

Abstract
Several products of the activated complement system are known to modulate endothelial cell function in vitro. It has been shown that the membrane attack complex (MAC) (C5b-C9) can enhance tumor necrosis factor alpha (TNF-α)-induced expression of P- and E-selectin and intercellular adhesion molecule type 1 in cell cultures of human umbilical vein endothelial cells. In the present study the potential role of this synegism for lung injury during endotoxin-mediated septic shock in vivo was examined using a model of C6-deficient PVG (C−) (RT1C) rats and the congenic PVG (C+) (RT1C) strain. Following administration of a high (5 mg/kg) or low (0.5 mg/kg) dose of lipopolysaccharide (LPS) (Escherichia coli O55:B5), we determined the expression of cytokines, chemokines, and adhesion molecules as well as the recruitment of leukocytes in the lung. Challenge with intraperitoneal i.p. injections of LPS resulted in a strong induction of TNF-α, interleukin-1α/β, cytokine-induced neutrophil chemoattractant, interferon-inducible protein 10, macrophage inflammatory proteins 1α and 2, macrophage chemotactic protein 1, and P-selectin. However, there were no significant differences between PVG (C−) and PVG (C+) rats. Immunoperoxidase staining showed a similar increase of lung infiltration by CD11b/c+ leukocytes in both rat strains. We therefore conclude that the described synergism between TNF-α and the MAC of the complement system on the induction of endothelial adhesion molecules is dispensable for inflammatory processes during endotoxin-mediated septic shock in vivo.
Systemic administration of lipopolysaccharide (LPS) results in acute inflammatory lung injury and septic shock (1, 6). This challenge of the immune system induces the systemic release of cytokines including tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), IL-2, IL-4, IL-6, and gamma interferon and induction of both endothelial and leukocyte adhesion molecules such as E-selectin, P-selectin, intercellular cell adhesion molecule type 1 (ICAM-1) and CD11b/c (32, 33). Components of the complement system like C3b, iC3b, and C5a are considered to play important roles in leukocyte-endothelial cell adhesive interactions and endothelial cell activation (9, 15). Both C5a and the membrane attack complex (MAC) promote expression of the adhesion molecule P-selectin by endothelial cells (15). In vitro experiments have demonstrated that C5a acts on human umbilical vein endothelial cells to induce rapid expression of the adhesion molecule P-selectin and to promote neutrophil adhesion (9). TNF-α acts in concert with numerous mediators like IL-1 and IL-6. Moreover, upregulation of lung vascular ICAM-1 induced by intratracheally administered TNF-α was prevented by complement depletion, which suggested an in vivo function of complement for the upregulation of vascular ICAM-1 (31). Using an in vitro system, it was demonstrated that endothelial cell activation with TNF-α followed by assembly of the MAC resulted in a marked increase of E-selectin and ICAM-1, suggesting a synergistic action of TNF-α and MAC (12) for the induction of leukocyte-endothelial cell interactions.
The induction of adhesion molecules and chemokines is considered important for the development of inflammatory lung injury. Thus, the concentrations of IL-8 in bronchioalveolar lavage fluid of acute respiratory distress syndrome patients was directly correlated with mortality and with neutralization of IL-8 receptor-binding chemokines protected from LPS-induced lung vascular damage in experimental animals (7, 10, 16, 25, 29). Following systemic application of LPS or intrapulmonary deposition of immune complexes, acute damage to the lungs was prevented by antibodies against macrophage inflammatory protein 1α (MIP-1α) (26, 27). In the immune complex model, E-selectin and VLA-4 were involved in the generation of lung injury, while blockade of P-selectin and Mac-1 protected the host from injury induced by cobra venom factor (18, 20–23). In contrast, inhibition of LFA-1, ICAM-1, or L-selectin showed protective effects in both models (17, 20, 22, 23). Thus, depending on the nature of the inflammatory stimulus or the route of application, specific subsets of chemokines and adhesion molecules may be involved in the pathogenesis of inflammatory lung injury.
In the present study, we investigated the possible synergism between TNF-α and the MAC for the development of lung inflammation in vivo using a model of PVG (C−) (RT1C) rats, which are entirely deficient in C6 and unable to assemble the MAC (C5b-C9), and the congenic PVG (C+) (RT1C) rats with normal hemolytic complement activity (3, 5, 14). Lung inflammation was induced by intraperitoneal (i.p.) injection of low (0.5 mg/kg) and high (5 mg/kg) doses of LPS. The induction of cytokines (TNF-α, IL-1α, IL-1β, and IL-6), chemokines (cytokine-induced neutrophil chemoattractant [KC], MIP-2, MIP-1α, macrophage chemotactic protein 1 [MCP-1], interferon-inducible protein 10 [IP-10]), and adhesion molecules (P-selectin, E-selectin, ICAM-1, and vascular cell adhesion molecule type 1 [VCAM-1]), as well as cellular infiltration by CD11b/c-positive leukocytes, in the lungs were determined.
MATERIALS AND METHODS
Animals.
Male 9- to 12-week-old homozygous C6-deficient rats PVG (C−) (RT1C) rats were obtained from Bantin & Kingman (Fremont, Calif.), and PVG (C+) (RT1c) rats were obtained from Bantin & Kingman Universal Limited (Grimston, United Kingdom). Sera of PVG (C−) and PVG (C+) rats were tested by hemolytic complement assays and gel electrophoresis to confirm C6 deficiency as described previously (5).
Endotoxin-mediated septic shock.
PVG (C−) and PVG (C+) rats (five per group) were injected i.p. with a single dose of either low-dose (0.5 mg/kg) or high-dose (5 mg/kg) LPS from Escherichia coli serotype O55:B5 (Sigma Chemical Co., St. Louis, Mo.) dissolved in 10 ml of phosphate-buffered saline (PBS), and 4, 8, or 12 h later the lungs were removed. The thorax was opened by bilateral thoracotomy. The right main bronchus was ligated, and the right lung was removed, snap frozen in liquid nitrogen, and stored at −80°C for RNA analysis. The cervical trachea was exposed and opened by horizontal incision, and a 17G Venflon catheter was inserted. The left lung was filled with medium consisting of a 1:1 mixture of Tissue-Tec (OCT compound) and 10% sucrose (Sigma) dissolved in PBS. The left main bronchus was ligated, and the left lung was removed, covered with Tissue-Tec, snap frozen in liquid nitrogen, and stored at −80°C for immunohistochemistry.
Antibodies.
The following monoclonal antibodies (MAbs) were used in this study: mouse anti-rat CD11b/c (Ox-42) MAb, rabbit anti-human CD62P (P-selectin) MAb (cross-reaction with rat), mouse anti-rat CD54 (ICAM-1) (1A29) MAb, and mouse anti-rat CD45 (Ox-1) MAb. Purified mouse immunoglobulin G1 kappa chain (IgG1κ) (MOPC-21) was used as an isotype control. All MAbs were purchased from PharMingen (San Diego, Calif.). Polyclonal goat anti-human C6 was obtained from Accurate (Westbury, N.Y.) and anti-human IgG was obtained from PharMingen.
Immunohistochemistry.
Cryostat sections 8 μm thick were prepared, fixed in cold acetone for 10 min, dried, and stored at −80°C. Endogenous peroxidase activity was blocked by preincubation of the sections with rat serum and H2O2. Thereafter, the sections were incubated for 30 min with 100 μl of the diluted MAbs (dilution, 1:100). They were washed three times in PBS, and 100 μl of secondary goat-anti mouse IgG Ab (dilution, 1:100) or goat-anti rabbit IgG Ab (dilution, 1:100) labeled with horseradish peroxidase (PharMingen) was added for 30 min. Nonspecific background staining was reduced by preincubation of the peroxidase-conjugated antiserum with normal rat serum (dilution, 1:100). All incubations were conducted in a moist, light-protected chamber at room temperature. The sections were rinsed again in PBS, fixed for 5 min in 0.1% glutaraldehyde, and stained for 10 min in 50 mM acetate buffer containing 0.01% H2O2 and 5 mg of 3-amino-9-ethylcarbazole (Sigma) per ml, which was dissolved in N,N′-dimethylformamide. After extensive washing in PBS, the slides were counterstained with Mayer's hematoxylin for 10 min and mounted with glycerol-gelatin. All of the infiltrating leukocytes were counted in 10 different high-power fields in three sections of every tissue sample. The number of infiltrating cells was expressed as the mean value with standard deviation.
Quantification of mRNA levels by RT-PCR.
After the rats were challenged with LPS, the right lung was removed and snap frozen in liquid nitrogen. Total cellular RNA was extracted using the acid guanidinium thiocyanate–phenol–chloroform extraction method (8). First-strand cDNA was synthesized from 20 μg of total RNA using a mixture of oligo(dT)12–18 and random hexamer primers and with Superscript reverse transcriptase (RT) (Gibco/BRL, Paisley, United Kingdom). The reaction mixture was incubated for 75 min at 37°C, and the reaction was terminated by heating to 95°C for 5 min. Thereafter, serial dilutions (1:3) were prepared from cDNA and used as a template in PCR. The primers used for amplification of specific cDNA fragments are listed in Table 1. A 460-bp fragment of rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as a control. Primer sequences for GAPDH were separated by introns to control for contamination with genomic DNA. The amplification reactions were allowed to proceed for 30 cycles, each consisting of a 1-min denaturation step at 94°C, a 30-s annealing step at 63°C, and a 90-s extension step at 72°C. The identity of the amplification products was confirmed in each case by restriction enzyme analysis. The final cDNA dilution yielding detectable amplification products was scored for each sample. To normalize mRNA levels, the cDNA titers for chemokines, cytokines, and adhesion molecules were divided by the GAPDH titers obtained from the same cDNA template.
TABLE 1.
Primers for RT-PCR analysis of lung adhesion molecule, chemokine, and cytokine expression
| Target | Size of product (bp) | Sequence |
|---|---|---|
| GAPDH sense | 460 | 5′-TGC-ATC-CTG-CAC-CAC-CAA-CT-3′ |
| GAPDH antisense | 460 | 5′-GCC-CCA-GCA-TCA-AAG-GTG-G-3′ |
| ICAM-1 sense | 485 | 5′-GCG-GCC-TTG-GAG-GTG-GAT-G3′ |
| ICAM-1 antisense | 485 | 5′-GGA-GGC-GGG-GCT-TGT-ACC-3′ |
| VCAM-1 sense | 471 | 5′-CCT-GTC-CCA-GAG-GAG-GGC-3′ |
| VCAM-1 antisense | 471 | 5′-CAA-CTG-CGA-GCC-GAC-TTC-G-3′ |
| P-selectin sense | 427 | 5′-CGG-GGC-TTC-AGG-ACA-ATG-G-3′ |
| P-selectin antisense | 427 | 5′-TGG-GTC-ATA-TGC-AGC-GTT-CG-3′ |
| E-selectin sense | 362 | 5′-GCA-CCA-CAG-CGA-GGC-CAC-3′ |
| E-selectin antisense | 362 | 5′-CGG-GGA-GGG-TTG-GCT-GGG-3′ |
| KC sense | 476 | 5′-ATG-GTC-TCA-GCC-ACC-CGC-T-3′ |
| KC antisense | 476 | 5′-CGA-TGA-AAC-GCA-TCC-ACA-TCA-3′ |
| IP-10 sense | 357 | 5′-AAC-TGT-CCC-TGT-TTC-TCC-TG-3′ |
| IP10 antisense | 357 | 5′-GGA-AGG-TGG-TGG-TAA-GTT-TG3′ |
| MIP-2 sense | 329 | 5′-GCT-CCT-CAA-TGC-TGT-ACT-GG-3′ |
| MIP-2 antisense | 329 | 5′-GAT-TCT-GCC-CGT-TGA-GGT-AC-3′ |
| MCP-1 sense | 440 | 5′-TGC-AGG-TCT-CTG-TCA-GGC-TT-3′ |
| MCP-1 antisense | 440 | 5′-CTC-TGT-CAT-ACT-GGT-CAC-TTC-3′ |
| MIP-1α sense | 275 | 5′-GGT-CTC-CAC-CAC-TGC-CCT-T-3′ |
| MIP-1α antisense | 275 | 5′-TCA-GGC-ATT-CAG-TTC-CAG-CTC-3′ |
| IL-12 p40 sense | 399 | 5′-AGC-ACC-AAA-CTA-CTC-CGG-AC-3′ |
| IL-12 p40 antisense | 399 | 5′GGA-GTG-CTC-CAG-GAG-TCA-G-3′ |
RNase protection assay.
For quantification of rat cytokine RNA (IL-1α, IL-1β, IL-6, and TNF-α) levels in the lungs, the RiboQuant multiprobe RNase protection assay (rCK-1; PharMingen) was performed as specified by the manufacturer. Briefly, RNA probes were synthesized for 1 h by T7 RNA polymerase in the presence of [α-32P]UTP and the reaction was terminated by adding DNase. From each lung sample, 10 μg of total RNA was hybridized with the labeled RNA probe. Nonhybridizing RNA was removed by RNase A digestion, and this reaction was terminated with proteinase K. After phenol-chloroform extraction, probes were run on an acrylamide gel together with a dilution of the probe set serving as size markers. The gels were dried and exposed on a phosphorimager screen. With the undigested probes as markers, a standard curve was plotted and used to establish the identity of RNase-protected bands in the experimental samples. Cytokine mRNA levels were normalized to the L32 standard.
Hemolytic complement activity.
The total hemolytic activity (CH50) was determined as previously described (3). Briefly, sheep red blood cells (SRBC) were sensitized with rabbit anti-SRBC serum (Accurate) diluted 1:100. SRBC were added to serially diluted serum samples in 96-well U-bottom plates in triplicate. After incubation at 37°C for 60 min, the plates were centrifuged at 1,100 × g at 4°C for 5 min. Supernatants were transferred to fresh 96-well plates to measure the hemoglobin (Hgb) release by determination of the optical density at 405 μm with an automated scanner. The percent maximal Hgb release was calculated for each dilution as follows:
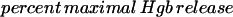 |
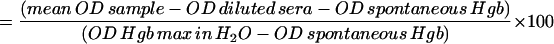 |
CH50 was defined as the serum dilution yielding 50% of maximal Hgb release.
Western blot analysis.
Nonreducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (7% polyacrylamide) was performed using the method of Laemmli (13). Serum samples were applied at at a dilution of 1:50. Proteins were electrophoretically transferred to nitrocellulose paper (Schleicher & Schuell, Keene, N.H.), and nonspecific antibody binding was blocked by overnight incubation in Blotto (20% milk powder, 0.05% NaN3). C6 was detected with goat anti-human C6 antiserum (Calbiochem) followed by biotinylated mouse anti-human Ab and alkaline phosphatase-streptavidin. Polyclonal goat anti-human IgG (Dianova, Hamburg, Germany) was used for the isotype control. Staining was performed with 64 λ BCIP (5-bromo-4-chloro-3-indolylphosphatase) in conjunction with 132 λ NBT (nitroblue tetrazolium [Promega, Madison, Wis.]) in 20 ml of substrate buffer (100 mM Trizma base, 100 mM NaCl, 5 mM MgCl2 [pH 9.5]). The color reaction was stopped with a buffer containing 20 mM Trizma base and 5 mM EDTA (pH 8).
Statistical analysis.
Data are represented as means and standard deviations. For statistical analysis, Student's t test was used. Statistical significance was defined as P < 0.01.
RESULTS
Complement deficiency in PVG (C−) rats.
Both PVG rat strains were tested for hemolytic complement activity. The CH50 titer of PVG (C+) ranged between 40 and 80. PVG (C−) rats had no detectable hemolytic complement activity in CH50 assays (Fig. 1). Western blot analysis of serum samples revealed a strong C6 signal at 85 kDa in PVG (C+) rats, whereas PVG (C−) rats had no detectable C6 expression (Fig. 2). These results therefore confirm the C6 complement deficiency of PVG (C−) rats.
FIG. 1.

Hemolytic complement activity of PVG (C+) and PVG (C−) serum. Hemolytic complement activity of serum from PVG (C+) rats was expressed as the CH50 titer, which ranged between 40 and 80 (50% of hemolysis of presensitized sheep RBC). Serum of PVG (C−) rats had no hemolytic complement activity.
FIG. 2.

C6 production in PVG (C+) and PVG (C−) rats. Western blot analysis of serum samples from PVG (C+) and PVG (C−) rats was performed using a goat anti-human C6 polyclonal Ab. Polyclonal goat anti-human IgG Ab was used as a control. A strong band corresponding to C6 in the serum of PVG (C+) rats was detectable at 95 kDa. The serum of PVG (C−) rats and the control was negative for C6.
Lung cytokine induction by LPS in C6-deficient rats.
Lung cytokine production was analyzed following i.p. injection of low-dose (0.5 mg/kg) and high-dose (5 mg/kg) LPS in PVG (C−) and PVG (C+) rats. Induction of mRNA for TNF-α, IL-1α, IL-1β, and IL-6 was determined 4 h after LPS injection by an RNase protection assay. The results for the 0.5- and 5-mg/kg groups are given in Fig. 3 and demonstrate a strong increase in the cytokine mRNA levels. Cytokine mRNA induction was about twofold stronger in the high-dose group than in the low-dose group. Importantly, we did not observe significant differences in lung cytokine induction between PVG (C−) and PVG (C+) rats in the low-dose or high-dose group (Fig. 3 and data not shown). Furthermore, TNF-α levels in serum, which transiently peaked at 1.5 h after LPS administration, were comparable in PVG (C−) and PVG (C+) rats (data not shown). We therefore conclude that the complement MAC is not required for LPS-stimulated cytokine synthesis in the lungs.
FIG. 3.

Lung cytokine production. Cytokine release of TNF-α, IL-6, IL-1β, and IL-1α in PVG (C−) and PVG (C+) rats was determined by an RNase protection assay 4 h after i.p. challenge with LPS (5 or 0.5 mg/kg LPS). Data are expressed as percent RNA levels relative to the L32 standard. There was no significant differences in cytokine induction between PVG (C−) and PVG (C+) rats (n = 4).
Induction of chemokines and adhesion molecules in lungs.
The production of the chemokines KC, MIP-2, MCP-1, MIP-1α, and IP-10 in the lung was determined by RT-PCR analysis 4 h after LPS injection into PVG (C−) or PVG (C+) rats. The challenge with 0.5 mg/kg caused only a weak induction of lung chemokines (data not shown), whereas the increase of KC, MCP-1, MIP-1α, and IP-10 mRNA levels was about 20- to 200-fold after injection of high-dose (5 mg/kg) LPS (Fig. 4). Stimulation of MIP-2 production was consistently lower than that of other chemokines. However, there were no significant differences between PVG (C−) and PVG (C+) rats at low or high LPS concentrations (Fig. 4 and data not shown).
FIG. 4.

Induction of lung chemokine expression after LPS (5 mg/kg) challenge. Total RNA was isolated from the lungs of PVG (C−) and PVG (C+) rats. Expression of MCP-1, MIP-1α, KC, IP-10, and MIP-2 was determined before (open symbols) or 4 h after (solid symbols) i.p. challenge with LPS. Expression of chemokines was examined by RT-PCR analysis. To normalize mRNA levels, the cDNA titer of each chemokine, defined as the final dilution yielding detectable amplification products, was divided by the GAPDH titer derived from the same cDNA template. Each symbol represents the result from the lungs of a single rat (n = 4).
In additional experiments, the mRNA expression of the adhesion molecules P-selectin, E-selectin, ICAM-1, and VCAM-1 was investigated by RT-PCR analysis 4 h after injection of LPS into PVG (C−) or PVG (C+) rats. Although a strong induction of P-selectin mRNA was observed after challenge with high-dose LPS (5 mg/kg), there were no significant differences between PVG (C−) and PVG (C+) rats (Figs. 5 and 6). P-selectin mRNA induction was not detected following injection of low-dose LPS 0.5 mg/kg (data not shown). In contrast, E-selectin, ICAM-1, and VCAM-1 mRNA levels were not significantly increased after challenge with either low- or high-dose LPS (Fig. 5 and 6 and data not shown). Together, our results indicate that the MAC of the complement system is not required for the induction of lung chemokines and adhesion molecules in response to systemic LPS administration.
FIG. 5.

RT-PCR analysis of lung adhesion molecule induction. Representative gels (n = 4) of the RT-PCR analysis of the adhesion molecules P-selectin, E-selectin, and VCAM-1, 4 h after challenge with 5 mg of LPS/kg, are shown. The GAPDH controls of the same samples are also depicted. Serial cDNA dilutions (1:3) were used as template for PCR analysis. The most intense induction was found for P-selectin.
FIG. 6.

Lung adhesion molecule induction. Induction of lung adhesion molecules 4 h after LPS challenge with 5 mg of LPS/kg was determined by RT-PCR as described in the legend to Fig. 4. Total RNA was isolated from the lungs of PVG (C−) and PVG (C+) rats. There was a significant induction of P-selectin after LPS challenge, but there was no difference between PVG (C−) and PVG (C+) rats (n = 4). The adhesion molecules E-selectin, ICAM-1, and VCAM-1 were not significantly induced by LPS challenge with 5 mg of LPS/kg.
Induction of lung infiltration by CD11b/c+ leukocytes.
CD11b/c is expressed on multiple leukocyte subsets, including neutrophils, mononuclear phagocytes, and NK cells, and it was detected by immunoperoxidase staining of lung sections. Challenge with high-dose (5 mg/kg) LPS resulted in a strong increase of CD11b/c+ infiltrating cells in lungs removed after 4, 8, and 12 h. However, there was no difference in the intensity of the CD11b/c+ cellular infiltrate after LPS administration between PVG (C−) and PVG (C+) rats. Representative immunohistochemistry samples are shown in Fig. 7. Challenge with low-dose (0.5 mg/kg) LPS also did not reveal any differences in lung leukocyte infiltration between PVG (C−) and PVG (C+) rats (data not shown). Thus, LPS-induced leukocyte recruitment to the lungs is also C6 independent.
FIG. 7.

Cellular infiltration of CD11b/c+ cells after LPS induction. LPS challenge is associated with an intense accumulation of CD11b/c+ leukocytes in the lungs. Lungs were removed 4 h following i.p. injection of 5 mg of LPS/kg. For immunohistochemical analysis of leukocyte infiltration, sections of snap-frozen tissue samples of PVG (C+) (B) or PVG (C−) rats (C) were incubated with MAb CD11b/c. Control sections were incubated with isotype-matched control Ab (A). MAb reactivity was detected using an immunoperoxidase technique. (Magnification, × 180).
DISCUSSION
It is well known that cytokines such as TNF-α and IL-1β play central roles in endothelial cell activation and leukocyte-endothelial cell interactions during inflammation (2, 24). Early activated components of the complement cascade, including the soluble anaphylatoxins C3a and C5a and surface-bound iC3b and C3b, are also important proinflammatory mediators (9, 15). Although the function of the distal complement product (C5b-C9) in cytolysis has been extensively studied, its role in inflammatory processes is less well defined. Previous studies have shown that activation of complement factors close to the surface of endothelial monolayers resulted in the rapid upregulation of P-selectin. Further analysis suggested that the MAC was responsible for P-selectin expression (11). In vitro studies with human umbilical vein endothelial cells showed an enhanced upregulation of ICAM-1 and E-selectin after treatment with TNF-α and preassembled complement MAC (12). Using complement depletion by CVF, Vaporciyan et al. (31) observed an in vivo relationship between complement factors and upregulation of lung vascular ICAM-1, which was dependent on the presence of TNF-α. However, the complement components responsible for ICAM-1 regulation could not be identified using this approach.
PVG (C−) and PVG (C+) rats are genetically identical except for the deficiency in C6, which was demonstrated in various skin, heart, and liver transplant models (4, 5). The C6 deficiency in PVG (C−) rats is probably caused by an unstable mRNA or a point mutation in the C6 gene, resulting in an aberrant transcription of the C6 gene (30). In the present study, a defined endotoxin-mediated septic shock was induced in the recently described model of C6-deficient PVG (C−) (RT1C) rats, which are unable to assemble the distal complement factors to the MAC. The congenic PVG (C+) rats (RT1C) (3, 5, 14) served as controls. These rat strains were used to investigate whether the suggested synergism between TNF-α and the MAC on the expression of P-selectin and ICAM-1 and possibly on other components of the inflammatory reaction is critical for the endotoxin-mediated lung injury.
The mRNA induction of the cytokines TNF-α, IL-1, and IL-6 in the lungs was nearly identical in PVG (C−) and PVG (C+) rats following LPS injection. Thus, it appears unlikely that differences in local cytokine concentrations between C6-deficient and control rats affect the results of chemokine and adhesion molecule analysis. Extensive analysis of the mRNA levels of P-selectin and E-selectin 4 h after challenge with LPS revealed only minor, statistically insignificant differences between PVG (C−) and PVG (C+) rats. Additional experiments demonstrated that chemokine upregulation in response to systemic LPS administration was also comparable in PVG (C−) and PVG (C+) rats. Consistent with these results, we were not able to detect any differences in lung leukocyte recruitment between these rat strains.
There are several possible reasons, why the recently described synergism (12, 31) between TNF-α and the MAC on the expression of the adhesion molecules was not confirmed in the endotoxic shock model. First, potential costimulatory effects of the MAC may be detectable only at submaximal LPS dosages. We have therefore analyzed C6-deficient rats injected with low-dose LPS. This treatment resulted in a partial induction of cytokines, chemokines, and adhesion molecules, demonstrating submaximal LPS stimulation. However, even under these conditions, there were no significant differences between PVG (C+) and PVG (C−) rats. Second, the synergy between the MAC and TNF-α for adhesion molecule induction was shown only in vitro and with the use of preassembled MAC (12, 31). It is therefore possible that this in vitro activity of the MAC either is not be operative in vivo or is replaced by other inflammatory mediators. Third, the role of complement for the upregulation of lung ICAM-1 was shown by complement depletion with CVF (28). It should be considered, however, that CVF treatment generates activated complement factors and numerous other potent inflammatory stimuli. Thus, mediators released in response to CVF may alter the reactivity to a secondary inflammatory stimulus, or complement components other than the MAC may be involved in lung inflammation. A recent report on CVF-mediated lung injury is consistent with this notion (19). Finaly, the role of complement or MAC may be dependent on the nature of the inflammatory stimulus. Studies showing different adhesion molecule requirements for immune complex-induced (18, 20) and CVF-induced (17, 23) lung injury support this hypothesis. It is therefore also conceivable that complement or the MAC is involved in IgG immune complex-dependent but not LPS-dependent lung inflammation.
In summary, we conclude that the MAC of the complement system is not required for the development of lung inflammation in response to a systemic endotoxin stimulus.
ACKNOWLEDGMENT
This work was supported by DFG grant BR1294/4-1.
REFERENCES
- 1.Beutler B, Cerami A. Tumor necrosis, cachexia, shock, and inflammation: a common mediator. Annu Rev Biochem. 1988;57:505–518. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 2.Bevilacqua M P, Pober J S, Mendrick D L, Cotran R S, Gimbrone M A., Jr Identification of an inducible endothelial-leukocyte adhesion molecule. Proc Natl Acad Sci USA. 1987;84:9238–9242. doi: 10.1073/pnas.84.24.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brauer R B, Baldwin III W M, Daha M R, Pruitt S K, Sanfilippo F. Use of C6-deficient rats to evaluate the mechanism of hyperacute rejection of discordant cardiac xenografts. J Immunol. 1993;151:7240–7248. [PubMed] [Google Scholar]
- 4.Brauer R B, Baldwin III W M, Ibrahim S, Sanfilippo F. The contribution of terminal complement components to acute and hyperacute allograft rejection in the rat. Transplantation. 1995;59:288–293. [PubMed] [Google Scholar]
- 5.Brauer R B, Baldwin III W M, Wang D, Horwitz L R, Hess A D, Klein A S, Sanfilippo F. Hepatic and extrahepatic biosynthesis of complement factor C6 in the rat. J Immunol. 1994;153:3168–3176. [PubMed] [Google Scholar]
- 6.Brigham K L, Meyrick B. Endotoxin and lung injury. Am Rev Respir Dis. 1986;133:913–927. [PubMed] [Google Scholar]
- 7.Broaddus V C, Boylan A M, Hoeffel J M, Kim K J, Sadick M, Chuntharapai A, Hebert C A. Neutralization of IL-8 inhibits neutrophil influx in a rabbit model of endotoxin-induced pleurisy. J Immunol. 1994;152:2960–2967. [PubMed] [Google Scholar]
- 8.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate- phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 9.Foreman K E, Vaporciyan A A, Bonish B K, Jones M L, Johnson K J, Glovsky M M, Eddy S M, Ward P A. C5a-induced expression of P-selectin in endothelial cells. J Clin Investig. 1994;94:1147–1155. doi: 10.1172/JCI117430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frevert C W, Huang S, Danaee H, Paulauskis J D, Kobzik L. Functional characterization of the rat chemokine KC and its importance in neutrophil recruitment in a rat model of pulmonary inflammation. J Immunol. 1995;154:335–344. [PubMed] [Google Scholar]
- 11.Hattori R, Hamilton K K, McEver R P, Sims P J. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem. 1989;264:9053–9060. [PubMed] [Google Scholar]
- 12.Kilgore K S, Shen J P, Miller B F, Ward P A, Warren J S. Enhancement by the complement membrane attack complex of tumor necrosis factor-α-induced endothelial cell expression of E-Selectin and ICAM-1. J Immunol. 1995;155:1434–1441. [PubMed] [Google Scholar]
- 13.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 14.Leenaerts P L, Stad R K, Hall B M, Van Damme B J, Vanrenterghem Y, Daha M R. Hereditary C6 deficiency in a strain of PVG/c rats. Clin Exp Immunol. 1994;97:478–482. doi: 10.1111/j.1365-2249.1994.tb06113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marks R M, Todd R F, Ward P A. Rapid induction of neutrophil-endothelial adhesion by endothelial complement fixation. Nature. 1989;339:314–317. doi: 10.1038/339314a0. [DOI] [PubMed] [Google Scholar]
- 16.Miller E J, Cohen A B, Nagao S, Griffith D, Maunder R J, Martin T R, Weiner-Kronish J P, Sticherling M, Christophers E, Matthay M A. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis. 1992;146:427–432. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- 17.Mulligan M S, Miyasaka M, Tamatani T, Jones M L, Ward P A. Requirements for L-selectin in neutrophil-mediated lung injury in rats. J Immunol. 1994;152:832–840. [PubMed] [Google Scholar]
- 18.Mulligan M S, Polley M J, Bayer R J, Nunn M F, Paulson J C, Ward P A. Neutrophil-dependent acute lung injury. Requirement for P-selectin (GMP-140) J Clin Investig. 1992;90:1600–1607. doi: 10.1172/JCI116029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulligan M S, Schmid E, Beck-Schimmer B, Till G O, Friedl H P, Brauer R B, Hugli T E, Miyasaka M, Warner R L, Johnson K J, Ward P A. Requirement and role of C5a in acute lung injury in rats. J Clin Investig. 1996;98:503–512. doi: 10.1172/JCI118818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulligan M S, Smith C W, Anderson D C, Todd R F d, Miyasaka M, Tamatani T, Issekutz T B, Ward P A. Role of leukocyte adhesion molecules in complement-induced lung injury. J Immunol. 1993;150:2401–2406. [PubMed] [Google Scholar]
- 21.Mulligan M S, Varani J, Dame M K, Lane C L, Smith C W, Anderson D C, Ward P A. Role of endothelial-leukocyte adhesion molecule 1 (ELAM-1) in neutrophil-mediated lung injury in rats. J Clin Investig. 1991;88:1396–1406. doi: 10.1172/JCI115446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulligan M S, Watson S R, Fennie C, Ward P A. Protective effects of selectin chimeras in neutrophil-mediated lung injury. J Immunol. 1993;151:6410–6417. [PubMed] [Google Scholar]
- 23.Mulligan M S, Wilson G P, Todd R F, Smith C W, Anderson D C, Varani J, Issekutz T B, Miyasaka M, Tamatani T, Rusche R J, Vaporciyan A A, Ward P A. Role of beta 1, beta 2 integrins and ICAM-1 in lung injury after deposition of IgG and IgA immune complexes. J Immunol. 1993;150:2407–2417. [PubMed] [Google Scholar]
- 24.Pober J S, Lapierre L A, Stolpen A H, Brock T A, Springer T A, Fiers W, Bevilacqua M P, Mendrick D L, Gimbrone M A., Jr Activation of cultured human endothelial cells by recombinant lymphotoxin: comparison with tumor necrosis factor and interleukin 1 species. J Immunol. 1987;138:3319–3324. [PubMed] [Google Scholar]
- 25.Schmal H, Shanley T P, Jones M L, Friedl H P, Ward P A. Role for macrophage inflammatory protein-2 in lipopolysaccharide-induced lung injury in rats. J Immunol. 1996;156:1963–1972. [PubMed] [Google Scholar]
- 26.Shanley T P, Schmal H, Friedl H P, Jones M L, Ward P A. Role of macrophage inflammatory protein-1 alpha (MIP-1 alpha) in acute lung injury in rats. J Immunol. 1995;154:4793–4802. [PubMed] [Google Scholar]
- 27.Standiford T J, Kunkel S L, Lukacs N W, Greenberger M J, Danforth J M, Kunkel R G, Strieter R M. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. 1995;155:1515–1524. [PubMed] [Google Scholar]
- 28.Till G O, Ward P A. Systemic complement activation and acute lung injury. Fed Proc. 1986;45:13–18. [PubMed] [Google Scholar]
- 29.Ulich T R, Howard S C, Remick D G, Wittwer A, Yi E S, Yin S, Guo K, Welply J K, Williams J H. Intratracheal administration of endotoxin and cytokines. VI. Antiserum to CINC inhibits acute inflammation. Am J Physiol. 1995;268:245–250. doi: 10.1152/ajplung.1995.268.2.L245. [DOI] [PubMed] [Google Scholar]
- 30.van Dixhoorn M G, Timmerman J J, Van Gijlswijk-Janssen D J, Muizert Y, Verweij C, Discipio R G, Daha M R. Characterization of complement C6 deficiency in a PVG/c rat strain. Clin Exp Immunol. 1997;109:387–396. doi: 10.1046/j.1365-2249.1997.4551354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaporciyan A A, Mulligan M S, Warren J S, Barton P A, Miyasaka M, Ward P A. Up-regulation of lung vascular ICAM-1 in rats is complement dependent. J Immunol. 1995;155:1442–1449. [PubMed] [Google Scholar]
- 32.Varki A. Selectins and other mammalian sialic acid-binding lectins. Curr Opin Cell Biol. 1992;4:257–266. doi: 10.1016/0955-0674(92)90041-a. [DOI] [PubMed] [Google Scholar]
- 33.Ward P A, Marks R M. The acute inflammatory reaction. Curr Opin Immunol. 1989;2:5–9. doi: 10.1016/0952-7915(89)90090-3. [DOI] [PubMed] [Google Scholar]