

Abstract
Although neurohormones and Renin‐Angiotensin‐Aldosterone‐System (RAAS) components are important predictors of cardiovascular mortality (CVM), their importance for predicting outcomes in patients with/without RAAS‐blockers and different degrees of arterial stiffness is less understood. We therefore analyzed long‐term data from the Ludwigshafen Risk and Cardiovascular Health (LURIC) study in 3316 patients subdivided according to pulse pressure (PP) and RAAS‐blocker use. Patients on RAAS‐inhibition had higher renin and noradrenaline, lower aldosterone and aldosterone/renin quotient (ARQ). Renin and noradrenaline significantly predicted CVM in patients without RAAS‐blocker (HR = 1.17, 1.15) and in patients receiving angiotensin‐converting‐enzyme (ACE) inhibitors (HR = 1.17, 1.29), whereas aldosterone predicted CVM only in patients receiving ACE‐inhibitors (HR = 1.13). CVM was predicted independently from PP by renin, noradrenaline and angiotensin II. Independently from RAAS inhibition renin decreased and ARQs increased with rising PP. Furthermore, noradrenaline increased with PP, but only without ACE‐inhibition. The HR for CVM in the ACE‐inhibitor group were 1.29, 1.28, 1.29 for renin in the first, second and third PP quartiles and 1.22, and 1.19 for aldosterone in the second and fourth quartile. Furthermore, we showed that noradrenaline predicts CVM in all PP quartiles in patients with ACE‐inhibition. In the RAAS‐blocker‐free group, the HR for renin for CVM were 1.36 and 1.18 in the third and fourth PP quartiles, but neither aldosterone nor noradrenaline were predictive for CVM within the PP quartiles. Renin and noradrenaline are strong predictors of CVM regardless of RAAS blockade, whereas aldosterone is predictive only in the ACE‐inhibitor group. Catecholamines but not renin are associated with rising PP.
Keywords: ACE‐inhibitors, arterial compliance, atherosclerosis, cardiovascular mortality, noradrenaline, pulse pressure
1. INTRODUCTION
The renin‐angiotensin‐aldosterone system (RAAS) is already known as a key player in maintaining blood pressure, circulation, water‐, and sodium balance as well as vascular tension. 1 Angiotensin II mediates most of the known pressor, profibrotic, and proliferative effects by binding to the angiotensin receptor type 1 (AT1). 1 High levels of renin, which catalyzes the conversion of angiotensinogen to angiotensin‐I, have been shown to be associated with higher cardiovascular mortality (CVM) in an earlier analysis of the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. 2 Furthermore in previous analyses of the LURIC study both low levels of 25(OH) vitamin D and 1,25(OH) vitamin D, as well as higher leukocyte‐derived myeloperoxidase and asymmetrical dimethyl‐arginine (ADMA) levels were independently associated with an upregulated circulating RAAS. 3 Moreover, the MESA (Multi‐Ethnic Study of Atherosclerosis) study demonstrated that higher aldosterone levels were linked to an elevated risk of subclinical coronary atherosclerosis and all‐cause mortality, especially if renin was suppressed. 4
Uromodulin, which was detected first in human urine by Tamm and Horsfall in 1950, 5 is produced in the thick ascending limb of Henle and is mainly secreted into the tubular lumen, where it plays an important role in ion transport as well as maintenance of electrolyte and water balance 6 and shows antibacterial effects. 7 Apart from that, higher serum uromodulin concentrations are associated with a significantly reduced risk of all‐cause and CVM, 8 and serum uromodulin concentrations have been linked with the amount of functioning renal mass. 9
In one study of 24,297 participants without chronic kidney disease (CKD) at baseline, a high cardio‐ankle vascular index, a marker of arterial stiffness, was significantly correlated with CKD events and proteinuria over a mean follow‐up period of 3.1 years. 10 Pulse pressure greater than 60 mmHg was also significantly associated with the prevalence of carotid plaque and left ventricular hypertrophy in 7336 hypertensive patients in the Campania Salute Network registry. These patients also showed a 57% increased risk for major cardiovascular events. 11
The predictive power of renin and other (neuro‐) hormones such as aldosterone and catecholamines for CVM in patient subgroups with or without RAAS blockers and with different degrees of arterial stiffness is less well known. Therefore, CVM prediction by RAAS components and neurohormones in relation to RAAS inhibition and arterial stiffness in different quartiles of pulse pressure was studied in the LURIC study. Thus, we extended our present analysis, beyond an earlier analysis, 2 to important patient subgroups, that is, without RAAS inhibitors and with ACE‐inhibitors or ARBs and patient subgroups according to the extent of arterial stiffness and have in addition further specified the effects of aldosterone and noradrenaline.
2. METHODS
2.1. Study cohort
The Ludwigshafen Risk and Cardiovascular Health (LURIC) study is a monocentric hospital‐based cohort study that recruited 3316 patients of German ancestry who were living in the surroundings of the German cities Ludwigshafen, Mannheim and Heidelberg and underwent coronary angiography between July 1997 and January 2000. The Landesärztekammer Rheinland‐Pfalz provided ethical approval for this study [#837.255.97(1394)]. The study protocol was in accordance with the Declaration of Helsinki, and all study participants provided written consent to participate in the study. The main indications for coronary angiography were acute chest pain or a positive result in a noninvasive cardiac stress test. Exclusion criteria were the presence of other acute cardiac diseases such as decompensated heart failure or decompensated valvular disease or acute non‐cardiac diseases such as infection, endocrine disease or any type of surgery within the previous three months, chronic polymorbid disease where non‐cardiac disease was predominant (i.e., chronic renal failure and hemodialysis, severe rheumatic arthritis, persistent incapacitation after accident/trauma), history of malignant disease within the previous five years, and inability to understand the purpose of the study. 12 After study inclusion, a detailed physical examination, including assessment of vital signs, and a detailed blood test were performed.
Using an automated oscillometric device (Omron MX4; Omron Healthcare GmbH, Hamburg, Germany), blood pressure was measured while being in a supine position for at least 10 min. At least three consecutive measurements of systolic and diastolic blood pressures were obtained at intervals of 30 s. If the systolic, diastolic, or heart rate measurements varied by > 10 mmHg, > 5 mmHg, or > 5 beats per minute (except during atrial fibrillation), they were regarded as invalid and then repeated. Both valid and invalid measurements were recorded. The invalid measurements were immediately identified as such. Only stable measurements that matched the reproducibility criteria were entered into the database. One part of the questionnaire investigated whether blood pressure had been measured according to protocol (supine, 10 min at rest). The study protocol and study procedures have been reported in some detail previously. 12
For the purpose of the present analyses, we subdivided the LURIC patient cohort first into three groups of patients either receiving no RAAS inhibitor, or receiving an ACE‐inhibitor or receiving an ARB. These subgroups were chosen because RAAS blockers markedly affect components of the RAAS and also of (neuro‐)hormones and may thereby affect the association between these markers and outcome. Since the effects of ACE‐inhibitors and ARBs on these hormones also vary considerably, we analyzed these subgroups separately. Patients with aldosterone antagonists were excluded from the analysis because aldosterone antagonists markedly affect the RAAS hormones in a different way than ACE‐inhibitors and ARBs and patients receiving aldosterone antagonists represented only a very small subgroup of 38 patients, precluding any meaningful subgroup analysis. For the same reasons we also excluded the even smaller subgroup of patients treated with a combination of ACE‐inhibitors and ARBs (n = 27) from our analysis. With regard to our analysis of arterial stiffness we subdivided the LURIC cohort according to quartiles of PP.
2.2. Laboratory measurements
Venous blood was drawn under standardized conditions after an overnight fasting period. Within 30 min after venipuncture, the obtained blood was centrifuged at 3000 × g for 10 min, immediately aliquoted, and frozen at −80°C until further analysis. A summary of the analytic methods has been reported previously. 12 Total cholesterol, high‐density lipoprotein (HDL) cholesterol, and triglyceride concentrations were determined enzymatically with a Synchron LX‐20 (Beckman Coulter, Munich, Germany). LDL and very low‐density lipoprotein (VLDL) cholesterol were separated by ultracentrifugation in a Beckman LM‐8 ultracentrifuge in 100‐μl volumes with a VT‐51.2 rotor (Beckman Coulter). The N‐terminal‐pro‐brain natriuretic peptide (NT‐pro‐BNP) level was measured by electrochemiluminescence on an Elecsys 2010 (Roche Diagnostics). Creatinine was measured using the CREA assay (Roche, Germany) on a Hitachi 717 analyzer. The estimated glomerular filtration rate was calculated using the MDRD formula. Renin was measured by an immunoradiometric assay (Active renin/Berthold Multi‐crystal counter LB2014). Adrenaline and noradrenaline were determined by electrochemical detection (Chromsystems HPLC Adrenaline, Noradrenaline/Waters Millennium chromatography with Waters detector 460). Aldosterone was measured by immunoradiometric assay (Coat‐a‐count aldosterone/Berthold Multi‐crystal counter LB2014).
2.3. Clinical definitions
Coronary artery disease (CAD) was defined according to the American Heart Association by visible luminal narrowing of 20% stenosis or more in at least one of 15 coronary segments. 12 Coronary 1‐vessel disease, coronary 2‐vessel disease, and coronary 3‐vessel disease were defined as stenosis of 20% or higher in one, two, or three of the major coronary arteries, namely, left anterior descending artery (LAD), ramus circumflexus (RCX), and right coronary artery (RCA). 12 The echocardiographic classification of heart failure has been already published in some detail in addition to a summary of further clinical definitions. 12
2.4. Follow‐up assessments
Patients were followed up for a median of 9.9 years. Information on vital status was obtained from local registries. Two experienced clinicians reviewed death certificates, medical records of local hospitals, and autopsy data independently. They were blinded to patient characteristics and classified the causes of death. In cases involving disagreement or uncertainty concerning the coding of a specific cause of death, the decision was made by a principal investigator (W.M.).
2.5. Statistical analysis
IBM SPSS® Statistics version 25.0 (SPSS Inc., IBM Corporation, NY, USA) and R v4.0.2 were used for all analyses. Hazard ratios were calculated by Cox proportional hazards regression. Variables were Z‐transformed before entering the analyses, so the hazard ratios were not calculated for an increase per unit but for an increase of one standard deviation in the respective marker. We performed multivariate analysis with adjustments for age and sex. All data were complete except for the missing eGFR data for three study participants. These were excluded from the adjusted analysis.
3. RESULTS
3.1. Comparison of patients with RAAS inhibitors vs. without RAAS inhibitors
Patients receiving ACE‐inhibitors were on average 3.1 years older, had significantly higher renin levels, lower aldosterone/renin quotients (ARQs), higher PP, higher noradrenaline and cystatin C levels, and lower uromodulin concentrations than patients receiving no RAAS blocker, while the creatinine levels of the two groups were comparable (Table 1). Patients on angiotensin‐II‐receptor blocker (ARB) therapy were even older, had higher PP and noradrenaline concentrations than patients on ACE‐inhibitors. Creatinine, cystatin C, renin and uromodulin were found to be at a similar level as in patients on ACE‐inhibitors (Table 1). Patients who received both ACE‐blockers and ARBs were 1 year older, had a significantly higher PP, creatinine and cystatin C levels as well as lower uromodulin levels than patients who received ACE‐inhibitors only. Furthermore, these patients showed the highest levels of renin and the lowest ARQ (Table 1).
TABLE 1.
Baseline parameters in patients receiving only ACE‐inhibitors (ACE‐I), ARB or none of these compounds
| No ACE‐I/no ARB | ACE‐I /no ARB | No ACE‐I /ARB | ||
|---|---|---|---|---|
| Variable | (N = 1416) | (N = 1717) | (N = 118) | P* |
| Age (years) | 60.8(11.4) | 63.9(9.94) | 66.2(8.96) | <0.001 |
| Female sex (%) | 32.1 | 28.3 | 35.6 | 0.070 |
| BMI (kg/m2) | 27(3.62) | 27.8(4.32) | 28.7(4.11) | <0.001 |
| LDL‐C (mg/dl) | 120(35.3) | 114(33.4) | 113(33.2) | <0.001 |
| HDL‐C (mg/dl) | 40.2(11) | 37.5(10.5) | 40.4(11.5) | <0.001 |
| TG (mg/dl) | 144(105‐200) | 148(112‐201) | 150(114‐193) | 0.024 |
| SBP (mmHg) | 139(21.8) | 143(24.7) | 148(24.7) | <0.001 |
| DBP (mmHg) | 80.5(10.8) | 81.4(11.8) | 82.6(12.5) | 0.044 |
| Creatinine | 0.953(0.466) | 0.987(0.263) | 0.978(0.236) | <0.001 |
| Cystatin C | 0.938(0.399) | 1.03(0.385) | 1.03(0.271) | <0.001 |
| Uromodulin | 171(77.4) | 148(70.1) | 148(73.1) | <0.001 |
| Central PP | 69.2(19.8) | 70.8(22.1) | 78.1(24.6) | 0.001 |
| PP | 58.5(16.9) | 61.3(19) | 65.2(18) | <0.001 |
| DP | 9530(2350) | 9820(2470) | 10200(2260) | <0.001 |
| MAP | 100(13.2) | 102(14.7) | 104(15.3) | <0.001 |
| Renin (pg/ml) | 8.98(4.79‐16.2) | 14.4(6.59‐33.5) | 14.1(7.34‐36.1) | <0.001 |
| Aldosterone (ng/l) | 83(52‐126) | 72.5(45‐116) | 84(52.2‐133) | <0.001 |
| Angiotensinogen (nnmol/l) | 1200(943‐1520) | 1130(845‐1450) | 1160(804‐1450) | <0.001 |
| Angiotensin‐I (ng/l) | 1550(1220‐1980) | 1460(1100‐1880) | 1500(1040‐1880) | <0.001 |
| Angiotensin‐II (ng/l) | 19(12‐32) | 20(13‐37) | 24(15‐36.5) | <0.001 |
| ACE (U/l) | 30(23‐39) | 14(8‐27) | 33(24.2‐41) | <0.001 |
| Adrenalin (ng/l) | 36(22‐57) | 34(22‐55) | 35(22‐54) | <0.001 |
| Noradrenalin (ng/l) | 292(205‐416) | 318(217‐454) | 360(254‐448) | <0.001 |
| Aldosterone / Renin | 5.12(2.84‐9.33) | 2.86(1.14‐6.24) | 3.01(1.41‐6.73) | <0.001 |
| NT‐proBNP (pg/ml) | 171(74‐458) | 437(160‐1230) | 310(145‐790) | <0.001 |
| Coronary artery disease (%) | 68.9 | 85.3 | 75.4 | <0.001 |
| Diabetes mellitus (%) | 32.6 | 44 | 52.5 | <0.001 |
| Smoking (current or ex, %) | 62.8 | 67 | 53.4 | 0.005 |
*ANOVA for continuous variables, non‐normally distributed variables were log‐transformed before entering analyses, and chi‐square test for categorical variables.
Abbreviations: SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; DP, double product; MAP, mean arterial pressure; ACE, angiotensin Converting Enzyme; CAD, Coronary artery disease.
Both renin and noradrenaline showed significant predictive value for CVM, with HRs of 1.17, and 1.15 for an increase of one standard deviation in patients without any RAAS blocker therapy (Figure 1(A)). In patients receiving ACE‐inhibitor therapy, renin, aldosterone, and noradrenaline levels also showed significant predictive values for CVM with HRs of 1.17, 1.13, and 1.29 for an increase of one standard deviation (Figure 1(B)). In patients receiving only ARBs, no significant HR could be detected. This was probably due to the small number of patients in this subgroup (n = 118) (Figure 1(C)). All HR values were calculated after adjustment for age and sex.
FIGURE 1.
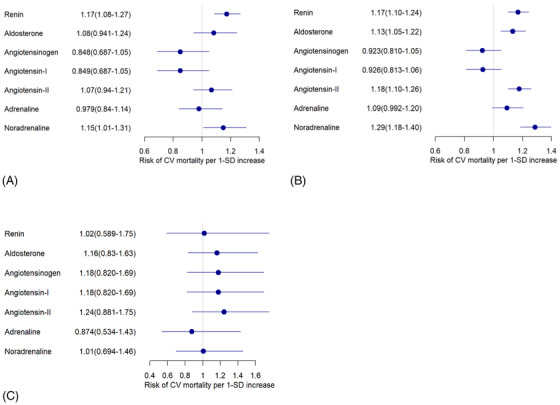
Hazard ratios for CV mortality for patients receiving (A) no ACE inhibitors, no ARB, no aldosterone blockers, (B) ACE inhibitors but no ARB, no aldosterone blockers and (C) no ACE inhibitors, no aldosterone blockers, but ARB. Cox proportional hazards regression for Z‐transformed variables with adjustment for age and sex
3.2. Comparison of patients in PP quartiles
First, we analyzed prediction of CVM in dependency of vascular stiffness, namely in PP quartiles, for all patients excluding only those taking aldosterone blockers (n = 38) and those taking ACE‐inhibitors and ARBs simultaneously (n = 27). We saw significant HRs for CVM of 1.22, 1.26 and 1.24 for renin in the first, second and third PP quartile, respectively, while noradrenaline provided HRs of 1.42, 1.25, 1.20 and 1.15 in the first until fourth PP quartile. In addition, aldosterone showed significant HR for CVM only in the 2nd and 4th quartile of 1.25 and 1.14, respectively. On the other hand, angiotensin II provided significant CMV prediction with HR of 1.16, 1.21, 1.14 and 1.14 in the first until fourth PP quartile (Figure 2).
FIGURE 2.
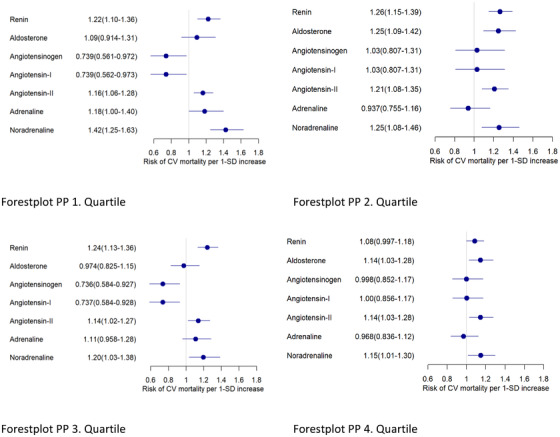
Hazard ratios for CVM for the entire patient cohort, excluding those taking aldosterone blockers or ACE‐inhibitors and ARBs simultaneously in pulse pressure (PP) quartiles. Cox proportional hazards regression for Z‐transformed variables with adjustment for age and sex
Next we analyzed prediction of CVM in subgroups according to both PP quartiles and with regard to RAAS use. In the RAAS‐blocker‐free group, with increasing PP, the renin concentration fell significantly from 10.8 pg/ml in the first PP quartile to 7.78 pg/ml in the fourth PP quartile. Conversely, the aldosterone/renin quotient increased significantly from 4.43 in the first PP quartile to 5.65 in the fourth PP quartile (Table 2).
TABLE 2.
Baseline parameters in patients receiving no RAAS inhibitors for different PP quartiles
| PP < 46.2 mmHg | PP 46.2 ‐ 57.0 mmHg | PP 57.0 ‐ 69.3 mmHg | PP > 69.3 mmHg | |||
|---|---|---|---|---|---|---|
| Variable | (N = 360) | (N = 355) | (N = 348) | (N = 353) | P* | P. linear** |
| Age (years) | 54(10.4) | 58.4(11) | 62.5(9.89) | 68.5(8.69) | <0.001 | <0.001 |
| Female sex (%) | 34.2 | 27.3 | 31.6 | 35.4 | 0.101 | 0.477 |
| BMI (kg/m2) | 26.5(3.77) | 26.7(3.52) | 27.3(3.47) | 27.2(3.66) | 0.002 | 0.002 |
| LDL‐C (mg/dl) | 121(35.4) | 121(34.6) | 122(35.5) | 116(35.3) | 0.086 | 0.085 |
| HDL‐C (mg/dl) | 40.3(10.5) | 40.2(10.7) | 40.3(11.1) | 40(11.6) | 0.704 | 0.704 |
| TG (mg/dl) | 144(101‐199) | 146(110‐202) | 142(109‐201) | 144(101‐199) | 0.530 | 0.530 |
| SBP (mmHg) | 116(11.8) | 131(11.1) | 145(10.5) | 165(15.7) | <0.001 | <0.001 |
| DBP (mmHg) | 77.1(10.5) | 79.6(10.5) | 82.1(9.65) | 83.4(11.5) | <0.001 | <0.001 |
| Creatinine | 0.945(0.568) | 0.933(0.324) | 0.938(0.308) | 0.997(0.586) | 0.143 | 0.143 |
| Cystatin C | 0.9(0.41) | 0.909(0.42) | 0.924(0.31) | 1.02(0.433) | <0.001 | <0.001 |
| Uromodulin | 181(82.2) | 175(78) | 172(75.8) | 155(71.1) | <0.001 | <0.001 |
| central PP | 58.1(15.3) | 61.9(16.1) | 72.6(17.7) | 83.9(18.9) | <0.001 | <0.001 |
| PP | 39.1(5.46) | 51.6(3.07) | 62.5(3.33) | 81.5(11) | <0.001 | <0.001 |
| DP | 7910(1700) | 9020(1930) | 9850(1790) | 11400(2410) | <0.001 | <0.001 |
| MAP | 90.2(10.6) | 96.8(10.6) | 103(9.81) | 111(12) | <0.001 | <0.001 |
| Renin (pg/ml) | 10.8(5.99‐19.8) | 9.58(5.39‐16.8) | 8.38(4.79‐14.4) | 7.78(4.79‐13.8) | <0.001 | <0.001 |
| Aldosterone (ng/l) | 83.5(52‐125) | 90(56‐131) | 80(49.2‐123) | 80(50.2‐121) | 0.074 | 0.073 |
| Angiotensinogen (nnmol/l) | 1190(905‐1490) | 1170(942‐1470) | 1230(965‐1580) | 1210(971‐1540) | 0.056 | 0.056 |
| Angiotensin‐I (ng/l) | 1540(1170‐1930) | 1510(1220‐1900) | 1590(1250‐2040) | 1560(1260‐2000) | 0.055 | 0.055 |
| Angiotensin‐II (ng/l) | 20(13‐33.2) | 18(12‐32) | 20(12‐31) | 18.5(12‐34) | 0.115 | 0.115 |
| ACE (U/l) | 32(24‐40) | 30(24‐38.5) | 30(23‐38) | 29(22‐40) | 0.003 | 0.003 |
| Adrenalin (ng/l) | 33(20‐51) | 33.5(21‐51) | 39(24‐60) | 39(23‐64) | 0.001 | 0.001 |
| Noradrenalin (ng/l) | 256(185‐384) | 278(196‐403) | 302(216‐419) | 324(236‐460) | <0.001 | <0.001 |
| Aldosterone / Renin | 4.43(2.5‐7.76) | 5.24(2.88‐8.85) | 5.48(2.99‐10.2) | 5.65(2.82‐10.4) | 0.001 | 0.001 |
| NT‐proBNP (pg/mL) | 131(51.2‐334) | 125(58‐352) | 170(79.2‐414) | 310(130‐756) | <0.001 | <0.001 |
| Coronary artery disease (%) | 58.6 | 65.6 | 70.4 | 81 | <0.001 | <0.001 |
| Diabetes mellitus (%) | 21.9 | 30.4 | 31.6 | 46.7 | <0.001 | <0.001 |
| Smoking (current or ex, %) | 68.1 | 65.4 | 63.2 | 54.4 | 0.001 | <0.001 |
*ANOVA for continuous variables, non‐normally distributed variables were log‐transformed before entering analyses, and chi‐square test for categorical variables; ** Linear trend, for categorical variable the Cochrane‐Armitage test was used.
Abbreviations: SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; DP, double product; MAP, mean arterial pressure; ACE, angiotensin Converting Enzyme; CAD: Coronary artery disease.
In the ACE‐inhibitor group, renin behaved in a similar way and fell significantly from 22.8 pg/ml in the first PP quartile to 11.4 pg/ml in the fourth PP quartile. Simultaneously, the aldosterone/renin quotient increased significantly from 1.71 to 3.76 across the four PP quartiles (Table 3).
TABLE 3.
Baseline parameters in patients receiving ACE‐inhibitors but no ARB for different PP quartiles
| PP < 47.0 mmHg | PP 47.0 ‐ 60.0 mmHg | PP 60.0 ‐ 73.4 mmHg | PP > 73.4 mmHg | |||
|---|---|---|---|---|---|---|
| Variable | (N = 432) | (N = 432) | (N = 426) | (N = 427) | P* | P. linear** |
| Age (years) | 58.5(10.4) | 62.9( 10 ) | 65.8(8.37) | 68.5(7.84) | <0.001 | <0.001 |
| Female sex (%) | 24.5 | 26.4 | 28.4 | 34 | 0.015 | 0.002 |
| BMI (kg/m2) | 27.5(4.24) | 27.7(4.61) | 28.1(4.22) | 27.9(4.19) | 0.131 | 0.131 |
| LDL‐C (mg/dl) | 114(34.8) | 114(31.4) | 114(32.3) | 116(34.9) | 0.286 | 0.286 |
| HDL‐C (mg/dl) | 35.5(9.45) | 37.5(10.1) | 37.8(10.6) | 39.4(11.3) | <0.001 | <0.001 |
| TG (mg/dl) | 144(112‐202) | 144(113‐190) | 157(114‐205) | 146(108‐198) | 0.508 | 0.508 |
| SBP (mmHg) | 115(12.9) | 134(12.2) | 150(12.5) | 171(16) | <0.001 | <0.001 |
| DBP (mmHg) | 76.7(11) | 80.5(11.3) | 83.7(11.4) | 84.6(11.9) | <0.001 | <0.001 |
| Creatinine | 0.982(0.232) | 0.972(0.222) | 0.981(0.245) | 1.01(0.337) | 0.079 | 0.079 |
| Cystatin C | 1.01(0.403) | 0.997(0.278) | 1(0.287) | 1.12(0.512) | <0.001 | <0.001 |
| Uromodulin | 158(71.9) | 157(70.9) | 141(63) | 137(71.9) | <0.001 | <0.001 |
| Central PP | 55.6(17.5) | 65.6(18.7) | 74.6(19) | 87.4(19.8) | <0.001 | <0.001 |
| PP | 38.5(5.98) | 53.6(3.69) | 66.5(3.86) | 86.8(11.1) | <0.001 | <0.001 |
| DP | 7990(1700) | 9270(1930) | 10300(2030) | 11700(2470) | <0.001 | <0.001 |
| MAP | 89.6(11.3) | 98.4(11.5) | 106(11.7) | 114(12.3) | <0.001 | <0.001 |
| Renin (pg/ml) | 22.8(8.98‐51.3) | 15(6.59‐33.5) | 12(6.59‐27.5) | 11.4(5.99‐24) | <0.001 | <0.001 |
| Aldosterone (ng/l) | 69(43‐120) | 73(43‐112) | 72.5(46‐115) | 79(47‐120) | 0.234 | 0.234 |
| Angiotensinogen (nnmol/l) | 1060(740‐1360) | 1100(830‐1390) | 1200(906‐1510) | 1170(905‐1450) | <0.001 | <0.001 |
| Angiotensin‐I (ng/l) | 1380(960‐1770) | 1420(1080‐1810) | 1550(1170‐1960) | 1520(1170‐1880) | <0.001 | <0.001 |
| Angiotensin‐II (ng/l) | 23(14‐44) | 20(11‐35) | 20(13‐37) | 18(11‐33) | 0.001 | 0.001 |
| ACE (U/l) | 15(8‐28) | 15(8‐30) | 14(8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26) | 14(8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25) | 0.022 | 0.022 |
| Adrenalin (ng/l) | 32(20‐50) | 33(21‐55) | 35(23‐56) | 35(24‐59.8) | 0.005 | 0.005 |
| Noradrenalin (ng/l) | 306(208‐444) | 310(214‐453) | 329(230‐459) | 324(219‐470) | 0.129 | 0.129 |
| Aldosterone / Renin | 1.71(0.782‐3.94) | 2.94(1.12‐6.2) | 3.24(1.51‐6.75) | 3.76(1.43‐8.14) | <0.001 | <0.001 |
| NT‐proBNP (pg/mL) | 456(170‐1390) | 397(130‐1100) | 415(152‐1000) | 498(213‐1400) | 0.285 | 0.284 |
| Coronary artery disease (%) | 82.4 | 84.5 | 84.5 | 89.7 | 0.020 | 0.004 |
| Diabetes mellitus (%) | 37 | 38.7 | 45.8 | 54.8 | <0.001 | <0.001 |
| Smoking (current or ex, %) | 72.5 | 69 | 62.7 | 63.9 | 0.007 | 0.002 |
*ANOVA for continuous variables, non‐normally distributed variables were log‐transformed before entering analyses, and chi‐square test for categorical variables; ** Linear trend, for categorical variable the Cochrane‐Armitage test was used.
Abbreviations: SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; DP, double product; MAP, mean arterial pressure; ACE, angiotensin Converting Enzyme, CAD: Coronary artery disease.
Apart from this, the catecholamine concentrations increased with increasing PP. In the RAAS blocker‐free group, the adrenaline level increased significantly from 33 ng/L in the first PP quartile to 39 ng/L in the fourth PP quartile, while the noradrenaline level increased significantly and markedly from 256 ng/L to 324 ng/L (Table 2).
Similar results were obtained for the ACE‐inhibitor group. Adrenaline increased significantly from 32 to 35 ng/L. Although the differences in noradrenaline concentrations missed the level of significance, they also showed a trend for rising levels (Table 3).
In the RAAS‐blocker free group, the z‐standardized hazard ratios for CVM were 1.36 and 1.18 for renin in the third and fourth PP quartiles, whereas no statistical significance was achieved in the first and second PP quartile. HR for noradrenaline, and aldosterone did not reach the significance level in any PP quartile (Figure 3).
FIGURE 3.
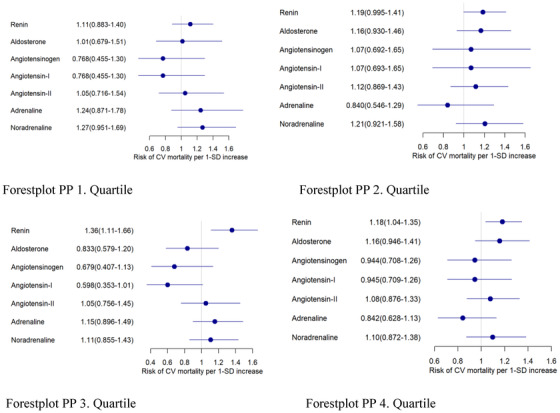
Hazard ratios for CV mortality in the RAAS ‐inhibitor free group in pulse pressure (PP) quartiles. Cox proportional hazards regression for Z‐transformed variables with adjustment for age and sex
In the first, second and third PP quartile, the z‐standardized hazard ratios for CVM in the ACE‐inhibitor group were 1.29, 1.28 and 1.29 for renin, whereas no statistical significance was reached in the fourth PP quartile, and the corresponding values were 1.53, 1.20, 1.25, 1.19 for noradrenaline in the first, second, third, and fourth PP quartiles, and the corresponding values were 1.22 and 1.19 for aldosterone in the second and fourth PP quartiles, respectively (Figure 4).
FIGURE 4.
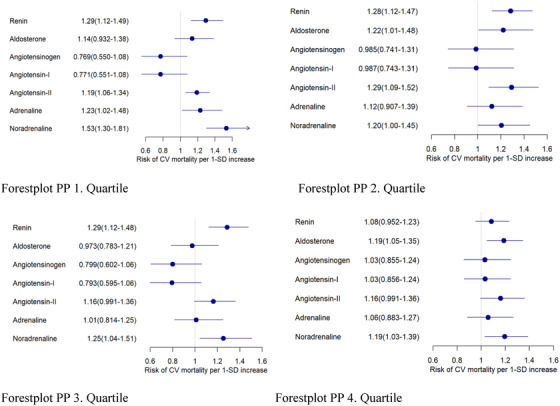
Hazard ratios for CV mortality in the ACE‐inhibitor group in pulse pressure (PP) quartiles. Cox proportional hazards regression for Z‐transformed variables with adjustment for age and sex
4. DISCUSSION
With RAAS blocker therapy we noticed as expected higher renin and lower ARQ levels, but also higher noradrenaline concentrations. This is most likely due to the missing negative feedback from angiotensin II for renin secretion. Plasma renin and noradrenaline are known as important prognostic indicators for CVM in untreated patients. 13 , 14 , 15 , 16 , 17 However, the association between renin or noradrenaline and CV risk in patients receiving antihypertensive treatment is inconsistent across various populations and in different clinical conditions. 18 One of the main findings of our study is that both renin and noradrenaline allow a reliable prediction of CVM in patients with and without RAAS blockers. This extends the current knowledge that renin and noradrenaline are generally independent predictors of CV mortality, which has been extensively discussed in the literature. 19 Interestingly, aldosterone does not provide significant CVM prediction in patients without RAAS blocker therapy, but it does predict CVM in patients on ACE‐inhibitor therapy. This result may fit with the well‐known aldosterone escape, possibly identifying a patient subgroup with worse prognosis. 20
With regard to arterial stiffness, we found, that renin and noradrenaline, but also angiotensin II significantly predict CVM in all PP quartiles. Qualitatively the predictive power seemed to be diminished in the 4th PP quartile, but still was statistically significant (Figure 2). Therefore, these above biomarkers can be used independently from the presence and extent of arterial stiffness. Finally, we also examined the CVM prediction in different subgroups depending both on the antihypertensive therapy (ACE inhibitors vs. no RAAS inhibitors) and additionally in relation to vascular stiffness, given in PP quartiles.
We found that noradrenaline allows a reliable CVM prediction in all PP quartiles in patients with ACE‐inhibition in contrast to patients without any RAAS blocker therapy. This difference needs to be confirmed in further studies and, even might be considered for treatment decisions in hypertension therapy. We could show that renin decreases and ARQ increases with increasing PP, a relationship which seems to be independent of any RAAS blocker therapy. The major explanation for this finding may be that with increasing PP, the SBP also increased considerably and caused a reduction in renin secretion via pressure control of renin release. This hypothesis fits well with the data on low‐renin hypertension. 21 , 22 In addition, adrenaline and noradrenaline increased significantly with increasing PP more in the RAAS inhibitor free group, with noradrenaline missing the significance level in the RAAS blocker group. Thus, catecholamines may increase vascular stiffness and the surrogate parameter PP mainly through their vasoconstrictor effects. 23 , 24 In addition, the reduction in serum uromodulin levels with an increasing PP indicates the onset of renal impairment in the context of hypertensive/vascular nephropathy. This effect appears independent of RAAS blocker therapy and starts in parallel with cystatin C in the “creatinine‐blind area.” This association gains further significance when considering that patients with lower serum uromodulin have a higher risk of developing end stage kidney disease, 25 higher risk for mortality, 8 cardiovascular events and kidney failure in white patients with CKD. 26
4.1. Limitations
In the LURIC study, blood pressure parameters and laboratory values were only evaluated upon inclusion in the study and were not assessed again during the follow‐up period.
CONFLICTS OF INTEREST
Prof. Dr. med. B. K. Krämer reports lecture fees and/or advisory board memberships and/or study participation from Astellas, Bayer, Boehringer Ingelheim, Chiesi, Riepharm, Pfizer, Sanofi, Servier, and Vifor Pharma, all outside the submitted work. He is the past president of the German Hypertension Society DHL. Dr. rer. nat. Marcus E. Kleber is employed by Synlab Holding Deutschland GmbH. Prof. Dr. med. W. März reports grants and personal fees from: Aegerion Pharmaceuticals, AMGEN, Sanofi, Alexion Pharmaceuticals, BASF, Abbott Diagnostics, Numares AG, Berlin‐Chemie, Akzea Therapeutics; grants from: Siemens Healthineers, Astrazeneca, Bayer Vital
GmbH, bestbion dx GmbH, Boehringer Ingelheim Pharma GmbH Co KG, Immundiagnostik
GmbH, Merck Chemicals GmbH, MSD Sharp and Dohme GmbH, Novartis Pharma GmbH, Olink
Proteomics, other from Synlab Holding Deutschland GmbH, all outside the submitted work. The remaining authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
Babak Yazdani and Bernhard K. Krämer designed the research, wrote the first draft of the manuscript, did the literature research and interpreted the results. Marcus Kleber and Graciela Delgado made statistical analyses. Gökhan Yücel and Jan Leipe helped to discuss the results and thereby made essential contributions to the manuscript. All other authors proofread the manuscript and made important intellectual contributions. Winfried März is principal investigator of the LURIC study and was instrumental in performing of the study and providing the data for analysis.
ACKNOWLEDGMENTS
We thank all participants of the LURIC study, as well as the study team who were either temporarily or permanently involved in patient recruitment as well as sample and data handling. We also thank the laboratory staff at the Ludwigshafen General Hospital and the Universities of Freiburg, Ulm, and Heidelberg in Germany. The work of G.E.D. was supported by the European Union's Horizon 2020 research and innovation programme under ERA‐Net Cofund action N° 727565 (OCTOPUS project) and the German Ministry of Education and Research (grant number 01EA1801A). The funding sources were not involved in the study design; collection, analysis and interpretation of data; writing of the report; or the decision to submit the article for publication.
Yazdani B, Delgado GE, Kleber ME, et al. The renin‐angiotensin‐aldosterone system, neurohumoral axis and cardiovascular mortality in LURIC. J Clin Hypertens. 2022;24:1587–1597. 10.1111/jch.14593
REFERENCES
- 1. Ferrario CM, Strawn WB. Role of the renin‐angiotensin‐aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98(1):121‐128. [DOI] [PubMed] [Google Scholar]
- 2. Tomaschitz A, Pilz S, Ritz E, et al. Associations of plasma renin with 10‐year cardiovascular mortality, sudden cardiac death, and death due to heart failure. Eur Heart J. 2011;32(21):2642‐2649. [DOI] [PubMed] [Google Scholar]
- 3. Tomaschitz A, Pilz S, Ritz E, et al. Independent association between 1,25‐dihydroxyvitamin D, 25‐hydroxyvitamin D and the renin‐angiotensin system: the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin Chim Acta. 2010;411(17‐18):1354‐1360. [DOI] [PubMed] [Google Scholar]
- 4. Inoue K, Goldwater D, Allison M, et al. Serum aldosterone concentration, blood pressure, and coronary artery calcium: the multi‐ethnic study of atherosclerosis. Hypertension. 2020;76(1):113‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tamm I, Horsfall FL. Characterization and separation of an inhibitor of viral hemagglutination present in urine. Proc Soc Exp Biol Med. 1950;74(1):106‐108. [PubMed] [Google Scholar]
- 6. Torffvit O, Melander O, Hultén UL. Urinary excretion rate of Tamm‐Horsfall protein is related to salt intake in humans. Nephron Physiol. 2004;97(1):p31‐6. [DOI] [PubMed] [Google Scholar]
- 7. Pak J, Pu Y, Zhang ZT, Hasty DL, Wu XR. Tamm‐Horsfall protein binds to type 1 fimbriated Escherichia coli and prevents E. coli from binding to uroplakin Ia and Ib receptors. J Biol Chem. 2001;276(13):9924‐9930. [DOI] [PubMed] [Google Scholar]
- 8. Delgado GE, Kleber ME, Scharnagl H, et al. Serum uromodulin and mortality risk in patients undergoing coronary angiography. J Am Soc Nephrol. 2017;28(7):2201‐2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dawnay AB, Cattell WR. Serum Tamm‐Horsfall glycoprotein levels in health and in renal disease. Clin Nephrol. 1981;15(1):5‐8. [PubMed] [Google Scholar]
- 10. Itano S, Yano Y, Nagasu H, et al. Association of arterial stiffness with kidney function among adults without chronic kidney disease. Am J Hypertens. 2020;33(11):1003‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mancusi C, Losi MA, Izzo R, et al. Higher pulse pressure and risk for cardiovascular events in patients with essential hypertension: the Campania Salute Network. Eur J Prev Cardiol. 2018;25(3):235‐243. [DOI] [PubMed] [Google Scholar]
- 12. Winkelmann BR, März W, Boehm BO, et al. Rationale and design of the LURIC study–a resource for functional genomics, pharmacogenomics and long‐term prognosis of cardiovascular disease. Pharmacogenomics. 2001;2(1):S1‐73. [DOI] [PubMed] [Google Scholar]
- 13. Reuben DB, Talvi SL, Rowe JW, Seeman TE. High urinary catecholamine excretion predicts mortality and functional decline in high‐functioning, community‐dwelling older persons: macArthur Studies of Successful Aging. J Gerontol A Biol Sci Med Sci. 2000;55(10):M618‐24. [DOI] [PubMed] [Google Scholar]
- 14. Christensen NJ, Schultz‐Larsen K. Resting venous plasma adrenalin in 70‐year‐old men correlated positively to survival in a population study: the significance of the physical working capacity. J Intern Med. 1994;235(3):229‐232. [DOI] [PubMed] [Google Scholar]
- 15. Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311(13): 819‐823. [DOI] [PubMed] [Google Scholar]
- 16. Daimon M, Konta T, Oizumi T, et al. Higher plasma renin activity is a risk factor for total mortality in older Japanese individuals: the Takahata study. Metabolism. 2012;61(4):504‐511. [DOI] [PubMed] [Google Scholar]
- 17. Parikh NI, Gona P, Larson MG, et al. Plasma renin and risk of cardiovascular disease and mortality: the Framingham Heart Study. Eur Heart J. 2007;28(21):2644‐2652. [DOI] [PubMed] [Google Scholar]
- 18. Volpe M, Unger T. Plasma renin and cardiovascular risk: what is the evidence for an association? Cardiology. 2013;125(1):50‐59. [DOI] [PubMed] [Google Scholar]
- 19. Wallentin L, Eriksson N, Olszowka M, et al. Plasma proteins associated with cardiovascular death in patients with chronic coronary heart disease: a retrospective study. PLoS Med. 2021;18(1). 10.1371/journal.pmed.1003513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rubel D, Zhang Y, Sowa N, Girgert R, Gross O. Organoprotective effects of spironolactone on top of ramipril therapy in a mouse model for Alport syndrome. J Clin Med. 2021;10(13):2958. 10.3390/jcm10132958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon RD, Laragh JH, Funder JW. Low renin hypertensive states: perspectives, unsolved problems, future research. Trends Endocrinol Metab. 2005;16(3):108‐113. [DOI] [PubMed] [Google Scholar]
- 22. Laragh JH. Two forms of vasoconstriction in systemic hypertension. Am J Cardiol. 1987;60(12):93g. [DOI] [PubMed] [Google Scholar]
- 23. Nardone M, Floras JS, Millar PJ. Sympathetic neural modulation of arterial stiffness in humans. Am J Physiol Heart Circ Physiol. 2020;319(6):H1338. [DOI] [PubMed] [Google Scholar]
- 24. Petrák O, Strauch B, Zelinka T, et al. Factors influencing arterial stiffness in pheochromocytoma and effect of adrenalectomy. Hypertens Res. 2010;33(5):454‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lv L, Wang J, Gao B, et al. Serum uromodulin and progression of kidney disease in patients with chronic kidney disease. J Transl Med. 2018;16(1):316. 10.1186/s12967-018-1693-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steubl D, Schneider MP, Meiselbach H, et al. Association of serum uromodulin with death, cardiovascular events, and kidney failure in CKD. Clin J Am Soc Nephrol. 2020;15(5):616‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]