

Abstract
Blockade of immune checkpoints as a strategy of cancer cells to overcome the immune response has received ample attention in cancer research recently. In particular, expression of PD‐L1 by various cancer cells has become a paradigm in this respect. Delivery of PD‐L1 to its site of action occurs either by local diffusion, or else by transport via small extracellular vesicles (sEVs, commonly referred to as exosomes). Many steps of sEVs formation, their packaging with PD‐L1 and their release into the extracellular space have been studied in detail. The likely dependence of release on Ca2+‐signaling, however, has received little attention. This is surprising, since the intracellular Ca2+‐concentration is known as a prominent regulator of many secretory processes. Here, we report on the roles of three Ca2+‐dependent proteins in regulating release of PD‐L1‐containing sEVs, as well as on the growth of tumors in mouse models. We show that sEVs release in cancer cell lines is Ca2+‐dependent and the knockdown of the gene coding the Ca2+‐channel protein ORAI1 reduces Ca2+‐signals and release of sEVs. Consequently, the T cell response is reinvigorated and tumor progression in mouse models is retarded. Furthermore, analysis of protein expression patterns in samples from human cancer tissue shows that the ORAI1 gene is significantly upregulated. Such upregulation is identified as an unfavorable prognostic factor for survival of patients with non‐small‐cell lung cancer. We show that reduced Ca2+‐signaling after knockdown of ORAI1 gene also compromises the activity of melanophilin and Synaptotagmin‐like protein 2, two proteins, which are important for correct localization of secretory organelles within cancer cells and their transport to sites of exocytosis. Thus, the Ca2+‐channel ORAI1 and Ca2+‐dependent proteins of the secretion pathway emerge as important targets for understanding and manipulating immune checkpoint blockade by PD‐L1.
Keywords: Ca2+ , ORAI1, PD‐L1, exosome, lung cancer
1. INTRODUCTION
Cancer immunotherapy has resulted in revolutionary advances in treating various cancers such as non‐small‐cell lung cancer (NSCLC), melanoma, renal cell cancer (Brahmer et al., 2012; Topalian et al., 2012), head and neck squamous cell carcinoma (HNSCC) (Ferris et al., 2016), and colorectal cancer (Overman et al., 2017). New checkpoint inhibitors targeting particular checkpoint molecules, CTLA4, PD‐1, and PD‐L1, are being developed continuously. The therapeutic strategy of checkpoint inhibitors shows considerable effectiveness in patients who have previously experienced the failure of traditional treatments. However, the survival rate for some cancer patients is only slightly advanced (Topalian et al., 2019). Also resistance can emerge during treatment (Wang et al., 2020). For some cancers, such as prostate cancer, anti‐PD‐L1 therapy has been shown to confer only little benefit (Goswami et al., 2016). The mechanisms regarding low response rate and acquired resistance to treatments of checkpoint inhibitors are not fully understood.
PD‐L1, one of the critical checkpoint molecules, binds to its receptor PD‐1 expressed on immune cells to mediate a T‐cell‐related immunosuppressive response, which is critical to maintaining the immune homeostasis and preventing cell death and tissue damage due to the overreaction of immune cells. However, tumor cells can trick the immune cells during cancer development and escape from proper immune surveillance by expressing PD‐L1. Therapeutic checkpoint inhibitors block the interaction between PD‐1 and PD‐L1, reinvigorating the exhausted T cell, therefore suppressing tumor progression. PD‐L1 is a transmembrane protein, primarily expressed at the plasma membrane. It also has a soluble form because an extracellular enzyme can cut its ectodomain (Hira‐Miyazawa et al., 2018; Romero et al., 2020). Furthermore, there are splice variants without a transmembrane domain. The expression level of soluble PD‐L1 in peripheral blood is closely related to the resistance against anti‐PD‐L1 treatment (Gong et al., 2019). Recently, several studies have shown that multiple cancer cell lines secrete PD‐L1, packaged within specific membrane‐enclosed extracellular vesicles (EVs), so‐called exosomal PD‐L1, or small extracellular vesicular PD‐L1 (sEV PD‐L1), which mediates suppression of the antitumor immune response (Chen et al., 2018; Poggio et al., 2019; Ricklefs et al., 2018; Yang et al., 2018). The function and regulation of sEV PD‐L1 for tumor development and the impact of sEV PD‐L1 on the tumor microenvironment are not fully understood. Great attention is drawn to the new form of PD‐L1 located on EVs (Daassi et al., 2020; Xie et al., 2019).
EVs are cell‐released lipid bilayer membranous structures of various subtypes, proposed to have distinct roles from cancer to obesity, from neurodegeneration to autoimmune disease. Given their heterogeneous population, EVs are categorized based on their size as small EVs (sEVs) and medium/large EVs (m/lEVs) with diameters <200 nm or >200 nm, respectively. In addition, other types of membrane vesicles are produced by cells, such as apoptotic bodies or other membrane fragments (Thery et al., 2018). sEVs, also referred to as exosomes, are a particular class of EVs carrying numerous informative biological molecules, including proteins, lipids, DNA, and diverse types of RNA (Yanez‐Mo et al., 2015). sEVs and their function as communication ambassadors to reshape ambient microenvironments, receive increasing interest and enthusiasm from various research fields. Recently discovered sEV PD‐L1 and MHC molecules are typical messengers transmitting the immune signal from producer cells to receptor cells in an autocrine or paracrine form of cell‐cell communication (Barros et al., 2018).
Multivesicular bodies (MVBs) are specialized endosomes containing multiple intraluminal vesicles (ILVs) that bud inwardly into the MVB lumen when MVBs mature. When MVBs fuse with the plasma membrane (PM), ILVs are released as sEVs. Several essential classes of proteins are involved in sEV biogenesis and release, such as Rab‐proteins and SNARES (Colombo et al., 2014), cytoskeletal proteins and proteins mediating the synthesis of ceramide and autophagic degradation (Doyle & Wang, 2019). Cytosolic calcium, as a universal second messenger, plays a leading role in many forms of vesicle exocytosis (fusion of a vesicular structure with the plasma membrane, thereby releasing its contents to the extracellular space) and the reverse process, endocytosis (Berridge et al., 2003). Sparked by the interest in the role of free intracellular calcium in synaptic transmission whereby neurotransmitter is released, there is an extensive body of literature on calcium‐effects on exocytosis, endocytosis and organelle trafficking, not only with regard to the extremely rapid triggering effect of calcium‐increases in neurotransmitter release (Del Castillo & Katz, 1954; Jahn & Scheller, 2006), but also on the calcium‐mediated modulation of slower vesicle trafficking steps before exocytosis (Neher and Brose, 2018) and on the recycling of vesicles following exocytosis (Ebner et al., 2019). Compared to that, the role of calcium in endosome‐like organelles ‐ although firmly established (Colombo et al., 2014) ‐ has received little attention. Notably, recent studies revealed that Ca2+‐stimulated MVB‐PM fusion mediates exosome release involving SNARE proteins (Messenger et al., 2018; Verweij et al., 2018). However, the issue of whether calcium also regulates the generation of sEV PD‐L1 by exocytosis from MVBs has not been addressed in detail.
In this paper, we show that the generation of sEV PD‐L1 occurs in a calcium‐dependent manner in a NSCLC mouse model. Among calcium‐influx pathways in non‐excitable cell types, the calcium release‐activated calcium channel ORAI1 plays a dominant role (Parekh, 2010). By knocking down the ORAI1 gene, we show that ORAI1 plays an essential role in the generation of sEV PD‐L1. We show that the reduction of sEV PD‐L1 levels by ORAI1‐KD inhibits tumor growth in an immune‐dependent manner. Rab27 effectors, melanophilin and Synaptotagmin‐like protein 2, contribute to the calcium‐dependent generation of sEV PD‐L1 in distinct ways. These findings may pave the way to a better understanding of molecular mechanisms underlying PD‐1/PD‐L1 tumor immunotherapy and its rapidly developing resistance to inhibitors.
2. MATERIALS AND METHODS
2.1. Materials
Cell Lines: H1299 (NCI‐H1299), A549, H2122 (NCI‐H2122), H1975 (NCI‐H1975), Lewis lung carcinoma (LLC1 or LL/2) and 293T cell lines were obtained from the American Typical Culture Collection (ATCC, Manassas, VA, USA). H1299, A549, H2122, and H1975 cell lines were non‐small cell lung cancer cell lines derived from patients. LLC1 cell line was derived from Lewis lung carcinoma in C57BL mice. The 293T cell line is a highly transfected derivative of human embryonic kidney 293 cells.
Animals: male C57 BL/6 mice (6‐8 weeks old) were purchased from SPF Biotechnology Co., Ltd. (Beijing, China).
Reagents and kits: RPMI‐1640 Medium (31800022, Thermo Fisher Scientific, Waltham, MA, USA); DMEM Medium (12800017, Thermo Fisher Scientific, Waltham, MA, USA); Fetal Bovine Serum (10270106, Thermo Fisher Scientific, Waltham, MA, USA); Penicillin Streptomycin Glutamine solution (A1113802, Thermo Fisher Scientific, Waltham, MA, USA); NheI (R3131M, New England Biolabs, Ipswich, MA, USA); EcorI (R3101M, New England Biolabs, Ipswich, MA, USA); HindIII (R3104M, New England Biolabs, Ipswich, MA, USA); BamHI (R3136M, New England Biolabs, Ipswich, MA, USA); polybrene (TR‐1003‐G, Sigma‐Aldrich, St. Louis, MO, USA); puromycin dihydrochloride (A1113803, Thermo Fisher Scientific, Waltham, MA, USA); G418 disulfate salt (108321‐42‐2, Sigma‐Aldrich, St. Louis, MO, USA); Hygromycin B (31282‐04‐9, Sigma‐Aldrich, St. Louis, MO, USA); exosome‐depleted Fetal Bovine Serum (A2720803, Thermo Fisher Scientific, Waltham, MA, USA); IFN‐γ human (SRP3058‐100UG, Sigma‐Aldrich, St. Louis, MO, USA); IFN‐γ Mouse (CM052‐100MP, Chamot Biotechnology, Shanghai, China); Calpain inhibitor III (88191‐84‐8, MedChemExpress, Monmouth Junction, NJ, USA); Bio‐Rad Protein Assay Kit (5000001, Bio‐Rad Laboratories, USA); Sucrose (57‐50‐1, Sigma‐Aldrich, St. Louis, MO, USA); FLIPR Calcium 6 Assay Kit (R8190, Molecular Devices, San Jose, CA, USA); TRIzol (15596026, Thermo Fisher Scientific, Waltham, MA, USA); Transcriptor Universal cDNA Master (5893151001, Sigma‐Aldrich, St. Louis, MO, USA); SYBRTM Green Master Mix (A25742, Thermo Fisher Scientific, Waltham, MA, USA); RIPA buffer (ab156034, Abcam, Cambridge, UK); Pierce Protease and Phosphatase Inhibitor (A32959, Thermo Fisher Scientific, Waltham, MA, USA); Nitrocellulose Transfer Membrane (66485, Pall Life Sciences, Ann Arbor, MI, USA); Western ECL Substrate (1705062, Bio‐Rad Laboratories, Hercules, CA, USA); Human PD‐L1 ELISA kit (CHE0312; 4A Biotech, Beijing, China); Mouse PD‐L1 ELISA kit(CME0129; 4A Biotech, Beijing, China); IL‐2 ELISA kit (CME0001; 4A Biotech, Beijing, China); Fixation/Permeabilization Buffer (424401, BioLegend, San Diego, CA, USA); Phosphotungstic acid (HT152, Sigma‐Aldrich, St. Louis, MO, USA); Adenosine 5′‐triphosphate magnesium salt (MgATP) (A9187, Sigma‐Aldrich, St. Louis, MO, USA) Thapsigargin (T7459, Thermo Fisher Scientific, Waltham, MA, USA); Immunohistochemical kit (FXP019; 4A Biotech, Beijing, China), Actin‐Tracker Green (C1033, Beyotime, Jiangshu, China).
Specific information on primers, plasmids, and antibodies is in Table 1.
TABLE 1.
Information of primers, plasmids, and antibodies
| Name | Sequence |
|---|---|
| pHluorinCD63‐PDL1‐SP_F | 5’‐AGGATATTTGCTGTCTTTATATTCATGACCTACTGGCATTTGCTGAACGCACTAGCCACCATGGGAAGTAAAGG‐3’ |
| pHluorinCD63_R | 5’‐GCTGCTTCCACCTCCACCGCTACCGCTACCGCTACCTCGAGCTCCACCGGTTTTG‐3’ |
| pHluorinCD63‐PDL1_NF | 5'‐ACGCTTTGCCGCCACCATGAGGATATTTGCTGTCTTTATATTCATG‐3’ |
| pHluorinCD63‐PDL1_NR | 5'‐GCTGCTTCCACCTCCACC‐3' |
| pHR_PD‐L1‐CLIP‐C_F | 5’‐GGTAGCGGTAGCGGTAGC‐3’ |
| pHR_PD‐L1‐CLIP‐C‐His_R | 5’‐GGGTTTAAACTCAATGGTGATGGTGATGATGACCGGTCGTCTCCTCCAAATGTGTATCAC‐3’ |
| pHR_PD‐L1‐pHluorin_F | 5’‐AGATCCGCTAGCGCCACCATGAGGATATTTG‐3’ |
| pHR_PD‐L1‐pHluorin‐His_R | 5’‐AACCTAGAATTCTCAATGGTGATGGTGATGATG‐3’ |
| MLPH_HindIII‐390_F | 5’‐GAGCTCAAGCTTCGAATTCAATGGGGAAGA‐3’ |
| MLPH_HindIII‐390_R | 5’‐AACTGATTTGTCCCTGTTCAGCTTCCTCCTCAGGGC‐3’ |
| MLPH_415‐BamHI_F | 5’‐GCCCTGAGGAGGAAGCTGAACAGGGACAAATCAGTT‐3’ |
| MLPH_415‐BamHI_R | 5’‐GTGGATCCTTAGGACTGGTGGGCCACCACA‐3’ |
| Slp2‐a_T614*_F | 5’‐TAAAGGAAATATTCAGTTTGCAATTGAATAGGTGGAGTCACTGAAGG‐3’ |
| Slp2‐a_T614*_R | 5’‐CCTTCAGTGACTCCACCTATTCAATTGCAAACTGAATATTTCCTTTA‐3’ |
| H‐ORAI1_F | 5’‐AGGTGATGAGCCTCAACGAG‐3’ |
| H‐ORAI1_R | 5’‐CTGATCATGAGCGCAAACAG‐3’ |
| M‐ORAI1_F | 5’‐ CATAAGACGGACCGGCAGTT‐3’ |
| M‐ORAI1_R | 5’‐ AGGAAGAGGTGTCCCTTGTG‐3’ |
| H‐Slp2‐a_R | 5’‐TCTTCTACAACGCCAGCCAG‐3’ |
| H‐Slp2‐a_F | 5’‐ AGGGCCGAAGAAGAGAGAGT‐3’ |
| H‐Mlph_F | 5’‐AAGAGGTTCGGAAGTGCCAAGG‐3’ |
| H‐Mlph_R | 5’‐ AGCCAGGTTCTCCATCCTCATC‐3’ |
| Name | Resource | Note |
|---|---|---|
| pCMV‐CD63‐pHluorin | Tom Martin at University of Wisconsin‐Madison | |
| pHR‐CLIP‐PD‐L1 | Enfu Hui at UC San Diego | |
| pLJM1_FLAG | Zheng Zhang at Xiangya School of Medicine, Central South University | |
| pLV[shRNA]‐Scamblem | Vectorbuilder | GCTTCGCGCCGTAGTCTTA |
| pLV[shRNA]‐hOrai1 | Vectorbuilder | GGTGAGCAACGTGCACAATCT |
| pLV[shRNA]‐mOrai1 | Vectorbuilder | TCTGCTGGGTCAAGTTCTTAC |
| pLV[shRNA]‐hMlph | Vectorbuilder | GTCTTCTGAGAGTCAGATCTT |
| pLV[shRNA]‐hSlp2‐a | Vectorbuilder | GCCTTGATCTACCACTGCTAA |
| pMD2.G | Addgene #12259 | |
| pMDLg/pRRE | Addgene #12251 | |
| pRSV‐Rev | Addgene #12253 | |
| DsRed_MLPH | Addgene #89231 |
| Antibody | Company | Cat. No. | ST con. |
|---|---|---|---|
| Anti‐Orai1 Antibody | Abcam, Cambridge, UK | ab175040 | 1:1000 |
| Anti‐CD63(E‐12) Antibody | Santa Cruz, TX, USA | sc‐365604 | 1:1000 |
| Anti‐HRS(D7T5N) Antibody | Cell Signaling Technology, MA, USA | 15087 | 1:1000 |
| Anti‐PD‐L1(E1L3N) Antibody | Cell Signaling Technology, MA, USA | 13684 | 1:1000 |
| Anti‐CD63 Antibody (MX‐49.129.5) | Santa Cruz, TX, USA | sc‐5275 | 1:1000 |
| Anti‐PD‐L1 Antibody | Proteintech Group, IL, USA | 66248‐1‐Ig | 1:1000 |
| Anti‐SLP2 Antibody | Proteintech Group, IL, USA | 12359‐1‐AP | 1:1000 |
| Anti‐Slac2‐a (G‐10) Antibody | Santa Cruz, TX, USA | sc‐515894 | 1:1000 |
| Anti‐Histone H3 antibody | Abcam, Cambridge, UK | ab201456 | 1:2000 |
| Anti‐Annexin A1/ANXA1 Antibody | Abcam, Cambridge, UK | ab214486 | 1:1000 |
| Anti‐GM130 Antibody | Abcam, Cambridge, UK | ab52649 | 1:1000 |
| Anti‐GAPDH | Santa Cruz, TX, USA | sc‐32233 | 1:1000 |
| Anti‐β‐actin | Santa Cruz, TX, USA | sc‐47778 | 1:2000 |
| Anti‐Rabbit HRP‐Conjugated Secondary Antibodies | Abmart, Shanghai, China | M21002 | 1:4000 |
| Anti‐Mouse HRP‐Conjugated Secondary Antibodies | Abmart, Shanghai, China | M21001 | 1:4000 |
| Animal experiments | |||
| GoInVivoTM Purified anti‐mouse CD274 Antibody | BioLegend, San Diego, CA, USA | 124328 | |
| GoInVivoTM Purified Rat IgG2b, κ Isotype Ctrl Antibody | BioLegend, San Diego, CA, USA | 400666 | |
| Flow cytometry analysis | |||
| Anti‐Mouse CD16/32 Antibody | BioLegend, San Diego, CA, USA | 101303 | |
| FITC Anti‐Mouse CD45 Antibody | BioLegend, San Diego, CA, USA | 103108 | |
| Percp/Cyanine5.5 Anti‐Mouse CD3 Antibody | BioLegend, San Diego, CA, USA | 100218 | |
| APC Anti‐Mouse CD4 Antibody | BioLegend, San Diego, CA, USA | 100412 | |
| PE/Cyanine7 Anti‐Mouse CD8a Antibody | BioLegend, San Diego, CA, USA | 100722 | |
| PE Anti‐Mouse CD366 (Tim‐3) Antibody | BioLegend, San Diego, CA, USA | 134003 | |
| PE/Dazzletm 594 Anti‐Mouse CD279 (PD‐1) Antibody | BioLegend, San Diego, CA, USA | 135228 | |
| PE Anti‐Human/Mouse Granzyme B Recombinant Antibody | BioLegend, San Diego, CA, USA | 396406 | |
| PE/Dazzletm 594 Anti‐Mouse Ki‐67 Antibody | BioLegend, San Diego, CA, USA | 652428 |
2.2. Cell culture
The base medium for H1299, H2122, and H1975 cells is RPMI‐1640 Medium. The base medium for 293T, A549 and LLC1 cells is DMEM Medium. All media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cells were incubated at 37°C in a humid atmosphere containing 5% CO2.
2.3. Construction of plasmids
To construct a pH‐sensitive pHluorin insert into the PD‐L1 expression vector: We first amplified the superecliptic pHluorin sequence out of a pCMV‐CD63‐pHluorin using the primers pHluorinCD63‐PDL1‐SP_F and pHluorinCD63_R. The PCR product contained partial PD‐L1 signal peptide (PDL1‐SP) at its N‐terminal and linker sequence at its C‐terminal. The PCR primers pHluorinCD63‐PDL1_N_F and pHluorinCD63‐PDL1_NR were used to add complete PDL1‐SP at pHluorin N‐terminal. Next, we amplified the PD‐L1 C‐terminal sequence out of a pHR‐CLIP‐PD‐L1 using the primer pHR_PD‐L1‐CLIP‐C_F and pHR_PD‐L1‐CLIP‐C‐His_R. The PD‐L1 C‐terminal was linked with linker sequence at N‐terminal. The two PCR fragments, pHluorin and PD‐L1 C‐terminal, were joined by fusion PCR with primers pHR_PD‐L1‐pHluorin_F and pHR_PD‐L1‐pHluorin‐His_R to form the pHluorin‐PD‐L1 sequence and add the NheI and EcoRI restriction sites at both ends. The final pLJM1_pHluorin‐PD‐L1 was obtained by insertion of pHluorin‐PD‐L1 into pLJM ‐FLAG vector after digestion of fusion PCR product with NheI/EcoRI.
To construct an expression vector for DsRed_MLPH Δ390‐415: Mutant MLPH ∆390‐415 lacking the PEST‐like sequence (amino acids 390–415) was generated by PCR from DsRed_MLPH using the following primers DsRed_MLPH HindIII‐390_F, DsRed_MLPH HindIII‐390_R, DsRed_MLPH 415‐BamHI_F, and DsRed_MLPH 415‐BamHI_R. After HindIII / BamHI digestion, the MLPH ∆390‐415 sequence was replaced by the MLPH WT sequence in DsRed_MLPH to obtain DsRed_MLPH ∆390‐415.
To construct an expression vector for mCherry_SLP2‐a ΔC2: We used primers SLP2‐a T614*_F and SLP2‐a T614*_R for targeted mutagenesis of mCherry_SLP2‐a. The resulting mCherry_SLP2‐a ∆C2 was generated with the substitution of the stop codon for tyrosine at amino acid position 614.
2.4. Stable cell line generation
293T cells were grown until fusion reached 60%–70% prior to transfection. The cells were co‐transfected using pLJM1_pHluorin‐PD‐L1 or pGIPZ‐PD‐L1‐EGFP or pLV[shRNA]‐Scamblem or pLV[shRNA]‐hORAI1 or pLV[shRNA]‐mORAI1 or pLV[shRNA]‐hMlph or pLV[shRNA]‐hSLP2‐a and the lentiviral packaging plasmids VSVG (Addgene Inc., Plasmid #12259), GAG (Addgene Inc., Plasmid #12251), REV (Addgene Inc. Plasmid #12253). Six hours after transfection, the medium was replaced. The lentivirus‐containing medium was collected during the next 48 and 72 h, followed by centrifugation at 1000 × g and filtration through a 0.45 μm filter to eliminate cell debris. Cells were seeded into 6‐well plates and 50%–60% cell confluence was reached 12 h later. Then the medium was replaced with 2 ml of medium containing lentivirus and 10 μg/ml polybrene. Cells were fed with fresh medium overnight. After 48 h of infection, cells were treated with 2 μg/ml puromycin or 800 μg/ml G418 or 300 μg/ml hygromycin B. After 2 weeks of selection, cells were identified by fluorescence microscopy (Olympus ORCA‐Flash 40 LT Plus Scientific CMOS Digital Camera), RT‐qPCR and Western blot.
2.5. EVs isolation
H1299 cells were plated at a density of 5 × 106 per T175 Flask. After 24 h of incubation, they were washed three times with Phosphate‐Buffered Saline (PBS) and then grown in RPMI‐1640 Medium containing 10% serum exosome‐depleted Fetal Bovine Serum. After 48 hours of incubation, the media from the two T175 Flasks were combined.
The LLC1 cells were plated at a density of 1 × 107 per T175 Flask. After 24 h of incubation, the cells were washed three times with phosphate buffer (PBS) and then grown in DMEM Medium containing 10% serum exosome‐depleted Fetal Bovine Serum. After 48 h of incubation, the media from the two T175 Flasks were combined.
For IFN‐γ or Calpain inhibitor III treatment, 10 ng/ml IFN‐γ or 10 μM Calpain inhibitor III was added to the medium 48 h prior to sEV and cell collection.
sEVs were isolated from conditioned culture media or C57 mouse serum samples by the following centrifugation steps (Consortium et al., 2017). The centrifugation is performed at 300 × g for 10 min to pellet and remove the cells at room temperature. Subsequent centrifugation steps are performed at 4°C. The supernatant was centrifuged at 2k × g for 20 min to remove cell debris and apoptotic bodies, followed by centrifugation at 10k × g for 60 min to remove medium/large EVs. Then, the supernatant was passed through a 0.22 μm PVDF filter (Millipore). The filtered medium supernatant (35 ml) was loaded into a PA Ultracrimp Tube (03141, Thermo Fisher Scientific) and subjected to the ultracentrifugation at 100k × g for 70 min with a Thermo Sorvall WX 100 and T‐865 Rotor (Thermo Fisher Scientific). For the mouse serum samples, the filtered supernatant after removal of medium/large EVs, was diluted to 6 ml with ice‐cold PBS in a PA Ultracrimp Tube (03945, Thermo Fisher Scientific), and subjected to the ultracentrifugation at 100k × g for 70 min with a Thermo Sorvall WX 100 and T‐8100 Rotor (Thermo Fisher Scientific) to precipitate pellets. The precipitated pellets, either from conditioned culture media or from mouse serum, were washed and resuspended with ice‐cold PBS, and experienced the ultracentrifugation step again to collect the final pellets, which were resuspended in ice‐cold PBS and measured using a Bio‐Rad Protein Assay Kit according to the instruction.
The isolation of sEVs by sucrose density gradient centrifugation was conducted when necessary. The sEVs collected by ultracentrifugation were loaded onto a sucrose gradient (20%‐60% sucrose). The sEVs were centrifuged 100k × g for 16 h at 4°C using a Thermo Sorvall WX 100 and T‐865 Rotor. Sucrose solutions of different densities were collected and diluted with cold PBS. The sEVs were further centrifuged at 100k × g for 70 min at 4°C using a Thermo Sorvall WX 100 and T‐8100 Rotor and resuspended in cold PBS again. Sucrose concentration was measured using a refractometer.
2.6. Negatively‐stained transmission electron microscopy (TEM)
After fixing the purified sEVs in 2% (v/v) paraformaldehyde for 5 min at room temperature, 10 μg of sEV suspension was dropped onto an EM copper grid. After waiting for 5 min, the suspension was removed using filter paper and s sEVs were stained with 2% phosphotungstic acid for 3 min. After removing excess phosphotungstic acid with filter paper, the grids were dried at room temperature for 10 min and examined under a transmission electron microscope (JEM‐1200EX, JEOL Ltd., Japan).
2.7. Nanoparticle tracking analysis (NTA)
Size distribution and particle concentration in isolated sEV samples were measured using a NanoSight NS300 instrument (Malvern Instruments, Ltd., UK). The sEV samples were diluted 20–100 times with PBS until the number of particles in the field of view was less than 100. The sEV sample detection was dependent on the number of sEV‐secreting cells present. All sEV samples used for comparison were obtained from the same number of cells and diluted simultaneously with the same dilution fraction. Samples were measured three times and analyzed using NanoSight NS300 NTA software (version 3.0, Malvern Instruments, Ltd., UK).
2.8. Analysis of intracellular Ca2+ dynamics
Intracellular calcium dynamics were measured using the FLIPR Calcium 6 Assay Kit and FLIPR Tetra Real‐time Kinetic Cellular Assays System (Molecular Devices, USA) according to the manufacturer's instructions. Briefly, cells were cultured at a density of 1.5 × 105 cells per well in 96‐well analysis plates (black plates, clear bottom) with or without IFN‐γ treatment for 18 h prior to testing. Calcium 6 was then added to the cells and incubated for 2 h. The specified concentrations of ATP or Tg were added to the wells and the data were collected via the FLIPR Tetra Real‐time Kinetic Cellular Assays System.
2.9. Reverse transcriptase‐quantitative PCR
Total RNA was extracted from cells by TRIzol. Determination of RNA concentration by Nano Drop 2000c spectrophotometer (Thermo Science). Complementary DNAs (cDNAs) were reverse transcribed using Transcriptor Universal cDNA Master. The cDNA samples were run on ViiATM7 Real‐Time PCR System (Applied Biosystems) using SYBRTM Green Master Mix. The primers used are shown in Table 1.
2.10. Protein immunoblot analysis
Cells and sEV samples were lysed in RIPA buffer with Pierce Protease and Phosphatase Inhibitor. The sEV sample detection was dependent on the number of sEV‐secreting cells present. All sEV samples used for comparison were obtained from the same number of cells and diluted simultaneously with the same dilution fraction. Protein samples were electrophoresed on 8%–12% SDS‐PAGE gels and transferred to Nitrocellulose Transfer Membrane. After blocking with 5% lipid milk and 0.1% Tween 20 in Tris‐buffered saline solution (TBS) for 1 h at room temperature, the membranes were incubated with primary antibodies overnight at 4°C. The membranes were then incubated with secondary antibodies for 2 h at room temperature. Protein bands were visualized using ECL reagents and Amersham Imager 800 (GE Healthcare, USA) and quantified using the ImageJ software (NIH, Bethesda, USA). The antibodies used are shown in Table 1.
2.11. ELISA measurement of sEV PD‐L1 and interleukin 2 (IL‐2)
To detect PD‐L1 in sEVs, sEVs extracted from culture media and C57 mouse serum were analyzed by ELISA. The PD‐L1 protein content of the extracted sEVs was determined using the PD‐L1 ELISA kit (human/mouse) as shown in Figure 2f. The PD‐L1 concentration in each sample was calculated from a standard curve made from recombinant PD‐L1 with confirmed concentration following the manufacturer's protocol.
FIGURE 2.
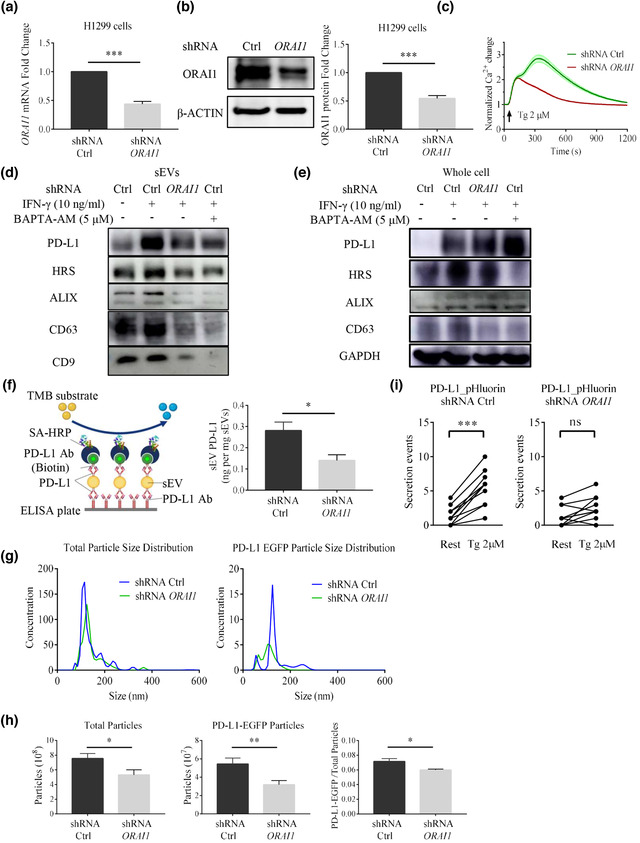
ORAI1 KD reduces sEV PD‐L1 secretion in H1299 cells. (a) The ORAI1 mRNA level is reduced in shRNA knockdown (KD) against the ORAI1 gene in H1299 cells (n = 3). (b) Immunoblotting confirms that ORAI1 protein expression is reduced after knocking down ORAI1 gene in H1299 cells. The right panel shows the quantification of each blot normalized to β‐actin (n = 3). (c) ORAI1‐KD cells show the reduction of Tg‐ (2 μM) mediated Ca2+ responses in H1299 cells. Values of fluorescent intensity represent mean ± SEM, which is normalized to the basic fluorescence intensity. All data is derived from more than 4 wells in 3 individual experiments. Immunoblotting analysis of PD‐L1 and sEV markers in sEVs (d) and whole‐cell lysate (e). IFN‐γ (10 ng/ml) or Bapta‐AM (5 μM) treatment are indicated in control H1299 cells and ORAI1‐KD H1299 cells (n = 3). (f) Schematic diagram of ELISA for the measurement of the concentration of sEV PD‐L1 (left). Comparison of the amounts of PD‐L1 in sEVs derived from control cells and ORAI1‐KD cells by ELISA experiments (right, n = 3). (g) Comparison of total particle size distribution (left) and PD‐L1_EGFP particle size distribution (right) in medium derived from control H1299 cells and ORAI1‐KD H1299 cells by NTA. Both cell types have been transfected with PD‐L1_EGFP plasmids. Units are in millions of particles per milliliter. (h) Comparison of total secreted particles (left), PD‐L1_EGFP particles (middle), and the fraction of PD‐L1 EGFP particles among total secreted particles (right) by NTA (n = 3). (i) Measurement of fusion events in individual H1299 cells (n = 10) transfected with PD‐L1_pHluorin in control cells (left) and ORAI1‐KD cells (right) before and during the Tg (2 μM) stimulation. For all of Figure 2, data are representative of at least three independent experiments. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
The cytokines IL‐2 content in the serum of C57 mice was determined using the IL‐2 ELISA kit according to the manufacturer's instructions. The concentration of IL‐2 in each sample was calculated from a standard curve prepared using recombinant IL‐2 from a standard.
2.12. Kaplan‐Meier Plotter analysis
The prognostic value of ORAI1 mRNA expression was assessed using the online analysis tool Kaplan‐Meier Plotter (www.kmplot.com) (Gyorffy et al., 2013). The analysis contained gene expression data and survival information for 1144 lung cancer patient samples from 10 independent datasets (GSE14814, GSE19188, GSE29013, GSE30219, GSE31210, GSE3141, GSE31908, GSE37745, GSE4573, GSE50081, and TCGA). To better analyze Overall Survival in lung cancer patients, patient samples were divided into two groups (high and low expression) based on hazard ratio (HR) values and log‐rank p‐values using Auto select best cut‐off (Figure S4). The Kaplan‐Meier survival plots were evaluated with a hazard ratio (HR) of 95% confidence interval (CI) and a log‐rank P‐value.
2.13. Flow cytometry analysis
To assess the subpopulation of T cells in the spleen of C57 mice, flow cytometry assays were performed. Single cells were isolated from the spleens of mice. 1 × 107 splenocytes were blocked with anti‐mouse CD16/32 Antibody for 10 min and then incubated with the antibody for 30 min, followed by washing with staining buffer. Fixation and permeabilization treatments were performed using Fixation/Permeabilization Buffer according to the manufacturer's protocol, followed by intracellular staining for Ki67 and Granzyme B. Flow cytometry and data analysis were performed on CytExpert software (Beckman Coulter, USA). The gating strategy for flow analysis is shown in Figure S5. Details of the flow cytometry antibodies used in this study can be found in Table 1.
2.14. Animal experiments
To establish a xenograft model, C57 BL/6 mice (four per cage) were housed in a thermostatically controlled room at the Animal Centre of the Macau University of Science and Technology with a 12‐h light/dark cycle and free access to food and water. All experimental procedures were carried out following the Institutional Animal Care and User Committee Guidelines of the Macau University of Science and Technology.
Control or Orai1‐KD LLC‐1 cells (1 × 106 cells per 0.1 ml) were injected subcutaneously into the right hypochondrium of mice (n = 8 mice per group) to grow tumors. To estimate tumor size, the length (L) and width (W) of each tumor were measured daily from day 7 after tumor injection using Vernier calipers, and tumor volume was calculated according to the following equation: (L×W^2)/2. Mice were considered ‘terminal’ when tumors reached 2 cm in at least one dimension. Individual tumor sizes and mouse survival curves were recorded.
Control or Orai1‐KD LLC‐1 cells (5 × 105 cells per 0.1 ml) were injected subcutaneously into the right hypochondrium of mice (n = 8 mice per group) to grow tumors. Mice were subsequently treated with 200 μg of antibody (anti‐PD‐L1 antibody clone 10F.9G2 or isotype control) in PBS per dose (8–10 mg/kg) intravenously every 3 days for a total of five injections. Tumor size was measured as described above. The experimental plan is shown in Figure 4a. Tissue samples and serum were collected on day 28 after the LLC1 cells injection. The tumor and spleen were weighed. Tumor tissue was taken and used for paraffin sections for immunohistochemical analysis. The spleen tissue was taken and dispersed into a single cell suspension for flow cytometric analysis.
FIGURE 4.
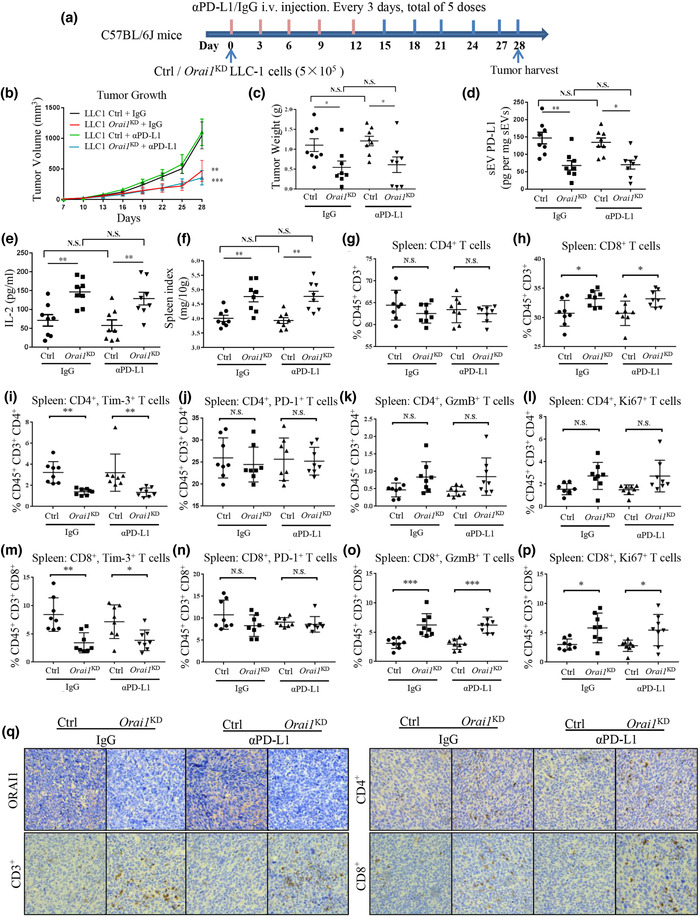
Orai1 KD suppresses tumor progression by enhancing antitumor immunity. (a) Schematic experimental design for animal model experiments. (b) Comparison of tumor growth (volume) over time in mouse lung cancer models. Each C57BL/6J mouse was injected subcutaneously with 5 × 105 Ctrl or Orai1‐KD LLC‐1 cells and treated as indicated (n = 8). (c) Comparison of tumor weights in mouse lung cancer models. (d) Comparison of PD‐L1 protein levels in serum sEVs derived from mouse lung cancer models by ELISA measurement. (e) ELISA quantification of serum IL‐2 expression in mouse lung cancer models. (f) Comparison of spleen index of mouse lung cancer models. (g & h) Flow cytometric quantification of the percentage of CD4+ (g) and CD8+. (h), respectively, among CD45+, CD3+ cells in the spleen. (i & m) Quantification of Tim3+ cells among CD4+ (i) and CD8+ (m) T cells. (j & n) Quantification of PD‐1+ cells among CD4+ (j) and CD8+ (n) T cells. (k & o) Quantification of granzyme B (GzmB) CD4+ (k) and CD8+ (o) T cells. (l & p) Quantification of Ki67 CD4+ (l) and CD8+ (p) T cells. (q) ORAI1, CD3, CD4+ and CD8+ immunohistochemistry (IHC) staining in mice tumor tissues (original magnification 40×). For all of Figure 4, data are representative of 8 experimental animals per group. ns, not significant; *P < 0.05; **P < 0.01;*** P < 0.001. See also Figure S5.
2.15. Live cell fluorescence microscopy assay
Coverslips containing cells were placed in the imaging chamber and perfused with normal ringer solution (2 mM CaCl2, 2.5 mM KCl, 145 mM NaCl, 1 mM MgCl2, 10 mM glucose, 10 mM Hepes, pH 7.4). Stimulation was performed using calcium‐free Ringer solution (0.5 mM EGTA, 2.5 mM KCl, 145 mM NaCl, 1 mM MgCl2, 10 mM glucose, 10 mM Hepes, pH 7.4) or the normal ringer solution containing 2 μM Thapsigargin (Tg). Images were taken on the API Delta Vision Live‐cell Imaging System (GE Healthcare Company, USA) using a 40× flat‐field apochromatic mirror and the Leica TCS SP8 Confocal Laser Scanning Microscope System (Leica Biosystems Imaging, USA) using a 40× oil objective.
2.16. Immunohistochemical (IHC) staining
Tumor sections were dewaxed and antigenically repaired in xylene followed by the inactivation of endogenous peroxidase in 3% H2O2/MeOH. IHC was performed to assess the expression of ORAI1, CD3+, CD4+ and CD8+ using an immunohistochemical kit (FXP019; 4A Biotech, Beijing, China) according to the manufacturer's protocol. Images were acquired using a Leica optical microscope (Leica Biosystems Imaging, USA).
2.17. Immunocytochemistry (ICC) staining
Cells were fixed in 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X‐100 for 5 min. After blocking in 5% BSA, they were incubated with Actin‐Tracker Green (phalloidin‐FITC) for 30 min. Fluorescent images were captured with a × 40 objective on the API Delta Vision Live‐cell Imaging System (GE Healthcare Company, USA).
2.18. Statistical analysis
Statistical analysis was performed using GraphPad online software (https://www.graphpad.com/quickcalcs/). The results were expressed as means ± SEM as indicated. The difference was considered statistically significant when the p‐value was less than 0.05. Student's t‐test, two‐way ANOVA analysis or Wilcox test was used to compare different groups.
3. RESULTS
3.1. Ca2+ is critical for sEV PD‐L1 secretion
According to current knowledge, EVs are cell‐released membrane structures, including exosomes, ectosomes, microvesicles (MVs), microparticles, oncosomes, apoptotic bodies and many other EV subsets (Doyle & Wang, 2019). They act as special mediators to communicate with and affect the cellular environment (Kalluri, 2016). Tumorigenesis and progression are closely related to the tumor microenvironment, which is significantly reshaped by EVs. Exosomes carrying PD‐L1 were reported to promote tumor growth in NSCLC (Chen et al., 2018; Kim et al., 2019; Poggio et al., 2019; Yang et al., 2018), which may be the reason for the low response rates of patients receiving PD‐1/PD‐L1 immunotherapy. We, therefore, studied the roles of intracellular calcium in such sEVs release in NSCLC. We used differential centrifugation to isolate 100k g pellets (Figure S1a) from the supernatant of medium for NSCLC H1299 cell, which was reported to release sEVs carrying PD‐L1. (Colombo et al., 2014; Melo et al., 2015; Peinado et al., 2012; Robbins & Morelli, 2014). We examined the 100k g pellets by transmission electron microscopy (TEM) (Figure 1a and Figure S1b) and nanoparticle tracking analysis (NTA) (Figure 1c). Considering the size, shape, and other characteristics of different EVs (van Niel et al., 2018), the majority of vesicles pelleted by our differential centrifugation method displayed properties similar to those generally assigned to sEVs (i.e., 30–200 nm). PD‐L1 is expressed on the surface of tumor cells, and it is also contained in sEVs. Our experiments using density gradient centrifugation showed that PD‐L1 was co‐localized with the standard exosome‐enriched markers CD63 and HRS (Figure 1b). Both markers were present mainly in the 20%–40% sucrose band, confirming that PD‐L1 release and transport occur mainly through sEVs (Chen et al., 2018; Poggio et al., 2019). Furthermore, PD‐L1, HRS, ALIX, CD63 and CD9 were significantly enriched in the isolated sEVs fraction (Figure 1e), compared to the whole‐cell lysate and the m/lEV fraction. Meanwhile, signals from GM130 (a marker of Golgi) and H3 (markers of nucleus) in the sEVs fraction were almost undetectable, further supporting the high purity of our preparation. These data show that we had successfully isolated sEVs, which contained significant amounts of PD‐L1.
FIGURE 1.
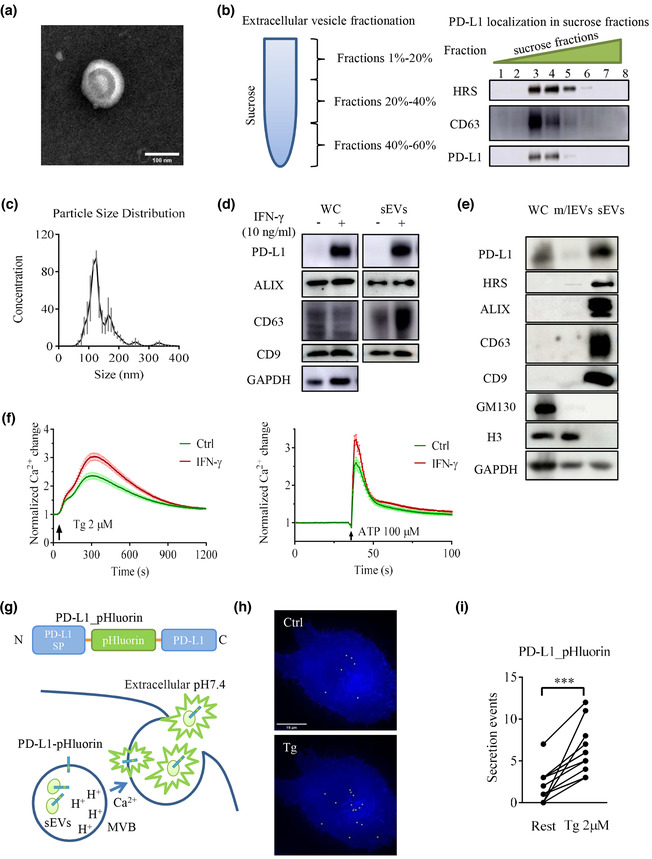
IFN‐γ and Ca2+ promote sEV PD‐L1 secretion in H1299 cells. (a) A representative transmission electron microscopy (TEM) image of a purified sEV from H1299 cells. Scale bar, 100 nm. (b) sEVs were isolated by sucrose gradient density ultracentrifugation, followed by the immunoblotting for PD‐L1 and sEVs markers CD63 and HRS. (c) Nanoparticle tracking analysis (NTA) was conducted on H1299 cells to determine the size distribution of sEVs isolated by ultracentrifugation. Traces represent mean values in units of million particles per ml from 3 experiments. (d) Immunoblots for PD‐L1 and sEVs markers ALIX, CD63 and CD9 in whole‐cell lysates and sEVs from H1299 cells treated with or without IFN‐γ (10 ng/ml). (e) Immunoblotting analysis of PD‐L1, EVs markers and organelle makers performed on whole‐cell lysates (WC), m/lEVs and sEVs from H1299 cells. (f) Effect of IFN‐γ pretreatment on thapsigargin‐(Tg 2 μM) and ATP‐(100 μM) mediated Ca2+ dynamics. Values of fluorescent intensity represent mean ± SEM, which is normalized to the basic fluorescence intensity. All data is derived from more than 4 wells in 3 individual experiments. (g) Schematic illustration of PD‐L1_pHluorin plasmid (upper) and proposed sEVs releasing model (below) through visualization of PD‐L1_pHluorin. sEV PD‐L1_pHluorins are quenched while resident in the acidic lumen of the multivesicular body (MVB). Immediately after sEVs secretion, the pHluorin is exposed to the neutral extracellular medium, causing a sudden increase in fluorescence intensity. (h) Total projection of secretion events over a 180 s was recorded from a cell before (top) and after (bottom) stimulation with Tg (2 μM). (i) Counts of fusion events within a time window of 180 s in individual H1299 cells (n = 11) transfected with PD‐L1_pHluorin. Two points connected by a line represent the fusion events numbers from the same cell but under different conditions (before Tg vs. after Tg). For all of Figure 1, data are representative of at least three independent experiments. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Recent studies have demonstrated that IFN‐γ released by immune cells can upregulate PD‐L1 expression in cancer cells. This may cause adaptive immune resistance and suppresses the anti‐tumor immune response (Garcia‐Diaz et al., 2017). In prostate cancer, such upregulation of PD‐L1 was also shown in exosomes, suggesting the critical roles of sEV PD‐L1 in mediating the escape from the systemic anti‐tumor immunity (Poggio et al., 2019), which may compromise the final results in checkpoint inhibitor therapy. In our experiments, IFN‐γ upregulated PD‐L1 levels in many NSCLC cells (Figure S2). In H1299 cells, IFN‐γ increased the PD‐L1 protein level in the whole‐cell lysate and sEVs (Figure 1d). Thapsigargin (Tg) and ATP can increase intracellular calcium levels. Interestingly, such responses in H1299 cells were boosted after treatment with IFN‐γ (Figure 1f). Calcium is the key player for exocytosis and endocytosis. The fusion of MVBs with the plasma membrane is a typical form of exocytosis. Previous studies have shown that elevated intracellular calcium promotes secretion (Messenger et al., 2018). We hypothesized that the IFN‐γ‐mediated increase in sEV PD‐L1 is associated with high intracellular calcium. To test this hypothesis, we used a PD‐L1_pHluorin construct containing a pH‐sensitive pHluorin at the luminal/extracellular N‐terminus of PD‐L1 (Figure 1g Top) to observe the sEV PD‐L1 release in real‐time by fluorescence microscopy. PD‐L1_pHluorin fluorescence is quenched in the lumen of the MVB due to the intra‐MVB acidic milieu. Thus, its fluorescent signal can hardly be detected inside MVBs. However, after the fusion of MVBs with the plasma membrane, exosomes are released from MVBs, and pHluorin tagged to PD‐L1 is neutralized by the extracellular solution, resulting in a sudden increase in fluorescence intensity of pHluorin (Figure 1g Bottom). Such localized sudden fluorescence increases represent the release of several sEVs from a single MVB (Messenger et al., 2018; Verweij et al., 2018). In H1299 cells transfected with PD‐L1_pHluorin, only few vesicles were released under resting conditions (Figure 1h Top, see movie1). However, secretion events increased markedly in frequency after stimulation by Tg (Figure 1h Bottom, see movie2). Analyzing signals from several single cells (Figure 1i), we observed a significant increase in sEV‐mediated fusion activity within 3 min after the onset of Tg stimulation. Similarly, increased secretion events were also observed in H1975 cells (Figure S3a). These results confirm that elevated intracellular calcium can promote sEV PD‐L1 secretion.
3.2. ORAI1 can regulate the release of sEV PD‐L1
ORAI1 is a pore‐forming ion channel that is highly selective for calcium ions (Vig et al., 2006). It can be activated by ER‐localized STIM proteins that sense the depletion of ER calcium stores to increase the intracellular calcium level. In many types of cancer, ORAI1 plays a prominent role in regulating tumor proliferation and migration (McAndrew et al., 2011; Villalobos et al., 2019; Zhan et al., 2015). It is also the downstream target for some GPCR signaling, such as histamine and other inflammatory cytokines (Sun & Ye, 2012). To explore the role of ORAI1 in sEV PD‐L1 secretion, we used lentiviral shRNA to knock down (KD) the ORAI1 gene expression in NSLCC H1299 cells. For control experiments, we made WT H1299 cells with control shRNA. KD treatment significantly reduced the levels of both ORAI1 mRNA and protein (Figure 2a, 2b). It also considerably reduced the response of H1299 cells to Tg application (Figure 2c). Compared with WT H1299 cells, the IFN‐γ‐induced upregulation of sEV PD‐L1 was significantly suppressed in ORAI1‐KD H1299 cells. We observed a significant decrease in HRS, ALIX, CD63 and CD9 protein levels as well in sEVs from ORAI1‐KD H1299 cells. In the presence of the calcium chelator Bapta‐AM, IFN‐γ could not upregulate sEV PD‐L1 and other markers for sEVs, which are enriched in WT H1299 cells (Figure 2d). Notably, in the whole‐cell lysate, the IFN‐γ‐induced upregulation of sEV PD‐L1 was not inhibited by ORAI1 KD or Bapta‐AM treatment. Rather in both cases, an upregulation was observed. However, ORAI1 KD or Bapta‐AM treatment showed suppressive effects on HRS and CD63 (Figure 2e). We also measured sEV PD‐L1 by using an ELISA kit of PD‐L1 antibody. In these experiments, the amount of sEV PD‐L1 secreted from ORAI1‐KD cells was significantly smaller than that from WT cells when both cell types were cultured in the presence of INF‐γ (Figure 2f). To further assess the effect of ORAI1 KD on sEV PD‐L1 secretion, we ectopically expressed PD‐L1_EGFP in H1299 cells. When sEVs extracted by ultracentrifugation were subjected to NTA, a decreased total particle number was observed in ORAI1‐KD H1299 cells, as compared to WT H1299 cells (Figure 2g). Furthermore, PD‐L1_EGFP+ particles decreased to a larger extent, lowering the ratio of PD‐L1_EGFP+ particles number over the total particle number (Figure 2h). Using PD‐L1_pHluorin for measuring real‐time sEV PD‐L1 secretion, as described above, there was no significant increase of fusion events in single cells recorded in the ORAI1‐KD group after Tg stimulation (Figure 2i), unlike the case for WT cells (Figure 1i). To explore the regulation of ORAI1 on the release of sEV PD‐L1 in other NSLCC, we repeated the experiments in H1975 cells and found that ORAI1‐KD H1975 cells had less sEV PD‐L1 secretion (Figure S3). The results suggest that ORAI1 plays an essential role in calcium‐dependent sEV PD‐L1 release.
3.3. ORAI1 loss inhibits tumor progression by reducing sEV PD‐L1 secretion
Considering that ORAI1 loss can reduce sEV PD‐L1 secretion, we want to know whether ORAI1 loss can inhibit tumor progression in vivo. For this purpose we used a syngeneic model for lung cancer, the LLC1 model (Kellar et al., 2015), which shows resistance to PD‐1/PD‐L1 antibody blockade (Li et al., 2017; Lin et al., 2018). In the LLC1 cells, we used lentiviral shRNA to knock down the mouse Orai1 gene. The qPCR and western blotting verified that Orai1 mRNA and ORAI1 protein levels were significantly decreased (Figure 3a, b). Orai1 KD suppressed the Tg‐mediated intracellular calcium response and reduced the sEV PD‐L1 protein level, compared with cells treated with control shRNA (WT LLC1 cells) (Figure 3c, d). However, the KD treatment did not affect PD‐L1 protein level in whole cells (Figure 3d). Results with LLC1 cells were similar to those obtained with H1299 cells (Figure 2d, e).
FIGURE 3.
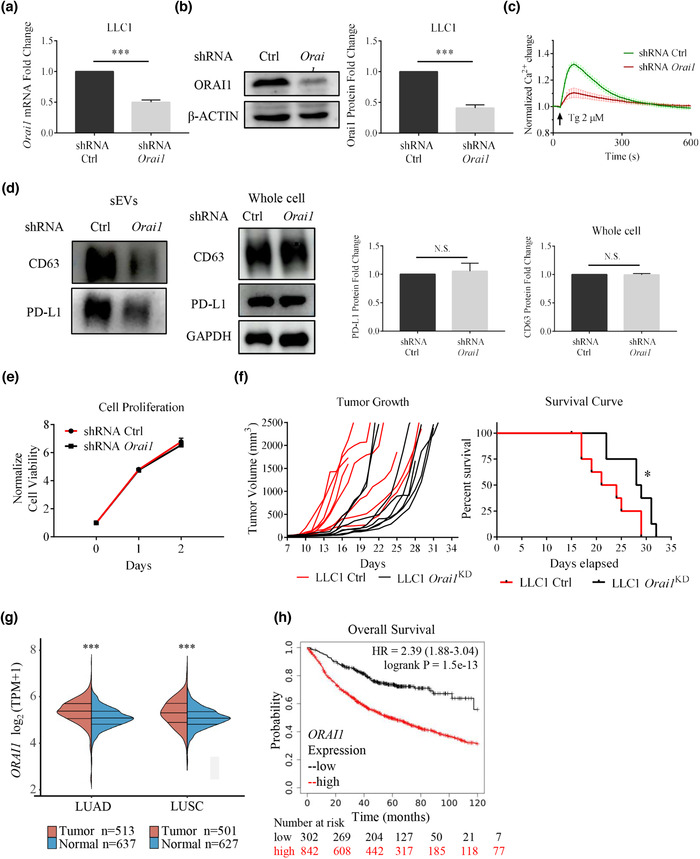
Orai1 KD suppresses tumor progression in the LLC‐1 lung cancer model by reducing sEV PD‐L1 secretion. (a) shRNA efficiently reduced Orai1 mRNA levels in mouse LLC‐1 cells (n = 3). (b) Immunoblotting analysis confirms Orai1 KD; quantification of western blotting data (right, n = 3). (c) Orai1 KD inhibits Tg‐ (2 μM) mediated Ca2+ increases in LLC‐1 cells. Values of fluorescent intensity represent mean ± SEM, which is normalized to the basic fluorescence intensity. All data is derived from more than 4 wells in three‐five individual experiments. (d) Immunoblotting analysis of sEVs (left) and whole cell (right) PD‐L1 protein in control and Orai1‐KD LLC‐1 cells. The normalization analysis is shown on the right (n = 3). (e) Cell counts over time for control and Orai1‐KD LLC‐1 cells. The number is normalized to the initial cell number. (n = 4) (f) Tumor volume graphs from individual animals (left) and mouse survival curves (right) are shown. Tumors are implanted in C57BL/6J mice by subcutaneously injecting 1×106 wild type (control) or Orai1‐KD LLC‐1 cells (n = 8 for each genotype). The significance of the difference between the two groups of samples passed the Log‐rank (Mantel‐Cox) test. (g) The expression of ORAI1 gene in tumor tissues (red) and normal tissues (blue) from TCGA data and GTEx data, where the horizontal axis represents tumor tissues of different NSCLCs, and the vertical axis represents the gene expression distribution. The significance of the difference between the two groups of samples passed the Wilcox test. (h) Kaplan‐Meier survival curves for the ORAI1 gene in NSCLC patients. Overall survival (OS) of patients is plotted both for the low (black) and high (red) ORAI1 expression groups. The ‘number‐at‐risk’ is indicated below the plot. For all of Figure 3, data are representative of at least three independent experiments. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Before performing animal experiments, we examined cell proliferation. Orai1‐KD LLC1 cells did not show any difference compared with WT LLC1 cells (Figure 3e). These two types of lung cancer cells, WT LLC1 cells and Orai1‐KD LLC1 cells, were injected into the ventral axilla of C57BL6/J syngeneic mice, respectively. All mice injected with LLC1 cells developed visible tumors at around seven days, and they were euthanized between 17 and 29 days due to the larger and fast‐growing tumors. In contrast, mice injected with Orai1‐KD LLC1 cells had smaller and slow‐growing tumors, together with a significantly extended lifespan (Figure 3f). These data show that ORAI1 is strongly correlated with tumor genesis and development in the lung cancer mouse model.
After these animal experiments, we addressed the lung cancer disease in humans. An analysis of ORAI1 gene expression in 1700 normal and NSCLCs human tissues listed in the TCGA and GTEx databases showed that the ORAI1 gene is significantly upregulated in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) samples (Figure 3g). We hypothesized that ORAI1 gene expression might influence the prognostic factors in patients of NSCLCs. Using the Kaplan‐Meier survival analysis platform, we found that increased ORAI1 mRNA level is an unfavorable prognostic factor for overall survival in patients with NSCLCs (hazard ratio, HR = 2.39; 95% CI: 1.88‐3.04; P = 1.5e‐13; n = 1144) among homologous proteins (Figure 3h, Figure S4). Therefore, ORAI1 seems essential for lung cancer, and its role is worth further exploration.
3.4. ORAI1 loss activates anti‐tumor immunity by reducing sEV PD‐L1 secretion
Next, we asked whether ORAI1 loss inhibits tumor progression by affecting the immune response against the tumor. To address this question, we examined tumor tissue, plasma, and spleen tissue from two groups of mice injected with WT and Orai1‐KD LLC1 cells, respectively. To study the effects of checkpoint inhibitors, anti‐PD‐L1 antibody and its isotype control (IgG) were also used (Figure 4a). On day 28, the tumors of mice injected with Orai1‐KD LLC1 cells were significantly smaller than those of mice injected with WT cells (Figure 4b, 4c). The anti‐PD‐L1 antibody had no significant effect on tumors in both groups of mice (Figure 4b, 4c), as has been reported (Li et al., 2017; Lin et al., 2018). Compared with the mice injected with WT LLC1 cells, mice injected with Orai1‐KD LLC1 cells had significantly reduced plasma sEV PD‐L1 (Figure 4d) and higher plasma IL‐2 level (Figure 4e) measured by ELISA. Their spleens were considerably larger (Figure 4f), indicating a more robust immune response in mice bearing the Orai1‐KD LLC1 cells.
sEVs, released into the extracellular space, are likely not only to influence the tumor microenvironment, but rather to have long‐range actions in addition. In order to test this expectation, we performed immune‐cell typing in the spleen, an organ far away from the original tumor xenograft site initiating adaptive immune responses. The highly vascularized spleen has strong power to filter the blood stream and monitor its composition, including sEVs released from other sites. Flow cytometric quantification showed striking differences of cell types between the two groups of mice under study (Figure S5). Injection of Orai1‐KD LLC1 cells resulted in an increased proportion of CD8+ T cells relative to WT LLC1 cell injection (Figure 4h). In contrast, CD4+ T cells remained unchanged (Figure 4g). We further evaluated T cell depletion and activation markers in CD8+ and CD4+ T cell populations. The proportion of cells expressing Tim‐3, a T cell exhaustion marker, was significantly decreased in CD8+ and CD4+ cells from mice injected with Orai1‐KD LLC1 cells (Figure 4i, 4m). Meanwhile, in CD8+ T cells, the proportions of cells expressing the activation marker Granzyme B and cells expressing the proliferation marker Ki67 were significantly increased in number (Figure 4o, 4p). Their abundance had an upward trend in CD4+ T cells (Figure 4k, 4l). In contrast, the fraction of PD‐1‐positive CD8+ and CD4+ cells did not show significant differences in the two groups of mice (Figure 4j, 4n).
Based on these results, we further looked into the tumor tissues and investigated the interactions between immune cells and tumor cells. The immunohistochemical staining showed a lower level of ORAI1 protein in the tumor tissue generated from the Orai1‐KD LLC1 cells. At the same time, we observed significantly more tumor‐infiltrating T‐lymphocytes in mice injected with Orai1‐KD LLC1 cells than in mice injected with WT LLC1 cells. The infiltrating lymphocytes included CD3+, CD4+, and CD8+ cells. In contrast, there appeared to be no significant effect of αPD‐L1 treatment on infiltrating lymphocytes (Figure 4q). These data suggest that the removal of ORAI1 inhibits tumor progression by reducing the tumor‐secreted sEV PD‐L1 and activating T‐cells. This process takes place not only at the tumor site but also at other sites, such as the spleen.
3.5. Melanophilin is required for the release of sEV PD‐L1
Several Rab GTPase family members, such as Rab2, 5, 11, 27, and 35, have been implicated in sEV release (Abels & Breakefield, 2016; Kowal et al., 2014; Vader et al., 2014). During endosomal positioning, maturation, and fusion of MVBs with the plasma membrane, Rab27a plays an essential role. It cooperates with its effectors regulating particular steps during sEV secretion (Messenger et al., 2018; Ostrowski et al., 2010). For example, its effector, Munc13‐4, binds to Rab27a forming a vital complex for secretory vesicle transport to the plasma membrane and degranulation (Elstak et al., 2011). Another effector of Rab27a, melanophilin (MLPH), is required to release mast cell granules and melanosomes (Fukuda et al., 2002; Singh et al., 2013). Furthermore, MLPH has multiple proteolysis sequences for calpain, which is a calcium‐dependent proteolytic enzyme. We hypothesized that MLPH also regulates sEV PD‐L1 release. To validate this conjecture, we used lentiviral shRNA to knock down the MLPH gene in H1299 cells. Both mRNA and protein levels were significantly decreased in MLPH‐KD H1299 cells (Figure 5a, 5b). These cells secreted a substantially lower amount of sEV PD‐L1 (Figure 5c) in the presence of IFN‐γ. sEVs extracted from MLPH‐deficient H1299 cells and examined by ELISA led to similar results (Figure 5d). Measurements of extracted sEVs by NTA showed a significant decrease in both total granule number and PD‐L1_EGFP+ granules in MLPH‐KD H1299 cells compared to WT H1299 cells (Figure 5e, 5f). Likewise, in H1975 cells, MLPH‐KD treatment significantly reduced sEV PD‐L1 secretion (Figure S6). The results suggest that MLPH also plays a vital role in the IFN‐γ‐mediated release of sEV PD‐L1.
FIGURE 5.
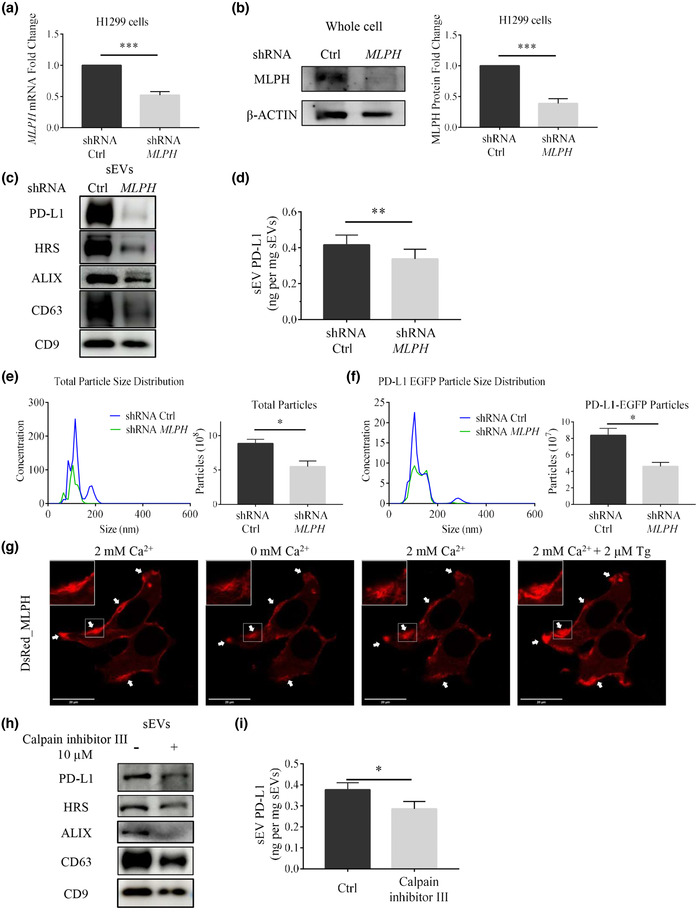
MLPH KD reduces sEV PD‐L1 secretion in H1299 cells. (a) shRNA efficiently reduces MLPH mRNA levels in H1299 cells (n = 3). (b) Immunoblotting analysis confirms MLPH KD (left) and its immunoblotting quantification (right, n = 3). (c) Immunoblotting Comparison of sEVs protein in H1299 cells with or without MLPH‐KD treatment. (d) ELISA measurements of PD‐L1 abundance in sEVs derived from H1299 cells with or without MLPH‐KD treatment (n = 3). (e) NTA comparison of total particle size distribution in H1299 cells with or without MLPH‐KD treatment (n = 3). Units are in millions of particles per milliliter. (f) NTA comparison of PD‐L1 EGFP particle size distribution in H1299 cells transfected with PD‐L1_EGFP with or without MLPH‐KD treatment (n = 3). Units are in millions of particles per milliliter. (g) Distribution of DsRed_MLPH in H1299 cells when cells are exposed to 0 mM Ca2+, 2 mM Ca2+ and 2 mM Ca2+ plus 2 μM Tg treatment. White arrows in the panels point to puncta formed by the MLPH aggregates. (h) Immunoblotting analysis of sEVs protein in H1299 cells in the absence or presence of Calpain inhibitor III (10 μM). (i) Comparison between amounts of PD‐L1 in sEVs derived from H1299 cells with or without Calpain inhibitor III (10 μM) by ELISA (n = 3). For all of Figure 5, data are representative of at least three independent experiments. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
To further investigate the functions of MLPH, we overexpressed DsRed_MLPH in H1299 cells (Figure 5g). We observed lamellipodia‐like protrusions along the edge of the plasma membrane. Frequently, fluorescent signals showed a cytoskeleton fiber bundle or aggregated puncta. Interestingly, the MLPH fluorescent signals dynamically changed their distribution following changes of intracellular calcium levels. The extracellular calcium chelator EGTA decreased the peripheral MLPH fluorescent intensity and marginally increased the intracellular signal. It seems that fluorescent material close to the plasma membrane moved towards the intracellular space. When raising the extracellular calcium level to 2 mM, the peripheral MLPH fluorescence increased. The MLPH signal became stronger and stronger near points of contact with the plasma membrane after continually increasing the calcium level by Tg. MLPH contains multiple potential PEST‐like sequences, which are rich in proline (P), glutamic acid (E), serine (S), and threonine (T), and are recognized by calpain for cleaving the protein in a calcium‐dependent way (Fukuda & Itoh, 2004). The MLPH fluorescent responses to calcium suggested calpain activities in the dynamic regulation of MLPH. We treated H1299 cells with calpain inhibitor III and found that this inhibitor could block the secretion of sEV PD‐L1 in the presence of IFN‐γ (Figure 5h, 5i, S7a, b). Significant reduction of sEV PD‐L1 secretion by calpain inhibitor III was also found in H1975 cells (Figure S8a‐c). Entries in the Modulatory Proteolysis Database and other studies (Fukuda & Itoh, 2004) showed that amino acids 390–415 of MLPH constitute PEST‐like cleavage sites for calpain. This motif is at the interface between the myocardin Va‐binding domain (MBD) and an actin‐binding domain (ABD). Deletion of the PEST‐like sequence does not affect myocardin Va‐binding or actin‐binding activity but affects the MLPH's cleavage by calpains (Fukuda & Itoh, 2004). We made a MLPH mutant without a.a. 390.‐415 (Figure S7c, d) and overexpressed it in H1299 cells. WT MLPH co‐localized significantly with actin filaments while aggregating into clusters. MLPH Δ390‐415 mutant partially co‐localized with actin filaments but showed mainly a diffuse distribution without any apparent specificity. Notably, after treatment with calpain inhibitor III or 0 mM calcium plus EGTA, the WT MLPH localization was significantly altered with clusters disappearing. Co‐localization with actin filaments was reduced considerably. However, these changes were not observed, when cells overexpressing MLPH Δ390‐415 mutant were used (Figure 6a). We also examined the expression of MLPH and PD‐L1 (Figure 6b) as well as the expression of MLPH and myosin V (Figure S7e). These combinations of proteins did not show as strong co‐localization as seen with MLPH and actin. But WT MLPH still showed a similar distribution and corresponding alterations after treatments of calpain inhibitor III or removal of calcium. The PEST‐like sequences of MLPH were also found to be essential for sEV PD‐L1 secretion in H1975 cells (Figure S8d, e). Therefore, it is reasonable to assume that calcium may influence sEV PD‐L1 secretion by regulating the cleavage of MLPH by calpain.
FIGURE 6.

Truncation of a PEST‐like sequence alters the distribution of MLPH in H1299 cells. (a) H1299 cells transfected with WT DsRed_MLPH (left) or DsRed_MLPH Δ390‐415 (right) were analyzed by immunofluorescence of FITC_ACTIN. Cells were treated with 0 mM Ca2+ or 10 μM Calpain inhibitor III. White arrows in the panels point to puncta formed by MLPH aggregates. FITC phalloidin staining for visualization of the actin cytoskeleton; blue arrows point to actin aggregates in the cell cortex. (b) H1299 cells transfected with WT DsRed_MLPH (left) or DsRed_MLPH Δ390‐415 (right) were cotransfected with PD‐L1_EGFP. Cells were treated with 0 mM Ca2+ or 10 μM Calpain inhibitor III. White arrows in the panels point to puncta formed by MLPH aggregates. For all of Figure 6, data are representative of at least three independent experiments.
3.6. SLP2‐a regulates the release of sEV PD‐L1
Synaptotagmin‐like protein 2 (SLP2‐a) is playing a role in vesicle trafficking and secretion (Menasche et al., 2008). It has a SLP homology domain (SHD) at the N‐terminal for binding to Rab27a and C2 domains at the C‐terminal for vesicle targeting, which may confer calcium‐sensitivity (Yu et al., 2007). Could the SLP2‐a protein also be involved in the sEV PD‐L1 secretion? We used lentiviral shRNA to knock down the expression of the SLP2‐a gene in H1299 cells. Both SLP2‐a mRNA and protein levels were significantly reduced (Figure 7a, b) in SLP2‐a‐KD H1299 cells. The cells secreted significantly lower levels of sEV PD‐L1, similar to that of MLPH‐KD H1299 cells (Figure 7d, e). Similar results were obtained using the NTA assay of extracted sEVs (Figure 7f, g). All these results demonstrate that SLP2‐a is also involved in the release of sEV PD‐L1. We checked the distributional responsiveness to EGTA and Tg after overexpression of mCherry_SLP2‐a in H1299 cells. We did not observe any variation in the SLP2‐a distribution when altering intracellular calcium levels this way. Surprisingly, we found that the knockdown of SLP2‐a gene in H1299 cells appeared to decrease the intracellular calcium response to Tg (Figure 7c). SLP2‐a has C2 domains, which are usually responsive to intracellular calcium changes in distinct manners. Also, the C2 domains are essential structures for SLP2‐a localization (Galvez‐Santisteban et al., 2012). To further investigate the function of SLP2‐a, we made a SLP2‐a mutant, SLP2‐a ΔC2, which lacks the C2 domain (Figure S9a, b). When overexpressing SLP2‐a and PD‐L1 in H1299 cells, WT SLP2‐a showed strong co‐localization with PD‐L1, whereas SLP2‐a ΔC2 had a diffuse distribution with no clear co‐localization with PD‐L1 (Figure 7h). We extended these experiments by overexpressing SLP2‐a and Rab27a in H1299 cells. It seemed that SLP2‐a and its mutant SLP2‐a ΔC2 co‐localized well with Rab27a. However, SLP2‐a ΔC2 did not redistribute to the plasma membrane. SLP2‐a ΔC2 was mainly found in the cytoplasm and nucleus (Figure 7i). By looking at the real‐time sEV PD‐L1 secretion, we found that H1299 cells overexpressing SLP2‐a ΔC2 had fewer fusion events than cells overexpressing the WT SLP2‐a (Figure S9c, d). In H1975 cell, we also found that SLP2‐a plays an essential role in sEV PD‐L1 secretion and its regulation is associated with the C2 domain at the SLP2‐a C‐terminal (Figure S10). Therefore, SLP2‐a is involved in sEV PD‐L1 secretion probably by affecting intracellular calcium dynamics and Rab27a distribution.
FIGURE 7.
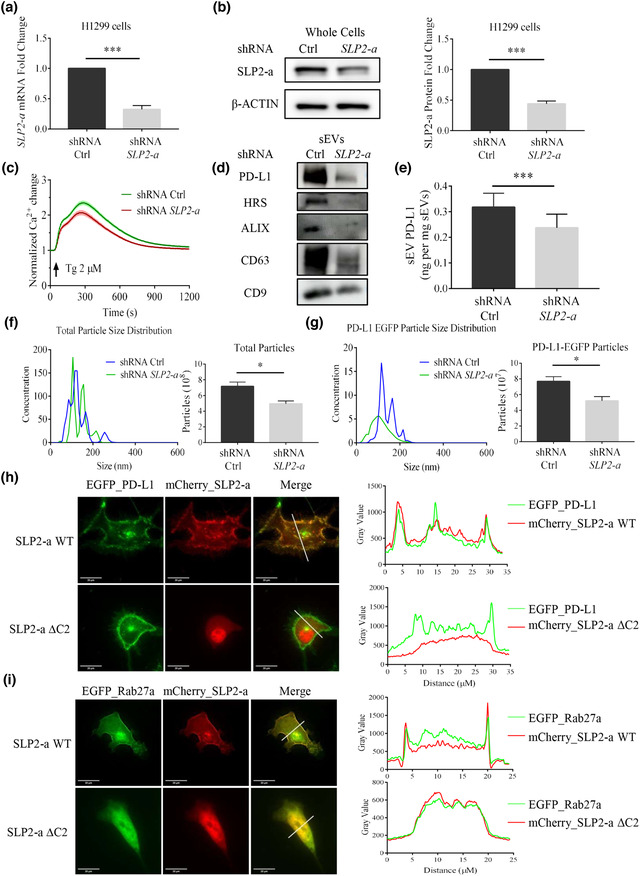
SLP2‐a KD reduces sEV PD‐L1 secretion in H1299 cells. (a) shRNA efficiently reduces SLP2‐a mRNA levels in H1299 cells. (n = 3). (b) Immunoblotting analysis confirms SLP2‐a KD (left). Its quantification by western blots is shown in the right panel (n = 3). (c) SLP2‐a KD inhibits Tg‐ (2 μM) mediated Ca2+ dynamics in H1299 cells. Values of fluorescent intensity represent mean ± SEM, which is normalized to the basic fluorescence intensity. All data is derived from more than 4 wells in 3 individual experiments. (d) Immunoblotting analysis of sEVs protein in H1299 cells with or without SLP2‐a‐KD treatment. (e) Comparison by ELISA between amounts of PD‐L1 in sEVs derived from H1299 cells with or without SLP2‐a‐KD treatment. (n = 3). (f). Comparison of the total particle size distribution (left) and its quantification with or without SLP2‐a‐KD treatment in H1299 cells by NTA (n = 3). Units are in millions of particles per milliliter. (g) Comparison between PD‐L1 EGFP particle size distributions and quantification with or without SLP2‐a‐KD treatment in H1299 cells transfected with PD‐L1_EGFP by NTA (n = 3). Units are in millions of particles per milliliter. (h) Fluorescence images show the coexpression of PD‐L1_EGFP with mCherry_SLP2‐a (upper), or with mCherry_SLP2‐a ΔC2 (below). Quantitative fluorescent intensities along the white lines, as plotted in the original images. (i) Fluorescence imagines show the coexpression of EGFP_Rab27a with mCherry_SLP2‐a (upper), or with mCherry_SLP2‐a ΔC2 (below). Quantitative fluorescent intensities along the white lines, as plotted in the original images (right). For all of Figure 7, data are representative of at least three independent experiments.
4. DISCUSSION
Interfering with the PD‐1/PD‐L1 pathway can induce specific anti‐tumor immune responses and has shown significant therapeutic effects in the clinic for various cancers, including non‐small cell lung cancer (Garon et al., 2015; Rajan & Gulley, 2014). However, the mechanisms underlying the immune escape resulting in low response rates remain unclear (Topalian et al., 2019). PD‐L1 is a single transmembrane glycoprotein distributed on the tumor cell membrane surface. It can also be present in soluble form due to differentially spliced variants or proteolytic cleavage (Zhou et al., 2017; Zhu & Lang, 2017). Recently, many studies have shown that PD‐L1 is expressed on sEVs. The sEV PD‐L1 plays a significant role in immunomodulation, which may be related to immune escape (Chen et al., 2018; Kim et al., 2019; Poggio et al., 2019; Yang et al., 2018). The current study focused on the mechanism associated with the secretion of sEV PD‐L1 from non‐small cell lung cancer cells. We found that calcium ions and calcium channels ORAI11 could regulate the secretion of sEV PD‐L1 and modulate the tumor immune response.
The sEV PD‐L1 has the same extracellular topology as PD‐L1 expressed on the cell membrane and thus shares similar immunosuppressive functions, which can induce apoptosis of CD8+ T‐cells. It inhibits the secretion of IFN‐γ, IL‐2, and TNF‐α from CD8+ cells and diminishes the expression of activation markers for immune response (Chen et al., 2018; Cordonnier et al., 2020; Kim et al., 2019; Poggio et al., 2019; Ricklefs et al., 2018; Yang et al., 2018). The immunomodulatory function of sEV PD‐L1 exerts unique complex effects on the whole system. It may act on the tumor microenvironment in a combination of autocrine and paracrine ways. It may also work remotely on other targeted cells and targeted organs. In LLC1 lung cancer tumors of our study, the number of tumor‐infiltrating cells was low. However, within the tumor tissue generated by Orai1‐KD cells, the number of immune cells increased. We conclude that inhibition of ORAI1 calcium channels, which significantly decreased the sEV PD‐L1 release from cancer cells, is the cause of increased immune activation. We show that the Orai1‐KD tumor microenvironment is very distinct with respect to the WT one. Effects of sEV PD‐L1 at the draining lymph nodes have been investigated in detail. In the TRAMP‐C2 prostate cancer cell model, reduced secretion of sEV PD‐L1 induced proliferation (Ki67) and functional activation (GzmB) of lymphocytes and reduced lymphocyte exhaustion (TIM‐3) (Poggio et al., 2019). In the same cancer model, decreasing sEV PD‐L1 increased spleen size. Our study found an increase in spleen size in the Orai1‐KD LLC1 tumor model compared to the WT LLC1 tumor model. We further investigated the number and function of lymphocytes in the spleen. The markers of immune cell proliferation (Ki67), activation (GzmB), and exhaustion (TIM‐3) all displayed properties in favor of immune activation. The results from the tumor growth status, survival curve, and the plasma IL‐2 levels of the animals also indicated a more robust immune activation in the Orai1‐KD tumor animals. Therefore, we conclude that inhibition of sEV PD‐L1 secretion by intervention with ORAI11 enhances anti‐tumor immunity in the tumor microenvironment, in the remote immune targets, and in the whole animal body.
Calcium and ORAI1 calcium channels affect the sEV PD‐L1 secretion of tumor cells. Here we provide the following four pieces of evidence. First, IFN‐γ is a typical cytokine that stimulates the secretion of sEV PD‐L1 (Chen et al., 2018). In our studies, IFN‐γ promoted the secretion of sEV PD‐L1 and increased the intracellular calcium level in H1299 cells. Second, we used pHluorin tagged to PD‐L1 as an optical reporter to monitor sEV PD‐L1 secretion. Real‐time imaging experiments on single cells showed that increasing intracellular calcium concentration significantly increased the release of sEVs carrying PD‐L1. This increase was not evident in ORAI1‐KD cells, suggesting that calcium influx through ORAI1 channels is required for sEV PD‐L1 secretion. Third, treatment with calcium chelator or shRNA KD against ORAI1 significantly reduced the expression level of sEV PD‐L1. Fourth, in H1299 cells overexpressing PD‐L1 EGFP, ORAI1‐KD treatment significantly reduced the total number of sEVs, the number of PD‐L1 EGFP sEVs, and the ratio of PD‐L1 sEVs relative to total sEVs. We observed similar results in mouse LLC1 lung cancer cells. These findings together demonstrate that calcium influx through ORAI1 calcium channels is required for the secretion of sEV PD‐L1 from tumor cells.
In our study, we concentrate on the role of intracellular calcium in the generation and trafficking of extracellular vesicles carrying PD‐L1. We provide several pieces of evidence, that such vesicles are likely to be sEVs in the sense of recent classifications (Meldolesi, 2018; Thery et al., 2018; Yanez‐Mo et al., 2015). However, other types of extracellular vesicles might contribute to some of our findings. We would like to point out, though, that intracellular calcium is a universal second messenger regulating many cellular functions, such as metabolism and gene expression, as well as cell growth, proliferation and exocytosis (Berridge et al., 2003; Parekh, 2010). Exocytosis, as well as fusion between endosome‐like organelles involves the merging of two membranes in an environment of very low calcium (μmolar). For such processes influx of calcium through ion channels as well as calcium‐release from intracellular stores can exert a strong triggering or modulating effect (Clapham, 2007). At least two steps in the sEV trafficking of soluble or membrane‐bound proteins between cells occur in such a low‐calcium environment: maturation of MVBs (Messenger et al., 2018) and fusion of MVBs with the PM (Verweij et al., 2018) (our imaging with pHluorin tagged to PD‐L1). Extensive literature deals with the physiological and molecular nature of such membrane fusion in both electrically excitable and non‐excitable cell types. Membrane fusion is accomplished by common families of proteins (SNARES, Rab‐proteins, Munc‐proteins, and Synaptotagmin‐like proteins). Ion channels mediating calcium‐influx comprise voltage‐dependent calcium‐permeable channels (mainly in neurons and other excitable cells), TRP‐channels (in many different cell types), and ORAI1 channels, the main mediators of calcium‐influx in many non‐excitable cells (Clapham, 2007). Their opening leads to order‐of‐magnitude changes in intracellular calcium, which, in turn, causes rapid and large responses, such as neurotransmitter release. In contrast, trafficking events in which two membranes merge in an extracellular milieu, such as the fusion of extracellular vesicles with the plasma membrane, are less likely to be influenced by calcium‐permeable channels in a major way, since typical fluxes across channels are not on the order of magnitude, which would cause major changes in the millimolar concentrations of calcium, which prevail in the extracellular fluid. These kinds of fusion events are regulated by a very different set of proteins (Colombo et al., 2014; Hessvik & Llorente, 2018).
By targeting ORAI1 and two other proteins involved in calcium‐dependent regulation of membrane fusion and trafficking, we expect that at least some of our findings relate to PD‐L1 carrying MVB‐derived exosomes. Their biogenesis, transport, and release have been important topics in cancer research. However, still, many questions are waiting for answers. Rab27a and Neutral sphingomyelinase type 2 (nSMase2) are two essential proteins involved in sEV trafficking (Hessvik & Llorente, 2018; Ostrowski et al., 2010; Trajkovic et al., 2008). Genetic and pharmacological manipulations of these proteins provide substantial knowledge regarding the various functions of sEVs. There are three distinct types of mammalian Rab27a effectors, including Munc13‐4, synaptotagmin‐like protein, and a Slp homolog lacking C2 domains (Slac2) (Fukuda, 2013). The sEV release is regulated by Munc13‐4, a Rab11 binding protein required for calcium‐dependent MVB‐PM fusion (Messenger et al., 2018). Slac2s are essential participants in the formation of a transport complex and a docking complex. In melanocytes, MLPH (Slac2‐a) forms a complex with Rab27a and myosin‐Va for transporting melanosomes along actin filaments (Fukuda et al., 2002; Hume et al., 2006; Provance et al., 2002; Strom et al., 2002), and SLP2‐a is responsible for docking of melanosomes to phosphatidylserine in the plasma membrane via its C2A domain (Kuroda and Fukuda, 2004). We studied the MLPH protein and SLP2‐a protein as well. And we noticed that manipulating intracellular calcium concentrations could change the MLPH distribution. At rest, MLPH was found in actin bundles along the edge of the lamellipodium and at sites of membrane ruffling. Gradually it moved into the cytoplasm as intracellular calcium concentration decreased, and it reassembled near the cell membrane after calcium concentration increased, suggesting that calcium can dynamically regulate the transport of actin‐based sEVs associated with the MLPH‐Rab27 complex. This regulation is dependent on calpain, a calcium‐dependent proteolytic enzyme, because treatment of cells with calpain inhibitors as well as genetic removal of the proteolytic site in MLPH could disrupt the dynamic response induced by manipulations of the intracellular calcium level. Likewise, such treatments attenuated the aggregation of MLPH at the edge of the cell membrane. After MLPH KD in H1299 cells, cells produced significantly fewer sEVs, and fewer sEVs carried PD‐L1. We did not observe that manipulating intracellular calcium concentrations could alter the distribution of SLP2‐a, although SLP family proteins have a synaptotagmin‐like C2domain. Studies have shown that the family members of SLP respond differently to calcium ions and that their C2 domains bind membrane phospholipids differently (Fukuda, 2006; Fukuda, 2013). In MDCK cells, SLP2‐a is expressed on the apical surface. It plays an essential role in the establishment of cell polarization (Yasuda et al., 2012). The phospholipid‐binding activity of the tandem C2 domains of SLP2‐a is required for its apical localization (Kuroda & Fukuda, 2004). We found overexpressed SLP2‐a co‐localized well with overexpressed PD‐L1 and Rab27a, respectively, especially on the cell membrane. After we overexpressed a SLP2‐a mutant lacking C2 domains, there was no obvious change in the localization of overexpressed PD‐L1, but overexpressed Rab27a showed a diffuse distribution throughout the cytoplasm similar to that of SLP2‐a ΔC2 mutant and it partially entered the nucleus. After SLP2‐a KD in H1299 cells, the numbers of total sEVs and PD‐L1 sEVs were decreased. Furthermore, the calcium response to Tg was partially inhibited in SLP2‐a‐KD cells compared to that in WT cells, suggesting that SLP2‐a elicits its effects on sEV secretion by regulating the localization of Rab27a in conjunction with intracellular calcium signaling. In general, both SLP2‐a and MLPH regulate the sEV PD‐L1 secretion in a calcium‐related manner.
In conclusion, sEV PD‐L1 is a critical target for immune therapy research. The current study promotes a more profound understanding of the tumor immune response, which may help in improving the response rate of PD‐L1 treatment As shown in Figure 8, ORAI1 calcium channels provide the calcium signal for regulating the secretion of sEV PD‐L1, while MLPH and SLP2‐a together promote the precise transport of organelles involved in the release of sEV PD‐L1. All the proteins examined in our study are candidates for future in‐depth studies targeting the action of sEV PD‐L1 in immune escape.
FIGURE 8.
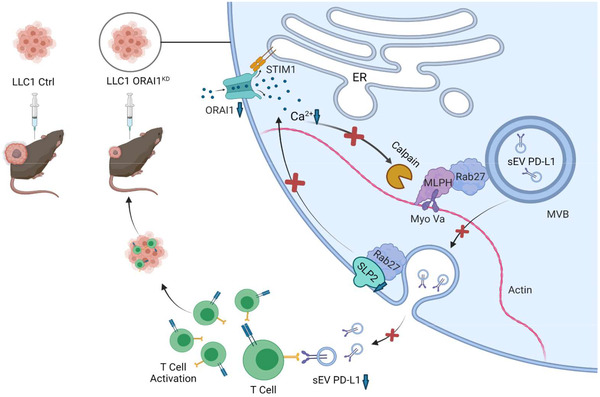
A schematic diagram for the proposed role of ORAI1 calcium channel in regulating the sEV PD‐L1 release of lung cancer cells and suppressing tumor growth through upregulation of anti‐tumor immunity. Upon influx through ORAI1 channels, intracellular calcium is critical for calpain proteolytic enzyme activities, which sustain the MLPH functions to cooperate with Rab27 transporting the sEV‐contained MVB to the plasma membrane. When MVB fuse with the PM, the intraluminal PD‐L1 vesicles will be released. During the Rab27‐dependent vesicle trafficking, SLP2‐a also plays a key role in correct MVB transport and intracellular calcium dynamics. Thus, the ORAI1 calcium channel is essential for sEV PD‐L1 release and tumor growth. Interruption of the intracellular calcium signal by inhibition of ORAI1 channel can suppress the sEV PD‐L1 release and tumor growth.
CONFLICT OF INTEREST
We declare no conflict of interest in this work.
ETHICS STATEMENT
Ethical approval was given by the Medical Ethics Committee of Macau University of Science and Technology. Animal experiments were permitted by the Animal Ethics Committees of Macau University of Science and Technology. All animal work was conducted according to approved protocols and institutional animal welfare guidelines.
AUTHOR CONTRIBUTIONS
Xi Chen, Erwin Neher and Haijie Yu designed this study; Xi Chen, Jiaqi Li, Ren Zhang, Yao Zhang, Xiaoxuan Wang, Elaine Lai‐Han Leung, Lijuan Ma performed the experiments; Xi Chen, Erwin Neher and Haijie Yu analyzed the data; Xi Chen and Haijie Yu drew the figures and tables; Xi Chen, Lijuan Ma, Erwin Neher and Haijie Yu drafted the initial manuscript; Vincent Kam Wai Wong, Liang Liu, Erwin Neher and Haijie Yu revised this manuscript; All authors read and approved the final manuscript.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supplemental Video 1
Supplemental Video 2
Supporting Information
ACKNOWLEDGEMENTS
We acknowledge Dr. Michiel Pegtel at VU University Medical Center, Dr. Tom Martin at University of Wisconsin‐Madison, Dr. Enfu Hui at UC San Diego and Dr. Zheng Zhang at Xiangya School of Medicine, Central South University for sharing their plasmids. This work was supported by Faculty Research Grant Projects of Macau University of Science and Technology, Macau, China (Project code: FRG‐18‐002‐SKL to L.M. and FRG‐18‐003‐SKL to H.Y.) and grants from the Macau Science and Technology Development Fund, Macau, China (Project code 0020/2019/A1, 0062/2021/A2 & 001/2020/ALC to E.N. & H.Y.)
Chen, X. , Li, J. , Zhang, R. , Zhang, Y. , Wang, X. , Leung, E. L.‐H. , Ma, L. , Wong, V. K. W. , Liu, L. , Neher, E. , & Yu, H. (2022). Suppression of PD‐L1 release from small extracellular vesicles promotes systemic anti‐tumor immunity by targeting ORAI1 calcium channels. Journal of Extracellular Vesicles, 11, e12279. 10.1002/jev2.12279
Contributor Information
Vincent Kam Wai Wong, Email: kawwong@must.edu.mo.
Liang Liu, Email: lliu@must.edu.mo.
Erwin Neher, Email: eneher@gwdg.de.
Haijie Yu, Email: hjyu@must.edu.mo.
REFERENCES
- Abels, E. R. , & Breakefield, X. O. (2016). Introduction to extracellular vesicles: Biogenesis, RNA cargo selection, content, release, and uptake. Cellular and Molecular Neurobiology, 36, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros, F. M. , Carneiro, F. , Machado, J. C. , & Melo, S. A. (2018). Exosomes and immune response in cancer: Friends or Foes? Frontiers in Immunology, 9, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge, M. J. , Bootman, M. D. , & Roderick, H. L. (2003). Calcium signalling: Dynamics, homeostasis and remodelling. Nature Reviews Molecular Cell Biology, 4, 517–529. [DOI] [PubMed] [Google Scholar]
- Brahmer, J. R. , Tykodi, S. S. , Chow, L. Q. , Hwu, W. J. , Topalian, S. L. , Hwu, P. , Drake, C. G. , Camacho, L. H. , Kauh, J. , Odunsi, K. , Pitot, H. C. , Hamid, O. , Bhatia, S. , Martins, R. , Eaton, K. , Chen, S. , Salay, T. M. , Alaparthy, S. , Grosso, J. F. , … Wigginton, J. M. (2012). Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. New England Journal of Medicine, 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G. , Huang, A. C. , Zhang, W. , Zhang, G. , Wu, M. , Xu, W. , Yu, Z. , Yang, J. , Wang, B. , Sun, H. , Xia, H. , Man, Q. , Zhong, W. , Antelo, L F. , Wu, B. , Xiong, X. , Liu, X. , Guan, L. , Li, T. , … Guo, W. (2018). Exosomal PD‐L1 contributes to immunosuppression and is associated with anti‐PD‐1 response. Nature, 560, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham, D. E. (2007). Calcium signaling. Cell, 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Thery, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Review of Cell and Developmental Biology, 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Consortium, E.‐T. , Van Deun, J. , Mestdagh, P. , Agostinis, P. , Akay, O. , Anand, S. , Anckaert, J. , Martinez, Z. A. , Baetens, T. , Beghein, E. , Bertier, L. , Berx, G. , Boere, J. , Boukouris, S. , Bremer, M. , Buschmann, D. , Byrd, J. B. , Casert, C. , Cheng, L. , … Hendrix, A. (2017). EV‐TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods, 14, 228–232. [DOI] [PubMed] [Google Scholar]
- Cordonnier, M. , Nardin, C. , Chanteloup, G. , Derangere, V. , Algros, M. P. , Arnould, L. , Garrido, C. , Aubin, F. , & Gobbo, J. (2020). Tracking the evolution of circulating exosomal‐PD‐L1 to monitor melanoma patients. Journal of Extracellular Vesicles, 9, 1710899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daassi, D. , Mahoney, K. M. , & Freeman, G. J. (2020). The importance of exosomal PDL1 in tumour immune evasion. Nature Reviews Immunology, 20, 209–215. [DOI] [PubMed] [Google Scholar]
- Del Castillo, J. , & Katz, B. (1954). Quantal components of the end‐plate potential. Journal of Physiology, 124, 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, L. M. , & Wang, M. Z. (2019). Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells, 8, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner, M. , Koch, P. A. , & Haucke, V. (2019). Phosphoinositides in the control of lysosome function and homeostasis. Biochemical Society Transactions, 47, 1173–1185. [DOI] [PubMed] [Google Scholar]
- Elstak, E. D. , Neeft, M. , Nehme, N. T. , Voortman, J. , Cheung, M. , Goodarzifard, M. , Gerritsen, H. C. , van Bergen En Henegouwen, P. M. , Callebaut, I. , de Saint Basile, G. , & van der Sluijs, P. (2011). The munc13‐4‐rab27 complex is specifically required for tethering secretory lysosomes at the plasma membrane. Blood, 118, 1570–1578. [DOI] [PubMed] [Google Scholar]
- Ferris, R. L. , Blumenschein, G., Jr. , Fayette, J. , Guigay, J. , Colevas, A. D. , Licitra, L. , Harrington, K. , Kasper, S. , Vokes, E. E. , Even, C. , Worden, F. , Saba, N. F. , Docampo, L. C. I. , Haddad, R. , Rordorf, T. , Kiyota, N. , Tahara, M. , Monga, M. , Lynch, M. , … Gillison, M. L. (2016). Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. New England Journal of Medicine, 375, 1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, M. (2006). Rab27 and its effectors in secretory granule exocytosis: a novel docking machinery composed of a Rab27.effector complex. Biochemical Society Transactions, 34, 691–695. [DOI] [PubMed] [Google Scholar]
- Fukuda, M. (2013). Rab27 effectors, pleiotropic regulators in secretory pathways. Traffic (Copenhagen, Denmark), 14, 949–963. [DOI] [PubMed] [Google Scholar]
- Fukuda, M. , & Itoh, T. (2004). Slac2‐a/melanophilin contains multiple PEST‐like sequences that are highly sensitive to proteolysis. Journal of Biological Chemistry, 279, 22314–22321. [DOI] [PubMed] [Google Scholar]
- Fukuda, M. , Kuroda, T. S. , & Mikoshiba, K. (2002). Slac2‐a/melanophilin, the missing link between Rab27 and myosin Va: implications of a tripartite protein complex for melanosome transport. Journal of Biological Chemistry, 277, 12432–12436. [DOI] [PubMed] [Google Scholar]
- Galvez‐Santisteban, M. , Rodriguez‐Fraticelli, A. E. , Bryant, D. M. , Vergarajauregui, S. , Yasuda, T. , Banon‐Rodriguez, I. , Bernascone, I. , Datta, A. , Spivak, N. , Young, K. , Slim, C. L. , Brakeman, P. R. , Fukuda, M. , Mostov, K. E. , & Martín‐Belmonte, F. (2012). Synaptotagmin‐like proteins control the formation of a single apical membrane domain in epithelial cells. Nature Cell Biology, 14, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Diaz, A. , Shin, D. S. , Moreno, B. H. , Saco, J. , Escuin‐Ordinas, H. , Rodriguez, G. A. , Zaretsky, J. M. , Sun, L. , Hugo, W. , Wang, X. , Parisi, G. , Saus, C. P. , Torrejon, D. Y. , Graeber, T. G. , Comin‐Anduix, B. , Hu‐Lieskovan, S. , Damoiseaux, R. , Lo, R. S. , & Ribas, A. (2017). Interferon receptor signaling pathways regulating PD‐L1 and PD‐L2 expression. Cell Reports, 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon, E. B. , Rizvi, N. A. , Hui, R. , Leighl, N. , Balmanoukian, A. S. , Eder, J. P. , Patnaik, A. , Aggarwal, C. , Gubens, M. , Horn, L. , Carcereny, E. , Ahn, M. J. , Felip, E. , Lee, J. S. , Hellmann, M. D. , Hamid, O. , Goldman, J. W. , Soria, J. C. , Dolled‐Filhart, M. , … Gandhi, L. (2015). Pembrolizumab for the treatment of non‐small‐cell lung cancer. New England Journal of Medicine, 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Gong, B. , Kiyotani, K. , Sakata, S. , Nagano, S. , Kumehara, S. , Baba, S. , Besse, B. , Yanagitani, N. , Friboulet, L. , Nishio, M. , Takeuchi, K. , Kawamoto, H. , Fujita, N. , & Katayama, R. (2019). Secreted PD‐L1 variants mediate resistance to PD‐L1 blockade therapy in non‐small cell lung cancer. Journal of Experimental Medicine, 216, 982–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami, S. , Aparicio, A. , & Subudhi, S. K. (2016). Immune checkpoint therapies in prostate cancer. Cancer Journal (Sudbury, Mass.), 22, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy, B. , Surowiak, P. , Budczies, J. , & Lanczky, A. (2013). Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non‐small‐cell lung cancer. Plos One, 8, e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessvik, N. P. , & Llorente, A. (2018). Current knowledge on exosome biogenesis and release. Cellular and Molecular Life Sciences, 75, 193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hira‐Miyazawa, M. , Nakamura, H. , Hirai, M. , Kobayashi, Y. , Kitahara, H. , Bou‐Gharios, G. , & Kawashiri, S. (2018). Regulation of programmed‐death ligand in the human head and neck squamous cell carcinoma microenvironment is mediated through matrix metalloproteinase‐mediated proteolytic cleavage. International Journal of Oncology, 52, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume, A. N. , Tarafder, A. K. , Ramalho, J. S. , Sviderskaya, E. V. , & Seabra, M. C. (2006). A coiled‐coil domain of melanophilin is essential for Myosin Va recruitment and melanosome transport in melanocytes. Molecular Biology of the Cell, 17, 4720–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn, R. , & Scheller, R. H. (2006). SNAREs–engines for membrane fusion. Nature Reviews Molecular Cell Biology, 7, 631–643. [DOI] [PubMed] [Google Scholar]
- Kalluri, R. (2016). The biology and function of exosomes in cancer. Journal of Clinical Investigation, 126, 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellar, A. , Egan, C. , & Morris, D. (2015). Preclinical murine models for lung cancer: clinical trial applications. BioMed Research International, 2015, 621324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. H. , Kim, H. , Choi, Y. J. , Kim, S. Y. , Lee, J. E. , Sung, K. J. , Sung, Y. H. , Pack, C. G. , Jung, M. K. , Han, B. , Kim, K. , Kim, W. S. , Nam, S. J. , Choi, C. M. , Yun, M. , Lee, J. C. , & Rho, J. K. (2019). Exosomal PD‐L1 promotes tumor growth through immune escape in non‐small cell lung cancer. Experimental & Molecular Medicine, 51, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J. , Tkach, M. , & Thery, C. (2014). Biogenesis and secretion of exosomes. Current Opinion in Cell Biology, 29, 116–125. [DOI] [PubMed] [Google Scholar]
- Kuroda, T. S. , & Fukuda, M. (2004). Rab27A‐binding protein Slp2‐a is required for peripheral melanosome distribution and elongated cell shape in melanocytes. Nature Cell Biology, 6, 1195–1203. [DOI] [PubMed] [Google Scholar]
- Li, H. Y. , McSharry, M. , Bullock, B. , Nguyen, T. T. , Kwak, J. , Poczobutt, J. M. , Sippel, T. R. , Heasley, L. E. , Weiser‐Evans, M. C. , Clambey, E. T. , & Nemenoff, R. A. (2017). The tumor microenvironment regulates sensitivity of murine lung tumors to PD‐1/PD‐L1 antibody blockade. Cancer Immunology Research, 5, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H. , Wei, S. , Hurt, E. M. , Green, M. D. , Zhao, L. , Vatan, L. , Szeliga, W. , Herbst, R. , Harms, P. W. , Fecher, L. A. , Vats, P. , Chinnaiyan, A. M. , Lao, C. D. , Lawrence, T. S. , Wicha, M. , Hamanishi, J. , Mandai, M. , Kryczek, I. , & Zou, W. (2018). Host expression of PD‐L1 determines efficacy of PD‐L1 pathway blockade‐mediated tumor regression. Journal of Clinical Investigation, 128, 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAndrew, D. , Grice, D. M. , Peters, A. A. , Davis, F. M. , Stewart, T. , Rice, M. , Smart, C. E. , Brown, M. A. , Kenny, P. A. , Roberts‐Thomson, S. J. , & Monteith, G. R. (2011). ORAI1‐mediated calcium influx in lactation and in breast cancer. Molecular Cancer Therapeutics, 10, 448–460. [DOI] [PubMed] [Google Scholar]
- Meldolesi, J. (2018). Exosomes and ectosomes in intercellular communication. Current Biology, 28, R435–R444. [DOI] [PubMed] [Google Scholar]
- Melo, S. A. , Luecke, L. B. , Kahlert, C. , Fernandez, A. F. , Gammon, S. T. , Kaye, J. , LeBleu, V. S. , Mittendorf, E. A. , Weitz, J. , Rahbari, N. , Reissfelder, C. , Pilarsky, C. , MFraga, M. F. , Piwnica‐Worms, D. , & Kalluri, R. (2015). Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. Nature, 523, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menasche, G. , Menager, M. M. , Lefebvre, J. M. , Deutsch, E. , Athman, R. , Lambert, N. , Mahlaoui, N. , Court, M. , Garin, J. , Fischer, A. , & de Saint Basile, G. (2008). A newly identified isoform of Slp2a associates with Rab27a in cytotoxic T cells and participates to cytotoxic granule secretion. Blood, 112, 5052–5062. [DOI] [PubMed] [Google Scholar]
- Messenger, S. W. , Woo, S. S. , Sun, Z. , & Martin, T. F. J. (2018). A Ca(2+)‐stimulated exosome release pathway in cancer cells is regulated by Munc13‐4. Journal of Cell Biology, 217, 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher, E. , & Brose, N. (2018). Dynamically primed synaptic vesicle states: key to understand synaptic short‐term plasticity. Neuron, 100, 1283–1291. [DOI] [PubMed] [Google Scholar]
- Ostrowski, M. , Carmo, N. B. , Krumeich, S. , Fanget, I. , Raposo, G. , Savina, A. , Moita, C. F. , Schauer, K. , Hume, A. N. , Freitas, R. P. , Goud, B. , Benaroch, P. , Hacohen, N. , Fukuda, M. , Desnos, C. , Seabra, M. C. , Darchen, F. , Amigorena, S. , Moita, L. F. , & Thery, C. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nature Cell Biology, 12, 19–30. sup pp 11–13. [DOI] [PubMed] [Google Scholar]
- Overman, M. J. , McDermott, R. , Leach, J. L. , Lonardi, S. , Lenz, H. J. , Morse, M. A. , Desai, J. , Hill, A. , Axelson, M. , Moss, R. A. , Goldberg, M. V. , Cao, Z. A. , Ledeine, J. M. , Maglinte, G. A. , Kopetz, S. , & André, T. (2017). Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): an open‐label, multicentre, phase 2 study. The Lancet Oncology, 18, 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh, A. B. (2010). Store‐operated CRAC channels: function in health and disease. Nature Reviews Drug Discovery, 9, 399–410. [DOI] [PubMed] [Google Scholar]
- Peinado, H. , Aleckovic, M. , Lavotshkin, S. , Matei, I. , Costa‐Silva, B. , Moreno‐Bueno, G. , Hergueta‐Redondo, M. , Williams, C. , Garcia‐Santos, G. , Ghajar, C. , Nitadori‐Hoshino, A. , Hoffman, C. , Badal, K. , Garcia, B. A. , Callahan, M. K. , Yuan, J. , Martins, V. R. , Skog, J. , Kaplan, R. N. , … Lyden, D. (2012). Melanoma exosomes educate bone marrow progenitor cells toward a pro‐metastatic phenotype through MET. Nature Medicine, 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggio, M. , Hu, T. , Pai, C. C. , Chu, B. , Belair, C. D. , Chang, A. , Montabana, E. , Lang, U. E. , Fu, Q. , Fong, L. , & Blelloch, R. (2019). Suppression of exosomal PD‐L1 induces systemic anti‐tumor immunity and memory. Cell, 177, 414–427. e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provance, D. W. , James, T. L. , & Mercer, J. A. (2002). Melanophilin, the product of the leaden locus, is required for targeting of myosin‐Va to melanosomes. Traffic (Copenhagen, Denmark), 3, 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan, A. , & Gulley, J. L. (2014). Nivolumab (anti‐PD‐1, BMS‐936558, ONO‐4538) in patients with advanced non‐small cell lung cancer. Translational Lung Cancer Research, 3, 403–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklefs, F. L. , Alayo, Q. , Krenzlin, H. , Mahmoud, A. B. , Speranza, M. C. , Nakashima, H. , Hayes, J. L. , Lee, K. , Balaj, L. , Passaro, C. , Rooj, A. K. , Krasemann, S. , Carter, B. S. , Chen, C. C. , Steed, T. , Treiber, J. , Rodig, S. , Yang, K. , Nakano, I. , … Lee, H. (2018). Immune evasion mediated by PD‐L1 on glioblastoma‐derived extracellular vesicles. Science Advances, 4, eaar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, P. D. , & Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nature Reviews Immunology, 14, 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, Y. , Wise, R. , & Zolkiewska, A. (2020). Proteolytic processing of PD‐L1 by ADAM proteases in breast cancer cells. Cancer Immunology, Immunotherapy, 69, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, R. K. , Mizuno, K. , Wasmeier, C. , Wavre‐Shapton, S. T. , Recchi, C. , Catz, S. D. , Futter, C. , Tolmachova, T. , Hume, A. N. , & Seabra, M. C. (2013). Distinct and opposing roles for Rab27a/Mlph/MyoVa and Rab27b/Munc13‐4 in mast cell secretion. Febs Journal, 280, 892–903. [DOI] [PubMed] [Google Scholar]
- Strom, M. , Hume, A. N. , Tarafder, A. K. , Barkagianni, E. , & Seabra, M. C. (2002). A family of Rab27‐binding proteins. Melanophilin links Rab27a and myosin Va function in melanosome transport. Journal of Biological Chemistry, 277, 25423–25430. [DOI] [PubMed] [Google Scholar]
- Sun, L. , & Ye, R. D. (2012). Role of G protein‐coupled receptors in inflammation. Acta Pharmacologica Sinica, 33, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, S. L. , Hodi, F. S. , Brahmer, J. R. , Gettinger, S. N. , Smith, D. C. , McDermott, D. F. , Powderly, J. D. , Carvajal, R. D. , Sosman, J. A. , Atkins, M. B. , Leming, P. D. , Spigel, D. R. , Antonia, S. J. , Horn, L. , Drake, C. G. , Pardoll, D. M. , Chen, L. , Sharfman, W. H. , Anders, R. A. , … Sznol, M. (2012). Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. New England Journal of Medicine, 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, S. L. , Hodi, F. S. , Brahmer, J. R. , Gettinger, S. N. , Smith, D. C. , McDermott, D. F. , Powderly, J. D. , Sosman, J. A. , Atkins, M. B. , Leming, P. D. , Spigel, D. R. , Antonia, S. J. , Drilon, A. , Wolchok, J. D. , Carvajal, R. D. , McHenry, M. B. , Hosein, F. , Harbison, C. T. , Grosso, J. F. , & Sznol, M. (2019). Five‐year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non‐small cell lung cancer treated with nivolumab. JAMA Oncology, 5, 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic, K. , Hsu, C. , Chiantia, S. , Rajendran, L. , Wenzel, D. , Wieland, F. , Schwille, P. , Brugger, B. , & Simons, M. (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science, 319, 1244–1247. [DOI] [PubMed] [Google Scholar]
- Vader, P. , Breakefield, X. O. , & Wood, M. J. (2014). Extracellular vesicles: emerging targets for cancer therapy. Trends in Molecular Medicine, 20, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel, G. , D'Angelo, G. , & Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology, 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Verweij, F. J. , Bebelman, M. P. , Jimenez, C. R. , Garcia‐Vallejo, J. J. , Janssen, H. , Neefjes, J. , Knol, J. C. , de Goeij‐de Haas, R. , Piersma, S. R. , Baglio, S. R. , Verhage, M. , Middeldorp, J. M. , Zomer, A. , van Rheenen, J. , Coppolino, M. G. , Hurbain, I. , Raposo, G. , Smit, M. J. , Toonen, R. F. G. , … Pegtel, D. M. (2018). Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. Journal of Cell Biology, 217, 1129–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig, M. , Peinelt, C. , Beck, A. , Koomoa, D. L. , Rabah, D. , Koblan‐Huberson, M. , Kraft, S. , Turner, H. , Fleig, A. , Penner, R. , & Kinet, J. P. (2006). CRACM1 is a plasma membrane protein essential for store‐operated Ca2+ entry. Science, 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos, C. , Hernandez‐Morales, M. , Gutierrez, L. G. , & Nunez, L. (2019). TRPC1 and ORAI1 channels in colon cancer. Cell Calcium, 81, 59–66. [DOI] [PubMed] [Google Scholar]
- Wang, F. , Wang, S. , & Zhou, Q. (2020). The Resistance Mechanisms of Lung Cancer Immunotherapy. Frontiers in Oncology, 10, 568059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, F. , Xu, M. , Lu, J. , Mao, L. , & Wang, S. (2019). The role of exosomal PD‐L1 in tumor progression and immunotherapy. Molecular Cancer, 18, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez‐Mo, M. , Siljander, P. R. , Andreu, Z. , Zavec, A. B. , Borras, F. E. , Buzas, E. I. , Buzas, K. , Casal, E. , Cappello, F. , Carvalho, J. , Colás, E. , Cordeiro‐da Silva, A. , Fais, S. , Falcon‐Perez, J. M. , Ghobrial, I. M. , Giebel, B. , Gimona, M. , Graner, M. , Gursel, I. , … De Wever, O. (2015). Biological properties of extracellular vesicles and their physiological functions. Journal of Extracellular Vesicles, 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Li, C. W. , Chan, L. C. , Wei, Y. , Hsu, J. M. , Xia, W. , Cha, J. H. , Hou, J. , Hsu, J. L. , Sun, L. , & Hung, M. C. (2018). Exosomal PD‐L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Research, 28, 862–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, T. , Saegusa, C. , Kamakura, S. , Sumimoto, H. , & Fukuda, M. (2012). Rab27 effector Slp2‐a transports the apical signaling molecule podocalyxin to the apical surface of MDCK II cells and regulates claudin‐2 expression. Molecular Biology of the Cell, 23, 3229–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, M. , Kasai, K. , Nagashima, K. , Torii, S. , Yokota‐Hashimoto, H. , Okamoto, K. , Takeuchi, T. , Gomi, H. , & Izumi, T. (2007). Exophilin4/Slp2‐a targets glucagon granules to the plasma membrane through unique Ca2+‐inhibitory phospholipid‐binding activity of the C2A domain. Molecular Biology of the Cell, 18, 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, Z. Y. , Zhong, L. X. , Feng, M. , Wang, J. F. , Liu, D. B. , & Xiong, J. P. (2015). Over‐expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non‐small cell lung carcinoma. International Journal of Clinical and Experimental Pathology, 8, 5080–5088. [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Mahoney, K. M. , Giobbie‐Hurder, A. , Zhao, F. , Lee, S. , Liao, X. , Rodig, S. , Li, J. , Wu, X. , Butterfield, L. H. , Piesche, M. , Manos, M. P. , Eastman, L. M. , Dranoff, G. , Freeman, G. J. , & Hodi, F. S. (2017). Soluble PD‐L1 as a biomarker in malignant melanoma treated with checkpoint blockade. Cancer Immunology Research, 5, 480–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , & Lang, J. (2017). Soluble PD‐1 and PD‐L1: predictive and prognostic significance in cancer. Oncotarget, 8, 97671–97682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supplemental Video 1
Supplemental Video 2
Supporting Information