

Abstract
Acute myeloid leukemia (AML) is driven by numerous molecular events that contribute to disease progression. Herein, we identify hnRNP K overexpression as a recurrent abnormality in AML that negatively correlates with patient survival. Overexpression of hnRNP K in murine fetal liver cells results in altered self-renewal and differentiation potential. Further, murine transplantation models reveal that hnRNP K overexpression results in myeloproliferation in vivo. Mechanistic studies expose a direct functional relationship between hnRNP K and RUNX1—a master transcriptional regulator of hematopoiesis often dysregulated in leukemia. Molecular analyses show that overexpression of hnRNP K results in an enrichment of an alternatively spliced isoform of RUNX1 lacking exon 4. Our work establishes hnRNP K’s oncogenic potential in influencing myelogenesis through its regulation of RUNX1 splicing and subsequent transcriptional activity.
INTRODUCTION
Acute myeloid leukemia (AML) is an often-devastating hematologic malignancy wherein normal hematopoiesis is superseded by rapid proliferation of incompletely differentiated myeloid cells. Identification of recurrent genomic events in AML (e.g. mutations in FLT3 or IDH1/2) has led to development of targeted therapeutic agents that improve patient outcomes and quality of life; however, many patients lack these genomic alterations, rendering them ineligible for such treatments. Furthermore, despite prolonging life, patients treated with these agents are often still at risk for relapse (1–4), highlighting the need to understand the molecular underpinnings of AML such that alternative, effective therapeutic options can be developed.
Myeloid malignancies, including AML and its frequently associated precursor condition, myelodysplastic syndrome (MDS), often have alterations in RNA-binding proteins (RBPs). Splicing factors such as SRSF2, SF3B1 and U2AF1 are widely known RBPs in this context, and are often mutated (5–7). Importantly, other RBPs such as MUSASHI2 and METTL3 are also aberrantly expressed, though infrequently mutated, in hematologic malignancies, and have been identified as critical to the pathogenesis of AML (8–11). However, the roles of other RBPs in myeloid malignancies, including AML, have not been extensively deciphered.
An RBP of accumulating interest in hematologic and solid malignancies is heterogeneous nuclear ribonucleoprotein K (hnRNP K). Overexpression of hnRNP K has been associated with adverse pathology in a handful of clinical studies evaluating solid tumors (12–16) and increased expression has been observed in cells in blast crisis in chronic myeloid leukemia (17). Further, we have previously shown that elevated hnRNP K expression is commonly observed in B-cell lymphoma (18). These findings led to the consideration that hnRNP K aberrancies may contribute to a broader array of hematologic malignancies.
In this study, we address the hypothesis that hnRNP K is overexpressed in AML which impacts clinical outcomes, and that such overexpression contributes to myeloid aberrations in a murine model. We find that hnRNP K overexpression leads to extramedullary hematopoiesis and gross hematopoietic abnormalities in mice. Mechanistically, we identified that hnRNP K modulates the expression of RUNX1, a crucial hematopoietic transcription factor that is commonly mutated or aberrantly expressed in leukemias including AML (19–21). hnRNP K interacts with RUNX1 RNA in a sequence-specific manner in humans and mice, and causes exclusion of RUNX1 exon 4, ultimately leading to expression of a more stable RUNX1 isoform (RUNX1 ΔEx4) with altered transcriptional activity. Furthermore, expression of RUNX1 ΔEx4 recapitulates an in vitro phenotype associated with hnRNP K overexpression, supporting the notion that hnRNP K mediates its hematopoietic alterations, at least in part, via altered RUNX1 splicing.
Taken together, these studies identify hnRNP K as a potential driver alteration in AML. In addition, hnRNP K-mediated alternative splicing of RUNX1 provides an alternate mechanism whereby RUNX1 expression is impacted in AML in the absence of mutations or translocations.
MATERIALS AND METHODS
Reverse phase protein array (RPPA) data
Data are publicly available at www.leukemiaatlas.org (22). High hnRNP K expression was defined as ≥1 standard deviation above the median hnRNP K expression of healthy controls.
Plasmids
For generation of stable cell lines, lentiviral plasmids containing full-length HNRNPK, full-length RUNX1(b) or RUNX1 ΔEx4 cDNA were cloned into the XhoI/NotI sites in the all-in-one tetracycline inducible lentiviral vector TRE3G-ORF-P2A-eGFP-PGK-Tet3G-bsd (TLO2026, transOMIC Technologies, Huntsville, AL) containing a C-terminal Flag tag. For retroviral plasmids used in fetal liver cells, the MSCV-AML1/ETO-IRES-GFP plasmid was obtained from Addgene (Addgene plasmid #60832; http://n2t.net/addgene:60832; RRID:Addgene_60832 (23)) and AML1/ETO cDNA was replaced with HNRNPK, RUNX1(b), or RUNX1 ΔEx4 amplified from cDNA obtained from 293T cells and the pINDUCER-21-RUNX1 plasmid (Addgene plasmid #97043; http://n2t.net/addgene:97043; RRID:Addgene_97043 (24)), respectively. cDNAs were cloned into XhoI/EcoRI sites in the MSCV-IRES-GFP plasmid. For generation of stably knocked-down cell lines, tetracycline-inducible human HNRNPK PGK-TurboRFP shRNAs were purchased from Dharmacon (clone ID: V3IHSPGR_10844995 mature antisense TCGACGAGGGCTCATATCA, targeting exon 10), and one targeting the 3’ UTR, referred to as shHNRNPK ex16-2 (clone ID: V3IHSPGR_9103684, mature antisense ATAAAATCCACTCACTCTG), and control PGK-TurboRFP (VSC11656, mature antisense TGGTTTACATGTTGTGTGA; Lafayette, CO).
Reporter assays to assess RUNX1 transcriptional activity were performed using the pMCSF-R-luc plasmid (Addgene plasmid #12420; http://n2t.net/addgene:12420; RRID:Addgene_12420) (25). The pCMV β-galactosidase plasmid was a gift from Dr. Vrushank Davé (University of South Florida).
Stable cell line generation
Cell lines were spun for 90 min at 600 × g with filtered viral supernatant from 293T cells transfected with indicated plasmids (see above), and pCMV-VSV-G/pCMV-dR8.2 for human cell lines (Addgene plasmid #8454; http://n2t.net/addgene:8454; RRID:Addgene_8454 and Addgene plasmid #8455; http://n2t.net/addgene:8455; RRID:Addgene_8455, respectively (26)) or pCL-ECO for fetal liver cells (Addgene plasmid #12371; http://n2t.net/addgene:12371; RRID:Addgene_12371 (27)). Human cells were selected in media containing antibiotics (puromycin or blasticidin, Fisher Scientific, Waltham, MA) followed by appropriate fluorescence (>90% required) prior to downstream assays. Cells were maintained in strict tetracycline-free conditions until induction with 0.2 μg/ml doxycycline (Millipore Sigma, Burlington, MA) for shRNA and 0.4 μg/ml doxycycline for overexpression. FLCs were sorted for GFP positivity 72 h after infection.
FLC isolation, transduction, and transplantation
All mouse studies were performed with approval from the Institutional Animal Care and Use Committee at MD Anderson under protocol 0000787-RN01/2. Pregnant wildtype CD45.2+ C57Bl/6 females were euthanized via CO2 exposure and cervical dislocation between days 13.5 and 16.5 of gestation, fetal livers were then dissected, and disrupted on a 70 μm filter into single-cell suspension. Cells were briefly subjected to red blood cell lysis (BDPharm Lyse, BD Biosciences, San Jose, CA) and resuspended in medium containing 37% DMEM (Corning Inc, Corning, NY), 37% Iscove's modified Dulbecco's Medium (Corning Inc, Corning, NY), 20% fetal bovine serum, 2% l-glutamine (200 mM; Corning Inc, Corning, NY), 100 U/ml penicillin/streptomycin (Millipore Sigma, Burlington, MA), 5 × 10−5 M 2-mercaptoethanol (Millipore Sigma, Burlington, MA), recombinant murine IL-3 (0.2 ng/ml), IL-6 (2 ng/ml), and SCF (20 ng/ml; Stem Cell Technologies, Vancouver, BC) at high density overnight at 37°C prior to retroviral transduction (23,28). For transplantation assays, NOD-scid-IL2R-gamma (NSG) mice were irradiated with 2.5 Gy prior to injection of 50 000 sorted cells into the retro-orbital sinus.
Immunoblotting
Cells were homogenized in NP40 lysis buffer and standard immunoblotting procedures were performed as previously described (18) using antibodies against hnRNP K (3C2), RUNX1 (EPR3099, both from Abcam, Cambridge, MA), β-actin (AC-15, Santa Cruz Biotechnology, Dallas, TX), Flag (F1804, Millipore Sigma, Burlington, MA), GFP (D5.1, Cell Signaling, Danvers, MA) and HSP90 (ADI-SPA-836-D, Enzo Life Sciences, East Farmingdale, NY).
Colony formation assay
GFP-sorted FLCs were cultured in Methocult (M3434, StemCell Technologies, Vancouver, BC). Colonies were counted after 7 days then gently disrupted in PBS and subjected to cytospin or flow cytometry.
Flow cytometry
Cells were pre-treated with murine Fc block (TruStain FcX, BioLegend, San Diego, CA) at room temperature for 15 min then incubated with antibodies Gr1 [RB6-8C5], CD11b [M1/70;], c-kit [2B8], CD45 [30F11] (all from BD Biosciences, East Rutherford, NJ) and Sca-1 (D7; eBioscience, San Diego, CA). Flow cytometry was performed on a Gallios flow cytometer (Beckman Coulter, Brea, CA). Data was analyzed using FlowJo (Beckton Dickinson, Franklin Lakes, NJ).
Tissue harvest
All tissue harvesting was performed in moribund mice. Spleen, liver, and sternum were collected and immediately fixed in 10% neutral-buffered formalin. Paraffin-embedded blocks were sectioned and stained with standard hematoxylin/eosin.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissues were deparaffinized in xylene and rehydrated in an alcohol gradient. Following antigen retrieval with citrate (pH 6.0), slides were incubated with 3% hydrogen peroxide/methanol prior to incubation with primary antibody at 4°C overnight. Primary antibodies: hnRNP K [3C2], CD3 [SP162], MPO [ab9535], CD14 [4B4F12] (Abcam, Cambridge, MA), CD34 [MEC14.7] and c-kit [2B8] (ThermoFisher Scientific, Waltham, MA). Antibody-protein interactions were visualized with Vectastain Elite ABC and DAB peroxidase substrate kits (Vector Laboratories, Burlingame, CA) and counterstained with nuclear fast red.
Peripheral blood analysis
Complete blood counts were performed with an ABX Pentra analyzer (Horiba, Kyoto, Japan) using blood from moribund mice.
RNA-sequencing
RNA was extracted and purified from GFP+ sorted FLCs, 72 h post-infection, using Zymo Quick-RNA columns (Zymo Research, Irvine, CA). Barcoded, Illumina-stranded total RNA libraries were prepared using the TruSeq Stranded Total RNA Sample Preparation Kit (Illumina, San Diego, CA). Briefly, 250 ng of DNase I-treated total RNA was depleted of cytoplasmic and mitochondrial ribosomal RNA (rRNA) using Ribo-Zero Gold (Illumina, San Diego, CA). After purification, RNA was fragmented using divalent cations and double stranded cDNA was synthesized using random primers. The ends of the resulting double stranded cDNA fragments were repaired, 5′-phosphorylated, 3’-A tailed, and Illumina-specific indexed adapters were ligated. The products were purified and enriched by 12 cycles of PCR to create the final cDNA library. The libraries were quantified by qPCR and assessed for size distribution using the 4200 TapeStation High Sensitivity D1000 ScreenTape (Agilent Technologies, Santa Clara, CA) then multiplexed three libraries per lane and sequenced on the Illumina HiSeq4000 sequencer (Illumina, San Diego, CA) using the 75 bp paired end format. The RNA-sequencing experiments were done in 3 biological replicates of FLCs infected with EV (n = 3) and hnRNP K plasmid (n = 3). The data has been deposited to Array Express (E-MTAB-11886).
RNA-seq and splicing analysis
Fastq files were pseudoaligned using Kallisto v0.44.0 (29) with 30 bootstrap samples to a transcriptome index based on the Mus musculus GRCm38 release (Ensembl). The resulting abundance data was further analyzed with Sleuth v0.30.0 (30) using models with covariates for both batch and condition. Gene-level 80 abundance estimates were calculated as the sum of transcripts per million (TPM) estimates of all transcripts mapped to a given gene. Differential transcript abundance was used to inform splice isoform changes. Wald tests were performed at both the gene and transcript level for the ‘condition’ covariate with a significance threshold of FDR <10%. Percent spliced in index (PSI) was calculated as previously described (31).
fRIP analysis
Previously published data deposited to the Gene Expression Omnibus (GSE126479) was cross-referenced with known tumor suppressors and oncogenes as identified in the COSMIC database.
Identification of putative hnRNP K binding sites
As described previously (18), a computer algorithm was used to scan transcripts of interest for two or more binding motifs within 19 nucleotides, which are defined as ‘TCCCG’, ‘TCCCT’, ‘TCCCA’, ‘ACCCT’, ‘ACCCA’ or ‘CCCC’.
Fluorescence anisotropy (FA)
Recombinant hnRNP K protein, produced in Escherichia coli as described previously (18), was serially diluted in PBS (0.1 nM to 10 μM) and incubated with 6-FAM labelled RNA oligonucleotides. FA values were measured with excitation wavelength 485 nm and emission wavelength 528 nm on a Synergy Neo multi-mode plate reader (BioTek, Winooski, VT). Data was fit to the following equation:  , where initial
, where initial  is represented by
is represented by 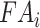 and the overall change in
and the overall change in  is represented by
is represented by  . Oligos: hRUNX1 int3-4 UCUCUUCCCUCCCUCCUUCCCUCCCCCCAU, hRUNX1 int3-4(mut) UCUGUUCGCUCGCUCGUUCGCUCGCGCCAU, mRunx1 int3-4 UCCUCCUCCCUUCCCCUCCCGGUCCCUA, mRunx1 int3-4(mut) UCCUCCUCGCUUCGCCUCGCGGUCGCUA.
. Oligos: hRUNX1 int3-4 UCUCUUCCCUCCCUCCUUCCCUCCCCCCAU, hRUNX1 int3-4(mut) UCUGUUCGCUCGCUCGUUCGCUCGCGCCAU, mRunx1 int3-4 UCCUCCUCCCUUCCCCUCCCGGUCCCUA, mRunx1 int3-4(mut) UCCUCCUCGCUUCGCCUCGCGGUCGCUA.
Thermal shift assays
Recombinant hnRNP K protein (18) was incubated with SYPRO orange dye in the presence of DNA oligos (the DNA equivalent to RNA oligos used in FA assays). Samples were heated from 25°C to 99°C in PBS buffer and fluorescence was measured at each temperature increment using a StepOne Plus Real Time PCR System (Applied Biosystem, Foster City, CA). No oligo and no protein controls were run on each plate. The melting temperature (Tm) of the protein sample was determined by calculating the first derivative of fluorescence at each temperature. The temperature at which the change in fluorescence was the lowest was determined to be the Tm of the sample as in (32).
qPCR for RUNX1 isoforms
RNA from OCI-AML3 cells was extracted using Zymo Quick-RNA columns (Zymo Research, Irvine, CA). Purified RNA was reverse transcribed using the iScript cDNA Synthesis kit (BioRad) to generate cDNA. qRT-PCR was performed with iTaq Universal SYBR Green SMX as per instructions on an ABI StepOnePlus Real Time PCR System. All assays were performed in triplicate. Changes in expression were compared using the Pfaffl method (33) by comparing expression changes between target genes and the housekeeping control (PPIA and RPLP0). Primers used were RUNX1 Exon4/5 forward primer (detects full length): CAGATGCAGGATACAAGGCAGATC, RUNX1 Exon3/5 forward primer (detects RUNX1 ΔEx4 isoform): AACCTCGAAATACAAGGCAGATC, RUNX1 universal reverse primer: TCGACTGGAAAGTTCTGC, PPIA forward: CCCACCGTGTTCTTCGACATT, PPIA reverse: GGACCCGTATGCTTTAGGATGA, RPLP0 forward: CCTTCTCCTTTGGGCTGGTCATCCA and RPLP0 reverse: CAGACACTGGCAACATTGCGGACAC.
Protein stability assays
293T cells were stably transduced with tetracycline-inducible constructs to overexpress RUNX1, either full-length or lacking exon 4. 0.4 μg/ml doxycycline was added to cells prior to the addition of cycloheximide (10 μM, Millipore Sigma, Burlington, MA) with or without MG-132 (10 μM, SelleckChem, Houston, TX) for 1–8 h. Cells were collected and lysed in NP40 lysis buffer with protease and phosphatase inhibitors prior to western blot.
Reporter assays
The M-CSF promoter reporter (pMCSF-R-luc Addgene plasmid #12420; http://n2t.net/addgene:12420; RRID: Addgene_12420 (25)) was used to assess RUNX1 transcriptional activity. 293T cells were transiently transfected with luciferase-based reporter plasmids and expression plasmids using jetPRIME (Polyplus, New York, NY). Total DNA quantity was constant across all wells. 48 h post-transfection, luciferase assay reagent was mixed in a 1:1 ratio with cell lysate (Luciferase Assay System kit, Promega, Madison, WI). Luciferase activity was measured with Synergy H4 Hybrid Reader (BioTek, Winooski, VT). Transfection efficiency for each well was normalized using 62.5 ng of a pCMV β-galactosidase plasmid, which was co-expressed in each experiment.
RESULTS
hnRNP K is overexpressed in many AML patient samples and correlates with poor clinical outcomes
We first assessed hnRNP K protein expression in AML using a publicly available reverse phase protein array (RPPA) dataset (22). While hnRNP K expression varied, AML cases had significantly higher median hnRNP K expression than healthy human bone marrow (P = 0.0056, Figure 1A). As expected, a small percentage of cases had decreased hnRNP K expression, consistent with previous descriptions of HNRNPK haploinsufficiency corresponding with del(9q) (34–36). Stratification of patients into high and low hnRNP K expression groups revealed that high hnRNP K expression correlated with a statistically significant decrease in overall survival (OS; 24.3 months versus 48.7 months; HR 1.9; 95% CI 1.3–2.7; P = 0.00034, Figure 1B), suggesting that hnRNP K overexpression may be involved in the pathology of AML.
Figure 1.

hnRNP K expression in de novo AML. (A) Relative hnRNP K protein expression as quantified by RPPA. CD34+ cells from healthy human donor bone marrows (n = 10) are indicated in black. Cells from bone marrows of patients with AML (n = 205) are shown in red and blue. Corresponding hnRNP K IHC from the bone marrow of healthy donors and AML patients is shown. Scale bar represents 50 μm. P-values were determined using a two-sided Mann-Whitney test. (B) Overall survival of AML patients with either high hnRNP K protein expression as determined by RPPA (red; n = 45) or normal/low hnRNP K expression (blue; n = 160). P-values were determined using a Log rank test. Data is publicly available at www.leukemiaatlas.org.
We considered the possibility that increased levels of hnRNP K could be a consequence of mutation. In line with the findings of others, only 2.9% of AML cases at our institution were found to have an HNRNPK mutation (Supplemental Figure S1A and B) (37–39). Given the discordance between this small mutational occurrence and the relatively large proportion of AML cases with elevated hnRNP K protein levels, we therefore posited that increased hnRNP K is of the wildtype form.
hnRNP K overexpression impacts the differentiation potential of fetal liver cells (FLCs)
To evaluate the role of hnRNP K overexpression in myeloid disease, we harvested murine fetal liver cells (FLCs) collected between embryonic days 13.5 and 16.5, when fetal livers are heavily enriched in hematopoietic stem and progenitor cells (40). Using a well-established retroviral transduction methodology (23), we infected FLCs with viral vectors overexpressing wild-type hnRNP K or an empty vector control (Figure 2A and Supplemental Figure S2A). Both viral vectors contained an IRES element followed by GFP allowing for fluorescence-based cell sorting. These vectors also allowed for expression of GFP protein in the empty vector samples facilitating a meaningful and appropriate comparison to cells with hnRNP K overexpression (Supplemental Figure S2B). Colony formation assays revealed that hnRNP K-overexpressing FLCs formed more colonies than empty vector expressing counterparts (P = 0.004, Figure 2B), suggesting that elevated hnRNP K influences the self-renewal capacity of FLCs. Flow cytometry revealed that hnRNP K-overexpressing colonies were composed of fewer Gr1+CD11b+ mature myeloid cells compared to controls (P = 0.0286, Figure 2C). However, immature c-kit+Sca-1+ cells were more prominent in hnRNP K-overexpressing colonies compared to controls (P = 0.0286, Figure 2D). These data suggested that hnRNP K overexpression hinders FLC differentiation into mature myeloid cells and may be involved in myeloid differentiation.
Figure 2.

hnRNP K overexpression in murine FLCs. (A) Western blot showing hnRNP K protein levels in FLCs infected with empty vector or hnRNP K plasmids with quantification (n = 3). Data are represented as mean ± SEM. (B) Representative brightfield images of colonies from FLCs infected with empty vector or hnRNP K plasmid. Scale bar represents 500 μm. Bar graph depicts quantification of the number of colonies per well in FLCs infected with empty vector (n = 14) and hnRNP K plasmids (n = 10). Data are represented as mean ± SD. P-values were determined using two-sided Mann–Whitney test. (C) Flow cytometry analysis of the percentage of GR1+CD11b+ FLCs infected with empty vector and hnRNP K plasmids with quantification (n = 4 per group). Data are represented as mean ± SEM. P-values were determined using a two-sided Mann–Whitney test. (D) Flow cytometry analysis of the percentage of c-kit+Sca-1+ cells in FLCs infected with quantification (n = 4 per group). Data are represented as mean ± SEM. P-values were determined using a two-sided Mann–Whitney test.
Overexpression of wildtype hnRNP K is sufficient to drive myeloproliferation in mice
To determine if overexpression of hnRNP K alone was sufficient to affect myeloproliferation, we injected murine FLCs overexpressing wild type hnRNP K or an empty vector control into sub-lethally irradiated NSG mice. A schematic of the murine experiment is presented in Supplemental Figure S3A. Recipients of hnRNP K-overexpressing FLCs had shortened survival compared to recipients of control FLCs (median survival 8.1 weeks versus median not reached at 36 weeks, HR 3.0, 95% CI 1.2–7.3, P = 0.018, Figure 3A). Recipients of hnRNP K-overexpressing FLCs had marked splenomegaly (P = 0.0039, Figure 3B and C), and histologic evaluation revealed starkly disrupted splenic architecture (Figure 3D).
Figure 3.

Phenotypes observed in mice transplanted with hnRNP K overexpressing FLCs. (A) Kaplan-Meier curves indicating survival of mice transplanted with FLCs infected with an empty vector (n = 13) and hnRNP K overexpression plasmid (n = 22). P-values were determined using a Log rank test. (B) Representative photos of spleens from mice transplanted with empty vector and hnRNP K overexpressing FLCs. (C) Bar graph depicting spleen weights from mice transplanted with empty vector (n = 12) and hnRNP K overexpressing (n = 21) FLCs. Data are represented as mean ± SD. P-values were determined using a two-sided Mann-Whitney test. (D) H&E and immunohistochemical analyses for hnRNP K in spleen samples from mice transplanted with empty vector or hnRNP K-overexpressing FLCs. The scale bar represents 200 μm. (E) H&E staining in bone marrow samples obtained from mice transplanted with empty vector and hnRNP K-overexpressing FLCs. The scale bar represents 50 μm.
Recipients of hnRNP K-overexpressing FLCs also harbored infiltrating leukocytes in the hepatic parenchyma that were not present in mice transplanted with empty vector-containing FLCs (Supplemental Figure S3B). These hepatic infiltrates observed in recipients of hnRNP K-overexpressing FLCs were largely negative for CD3, but positive for c-kit, CD14, and myeloperoxidase (MPO; Supplemental Figure S3C), indicating that immature hematopoietic cells and cells of myeloid origin were present in this organ. Notably, lack of CD3 expression largely ruled out a graft versus host effect.
Analysis of peripheral blood revealed that recipients of hnRNP K-overexpressing FLCs had leukocytosis without significant changes in hemoglobin and platelet counts (Supplemental Figure S3D–F), suggesting hnRNP K-overexpression appears to selectively affect the leukocyte compartment in our murine model. Indeed, mature neutrophils were less abundant in peripheral blood of mice transplanted with hnRNP K-overexpressing FLCs, while circulating monocytes or eosinophils did not differ between groups (Supplemental Figure S3G–I). On the other hand, hnRNP K-overexpression caused an increase in lymphocytes (Supplemental Figure S3J).
Analysis of bone marrow revealed that recipients of hnRNP K-overexpressing FLCs had more cellular bone marrow, as well as an enrichment of myeloid cells when compared to control recipients (Figure 3E). Taken together, these data indicate that hnRNP K overexpression in hematopoietic stem and progenitor cells is sufficient to drive myeloproliferation in mice.
hnRNP K binds to and influences the alternative splicing of the RUNX1 transcript
hnRNP K is an RNA-binding protein capable of influencing many facets of cellular biology including transcription, translation, and splicing. To elucidate the mechanism by which hnRNP K affects myeloid development, we first performed RNA-Seq on hnRNP K-overexpressing FLCs (Supplemental Figure S4A, E-MTAB-11886). Pathway analysis uncovered several pathways implicated in cancer, but splicing emerged as a highly differentially regulated pathway (Supplemental Figure S4B). Analysis of alternatively spliced isoforms resulted in a list of ∼900 differentially spliced genes (q<0.01), which we cross-referenced against known oncogenes implicated in hematological malignancies (obtained from COSMIC (41), cancer.sanger.ac.uk). This resulted in a list of 15 differentially spliced oncogenes which we then cross-referenced against our previously published RNA-immunoprecipitation dataset in OCI-AML3 cells to identify transcripts that hnRNP K directly interacted with (GSE126479).
Here, we identified three putative candidates: XPO1, RNF213 and RUNX1 (Figure 4A). Given RUNX1’s known role in leukemogenesis, we further explored its relationship with hnRNP K. Our splicing data revealed an enrichment in an isoform of the Runx1 transcript, which lacks exon 4, when hnRNP K is overexpressed (Supplemental Figure S4C, D). Runx1 isoforms lacking exon 4 (RUNX1 ΔEx4) have also been referred to as Exon 6 in the context of Runx1c isoform in the literature (31,42,43).
Figure 4.

Mechanistic basis for the oncogenicity of hnRNP K. (A) Venn diagram depicting the intersection of differentially spliced genes from RNA-Seq in FLCs, hnRNP K targets previously identified in our fRIP dataset (GSE126479), and oncogenes in hematological malignancies (COSMIC database). (B) Identification of putative hnRNP K binding sites in human and murine RUNX1 transcripts using a previously generated computer algorithm. (C, D) Fluorescence anisotropy binding curves for purified full-length hnRNP K with FAM-labeled human (C) and murine (D) RUNX1 intron 3–4 wild-type and mutant oligos. Binding assays were performed in triplicate. Data are represented as mean ± SD.
In order to assess the potential of a direct hnRNP K-RUNX1 interaction, we looked for hnRNP K binding sites in both mouse and human RUNX1 transcripts. hnRNP K interacts with RNA enriched in poly(C) residues through its three K-homology (KH) domains (44). We previously developed a computer algorithm (18) to detect triple-C sequences flanked by pre-specified nucleotides spaced less than 19 nucleotides apart, a sequence motif known to be enriched in hnRNP K targets. Using this algorithm, we identified a conserved hnRNP K binding site near the 3’ splice site of the intron 3- exon 4-junction in both human and murine sequences (Figure 4B), which is consistent with a previous report of a similar site in the rat homolog (45).
To assess whether hnRNP K directly bound these sites in RUNX1 RNA, we performed fluorescence anisotropy assays. Purified hnRNP K protein stringently bound the intron 3–4 site in both human and mouse RUNX1 (Kd = 13.5 nM and 14.5 nM respectively, Figures 4C-D). This hnRNP K-RUNX1 interaction was abrogated when the hnRNP K consensus binding site was mutated (Kd = 3710 and 425.9 nM respectively, Figures 4C-D), indicating that hnRNP K binds RUNX1 directly and in a sequence-specific manner. These findings were confirmed in thermal shift assays, where hnRNP K was stabilized (i.e. had an increased melting temperature which is indicative of binding) in the presence of an oligo derived from the human RUNX1 intron 3–4 sequence (Supplemental Figure S4E). Consistent with the fluorescence anisotropy assays, mutations in the hnRNP K binding site in the RUNX1 oligos resulted in an attenuated increase in the melting temperature compared to WT RUNX1, indicating a loss in protein/oligo binding (Supplemental Figure S4E).
Taken together, these findings indicate that hnRNP K directly associates with the RUNX1 transcript near 3’ splice site of the intron 3-exon 4 junction in a sequence-specific manner resulting in enriched expression of the RUNX1 ΔEx4 isoform.
hnRNP K expression levels impact the generation of the RUNX1Δex4 splice isoform
To explicitly understand the functional impact of hnRNP K levels on cell viability and RUNX1 splicing, we developed OCI-AML3 cell lines wherein we could inducibly knockdown or overexpress hnRNP K upon doxycycline addition. To determine the functional consequence of altering hnRNP K levels in human AML cell lines, we performed viability assays in OCI-AML3 cells with inducible knock-down of HNRNPK. Our results indicated that knockdown of hnRNP K results in a reduced viability of the OCI-AML3 cells using two different hairpins (targeting Exon 10 in the coding sequence of HNRNPK, shHNRNPK Ex10, and targeting Exon 16 in the 3’UTR of HNRNPK, shHNRNPK Ex16-2) (Supplemental Figure S5A). To quantitate changes in RUNX1 splicing upon alterations in hnRNP K levels, we designed qRT-PCR primers spanning the exon 4–5 junction (for full length RUNX1) and the exon 3–5 boundary (for RUNX1 ΔEx4). Doxycycline-mediated knockdown of HNRNPK for 72 h using two hairpins targeting HNRNPK revealed that decreased hnRNP K expression reduced the expression ratio of the ΔEx4 isoform relative to the full-length isoform of RUNX1 (RUNX1 FL) by about 30–40% (Figure 5A and Supplemental Figure S5A-B). This observation correlated with a corresponding decrease in the ratio of RUNX1 ΔEx4 to RUNX1 FL on the protein level as well (Figure 5B).
Figure 5.

hnRNP K impacts RUNX1 alternative splicing. (A) Ratio of RUNX1 ΔEx4 to RUNX1 full length isoforms as determined by RT-PCR in OCI-AML3 cells with inducible knock-down of HNRNPK (72 h post-induction, n = 3 per group). Data are represented as mean ± SEM. P-values were calculated using an ANOVA test with a post-hoc Dunnett's multiple comparison test. (B) Western blot of hnRNP K and RUNX1 expression in inducible OCI-AML3 cells with inducible knock-down of HNRNPK, with quantification (72 h post-induction, n = 3 per group). Data are represented as mean ± SEM. P-values were calculated using an ANOVA test with a post-hoc Dunnett's multiple comparison test. (C) Ratio of RUNX1 ΔEx4 to RUNX1 full length isoforms as determined by RT-PCR in OCI-AML3 cells with inducible overexpression of HNRNPK (72 h post-induction, n = 3 per group). Data are represented as mean ± SEM. P-values were determined using a two-sided Mann-Whitney test. (D) Western blot of hnRNP K and RUNX1 expression in OCI-AML3 cells with inducible overexpression of hnRNP K with quantification (72 h post-induction, n = 3 per group). Data are represented as mean ± SEM. P-values were determined using a two-sided Mann–Whitney test.
Conversely, doxycycline-mediated overexpression of hnRNP K had a relative enrichment of RUNX1 ΔEx4 transcript compared to cells expressing an empty vector control (Figure 5C and Supplemental Figure S5D-F). Again, this alteration correlated with an increase in RUNX1 ΔEx4: RUNX1 FL expression at the protein level (Figure 5D, P = 0.011).
We next explored protein levels of hnRNP K in different human AML cell lines: OCI-AML2, AML-193, Nomo-1 and MOLM13. Here, we observed varying amounts of hnRNP K protein in these cells and these levels correlated with the ratio of RUNX1 ΔEx4 to RUNX1 FL transcript in these cell lines (Supplemental Figure S5G-H).
The RUNX1 ΔEx4 splice isoform has increased protein stability and differential function
Since hnRNP K overexpression leads to relative enrichment of RUNX1 ΔEx4, we next sought to evaluate functional consequences of this enrichment. Cycloheximide chase assays in cells with inducible expression of RUNX1 FL or RUNX1 ΔEx4 demonstrated that the RUNX1 ΔEx4 protein was substantially more stable than RUNX1 FL protein (Figure 6A) (42,46). This effect was almost completely abrogated by addition of MG132 (Figure 6B), indicating that exon 4 is a critical mediator of RUNX1 protein stability which is consistent with reports that identified domains required for RUNX1 stability (47).
Figure 6.

Functional relevance of the RUNX ΔEx4 isoform. (A) Western blot of RUNX1 expression following a cycloheximide chase experiment in 293T cells that stably express RUNX1 full length or RUNX ΔEx4. HSP90 serves as loading control. (B) Western blot of RUNX1 following cycloheximide + MG132 treatment in 293T cells that stably express RUNX1 full-length or RUNX1 ΔEx4. HSP90 serves as the loading control. (C) Bar graph representing number of colonies formed per well for FLCs infected with empty vector (n = 7), RUNX1 FL (n = 4) or RUNX1 ΔEx4 (n = 4) plasmid. Data are represented as mean ± SEM. P-values were determined using a two-sided Mann–Whitney test. (D) Luciferase-based reporter assay to assess transactivation from a CSF-1R promoter reporter in 293T cells that stably express either empty vector, RUNX1 full length, or RUNX1 ΔEx4. The experiment was independently done three times in triplicate. Data are represented as mean ± SEM. P-values were determined using a two-sided Mann–Whitney test.
To understand whether this isoform of RUNX1 ΔEx4 is in part responsible for mediating the hnRNP K-overexpression phenotype, we repeated colony formation assays using FLCs. FLCs overexpressing RUNX1 FL had a slight, non-significant decrease in colonies/well, whereas FLCs overexpressing RUNX1 ΔEx4 formed significantly more colonies in vitro than FLCs infected with empty vector (Figure 6C). Both of these observations are consistent with results from (42). This increase in number of colonies for the RUNX1 ΔEx4 isoform corresponds to our observations in colony formation assays when overexpressing hnRNP K (Figure 2B), suggesting that increased expression of RUNX1 ΔEx4 may be partly responsible for this hnRNP K-mediated effect.
As RUNX1 is a well-defined transcription factor, we then queried whether lack of exon 4 would affect the transcriptional capabilities of this protein. Indeed, luciferase assays indicated that RUNX1 ΔEx4 has decreased ability to transactivate a promoter-reporter derived from the CSF1R promoter, which is known to have RUNX1 binding sites (25), compared to RUNX1 FL (Figure 6D). Together, our data demonstrate that RUNX1 FL and RUNX1 ΔEx4 are phenotypically and functionally distinct isoforms.
DISCUSSION
Recent advances in our understanding of RNA-binding proteins (RBPs) have highlighted their importance in regulating hematopoiesis and initiating disease (8,18,48,49). In this paper, we focused on the RBP hnRNP K, whose expression is frequently altered in cancers. Using RPPA, we identified novel changes in protein expression that associated with poor outcomes in AML. Of particular interest was hnRNP K, an RNA binding protein which has been previously implicated in the progression of diffuse large B-cell lymphoma, chronic myeloid leukemia, and associated with poor outcomes in some solid tumors. Our work demonstrates that the wild-type form of hnRNP K is overexpressed in AML, and that this overexpression correlates with poor patient outcomes. Overexpression of hnRNP K in murine fetal liver cells (FLCs) resulted in increased proliferative potential, and transplantation of hnRNP K-overexpressing FLCs based on a model developed by the Lowe laboratory (23) resulted in myeloproliferation. The aberrant myelopoiesis observed in our mouse model is intriguing for several reasons. We recently described a mouse model wherein B-cell lymphomas develop secondary to hnRNP K overexpression in B-cells (18). However, in the current study, no lymphomas were observed when hnRNP K was overexpressed in FLCs. This suggests that hnRNP K overexpression earlier in hematopoiesis promotes a myeloid bias and/or inhibits lymphoid differentiation. Consistent with this, microarray data in human hematopoiesis revealed that HNRNPK transcript expression is higher in myeloid-biased compared to lymphoid-biased progenitor cells (50,51). This observation, coupled with our previous data showing shortened survival and myeloid hyperplasia in mice haploinsufficient for Hnrnpk (36), suggests that normal hematopoiesis, particularly in the myeloid lineage, requires exquisitely tight regulation of hnRNP K expression.
In our data, we observed that FLCs overexpressing hnRNP K had higher expression of the RUNX1 isoform lacking exon 4 (RUNX1 ΔEx4). Similarly, we observed an association of altered hnRNP K expression in human AML cells with changes in relative expression of RUNX1 ΔEx4. Though the magnitude of the effect on the RUNX1 ΔEx4 transcript levels is modest, it is likely physiologically relevant for two reasons. First, RUNX1 ΔEx4 is a more stable protein, thus, subtle increases in RUNX1 ΔEx4 transcript may have a disproportional impact on protein expression. Secondly, expression of RUNX1 ΔEx4 results in alterations in its transcriptional activity, as this domain is responsible for its interaction with the transcriptional co-repressor SIN3A. Furthermore, this domain also contains arginine methylation sites, which have been implicated in regulation of transcriptional activity (52). These mechanisms may in part explain why overexpression of RUNX1 ΔEx4 alone in FLCs recapitulates the increase in self-renewal capacity seen with hnRNP K overexpression (Figures 2B and 6C) (42,46).
Beyond its role in regulating RUNX1 splicing, hnRNP K is also known to impact the function of Core-Binding Factor Subunit Beta (CBFB), the transcriptional partner of RUNX1. A recent study has shown that CBFB associates with several transcripts, including RUNX1, through the RNA-binding properties of hnRNP K to impact their translation into protein (53). It is plausible that the role of hnRNP K in regulating the translation of its target transcripts, such as RUNX1, plays a role in this model system. Additionally, hnRNP K, like many RBPs, is highly multifunctional, and alterations to splicing of the RUNX1 transcript are unlikely to be the sole mechanism driving its oncogenic functions. Indeed, there are several functions of hnRNP K, outside of its impact on RUNX1, that can contribute to its oncogenicity including, but not limited to, its regulation of SET, PU.1 and MYC (17,18,54–56). The data presented here provide evidence and lend credence to the idea that aberrant hnRNP K expression alters the proper function of the spliceosome and has far-reaching cellular implications. However, the full impact of such altered expression warrants further investigation.
DATA AVAILABILITY
RNA-Seq datasets associated with this manuscript have been deposited to Array Express (E-MTAB-11886).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Kendra Allton, Sabrina Stratton, and other members of Dr. Michelle Barton's laboratory for helpful conversations, Taghi Manshouri, Christopher Mill, Warren Fiskus and Vera Adema for technical assistance and collaborative efforts.
Contributor Information
Marisa J L Aitken, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, USA.
Prerna Malaney, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; Department of Biochemistry and Cell Biology, Geisel School of Medicine at Dartmouth, Hanover, NH, USA; Norris Cotton Cancer Center, Geisel School of Medicine at Dartmouth, Lebanon, NH, USA.
Xiaorui Zhang, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Shelley M Herbrich, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, USA; Department of Genitourinary Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Lauren Chan, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Oscar Benitez, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Ashley G Rodriguez, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Huaxian Ma, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Rodrigo Jacamo, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Ruizhi Duan, School of Health Professions, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; Baylor College of Medicine, Houston, TX, USA.
Todd M Link, Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Steven M Kornblau, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Rashmi Kanagal-Shamanna, Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Carlos E Bueso-Ramos, Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Sean M Post, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
This study has been supported by funding from a National Cancer Institute Cancer Center Support grant (CA016672) to core facilities at MD Anderson Cancer Center. MJLA is a recipient of the Dr. John J. Kopchick Fellowship. This research has been supported by a grant from the Jane Coffin Childs Memorial Fund for Medical Research (PM); American Society of Hematology (PM); a National Cancer Institute/National Institutes of Health Award (R01CA207204, SMP); and Leukemia and Lymphoma Society (6577–19, SMP).
Conflicts of interest statement. None declared.
REFERENCES
- 1. Stone R.M., Mandrekar S.J., Sanford B.L., Laumann K., Geyer S., Bloomfield C.D., Thiede C., Prior T.W., Dohner K., Marcucci G.et al.. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017; 377:454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perl A.E., Martinelli G., Cortes J.E., Neubauer A., Berman E., Paolini S., Montesinos P., Baer M.R., Larson R.A., Ustun C.et al.. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019; 381:1728–1740. [DOI] [PubMed] [Google Scholar]
- 3. Stein E.M., DiNardo C.D., Fathi A.T., Pollyea D.A., Stone R.M., Altman J.K., Roboz G.J., Patel M.R., Collins R., Flinn I.W.et al.. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019; 133:676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DiNardo C.D., Stein E.M., de Botton S., Roboz G.J., Altman J.K., Mims A.S., Swords R., Collins R.H., Mannis G.N., Pollyea D.Aet al.. Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018; 378:2386–2398. [DOI] [PubMed] [Google Scholar]
- 5. Papaemmanuil E., Cazzola M., Boultwood J., Malcovati L., Vyas P., Bowen D., Pellagatti A., Wainscoat J.S., Hellstrom-Lindberg E., Gambacorti-Passerini C.et al.. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011; 365:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Graubert T.A., Shen D., Ding L., Okeyo-Owuor T., Lunn C.L., Shao J., Krysiak K., Harris C.C., Koboldt D.C., Larson D.E.et al.. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011; 44:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M.et al.. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478:64–69. [DOI] [PubMed] [Google Scholar]
- 8. Vu L.P., Pickering B.F., Cheng Y., Zaccara S., Nguyen D., Minuesa G., Chou T., Chow A., Saletore Y., MacKay M.et al.. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017; 23:1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kharas M.G., Lengner C.J., Al-Shahrour F., Bullinger L., Ball B., Zaidi S., Morgan K., Tam W., Paktinat M., Okabe R.et al.. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 2010; 16:903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saha S., Murmu K.C., Biswas M., Chakraborty S., Basu J., Madhulika S., Kolapalli S.P., Chauhan S., Sengupta A., Prasad P.. Transcriptomic analysis identifies RNA binding proteins as putative regulators of myelopoiesis and leukemia. Front. Oncol. 2019; 9:692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang E., Lu S.X., Pastore A., Chen X., Imig J., Chun-Wei Lee S., Hockemeyer K., Ghebrechristos Y.E., Yoshimi A., Inoue D.et al.. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell. 2019; 35:369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carpenter B., McKay M., Dundas S.R., Lawrie L.C., Telfer C., Murray G.I.. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br. J. Cancer. 2006; 95:921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L.C., Chung I.C., Hsueh C., Tsang N.M., Chi L.M., Liang Y., Chen C.C., Wang L.J., Chang Y.S.. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ. 2010; 17:1463–1473. [DOI] [PubMed] [Google Scholar]
- 14. Wen F., Shen A., Shanas R., Bhattacharyya A., Lian F., Hostetter G., Shi J.. Higher expression of the heterogeneous nuclear ribonucleoprotein k in melanoma. Ann. Surg. Oncol. 2010; 17:2619–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barboro P., Repaci E., Rubagotti A., Salvi S., Boccardo S., Spina B., Truini M., Introini C., Puppo P., Ferrari N.et al.. Heterogeneous nuclear ribonucleoprotein K: altered pattern of expression associated with diagnosis and prognosis of prostate cancer. Br. J. Cancer. 2009; 100:1608–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou R., Shanas R., Nelson M.A., Bhattacharyya A., Shi J.. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int. J. Cancer. 2010; 126:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Notari M., Neviani P., Santhanam R., Blaser B.W., Chang J.S., Galietta A., Willis A.E., Roy D.C., Caligiuri M.A., Marcucci G.et al.. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood. 2006; 107:2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gallardo M., Malaney P., Aitken M.J.L., Zhang X., Link T.M., Shah V., Alybayev S., Wu M.H., Pageon L.R., Ma H.et al.. Uncovering the role of hnRNP K, an RNA-binding protein, in B-cell lymphomas. J. Natl. Cancer Inst. 2019; 112:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Braekeleer E., Douet-Guilbert N., Morel F., Le Bris M.J., Ferec C., De Braekeleer M.. RUNX1 translocations and fusion genes in malignant hemopathies. Future Oncol. 2011; 7:77–91. [DOI] [PubMed] [Google Scholar]
- 20. Gaidzik V.I., Teleanu V., Papaemmanuil E., Weber D., Paschka P., Hahn J., Wallrabenstein T., Kolbinger B., Köhne C.H., Horst H.A.et al.. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016; 30:2160–2168. [DOI] [PubMed] [Google Scholar]
- 21. Said F., Shafik R.E., Hassan N.M.. RUNX1 gene expression in Egyptian acute myeloid leukemia patients: may it have therapeutic implications. Egypt. J. Med. Hum. Genet. 2021; 22:58. [Google Scholar]
- 22. Hu C.W., Qiu Y., Ligeralde A., Raybon A.Y., Yoo S.Y., Coombes K.R., Qutub A.A., Kornblau S.M.. A quantitative analysis of heterogeneities and hallmarks in acute myelogenous leukaemia. Nat. Biomed. Eng. 2019; 3:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zuber J., Radtke I., Pardee T.S., Zhao Z., Rappaport A.R., Luo W., McCurrach M.E., Yang M.M., Dolan M.E., Kogan S.C.et al.. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009; 23:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sugimura R., Jha D.K., Han A., Soria-Valles C., da Rocha E.L., Lu Y.F., Goettel J.A., Serrao E., Rowe R.G., Malleshaiah M.et al.. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017; 545:432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang D.E., Hetherington C.J., Chen H.M., Tenen D.G.. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol. Cell. Biol. 1994; 14:373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S., Hahn W.C., Sharp P.A.et al.. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003; 9:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naviaux R.K., Costanzi E., Haas M., Verma I.M.. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 1996; 70:5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmitt C.A., Fridman J.S., Yang M., Baranov E., Hoffman R.M., Lowe S.W.. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002; 1:289–298. [DOI] [PubMed] [Google Scholar]
- 29. Bray N.L., Pimentel H., Melsted P., Pachter L.. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- 30. Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L.. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017; 14:687–690. [DOI] [PubMed] [Google Scholar]
- 31. Ghanem L.R., Kromer A., Silverman I.M., Ji X., Gazzara M., Nguyen N., Aguilar G., Martinelli M., Barash Y., Liebhaber S.A.. Poly(C)-binding protein Pcbp2 enables differentiation of definitive erythropoiesis by directing functional splicing of the Runx1 transcript. Mol. Cell. Biol. 2018; 38:e00175-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malaney P., Benitez O., Zhang X., Post S.M.. Assessing the role of intrinsic disorder in RNA-binding protein function: hnRNP K as a case study. Methods. 2022; 208:59–65. [DOI] [PubMed] [Google Scholar]
- 33. Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001; 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peniket A., Wainscoat J., Side L., Daly S., Kusec R., Buck G., Wheatley K., Walker H., Chatters S., Harrison C.et al.. Del (9q) AML: clinical and cytological characteristics and prognostic implications. Br. J. Haematol. 2005; 129:210–220. [DOI] [PubMed] [Google Scholar]
- 35. Kronke J., Bullinger L., Teleanu V., Tschurtz F., Gaidzik V.I., Kuhn M.W., Rucker F.G., Holzmann K., Paschka P., Kapp-Schworer S.et al.. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013; 122:100–108. [DOI] [PubMed] [Google Scholar]
- 36. Gallardo M., Lee H.J., Zhang X., Bueso-Ramos C., Pageon L.R., McArthur M., Multani A., Nazha A., Manshouri T., Parker-Thornburg J.et al.. hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell. 2015; 28:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tyner J.W., Tognon C.E., Bottomly D., Wilmot B., Kurtz S.E., Savage S.L., Long N., Schultz A.R., Traer E., Abel M.et al.. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018; 562:526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Research CancerGenomeAtlas, Ley N., Miller T.J., Ding C., Raphael L., Mungall B.J., Robertson A.J., Hoadley A., Triche K., T.J. Jr.et al.. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Papaemmanuil E., Dohner H., Campbell P.J.. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016; 375:900–901. [DOI] [PubMed] [Google Scholar]
- 40. Morrison S.J., Hemmati H.D., Wandycz A.M., Weissman I.L.. The purification and characterization of fetal liver hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:10302–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E.et al.. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Komeno Y., Yan M., Matsuura S., Lam K., Lo M.C., Huang Y.J., Tenen D.G., Downing J.R., Zhang D.E.. Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood. 2014; 123:3760–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nanjundan M., Zhang F., Schmandt R., Smith-McCune K., Mills G.B.. Identification of a novel splice variant of AML1b in ovarian cancer patients conferring loss of wild-type tumor suppressive functions. Oncogene. 2007; 26:2574–2584. [DOI] [PubMed] [Google Scholar]
- 44. Matunis M.J., Michael W.M., Dreyfuss G.. Characterization and primary structure of the poly(C)-binding heterogeneous nuclear ribonucleoprotein complex K protein. Mol. Cell. Biol. 1992; 12:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cao W., Razanau A., Feng D., Lobo V.G., Xie J.. Control of alternative splicing by forskolin through hnRNP K during neuronal differentiation. Nucleic Acids Res. 2012; 40:8059–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun W., Zeng J., Chang J., Xue Y., Zhang Y., Pan X., Zhou Y., Lai M., Bian G., Zhou Q.et al.. RUNX1-205, a novel splice variant of the human RUNX1 gene, has blockage effect on mesoderm-hemogenesis transition and promotion effect during the late stage of hematopoiesis. J. Mol. Cell Biol. 2020; 12:386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Biggs J.R., Peterson L.F., Zhang Y., Kraft A.S., Zhang D.E.. AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex. Mol. Cell. Biol. 2006; 26:7420–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kharas M.G., Lengner C.J., Al-Shahrour F., Bullinger L., Ball B., Zaidi S., Morgan K., Tam W., Paktinat M., Okabe R.. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 2010; 16:903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barbieri I., Tzelepis K., Pandolfini L., Shi J., Millan-Zambrano G., Robson S.C., Aspris D., Migliori V., Bannister A.J., Han N.et al.. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017; 552:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Novershtern N., Subramanian A., Lawton L.N., Mak R.H., Haining W.N., McConkey M.E., Habib N., Yosef N., Chang C.Y., Shay T.et al.. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011; 144:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Verhaak R.G., Wouters B.J., Erpelinck C.A., Abbas S., Beverloo H.B., Lugthart S., Lowenberg B., Delwel R., Valk P.J.. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009; 94:131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao X., Jankovic V., Gural A., Huang G., Pardanani A., Menendez S., Zhang J., Dunne R., Xiao A., Erdjument-Bromage H.et al.. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 2008; 22:640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Malik N., Yan H., Moshkovich N., Palangat M., Yang H., Sanchez V., Cai Z., Peat T.J., Jiang S., Liu C.et al.. The transcription factor CBFB suppresses breast cancer through orchestrating translation and transcription. Nat. Commun. 2019; 10:2071–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cheng J.X., Chen L., Li Y., Cloe A., Yue M., Wei J., Watanabe K.A., Shammo J.M., Anastasi J., Shen Q.J.et al.. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 2018; 9:1163–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Padovani K.S., Goto R.N., Fugio L.B., Garcia C.B., Alves V.M., Brassesco M.S., Greene L.J., Rego E.M., Leopoldino A.M.. Crosstalk between hnRNP K and SET in ATRA-induced differentiation in acute promyelocytic leukemia. FEBS Open Bio. 2021; 11:2019–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nika E., Brugnoli F., Piazzi M., Lambertini E., Grassilli S., Bavelloni A., Piva R., Capitani S., Bertagnolo V.. hnRNP K in PU.1-containing complexes recruited at the CD11b promoter: a distinct role in modulating granulocytic and monocytic differentiation of AML-derived cells. Biochem. J. 2014; 463:115–122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq datasets associated with this manuscript have been deposited to Array Express (E-MTAB-11886).