (trauma patients; no evidence of bacteria in blood cultures and no SIRS; 2) SIRS (trauma patients with SIRS but with no evidence of bacteria in blood cultures) and 3) sepsis (trauma patients with SIRS and with evidence of bacteria in blood cultures) [14,15]. No samples or data were collected from participants who had preexisting conditions that would affect the normal function of the immune system, including those with cancer, HIV or autoimmune disease. Permission to conduct this study was obtained from Augusta University institutional review board. Immunoblotting, described in detail below, was performed to confirm the presence of mitochondrial NADH dehydrogenase 6 (ND6) in the circulation. This protein has been shown to contain N-formylmethionine residues and be chemoattractant for neutrophil [12].
2.2. Animals
Twelve-week old male Wistar rats (Harlan) were maintained on a 12:12 h light–dark cycle with both standard chow and water ad libitum. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH) and were reviewed and approved by the Institutional Animal Care and Use Committee of the Augusta University.
In vivo experiments:
Male Wistar rats were anaesthetized with isoflurane 4% and intratracheally challenged with an equal volume of mitochondrial formylated peptides derived from NADH dehydrogenase 6 (F-MITs; sequence: N-formyl-Met-Met-Tyr-Ala-Leu-Phe; (0.02 mg/rat) or non-formylated peptide (control; sequence: Met-Met-Tyr-Ala-Leu-Phe; 0.02 mg/rat). For this, a midline incision was made above the sternum and the trachea was exposed. A 28-gauge needle was inserted into the trachea and 0.3 ml of F-MITs or vehicle was instilled. After 6 h, the rats were killed and the lungs, trachea, bronchus and bronchiole were removed. The treatment did not change lung weight (Control: 1.71 ± 0.10 vs. F-MIT: 1.60 ± 0.12, p > 0.05). The lung and airways (trachea, bronchus and bronchiole) were cleaned and stored at −80 °C for future analyses.
In vitro experiments:
Another group of Wistar rats were anaesthetized with isoflurane (5%) and killed. Airways (trachea, bronchi and bronchioles) were removed, cleaned of surrounding tissue, and mounted in a pin (trachea and bronchi) or wire (bronchioles) myograph to assess smooth muscle responsiveness to F-MITs and control.
2.3. Immunofluorescence and immunohistochemistry
After 6 h of intratracheal challenge with F-MITs or non-formylated peptides, the right lungs were collected and embedded in tissue medium frozen (OCT, Triangle Biomedical Sciences, NC), cut in a cryostat (10 μm section) and fixed. Conventional immunofluorescence was used to measure the presence of neutrophil elastase and total FPR protein expression. After blocking for 1 h in a solution of 1X phosphate buffered saline (PBS) with 0.01% Triton X-100 and 5% horse serum, sections were incubated with primary antibodies, rabbit anti-FPR (1:100; Abcam, Cambridge, MA) or rabbit anti-neutrophil elastase (1:100; Abcam, Cambridge, MA) for 24 h. Reactions with primary antibodies were followed by 1 h incubation in the presence of fluorescently labeled secondary antibody (anti-rabbit FITC-labeled antibody). Sections were then washed and visualized using a Zeiss LSM 780 upright confocal microscope (40X objective; single optical plane = 1 μm thick) (Carl Zeiss MicroImaging, Oberkochen, Germany). The presence of neutrophil elastase in lungs were confirmed by immunohistochemistry. For this, sections were heated for 5 min at 95 °C in Target Retrieval solution (Deko), followed by endogenous peroxidase activity blocking with 0.3% H2O2, non-specific binding blocking with competing serum and overnight incubation with anti-elastase antibody (1:100, Abcam) in a humidified chamber. Then, after incubation with biotinylated secondary antibody and Vectastain ABC Elite reagent, sections were exposed to diaminobenzidine (DAB)/H2O2 and staining was monitored and timed. Sections were then counterstained with hematoxylin, dried and mounted. Photographs were taken using an inverted Olympus microscope (20x magnification).
2.4. Preparation of single-cell suspension of lung and analytical flow cytometry
Lungs underwent mechanical disruption by a GentleMacs tissue dissociator (Miltenyi Biotec) for 1 min in 2 ml of intracellular fixative (eBioscience). The fixation reaction was stopped through the addition of 20 ml of ice-cold flow cytometry wash buffer (FWB)-PBS with 2% FCS, 0.1% sodium azide, and 5 mM EDTA. The suspension was subsequently sieved through a 40-μm nylon filter and washed again with 20 ml ice-cold FWB. The lung cell suspension was centrifuged at 4 °C at 2000 rpm. The cell pellet was resuspended in 1 ml of FWB and placed on ice [16]. The pellet then was subjected to flow cytometry analysis to detect and assess the neutrophil status in the lung tissues. Accordingly, commercially available antibodies against neutrophils (neutrophil elastase, LY6G and CD18) were used coupled with the use of a FACSCalibur flow cytometer (BD BioSciences, San Diego, CA) as described previously [17].
2.5. Airway responsiveness
To evaluate airway contractility, trachea, bronchi and bronchioles from naïve rats were removed and cleaned in cold Krebs-Henseleit solution containing (in mmol/l): 118 NaCl, 4.7 KCl, 25 NaHCO3, 2.5 CaCl2·2H2O, 1.2 KH2PO4, 1.2 MgSO4·7H2O, 0.01 EDTA, and 11 glucose. The tissues were cut in rings of 2–3 mm and mounted in a pin (trachea and bronchi) or wire (bronchiole) myograph. The segments were set to a basal force of 30 mN for trachea, 5 mN for bronchi and 2 mN for bronchiole. Following 30 min of equilibration, the contractility of the tissues was tested by exposure to a high-K+ (120 mmol/L KCl) solution. Some segments were incubated with FPR-1 antagonist (cyclosporin H, 1 μM), FPR-2 antagonist (WRW4, 10 μM) or mast cell degranulating agent (compound 48/80 25 μg/ml) for 30 min. Subsequently, the segments were contracted with acetylcholine (1 μmol/L) and cumulative concentrations–responses curves to F-MITs (1–30 μM) were performed. All results are presented as %KCl for contraction.
2.6. Biochemistry assays
Endotoxin detection assay (GenScript, USA) was used to confirm the absence of lipopolysaccharides (LPS) in non-formylated and formylated peptides (8 mg/ml; diluted in saline and 1% DMSO).
2.7. Immunoblotting
Proteins were extracted from the trachea, bronchus, bronchiole, lung or plasma. After loading (10–30 μg), proteins were separated by SDS-polyacrylamide electrophoresis as previously described [11]. PVDF membranes were incubated overnight at 4 °C with a primary antibody raised against iNOS (1:500), cell division control protein 42 (CDC42, 1:1000); NADH dehydrogenase 6 (ND6, 1:1000), neutrophil elastase (1:1000), FPR (1:1000), and β-actin (1:40,000).
2.8. Reagents and chemicals
Acetylcholine, protease inhibitor cocktail, C48/80 compound, DMSO and antibodies to β-actin and anti-NADH dehydrogenase 6 were purchased from Sigma–Aldrich (USA). WRW4 was purchased from Tocris Bioscience (UK). F-MITs, non-formylated peptides and endotoxin detection assay were purchased from GenScript (USA). Antibodies to iNOS and CDC42 were purchased from BD Biosciences (San Jose, CA). Cyclosporine H, and antibodies to FPR and neutrophil elastase were purchased from Abcam (USA). The primary antibodies against neutrophils (LY6G and CD18) were purchased from eBioscience.
2.9. Statistics
Results are presented as mean ± standard error of the mean (SEM). The statistical procedures used included Student’s unpaired t-test, one-way and two-way analysis of variance (ANOVA) and non-linear regression analysis. All analyses were performed using data analysis software GraphPad Prism 5.0 (USA). Statistical significance was set at p < 0.05.
3. Results
3.1. Traumatic injury induces the release of F-MITs and its expression is higher in SIRS and sepsis
We previously demonstrated that rats which underwent sterile trauma present circulating F-MITs, which was associated with FPR activation in lung [11]. Corroborating these data, in the present study we observed that F-MITs are released into the circulation of trauma patients (Fig. 1). However, F-MITs expression was higher in plasma samples from trauma patients with SIRS and sepsis when compared to control.
Fig. 1.
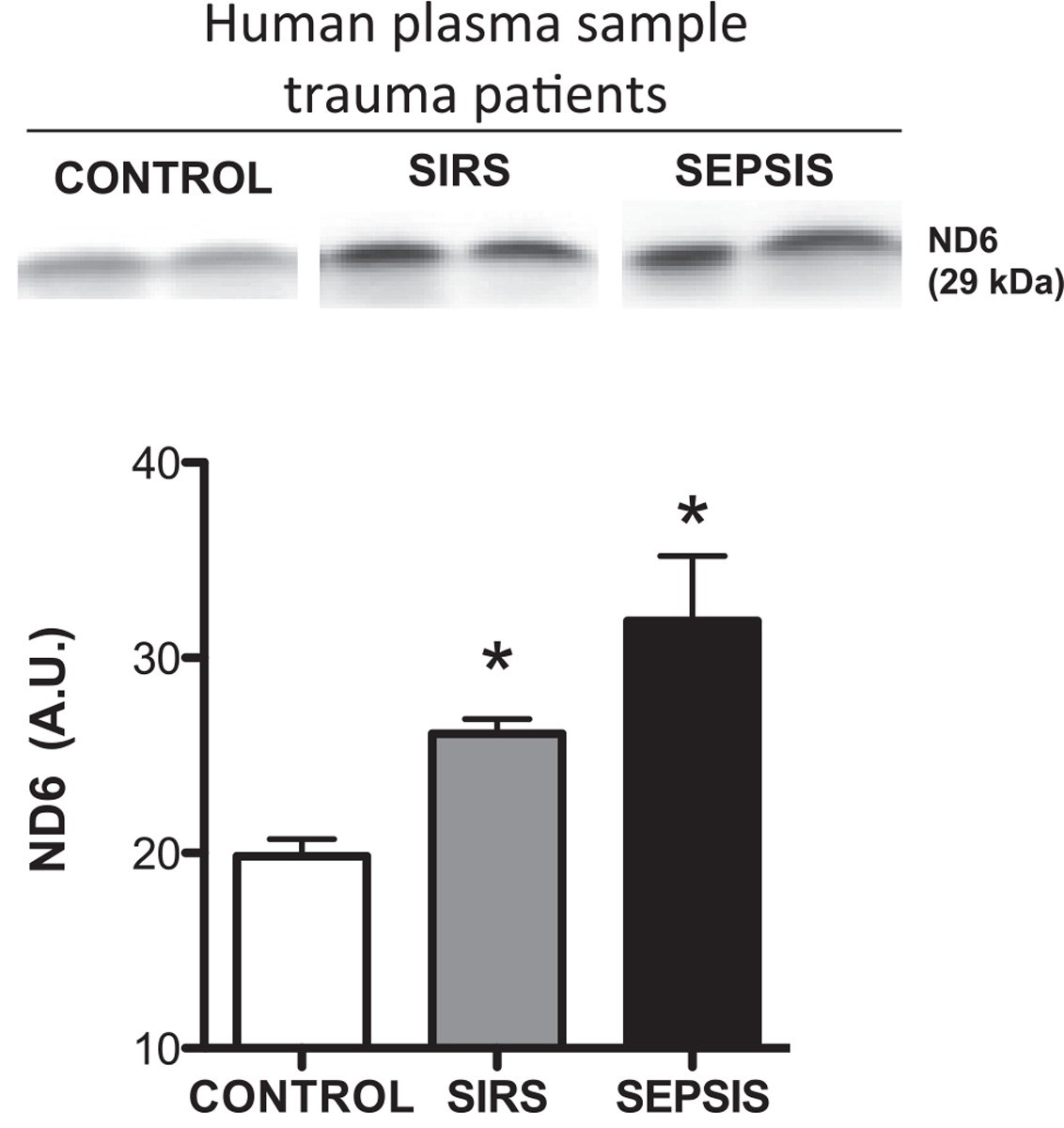
Representative blots and densitometric analyses from protein expression for mitochondrial NADH dehydrogenase 6 (ND6) (F-MITs) in plasma from trauma patients. Control (no evidence of bacteria in blood cultures and no signs of SIRS); SIRS (no evidence of bacteria in blood cultures) and SEPSIS (evidence of bacteria in blood cultures). Mean ± SEM, n = 3–5. One-way ANOVA *vs. control, p < 0.05.
3.2. Mitochondrial N-formyl peptides (F-MITs) induce lung neutrophil infiltration
To confirm that F-MIT was free of endotoxin contamination, an endotoxin detection assay was used on formylated and non-formylated peptides. Endotoxin was absent in all samples evaluated (data not shown). This result confirmed that F-MIT induces its effects in a sterile manner and independently of bacterial contamination.
It is known that neutrophils can release elastase and DNA following a danger signal. Together, elastase and DNA form a network of extracellular fibers called neutrophil extracellular traps, (NETs) to bind pathogens [18]. Here, we observed the presence of neutrophil elastase in lung tissue from Wistar rats that were intratracheally challenged with F-MITs (0.02 mg/rat) (Fig. 2B and E). To confirm the necessity of the N-formylated terminus for the observed lung and airway inflammation, some animals were instilled with a peptide fragment of the same amino acid sequence, but non-formylated (Met-Met-Tyr-Ala-Leu-Phe). The presence of neutrophil elastase in lungs from animals treated with non-formylated peptides (negative control) was not noteworthy (Fig. 2A). Our results suggest that formylated peptides, specifically mitochondrial N-formyl peptides, but not non-formylated peptides, are necessary to induce the release of neutrophil elastase. Also, using flow cytometry, we observed that the expression of neutrophil elastase and activated neutrophils are increased in lung tissue from rats treated with F-MIT (Fig. 2E). Corroborating the data observed by immunofluorescence, immunohistochemistry and flow cytometry (Fig. 2A, B and E), we demonstrated, by Western blot, that F-MITs instillation increased neutrophil elastase protein expression in lung, but not in airway segments (Fig. 2C). Additionally, F-MITs instillation increased iNOS protein expression in all airway segments and lung (Fig. 2D).
Fig. 2.
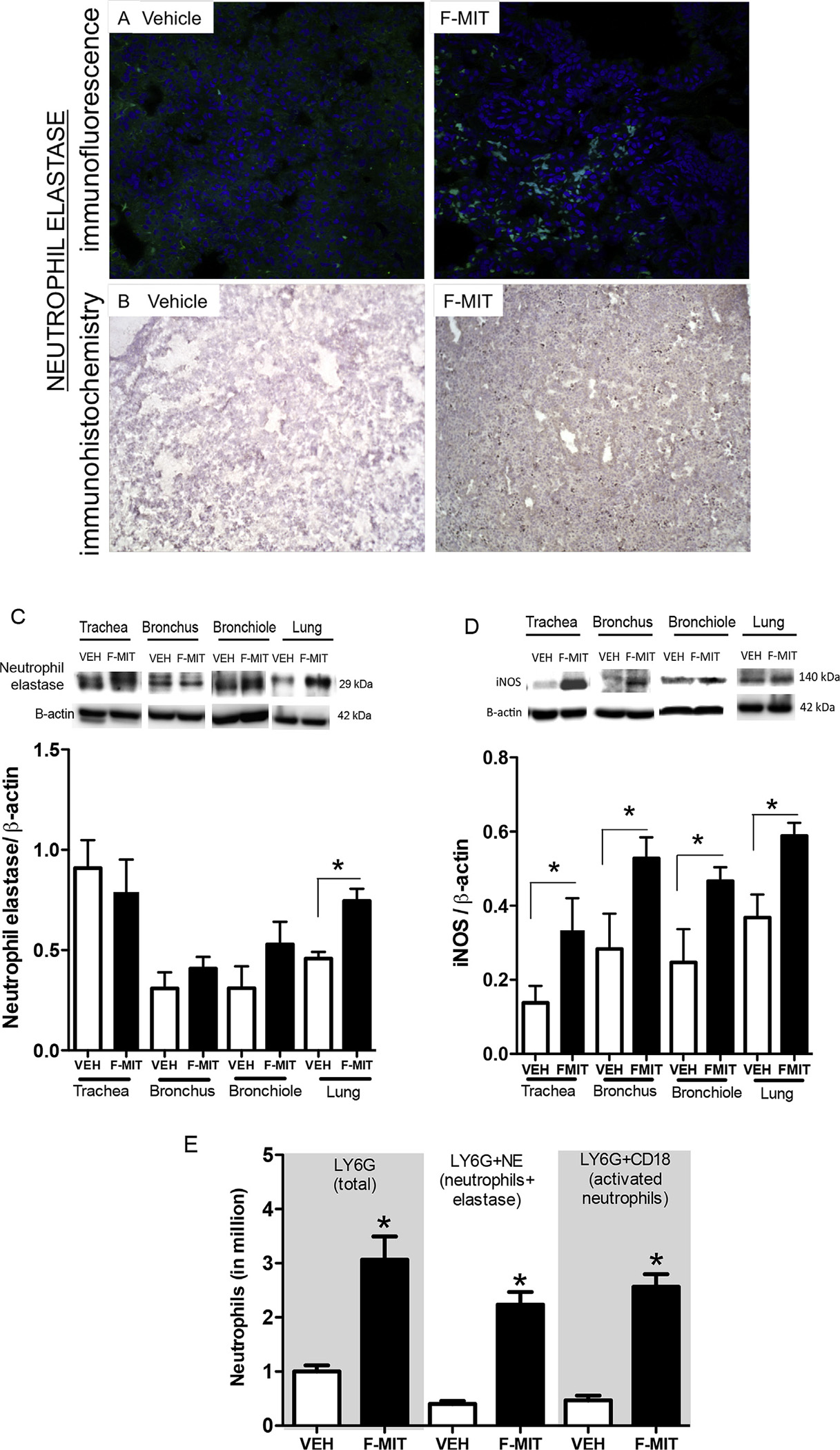
Immunofluorescence: (A) top image represents neutrophil elastase (confocal microscopy 40 X) in lung from Wistar rats that were intratracheally challenged with an equal volume of F-MITs (0.02 mg/rat) or non-formylated peptide control (vehicle, VEH) (B). Immunohistochemistry (optical microscopy 10 X) confirms the presence of neutrophil elastase in lung. (C) and (D) demonstrate representative blots and densitometric analyses from protein expression for neutrophil elastase and iNOS of lungs, trachea, bronchus and bronchiole from animals that were intratracheally challenged with F-MITs or VEH. (E) single and double-staining for neutrophils in lung tissue using flow cytometry. Cells positively stained with LY6G and neutrophil elastase were considered neutrophils, and LY6G and CD18 were considered activated neutrophil. Mean ± SEM, n = 3–5. T-test *vs. vehicle (VEH), p < 0.05.
3.3. Mitochondrial N-formyl peptides induce airway contraction via FPR and mast cells
To evaluate if F-MITs play a role in airway contractile function, a concentration–response curve to F-MITs (1–30 μmol/L) was performed in trachea, bronchi and bronchiole. Interestingly, F-MITs induced contraction in a concentration-dependent manner (Fig. 3A). The FPR-2 antagonist (WRW4, 10 μmol/L) (Fig. 3B), but not FPR-1 antagonist (cyclosporine H, 1 μmol/L) (Fig. 3C), decreased this response.
Fig. 3.
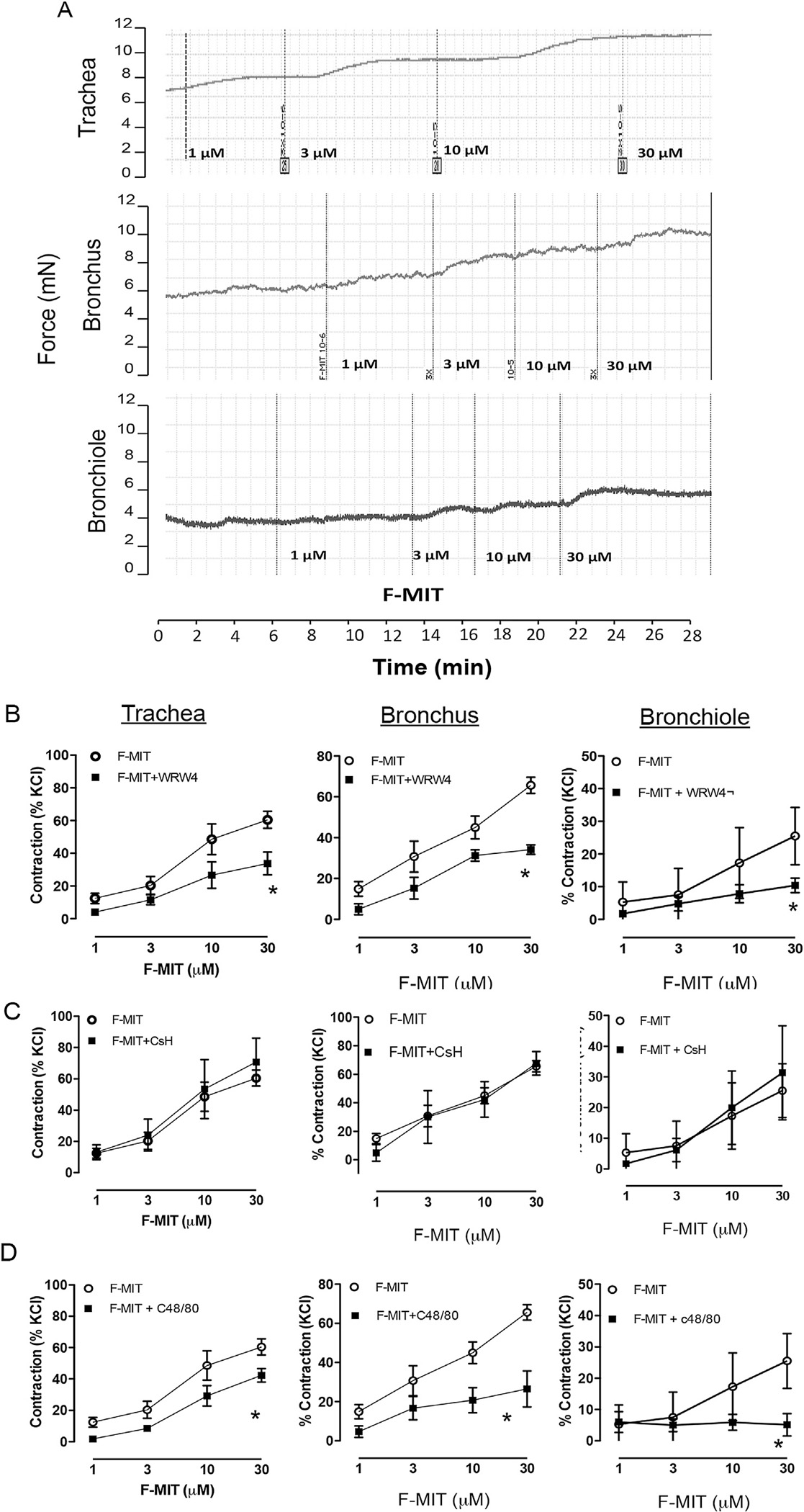
Typical trace for concentration–response curve to F-MITs (1–30 μM) in trachea, bronchus and bronchiole from naive Wistar rats (A). The rings were previously precontracted with acetylcholine (1 μM). Graphs represent concentration–response curve to F-MITs (1–30 μM) in the absence or presence of WRW4 (FPR-2 antagonist) (B), cyclosporin H (CsH, FPR-1 antagonist) (C) and C48/80 compound (mast cell degranulator) (D). Mean ± SEM, n = 4–8. Two-way ANOVA: *vs. F-MITs, p < 0.05.
In airways, mast cells are located adjacent to nerves, blood vessels and lymphatics. It is well known that, when activated, mast cells induce the release of smooth muscle contractile factors, including histamine [19,20]. Also, it has been shown that bacterial-N-formyl peptides activate FPR in mast cells [21]. Therefore, here we questioned if mast cells activation could also lead to F-MITs induced-contraction in airway segments. As observed in Fig. 3D, prior mast cell degranulation by compound 48/80 (25 μg/ml) decreased F-MITs-induced contraction.
3.4. Mitochondrial N-formyl peptides activate cell division control protein 42 (CDC42) in airways
Actin cytoskeletal remodeling is an important component of airway smooth muscle contraction. Up-regulation of a cytoskeletal recruitment in highly shortened airway smooth muscle has been shown to be an important mechanism of reduced airway distensibility [22]. CDC42 induces actin polymerization, either by stimulating de novo actin nucleation or by stimulating the uncapping or severing of filaments [23]. In Fig. 4, it was observed that intratracheal challenge with F-MITs (0.02 mg/rat) increased CDC42 protein expression in all airway segments evaluated. However, F-MITs did not activate CDC42 in lung.
Fig. 4.
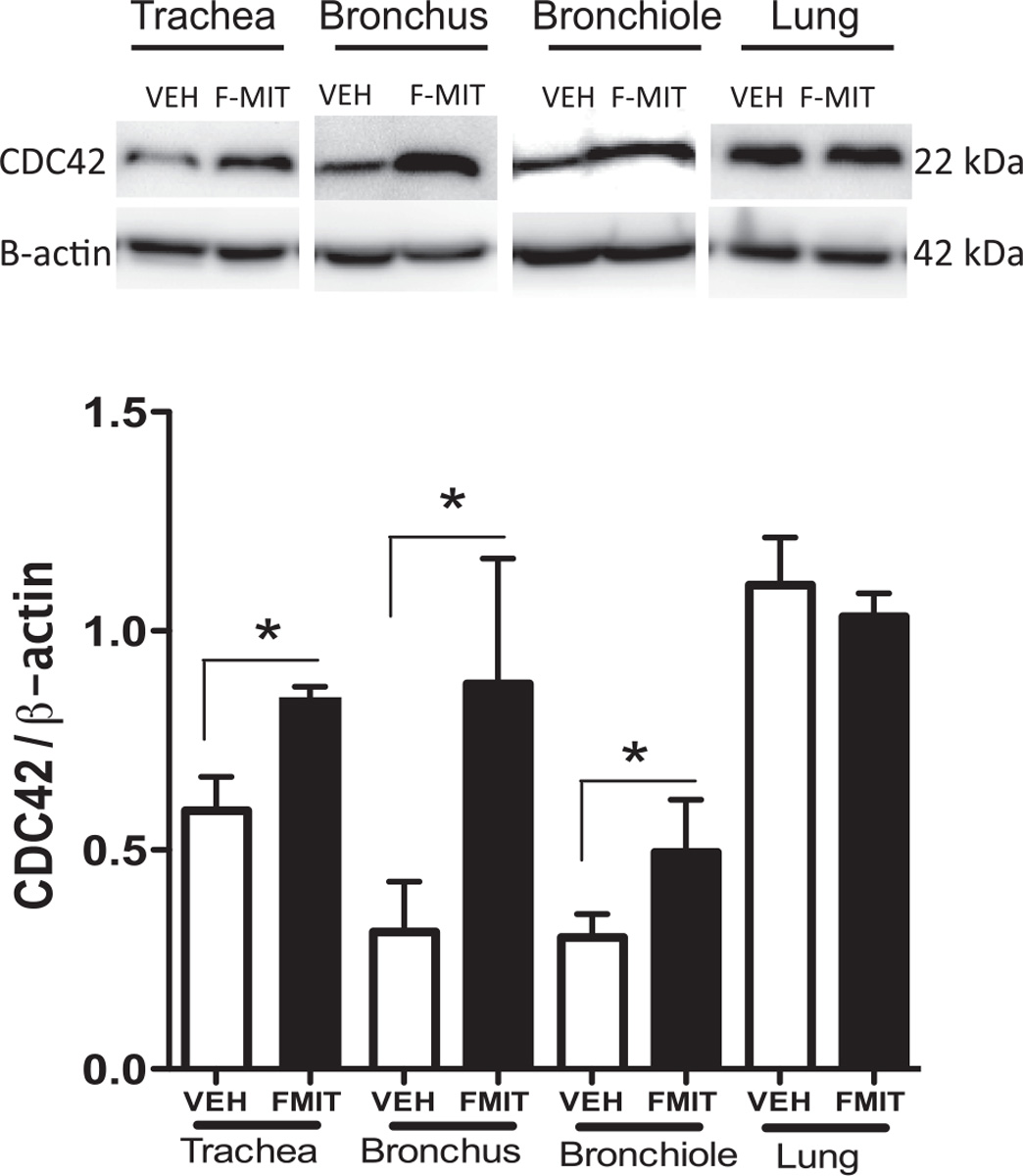
Representative blots and densitometric analyses from protein expression for CDC42 of lungs, trachea, bronchus and bronchiole from Wistar rats that were intratracheally challenged with an equal volume of F-MITs (0.02 mg/rat) or non-formylated peptide (VEH). Mean ± SEM, n = 4–5. t-test *vs. vehicle (VEH), p < 0.05.
4. Discussion
In normal conditions, fragments from mitochondria, including N-formylated peptides of mitochondrial origin, are supposed to be absent from plasma, given that mitochondria are located inside the cells. Also, this organelle, but not eukaryotic cells, uses N-formylmethionine to initiate all protein synthesis [9]. We observed that after cell trauma and injury in patients, these peptides are released into the circulation. However, only trauma patients that presented higher levels of F-MITs in the plasma demonstrated signs of SIRS and sepsis. Therefore, it is possible to infer that higher levels of F-MITs may be associated with the genesis and/or maintenance of systemic inflammation and multiple organ dysfunction, including respiratory failure, observed in patients with SIRS and sepsis. Since the worsening of lung function is correlated with the incidence of SIRS and sepsis [1,19] in trauma patients, it is possible that F-MITs may be the molecular factor released after cell damage that leads to respiratory failure. Supporting these results, we previously observed that rats which underwent hemorrhagic shock presented lung injury and high levels of F-MITs into the circulation [11]. However, prior treatment with FPR antagonist decreased hemorrhagic shock-induced lung damage [11].
Acute lung injury is present in sepsis and SIRS and is associated with increased pulmonary levels of proinflammatory cytokines and neutrophil extravasation into the alveolar space [19,24]. Hauser et al. [25] observed that fragmented mitochondria induced pulmonary inflammation via neutrophil activation. Accordingly, we recently demonstrated that F-MITs was able to cause edema, neutrophil infiltration and alveolar septal thickening associated with increased myeloperoxidase activity [11]. Further, hemorrhagic shock caused lung damage via FPR and MAPK activation [11]. In the present study, we observed the presence of neutrophil elastase in lungs from Wistar rats that were intratracheally challenged with F-MITs. Conventionally, it is known that the function of neutrophil elastase is the killing and degradation of pathogens and dangerous molecules ‘captured’ by neutrophils, whereas the main target for extracellular elastase is elastin [25]. However, it has been shown that neutrophils, upon activation, release elastase and DNA to form an extracellular fibril matrix known as neutrophil extracellular traps (NETs) to immobilize and kill pathogens [18]. Several studies demonstrated that neutrophil elastase is increased in clinical and animal models of acute lung injury and instillation or systemic application of neutrophil elastase induces respiratory failure [26]. In line with these studies, we have observed that F-MITs induced neutrophils activation and elastase release in lung, likely via FPR activation, since non-formylated peptide (control) did not reproduce this result. Additionally, it was observed that F-MITs instillation induced an increase of iNOS protein expression in lung. iNOS is extensively distributed in different tissues including airways and lung and this isoform is induced by different molecules associated with inflammation, infection and injury, e.g. cytokines and microbial products. In addition, iNOS is involved in several diseases, including pulmonary hypertension and asthma [27]. Interestingly, it has been shown that lung inflammation after allergen challenge in mice is partially dependent on NO produced mainly by iNOS [27]. Also, iNOS activation increases lung chemokine expression to facilitate the influx of inflammatory cells into the airways [27]. In the present study, although we observed an increase in iNOS expression in airways and lung, its expression was coupled with neutrophil infiltration and activation in lung tissue. This result corroborates our previous work [11], where rats which underwent hemorrhagic shock presented with increased iNOS protein expression in lung via FPR activation.
The FPR family has been most extensively investigated in the context of leukocyte recruitment and activation, where FPR promotes cell motility and mediates host defense [10,12]. It is known that the bacterial N-formyl peptides are a potent chemoattractant [10,12] with a high-affinity binding site for FPR. In an interesting study, Cardini et al. [28], reported that genetic ablation of the FPR1 gene (Fpr1) confers protection from smoking-induced lung emphysema in mice. Further, they observed that Fpr1 knockout mice displayed marked decreases in the lung migration of neutrophils and macrophages after cigarette smoke exposure [28]. Since FPR is expressed in airway segments [29] and its activation promotes cell motility, we questioned if F-MITs can bind to FPR and change airway contractility. We observed that F-MITs are able to induce contraction in a concentration-dependent manner in trachea, bronchi and bronchioles via FPR-2 activation in naive rats. Presently, the mechanism underlying selective activation of FPR2 by F-MITs in airway smooth muscle is unclear, however future studies will be performed to clarify this question. We also do not know how FPR-2 activation leads to contraction in airway segments. We can infer that FPR activation may induce actin polymerization leading to a slow contraction. This inference is based on our data showing that F-MITs instillation increased CDC42 protein expression in all airway segments evaluated. CDC42 is a member of Rho GTPase family, which regulates F-actin reorganization and induces actin polymerization, either by stimulating de novo actin nucleation or by stimulating the uncapping or severing of filaments [23]. Additionally, it is known that actin cytoskeletal remodeling is an important mechanism of airway smooth muscle contraction [30], therefore, in the present study we suggest that F-MITs bind FPR in airway segments leading to CDC42 activation and contraction. Supporting these data, it has been shown that FPR activation using bacterial N-formyl peptides significantly enhances epithelial cell restitution and migration via CDC42 [30]. Inhibition of Rac1 and CDC42 using pharmacological inhibitors and dominant negative mutants also inhibited the bacterial N-formyl peptides-induced increase in cell migration [30].
Stimulation of FPR by bacterial N-formyl peptides induces basophil and mast cells release of immunogenic compounds such as histamine [21,31]. Furthermore, we demonstrated that histamine plays a role in F-MITs-induced sepsis-like symptoms since cimetidine, a histamine H2-receptor antagonist, completely abolished cardiovascular collapse induced by F-MITs infusion [11]. It is well known that histamine causes the contraction of airway smooth muscle [20]. Further, in response to allergens, the release of histamine by mast cells induces a severe airway obstruction and, in some cases, can lead to anaphylaxis. Here, we observed that mast cell degranulation, using compound 48/80, also decreased F-MITs-induced contraction, probably through blocking histamine release.
Collectively, our findings provide a new and different way of considering the role of F-MITs in lung injury and airway contraction following trauma. As such, this pathway could be considered a putative target for the treatment of respiratory failure and sterile inflammation (Fig. 5).
Fig. 5.
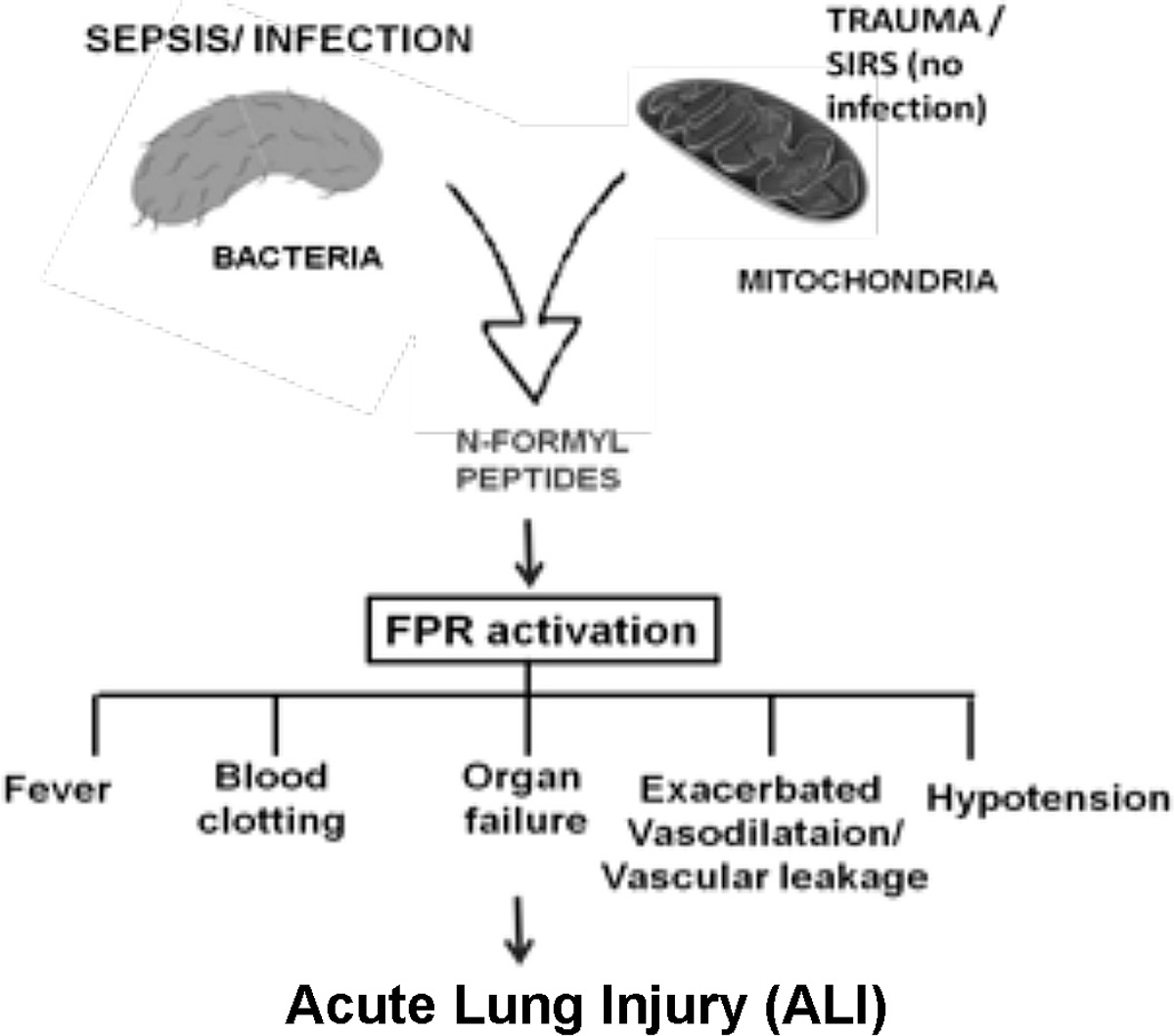
Scheme shows that trauma induces the release of mitochondrial N-formyl peptides leading to formyl peptide receptor (FPR) activation, airway contractility and lung injury.
Acknowledgments
This work was supported by grants from American Heart Association (14POST20490292 and #13PRE14080019) and National Institutes of Health.
Footnotes
Disclosure
none.
References
- [1].American College of Chest Physicians/Society of Critical Care Medicine, American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis, Crit. Care Med. 20 (1992) 864–874. [PubMed] [Google Scholar]
- [2].Dicker RA, Morabito DJ, Pittet JF, Campbell AR, Mackersie RC, Acute respiratory distress syndrome criteria in trauma patients: why the definitions do not work, J. Trauma 57 (2004) 522–526. [DOI] [PubMed] [Google Scholar]
- [3].Uhlig S, Nüsing R, von Bethmann A, Featherstone RL, Klein T, Brasch F, Müller KM, Ullrich V, Wendel A, Cyclooxygenase-2-dependent broncho-constriction in perfused rat lungs exposed to endotoxin, Mol. Med. 2 (1996) 373–383. [PMC free article] [PubMed] [Google Scholar]
- [4].Armour CL, Black JL, Johnson PR, Vincenc KS, Berend N, Formyl peptide-induced contraction of human airways in vitro, J. Appl. Physiol. 60 (1985) 141–146. [DOI] [PubMed] [Google Scholar]
- [5].Hongwei Y, Yang SR, Edirisinghe I, Rajendrasozhan S, Caito S, Adenuga D, O′Reilly MA, Rahman I, Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice, Am. J. Respir. Cell Mol. Biol. 39 (2008) 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wenceslau CF, McCarthy CG, Goulopoulou S, Szasz T, NeSmith EG, Webb RC, Mitochondrial-derived N-formyl peptides: novel links between trauma, vascular collapse and sepsis, Med. Hypotheses 81 (2013) 532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, Webb RC, Mitochondrial damage-associated molecular patterns and vascular function, Eur. Heart J. 35 (2014) 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ, Mitochondrial DAMPs cause inflammatory responses to injury, Nature 464 (2010) 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Carp H, Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils, J. Exp. Med. 155 (1982) 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Le Y, Murphy PM, Wang JM, Formyl peptide receptors revisited, Trends Immunol. 23 (2002) 541–548. [DOI] [PubMed] [Google Scholar]
- [11].Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC, Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome, Am. J. Physiol. Heart Circ. Physiol. 308 (2015) H768–H777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rabiet MJ, Huet E, Boulay F, Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR, Eur. J. Immunol. 35 (2005) 2486–2495. [DOI] [PubMed] [Google Scholar]
- [13].Shao G, Julian MW, Bao S, McCullers MK, Lai JP, Knoell DL, Crouser ED, Formyl peptide receptor ligands promote wound closure in lung epithelial cells, Am. J. Respir. Cell Mol. Biol. 44 (2011) 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brohi K, Injury severity score: overview and desktop calculator, Trauma.org Web site, http://www.trauma.org/index.php/main/article/383/, January 11, 2013 (2007) Accessed. [Google Scholar]
- [15].NeSmith EG, Weinrich SP, Andrews JO, Medeiros RS, Hawkins ML, Weinrich MC, Demographic differences in systemic inflammatory response syndrome score after trauma, Am. J. Crit. Care 21 (2012) 35–41. [DOI] [PubMed] [Google Scholar]
- [16].Patel BV, Tatham KC, Wilson MR, O′Dea KP, Takata M, In vivo compartmental analysis of leukocytes in mouse lungs, Am. J. Physiol. Lung Cell Mol. Physiol. 309 (2015) L639–L652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Baban B, Liu JY, Qin X, Weintraub NL, Mozaffari MS, Upregulation of programmed death-1 and Its ligand in cardiac injury models: interaction with GADD153, PLoS One 10 (2015) e0124059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Volker B, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A, Neutrophil extracellular traps kill bacteria, Science 303 (2004) 1532–1535. [DOI] [PubMed] [Google Scholar]
- [19].Ware LB, Matthay MA, The acute respiratory distress syndrome, N. Engl. J. Med. 342 (2000) 1334–1349. [DOI] [PubMed] [Google Scholar]
- [20].Shore S, Irvin CG, Shenkier T, Martin JG, Mechanisms of histamine-induced contraction of canine airway smooth muscle, J. Appl. Physiol. Respir. Environ. Exerc Physiol. 55 (1983) 22–26. [DOI] [PubMed] [Google Scholar]
- [21].Pundir P, Catalli A, Leggiadro C, Douglas SE, Kulka M, Pleurocidin, a novel antimicrobial peptide, induces human mast cell activation through the FPRL1 receptor, Mucosal Immunol. 7 (2014) 177–187. [DOI] [PubMed] [Google Scholar]
- [22].Kim HR, Liu K, Roberts TJ, Hai CM, Length-dependent modulation of cytoskeletal remodeling and mechanical energetics in airway smooth muscle, Am. J. Respir. Cell Mol. Biol. 44 (2011) 888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Macheskya LM, Insalla RH, Signaling to actin dynamics, J. Cell Biol. 146 (1999) 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schmidt EP, Tuder RM, Role of apoptosis in amplifying inflammatory responses in lung diseases, Cell Death 20 (2010) 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K, Mitochondrial DAMPs from femoral reamings activate neutrophils via formyl peptide receptors and P44/42 MAP kinase, J. Orthop. Trauma 24 (2010) 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kawabata K, Hagio T, Matsuoka S, The role of neutrophil elastase in acute lung injury, Eur. J. Pharmacol. 451 (2002) 1–10. [DOI] [PubMed] [Google Scholar]
- [27].Trifilieff A, Fujitani Y, Mentz F, Dugas B, Fuentes M, Bertrand C, Inducible nitric oxide synthase inhibitors suppress airway inflammation in mice through down-regulation of chemokine expression, J. Immunol. 165 (2000) 1526–1533. [DOI] [PubMed] [Google Scholar]
- [28].Cardini S, Dalli J, Fineschi S, Perretti M, Lungarella G, Lucattelli M, Genetic ablation of the fpr1 gene confers protection from smoking-induced lung emphysema in mice, Am. J. Respir. Cell Mol. Biol. 47 (2012) 332–339. [DOI] [PubMed] [Google Scholar]
- [29].Tae YM, Park HT, Moon HG, Kim YS, Jeon SG, Roh TY, Bae YS, Gho YS, Ryu SH, Kwon HS, Kim YK, Airway activation of formyl peptide receptors inhibits Th1 and Th17 cell responses via inhibition of mediator release from immune and inflammatory cells and maturation of dendritic cells, J. Immunol. 188 (2012) 1799–1808. [DOI] [PubMed] [Google Scholar]
- [30].Babbin BA, Jesaitis AJ, Ivanov AI, Kelly D, Laukoetter M, Nava P, Parkos CA, Nusrat A, Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42, J. Immunol. 179 (2007) 8112–8121. [DOI] [PubMed] [Google Scholar]
- [31].de Paulis A, Prevete N, Fiorentino I, Walls AF, Curto M, Petraroli A, Castaldo V, Ceppa P, Fiocca R, Marone G, Basophils infiltrate human gastric mucosa at sites of Helicobacter pylori infection, and exhibit chemotaxis in response to H. pylori-derived peptide Hp (2–20), J. Immunol. 172 (2004) 7734–7743. [DOI] [PubMed] [Google Scholar]