

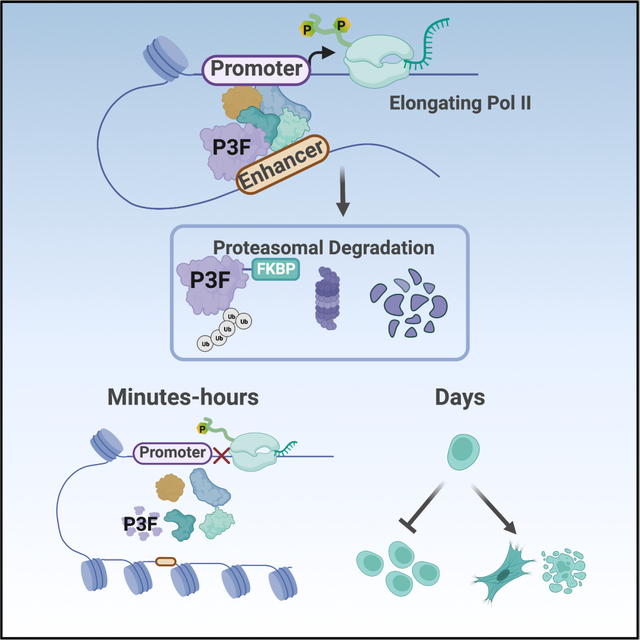
Summary
Transcriptional control is a highly dynamic process that changes rapidly in response to various cellular and extracellular cues, making it difficult to define the mechanism of transcription factor function using slow genetic methods. We used a chemical-genetic approach to rapidly degrade a canonical transcriptional activator, PAX3-FOXO1, to define the mechanism by which it disrupts gene expression programs. By coupling rapid protein degradation with the analysis of nascent transcription over short time courses and integrating CUT&RUN, ATAC-seq and eRNA analysis with deep proteomic analysis, we defined PAX3-FOXO1 function at a small network of direct transcriptional targets. PAX3-FOXO1 degradation impaired RNA polymerase pause release and transcription elongation at most regulated gene targets. Moreover, the activity of PAX3-FOXO1 at enhancers controlling this core network was surprisingly selective, affecting single elements in super-enhancers. This combinatorial analysis indicated that PAX3-FOXO1 was continuously required to maintain chromatin accessibility and enhancer architecture at regulated enhancers.
Keywords: PAX3, PAX3-FOXO1, enhancer, chromatin remodeling, ATAC-seq, PRO-seq, rhabdomyosarcoma, differentiation, therapeutics, t(2;13)(q35;q14)
In Brief
Zhang et al. use rapid protein degradation to define the mechanism of transcriptional control by, PAX3-FOXO1, a driver of aRMS. PAX3-FOXO1 functions at a limited number of enhancers to maintain chromatin accessibility and assemble critical co-regulatory complexes to trigger RNA polymerase II pause release and drive target gene expression.
Graphical Abstract:
Introduction
Master transcription factors establish gene expression networks to regulate cell fate decisions, and their deregulation is associated with cancer development. One common way that transcription factor activity is altered in cancer is through chromosomal translocation that gives rise to gain-of-function transcription factor fusion proteins. One example, PAX3-FOXO1, arises from the t(2;13)(q35;q14) translocation, and is the initiating and defining feature of a subset of highly aggressive, pediatric alveolar rhabdomyosarcoma (aRMS)1–5. In addition to the t(2;13) translocation, the mutational burden of these cancers is quite low, averaging only 6.4 somatic mutations per tumor6,7. This suggests that PAX3-FOXO1 is the critical driver of these tumors and an ideal therapeutic target. Thus, a thorough understanding of PAX3-FOXO1 function is critical to define aRMS etiology as well as to develop more innovative therapeutic strategies for this aggressive disease.
A critical component of understanding how oncogenic transcription factors such as PAX3-FOXO1 drive disease is defining their direct transcriptional targets. Moreover, defining with high certainty exactly where in the genome a transcription factor acts allows a detailed dissection of its mechanism of action. However, this seemingly simple task remains a challenge, because transcriptional control is a highly dynamic process, changing within the first hours in response to external stimuli 8. Despite this, the study of transcription factor functions has largely been limited to overexpression or genetic deletion approaches, which take days to alter transcription factor protein levels. These slow approaches make it impossible to distinguish direct from secondary or compensatory transcriptional effects9,10, and studies utilizing such methods consistently detect thousands of gene expression changes. In addition, these gene changes typically include equal numbers of genes up- and down-regulated. For instance, PAX3-FOXO1 was reported to up-regulate and down-regulate similar numbers of mRNAs as well as miRNAs, leading some to speculate it functions in both transcription activation and repression11–13.
Identification of direct targets can be further aided by assays of genome-wide transcription factor binding. However, these are only correlations, and ChIP-Seq for PAX3-FOXO1 identified many thousands of binding sites5,14,15. Thus, while it is clear from these studies that PAX3-FOXO1 overwhelmingly associates with distal enhancer elements, identifying which enhancer binding events drive changes in gene expression is a challenge. In addition, PAX3-FOXO1 is proposed to play a particular role in the establishment of clustered myogenic enhancer elements known as super-enhancers, which contain multiple PAX3-FOXO1 binding sites5,16. However, whether each of these binding events contributes equally to super-enhancer function and associated target gene expression remains unclear. While deep epigenetic analysis of xenografts has added to our knowledge base, even comparing alveolar versus embryonal RMS didn’t allow the identification of direct targets of PAX3-FOXO117.
Importantly, accurate identification of both direct gene targets as well as identification of regulated enhancers is critical for further elucidating the mechanism(s) by which PAX3-FOXO1 regulates gene expression. While both PAX3 and FOXO1 are considered transcriptional activators, early studies suggested that the fusion protein possessed an increased transactivation activity compared with the wild-type PAX3 protein. In addition, overexpression studies suggest that PAX3 and PAX3-FOXO1 induce disparate phenotypes and gene expression programs11,18. However, recent studies have proposed an enhancer hijacking model in which the translocation of enhancers at the 3’ end of the FOXO1 locus results in a failure of cells to silence the fusion locus, and therefore, failure to silence PAX3 target genes to promote normal cellular differentiation 19. Thus, it remains unclear whether the t(2;13) translocation drives tumorigenesis through the expression of a fusion transcription factor with altered transcriptional activities not possessed by the wild-type PAX3 protein, the loss of temporal control of PAX3 target gene expression, or some combination of the two. Moreover, it has been suggested that PAX3-FOXO1 can act as a pioneer factor to alter the local chromatin structure, even within heterochromatin, to allow transcription activation 15. Whether pioneer activity is unique to the fusion protein or is also possessed by the wild-type PAX3 protein requires further investigation.
Here, we used PAX3-FOXO1 as a model of a potent transcriptional activator to define how an oncogenic transcription factor reorganizes transcriptional networks to cause cancer. We used CRISPR-based engineering to integrate a degron tag (FKBP12) into the endogenous PAX3-FOXO1 locus20. This allowed us to rapidly degrade endogenous PAX3-FOXO1 to provide the temporal resolution required to identify direct gene targets. Nascent transcription analysis over a short time course following PAX3-FOXO1 degradation revealed that the fusion protein functioned overwhelmingly as a transcriptional activator and promoted RNA polymerase pause release at a small number of direct target genes. Moreover, while CUT&RUN detected PAX3-FOXO1 binding at over twenty thousand sites throughout the genome, degradation of the fusion protein reduced eRNA synthesis at only about 1% of these locations and affected chromatin accessibility at fewer than about 3% of these sites. Deep proteomic analysis of the endogenous fusion protein showed that PAX3-FOXO1 was in close proximity to many transcriptional regulatory complexes, but appeared to coordinate the activity of these complexes at only a few hundred sites. Perhaps most surprising, was that while PAX3-FOXO1 bound to multiple enhancer elements within super-enhancers, there was a time-resolved hierarchy of enhancers, as its continued presence was required at individual enhancer elements within these clusters to maintain chromatin accessibility and transcriptional complex assembly.
Overall, our studies identified just over a hundred high confidence PAX3-FOXO1 gene targets that exhibited rapid loss of expression and were associated with a nearby regulated enhancer. We note that while PAX3 bound most enhancers regulated by PAX3-FOXO1, its presence was not sufficient to maintain chromatin accessibility and gene expression in the absence of the fusion protein. This highlights how the fusion of the C-terminus of FOXO1 to the DNA binding domain of PAX3 creates a gain-of-function transcriptional activator, possibly with neomorphic activities.
Results
PAX3-FOXO1 degradation impairs RNA pol II pause release at gene targets
We used CRISPR-based genome editing to integrate a 2xHA-FKBP12F36V degron tag (abbreviated throughout as FKBP) into the last exon of the endogenous PAX3-FOXO1 locus of the Rh30 and Rh4 aRMS cell lines21–23. This yielded a model for rapid PAX3-FOXO1 protein degradation following the addition of a small molecule proteolysis-targeting chimera (PROTAC), dTAG-47 (Figure 1A and 1B)20,24. We generated single cell clones in which the wild-type FOXO1 locus, the PAX3-FOXO1 locus, or both were edited. To account for clonal variation, we generated four PAX3-FOXO1-2xHA-FKBP12F36V-expressing clones in Rh30 cells and two PAX3-FOXO1-2xHA-FKBP12F36V-expressing clones in Rh4 cells (e.g., Figure S1A). RNA-seq was performed from all four Rh30 PAX3-FOXO1-FKBP clones in the absence of dTAG-47, to demonstrate that the addition of the degron tag did not significantly alter gene expression patterns relative to unedited cells (Figure S1B–C).
Figure 1. Degradation of PAX3-FOXO1 triggers cell death and differentiation.
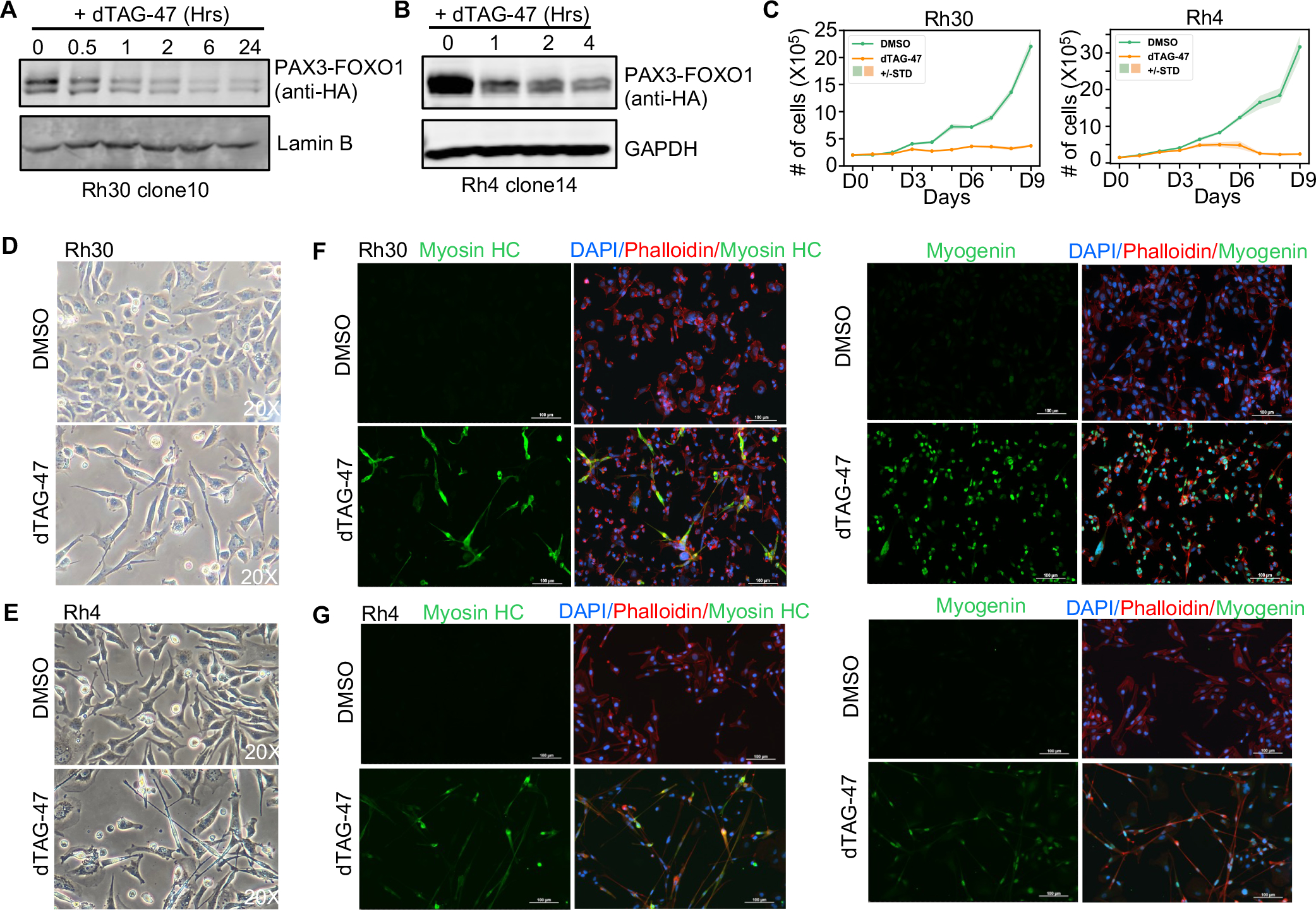
(A and B) Western blot analysis showing degradation of endogenous PAX3-FOXO1 in Rh30 clone 10 and Rh4 clone 14. (C) PAX3-FOXO1 is required to maintain cell growth. Rh30 PAX3-FOXO1-FKBP clone 10 and Rh4 PAX3-FOXO1-FKBP clone 14 were treated with 500 nM dTAG-47, and cell counts were determined using Trypan Blue dye exclusion. Shaded area shows the mean ± standard deviation (n=3). (D and E) Morphological analysis of PAX3-FOXO1-FKBP cells after treatment with dTAG-47 for 6 days (20X). (F and G) Immunofluorescence analysis of Myosin Heavy Chain (left) and Myogenin (right) expression in Rh30 PAX3-FOXO1-FKBP clone 10 (F) and Rh4 PAX3-FOXO1-FKBP clone 14 (G). PAX3-FOXO1-FKBP cells were treated with 500 nM dTAG-47 for 6 days. DAPI was used to label nuclei (blue). Alexa 568-labeled Phalloidin was used to mark actin filaments (red). Alexa 488 secondary antibody was used to visualize the primary antibody against skeletal muscle differentiation markers Myosin Heavy Chain and Myogenin (green; 20X).
We assessed the consequence of PAX3-FOXO1 removal from aRMS cells and found that all Rh30 and Rh4 clones expressing PAX3-FOXO1-FKBP exhibited growth inhibition following PAX3-FOXO1 degradation (Figure 1C and Figure S1D). We observed some variation in the degree to which growth was inhibited between the Rh30 clones with more consistent changes observed in the Rh4 clones. This variation could not be attributed to differences in PAX3-FOXO1 protein degradation (data not shown), suggesting cellular heterogeneity with regard to PAX3-FOXO1 dependence exists within the Rh30 cell line. Subsequent analyses focused on Rh30 clone 10 and Rh4 clone 4, where we also observed altered cell morphology, hallmarks of myogenic differentiation, G1 cell cycle arrest, increased cell death and reduced growth in soft agar following PAX3-FOXO1 degradation (Figure 1D–G and Figure S1E–I). In contrast, degradation of wild-type FOXO1 in Rh30 cells had no effect on cell growth, viability, or gene expression, likely due to the nuclear exclusion of FOXO1 in these cells (Figure S2A–F).
Next, we showed that the loss of PAX3-FOXO1 from chromatin as detected by CUT&RUN25 closely mirrored the kinetics of total protein loss detected by western blot analysis (Figure 2A and 2B). It is notable that essentially every peak was reduced in intensity within 4 hours of dTAG treatment. Moreover, there was a high degree of overlap in PAX3-FOXO1 bound sites between the two aRMS cell lines (Figure S2G). In addition, the intensity of the PAX3-FOXO1 signals was also highly consistent between Rh30 and Rh4 cell lines. K-means clustering was performed to partition the HA-PAX3-FOXO1 binding data into strong (cluster 1), moderate (cluster 2), and weak (cluster 3) peaks. The distribution of peaks between these clusters was similar in Rh30 and Rh4 cells (Figure 2A and 2B). Finally, we compared our HA-PAX3-FOXO1 CUT&RUN data to published ChIP-seq data and found that CUT&RUN identified roughly 80% of the previously published PAX3-FOXO1 peaks (Figure S2H and S2I). Thus, the presence of the FKBP12F36V tag did not dramatically alter PAX3-FOXO1 chromatin associations.
Figure 2. Degradation of PAX3-FOXO1 triggers a loss of expression of a small number of genes.
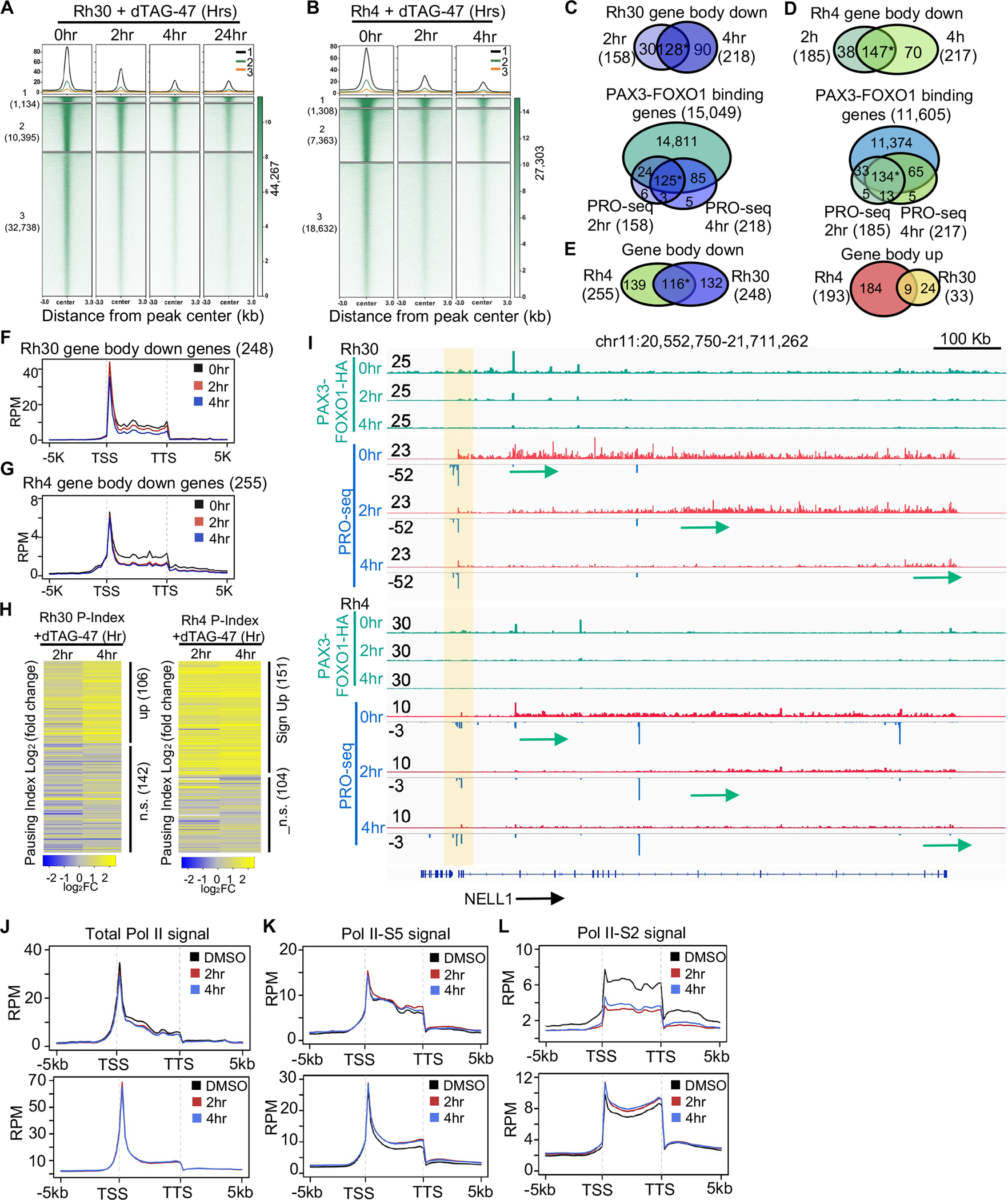
(A and B) K-means clustered heatmaps of PAX3-FOXO1 CUT&RUN peaks after treatment with dTAG-47 in Rh30 clone 10 (A) and Rh4 clone 14 (B). (C and D) Venn diagrams showing the PRO-seq signal quantified within the gene body with changes over time, and the overlap between CUT&RUN and changes in the PRO-seq signal o the gene body in Rh30 (C), or Rh4 (D) (*: p <= 1.0e-10). (E) Venn diagram showing the overlap between Rh30 and Rh4 gene body changes (*: p <= 1.0e-10). (F and G) Metagene plots of PRO-seq reads of all genes with decreased transcription in Rh30 (F) or Rh4 (G). (H) Heatmap of Log2 transformed fold change (log2FC) values of pausing indices of all the genes with changes in transcription in at least one time point. (I) Genome browser view of the NELL1 locus showing RNA polymerase pausing and the polymerase moving down the gene (arrows). Green tracks are the CUT&RUN signal before and after degradation of PAX3-FOXO1, which shows a peak at an enhancer within the 1st intron. (J-L) Metagene plots of the ChIP-seq signal for total Pol II (J), Pol II-S5 (K) and Pol II-S2 (L) of the 248 genes with decreased transcription (upper) and expressed genes with no transcription change (bottom).
We then performed precision nuclear run-on transcription sequencing (PRO-seq) at short time points following PAX3-FOXO1 degradation to determine how rapid loss of the PAX3-FOXO1 protein impacted gene expression. PRO-seq maps transcriptionally engaged RNA polymerase across the genome, and provides a readout of nascent transcription, as well as assessments of RNA polymerase pausing and elongation26,27. Relative changes in gene body transcription were quantified using the nascent RNA-sequencing analysis (NRSA) pipeline 28. In Rh30 cells, 158 genes exhibited decreased gene body transcription at 2hr following PAX3-FOXO1 degradation, and transcription of most of these genes remained reduced at 4 hours (Figure 2C), which is consistent with the established role for PAX3-FOXO1 in transcriptional activation. Moreover, the vast majority of down-regulated genes (94%) were associated with nearby PAX3-FOXO1 CUT&RUN peaks using a nearest neighbor algorithm (Figure 2C; 149 genes at 2hr and 210 at 4hr; Table S1A). Similar reductions in gene transcription were observed in Rh4 cells with 185 genes down-regulated at 2hr and 217 genes down-regulated 4hr after dTAG-47 addition with the majority of these genes associated with nearby PAX3-FOXO1 binding (Figure 2D, Table S1B). 116 genes were down-regulated in both cell lines (Figure 2E, Table S1C), suggesting that this is a commonly regulated core set of genes that requires the continued expression of PAX3-FOXO1 for expression. Among the high-confidence PAX3-FOXO1 gene targets were oncogenes and “stemness” factors such as JUN, RUNX2, PRDM12, FGFR2, FGF8, POU4F1, LGR5 and the co-repressor RUNX1T1, Table S1C)
Conversely, we found only 1 gene with increased transcription at 2hr and only 33 within 4hr of PAX3-FOXO1 degradation in Rh30 cells (Figure S2J). While Rh4 cells had a larger number of genes exhibiting increased gene expression than Rh30 cells, those changes were not consistent between timepoints (Figure S2K). Using nearest neighbor analysis, these genes were less likely to have an annotated PAX3-FOXO1 binding site (127 of 193, or 65%) and only 9 of these genes were common between Rh30 and Rh4 cells (Figure 2E). Therefore, we focused on those genes that appeared to be activated by PAX3-FOXO1 (Table S1C) for the subsequent analysis.
To cast the widest possible net for the mechanistic analysis of PAX3-FOXO1 activities, we plotted the PRO-Seq signal at all genes down-regulated in at least one timepoint following PAX3-FOXO1 degradation in both Rh30 and Rh4 cell lines (Figure 2F and G). Interestingly, down-regulated genes exhibited a clear reduction in gene body polymerase density, but maintained higher levels of paused polymerase just downstream of the transcription start site (Figure 2F and G). This pattern is indicative of a reduction in RNA polymerase pause release, rather than a significant change in transcription initiation. In fact, most down-regulated genes showed an increase in RNA polymerase pausing index, which reached statistical significance at roughly half of the down-regulated genes in both cell lines (Figure 2H, Table S1D and S1E). Examination of long genes such as NELL1 further illustrates this phenomenon (Figure 2I). By 2hr following PAX3-FOXO1 degradation, there was a specific reduction of elongating RNA polymerase at the 5’end of the gene, and by 4hr the elongating polymerase had reached the 3’ end and the locus was maximally silenced (Figure 2I, arrows). During this time, levels of paused polymerase downstream of the TSS remained relatively unchanged for both sense and antisense transcripts (Figure 2I, shaded box). Thus, PAX3-FOXO1-regulated the transcription of many target genes by promoting RNA pol II pause release and transcription elongation.
PRO-seq is the ideal method for analysis of transcriptional elongation29, but to extend and confirm these results, we performed ChIP-seq analysis for total RNA polymerase II or polymerase phosphorylated at Ser2 or Ser5 of the C-terminal 52 heptad repeats. For this analysis, we focused on only Rh30 cells and found that RNA polymerase II ChIP-seq reads were reduced across the body of the genes down-regulated upon PAX3-FOXO1 degradation, but not genes with a PAX3-FOXO1 binding site nearby that were not regulated (Figure 2J). While there was no reduction in RNA polymerase II phospho-Ser5 (Figure 2K), there was a rapid and dramatic loss of RNA polymerase II phosphorylated on Ser2 in the absence of PAX3-FOXO1 (Figure 2L) from down-regulated genes. In contrast, RNA pol II phospho-Ser2 levels were unaffected at genes that were not regulated by PAX3-FOXO1 (Figure 2L, lower). Thus, the combination of PRO-seq and ChIP-seq for RNA polymerase II phospho-Ser2 demonstrates that PAX3-FOXO1 is required for efficient RNA polymerase II pause release and elongation.
We then performed RNA-seq to determine if the reduced polymerase activity detected by PRO-seq resulted in a reduction of mature mRNA (Figure S2L). Most PRO-seq identified targets showed a significant reduction in mRNA within 6 hours of dTAG-47 treatment, further confirming these genes as direct PAX3-FOXO1 gene targets. It is notable that several transcription factors were turned off by degradation of PAX3-FOXO1, including (e.g., JUN, RUNX2, ETS1, PRDM12 and the co-repressor RUNX1T1, Table S1C), which likely trigger cascades of transcriptional changes. For example, within 6hr of dTAG-47 addition, there were 717 significant changes by RNA-seq with roughly 40% of these mRNAs increased rather than decreased, and by 24hr there were nearly two thousand changes in mRNA levels (Figure S2M and S2N). These results emphasize the need to use nascent transcript analysis at early timepoints after inactivation of transcription factors to avoid detection of secondary transcriptional changes found by RNA-seq at later times.
Identification of PAX3-FOXO1-regulated enhancers.
Greater than 80% of PAX3-FOXO1 CUT&RUN peaks were localized to intergenic or intronic regions (Figure S3A), suggesting that PAX3-FOXO1 functions at enhancers to regulate gene expression. Therefore, the PRO-seq data was used to quantify intergenic enhancer RNA (eRNA) transcription following PAX3-FOXO1 degradation. Within the first 4hr of PAX3-FOXO1 degradation, 305 eRNAs were significantly down-regulated in Rh30 cells and 500 in Rh4 cells (Figure 3A and Figure S3B), and 289 (96%; Rh30) and 472 (94%, Rh4) of these overlapped with PAX3-FOXO1 binding sites identified by CUT&RUN (Figure 3B and Figure S3C). To begin to address the mechanism of regulation at PAX3-FOXO1-regulated enhancers, we performed ChIP-seq for histone H3K27ac (Figure S3D), CUT&RUN for BRD4 (Figure S3E), and ATAC-seq to identify accessible regions (Figure 3C and Figure S3F), all of which are hallmarks of active enhancers 30. When examining all PAX3-FOXO1 bound regions, there was no change in H3K27ac levels, BRD4 occupancy, or chromatin accessibility (Rh30, Figure 3C, left; Rh4, Figure S3F shows BRD4 and ATAC-seq, left). However, when we focused on PAX3-FOXO1 peaks associated with changes in eRNA production, we observed higher levels of PAX3-FOXO1 and BRD4 at these enhancers and a rapid and dramatic reduction in H3K27ac, BRD4 binding, and chromatin accessibility following PAX3-FOXO1 degradation (Rh30, Figure 3C, right panels; Rh4, Figure S3G shows BRD4 and ATAC-seq, right panels). This was despite a similar reduction of PAX3-FOXO1 binding at all sites (Figure 3C for Rh30, and Figure S3F, upper panels for Rh4). In all, these data indicate that while PAX3-FOXO1 associates with chromatin at many sites throughout the genome, only a small number of those sites displayed changes in enhancer transcription following PAX3-FOXO1 degradation.
Figure 3. Identification of PAX3-FOXO1-regulated enhancers.
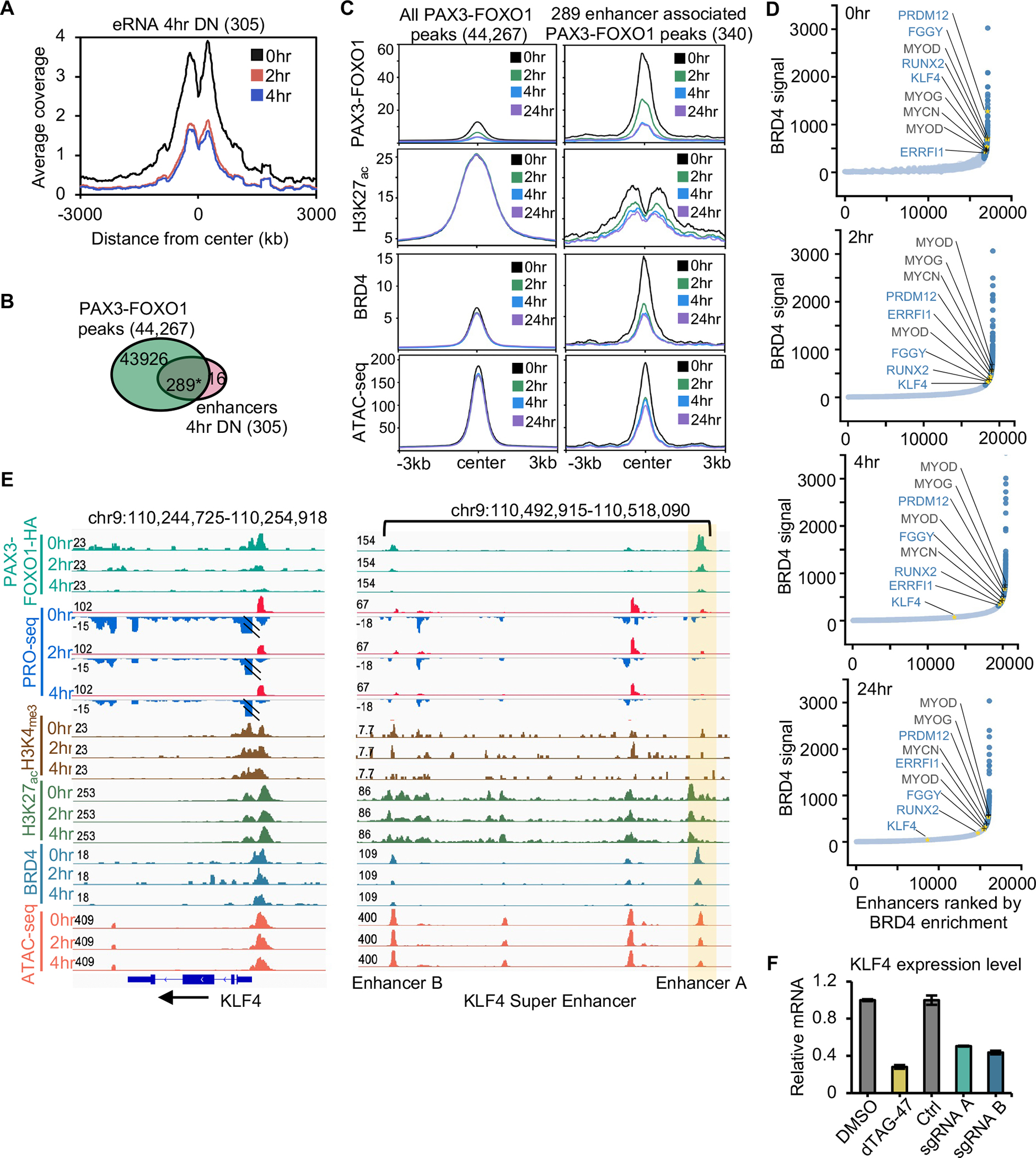
(A) Histogram of PRO-seq reads around enhancer centers after PAX3-FOXO1 degradation. (B) Venn diagram showing the overlap between PAX3-FOXO1 peaks and the down-regulated eRNA peaks at 4hr (*: p <= 1.0e-10). (C) Average signal of PAX3-FOXO1, H3K27ac, BRD4, and ATAC-seq over a 24hr time course of dTAG-47 treatment over the regions encompassing all PAX3-FOXO1 bound sites or those peaks associated with changes in eRNA transcription. (D) Distribution of BRD4 CUT&RUN density across the putative typical-enhancers (light blue) and super-enhancers (dark blue) in Rh30 cells at 0, 2, 4, 24 hrs following PAX3-FOXO1 degradation. Gold asterisks mark the examples of super-enhancers that are regulated by PAX3-FOXO1, and black type and + marks the examples of previously identified super-enhancers that were not regulated by PAX3-FOXO1 degradation. (E) IGV gene tracks showing a time course analysis of dTAG-47 treatment for PRO-seq and ATAC-seq coupled with localization of PAX3-FOXO1, BRD4, H3K27ac, and H3K4me3 at the KLF4 locus. Shaded area highlights a PAX3-FOXO1 regulated enhancer. (F) Bar graph reveals the relative mRNA expression of RUNX2 after deletion of either enhancer A or B.
PAX3-FOXO1 has been suggested to establish super-enhancers to drive myogenic transcription networks15,19,31. Therefore, we identified super-enhancers based the enrichment of BRD4 30,32–34 and asked whether continued PAX3-FOXO1 expression was required to maintain these regulatory structures (Figure 3D). Our analysis identified many of the super-enhancers previously associated with PAX3-FOXO1 function, but degradation of PAX3-FOXO1 did not affect the BRD4 recruitment, eRNA production, or ATAC-seq peaks at the super-enhancers of MYOD1, MYOG, and MYCN, which were previously reported to be regulated by PAX3-FOXO1 5 (Figure 3D and S4A). Moreover, while we did observe PAX3-FOXO1 binding at all three of these super-enhancers, we observed no change in MYCN transcription, an increase in MYOG transcription, and only a minimal decrease in MYOD1 transcription at 4 hours, which did not result in altered MYOD1 mRNA levels by RNA-seq (Table S1). Moreover, the MYOD1, MYOG and MYCN super-enhancers persisted even 24 hours after PAX3-FOXO1 degradation (Figure 3D), suggesting that PAX3-FOXO1 was not required for their maintenance. In contrast, we identified super-enhancers whose associated genes were rapidly down-regulated following PAX3-FOXO1 degradation, including RUNX2, KLF4, FGGY, and PRDM12 (Figure 3D). Interestingly, these super-enhancers fell lower on the BRD4-defined super-enhancer list over time, indicating reductions in BRD4 signal (e.g., FGGY, Figure S4B).
Manual inspection of the PAX3-FOXO1-regulated super-enhancers showed that they were bound by PAX3-FOXO1, but rather than a complete collapse of the super-enhancer, there was a disruption of select PAX3-FOXO1-bound enhancer elements upon degradation of PAX3-FOXO1. For instance, the regions upstream of KLF4 and RUNX2 and the intronic region of FGGY contained multiple enhancers marked by eRNA production, chromatin accessibility, H3K27ac, and BRD4 binding. While many of these putative enhancers showed a rapid reduction in eRNA production and BRD4 binding upon PAX3-FOXO1 degradation, only one displayed changed in accessibility (Figure 3E and Figure S4B–D). Therefore, to investigate whether individual enhancers within a super enhancer cluster contributed to target gene expression, we deleted the enhancer element that showed a rapid reduction in ATAC signal following PAX3-FOXO1 degradation and a second enhancer element that was not regulated by PAX3-FOXO1 (Figure S4C and D, Enhancer A and Enhancer B). CRISPR-mediated enhancer deletion was verified by genomic PCR (Figure S4E). RNA was isolated 72 hours following enhancer deletion and KLF4 and RUNX2 mRNA levels were quantified by qRT-PCR (Figure 3F and Figure S4F). Deletion of the PAX3-FOXO1-regulated enhancer was sufficient to significantly reduce both KLF4 and RUNX2 expression (Figure 3F and Figure S4F). In addition, deletion of the enhancer element not regulated by PAX3-FOXO1 resulted in a similar reduction in gene expression. Overall, these data suggest that each element was independently required to maintain gene expression, and disruption of a single element upon PAX3-FOXO1 depletion was sufficient to alter transcription.
Proteomics analyses reveal PAX3-FOXO1-associated protein complexes
Having identified PAX3-FOXO1-regulated enhancers and target genes, we next sought to define protein complexes that contribute to PAX3-FOXO1-mediated transcription activation at these loci. Previous proteomic analysis of PAX3-FOXO1 relied on over-expression of the fusion protein35. Therefore, we modified our CRISPR homology-directed repair vector to generate an endogenous PAX3-FOXO1-APEX2 protein fusion. APEX2 is an engineered peroxidase, which creates biotin-phenoxyl radicals that covalently modify nearby proteins (<20 nm)36–38. Thus, proteins in close proximity to PAX3-FOXO1 were purified using streptavidin beads and identified by liquid chromatography coupled with mass spectrometry (LC-MS). We identified over 500 significantly enriched proteins including components of multiple transcriptional complexes, such as FACT and SWI/SNF, transcription elongation factors (e.g. CDK9, CCNT1, NELFB), and components of a Mediator subcomplex that is associated with transcriptional elongation39 (MED12 and CDK8, Figure 4A–B and Table S1F). We also identified other sequence-specific transcription factors that may cooperate with PAX3-FOXO1 to regulate gene expression, including MYOD, HEB (TCF12), and RUNX1/CBFB (Figure 4A–B).
Figure 4. PAX3-FOXO1 recruits complexes involved in transcription.
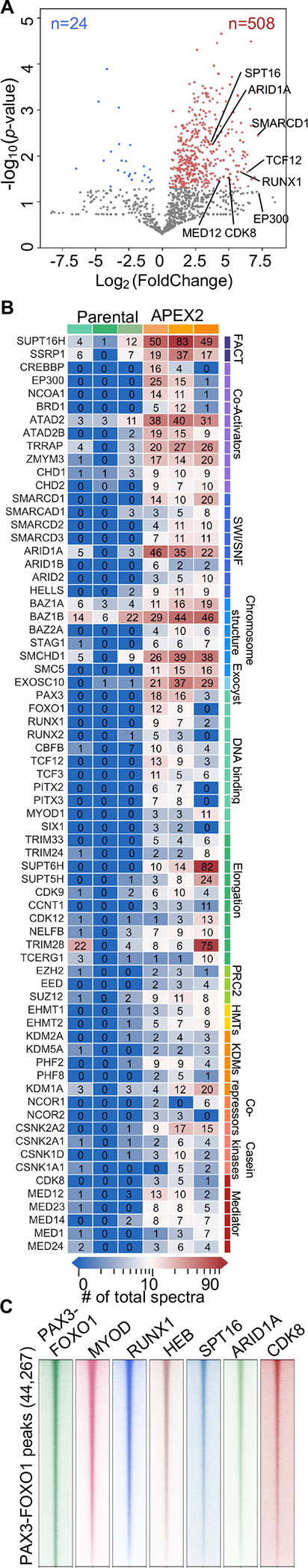
(A) Volcano plot showing log2-fold change (PAX3-FOXO1-APEX2/Parental) vs. −log10 of the p-value generated from the mass spectrometry results of PAX3-FOXO1-APEX2-mediated biotinylation (n=3, one-tail unpaired T-test). Blue dots, enriched in parental samples; red dots, enriched in PAX3-FOXO1-APEX2 samples. (B) Heatmaps of selected PAX3-FOXO1-APEX2 biotinylated proteins from the APEX2-mass spectrometry analysis. Total spectral count is shown within each box. (C) Heatmaps of CUT&RUN analysis of representative factors from the analysis in B plotted around all PAX3-FOXO1 binding sites.
Next, we used CRISPR/Cas9 to integrate a 3xFLAG tag into the endogenous PAX3-FOXO1 locus and confirmed a number of these associations by affinity purification using the 3xFLAG tag and LC-MS (Figure S5A–B and Table S1G). We also performed CUT&RUN for MYOD1, HEB (TCF12), RUNX1, ARID1A (SWI/SNF component), SPT16 (FACT component), and CDK8 (Mediator kinase module component). There was substantial co-localization of all of these factors with PAX3-FOXO1 in both Rh30 and Rh4 cells, further confirming the proteomic analysis (Figure 4C, Figure S5C).
PAX3-FOXO1-regulated enhancers are strongly bound by BRD4 and CDK8
PAX3-FOXO1 binds at tens-of-thousands of sites throughout the genome (Figure 2A and B), yet only a small fraction of those binding events control gene expression. Therefore, we asked whether there were any features unique to the small number of enhancers that were regulated by PAX3-FOXO1 binding (Figure 2–3). To address this, we first established a list of high-confidence PAX3-FOXO1 regulated enhancer elements. While eRNA was useful for quantifying changes in enhancer activity following PAX3-FOXO1 depletion (Figure 3), one limitation of this approach is that it does not include intronic enhancers, because coding transcription confounds their accurate quantification. This is particularly problematic, because PAX3-FOXO1 binding occurs equally at intergenic and intronic elements (Figure S3A). Therefore, given the rapid and robust reduction in chromatin accessibility observed upon PAX3-FOXO1 degradation, we identified both intergenic and intronic ATAC-seq peaks that changed following dTAG-47 treatment.
At 2hr following PAX3-FOXO1 degradation in Rh30 cells, loss of chromatin accessibility was observed at 1,129 regulatory elements, while only 6 elements exhibited an increase in accessibility (Figure 5A). We obtained similar results in Rh4 cells where 1,100 sites lost accessibility following degradation of the fusion protein (Figure S5D). 87% of the 1,129 ATAC-seq peaks that changed within 2hr were associated with the most robust PAX3-FOXO1 binding sites (cluster 1 and cluster 2, Figure 2A). By overlapping the ATAC-seq changes with the PRO-seq identified PAX3-FOXO1 gene targets that were deregulated in both Rh30 and Rh4 cell lines (116 genes), we identified a set of very high confidence PAX3-FOXO1-regulated enhancers (115 enhancers in Rh30 cells; 136 enhancers in Rh4 cells) (Table S1H and S1I, Figure 5B). Motif analysis was performed on all PAX3-FOXO1 bound loci common between Rh30 and Rh4 cells (21,401) and on the high confidence PAX3-FOXO1-regulated enhancers. While the PAX3-FOXO1 binding motif was only detected at 6% of the total PAX3-FOXO1-bound loci, that number increased to ~31% at the high-confidence regulated enhancers (a 5-fold enrichment over all binding sites; Figure 5C and B, Figure S5E). This result is consistent with these enhancers being associated with peaks that fell within the most robust PAX3-FOXO1 K means clusters.
Figure 5. Comparison of PAX3-FOXO1-regulated and non-regulated binding sites.
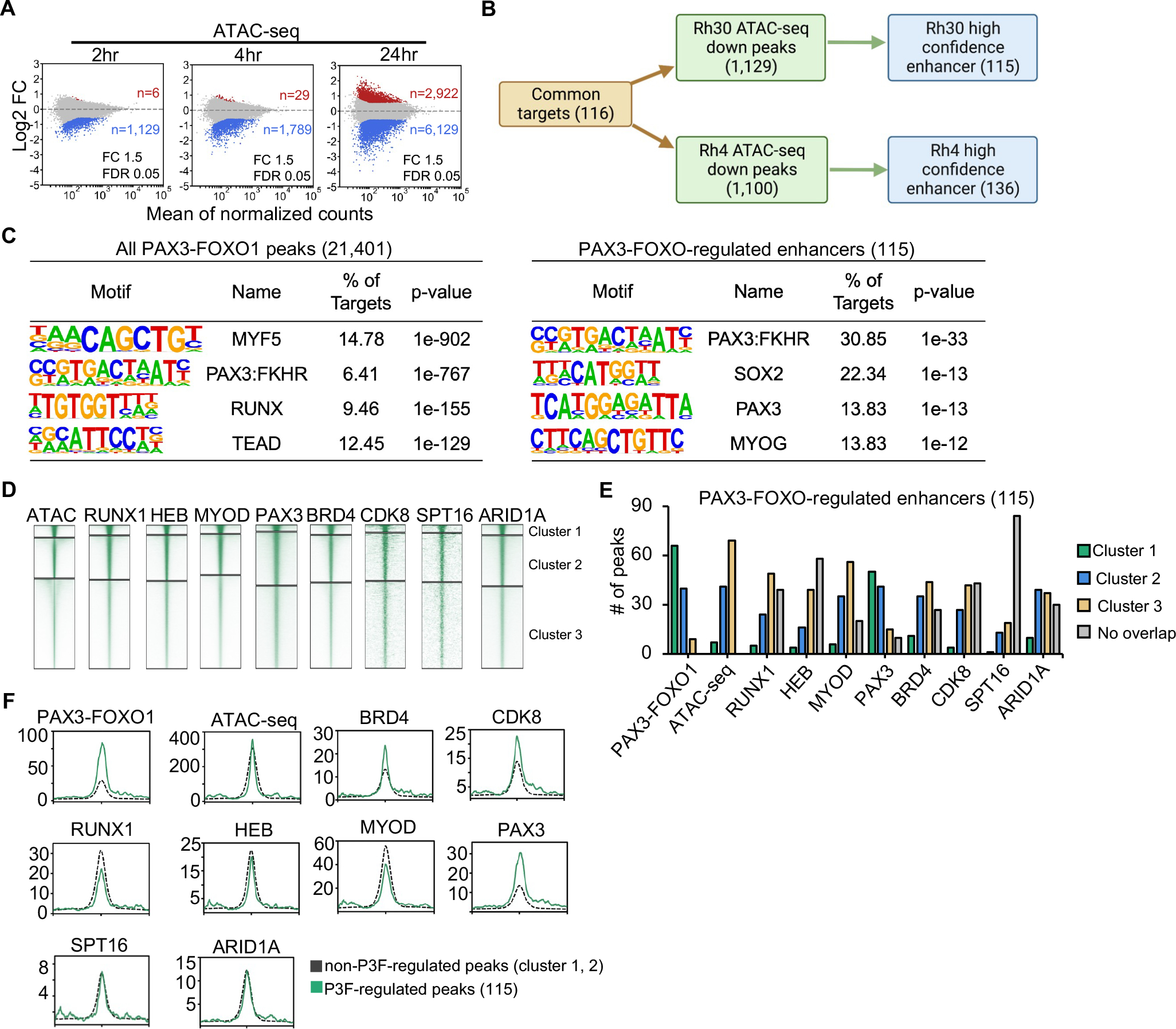
(A) MA plots showing ATAC-seq peak changes; red (up) or blue (down) (dTAG-47/DMSO, n=2). (B) Flow chart showing the identification of high confidence PAX3-FOXO1 regulate genes and their associated enhancers. (C) Motif analysis of the area under all PAX3-FOXO1 DNA binding peaks from the CUT&RUN analysis (left) or under the 115 ATAC-seq sites associated with genes down-regulated after PAX3-FOXO1 degradation. (D) Heatmap of the intensity of CUT&RUN or ChIP-seq signal after K means clustering of each of the indicated factors. (E) Peaks from the clusters in D that were associated with the regulated genes are shown as a bar graph. (F) Histograms of the CUT&RUN signal of the indicated factors around the regulated ATAC-seq peaks versus the remaining cluster 1 & 2 peaks from K means clustering.
In order to determine whether the small number of PAX3-FOXO1-regulated enhancers displayed particularly robust binding by any of the associated factors identified by mass spectrometry, we used K means clustering to segment the intensity of the signal for RUNX1, MYOD, HEB, PAX3, BRD4, SPT16, CDK8, and ARID1A (Rh30, Figure 5D; Rh4, Figure S5F). We partitioned the very high confidence enhancers (115 in Rh30 and 136 in Rh4) into these clusters to determine whether these were among the best binding sites for each of these factors. The high confidence, regulated enhancers were enriched for the strongest PAX3-FOXO1 and wild-type PAX3 binding events with a majority of these enhancers falling into cluster 1 and few in cluster 3 (Rh30, Figure 5E; Rh4, Figure S5G). In contrast, relatively few of these sites were enriched in cluster 1 for MYOD, RUNX1 or HEB co-localized to the regulated sites, but more cluster 2 peaks were associated with MYOD binding, especially in Rh4 cells (Figure 5E and Figure S5G). This suggests that PAX3-FOXO1 binds to these enhancers independently of these other DNA binding factors. At the same time, the non-DNA binding transcriptional regulatory complexes are expected to be localized to nearly every transcribed gene, yet BRD4, ARID1A, and CDK8 showed somewhat more cluster 1 and 2 binding sites (Figure 5E and Figure S5G). Interestingly, the PAX3-FOXO1 regulated elements were enriched for the weakest ATAC-seq peaks, suggesting that chromatin context may influence whether PAX3-FOXO1 binding can regulate enhancer activity.
Finally, we compared the intensity of the CUT&RUN signal at non-regulated versus regulated enhancers. Because most of the regulatory PAX3-FOXO1 binding sites fell into clusters 1 and 2 (Figure 5E, 2A), we compared these sites to the non-regulated peaks contained within these clusters (Figure 5F and Figure S5H). PAX3-FOXO1-associated factors did not show dramatic differences in their association with regulated versus non-regulated enhancers, with the exception of CDK8 and BRD4, which showed higher levels of binding to the regulated sites (Figure 5F). These data suggest regulatory PAX3-FOXO1 binding events are enriched in the most robust PAX3-FOXO1 peaks and those peaks enriched for the stringent PAX3-FOXO1 binding motif. In addition, complexes associated with transcriptional elongation (BRD4 and CDK8) were more prevalent at enhancers regulated by PAX3-FOXO1 binding. However, it is important to note that even intense PAX3-FOXO1 binds were not sufficient to trigger gene expression as many strong binding sites were not associated with changes in gene expression.
PAX3-FOXO1 maintains accessible chromatin to retain transcriptional regulatory factors
Finally, we performed CUT&RUN analysis over a time course after PAX3-FOXO1 degradation to determine if PAX3-FOXO1 was required to maintain the APEX2-identified complexes at regulated enhancers (Figure 6A, Figure S6A). First, we plotted histograms of the binding of these factors at all PAX3-FOXO1 binding sites (Figure 6A and Figure S6A, left panels). While PAX3-FOXO1 was lost across all DNA binding sites, there were no significant reductions of any associated factors following PAX3-FOXO1 degradation in either Rh30 or Rh4 cells when all binding sites were considered (Rh30, Figure 6A, left; Rh4, Figure S6A, left). However, when we focused on the very high confidence enhancers that could be associated with the 116 genes that were commonly down-regulated in both cell types (Rh30, Figure 6A, right panel; Rh4, Figure S6A, right panel), we observed the synchronous loss of the factors that associated with PAX3-FOXO1, including a robust loss of CDK8 and accessibility (Rh30 data shown in Figure 6A, right panels; data from Rh4 cells in Figure S6A, right panels). In contrast, PAX3 binding was only minimally affected by PAX3-FOXO1 degradation, indicating that PAX3 was not sufficient to maintain these complexes (Figure 6A).
Figure 6. PAX3-FOXO1 is required for maintaining open chromatin structure at the regulated enhancers.

(A) Histograms of the CUT&RUN signal of the indicated factors around all PAX3-FOXO1 peaks and the ATAC-seq peaks regulated by PAX3-FOXO1 in Rh30 cells. (B) MA plots showing ATAC-seq peak changes at 0.5 hr, 1hr and 2hr after PAX3-FOXO1 degradation; red (up) or blue (down) (dTAG-47/DMSO, n=2). (C) Heatmap of a short time course of dTAG-47 treatment that uses ATAC-seq changes at 2hr after PAX3-FOXO1 degradation from Rh30 cells. (D) Genome browser view of the RUNX2 “super” enhancer. The shaded box highlights an enhancer showing changes in factor binding, eRNAs, and accessibility over time compared to neighboring enhancers.
The substantial loss of ATAC-seq signal upon degradation of PAX3-FOXO1 suggested that changes in chromatin accessibility might precede the recruitment of other factors. Therefore, we performed ATAC-seq and PRO-seq analysis at 30, 60 and 120min after addition of dTAG-47 to degrade PAX3-FOXO1. Although there were few peaks that reached our significance cut offs within 30 min to 1hr, by two hours there were 909 changes (Figure 6B). However, a heat map of these 909 peaks showed that we could detect changes in accessibility in the PAX3-FOXO1-regulated enhancers beginning at the 30min time point (Figure 6C). These differences were observed even within clusters of enhancers such as the RUNX2 super-enhancer, which showed loss of PAX3-FOXO1 at multiple sites, but loss of ATAC-seq signal, and occupancy by most of the factors that were assessed, at only a single enhancer (Figure 6D and S6B, shaded area). Thus, using rapid degradation of PAX3-FOXO1 uncovered direct targets which in turn allowed us to determine that chromatin was rapidly remodeled following PAX3-FOXO1 degradation, leading to the synchronous loss of transcriptional complexes and the collapse of a small number of discrete enhancer elements such as observed in the RUNX2 super enhancer (Figure 7).
Figure 7. PAX3-FOXO1 is required for maintaining open chromatin structure at the regulated enhancers.
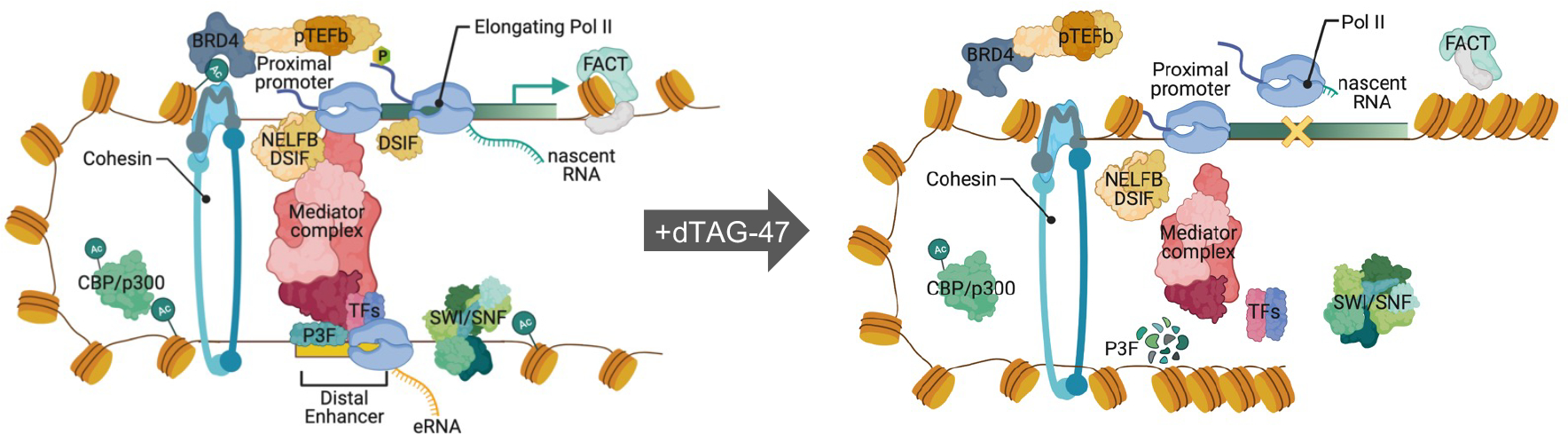
Model integrating the proteomic and genomic data into a hypothetical model by which PAX3-FOXO1 regulates transcription of its target genes. Created with BioRender.com.
Discussion
Given that transcriptional changes occur rapidly, traditional genetic approaches have failed to effectively define the direct targets of sequence-specific transcription factors, and therefore, have inadequately defined mechanisms of transcriptional control by these proteins40,41. Thus, CRISPR-based addition of degron tags to endogenous transcription factors has provided a technological breakthrough that is greatly aiding the study of transcription factor function 9,10,42,43. Here, we have applied this approach to an oncogenic fusion transcription factor, PAX3-FOXO1. While we identified over tens of thousands of PAX3-FOXO1 binding sites throughout the genome, by combining rapid protein degradation with nascent transcript analysis by PRO-seq and enhancer accessibility by ATAC-seq, we determined that PAX3-FOXO1 activates the transcription of approximately 116 high-confidence gene targets in Rh30 and Rh4 cells. Moreover, we found that many of these targets are regulated at the level of RNA polymerase pause release rather than at the stage of transcription initiation, which defines a new mechanism of PAX3-FOXO1 action. Further, by intersecting ATAC-seq and PRO-seq across Rh30 and Rh4 cells, we were able to identify 72 genes that were associated with high confidence ATAC-seq peaks (115 in Rh30 and 136 in Rh4) that changed upon PAX3-FOXO1 degradation, which could be used as a signature for PAX3-FOXO1 function in drug screens or CIRSPR-Cas9 screens.
Previous studies have postulated that PAX3-FOXO1 possesses pioneer activity, facilitating the establishment of de novo enhancer elements that drive myogenic transcriptional programs 5,15. Indeed, we observed the rapid loss of chromatin accessibility at regulated enhancers following PAX3-FOXO1 degradation. However, these regulated enhancers represented less than 3% of the PAX3-FOXO1 binding sites (Figure 6A and Figure S6A). Given the high degree of overlap between ATAC-seq peaks and PAX3-FOXO1 binding sites, it appears that PAX3-FOXO1 binds to nucleosome free regions throughout the genome, but that the vast majority of these sites do not rely on continued PAX3-FOXO1 expression to maintain accessibility. This could be indicative of a “hit and run” mechanism by which PAX3-FOXO1 establishes the accessible site and then other factors maintain the accessibility, but such a hypothesis is difficult to test. Another possibility is that genome-wide methods for identifying binding sites for transcription factors are too sensitive and pick up weak sites that are not regulatory. In fact, K means clustering and/or motif analysis of the CUT&RUN data suggested that most of the binding sites were of poor quality and/or low stability (i.e., cluster 3, Figure 2A). Interestingly, the sites that relied on continued expression of PAX3-FOXO1 did not display robust binding of other factors such as RUNX1/2, HEB, and MYOD, but did display a higher binding by CDK8 and BRD4, indicating that the recruitment of factors involved in transcription elongation is important for PAX3-FOXO1 mediated transcriptional control. Also, the recruitment of the SWI/SNF complex might be important to establish a nucleosome free region to allow the recruitment of the mediator complex and other general transcription factors.
We also detected the binding of PAX3-FOXO1 at super-enhancers (Figure 3E). However, we did not observe the broad collapse of these super-enhancers following PAX3-FOXO1 degradation, but rather a rapid loss of accessibility at specific enhancer elements within the enhancer cluster, which may then trigger collapse of the entire cluster of enhancers. That is, PAX3-FOXO1-regulated enhancer elements could nucleate the enhancer cluster to regulate its activity, yet deletion of other enhancer elements also appeared to impair the activity of the enhancer cluster, suggesting an interdependence of individual enhancer elements (e.g., RUNX2 and KLF4, Figure 3F and Figure S4C–F). These findings are consistent with CRISPR-mediated deletion of individual elements in super-enhancers that identified a hierarchical organization of super-enhancers in which a “hub” enhancer, in this case regulated by PAX3-FOXO1, was the major determinant of super-enhancer function44. Having identified a cohort of genes directly regulated by PAX3-FOXO1, we are poised to further define whether the fusion protein triggers the formation of higher order complexes (e.g., nuclear condensates or transcription factories) or acts through individual enhancer elements.
By engineering the endogenous PAX3-FOXO1 for proximity labeling, we identified DNA binding factors and transcriptional complexes that associated with PAX3-FOXO1 (Figure 4). Among the identified interacting proteins were components of the SWI/SNF complex such as ARID1A, which could suggest that the continued recruitment of SWI/SNF may be required to maintain the nucleosome free region at enhancers to allow full transcriptional complex assembly45. Our data also suggests that PAX3, which was bound at these same loci, was not sufficient to maintain open chromatin at these regulatory elements. These data indicate that PAX3-FOXO1, contains neomorphic activities; however, future studies are needed to more rigorously define these functions. Finally, these data indicate that PAX3-FOXO1 is continuously required to maintain the expression of genes critical for blocking terminal differentiation and to maintain cell viability such as RUNX2, KLF4, PRDM12, FGF8, and FGFR2 and further emphasize the utility of PAX3-FOXO1 as a therapeutic target in rhabdomyosarcoma.
Limitations:
Due to the genetic engineering constraints of adding a degron tag, we were limited to using cell lines that can be single cell cloned. Therefore, some direct targets of PAX3-FOXO1 that are critical to the initiation of tumorigenesis could be missed. However, we note that these cells rapidly differentiated or died upon degradation of PAX3-FOXO1 (Figure 1), indicating that the genes identified as being controlled by PAX3-FOXO1 were required to sustain cell viability and impair differentiation, even in established cancer cell lines. It is also possible that PAX3-FOXO1 could regulate genes that take longer than 4hr to be turned off. However, PAX3-FOXO1 regulates the expression of many other transcription factors (e.g. KLF4, RUNX2), which control additional transcriptional targets. Therefore, it is not possible to interpret events that occur after the initial changes in gene expression upon degrading PAX3-FOXO1.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Scott Hiebert (scott.hiebert@vanderbilt.edu).
Materials Availability
All the materials generated in this study are accessible upon request.
Data and code Availability
All genomic datasets are available at GEO accession GSE183281. Original data for figures in this study are accessible upon request and deposited to Mendeley Data: https://data.mendeley.com/datasets/64sn74fk56/draft?a=21d0d0f5-1c60-4f50-b4bd-d69ef94241f5
This paper does not report original code. Any code for analysis will be shared by the leading contact upon request.
Any Additional information required to reanalyze the data reported in this paper is available from the leading contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
The Rh30 cell line was obtained from ATCC. Rh30 cells were cultured in RPMI-1640 (Corning by Mediatech, Inc.) containing 10% FetalPlex (Gemini Bio-products.) and supplemented with 1% L-Glutamine (Corning by Mediatech, Inc.) and 1% penicillin/streptomycin (Corning by Mediatech, Inc.). Rh4 cells were a gift from Dr. Javed Khan (NCI). Rh4 cells were cultured in DMEM (Corning by Mediatech, Inc.) containing 10% FBS (R&D SYSTEMS.) and supplemented with 1% L-Glutamine (Corning by Mediatech, Inc.) and 1% penicillin/streptomycin (Corning by Mediatech, Inc.). Drosophila S2 cells (Schneider media supplemented with 10% FBS, 1% penicillin and streptomycin) were a gift from Dr. Emily Hodges.
dTAG-47
dTAG-47 was synthesized by the Vanderbilt University Medical Center (VUMC) Molecular Design and Synthesis Center (VICB, kk-25-065) as described20, and reconstituted in DMSO (Sigma).
METHOD DETAILS
Generation of endogenous PAX3-FOXO1-tagged Rh30 and Rh4 cell lines
The endogenous allele of PAX3-FOXO1 in Rh30 and Rh4 cells was engineered to express C-terminal FKBP12F36V-2xHA, APEX2-2xHA, or 3X-FLAG tags using homology-directed DNA repair 46. 180 bp upstream of the stop codon and 500 bp after the FOXO1 stop codon were cloned into pUC19 containing FKBP12F36V-2xHA-P2A-mCherry derived from Addgene #104370 (pAW6224) using the Gibson Assembly Cloning Kit (NEB #E5510S). The APEX2 sequence (Addgene #97421) or a 3xFLAG tag was cloned into the HDR donor plasmids to create the FOXO1-APEX2 and FOXO1-3xFLAG plasmids respectively. Cas9, gRNA and the HDR template plasmid were delivered into Rh30 and Rh4 cells by electroporation. mCherry positive cells were sorted and single cell cloning was performed to generate PAX3-FOXO1-tagged and FOXO1-tagged clones.
Primers used to construct template plasmid are listed below:
Primers to generate 5’ homology gene block:
F:GCCAAGTGGGTTGATGTCTGGTTTTTCCTTGAGAGAAGCTCCCAAGTGACTTGGATGGCATGTTC
R:GGGGAGATGGTTTCCACCTGCACTCCTCCGGATCCGCCTGACACCCAGCTATGTGTCGTTGTCTTG
Primers to generate 3’homology gene block:
Cherry-F:
ACTCCACCGGCGGCATGGACGAGCTGTACAAGTAAGGGTTAGTGAGCAGGTAAGTTCACCCCAAT
BFP-F:
ACCTCCCTAGCAAACTGGGGCACAAGCTTAATTAAGGGTTAGTGAGCAGGTAAGTTCACCCCAAT
R:CGGCCAGTGAATTCGAGCTCGGTACCCGGGGATCCCCAAGAAAACTAAAAGGGAGTTGGTGAAAG
5’Gibson Cloning Primers:
F:CAAGACAACGACACATAGCTGGGTGTCAGGCGGATCCGGAGGAGTGCAGGTGGAAACCATCTCCCC
R:GAACATGCCATCCAAGTCACTTGGGAGCTTCTCTCAAGGAAAAACCAGACATCAACCCACTTGGC
3’Gibson Cloning Primers:
Forward:
CTTTCACCAACTCCCTTTTAGTTTTCTTGGGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCG
Cherry-R:
ATTGGGGTGAACTTACCTGCTCACTAACCCTTACTTGTACAGCTCGTCCATGCCGCCGGTGGAGT
BFP-R:
ATTGGGGTGAACTTACCTGCTCACTAACCCTTAATTAAGCTTGTGCCCCAGTTTGCTAGGGAGGT
FOXO1 crRNA: 5’-CAGGCTGAGGGTTAGTGAGC
Western Blot
Cells were collected and lysed with RIPA buffer (50 mM Tris pH8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease inhibitor. After sonication, lysates were cleared by centrifugation and subjected to SDS-PAGE and electrophoretic transfer to membranes before incubation with antibodies directed against HA (Abcam, ab18181), FLAG (Sigma, F1804), GAPDH (Santa Cruz, sc-365062), Lamin B (Santa Cruz, sc-6217). Signal was visualized with secondary IR-Dye conjugated antibodies (Licor) and detected using the Licor Odyssey imaging system.
Cell growth
Cells were seeded at a density of 2 × 105 cells/ml on day 0 in 6-well culture plates and treated with DMSO or 500 nM dTAG-47. The cells were reseeded every 3 days at 2 × 105 cells/ml and maintained in DMSO/dTAG-47 treatment for the duration of the assay. Viable cells were counted with Trypan Blue dye exclusion every day for 9 days consecutive. Quantification was performed in triplicate and the values averaged and shown with standard deviations.
Cell cycle analysis
Cells were treated with DMSO or dTAG-47 for 3, 6, and 9 days. Before staining, cells in a 10-mm-dish at 80% confluence were pulsed with 20μM 5-bromo-2’-deoxyuridine (BrdU) for 2 hours and fixed overnight with 70% ethanol at 4°C. Cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-BrdU and counterstained with propidium iodide (PI) before analysis by flow cytometry. All flow cytometry figures were generated using FlowJo software.
Soft Agar colony formation assay
Cells were treated with DMSO or dTAG-47 for 3, 6, and 9 days, and then were plated in 0.3% agarose medium. Cells were fed with culture media with DMSO/dTAG-47 once per week. After 4 weeks, plates were stained with 0.005% Crystal Violet (Sigma), and colonies were counted using a dissecting microscope.
Immunofluorescence
Cells were seeded on coverslips and treated with DMSO or dTAG-47 for 3, 6, and 9 days. Cells were then fixed with 3.7% paraformaldehyde in PBS at room temperature for 15 minutes. The coverslips were then washed three times with phosphate buffered saline (PBS) and the cells were permeabilized using 0.3% Triton X-100 in PBS at room temperature for 30 minutes. In a humidified chamber, cells were blocked by adding 1% serum in PBS for 30 minutes, and then incubated with primary antibodies (Myogenin, Abcam, ab1835; Myosin Heavy Chain, R&D, MAB4470; HA, Cell Signaling, (C29F4) #3724) with appropriate dilution in 0.5% NP-40 and 1% serum in PBS at 37 °C for 1 hour. After washing 3 times with PBS, cells were incubated in Alexa Fluor secondary antibody (Abcam ab150117, Invitrogen A-11034) with Phalloidin (Thermo Fisher Scientific, A12380) and DAPI diluted in 0.5% NP-40, and 1% serum in PBS at 37 °C for 45 minutes. Coverslips were mounted on slides with Prolong Gold Antifade reagent (Thermo Fisher Scientific, P36930) and dried overnight in the dark. Images were collected using a Nikon fluorescent microscope.
Guide RNAs for enhancer knockout:
KLF4 enhancer_A:
singe guide RNA_1: gcatttgggaaaaggtgagg
singe guide RNA_2: cttaagtaaaggaaagaact
KLF4 enhancer_B:
singe guide RNA_1: CTCTGCAGTTGGGCACACCC
singe guide RNA_2: cacacagctactaaaactcg
RUNX2 enhancer_A:
singe guide RNA_1: caccGCGGGCGGTGAGCTAACACAT
singe guide RNA_2: aaacATGTGTTAGCTCACCGCCCGC
singe guide RNA_3: caccGCAGTTGGATGAGATCAAGCA
singe guide RNA_4: aaacTGCTTGATCTCATCCAACTGC
RUNX2 enhancer_B:
singe guide RNA_1: caccGGCAGCTGTAGCCCGCGGTT
singe guide RNA_2: aaacAACCGCGGGCTACAGCTGCC
singe guide RNA_3: caccGCTGATTCTGACGCCATCTG
singe guide RNA_4: aaacCAGATGGCGTCAGAATCAGC
Real-Time PCR (quantitative PCR, qPCR)
All qPCR experiments were performed in biological triplicate using SsoAdvanced Universal SYBR Green Supermix kit (BIO-RAD, #1725270). Total RNA was extracted using TRIzol. 1ug of RNA was used for reverse transcription after being treated with DNase I to digest DNA. Quantitative PCR measured the relative quantity of KLF4 and RUNX2 after 72 hours of dTAG-47 treatment or enhancer deletion. qPCR primers: KLF4 F- CAAGCCAAAGAGGGGAAGAC, KLF4 R- CGTCCCAGTCACAGTGGTAA. RUNX2 F- TGGTTACTGTCATGGCGGGTA, RUNX2 R- TCTCAGATCGTTGAACCTTGCTA. ACTB F-ACCTTCTACAATGAGCTGCG, ACTB R- CCTGGATAGCAACGTACATGG.
Nuclei isolation
30 million Rh30 cells were treated with DMSO or dTAG-47 and collected at indicated time points. Cells were washed with ice cold PBS and lysed with cell lysis buffer (10mM Tris-Cl pH7.4, 300mM sucrose, 3mM CaCl2, 2mM MgCl2, 0.5% NP-40, 5mM DTT, 1mM PMSF, EDTA free protease cocktail inhibitor tablet) using dounce homogenization, nuclei were pelleted by centrifugation and washed with nuclei storage buffer (50mM Tris-Cl pH8.3, 40% glycerol, 5mM MgCl2, 5mM DTT, 0.1 mM EDTA, 1mM PMSF, EDTA free protease cocktail inhibitor tablet). After counting, pelleted nuclei were resuspended in storage buffer, and stored at −80 °C.
Precision nuclear run-on and sequencing (PRO-seq)
PRO-seq was performed in biological replicates as previously described using approximately 20 million nuclei per run on with GTP, ATP, UTP, and biotin-11-CTP (PerkinElmer) using 0.5% Sarkosyl (Fisher Scientific) to prevent transcription initiation 27,47. RNA was reversed transcribed and amplified to make the cDNA library for sequencing by the Vanderbilt University Medical Center (VUMC) VANTAGE Genome Sciences Shared Resource on an Illumina Nextseq 500 (SR-75, 50 million reads). The sequences were aligned using bowtie2 (v2.2.2) before using the Nascent RNA Sequencing Analysis (NRSA) pipeline 28 to determine the gene body and eRNA changes.
RNA sequencing and data processing
All RNA-seq experiments were performed in biological duplicate. Total RNA was extracted using TRIzol. RNA was submitted to the VUMC VANTAGE core for library preparation and sequencing (Illumima NovaSeq, PE-150, 30 million reads). Adaptors were trimmed using Trimmomatic-0.32 and aligned to the human genome (hg19) using TopHat (v. 2.0.11) 48. Differential analysis was performed using CuffDiff (v. 2.1.1) 49.
Cleavage under targets and release using nuclease (CUT&RUN)
CUT&RUN experiments were performed in biological duplicate as described 25. Briefly, cells were treated with DMSO or dTAG-47 for the indicated times and 250,000 cells were collected and incubated with Concanavalin A-coated beads (Bangs Laboratories, BP531) for 10 minutes at room temperature, and incubated with 0.01% freshly dissolved digitonin. Anti-HA (Cell Signaling, C29F4 #3724), anti-BRD4 (Rh30: BETHYL, #A301-985A50; Rh4: Cell Signaling Technology, #13440), anti-H3K4me3 (Abcam, ab12209), anti-MYOD (Santa Cruz, sc-377460), anti-RUNX1 (Santa Cruz, sc-365644), anti-HEB (Santa Cruz, sc-357), anti-ARID1A (Cell Signaling, (D2A8U) #12354), anti-SPT16 (Cell Signaling, D7I2K #12191), anti-CDK8 (Santa Cruz, sc-13155), and anti-PAX3 (Abcam, ab69856) primary antibodies were added and incubated at 4 °C overnight, before washing and binding of secondary antibody (anti-Rabbit, #ab31238, anti-Mouse, #ab46540) for 1hr. After washing, CUTANA pAG-MNase (EpiCypher, #15-1116) fusion protein was added and incubated at 0 °C for 60–90 mins to digest targeted regions of the genome. DNA was then extracted using phenol-chloroform 25 and sequencing libraries were generated using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB #E7645S/L). Sequencing was performed by the VUMC VANTAGE core Illumina NovaSeq (PE-150, 10 million reads).
Chromatin Immunoprecipitation Sequencing (ChIP-seq)
Cells were treated with dTAG-47 for 0, 2, 4, and 24 hours. Five million cells were used to perform anti-H3K27ac, anti-Pol II, anti-PolII Ser2, and anti-PolII Ser5 ChIP-seq with Drosophila S2 cell spike-in (5%). Cells were cross-linked with 1% formaldehyde for 8 minutes and quenched with 125mM Glycine. Following nuclei isolation, chromatin fragments within 300~600 bp range were generated by sonication for 25 cycles (30s-on, 30s-off) for 25 cycles with a Biorupter (Diagenode). Chromatin fragments were immunoprecipitated with anti-H3K27ac (Abcam, #ab4729), anti-PolII (Santa Cruz, sc-899), anti-Pol II pSer2 (Abcam, #ab5095), and anti-Pol II pSer5 (Abcam, #5131) plus Protein A beads. NEBNext Ultra II DNA Library Prep Kit for Illumina was used to make the DNA libraries (NEB, #E7645S/L), which were sequenced on the Illumina NovaSeq (PE-150, 30 million reads) at the VUMC VANTAGE Shared Resource.
CUT&RUN and ChIP-seq data analysis
Raw FASTQ data were trimmed using Trimmomatic50 (v0.39) and paired end reads were aligned to a concatenated human and E.coli genome (hg19 and ecK12MG1655) for CUT&RUN, or Drosophila genome (hg19 and dm3) genome for ChIP-seq using Bowtie2 in very sensitive local mode (--local --very-sensitive-local --no-unal --no-mixed --no-discordant --phred33 -I 10 -X 700) 51. Peaks were called using MACS252 with a threshold of q<0.05. Peaks were annotated to the nearest TSS using HOMER53. Differential analysis was performed using DiffBind54 and DESeq255. Significantly changed peaks were defined by a 1.5-fold change threshold and FDR<0.05. BigWig files for PAX3-FOXO1-HA were normalized with E.coli reads and generated using Deeptools. BigWig files for other factors were generated and normalized with the DESeq2 size factors using Deeptools. Heatmaps were created by Deeptools56 using the normalized bigwig files.
Assay for Transposase Accessible Chromatin using sequencing (ATAC-seq)
ATAC-seq was performed in biological duplicate using the ATAC-Seq Kit (Active Motif, catalog No.53150)57. Briefly, 100,000 Rh30 cells and 2,000 Drosophila S2 cells were used to isolate nuclei. Nuclei were incubated with tagmentation Master Mix at 37 °C for 30 minutes after lysing the cells in ice-cold ATAC Lysis Buffer and the DNA was purified with the DNA purification column. PCR amplification of tagmented DNA was performed to make libraries with the appropriate indexed primers. After SPRI bead clean-up, the DNA libraries were sequenced on the Illumina NovaSeq (PE-150, 50 million reads) at the VUMC VANTAGE core.
ATAC-seq data analysis
Raw FASTQ data were trimmed by Trimmomatic (v0.32), and paired end reads were aligned to a concatenated human and Drosophila genome (hg19 and dm3) using Bowtie2 (-X 2000 -q --no-mixed --no-discordant). Peaks were called using Genrich (v. 0.6.1) (https://github.com/jsh58/Genrich) with the following options -j -y -r -e chrM -q 0.05 -a 20. Peaks were annotated to the nearest TSS using HOMER. Differential analysis was performed using DiffBind and DESeq2. Significantly changed peaks were defined by a 1.5-fold change threshold and FDR<0.05. BigWig files were generated and normalized using the DESeq2 size factors using Deeptools. Heatmaps were created by Deeptools using the DESeq2 size factor normalized bigwig files.
APEX2 proximity labeling
APEX2 proximity labeling was performed as described 58. Briefly, 100 million cells were incubated with 500 μm biotin phenol (Iris Biotech) dissolved in DMSO at 37 °C for 1hr. Hydrogen peroxide (H2O2) was added for 1 minute. After quenching with Trolox Cayman chemical, nuclei were isolated, lysed, and nuclear protein was harvested in RIPA buffer. Protein concentration was determined using DC Protein Assay (BioRad). Biotinylated proteins were purified with streptavidin beads (Thermo Fisher Scientific, #88817) and eluted by boiling in Laemmli sample buffer. Parental Rh30 cells were used as negative control to determine proteins specifically identified in Rh30-PAX3-FOXO1-APEX2 samples. Proteins from parental Rh30 cells and Rh30-PAX3-FOXO1-APEX2 cells were analyzed as biological triplicates that were processed independently.
FLAG affinity purification
Nuclei were isolated from 100 million cells per sample. The nuclei were then “extracted” by incubating with Pierce Universal Nuclease (Thermo Fisher Scientific, #88701) in co-IP buffer (20 mM Tris pH 8, 150 mM NaCl, 2 mM MgCl2, 0.1% NP-40, with protease inhibitors) on ice for 1 hour. Cleared lysates were passed over a 0.45 um cellulose acetate column (Corning) to remove any remaining particulates. Samples were incubated with 60 μl of equilibrated EZ Red Flag M2 bead slurry (EZview by Millipore Sigma, F2426) at 4°C for 2hr in co-IP buffer, after washing, purified proteins were eluted twice with 50 ul of 1 mg/ml 3X flag peptide (Sigma) in co-IP buffer on the nutator for 10 minutes at room temperature.
Mass Spectrometry
Eluents from Apex or FLAG purifications were prepared for analysis via S-trap trypsin digests using manufacturer’s protocol (S-Trap™ – ProtiFi). The peptides were separated on a self-packed 100 μm × 20 cm reversed phase (Phenomonex - Jupiter 3 micron, 300A) column from which peptides were ionized directly via nano-electrospray into an Exploris 480 (Thermo-Fisher) mass spectrometer. Both full-scan and peptide fragmentation (MS/MS) were collected over the course of a 70-minute aqueous to organic gradient elution in a data-dependent manner using dynamic exclusion to reduce redundancy of peptide acquisition. Resulting MS/MS spectra were searched using SEQUEST 59 against a human database containing common contaminants and reversed copies of each entry. Resulting identifications were filtered to a 5% false-discovery threshold, collated back to the protein level, and compared across samples using Scaffold (Proteome Software). Filtered total spectral count values were used for fold-change comparisons and p-value estimations.
QUANTIFICATION AND STATISTICAL ANALYSIS
N/A
ADDITIONAL RESOURCES
N/A
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-HA tag [HA.C5] | Abcam | CAT# ab18181 |
| α-HA tag (C29F4) | Cell Signaling Technology | CAT# 3724s |
| α-GAPDH (G9) | Santa Cruz | CAT# sc-365062 |
| α-Lamin B (M-20) | Santa Cruz | CAT# sc-6217 |
| α-FLAG®M2 | MilliiporeSigma | CAT# F1804 |
| α-Myogenin [F5D] | Abcam | CAT# ab1835 |
| α-Myosin Heave Chain | R&D SYSTEM | CAT# MAB4470 |
| Goat anti-Mouse IgG H&L (Alexa Fluor®488) | Abcam | CAT# ab150117 |
| Goat anti-Rabbit IgG H&L (Alexa Fluor®488) | Invitrogen | CAT# A-11034 |
| α-BRD4 | BETHYL | CAT# A301–985A50 |
| α-BRD4 (E2A7X) | Cell Signaling Technology | CAT# 13440 |
| α-Histone H3 (tri methyl K4) | Abcam | CAT# ab12209 |
| α-MYOD (G-1) | Santa Cruz | CAT# sc-377460 |
| α-RUNX1 (A-2) | Santa Cruz | CAT# sc-365644 |
| α-HEB (A-20) | Santa Cruz | CAT# sc-357 |
| α-ARID1A (D2A8U) | Cell Signaling Technology | CAT# 12354 |
| α-SPT16 (D7I2K) | Cell Signaling Technology | CAT# 12191 |
| α-CDK8 (D-9) | Santa Cruz | CAT# sc-13155 |
| α-PAX3 [C2] | Abcam | CAT# ab69856 |
| Donky anti-Rabbit IgG H&L | Invitrogen | CAT# 31238 |
| Rabbit Anti-Mouse IgG H&L | Abcam | CAT# ab46540 |
| α-H3(acetyl K27) | Abcam | CAT# ab4729 |
| α-Pol II (N-20) | Santa Cruz | CAT# sc-899 |
| α-Pol II (phospho S2) | Abcam | CAT# ab5095 |
| α-Pol II (phospho S5) | Abcam | CAT# ab5131 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| 5-bromo-2’-deoxyuridine | MilliiporeSigma | CAT# B5002 |
| BrdU Mouse anti-Human, FITC, Clone: 3D4 | Thermo Fisher Scientific | CAT# BDB556028 |
| Crystal Violet Acetate | MilliiporeSigma | CAT# C5042-10G |
| DMSO | MilliiporeSigma | CAT# D8481 |
| dTAG-47 | VICB | CAT# kk-25-065 |
| Digitonin | MilliiporeSigma | CAT# 300410 |
| Formaldehyde, 37% by Weight | Fisher Chemical™ | CAT# F79-500 |
| Triton™ X-100 | MilliiporeSigma | CAT# T8787 |
| BSA | MilliiporeSigma | CAT# A3059 |
| IGEPAL® CA-630 | MilliiporeSigma | CAT# 18896 |
| Alexa Fluor™ 568 Phalloidin | Thermo Fisher Scientific | CAT# A12380 |
| DAPI | Invitrogen | CAT# D1306 |
| Prolong™ Gold Antifade Mountant | Thermo Fisher Scientific | CAT# P36930 |
| TRIzol™ Reagent | Invitrogen | CAT# 15596-026 |
| TRIzol™ LS Reagent | Invitrogen | CAT# 10296010 |
| biotin-11-CTP | PerkinElmer | CAT# NEL542001EA |
| Sarkosyl | Fisher Scientific | CAT# IB07080 |
| Concanavalin A-coated beads | Bangs Laboratories | CAT# BP531 |
| CUTANA pAG-MNase | EpiCypher | CAT# 15-1116 |
| Hydrogen peroxide solution | MilliiporeSigma | CAT# 216763 |
| Biotin Tyramide (Biotin-Phenol) | Iris | CAT# 41994-02-9 |
| Trolox | CAYMAN Chemical | CAT# 10011659 |
| 2xLaemmli sample buffer | Bio-Rad | CAT# 161-0737 |
| Pierce™ Streptavidin Magnetic beads | Thermo Fisher Scientific | CAT# 88817 |
| Pierce™ Universal Nuclease for Cell Lysis | Thermo Fisher Scientific | CAT# 88701 |
| Purified 3xFLAG® peptide | MilliiporeSigma | CAT# SAE0194 |
| EZview™ Red ANTI-FLAG M2 Affinity Gel | EZview by Millipore Sigma | CAT# F2426 |
| Corning™ RPMI1640 Medium 1X without L-Glutamine | Thermo Fisher Scientific | CAT# MT15040CV |
| Gibco™ Schneider’s Drosophila Medium | Thermo Fisher Scientific | CAT# 21-720-024 |
| FetalPlex | Gemini Bio-Products | CAT# 100-602 |
| Fetal Bovine Serum | R&D Systems | CAT# S11150 |
| Corning™ Penicillin/Streptomycin | Thermo Fisher Scientific | CAT# MT30002CI |
| Corning™ L-glutamine Solution | Thermo Fisher Scientific | CAT# MT25005CI |
| DC Protein Assay | BioRad | CAT# 50001112 |
| Critical commercial assays | ||
| FITC Annexin V Apoptosis DetectionKit I | DB Pharmingen™ | CAT# 556547 |
| SsoAdvanced Universal SYBR Green Supermix | BIO-RAD | CAT# 1725270 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | CAT# 4368814 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | NEB | CAT# E7645S/L |
| ATAC-Seq Kit | Active Motif | CAT# 53150 |
| Gibson Assembly™ Cloning Kit | NEB | CAT# E5510S |
| QIAfilter Plasmid Maxi Kit | QIAGEN | CAT# 12263 |
| QuickChange Lightning | Agilent | CAT# 210518 |
| Deposited data | ||
| Raw and processed sequencing data | This paper | GEO: GES183281 |
| Raw data of cell images | This paper | https://data.mendeley.com/datasets/64sn74fk56/draft?a=21d0d0f5-1c60-4f50-b4bd-d69ef94241f5 |
| Experimental models: Cell lines | ||
| Rh30 | ATCC | CAT# CRL-2061 |
| Rh4 | Gifted | N/A |
| Rh30-PAX3-FOXO1-FKBP12F36V | This paper | N/A |
| Rh30-FOXO1-FKBP12F36V | This paper | N/A |
| Rh4-PAX3-FOXO1-FKBP12F36V | This paper | N/A |
| Drosophila S2 | Gifted | N/A |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Alt-R CRISPR-Cas9 tracrRNA | Integrated DNA Technologies | CAT# 1072534 |
| Recombinant DNA | ||
| pAW62.YY1.FKBP.knockin.mCherry | Addgene | Plasmid #104370 |
| pAW62-mCherry-PAX3-FOXO1-FKBP12F36V-2xHA | This paper | N/A |
| pAW62-mCherry-FOXO1-FKBP12F36V-2xHA | This paper | N/A |
| pAW62-mCherry-PAX3-FOXO1-APEX2-2xHA | This paper | N/A |
| pAW62-mCherry-PAX3-FOXO1-3xFLAG | This paper | N/A |
| Software and algorithms | ||
| Bowtie2 (v. 2.4.2) | Langmead and Salzberg, 2012 | N/A |
| MACS2 peak caller | Feng et al., 2012 | N/A |
| Genrich (V. 0.6.1) | https://informatics.fas.harvard.edu/atac-seq-guidelines.html | https://github.com/jsh58/Genrich |
| DESeq2 | Love et al., 2014 | N/A |
| DiffBind | Ross-Innes et al., 2012 | N/A |
| HOMER | Heinz et al., 2010 | N/A |
| deepTOOLS (v. 3.5.0) | Ramirez et al., 2016 | N/A |
| TopHat (v. 2.0.11) | Trapnell et al., 2012 | N/A |
| Cuffdiff (C. 2.1.1) | Trapnell et al., 2013 | N/A |
| Nascent RNA Sequencing Analysis (NRSA) | Wang et al., 2018 | N/A |
| Trimmomatic-0.39 | Bolger et al., 2014 | N/A |
| Other | ||
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | CAT#1081059 |
| CUTANA pAG-MNase | Epicypher | CAT#151-1016 |
Highlights.
PAX3-FOXO1 degradation leads to cell death and myogenic differentiation
PAX3-FOXO1 degradation causes loss of transcription of a small number of target genes
PAX3-FOXO1 regulates RNA polymerase II pause release at regulated genes
PAX3-FOXO1 controls co-regulatory complex assembly at regulated enhancers
Acknowledgements
We especially thank members of the Hiebert lab for helpful discussions, reagents and advice. We thank the Flow Cytometry, Chemical Synthesis, and Genome Sciences Shared Resources for services and support. We thank Dr. Javed Khan (NCI) for the gift of Rh4 cells. This work was supported by a St. Baldrick’s Research Grant with generous support from Rachael Chaffin’s Research Fund, The V Foundation Grant ID #: T2021-005, the T. J. Martell Foundation, the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation, the Edward P. Evans Foundation, National Institutes of Health grants (RO1-CA164605, R01-CA255446-01A1, T32-CA009582-33 to SWH, and R35-GM147213 to KRS), and as well as core services performed through Vanderbilt Digestive Disease Research grant (NIDDK P30DK58404), the Vanderbilt-Ingram Cancer Center support grant (NCI P30CA68485), and a grant from the National Center for Advancing Translational Sciences (2 UL1 TR000445-06). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of Interests
The authors declare no competing interests, although Scott Hiebert received research funding from Incyte Inc. through the Vanderbilt-Incyte Alliance. These funds did not support this work. Scott Hiebert is also a scientific advisor for the Edward P. Evans Foundation.
SUPPLEMENTAL INFORMATION
Supplementary Table S1. List of PAX3-FOXO1 target genes, enhancers and PAX3-FOXO1 binding proteins, Related to Figure 2, Figure 4, Figure 5, Figure S5.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barr FG, Galili N, Holick J, Biegel JA, Rovera G, and Emanuel BS (1993). Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet 3, 113–117. 10.1038/ng0293-113. [DOI] [PubMed] [Google Scholar]
- 2.Turc-Carel C, Lizard-Nacol S, Justrabo E, Favrot M, Philip T, and Tabone E (1986). Consistent chromosomal translocation in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 19, 361–362. 10.1016/0165-4608(86)90069-5. [DOI] [PubMed] [Google Scholar]
- 3.Bennicelli JL, Fredericks WJ, Wilson RB, Rauscher FJ 3rd, and Barr FG (1995). Wild type PAX3 protein and the PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma contain potent, structurally distinct transcriptional activation domains. Oncogene 11, 119–130. [PubMed] [Google Scholar]
- 4.Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, and Rauscher FJ 3rd (1995). The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol 15, 1522–1535. 10.1128/MCB.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, Schaefer B, Sen N, Song Y, Gualtieri A, et al. (2017). PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov 7, 884–899. 10.1158/2159-8290.CD-16-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Shern JF, Wei JS, Yohe ME, Song YK, Hurd L, Liao H, Catchpoole D, Skapek SX, Barr FG, et al. (2015). Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet 11, e1005075. 10.1371/journal.pgen.1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et al. (2014). Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4, 216–231. 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swift J, and Coruzzi GM (2017). A matter of time - How transient transcription factor interactions create dynamic gene regulatory networks. Biochim Biophys Acta Gene Regul Mech 1860, 75–83. 10.1016/j.bbagrm.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B, et al. (2018). SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805. 10.1126/science.aao2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stengel KR, Ellis JD, Spielman CL, Bomber ML, and Hiebert SW (2021). Definition of a small core transcriptional circuit regulated by AML1-ETO. Mol Cell 81, 530–545 e535. 10.1016/j.molcel.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Begum S, Emami N, Cheung A, Wilkins O, Der S, and Hamel PA (2005). Cell-type-specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene 24, 1860–1872. 10.1038/sj.onc.1208315. [DOI] [PubMed] [Google Scholar]
- 12.Hanna JA, Garcia MR, Lardennois A, Leavey PJ, Maglic D, Fagnan A, Go JC, Roach J, Wang YD, Finkelstein D, and Hatley ME (2018). PAX3-FOXO1 drives miR-486–5p and represses miR-221 contributing to pathogenesis of alveolar rhabdomyosarcoma. Oncogene 37, 1991–2007. 10.1038/s41388-017-0081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudjadi S, Pandey PR, Chatterjee B, Nguyen TH, Sun W, and Barr FG (2021). A Fusion Transcription Factor-Driven Cancer Progresses to a Fusion-Independent Relapse via Constitutive Activation of a Downstream Transcriptional Target. Cancer Res 81, 2930–2942. 10.1158/0008-5472.CAN-20-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, Yang F, Pineda M, Helman LJ, and Meltzer PS (2010). Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 70, 6497–6508. 10.1158/0008-5472.CAN-10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunkel BD, Wang M, LaHaye S, Kelly BJ, Fitch JR, Barr FG, White P, and Stanton BZ (2021). Evidence of pioneer factor activity of an oncogenic fusion transcription factor. iScience 24, 102867. 10.1016/j.isci.2021.102867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gryder BE, Pomella S, Sayers C, Wu XS, Song Y, Chiarella AM, Bagchi S, Chou HC, Sinniah RS, Walton A, et al. (2019). Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat Genet 51, 1714–1722. 10.1038/s41588-019-0534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, Gordon B, Dapper J, Blankenship K, Yang Y, et al. (2018). Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 34, 411–426 e419. 10.1016/j.ccell.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kendall GC, Watson S, Xu L, LaVigne CA, Murchison W, Rakheja D, Skapek SX, Tirode F, Delattre O, and Amatruda JF (2018). PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. Elife 7. 10.7554/eLife.33800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gryder BE, Wachtel M, Chang K, El Demerdash O, Aboreden NG, Mohammed W, Ewert W, Pomella S, Rota R, Wei JS, et al. (2020). Miswired Enhancer Logic Drives a Cancer of the Muscle Lineage. iScience 23, 101103. 10.1016/j.isci.2020.101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 14, 431–441. 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis RJ, and Barr FG (1997). Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A 94, 8047–8051. 10.1073/pnas.94.15.8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linardic CM (2008). PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett 270, 10–18. 10.1016/j.canlet.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, Concordet JP, Thway K, Oberlin O, Pritchard-Jones K, et al. (2012). PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 30, 1670–1677. 10.1200/JCO.2011.38.5591. [DOI] [PubMed] [Google Scholar]
- 24.Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588 e1528. 10.1016/j.cell.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. 10.1038/nprot.2016.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Zhao Y, Zhou X, Hiebert SW, Liu Q, and Shyr Y (2018). Nascent RNA sequencing analysis provides insights into enhancer-mediated gene regulation. BMC Genomics 19, 633. 10.1186/s12864-018-5016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jonkers I, and Lis JT (2015). Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16, 167–177. 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gryder BE, Wu L, Woldemichael GM, Pomella S, Quinn TR, Park PMC, Cleveland A, Stanton BZ, Song Y, Rota R, et al. (2019). Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat Commun 10, 3004. 10.1038/s41467-019-11046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pott S, and Lieb JD (2015). What are super-enhancers? Nat Genet 47, 8–12. 10.1038/ng.3167. [DOI] [PubMed] [Google Scholar]
- 33.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bohm M, Wachtel M, Marques JG, Streiff N, Laubscher D, Nanni P, Mamchaoui K, Santoro R, and Schafer BW (2016). Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J Clin Invest 126, 4237–4249. 10.1172/JCI85057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54. 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA, and Ting AY (2016). Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11, 456–475. 10.1038/nprot.2016.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, and Ting AY (2012). Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30, 1143–1148. 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donner AJ, Ebmeier CC, Taatjes DJ, and Espinosa JM (2010). CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol 17, 194–201. 10.1038/nsmb.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prozzillo Y, Fattorini G, Santopietro MV, Suglia L, Ruggiero A, Ferreri D, and Messina G (2020). Targeted Protein Degradation Tools: Overview and Future Perspectives. Biology (Basel) 9. 10.3390/biology9120421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaeger MG, and Winter GE (2021). Fast-acting chemical tools to delineate causality in transcriptional control. Mol Cell 81, 1617–1630. 10.1016/j.molcel.2021.02.015. [DOI] [PubMed] [Google Scholar]
- 42.Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944 e922. 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luan J, Xiang G, Gomez-Garcia PA, Tome JM, Zhang Z, Vermunt MW, Zhang H, Huang A, Keller CA, Giardine BM, et al. (2021). Distinct properties and functions of CTCF revealed by a rapidly inducible degron system. Cell Rep 34, 108783. 10.1016/j.celrep.2021.108783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang J, Li K, Cai W, Liu X, Zhang Y, Orkin SH, Xu J, and Yuan GC (2018). Dissecting super-enhancer hierarchy based on chromatin interactions. Nat Commun 9, 943. 10.1038/s41467-018-03279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laubscher D, Gryder BE, Sunkel BD, Andresson T, Wachtel M, Das S, Roschitzki B, Wolski W, Wu XS, Chou HC, et al. (2021). BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat Commun 12, 6924. 10.1038/s41467-021-27176-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Layden HM, Eleuteri NA, Hiebert SW, and Stengel KR (2021). A protocol for rapid degradation of endogenous transcription factors in mammalian cells and identification of direct regulatory targets. STAR Protoc 2, 100530. 10.1016/j.xpro.2021.100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Liu Q, Acharya P, Stengel KR, Sheng Q, Zhou X, Kwak H, Fischer MA, Bradner JE, Strickland SA, et al. (2016). High-Resolution Mapping of RNA Polymerases Identifies Mechanisms of Sensitivity and Resistance to BET Inhibitors in t(8;21) AML. Cell Rep 16, 2003–2016. 10.1016/j.celrep.2016.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31, 46–53. 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat Protoc 7, 1728–1740. 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29. 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK, Carr SA, and Ting AY (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6. 10.7554/eLife.24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic datasets are available at GEO accession GSE183281. Original data for figures in this study are accessible upon request and deposited to Mendeley Data: https://data.mendeley.com/datasets/64sn74fk56/draft?a=21d0d0f5-1c60-4f50-b4bd-d69ef94241f5
This paper does not report original code. Any code for analysis will be shared by the leading contact upon request.
Any Additional information required to reanalyze the data reported in this paper is available from the leading contact upon request.