

Abstract
Elevated levels of cytokines IL-1β and IL-6 are associated with severe COVID-19. Investigating the underlying mechanisms, we find that while primary human airway epithelia (HAE) have functional inflammasomes and support SARS-CoV-2 replication, they are not the source of IL-1β released upon infection. In leukocytes, the SARS-CoV-2 E protein upregulates inflammasome gene transcription via TLR2 to prime, but not activate, inflammasomes. SARS-CoV-2-infected HAE supply a second signal, which includes genomic and mitochondrial DNA, to stimulate leukocyte IL-1β release. Nuclease treatment, STING, and caspase-1 inhibition but not NLRP3 inhibition blocked leukocyte IL-1β release. After release, IL-1β stimulates IL-6 secretion from HAE. Therefore, infection alone does not increase IL-1β secretion by either cell type. Rather, bi-directional interactions between the SARS-CoV-2-infected epithelium and immune bystanders stimulates both IL-1β and IL-6, creating a pro-inflammatory cytokine circuit. Consistent with these observations, patient autopsy lungs show elevated myeloid inflammasome gene signatures in severe COVID-19.
Keywords: inflammasome, SARS-CoV-2, IL-6, IL-1β, NLRP3, NLRP1, AIM2, STING, TLR2
Graphical abstract
IL-1β and IL-6 are increased in severe COVID-19. Examining the underlying mechanisms, Barnett et al. show that SARS-CoV-2 E protein primes, and DNA from infected epithelial cells activates, inflammasome-dependent IL-1β secretion from leukocytes, which in turn stimulates IL-6 release from epithelial cells.
Introduction
The emergence of SARS-CoV-2 caused the COVID-19 pandemic that continues to cause global economic disruption, morbidity, and mortality. COVID-19 presentations vary widely, ranging from asymptomatic to fatal disease. Patient characteristics, including age and underlying medical conditions, increase the risk for severe COVID-19,1 a disease that reflects significant immune dysfunction2 , 3 , 4 and can lead to fatalities through a hyperinflammatory cytokine storm.5 , 6 Severe COVID-19 is associated with high levels of pro-inflammatory cytokines7 , 8 and an influx of blood-derived myeloid populations into the lung.9 , 10 Recent clinical trial data underscore the relevance of interleukin (IL)-1 and IL-6 in severe COVID-19 because treatment with either IL-1 or IL-6 receptor antagonists increases patient survival and decreases the duration of hospital and intensive care unit stays.11 , 12 , 13 Therefore, identifying how SARS-CoV-2 stimulates these cytokines is essential to understanding and treating severe COVID-19.
One source of IL-1 in COVID-19 is activation of the inflammasome, an innate immune sensory machinery that initiates IL-1β and IL-18 secretion and pyroptotic cell death.14 IL-1β, IL-18, and lactate dehydrogenase (LDH), a protein released from pyroptotic cells, are increased in severe COVID-19 patients.15 , 16 These cytokines may also contribute to COVID-19-associated neurological symptoms.17 , 18 While some suggested targeting the inflammasome in severe COVID-19,19 this pathway may have a protective role, as its loss increases morality to other respiratory viruses.20 , 21 , 22 Such a contrast in potential functions warrants investigation of how the inflammasome is activated by SARS-CoV-2 and how its activity is altered between COVID-19 disease states.
Many viruses induce inflammasome formation,23 a two-step process. A signal I, typically provided by Toll-like receptor (TLR) agonists or cytokines, primes a cell for inflammasome activation by upregulating inflammasome-related genes and inducing post-translational modifications to component proteins.24 During infection, viral nucleic acids or surface proteins may activate TLRs to supply this signal I.25 , 26 Then, a cytosolic signal II nucleates the inflammasome through activation of one of many sensors, including nucleotide-binding leucine-rich-repeat (NLR) family pyrin-domain-containing 3 (NLRP3), and absent in melanoma 2 (AIM2).14 Activated inflammasome sensors interact with the adaptor protein apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD) (ASC) and caspase-1 to form the canonical inflammasome. Then, caspase-1 mediates maturation and secretion of IL-1β and IL-18 and processing of gasdermin D (GSDMD) to form a plasma membrane pore, facilitating pyroptotic cell death.27 , 28 , 29 , 30
Inflammasome activation occurs in the cytosol, therefore whether a virus acts as a signal II depends on its ability to enter the responding cell and replicate. Some viruses, like influenza A virus (IAV), infect a wide range of cell types, including myeloid cells enriched in inflammasome pathway expression,31 and indeed, IAV replication activates the NLRP3 inflammasome in myeloid cells.20 , 21 , 22 SARS-CoV-2, however, requires angiotensin converting enzyme 2 (ACE2) for entry,32 , 33 limiting its cellular tropism. Because myeloid cells do not express ACE2,34 , 35 , 36 inflammasome activation by SARS-CoV-2 replication may be limited to ACE2+ cell types or infection may activate the inflammasome indirectly with damage-associated molecular patterns (DAMPs) generated as a by-product of viral replication acting as an inflammasome-nucleating signal II.
Several studies addressed a third possibility, in which SARS-CoV-2 replication directly activates the inflammasome in monocytes despite these hindrances.37 , 38 , 39 , 40 , 41 Reported mechanisms by which SARS-CoV-2 activates the inflammasome vary between studies as does the mechanism of viral entry and replication. One group suggests SARS-CoV-2 is a signal II, requiring an exogenous signal I,38 while another suggests the SARS-CoV-2 spike (S) protein primes inflammasomes, requiring an exogenous signal II.39 Yet, another report indicated SARS-CoV-2 enters monocytes upon addition of anti-SARS-CoV-2 antibodies through Fc receptor Ɣ, initiating inflammasome activation through abortive replication. Monocyte entry and inflammasome activation was limited to a small percentage of cells in culture and not observed with antibodies from vaccinated patients or healthy individuals.40 Two other studies suggest SARS-CoV-2 is both signals I and II in myeloid cells. One did not describe a viral entry mechanism,37 while another reported ACE2 on human lung macrophages mediates viral entry,41 an observation contrasting the findings of several other groups.34 , 35 , 36 The conflicting nature of these reports mandates further investigation. Though previous work focused on SARS-CoV-2 infection in monocytes, viral replication occurs predominantly in the airway and nasal epithelium,42 and one group suggests viral protease activity both activates and inhibits inflammasomes in the airway epitheilum.43 However, to date, no studies have addressed contributions of the epithelium in concert with immune cells to generate an inflammatory milieu, despite enhanced immune-epithelial interactions in severe COVID-19.44
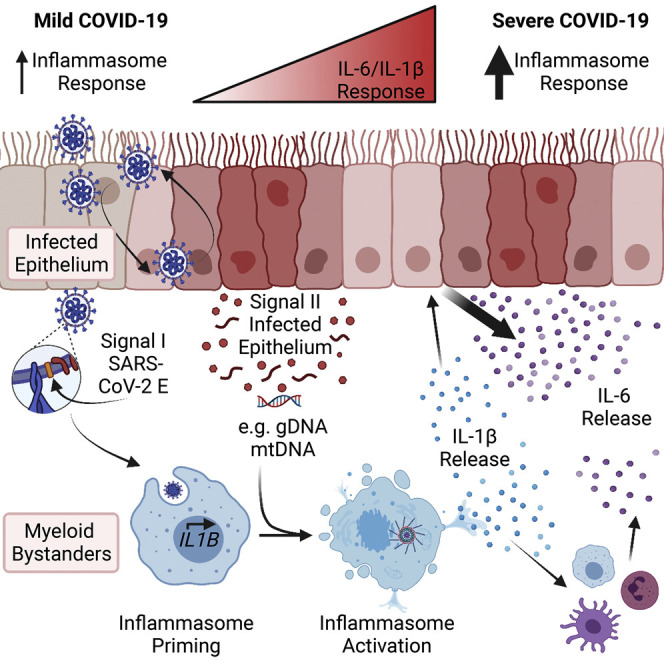
Here, we report primary human airway epithelia (HAE) have functional inflammasomes, support SARS-CoV-2 replication but do not release IL-1β during infection. Instead, myeloid bystanders secrete IL-1β in the presence of infected epithelial cells. The envelope (E) protein on the virion surface primes myeloid cells for inflammasome activation. The infected epithelium releases double-stranded DNA (dsDNA), a well-characterized inflammasome agonist,45 , 46 , 47 supplying signal II. Co-culture of primary human leukocytes with SARS-CoV-2-infected cells, but not each cell type in isolation, stimulates IL-1β release. Thus, interactions between the infected epithelium and leukocytes are required for IL-1β production in COVID-19. Supporting these findings, COVID-19 autopsy lungs revealed inflammasome priming and activation in myeloid cells lacking SARS-CoV-2 antigens. Additionally, meta-analysis of bronchioalveolar lavage fluid (BALF) scRNA-seq data revealed that severe COVID-19 patients have high variation in inflammasome gene expression, suggesting loss of regulation of inflammasome priming. These in vivo data corroborate an indirect model of inflammasome activation in COVID-19 and show divergence of inflammasome responses in different COVID-19 patients. Epithelial-immune communication is not limited to inflammasome activation. IL-1β promoted IL-6 release from human leukocytes and HAE, where HAE showed strikingly higher IL-6 production than peripheral blood mononuclear cells (PBMCs). Co-culture of HAE and PBMC during infection promoted IL-1β and IL-1-dependent IL-6 release. Thus, we find that infected epithelial cells drive inflammasome activation in myeloid bystander cells to secrete IL-1β, which in turn promotes IL-6 release from both cell types, creating a pro-inflammatory circuit in COVID-19.
Results
HAE can activate inflammasome-mediated IL-1β release but do not in response to SARS-CoV-2 infection
We first evaluated inflammasome activation by SARS-CoV-2 infection in ACE2+ epithelial cells. Because SARS-CoV-2 infection is species specific and immortalized cell lines often lack innate immune sensors,48 we utilized well-differentiated, primary HAE grown at an air-liquid interface (ALI), a culture system that recapitulates the pseudostratified epithelium of human airways.49 Previous work demonstrated SARS-CoV-2 replication in the ciliated cells of these cultures,42 making them an ideal model to study the intracellular innate immune response to SARS-CoV-2 infection.
While inflammasomes are characterized thoroughly in myeloid cells, mechanisms of inflammasome activation in the human airway epithelium are not well understood. To characterize inflammasomes in HAE, we exposed these cultures to multiple priming and activation stimuli and assessed IL-1β secretion (Figure 1 A). HAE were primed by exposure to the triacylated lipopeptide Pam3CysSerLys4 (Pam3-CSK4), a TLR1/2 agonist, or the dsRNA analog polyinosinic-polycytidylic acid (p(I:C)), a TLR3 agonist,25 and then were exposed to signal II agonists, including nigericin, an NLRP3 inflammasome agonist, the DPP8/9 inhibitor Val-boroPro, an NLRP1 and CARD8 agonist, transfected poly(deoxyadenylic-deoxythymidylic) acid (p(dA:dT)), an AIM2 ligand, or transfected flagellin, a NAIP/NLRC4 agonist.14 , 24 , 50 Val-boroPro but not nigericin stimulated IL-1β release from HAE (Figure 1A), while p(dA:dT) and flagellin stimulated modest responses slightly above baseline. Measurement of cell membrane rupture showed LDH release in conditions stimulating IL-1β secretion, suggesting pyroptotic cell death occurs upon inflammasome activation in HAE (Figure S1A). Apical secretion of both IL-1β and IL-18 was observed after stimulation with Pam3-CSK4 and Val-boroPro (Figure 1B). These results demonstrate HAE can execute pyroptosis and IL-1β secretion and highlight the role of the NLRP1/CARD8 inflammasome in these cultures.
Figure 1.

Primary, well-differentiated HAE form inflammasomes and secrete IL-1β but not in response to SARS-CoV-2 infection
(A) IL-1β in pooled HAE supernatants following treatment with indicated stimuli ± transfection (TFX).
(B) IL-1β and IL-18 in HAE supernatants following stimulation.
(C) IL-1β in HAE supernatants following SARS-CoV-2 infection.
(D) Viral titer from HAE apical wash following SARS-CoV-2 infection.
(E) Viral N RNA expression relative to ACTB.
(F) IL1B and IL18 expression relative to ACTB.
(G) Apical IL-1β secretion from HAE following SARS-CoV-2 infection or stimulation.
(H) Viral titer from HAE apical wash following infection.
(I) LDH in HAE apical wash following SARS-CoV-2 infection. Data points represent individual HAE donors with bars shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, or ∗∗∗∗p < 0.0001 calculated by ordinary one-way (A and I) or two-way (B and D) ANOVA followed by Dunnett’s (A and I) or Tukey’s (B and D) multiple comparison test.
See also Figure S1.
We next assessed whether SARS-CoV-2 infection stimulates IL-1β secretion from HAE. HAE were infected with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.5, and IL-1β secretion was measured after infection. IL-1β release above baseline was not detected in HAE apical washes or basolateral supernatants at 24 or 72 h post-infection (HPI) (Figure 1C). However, viral titers measured in parallel indicated viral replication (Figure 1D), as did measurement of viral RNA transcripts encoding the SARS-CoV-2 nucleocapsid (N) protein (Figure 1E). Additionally, inflammasome cytokines or components expression did not change with infection (Figures 1F, S1B, and S1C). Increasing the MOI or priming HAE cultures prior to infection did not alter this response (Figure 1G). No IL-1β secretion occurred under these conditions, despite viral replication at all time points (Figure 1H). However, SARS-CoV-2 infection did stimulate lytic cell death, as measured LDH release (Figure 1I). Thus, SARS-CoV-2 infection alone or with priming does not promote IL-1β release from ACE2+ HAE.
SARS-CoV-2 infection in the epithelium indirectly activates the inflammasome in leukocytes
SARS-CoV-2 may activate the inflammasome in monocytes and other myeloid cells, despite not stimulating this response in HAE. This may occur directly, through viral replication, or indirectly, through detection of virions and DAMPs generated by viral replication in the epithelium. While leukocytes are not thought to support viral replication, recent reports have suggested various models of inflammasome activation in leukocytes,38 , 39 , 40 including direct activation of the inflammasome through viral replication.37 , 41
Thus, we first assessed whether SARS-CoV-2 productively replicated in human PBMCs following infection at MOI 0.5. Between 4 and 24 HPI, viral titers present in infected PBMC culture supernatants decreased on average 10-fold (Figure 2 A). Similarly, viral RNA transcripts decreased over time (Figure 2B). In contrast, infection in ACE2+ HAE show log-fold increases in viral titer and RNA following infection (Figures 1D and 1E). Decreases in viral titer and RNA over time suggest productive viral replication does not occur in human PBMCs.
Figure 2.

SARS-CoV-2 primes the inflammasome in PBMCs, while co-culture of PBMCs with infected epithelial cells fully activates inflammasome-mediated IL-1β secretion
(A) Viral titer from PBMC supernatants.
(B) Viral N RNA expression in PBMC relative to ACTB.
(C) IL-1β release from PBMC supernatants following SARS-CoV-2 infection or stimulation.
(D) IL1B transcript expression relative to ACTB.
(E) Indicated transcript expression relative to ACTB after 4-h SARS-CoV-2 exposure.
(F) IL-1β in PBMC supernatants following SARS-CoV-2 exposure alone or in combination with indicated signal I or II stimuli.
(G) IL1B transcript expression relative to ACTB in PBMC at 4 HPI with SARS-CoV-2 ± Bay11-7082 (10 μM), chloroquine (10 μM), and C29 (25 μM).
(H) IL1B expression relative to ACTB following 3-h treatment with 1 μg/mL S, E, or Pam3-CSK4.
(I) IL-1β secretion from PBMCs following 3-h treatment described in (H) and subsequent ATP treatment (5 mM; 1 h).
(J) PCR-amplified detection of dsDNA in HAE supernatants ± SARS-CoV-2 (MOI 0.5) at 24 HPI.
(K and L) IL-1β in supernatants after infection in PBMCs, Vero-E6 cells, or co-culture of PBMCs with infected Vero-E6 cells (K) ± benzonase (20 units/mL) or H151 (10 μM) and (L) ± VX-765 (20 μg/mL) or MCC950 (5 μM). Data point color indicates specific PBMC donors and matched Vero-E6 values across conditions. Data points on graphs represent individual PBMC donors with bars or lines shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, or ∗∗∗∗p < 0.0001 as calculated by ordinary one-way (E, F, H, and I) or two-way (A, D, G, K, and L) ANOVA followed by Tukey’s (A, D, F, K, and L), Dunnet’s (E, H, and I), or Sidak’s (G) multiple comparison test.
See also Figure S2.
While NLRP3 stimulation promoted IL-1β secretion in PBMCs, SARS-CoV-2 infection did not (Figure 2C). IL1B mRNA expression, however, was increased at 4 HPI, indicating inflammasome priming (Figure 2D). IL1B mRNA expression stimulated by SARS-CoV-2 was more pronounced at MOI 2, and measurement of other inflammasome-related genes showed increases in AIM2 and NLRP3 transcripts (Figures 2E and S2A). Thus, SARS-CoV-2 uptake by PBMCs primes multiple inflammasomes.
Next, we systematically tested whether SARS-CoV-2 was a signal I, signal II, or both for inflammasome activation in human PBMCs, measuring IL-1β secretion after SARS-CoV-2 exposure alone or in combination with exogenous signal I or signal II stimuli. Accordingly, PBMCs were pre-treated with a signal I (Pam3-CSK4) and then exposed to SARS-CoV-2, exposed to SARS-CoV-2 alone, or pre-exposed to SARS-CoV-2 and then treated with a signal II (ATP or nigericin). SARS-CoV-2 exposure alone or with a signal I resulted in little IL-1β secretion (Figure 2F). By contrast, infection followed by signal II treatment led to high levels of IL-1β (>2,000 pg/mL on average) secretion. Therefore, SARS-CoV-2 predominantly serves as a signal I for inflammasome activation in primary human PBMCs.
We next investigated which SARS-CoV-2 virion components mediate inflammasome priming. Priming in PBMCs, measured by IL1B transcript upregulation, was sensitive to Bay11-7082, an NF-κΒ inhibitor, and chloroquine, an inhibitor of endosomal/phagosomal acidification, suggesting that these pathways are required for priming (Figure 2G). IL1B transcript upregulation was also inhibited by the TLR2 inhibitor C29 (Figure 2G), consistent with reports suggesting TLR2 is required for the pro-inflammatory response to SARS-CoV-2.26 , 39 , 51 Because TLR2 blockade decreased IL1B transcript upregulation, we determined whether the SARS-CoV-2 structural proteins S or E stimulated this response, as both are reported to stimulate TLR2.26 , 39 , 51 We observed upregulation of IL1B mRNA by E, while S did not (Figure 2H). Consistent with a role in priming, E, but not S, stimulated IL-1β secretion in combination with subsequent ATP treatment (Figure 2I). Thus, E primes inflammasomes via TLR2 stimulation, consistent with another report.26
If SARS-CoV-2 is primarily a signal I in PBMCs, then inflammasome activation in PBMCs requires an additional signal II. DAMPs released from infected epithelial cells may provide this signal. Genomic DNA encoding long-interspersed nuclear elements (LINE-1) and mitochondrial (mt)DNA encoding mitochondrial genes (MT-ND4 and MT-CYB) were increased in SARS-CoV-2-infected HAE supernatants (Figures 2J and S2B). dsDNA is a well-characterized inflammasome agonist that stimulates the AIM2 and/or NLRP3 inflammasomes, mediated by the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway in humans.45 , 46 , 47 Its presence in infected HAE supernatants confirms infected cells release inflammasome-stimulatory DAMPs. Therefore, infected epithelial cells produce both signal I (virions) and signal II (dsDNA) to activate the inflammasome in leukocytes.
To model inflammasome activation through cell-cell interaction, we infected or mock-infected the epithelial cell line Vero-E6 with SARS-CoV-2, and 72 HPI, PBMCs were co-cultured with infected or mock-infected cells for 24 h (Figure S2C). Similar to HAE, Vero-E6 cells support SARS-CoV-2 replication (Figure S2D). As a negative control, PBMCs were exposed or mock-exposed to SARS-CoV-2 for 24 h. Only PBMCs co-cultured with infected Vero-E6 cells secreted IL-1β (Figure 2K), indicating infected cells, not virions alone, promote de novo inflammasome activation in PBMCs. The extent of this response varied between PBMC donors. Addition of benzonase, a nuclease with activity in a wide range of buffers, abrogated IL-1β release (Figure 2K). This response was also sensitive to H151, an inhibitor of STING, an adaptor protein critical for the interferon and inflammasome responses to intracellular DNA.45 , 46 Thus, DNA DAMPs are essential for SARS-CoV-2-mediated inflammasome activation in co-culture.
We then tested the requirement of specific inflammasome components in this system. This response was sensitive to VX-765, a human caspase-1/4 inhibitor, confirming inflammasome-dependent secretion (Figure 2L). However, this response was reduced modestly but not significantly by NLRP3 inhibition, suggesting other or multiple inflammasomes mediate IL-1β release in co-culture. Taken together, these data suggest interactions with SARS-CoV-2-infected epithelial cells, rather than virions alone, stimulate inflammasomes in leukocytes through sensing of DNA DAMPs.
Myeloid cells from COVID-19 autopsy lungs have active inflammasomes but not SARS-CoV-2 antigens
Next, we investigated COVID-19 patient autopsy lungs for inflammasome priming and activation in the alveoli. First, we examined inflammasome priming by RNA in situ hybridization (RNA-ISH). Compared with control lungs from donors without pre-existing pulmonary disease, COVID-19 patient lungs exhibited increased expression of NLRP3, AIM2, and CASP1 (Figure 3 A), driven in part by the influx on myeloid populations into COVID-19 lungs. Consistent with a role in the pulmonary epithelium, NLRP1 expression in COVID-19 autopsy lungs was comparable to controls (Figure 3A). To assess which cell types expressed these inflammasome components, we utilized fluorescent RNA-ISH to label these transcripts in combination with CD68, expressed by macrophages,52 and MPO, expressed by neutrophils.53 CASP1, NLRP3, and AIM2 expressions were primarily associated with CD68+ MPO − cells, suggesting that these genes are predominantly expressed in macrophage populations in COVID-19 autopsy lungs (Figure 3B). NLRP1, however, was enriched in CD68− MPO − cells (Figure 3B). Multiple epithelial cell types have the NLRP1 inflammasome,54 , 55 and these data indicate its predominance in non-myeloid cell types in the lung. Overall, this increase in inflammasome component gene expression suggests the NLRP3 and AIM2 inflammasomes are primed in myeloid populations in COVID-19 autopsy lungs.
Figure 3.

Myeloid cells in COVID-19 autopsy lungs show hallmarks of inflammasome priming and activation primarily in cells lacking SARS-CoV-2 antigen
(A) Representative images (left) and quantification (right) of RNA-ISH in control and COVID-19 autopsy lungs.
(B) Representative images (left) and quantification (right) of fluorescent dual RNA-ISH labeling indicated inflammasome genes with CD68 or MPO.
(C) Representative maximum Z projection (left) and quantification (right) of ASC immunostaining in control or COVID-19 autopsy lungs.
(D) Representative images of ASC specks with cell-type markers in COVID-19 autopsy lungs (left) and quantifications (right).
(E) Representative images of COVID-19 autopsy lungs of ASC specks in combination with SARS-CoV-2 antigen and quantification (right). All scale bars represent 20 μm. Data points on graphs represent individual donor lungs with bars shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, or ∗∗∗∗p < 0.0001 calculated by Student’s two-tailed, unpaired t test.
See also Figure S3.
Increased inflammasome gene expression indicates inflammasome priming by signal I, but not activation by signal II. Therefore, we investigated ASC speck formation in these lungs by immunohistochemistry because this adaptor protein forms a large, perinuclear speck upon inflammasome activation and oligomerization.56 Compared with controls, COVID-19 autopsy lungs had an increased frequency of ASC specks, showing inflammasome activation in vivo (Figure 3C). These specks were present in several myeloid populations, including CD68+ and MPO+ cells, but most predominantly in CD11c+ cells, a marker of many myeloid subpopulations including dendritic cells (DCs) and alveolar macrophages57 (Figure 3D). To assess contributions of the alveolar epithelium, we investigated ASC speck formation in ABCA3+ alveolar type II cells, which support SARS-CoV-2 replication in the alveoli,42 and found ASC specks were largely absent from ABCA3+ cells (Figure S3A).
Previous reports note SARS-CoV-2 antigens in myeloid cells.38 , 40 , 41 However, the relative proportions of ASC speck+ myeloid cells containing viral antigens at the site of infection in humans remains unclear. Therefore, we determined whether cells with ASC specks also contained SARS-CoV-2 antigens in COVID-19 autopsy lungs. On average, over 80% ASC speck+ cells lacked SARS-CoV-2 antigens (Figure 3E). Furthermore, CD68+ and MPO+ cells infrequently colocalized with SARS-CoV-2 antigen (Figure S3B), though some myeloid cells do contain SARS-CoV-2, corroborating previous observations.38 , 40 , 41 Because many myeloid cells are professional phagocytes, uptake of infected cells and virions containing SARS-CoV-2 antigens during infection is expected and not confirmatory of viral infection or replication. These data suggest uninfected myeloid bystanders mediate a majority of the inflammasome response in fatal COVID-19.
scRNA-seq analysis of patient BALF samples reveals a heightened but discordant inflammasome response in severe COVID-19
Active inflammasomes in COVID-19 autopsy lungs demonstrates responses in fatal disease but does not allow for comparison with mild disease. Therefore, we conducted a meta-analysis of COVID-19 patient scRNA-seq data to determine whether COVID-19 disease states showed differential inflammasome priming.9 , 58 After filtering, we analyzed 117,497 high-quality transcriptomes from the BALF of three healthy donors, five mild/moderate COVID-19 patients, and 26 severe COVID-19 patients. We utilized shared nearest neighbor, a graph-based clustering method, to identify unique cell populations and visualized the results via uniform manifold approximation and projection (UMAP) analysis (Figure 4 A). To boost statistical power and incorporate new studies, we projected cells from additional COVID-19 and healthy patient scRNA-seq datasets onto this reference space (Figure S4A).59 , 60 , 61 In total, we analyzed over 300,000 high-quality transcriptomes from the BALF of 13 healthy donors, five mild/moderate COVID-19 patients, and 50 severe COVID-19 patients.
Figure 4.

Meta-analysis of COVID-19 patient BALF scRNA-seq reveals differential expression of inflammasome pathway by disease state
(A) UMAP of healthy and COVID-19 patient BALF scRNA-seq from pooled studies (healthy: n = 3; mild/moderate COVID-19: n = 5; severe COVID-19: n = 26). Each dot represents a cell, and each dot is colored according to cluster identity. Clusters are annotated by cell type. ‡ denotes the HLA-DRlow myeloid population.
(B) UMAP shown in (A) but colored by patient disease state (healthy, mild/moderate, or severe).
(C) Inflammasome scores for each cluster of the UMAP shown in (A). General cluster identity indicated below cluster number: M, myeloid cells; T, T cells; B, B cells, and E, epithelial cells.
(D) Inflammasome scores for all myeloid cells in COVID-19 disease states and healthy patients. Each dot represents a cell.
(E) Inflammasome scores for individual cells (individual points) within each myeloid cluster across COVID-19 disease states.
(F) Inflammasome score variance within each myeloid cluster by disease state. (C)–(F) include projected datasets. Statistical calculations are described in the STAR Methods and text, except for (F) which was determined by paired two-sided t test ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, or ∗∗∗∗p < 0.0001.
See also Figure S4.
We identified 22 unique cell clusters with varied distribution by disease state (Figures 4B and S4B), including 11 unique myeloid populations (Figure 4A). Epithelial cells were predominantly observed in COVID-19 patients, not healthy individuals, and present mostly in severe patients (Figure 4B), likely due to dissociation of infected cells from the lung epithelium. Distribution of myeloid cells within specific clusters shifted between disease states, suggesting myeloid characteristics change with COVID-19 disease status. HLA-DRlow myeloid cells were enriched (cluster 2; Figures 4A and 4B) in severe patients, a population previously observed in severe COVID-19.4 , 10 , 62
To gain a general understanding of the inflammasome pathway, we assigned each cell an “inflammasome score” based inflammasome-related gene expression (lL1B, IL18, NLRP1, NLRP3, NLRC4, NLRP6, AIM2, NAIP, MEFV, CASP1, CASP4, CASP5, PYCARD, and GSDMD), a technique used to define cell pathway programs in other scRNA-seq datasets63 (Figure 4C). To control for cell-specific effects, these scores were normalized by subtracting a control score defined by a gene set with comparable expression distribution, creating log10 inflammasome scores ranging between −0.84 and 1.51.64 Across all clusters, the mean inflammasome score was −0.05. The myeloid cluster 14 had the highest mean inflammasome score of 0.03926 with increased expression levels of IL1B, CASP1, and CASP4 compared with other clusters (Figures 4C and S4C). Other myeloid clusters with heightened IL1B expression, including the HLA-DRlow cluster 2, also had high inflammasome scores. In contrast, clusters 18, ciliated epithelial cells, 12, secretory epithelial cells, and 9, T cells, had the lowest inflammasome scores of −0.262, −0.163, and −0.193, respectively. Myeloid cells from both mild/moderate and severe COVID-19 patients tended to have increased inflammasome scores compared with healthy donors, indicating inflammasome priming occurs universally in COVID-19 (Figure 4D). Thus, myeloid populations, including those expanded in severe disease, dictate the inflammasome response in COVID-19.
The range of inflammasome scores within individual clusters in severe disease varied widely (Figure 4E). This was most apparent in the HLA-DRlow cluster 2 where inflammasome scores in severe patients encompassed the range of the entire dataset. We found inflammasome score variance increased stepwise by disease state with a mean of 0.01007 in healthy donors, 0.02955 in mild/moderate COVID-19, and 0.04140 in severe COVID-19 (Figure 4F). In myeloid clusters 14 and 15, variance significantly increased between mild/moderate and severe COVID-19, and inflammasome score variance was significantly increased in every myeloid cluster between healthy donors and severe COVID-19 patients (Figure S4D). Thus, severe COVID-19 showed higher variance of inflammasome scores compared with other disease states. Many cells from severe patients showed dramatically higher or lower scores than observed in mild/moderate disease. This disparate inflammasome response may indicate a loss of regulation of the inflammasome in severe COVID-19.
Given such profound heterogeneity, comparisons of mean inflammasome scores between disease states using linear mixed models approach showed no apparent differences. However, the wide range of inflammasome scores in severe disease indicates subpopulations of myeloid cells may have increased inflammasome gene expression. To compare the 75th percentile of inflammasome scores in each group, we employed a linear quantile mixed model to determine if the 75th quantile of inflammasome score changed between disease states and found significant disease effects in the 75th quantile of inflammasome scores (Figure 4D). Overall, the differences observed in inflammasome score variance between disease states highlight the heterogeneity inflammasome gene expression in severe COVID-19, while increased expression of these genes in the 75th quantile of severe patient myeloid cells indicates a subset of cells in severe disease have exacerbated inflammasome gene expression not observed in other disease states. This priming was only partially dependent on SARS-CoV-2 transcripts, since only a modest correlation between viral genomes and inflammasome score was observed (Figure S4E).
We next examined expression of individual genes. As described above, severe COVID-19 has high variance in inflammasome gene expression levels. Variation within samples (intra-patient variance) and variation between samples (inter-patient variance) contribute to this phenomenon. A major factor contributing to variation within samples is large numbers of gene dropout (zero RNA read count for some genes in some cells), an occurrence universally present in scRNA-seq datasets. However, a myriad of clinical and environmental factors, including age, gender, pre-existing conditions, batch preparation conditions, and course of treatment, contribute to inter-patient variance. These differences cannot be properly controlled with the small number of patients surveyed at single-cell levels. Therefore, we utilized a linear quantile mixed model treating sample identity as a random effect to identify inflammasome pathway genes differentially regulated by COVID-19 disease state. Because severe COVID-19 pathology involves increased inflammation, we compared inflammasome gene expression levels at the 75th quantile, assessing differences between the highest-expressing cells.
IL1B expression was exacerbated in myeloid cells from severe COVID-19 patients at orders of magnitude higher than mild/moderate COVID-19 (Figure 5 A). While no differences were observed between healthy and mild/moderate COVID-19, the 75th quantile of IL1B expression was increased in severe disease versus healthy controls in several clusters and versus mild/moderate disease in clusters 2 and 14 (Figure 5A). Other genes shared among inflammasome pathways, IL18, CASP1, and GSDMD, had less variable expression though cells with extremely high levels of expression were observed in severe COVID-19 (Figures S5A–S5C). Thus, a subset of myeloid cells in severe COVID-19 express high levels of shared inflammasome genes.
Figure 5.

Enriched expression of inflammasome genes and cytokines in specific myeloid subpopulations in severe COVID-19
Violin plots of normalized gene expression in each myeloid cluster by disease status. Includes projected data. Each cell shown by an individual dot. Each panel shows a different gene: (A) IL1B, (B) NLRP3, (C) AIM2, and (D) MEFV. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, or ∗∗∗∗p < 0.0001. Statistical calculations described in STAR Methods and text.
See also Figure S5.
Specific inflammasome sensors followed a similar pattern across disease states. NLRP3 showed heightened expression in severe COVID-19 in several myeloid clusters compared with other disease states (Figure 5B). Similarly, the 75th quantile of AIM2 expression was increased in specific myeloid clusters in severe and mild/moderate COVID-19 compared with healthy donors (Figure 5C). MEFV, the gene encoding Pyrin, expression was aggravated in several myeloid clusters in severe COVID-19, showing dynamic changes in both COVID-19 disease states (Figure 5D). Likewise, only myeloid cells in severe COVID-19 showed high levels of NLRP6 expression (Figure S5D). Additionally, the non-canonical caspase CASP4 showed exacerbated expression in some myeloid clusters in severe COVID-19 (Figure S5E).
Our scRNA-seq meta-analysis revealed differential inflammasome pathway expression between COVID-19 disease states. Myeloid cells from patients with mild/moderate COVID-19 showed lower variance in inflammasome gene expression and uniform enrichment of these genes over a healthy baseline. Since the inflammasome is protective in other respiratory viral infections, this uniform response may promote disease resolution in mild/moderate disease. However, in severe disease, myeloid cells showed striking heterogeneity with a subset of cells exhibiting exacerbated expression to levels not observed in mild/moderate disease. Such cells from severe patients may be able to mount a powerful inflammasome cytokine response in severe COVID-19.
SARS-CoV-2 infection in HAE stimulates IL-1β release from PBMC causing IL-6 release from both cell types
Both L-1β and IL-6 are thought to contribute to COVID-19 fatalities. IL-6 secretion can be induced by multiple mechanisms,65 and COVID-19 patients receiving IL-1 receptor blockade therapy have lower levels of circulating IL-6.11 Therefore, we investigated whether inflammasome-mediated IL-1β secretion amplified IL-6 production, too.
We assessed whether SARS-CoV-2 infection induced IL6 expression in HAE and/or PBMCs. At 4 HPI, we saw no increase in IL6 expression in PBMCs (Figure S6A), unlike IL1B measured in parallel (Figure 2D). In HAE, IL6 transcripts did not increase in at 6, 48, or 72 HPI, similar to acute stimulation with Pam3-CSK4 (Figures S6B and S6C). Thus, neither viral replication in epithelial cells nor PBMC uptake of virions stimulates increases in IL6 transcripts at acute time points, suggesting other mechanisms promote IL6 upregulation.
We then investigated whether IL-1β stimulation induced IL6 expression. PBMCs or HAE were treated with 100 pg/mL or 1 ng/mL IL-1β and assessed IL6 expression at 4 and 24 h post-treatment (HPT). IL-1β increased IL6 expression in PBMCs (Figure 6 A), demonstrating that IL-1β, but not SARS-CoV-2 virions, induces IL6 expression in this cell type. However, increases in IL6 expression following IL-1β treatment in HAE were modest (Figure 6B).
Figure 6.

IL-1β stimulates IL-6 secretion from PBMCs and HAE
(A–C) PBMCs or HAE treated with 100 pg/mL or 1 ng/mL IL-1β. (A) IL6 transcripts relative to ACTB in PBMCs (B) IL6 expression relative to ACTB in HAE. (C) IL-6 secretion from pooled HAE supernatants or PBMCs.
(D) Schematic of HAE-PBMC co-culture experiment. Created with BioRender.com.
(E) IL-1β in supernatants from experiment in (D).
(F) IL-6 in supernatants from experiment in (D).
(G) Representative images of RNA-ISH in COVID-19 autopsy lungs. Scale bar represents 5 μm. Data points on graphs represent individual donors (HAE or PBMC) or donor pairs (HAE + PBMC) with bars shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, or ∗∗∗∗p < 0.0001 as calculated by two-way ANOVA followed by Dunnett’s (A and B) or Tukey’s multiple comparison test (C, E, and F).
See also Figure S6.
In parallel, we measured IL-6 secretion. Both PBMCs and HAE secreted IL-6 following IL-1β stimulation, but the extent differed between cell types (Figure 6C). PBMCs treated with IL-1β showed some IL-6 secretion at 24HPT with an average around 1,000 pg/mL. However, pooled supernatants from HAE demonstrated a strong IL-6 response at 24 HPT with an average of over 7, 000 pg/mL (Figure 6C).
These data suggest a model in which SARS-CoV-2 infection in the epithelium triggers inflammasome activation in myeloid bystanders, and in turn the IL-1β released stimulates IL-6 secretion from both cell types. To test this, we developed a primary, human co-culture system of HAE and PBMC and measured IL-1β and IL-6 production during SARS-CoV-2 infection with or without the IL-1 receptor antagonist (IL-1Ra) (Figure 6D). Consistent with our previous observations (Figure 2K), IL-1β secretion only increased upon co-culture of HAE and PBMC during SARS-CoV-2 infection (Figure 6E). Similar to IL-1β, IL-6 secretion only increased during SARS-CoV-2 infection in co-culture (Figure 6F). SARS-CoV-2-stimulated IL-6 production in the co-culture setting depended on IL-1, as IL-1Ra fully blocked IL-6 secretion. This intercellular communication circuit was observed in COVID-19 autopsy lungs, where an IL6+ ABCA3+ alveolar type II epithelial cell was adjacent to a non-epithelial IL1B+ or IL1B+IL6+ cell (Figure 6G, lower panels). Therefore, a bi-directional communication loop between the infected epithelium and immune bystanders promotes production of IL-1β and IL-6 during SARS-CoV-2 infection.
Discussion
Pro-inflammatory cytokines are critical to mount an adaptive immune response and contain infection, yet blockade of IL-1 or IL-6 signaling in patients with severe disease promotes positive outcomes in COVID-19.11 , 12 , 13 , 66 Such contrasting functions underscores the necessity to understand how these cytokines are produced during infection. Here, we report inflammation driven by inflammasome-mediated IL-1β secretion is a product not only of viral infection but also crosstalk between the infected epithelium and myeloid bystanders. Integrated detection of virions and epithelial DAMPs produced during infection activates the inflammasome in myeloid cells. IL-1β release then potentiates IL-6 secretion from both cell types with a potent IL-6 response from the epithelium. Such a circuit of bi-directional cytokine amplification can potentially result in the uncontrolled cytokine storm that accompanies COVID-19 fatalities.
Several groups reported that SARS-CoV-2 replication directly activates the inflammasome in leukocytes,37 , 38 , 39 , 40 , 41 but this requires additional exogenous stimuli like bacterial ligands or non-neutralizing antibodies and still only occurs in a small proportion of cells.38 , 39 , 40 Due to lack of ACE2 expression,34 , 35 , 36 SARS-CoV-2 replication events in leukocytes are infrequent and abortive.40 , 67 Indeed, in the absence of exogenous factors, we did not observe productive viral replication or IL-1β secretion upon SARS-CoV-2 infection in PBMCs. Similarly, HAE, which support productive viral replication and have functional inflammasomes, do not release IL-1β following infection, possibly due to viral antagonism of the pathway.43 Therefore, while SARS-CoV-2 replication can stimulate modest responses under certain conditions, it is insufficient to drive robust IL-1β release. Furthermore, disease kinetics indicate viral replication is not the lone driver of severe COVID-19 pathology, as viral titers wane in late disease while inflammation continues to mount.68 Thus, it is paramount to understand how SARS-CoV-2 infection promotes IL-1β release beyond the direct sensing of viral replicative activity within an infected cell.
Here, we show how cell-cell interactions and DAMPs amplify inflammasome responses during SARS-CoV-2 infection. SARS-CoV-2 virions prime the inflammasome pathway in myeloid cells through TLR2-dependent sensing of the SARS-CoV-2 E protein.26 Then, infected epithelial cells release DAMPs that supply a signal II to initiate IL-1β release. A key DAMP in this process is the well-characterized inflammasome agonist dsDNA. SARS-CoV-2-infected HAE release both genomic and mitochondrial dsDNA, and co-culture of leukocytes with infected epithelial cells promotes IL-1β release, while infection in either cell type in isolation does not. Addition of nuclease or inhibition of STING, an adaptor protein essential for the interferon and inflammasome responses to DNA in humans,45 , 46 abrogated IL-1β release in co-culture. Thus, DNA DAMPs are critical for IL-1β release in this system. Furthermore, while blockade of caspase-1 activity was blocked IL-1β release in this system, NLRP3 inhibition had a modest and insignificant effect. This suggests the AIM2 inflammasome, previously observed in COVID-19 patient samples,40 may also contribute to response. Thus, viral replication in the epithelium provides both signals I and II to stimulate the inflammasome in myeloid bystanders through production of virions and release of DNA DAMPs. Such a model explains how inflammasome activation occurs during early infection in naive individuals and how this response is exacerbated in late disease, as sustained infection leads to significant damage to the lung epithelium.69
Observations in COVID-19 patients corroborated these in vitro findings. Active inflammasomes, evidenced by ASC speck formation, were prominent in multiple myeloid cell types in COVID-19 autopsy lungs. However, these ASC specks were rarely found in epithelial cells and infrequently colocalized with viral antigens. Thus, the bulk of inflammasome activation in fatal COVID-19 occurs in myeloid bystanders, not cells with active viral replication.
Meta-analysis of patient BALF scRNA-seq data permitted high-resolution dissection of inflammasome priming in COVID-19. We found that inflammasome priming occurs universally in COVID-19, but the extent of priming varied between disease states and was exceptionally discordant in severe COVID-19. Mild/moderate COVID-19 patients show modest increases in inflammasome cytokine and component gene expression in myeloid cells. In contrast, priming in severe disease showed striking heterogeneity with high variance in inflammasome-related gene expression, indicative of a loss of regulation. Maximal expression of inflammasome-related genes was significantly increased in severe COVID-19 patients’ myeloid cells, suggesting some cells in severe COVID-19 are hyper-primed for inflammasome activation. Thus, while the inflammasome response is not unique to severe disease, a discordant, hyperactive response may contribute to its pathology. Clinical practices reflect this notion, as effective IL-1 receptor blockade therapy relies on patient indicators of disease severity.11
Epithelial-immune interactions do not only amplify IL-1β release in COVID-19. In COVID-19 patients, IL-1 receptor blockade therapy also decreases circulating IL-6,11 and in vitro, we find that co-culture of infected HAE and PBMCs stimulates IL-6 release in an IL-1-dependent manner. Though both PBMCs and HAE secrete IL-6, HAE secreted much higher amounts, despite less dynamic changes in IL6 mRNA expression. This cautions against relying solely on transcriptional data and indicates a need to understand cytokine release in non-immune cell types.
As a whole, our work underscores the importance of immune-stromal interaction during SARS-CoV-2 infection. Viral infection alone is insufficient to generate robust pro-inflammatory responses in either cell type in isolation, but innate immune sensing of infection in the epithelium by leukocytes promotes production of IL-1β and IL-6. This indirect model of cytokine production offers insight into the heterogeneity observed in patient responses, as patient-derived factors, namely DAMPs, contribute to both the IL-1β and IL-6 responses in COVID-19. Such a model may extend to other viral infections with differential pro-inflammatory responses in patients. Indeed, epithelial-immune crosstalk may govern many aspects of the local immune response to viruses, mandating further study of these interactions during infection and how they are altered in high-risk patient groups.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-ASC | Adipogen | RRID: AB_2885200 |
| Goat Anti-Myeloperoxidase | R&D Systems | RRID: AB_2250866 |
| Mouse Anti-human CD68 | Agilent Dako | RRID: AB_2074844 |
| Rabbit Anti-human CD68 | Cell Signaling Technology | RRID: AB_2799882 |
| Mouse Anti-ABCA3 | Seven Hills Bioreagents | RRID: AB_577285 |
| Mouse Anti-human CD11c | ThermoFisher | RRID: AB_2881379 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | RRID: AB_2535792 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | RRID: AB_2534102 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | ThermoFisher | RRID: AB_2762848 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 | ThermoFisher | RRID: AB_2762835 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 | ThermoFisher | RRID: AB_2762830 |
| Bacterial and virus strains | ||
| SARS-CoV-2 USA-WA1/2020 | BEI Resources | NR52281 |
| icSARS-CoV-2-WT | Hou et al.42 | Genbank: MT461669 |
| Biological samples | ||
| Mouse serum anti-SARS-CoV-2 Spike | Hou et al.42 | N/A |
| Human bronchial epithelial cells | Marsico Lung Institute, UNC | N/A |
| Human lung samples from previously healthy individuals unsuitable for transplant | Carolina Donor Services (Durham, NC), the National Disease Research Interchange (Philadelphia, PA), & the International Institute for Advancement of Medicine (Edison, NJ) | N/A |
| Human COVID-19 lung samples | Drs. Edana Stroberg (Office of the Chief Medical Examiner, Oklahoma City, OK) & Ross. E. Zumwalt (University of New Mexico, Albuquerque, NM) | N/A |
| Anonymous donor leukapheresis samples | Gulf Coast Blood | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pam3-CSK4 | Invivogen | Cat# tlrl-pms |
| Poly(I:C) HMW | Invivogen | Cat# tlrl-pic |
| Nigericin | Invivogen | Cat# tlrl-nig |
| ATP | Invivogen | Cat# tlrl-atpl |
| Poly(dA:dT) | Invivogen | Cat# tlrl-patn |
| Flagellin (recombinant, purified) | Gift from Dr. Ed Miao | N/A |
| Bay11-7082 | Invivogen | Cat# tlrl-b82 |
| Chloroquine | Invivogen | Cat# tlrl-chq |
| VX-765 | Invivogen | Cat# inh-vx765i-1 |
| C29 | MedChem Express | Cat# HY-100461 |
| MMG-11 | Tocris | Cat# 6858 |
| H151 | Invivogen | Cat# inh-h151 |
| MCC950 | Invivogen | Cat# inh-mcc |
| Benzonase® Nuclease | Millipore Sigma | Cat# 70664-3 |
| Recombinant human IL-1β | Peprotech | Cat# 200-01B |
| Recombinant SARS-CoV-2 E Protein (Avi Epitope Tag) His (N-term) Avi-tag Protein | Novus | Cat#NBP2-90986 |
| Recombinant SARS-CoV-2 S Protein | R&D Systems | Cat# 10639-CV-100 |
| Critical commercial assays | ||
| Ficoll-Paque™ | Sigma | Cat#GE17-1440-02 |
| TRIzol™ | ThermoFisher | Cat#15596026 |
| Direct-zol RNA Miniprep | ZYMO Research | Cat#R2052 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | Cat# 1708890 |
| iTaq™ Universal SYBR Green Supermix | Bio-Rad | Cat# 17125120 |
| RNAScope Multiplex Flourescent Reagent Kit V2 | ACD Bio | Cat#323100 |
| RNAScope 2.5 HD Reagent Kit-RED | ACD Bio | Cat#322430 |
| RNAScope Probe NLRP1 (channel 1) | ACD Bio | Cat# 436859 |
| RNAScope Probe NLRP3 (channel 1) | ACD Bio | Cat# 478021 |
| RNAScope Probe AIM2 (channel 1) | ACD Bio | Cat# 577931 |
| RNAScope Probe CASP1 (channel 1) | ACD Bio | Cat# 417631 |
| RNAScope Probe ABCA3 (channel 3) | ACD Bio | Cat# 555501-C3 |
| RNAScope Probe MPO (channel 3) | ACD Bio | Cat# 603091-C3 |
| RNAScope Probe CD68 (channel 2) | ACD Bio | Cat# 560591-C2 |
| RNAScope Probe IL1B (channel 2) | ACD Bio | Cat# 310361-C2 |
| RNAScope Probe IL6 (channel 1) | ACD Bio | Cat# 310371 |
| Vector® TrueVIEW® Autofluorescence Quenching Kit | Vector Laboratories | Cat# SP-8400-15 |
| VECTASHIELD Vibrance® Antifade Mounting Medium with DAPI | Vector Laboratories | Cat# H-1800 |
| Roche Cell Cytotoxicity Kit (LDH) | Sigma | Cat#11644793001 |
| QIAamp MinElute ccfDNA Mini Kit (50) | Qiagen | Cat# 55204 |
| MyTaq™ HS Red Mix | Meridian Bioscience | Cat# BIO-25048 |
| Human IL-1β ELISA Set II | BD Biosciences | Cat# 557953 |
| Human Total IL-18/IL-1F4 Quantikine ELISA Kit | R&D Systems | Cat# DL180 |
| ELISA MAX™ Deluxe Set Human IL-6 | Biolegend | Cat#430515 |
| Deposited data | ||
| Single-cell landscape of bronchoalveolar immune cells in COVID-19 patients | Liao et al.9 | GEO: GSE145926 |
| Single-cell RNA sequencing of bronchoalveolar lavages from COVID-19 patients | Wauters et al.58 | EGA: EGAS00001004717 |
| Deciphering the state of immune silence in fatal COVID-19 patients | Bost et al.59 | GEO: GSE157344 |
| Single-cell sequencing of BALF reveals immune response of COV2019 infection | He et al.61 | GEO: GSE147143 |
| Bronchoalveolar lavage of healthy human adults | Mould et al.60 | GEO: GSE151928 |
| Original Code for scRNAseq analysis | This study | https://github.com/yuyingxie/An-epithelial-immune-circuit-amplifies-inflammasome-and-IL-6-responses-to-SARS-CoV-2 |
| Experimental models: Cell lines | ||
| Vero-E6 | ATCC | Cat# CRL-1586 |
| Oligonucleotides | ||
| See Table S1 | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad | https://graphpad.com |
| Zen Black | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software.html |
| FIJI | Image J | https://fiji.sc/ |
| Olyvia V3.1.1 | Olympus | https://olympus-lifescience.com |
| Applied Biosystems QuantStudio V1.7.2 | Applied Biosystems | https://www.thermofisher.com/us/en/home/global/forms/life-science/quantstudio-6-7-flex-software.html |
| R V4.0.2 | R Project | https://cran.r-project.org/bin/windows/base/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jenny P.-Y. Ting (jenny_ting@med.unc.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
Human lungs from donors with no preexisting pulmonary disease that were unsuitable for transplantation were obtained from the University of North Carolina (UNC) Tissue Procurement and Cell Culture Core (institutional review board (IRB)-approved protocol #03-1396). Excised tissue specimens were dissected and fixed in 10% neutral buffered formalin for 48 hours followed by paraffin-embedding. Tissue blocks obtained from four COVID-19 autopsy lungs were obtained from Drs. Edana Stroberg (Office of the Chief Medical Examiner, Oklahoma City, OK), Ross. E. Zumwalt (University of New Mexico, Albuquerque, NM), and Leigh B. Thorne (University of North Carolina at Chapel Hill, Chapel Hill, NC). The paraffin blocks were cut to produce 5 μm serial sections for RNA in situ hybridization (RNA-ISH) and immunohistochemistry. Donor demographics are described as follows.
Donor 1
40-year-old male. Medical history: Diabetes mellitus. Clinical course: This donor had upper respiratory infection (URI) symptoms three days before he was found dead at home. No intubation occurred. Postmortem testing of the lung was positive for SARS-CoV-2.
Donor 2
91-year-old female. Medical history: coronary artery disease, hyperlipidemia, and hypertension. Clinical course: This donor was transferred to the ER because of URI symptoms, hypoxia, weakness, and shortness of breath. A nasal swab was positive for SARS-CoV-2. She was treated with high flow nasal canula but died from acute pneumonia due to SARS-CoV-2 complicated by an acute respiratory failure.
Donor 3
77-year-old male. Medical history: Acute pancreatitis, acute cholecystitis, and splenectomy. Clinical course: This donor was transferred to the ER because of fever and respiratory distress six days before death. He died shortly after arrival. A swab from the nasal cavity and the postmortem lung were positive for SARS-CoV-2.
Donor 4
42-year-old male. Medical history: Myotonic dystrophy, diabetes mellitus, hyperlipidemia, gastroesophageal reflux, cognitive deficits, dysarthria, and frequent falls. Clinical course: This donor was transferred to the ER because of cough and shortness of breath. He died shortly after arrival. No intubation occurred. SARS-CoV-2 was positive in nasopharyngeal swab, but negative in the postmortem lung.
Donor 5
No available clinical information.
Primary cell isolation and culture
Primary bronchial HAE were isolated and expanded as previously described.49 Briefly, human bronchial epithelial cells were isolated from previously normal human lungs obtained from anonymous organ donors with lungs unsuitable for transplantation under UNC IRB-approved protocol (#03-1396). In brief, at two-passages post-isolation from human lungs, cells were plated onto 12mm 0.4μm polyester Transwell supports (Corning) coated with human placental collagen type IV (Sigma). Cells were maintained in UNC ALI media49 at 37°C in 5% CO2. Once cells were fully confluent, media from the apical chamber was removed, and cells were maintained at ALI and allowed to differentiate for 28 days after seeding. Media in the basolateral compartment was changed 3x/week. Well-differentiated HAE ALI cultures were utilized for experiments at 28-90 days after seeding.
Primary human PBMCs from anonymous donors (Gulf Coast Blood) were isolated from leukapheresis samples using a Ficoll-Paque™ density gradient (Sigma), frozen, and immediately used in experiments upon thawing. PBMCs were maintained in RPMI-1640 (Thermo) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin-streptomycin, L-glutamine, sodium pyruvate, and non-essential amino acids. Cells were maintained in an incubator at 37°C in 5% CO2. For studies involving PBMCs with or without co-culture with Vero-E6 cells, PBMCs were maintained in MEM (Thermo) with 2% fetal bovine serum (FBS), 100 U/mL penicillin-streptomycin, L-glutamine, sodium pyruvate, and non-essential amino acids.
Vero cell line culture
Vero-E6 were maintained in MEM supplemented with 10% (maintenance media) or 2% (infection media) fetal bovine serum (FBS), 100 U/mL penicillin-streptomycin, L-glutamine, sodium pyruvate, and non-essential amino acids. For passage, confluent cells were rinsed with phosphate buffered saline (PBS), lifted with 0.25% trypsin, and plated at a 1:10 dilution factor. Cells were maintained in an incubator at 37°C in 5% CO2.
Method details
Inflammasome stimulation
Inflammasome stimulation was performed using previously established protocols.70 Briefly, cells were primed both basolateral and apical application of TLR ligands in ALI media 10μg/mL Pam3-CSK4 (Invivogen) or 0.5μg/mL Poly(I:C) (Invivogen). All apical ALI stimulations were delivered in a small volume of ALI media (75μL) to allow for continued air contact. After 3 hours, priming signals were removed, and inflammasome stimuli were added to the cultures. To activate the NLRP3 inflammasome, 40μM Nigericin (Invivogen) in ALI media was added to HAE culture apical and basolateral chambers for 1 hour. To activate the NLRP1/CARD8 inflammasome, 10μM Val-boroPro (R&D) was added to HAE culture apical and basolateral chambers for 18 hours. To activate the AIM2 inflammasome, poly(dA:dT) (Invivogen) was transfected into HAE cultures apically using Lipofectamine 2000 (Thermo) in Opti-MEM media (Thermo) and stimulated for 3 hours. To activate the NLRC4 inflammasome, purified flagellin (a generous gift from Dr. Ed Miao) was transfected into HAE cultures apically using DOTAP (Sigma) in Opti-MEM media and stimulated for 2 hours. To maximize transfection efficiency of p(dA:dT) and flagellin, the epithelial barrier was disrupted immediately prior to transfection by apical treatment with 30mM sodium caprate (Sigma) for 5 minutes at 37°C 5% CO2. For NLRP3 inflammasome activation in human PBMCs, cells were primed with 1μg/mL Pam3-CSK4 (Invivogen) for 3 hours and then stimulated with 40μM Nigericin or 5mM ATP for 1 hour. For experiments looking at SARS-CoV-2 protein stimulation or SARS-CoV-2 infection in combination with known inflammasome stimulation, these treatments were applied as described in the text, preceded or followed by stimulations described above.
Following inflammasome activation, supernatants were collected and immediately frozen to measure IL-1β or IL-18 secretion by ELISA (BD OptEIA IL-1β or R&D IL-18) or LDH release (Roche/Sigma Cytotoxicity Detection Kit (LDH)). To collect apical supernatants from HAE ALI cultures, the apical surface was incubated with 250μL PBS for 30 minutes at 37°C 5% CO2, gently mixed and removed, and then frozen until analysis by ELISA. ELISA values from apical wash samples (250μL) were normalized to the volume of basolateral media (1mL) to allow for direct comparison between the compartments. In some instances, these supernatants were pooled.
IL-1β or Protein/Ligand stimulation
Human PBMCs were treated in suspension in culture medium with recombinant human IL-1β (100pg/mL or 1ng/mL) or a vehicle control for indicated timepoints. Similarly, human PBMCs were treated in suspension in culture medium with recombinant SARS-CoV-2 E protein (1μg/mL), S protein trimer (1μg/mL) or Pam3-CSK4 (1μg/mL). HAE cultures were stimulated at the apical surface and in the basolateral compartment with recombinant human IL-1β (100pg/mL or 1ng/mL), Pam3-CSK4 (1μg/mL), or a vehicle control for indicated timepoints. Following treatment, cells were lysed in Trizol reagent for RNA isolation and subsequent RT-qPCR analysis. Cell supernatants were collected and frozen until assessed by ELISA (Biolegend ELISA MAX™ human IL-6). Washes from the apical surface were collected as described above, stored frozen, and analyzed by ELISA.
Viral infections
SARS-CoV-2 USA-WA1/2020 or the synthetic clone of this strain icSARS-CoV-2 WT42 were both utilized in these experiments due to the need to utilize two collaborating BSL3 facilities because of scheduling difficulties. HAE ALI cultures were rinsed twice with PBS for 30 minutes at 37°C to remove mucus prior to infection. Cells were infected apically with viral stocks diluted in PBS for 2 hours at 37°C in 5% CO2 with frequent agitation, as described previously.42 Following infection, cells were washed 3x with PBS for 5 minutes and kept at an ALI for the duration of the experiment at 37°C in 5% CO2. Infection of PBMCs or Vero cell lines with SARS-CoV-2 occurred as described above with a few key differences. To infect PBMCs or Vero cell lines, culture media was removed and cells were incubated with diluted viral stocks in infection media (complete MEM 2%FBS, as described above) for 1 hour at 37°C in 5% CO2 with frequent agitation. Following the infection period, inoculum was removed and wells were given fresh infection media for the experiment duration. WT SARS-CoV-2 stocks were propagated in Vero-E6 cells. All SARS-CoV-2 infections, propagation, and assays requiring samples with live virus (ELISA, plaque assay) were performed under biosafety level 3 (BSL-3) containment at negative pressure and in Tyvek suits with personal respirators.
Following infection, HAE ALI apical wash and basolateral supernatants were collected and frozen for analysis, as described above. For PBMCs, cells were pelleted at 500xg for 5 minutes, supernatants were frozen and collected for analysis by ELISA. Cell lysates were collected in TRIzol reagent (Thermo) for measurement of gene expression by reverse transcription quantitative PCR (RT-qPCR).
RT-qPCR
Following lysis in TRIzol reagent, RNA was isolated according to the manufacturer’s protocol. RNA was converted into cDNA through reverse transcription using the iScript™ cDNA Synthesis Kit (Bio-Rad). Relative mRNA and viral RNA expression was then measured using the iTaq Universal SYBR Green Mastermix (Bio-Rad) on the Quantstudio 6 Real Time PCR System (Applied Biosystems). Primers used are listed in the key resources table.
PCR detection of DNA in supernatants
HAE supernatants were heat inactivated for 60 minutes and 60°C for removal from BSL3, dsDNA was isolated from these supernatants through spin column purification (Qiagen QIAamp MinElute ccfDNA Mini Kit), and then PCR amplification of genomic or mitochondrial DNA amplicons was performed (Meridian Bioscience MyTaq™ HS Red Mix). Cycle conditions are as follows: Denaure: 15s 98°C, Anneal: 15s 55°C, and Extend: 30s 72°C for 25 cycles followed by 5 minute extension period. Amplified dsDNA products were visualized on an agarose gel using SYBR green dye and analyzed in FIJI. Primers used are listed in the key resources table.
Plaque assay
Confluent Vero-E6 cells were infected with ten-fold serial dilutions of cell culture supernatants (PBMC/Vero) or apical washes (HAE cultures) to measure viral titer. Diluted samples were assayed in duplicate. Infections were performed for one hour as described above with frequent agitation. After 1 hour, overlay (1.25% carboxy methylcellulose and 1x alpha-MEM) was placed over the infected cells. Four days post infection, wells were fixed by adding 4% paraformaldehyde or formalin to each well. The overlay/fixative solution was removed, plates were submerged in fixative, rinsed with water and then stained for a minimum of 10 minutes with.25% crystal violet in water. Excess stain was removed, plates rinsed, and plaques were enumerated.
Integrated scRNA-seq meta-analysis
We retrieved single-cell RNA-Seq data from the following published datasets: GEO: GSE145926,9 GEO: GSE147143,61 GEO: GSE157344,59 GEO: GSE151928,60 and EGA: EGAS00001004717.58 Collectively, 26 patients with severe COVID-19 disease, 5 patients with moderate or mild symptoms, and 3 healthy controls were included in the initial analysis, while 10 healthy donors and 24 severe patients were added in the projected data. In total, cells from 13 healthy donors, 5 mild/moderate patients, and 50 severe patients were analyzed. We only kept high-quality cells that had unique feature counts between 200 and 10,000 and mitochondrial reads of less than 20%. Cells with unique molecular identifiers counts below 500 were filtered out. After processing, a total of 117,497 high-quality transcriptomes of PBMCs were available for integration. We implemented the sctransform algorithm for the integrated data set using Seurat v.3 to normalize and remove the library size effect.71 Then, we utilized the Mutual Nearest Neighbors algorithm to remove the batch effect between studies and between patients.72 To concentrate biology signals, we selected the top 2,000 genes with the highest cell-to-cell variation, in conjunction with a set of immune cell subsets signature genes to stabilize the lineage segregation. These additional anchor features include CXCR5, CD69, AIM2, IRF5, IRF1, IRF3, LGALS9, LY6E, NOS2, IL6, TNF, ISG15, GSDMD, CD8A, CD40, CD80, CD86, IL10, TGFB1, TMEM173, CD274, CXCL9, MRC1, SIGLEC15, TRDC, CD2, TRAC, CD4, CD8B, FOXP3, TRBC1, TRBC2, GZMB, EOMES, ICOS, CD3G, CD3E, IFNG, NCR1, CD19, CD79A, CD79B, ITGAM, ITGAX, BATF3, XCR1, GATA3, CTLA4, RORC, IL17A, BCL6, HAVCR2, TNFRSF4, TIGIT, CXCL10, MX1, IFNB1, IL3RA, NRP1, CLEC4C, FCER1A, TBX21, LAG3, IFNL3, PDCD1, CD14, and CD33. We obtained the top 50 principal components (PCs) from the integrated data. Then, we employed the Shared Nearest Neighbor method to group cells, informing 22 distinct clusters with a resolution of 0.8.73
Immunohistochemistry
Immunohistochemical staining was performed on non-disease explant lung (control) and COVID-19 autopsy lung sections, according to previously described protocols.42 Briefly, paraffin-embedded sections were baked at 60 °C for 2–4 hours and deparaffinized with xylene (2 changes × 10 min) and graded ethanol (100% 2 × 5 min, 95% 1 × 5 min, 70% 1 × 5 min). After rehydration, antigen retrieval was performed by boiling the slides in 0.1 M sodium citrate pH 6.0 (3 cycles with microwave settings: 100% power for 6.5 min, 60% for 6 min, and 60% for 6 min, refilling the Coplin jars with distilled water after each cycle). After cooling and rinsing with distilled water, slides were washed in PBS and blocked with 4% normal donkey serum for one hour at RT. Primary antibodies (ASC:1:500, MPO: 1:200, CD68: 1:100, CD11c: 1:500, Anti-SARS mouse antiserum: 1:4000) were diluted in 4% normal donkey serum in PBST (phosphate-buffered saline containing 0.1% Triton X-100) and incubated overnight at 4 °C. Sections were washed in PBST, and secondary antibodies (Alexa Fluor 488 donkey anti-rabbit IgG, at 1:1000 dilution; Alexa Fluor 647 donkey anti-mouse IgG, at 1:1000 dilution and Alexa Fluor 750 donkey anti-goat IgG, at 1:1000 dilution) were applied for 60 min at RT. After washing in PBST, the Vector® TrueVIEW Autofluorescence Quenching Kit (Vector laboratories) was used to reduce background staining, and glass coverslips were placed over tissue sections with the VECTASHIELD® Vibrance™ Antifade Mounting Medium with DAPI (Vector laboratories). Cover slipped slides were scanned and digitized using an Olympus VS200 with a 20X 0.75 NA objective. Representative images were generated from these digitized composite images using Olyvia software or captured on a Zeiss LSM 710 inverted laser scanning confocal microscope with a 40X/1.4 Oil Plan Apo objective using Zen software.
RNA in situ hybridization (RNA-ISH)
RNA-ISH was performed on paraffin-embedded 5μm tissue sections of control and COVID-19 autopsy lung sections using the RNAscope 2.5 HD Reagent Kit-RED (chromogenic images) and Multiplex Fluorescent Assay Kit v2 (fluorescent images) according to the manufacturer’s instructions (Advanced Cell Diagnostics). Sections were deparaffinized with xylene (2 changes × 5 min) and 100% ethanol (2 changes × 1 min), incubated with hydrogen peroxide for 10 min followed by target retrieval in boiling water for 15 min, and incubation with Protease Plus (Advanced Cell Diagnostics) for 15 min at 40°C. Slides were hybridized with custom probes at 40 °C for 2 hours, and signals were amplified according to the manufacturer’s instructions. Cover slipped slides were scanned and digitized using an Olympus VS200 with a 20X 0.75 NA objective. Representative images were generated from these digitized composite images using Olyvia software or captured on a Zeiss LSM 710 inverted laser scanning confocal microscope with a 40X/1.4 Oil Plan Apo objective using Zen software.
Quantification and statistical analysis
Image quantification
Stained areas of CASP1, NLRP1, NLRP3, and AIM2 mRNAs by chromogenic RNA-ISH in the alveolar regions were quantitated using FIJI software as previously described.74 Briefly, alveolar regions were cropped from the field, and staining of each mRNA was converted to a gray-scale image. Optimized threshold value was determined by adjusting the threshold to accurately represent the original images. We applied the optimized threshold value to all the sections. The stained-areas were normalized to alveolar areas. Fluorescent staining of lung samples was counted manually from randomly selected fields of view with an n≥500 for each biological replicate. ASC speck frequency was normalized to the frequency of nuclei, as indicated by DAPI staining. Co-incidence of ASC specks with other markers was normalized to the total number of nuclei-associated ASC specks, as indicated by DAPI staining. Co-incidence of genes with other markers was normalized to the total number of gene-associated nuclei, as indicated by DAPI staining.
Statistical analysis
With the exception of scRNAseq data, all statistical analysis was performed using Graphpad Prism 8 software. All experiments were performed in triplicate and represent 3 independent biological replicates. For comparisons in which multiple variables were tested with multiple conditions, two-way ANOVA analysis with Tukey’s, Dunnet’s or Sidak’s multiple comparison test correction was performed, as indicated in figure legends. For multiple comparisons of groups with a single independent variable, one-way ANOVA analysis with Tukey’s or Dunnet’s multiple comparison test correction was performed, as indicated in figure legends. Unpaired two-tailed t tests were used to compare two groups. Asterisk coding is as follows: ∗ p<= 0.05; ∗∗ p<=0.01; ∗∗∗ p<=0.001; ∗∗∗∗ p<=0.0001. Data with error bars depict the average with the SEM.
scRNAseq Statistical analysis
Cell type differential abundance analyses were performed using the R package ALDEx2, which utilize centered log-ratio transformation to ensure the data to be scale invariant and sub-compositionally coherent. To test the significance of Inflammasome score and gene expression quantile across disease statuses, we employed a linear quantile mixed model via the R package lqmm, where patient IDs were treated as a random effect. To enhance the rigor of the model, we removed samples that only had fewer than 5 cells in the given clusters. To test the significance of Inflammasome score and inflammasome genes on pool cells among disease status, we performed the non-parametric Kruskal-Wallis test taking account the fact that the data distributions are not Gaussian. Post-hoc Dunn’s tests were conducted to determine pair-wise differences between disease status.
Acknowledgments
This work is supported by NIH grants R56 AI158314, R01 AI158314, AI029564, AI141333, and CA232109 to J.P.-Y.T.; NCI T32CA071341 to K.C.B.; R.C.B. and T.A. are supported by UH3 HL123645, R01 HL136961, P30 DK 065988, P01 HL108808, and BOUCHE19R0; K.O. and R.C.B. are supported by BOUCHE19XX0; K.O. is supported by OKUDA20G0 from the Cystic Fibrosis Foundation; M.T.H., S.A.T.-B., and J.F.L. are supported by NC Policy Collaboratory, R01AI157253, and U19 AI100625; NSF GRFP 2019255197 to J.F.L. T.A. is a Japan Society for the Promotion of Science Overseas Research Fellow. Y.X. is supported by NSF DMS-191290 and NSF IOS 2107215. Y.X. and Y.L.L. are supported by NIH grants R01DE026728 and R01DE031951. HAE culture reagents were provided by the Marsico Lung Institute Tissue Procurement and Cell Culture Core supported by NIH grant DK065988 and Cystic Fibrosis Foundation grant BOUCHR19R0. Some BSL3 work was performed in the Duke Regional Biocontainment Laboratory (Duke RBL), which received partial support for construction from NIH/NIAID (UC6AI058607). Graphical abstract and Figure 6D created using Biorender.com. We would like to thank Ting lab members for helpful discussion.
Author contributions
K.C.B., Y.X., and T.A. contributed equally to this manuscript. Y.L.L., R.C.B., and J.P.-Y.T. also contributed equally to this manuscript. J.P.-Y.T. and K.C.B. conceived of the study. K.C.B. and J.P.-Y.T. wrote the manuscript. R.C.B., T.A., Y.L.L., and Y.X. edited the manuscript. K.C.B., T.A., Y.X., R.C.B., Y.L.L., and J.P.-Y.T. designed experiments. T.H.O., G.D.S., S.H.R., M.T.H., and S.A.T.-B. contributed input to experimental design. K.C.B., S.A.T.-B., J.F.L., D.S., and T.H.O. performed the BSL3 experiments. T.A., K.O., and R.C.G. contributed lung samples, performed RNA-ISH, and immunofluorescent staining. K.C.B. and T.A. captured images of stained lung samples. K.C.B., H.G., K.L., and S.Y. executed the other experiments and analyzed data. K.C.B., S.H.R., and K.L. established and maintained HAE cultures. Y.X., Y.L.L., and K.C.B. performed the scRNA-seq meta-analysis.
Declaration of interests
J.P.-Y.T. is a cofounder of IMMvention Therapeutix, which works on inflammasome inhibitors.
Published: December 9, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2022.12.005.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
-
•
All original code is deposited at GitHub and is publicly available. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181 doi: 10.1016/j.cell.2020.04.026. 1036–1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Channappanavar R., Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017;39:529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yifan Q., Chao H., Kun W., Peng H., Runsheng W., Jiang L., Tianzhi L., Rongyu P., Qinyong H., Yu S., et al. Cytokine release syndrome in COVID-19: a major mechanism of morbidity and mortality. Int. Rev. Immunol. 2021;41:217–230. doi: 10.1080/08830185.2021.1884248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGonagle D., Sharif K., O’Regan A., Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun. Rev. 2020;19:102537. doi: 10.1016/j.autrev.2020.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulchandani R., Lyngdoh T., Kakkar A.K. Deciphering the COVID-19 cytokine storm: systematic review and meta-analysis. Eur. J. Clin. Invest. 2021;51:e13429. doi: 10.1111/eci.13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 10.Silvin A., Chapuis N., Dunsmore G., Goubet A.-G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2021;182 doi: 10.1016/j.cell.2020.08.002. 1401–1418.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyriazopoulou E., Poulakou G., Milionis H., Metallidis S., Adamis G., Tsiakos K., Fragkou A., Rapti A., Damoulari C., Fantoni M., et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat. Med. 2021;27:1752–1760. doi: 10.1038/s41591-021-01499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kyriazopoulou E., Huet T., Cavalli G., Gori A., Kyprianou M., Pickkers P., Eugen-Olsen J., Clerici M., Veas F., Chatellier G., et al. Effect of anakinra on mortality in patients with COVID-19: a systematic review and patient-level meta-analysis. Lancet Rheumatol. 2021;3:e690–e697. doi: 10.1016/S2665-9913(21)00216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.REMAP-CAP Investigators. Gordon A.C., Mouncey P.R., Al-Beidh F., Rowan K.M., Nichol A.D., Arabi Y.M., Annane D., Beane A., Bentum-Puijk W. van, et al. Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N. Engl. J. Med. 2021;384:1491–1502. doi: 10.1056/NEJMoa2100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broz P., Dixit V.M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 15.Chi Y., Ge Y., Wu B., Zhang W., Wu T., Wen T., Liu J., Guo X., Huang C., Jiao Y., et al. Serum cytokine and chemokine profile in relation to the severity of coronavirus disease 2019 in China. J. Infect. Dis. 2020;222:746–754. doi: 10.1093/infdis/jiaa363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McElvaney O.J., McEvoy N.L., McElvaney O.F., Carroll T.P., Murphy M.P., Dunlea D.M., Ní Choileáin O.N., Clarke J., O’Connor E., Hogan G., et al. Characterization of the inflammatory response to severe COVID-19 illness. Am. J. Respir. Crit. Care Med. 2020;202:812–821. doi: 10.1164/rccm.202005-1583OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alnefeesi Y., Siegel A., Lui L.M.W., Teopiz K.M., Ho R.C.M., Lee Y., Nasri F., Gill H., Lin K., Cao B., et al. Impact of SARS-CoV-2 infection on cognitive function: a systematic review. Front. Psychiatry. 2020;11:621773. doi: 10.3389/fpsyt.2020.621773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kempuraj D., Selvakumar G.P., Ahmed M.E., Raikwar S.P., Thangavel R., Khan A., Zaheer S.A., Iyer S.S., Burton C., James D., Zaheer A. COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist. 2020;26:402–414. doi: 10.1177/1073858420941476. [DOI] [PubMed] [Google Scholar]
- 19.Yap J.K.Y., Moriyama M., Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J. Immunol. 2020;205:307–312. doi: 10.4049/jimmunol.2000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen I.C., Scull M.A., Moore C.B., Holl E.K., McElvania-TeKippe E., Taxman D.J., Guthrie E.H., Pickles R.J., Ting J.P.Y. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas P.G., Dash P., Aldridge J.R., Ellebedy A.H., Reynolds C., Funk A.J., Martin W.J., Lamkanfi M., Webby R.J., Boyd K.L., et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichinohe T., Lee H.K., Ogura Y., Flavell R., Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lupfer C., Malik A., Kanneganti T.-D. Inflammasome control of viral infection. Curr. Opin. Virol. 2015;12:38–46. doi: 10.1016/j.coviro.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swanson K.V., Deng M., Ting J.P.Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzgerald K.A., Kagan J.C. Toll-like receptors and the control of immunity. Cell. 2020;180:1044–1066. doi: 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng M., Karki R., Williams E.P., Yang D., Fitzpatrick E., Vogel P., Jonsson C.B., Kanneganti T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021;22:829–838. doi: 10.1038/s41590-021-00937-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., Sun H., Wang D.-C., Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 28.Kayagaki N., Stowe I.B., Lee B.L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T., et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 29.Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V.G., Wu H., Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 31.Ramos I., Fernandez-Sesma A. Cell receptors for influenza A viruses and the innate immune response. Front. Microbiol. 2012;3:117. doi: 10.3389/fmicb.2012.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181 doi: 10.1016/j.cell.2020.02.052. 271.e8–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 34.Rockx B., Kuiken T., Herfst S., Bestebroer T., Lamers M.M., Oude Munnink B.B.O., Meulder D. de, Amerongen G. van, Brand J. van den, Okba N.M.A., et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science. 2020;368:1012–1015. doi: 10.1126/science.abb7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y., Zhao Z., Wang Y., Zhou Y., Ma Y., Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020;202:756–759. doi: 10.1164/rccm.202001-0179LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferreira A.C., Soares V.C., Azevedo-Quintanilha I.G., Dias S.da S.G., Fintelman-Rodrigues N., Sacramento C.Q., Mattos M., Freitas C.S., Temerozo J.R., Teixeira L., et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021;7:43. doi: 10.1038/s41420-021-00428-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodrigues T.S., Sá K.S.G. de, Ishimoto A.Y., Becerra A., Oliveira S., Almeida L., Gonçalves A.V., Perucello D.B., Andrade W.A., Castro R., et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021;218:e20201707. doi: 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theobald S.J., Simonis A., Georgomanolis T., Kreer C., Zehner M., Eisfeld H.S., Albert M.C., Chhen J., Motameny S., Erger F., et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in COVID-19. EMBO Mol. Med. 2021;13:e14150. doi: 10.15252/emmm.202114150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junqueira C., Crespo Â., Ranjbar S., de Lacerda L.B., Lewandrowski M., Ingber J., Parry B., Ravid S., Clark S., Schrimpf M.R., et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature. 2022;606:576–584. doi: 10.1038/s41586-022-04702-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sefik E., Qu R., Junqueira C., Kaffe E., Mirza H., Zhao J., Brewer J.R., Han A., Steach H.R., Israelow B., et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature. 2022;606:585–593. doi: 10.1038/s41586-022-04802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hou Y.J., Okuda K., Edwards C.E., Martinez D.R., Asakura T., Kenneth H.D., 3rd, Kato T., Lee R.E., Yount B.L., Mascenik T.M., et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182 doi: 10.1016/j.cell.2020.05.042. 429–446.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Planès R., Pinilla M., Santoni K., Hessel A., Passemar C., Lay K., Paillette P., Valadão A.-L.C., Robinson K.S., Bastard P., et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell. 2022;82 doi: 10.1016/j.molcel.2022.04.033. 2385–2400.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 45.Gaidt M.M., Ebert T.S., Chauhan D., Ramshorn K., Pinci F., Zuber S., O’Duill F., Schmid-Burgk J.L., Hoss F., Buhmann R., et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171 doi: 10.1016/j.cell.2017.09.039. 1110–1124.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swanson K.V., Junkins R.D., Kurkjian C.J., Holley-Guthrie E., Pendse A.A., El Morabiti R.E., Petrucelli A., Barber G.N., Benedict C.A., Ting J.P.Y. A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med. 2017;214:3611–3626. doi: 10.1084/jem.20171749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D.R., Latz E., Fitzgerald K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hare D., Collins S., Cuddington B., Mossman K. The importance of physiologically relevant cell lines for studying virus–host interactions. Viruses. 2016;8:297. doi: 10.3390/v8110297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fulcher M.L., Randell S.H. Second Edition. Vol. 945. Humana Press; 2012. Epithelial Cell Culture Protocols (Methods in Molecular Biology) pp. 109–121. [DOI] [PubMed] [Google Scholar]
- 50.Taabazuing C.Y., Griswold A.R., Bachovchin D.A. The NLRP1 and CARD8 inflammasomes. Immunol. Rev. 2020;297:13–25. doi: 10.1111/imr.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan S., Shafiei M.S., Longoria C., Schoggins J.W., Savani R.C., Zaki H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife. 2021;10:e68563. doi: 10.7554/eLife.68563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holness C.L., Simmons D.L. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood. 1993;81:1607–1613. doi: 10.1182/blood.v81.6.1607.1607. [DOI] [PubMed] [Google Scholar]
- 53.Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018;640:47–52. doi: 10.1016/j.abb.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Robinson K.S., Teo D.E.T., Tan K.S., Toh G.A., Ong H.H., Lim C.K., Lay K., Au B.V., Lew T.S., Chu J.J.H., et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science. 2020;370:1–14. doi: 10.1126/science.aay2002. [DOI] [PubMed] [Google Scholar]
- 55.Zhong F.L., Mamaï O., Sborgi L., Boussofara L., Hopkins R., Robinson K., Szeverényi I., Takeichi T., Balaji R., Lau A., et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. 2016;167 doi: 10.1016/j.cell.2016.09.001. 187.e17–202.e17. [DOI] [PubMed] [Google Scholar]
- 56.Stutz A., Horvath G.L., Monks B.G., Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol. 2013;1040:91–101. doi: 10.1007/978-1-62703-523-1_8. [DOI] [PubMed] [Google Scholar]
- 57.Patel V.I., Metcalf J.P. Airway macrophage and dendritic cell subsets in the resting human lung. Crit. Rev. Immunol. 2018;38:303–331. doi: 10.1615/CritRevImmunol.2018026459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wauters E., Mol P., Garg A.D., Jansen S., Herck Y., Vanderbeke L., Bassez A., Boeckx B., Malengier-Devlies B., Timmerman A., et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021;31:272–290. doi: 10.1038/s41422-020-00455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bost P., De Sanctis F.D., Canè S., Ugel S., Donadello K., Castellucci M., Eyal D., Fiore A., Anselmi C., Barouni R.M., et al. Deciphering the state of immune silence in fatal COVID-19 patients. Nat. Commun. 2021;12:1428. doi: 10.1038/s41467-021-21702-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mould K.J., Moore C.M., McManus S.A., McCubbrey A.L., McClendon J.D., Griesmer C.L., Henson P.M., Janssen W.J. Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am. J. Respir. Crit. Care Med. 2021;203:946–956. doi: 10.1164/rccm.202005-1989OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He J., Cai S., Feng H., Cai B., Lin L., Mai Y., Fan Y., Zhu A., Huang H., Shi J., et al. Single-cell analysis reveals bronchoalveolar epithelial dysfunction in COVID-19 patients. Protein Cell. 2020;11:680–687. doi: 10.1007/s13238-020-00752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182 doi: 10.1016/j.cell.2020.08.001. 1419–1440.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2019;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184 doi: 10.1016/j.cell.2021.04.048. 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanaka T., Narazaki M., Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014;6:a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abani O., Abbas A., Abbas F., Abbas M., Abbasi S., Abbass H., Abbott A., Abdallah N., Abdelaziz A., Abdelfattah M. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397:1637–1645. doi: 10.1016/S0140-6736(21)00676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng J., Wang Y., Li K., Meyerholz D.K., Allamargot C., Perlman S. Severe acute respiratory syndrome coronavirus 2–induced immune activation and death of monocyte-derived human macrophages and dendritic cells. J. Infect. Dis. 2021;223:785–795. doi: 10.1093/infdis/jiaa753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lowery S.A., Sariol A., Perlman S. Innate immune and inflammatory responses to SARS-CoV-2: implications for COVID-19. Cell Host Microbe. 2021;29:1052–1062. doi: 10.1016/j.chom.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bryce C., Grimes Z., Pujadas E., Ahuja S., Beasley M.B., Albrecht R., Hernandez T., Stock A., Zhao Z., AlRasheed M.R., et al. Pathophysiology of SARS-CoV-2: the Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021;34:1456–1467. doi: 10.1038/s41379-021-00793-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo H., Ting J.P.-Y. Inflammasome assays in vitro and in mouse models. Curr. Protoc. Immunol. 2020;131:e107. doi: 10.1002/cpim.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177 doi: 10.1016/j.cell.2019.05.031. 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir el-A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okuda K., Dang H., Kobayashi Y., Carraro G., Nakano S., Chen G., Kato T., Asakura T., Gilmore R.C., Morton L.C., et al. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med. 2021;203:1275–1289. doi: 10.1164/rccm.202008-3198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
-
•
All original code is deposited at GitHub and is publicly available. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.