

Abstract
Background
Severity of coronavirus disease 2019 (COVID‐19) is often associated with thrombotic complications and cytokine storm leading to intensive are unit (ICU) admission. Platelets are known to be responsible for abnormal hemostasis parameters (thrombocytopenia, raised D‐dimers, and prolonged prothrombin time) in other viral infections through the activation of the nucleotide‐binding domain leucine repeat rich containing protein 3 inflammasome induced by signaling pathways driven by Bruton tyrosine kinase (BTK) and leading to caspase‐1 activation.
Objectives
We hypothesized that caspase‐1 activation and the phosphorylation of BTK could be associated with the severity of the disease and that ibrutinib, a BTK inhibitor, could inhibit platelet activation.
Methods and Results
We studied caspase‐1 activation by flow cytometry and the phosphorylation of BTK by Western blot in a cohort of 51 Afro‐Carribean patients with COVID‐19 disease (19 not treated in ICU and 32 treated in ICU). Patients with a platelet count of 286.7 × 109/L (69–642 × 109/L) were treated by steroids and heparin preventive anticoagulation. Caspase‐1 and BTK activation were associated with the severity of the disease and with the procoagulant state of the patients. Furthermore, we showed in vitro that the plasma of ICU patients with COVID‐19 was able to increase CD62P expression and caspase‐1 activity of healthy platelets and that ibrutinib could prevent it.
Conclusions
Our results show that caspase‐1 and BTK activation are related to disease severity and suggest the therapeutic hope raised by ibrutinib in the treatment of COVID‐19 by reducing the procoagulant state of the patients.
Keywords: Bruton tyrosine kinase, caspase‐1, COVID‐19, ibrutinib, platelets
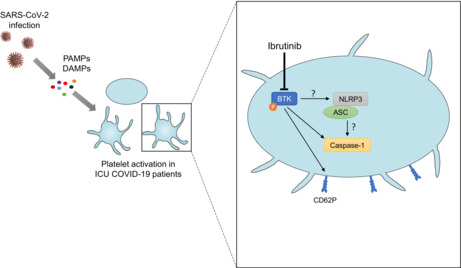
Essentials.
Platelet hyperreactivity and thrombotic complications are observed in coronavirus disease 2019 (COVID‐19).
Platelet caspase‐1 and BTK activation is related to disease severity in patients with COVID‐19.
BTK (an activator of caspase‐1) and caspase‐1 activation are reversed in vitro by ibrutinib.
Ibrutinib is a therapeutic hope in the treatment of COVID‐19.
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is responsible for coronavirus disease 2019 (COVID‐19), characterized in its typical form by pneumonia with fever, cough, and dyspnea. Among the patients, about 14% develop the severe form of the disease and 5% its critical form (with respiratory and multiorgan failures or systemic shock) due to an excessive inflammatory reaction known as “cytokine storm.” 1 It is characterized by a vast release of several proinflammatory cytokines such as interleukin (IL)‐6, tumor necrosis factor‐α (TNFα), interferon‐γ (IFNγ) and IL‐1β, leading to an inappropriate and excessive activation of the macrophages and the immune system as a whole. 2 , 3 Thrombotic complications are frequent in the severe forms of the disease and are associated with abnormal hemostasis parameters (thrombocytopenia, raised D‐dimers, and prolonged prothrombin time) mimicking disseminated intravascular coagulation. 4 However, thrombocytopenia observed during COVID‐19 infection remains moderate. Five percent to 42% of patients with COVID‐19 exhibited mild thrombocytopenia (100–150 × 109/L). 5 , 6 , 7 , 8 Patients with severe COVID‐19 have a lower platelet count (23–31 × 109/L) than in mild or moderate forms of COVID‐19. 9 , 10 Several studies reported platelet activation in patients with COVID‐19, suggesting that this blood cell type may be due to the occurrence of these devastating conditions. 11 , 12 Nevertheless, coagulopathy observed during COVID‐19 still appears to be the result of the exacerbated inflammatory response and activation/alteration of endothelial tissue. 13
Platelets play a well‐known role in hemostasis and thrombosis, but they also mediate inflammatory and immune processes in several infectious illnesses such as dengue, HIV‐1, and influenza. 14 , 15 , 16 , 17 , 18 These processes, referred to as immunothrombosis, can drive adverse clinical outcomes such as vascular thrombosis, organ failure, and death, as observed in COVID‐19. 19
Several signaling pathways may lead to platelet activation, including those related to toll‐like‐receptors (TLRs). Platelets express several TLRs, which can be stimulated by pathogen‐associated molecular patterns inducing signaling pathways driven by Bruton tyrosine kinase (BTK). 20 BTK is a nonreceptor signaling kinase downstream of several platelet receptors like glycoprotein (GP) VI, 21 , 22 GPIb/IX/V, 23 and C‐type lectin domain family 2. 24 BTK is able to activate the nucleotide‐binding domain leucine repeat rich containing protein 3 (NLRP3) inflammasome in platelets, 25 which results in caspase‐1 activation and the release of bioactive IL‐1β as well as other proinflammatory cytokines such as IL‐18. It is worth noting that a major role of NLRP3 platelet inflammasome in the cytokine storm has been highlighted in the case of dengue virus infection. 26
In the present study, our main hypothesis is that blood platelets may participate in the inflammatory response observed during COVID‐19 infection through the activation of caspase‐1. Therefore, we investigated caspase‐1 and BTK activation in platelets isolated from patients with COVID‐19 in different clinical conditions. In addition, we evaluated in vitro the impact of plasma from patients with severe COVID‐19 on platelets from healthy subjects and showed that ibrutinib, a BTK inhibitor, reverses the induced platelet activation.
2. METHODS
2.1. Human subjects and study design
In this cross‐sectional study, 51 patients with SARS‐CoV‐2 infection were recruited from the University Hospital of Guadeloupe from September 8 to October 20, 2020: 19 hospitalized in medicine departments (non‐ICU COVID‐19) and 32 hospitalized in the ICU. Before the start of the study, a sample size calculation was performed. Few data were available in the literature. For this calculation, we used preliminary (unpublished) data on platelet caspase‐1 activation in intubated (8.7 ± 6.9%; n = 6) and nonintubated patients with COVID‐19 (2.1 ± 1.4%; n = 4). From these figures, we calculated that 17 individuals per group were necessary to highlight a difference in means among our 3 groups with an alpha risk of 5% and a power of 80% (calculations performed with the software nQuery Advisor 7.0). All patients were included after legal authorization and as soon as possible after hospital admission. Given the constraints of platelet handling, we could propose the study to a maximum of only two patients per day. Their recruitment also depended on hospital admissions. Despite these factors, all of the patients we proposed to the study agreed to be a part of it. The median time for blood sampling from the beginning of the symptom was 9 days. Patient eligibility criteria were SARS‐CoV‐2 positivity and acceptance to participate in the study.
SARS‐CoV‐2 infection was confirmed by reverse transcription polymerase chain reaction (RT‐PCR) in accordance with current standards. Eight healthy donors who were not infected by SARS‐CoV‐2 (negative RT‐PCR) were also included in the same period. These donors were Guadeloupeans working at the University Hospital of Guadeloupe and gave their consent to participate in the study.
Patients with COVID‐19 and healthy control subjects were recruited after informed written consent, in accordance with the local institutional review boards (Nos. IDRCB: 2020–401,908‐31 and CNRIPH: 19–11–26‐67,312, respectively) and the Declaration of Helsinki. All patients with COVID‐19 were integrated in the following protocol: corticosteroid, preventive anticoagulation with heparin, ivermectin, and oxygen therapy.
No vaccine was available at this time, so none of the patients and healthy donors included in the study were vaccinated against SARS‐CoV‐2.
2.2. Standard biological and biochemical variables
Cellular blood counts were delivered by the XN‐1000 analyzer (Sysmex), coagulation parameters released by STAR‐R Evolution Expert Series analyzer (Diagnostica Stago S.A.S.) and biochemical parameters from COBAS 4800 analyzer (Roche Diagnostics).
2.3. Isolation, washing, and lysis of platelets
Blood samples were collected using a 21‐gauge needle into a 3.2% trisodium citrate tube and centrifuged (170 g, 10 min, room temperature, brake‐free) within 1 h after collection to obtain the platelet‐rich plasma (PRP). An aliquot of PRP was used for flow cytometry, and the remaining PRP was used for protein lysates. For this latter, PRP was incubated in the presence of prostaglandin E1 (1 μg/ml) and then was centrifuged. The supernatant was discarded, and platelets were incubated in Tyrode albumin buffer (137 mM NaCl, 2.7 mM KCl, 12 mM NaHCO3, 0.42 mM NaH2PO4‐H2O, 1 mM MgCl2‐6H2O, 2 mM CaCl2‐2H2O, 5 mM HEPES, 0.05 g glucose, 20% bovine serum albumin, pH 7.3). Platelets were then washed and incubated in Tyrode buffer (137 mM NaCl, 0.3 mM Na2HPO4, 2 mM KCl, 12 mM NaHCO3, 5 mM HEPES, 5 mM dextrose, 1 mM MgCl2, pH 7.3). 26 Finally, platelets were lysed with lysis buffer (10 mM Tris Base, 0.5% Triton‐X100, 25 mM NaCl, 125 mM sucrose, 25 mM NaF, 5 mM Na4P2O7‐10H2O, pH 7.4), 5 mM vanadate, 1 mM phenylmethylsulfonyl fluoride, 2 mM NaF, and 1/100 of protease cocktail (Sigma‐Aldrich). Proteins were stored at −20°C until used.
2.4. Western blot analysis
Phosphorylation level of BTK at Tyr223 was determined by western blot. Autophosphorylation at Tyr223 within the SH3 domain is necessary for full activation of BTK. Platelet lysates (50 μg) were separated by SDS‐PAGE and transferred to nitrocellulose membranes (Thermo Fisher Scientific). The membranes were then blocked and incubated with anti‐human phosphorylated BTK (p‐BTK) (5082S; Cell Signaling Technology), anti‐human BTK (3533S; Cell Signaling Technology) and anti‐human glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) (MAB374; Merck Millipore). Immune complexes were revealed with secondary peroxidase‐conjugated antibodies (Dako A/S). Western blots were analyzed using an Amersham Imager 600 (GE Healthcare). For quantification, densitometry was performed with ImageJ software (National Institutes of Health). The results are shown as the ratio of p‐BTK to total BTK.
2.5. Platelet phenotyping
Platelet characteristics were determined by flow cytometry (Cytoflex, Beckman Coulter). PRP was labeled with antibodies (anti‐CD41‐ECD and anti‐CD62P‐APC from Beckman Coulter) or a fluorescent probe green fluorescent labeled inhibitor of caspase‐1 (FLICA) that irreversibly binds to activated caspase‐1, according to the manufacturer's instructions (Immunochemistry Technologies). A minimum of 15,000 events was acquired. Platelets were distinguished by specific binding of anti‐CD41 and characteristic forward and side scattering. First, platelets were gated according to size/structure (forward and side scattering) and then selected on the CD41 labeling. Anti‐CD62P antibody and FAM‐FLICA probe were used to evaluate P‐selectin surface expression and active caspase‐1, reflecting platelet and caspase‐1 activation, respectively.
2.6. Plasma analysis
Plasma samples were obtained from an EDTA tube of blood from patients and healthy donors by centrifugation, then stored at −80°C until inflammatory mediator quantification. IL‐2, IL‐12 p70, IFNγ, TNFα, IL‐6, and IL‐1β quantification was performed using the Luminex technique in accordance with the manufacturer's instructions. Each plasma sample was tested in duplicate and data collected on a MAGPIX system (Thermo Fisher Scientific) and analyzed with ProcartaPlex Analyst 1.0 Software (ThermoFisher Scientific).
2.7. Platelet stimulation with plasma from patients with COVID‐19
The platelet‐poor plasma of each donor was obtained from a 3.2% trisodium citrate tube of blood after double centrifugation: a centrifugation at 170 g, 10 min (obtaining PRP) then centrifugation at 1500 g, 16 min at room temperature, and was then stored at −80°C until used. Platelets from healthy controls were incubated for 1 h with plasma from ICU patients with COVID‐19 or control subjects pretreated or not, during 10 min with a BTK inhibitor (ibrutinib, 5 μM; purchased from Selleck Chemicals). Thereafter, platelets were labeled using antibodies (anti‐CD41 ECD, anti‐CD62P APC) and a FAM‐FLICA probe or washed and lysed to assess BTK phosphorylation state as described above. For these indirect assays, the percentage of CD41+ CD62P+ and FLICA+ platelets were evaluated, respectively, by stimulation of platelets from 7 and 4 healthy subjects, respectively.
2.8. Statistical analysis
For all experiments, normality of distributions was assessed, and quantitative variables were expressed as medians and interquartile ranges (IQRs) or means and standard deviations (±SD). Patient characteristics were analyzed using the Student t test or Mann–Whitney nonparametric test, analysis of variance (ANOVA; followed by Tukey's post hoc test for multiple comparisons), or Kruskal–Wallis nonparametric test (followed by Dunn's post hoc test for multiple comparisons) for quantitative variables and Khi‐2 test or Fisher's exact test for categorical variables. Statistical analyses of flow cytometry data, western blot data, and cytokine concentrations were performed with ANOVA (followed by Tukey's post hoc test for multiple comparisons) or Kruskal–Wallis nonparametric test (followed by Dunn's post hoc test for multiple comparisons) according to the normality of the distributions. Pearson correlations were performed to test the associations between the parameters investigated. Paired t test or Wilcoxon test was used to evaluate the effects of ibrutinib treatment and plasma of ICU patients with COVID‐19 on healthy platelets. Statistical analyses were performed by using Prism version 8 (GraphPad Software). p value <0.05 was considered statistically significant.
3. RESULTS
3.1. COVID‐19 patient cohort
Fifty‐one patients with COVID‐19 were enrolled in our study: 19 non‐ICU COVID‐19 and 32 ICU COVID‐19 (Table 1). In our cohort, the mean age was not different between non‐ICU patients with COVID‐19 and ICU patients with COVID‐19, although the latter tended to be older (57 years vs. 64 years, respectively; p = 0.1460). Patients were mostly men in the two groups (79.0% and 71.9%, respectively). As expected, hospital duration was significantly longer in the ICU group (18 days vs. 4 days; p = 0.0004), while survival was significantly reduced in this group (50.0% vs. 94.7%; p = 0.0016). Compared to the non‐ICU group, ICU patients with COVID‐19 had significantly higher levels of leukocytes (13.00 × 109/L vs. 7.00 × 109/L; p = 0.0005), neutrophils (10.76 × 109/L vs. 5.47 × 109/L; p < 0.0001), C‐reactive protein (181.0 mg/L vs. 73.8 mg/L; p = 0.0177), D‐dimer (1.94 μg/ml vs. 0.76 μg/ml; p = 0.0022), fibrinogen (8.1 g/L vs. 6.4 g/L; p = 0.0413), creatinine (118 μmol/L vs. 85 μmol/L; p = 0.0114) and higher prothrombin time (15.6 s vs. 14.0 s; p < 0.0001). We also observed a trend for higher values of total bilirubin in ICU group (10 μmol/L vs. 7 μmol/L; p = 0.0765). Platelet, lymphocyte, and monocyte counts were not statistically different between these two groups, and few patients were thrombocytopenic (one ICU patient with COVID‐19 with a platelet count less than 100 × 109/L, seven less than 150 × 109/L; two non‐ICU patients with COVID‐19 less than 150 × 109/L; and none had a platelet count less than 50 × 109/L). Surprisingly, we did not observe differences in the classical comorbidities associated with the severity of COVID‐19 in the two groups (Table 1).
TABLE 1.
Clinical characteristics of patients with COVID‐19 and healthy donors
| Healthy donors (n = 8) | Patients with COVID‐19 (n = 51) | ||
|---|---|---|---|
| Non‐ICU COVID‐19 (n = 19) | ICU COVID‐19 (n = 32) | ||
| Age, mean, years (±SD) | 44 (16) | 57 (12) | 64 (11) |
| Male, n (%) | 2 (25.0) | 15 (79.0) | 23 (71.9) |
| Race/ethnicity | |||
| Afro‐Carribeans, n (%) | 8 (100) | 19 (100) | 32 (100) |
| Days of hospitalization, median (IQR) | NA | 4 (3–14) | 18 (9–22)* |
| Survivors, n (%) | NA | 18 (94.7) | 16 (50.0)** |
| SAPS II score, median (IQR) | NA | NA | 37 (29–45) |
| SOFA score, median (IQR) | NA | NA | 5 (3,5–8) |
| SOFA score <5, n (%) | NA | NA | 13 (41) |
| Respiratory assistance | |||
| Mechanical ventilation, n (%) | NA | 0 (0) | 23 (72) |
| High‐flow oxygen therapy, n (%) | NA | 0 (0) | 9 (28) |
| Extracorporeal membrane oxygenation, n (%) | NA | 0 (0) | 0 (0) |
| Laboratory results | |||
| Hemoglobin, g/dl, mean (±SD) | 13.1 (1.1) | 13.0 (1.4) | 9.6 (2.7)* |
| Leukocytes, ×109/L, median (IQR) | 5.60 (4.10–8.00) | 7.00 (5.30–9.90) | 13.00 (8.93–16.13)* |
| Lymphocytes, ×109/L, median (IQR) | 1.87 (1.38–2.30) | 1.13 (0.92–2.02) | 1.05 (0.58–1.58) |
| Monocytes, ×109/L, median (IQR) | 0.54 (0.41–0.63) | 0.56 (0.35–0.73) | 0.59 (0.31–0.97) |
| Neutrophils, ×109/L, median (IQR) | 3.03 (2.06–4.97) | 5.47 (3.08–7.14) | 10.76 (7.87–13.89)* |
| Platelets, ×109/L, median (IQR) | 291 (283–323) | 260 (202–334) | 236 (167–415) |
| Total bilirubin, μmol/L, median (IQR) | 7 (4–20) | 7 (6–10) | 10 (7–16) |
| C‐reactive protein, mg/L, median (IQR) | 0.9 (0.7–1.4) | 73.8 (41.9–106.8) | 181.0 (94.5–295.3)** |
| D‐dimer, μg/ml, median (IQR) | – | 0.76 (0.53–1.40) | 1.94 (1.01–5.52)** |
| Prothrombin time, s, mean (±SD) | – | 14.0 (0.61) | 15.6 (1.29)* |
| Fibrinogen, g/L, mean (±SD) | – | 6.4 (2.3) | 8.1 (1.7)** |
| Lactate, mmol/L, median (IQR) | – | 1.10 (0.80–1.50) | 1.40 (1.10–1.90) |
| Creatinine, μmol/L, median (IQR) | – | 85 (59–99) | 118 (93–530)** |
| Troponin, ng/ml, median (IQR) | – | 0.011 (0.008–0.015) | 0.026 (0.010–0.042) |
| Comorbidity | |||
| Hypertension, n (%) | – | 7 (36.8) | 12 (37.5) |
| Diabetes, n (%) | – | 3 (15.8) | 10 (31.3) |
| Obesity, n (%) | – | 6 (31.6) | 10 (31.3) |
| Body mass index, kg/m2, median (IQR) | – | – | 28.4 (26.8–30.2) |
| No comorbidity | – | 10 (52.6) | 14 (43.8) |
| Medications on day of blood collection | |||
| Steroids, n (%) | NA | 12 (75) | 23(71.9) |
| Heparin preventive anticoagulation, n (%) | NA | 12 (75) | 32 (100)** |
| Antiplatelet therapy, n (%) | NA | 1 (6.2) | 6 (18) |
Note: Data are median (IQR), mean (±SD) or n (%).
Abbreviations: ICU, intensive care unit; IQR, interquartile range; NA, not applicable; SD, standard deviation.
*p < 0.001 for the comparison of non‐ICU versus ICU patients with COVID‐19.
**p ≤ 0.05 and p ≥ 0.001 for the comparison of non‐ICU versus ICU patients with COVID‐19.
3.2. Platelet activation is increased in COVID‐19 patients and is dependent on the severity of the disease
We measured the membrane expression of platelet CD62P (P‐selectin), a marker of platelet α granule secretion. As shown in Figure 1A, platelets are more activated in ICU patients with COVID‐19 compared to non‐ICU patients with COVID‐19 and healthy controls. Therefore, the level of platelet activation is associated with the COVID‐19 severity. Platelet expression of CD62P is higher in ICU patients with COVID‐19 (Figure 1A) compared to non‐ICU patients with COVID‐19 (median, 19.76%; IQR, 14.46–27.46 vs. median 14.02%; IQR, 8.680–15.91; p = 0.0088) and healthy controls (median, 6.23%; IQR, 4.78–9.49; p < 0.0001). There was no significant difference between healthy controls and non‐ICU patients with COVID‐19 (p = 0.109).
FIGURE 1.
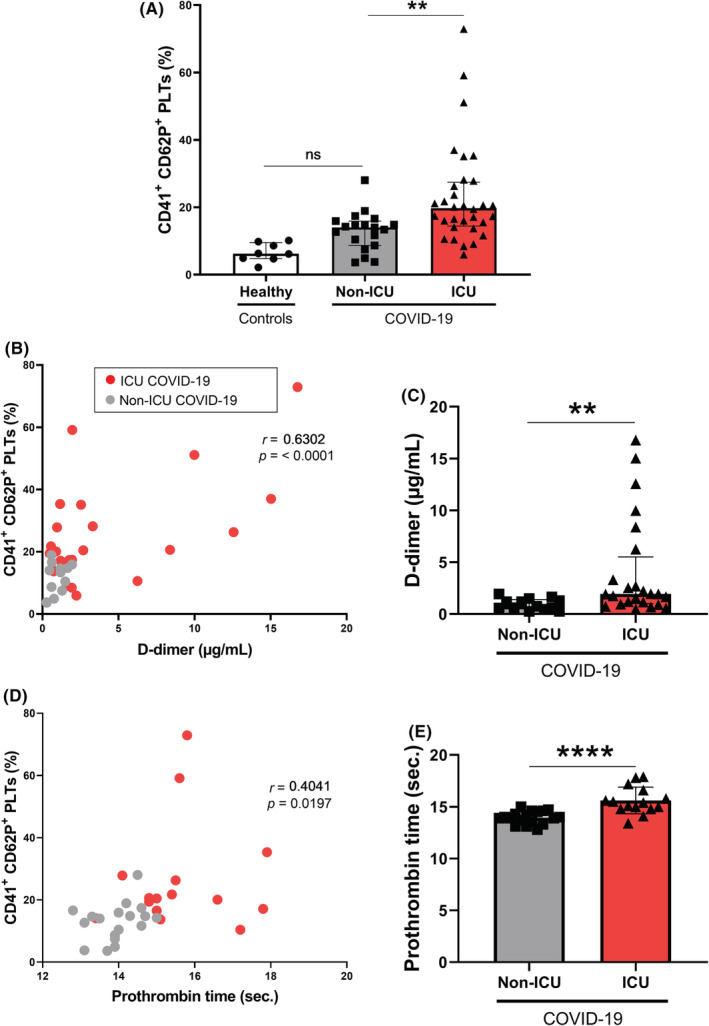
Platelet activation in patients with COVID‐19 and correlations with coagulation markers. (A) Platelet P‐selectin expression (CD41+ CD62P+ PLTs) was measured by flow cytometry in 8 healthy donors, 19 non‐ICU patients with COVID‐19 and 32 ICU patients with COVID‐19. Platelets were gated using CD41 expression and the percentage of CD62P+ platelets was evaluated among CD41+ platelets. (B) Strong positive correlation was observed between CD41+ CD62P+ PLTs percentage and D‐dimer concentration (r = 0.6302) and (D) weak positive correlation was observed between CD41+ CD62P+ PLTs percentage and prothrombin time (r = 0.4041). (C) D‐dimer concentration and (E) prothrombin time were evaluated in non‐ICU and ICU COVID‐19 groups. (A) Multiple comparisons were realized between healthy subjects, non‐ICU patients, and ICU patients with COVID‐19 and (C,E) were carried out between non‐ICU patients with COVID‐19 and ICU patients with COVID‐19. (A,C,E) Data are shown (A,C) as median with interquartile ranges or (E) mean with standard deviation. p values were calculated with (A) Kruskal–Wallis test followed by Dunn's post hoc test, (C) Mann–Whitney U test and (E) Student‐t test. (B,D) Pearson's correlation coefficients (r) were calculated and are shown in the panels. Not significant, *p < 0.05, **p < 0.01, ****p < 0.0001. COVID‐19, coronavirus disease 2019; ICU, intensive care unit; PLT, platelet
Because D‐dimer levels were associated with disease severity, 27 we analyzed the correlations between D‐dimer rate and CD62P expression. We showed that platelet activation was positively correlated with the D‐dimer rate of patients with COVID‐19 (r = 0.63, p = <0.0001) (Figure 1B) and that platelet activation was positively correlated with prothrombin time (r = 0.40, p = 0.0197) (Figure 1D).
3.3. Activation of the platelet caspase‐1 is increased in patients with COVID‐19 and associated with the severity of the disease
We tested the caspase‐1 activation using FAM‐FLICA probe. As shown in Figure 2, caspase‐1 is significantly more activated in ICU patients with COVID‐19, compared to healthy subjects (median, 8.02%; IQR, 4.57–13.97 vs. median, 3.11%; IQR, 1.66–4.93; p = 0.0048). There was no significant difference between healthy controls and non‐ICU patients with COVID‐19 (median, 3.11%; IQR, 1.66–4.93 vs. median, 5.80%; IQR, 3.26–9.95, p = 0.182).
FIGURE 2.
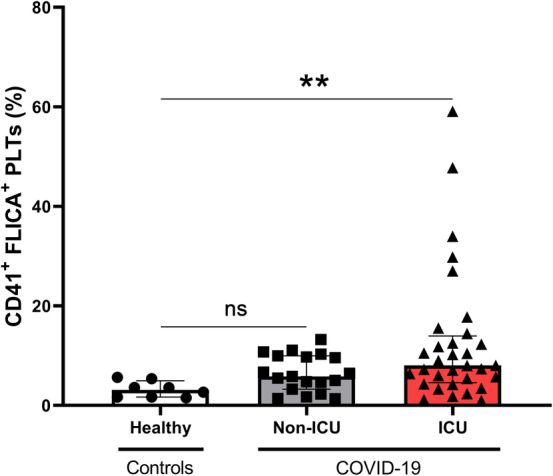
Caspase‐1 activation in platelets of patients with COVID‐19. Platelet caspase‐1 activation was measured by flow cytometry using FAM‐FLICA probe (CD41+ FLICA+ PLTs) in 8 healthy donors, 19 non‐ICU patients with COVID‐19 and 32 ICU patients with COVID‐19. Multiple comparisons were realized between healthy subjects, non‐ICU patients with COVID‐19, and ICU patients with COVID‐19. Data are shown as median with interquartile ranges and minimum and maximum values. p values were calculated with Kruskal–Wallis test followed by Dunn's post hoc test. Not significant, **p < 0.01. FLICA, fluorescent labeled inhibitor of caspase‐1; PLT, platelet
3.4. BTK is activated in platelets of patients with COVID‐19
We assessed the underlying mechanism of platelet caspase‐1 activation in patients with COVID‐19 by evaluating BTK activation through p‐BTK level in platelets of patients with COVID‐19 using western blot analysis (Figure 3). BTK phosphorylation level was significantly increased in platelets from ICU patients with COVID‐19 (median, 0.68; IQR, 0.55–1.21) compared to healthy subjects (median, 0.19; IQR, 0.05–0.30; p = 0.0004) and to non‐ICU patients with COVID‐19 (median, 0.48; IQR, 0.41–0.55; p = 0.0190) (Figure 3). There was no significant difference between healthy controls and non‐ICU patients with COVID‐19 (median, 0.19; IQR, 0.05–0.30 vs. median, 0.48; IQR, 0.41–0.55; p = 0.1681).
FIGURE 3.
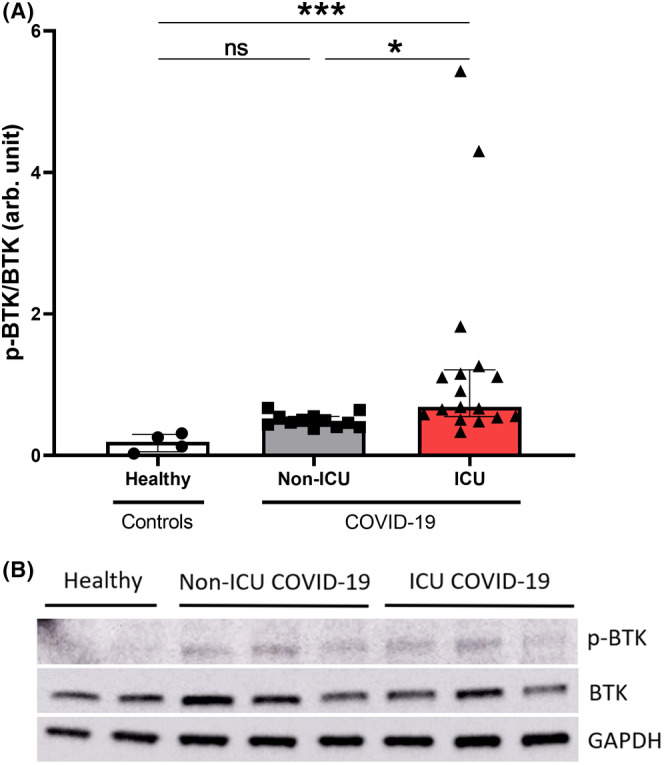
BTK phosphorylation in platelets of patients with COVID‐19. Phosphorylation state of BTK was measured in 4 healthy donors, 12 non‐ICU patients with COVID‐19, and 17 ICU patients with COVID‐19 by the densitometric analysis of western blots and normalized to total BTK. Multiple comparisons between healthy subjects, non‐ICU and ICU patients with COVID‐19 were realized. Data are shown as median with interquartile ranges and minimum and maximum values. p values were calculated with Kruskal–Wallis test followed by Dunn's post hoc test. Not significant, *p < 0.05, ***p < 0.001. BTK, Bruton tyrosine kinase; COVID‐19, coronavirus disease 2019; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; ICU, intensive care unit; p‐BTK, phosphorylated Bruton tyrosine kinase
3.5. Plasma from patients with COVID‐19 induces platelet activation in healthy individuals
Next, we examined if plasma from patients with COVID‐19 could affect the CD62P expression, caspase‐1 activation, and BTK phosphorylation in platelets isolated from healthy subjects. Our results showed that plasma from ICU patients with COVID‐19 significantly increased platelet CD62P expression (Figure 4A–C) (median, 8.74%; IQR, 5.61–21.97 vs. median, 14.11; IQR, 9.64–27.14; p = 0.0017) and caspase‐1 activity (mean, 5.79%; ±SD, 3.90 vs. mean, 11.49%; ±SD, 6.28; p = 0.0097) (Figure 4D–F) compared to plasma from healthy subjects. These data indicate that circulating factors are at least responsible for increased platelet activation and caspase‐1 activation in severe patients with COVID‐19.
FIGURE 4.
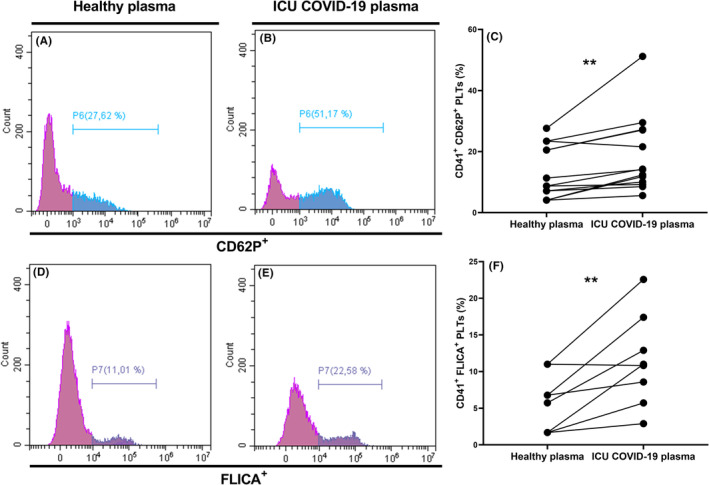
Plasma of ICU patients with COVID‐19 induce platelet and caspase‐1 activation. (A–F) Platelets of healthy subjects were incubated with plasma from healthy (A,D) or ICU patients with COVID‐19 (B,E) during 1 h. (A–C) P‐selectin expression and (D‐F) caspase‐1 activations of platelets were measured by flow cytometry. Platelets were distinguished by specific binding of anti‐CD41 and characteristic forward and side scattering. Expressions of P‐selectin (A,B) and caspase‐1 (D,E) on CD41+ platelets were then analyzed on specific histograms. Differences in the expression of P‐selectin (CD62P) and caspase‐1 (FLICA) on platelets of healthy subjects incubated with healthy subject plasma or ICU COVID‐19 plasma are illustrated in graphs C and F respectively. p value was calculated with (F) paired t test or (C) Wilcoxon test. **p < 0.01. COVID‐19, coronavirus disease 2019; FLICA, fluorescent labeled inhibitor of caspase‐1; ICU, intensive care unit; PLT, platelet
3.6. BTK inhibitor ibrutinib rescue the platelet activation induced by plasma of patients with COVID‐19
We then tested the ability of ibrutinib (Selleck Chemicals), one of the available BTK inhibitors, to reverse the platelet activation induced by plasma of ICU patients with COVID‐19.
Incubation of platelets with ibrutinib significantly suppressed platelet CD62P enhanced expression (Figure 5A–C) (median, 14.28%; IQR, 9.64–27.14 vs. median, 10.13%; IQR, 6.37–13.97; p = 0.0017) and caspase‐1 activity (Figure 5D–F) (mean, 11.49%; ±SD, 6.28 vs. mean, 5.67%; ±SD, 2.18; p = 0.0123) induced by plasma of ICU patients with COVID‐19, indicating that platelet CD62P expression and caspase‐1 activation depend, at least in part, on BTK activation. To confirm the ibrutinib action, protein lysates of stimulated healthy platelets were analyzed by western blot. Ibrutinib completely abolished BTK phosphorylation in healthy patient platelets stimulated or not by the plasma of ICU patients with COVID‐19 (Figure 5G).
FIGURE 5.
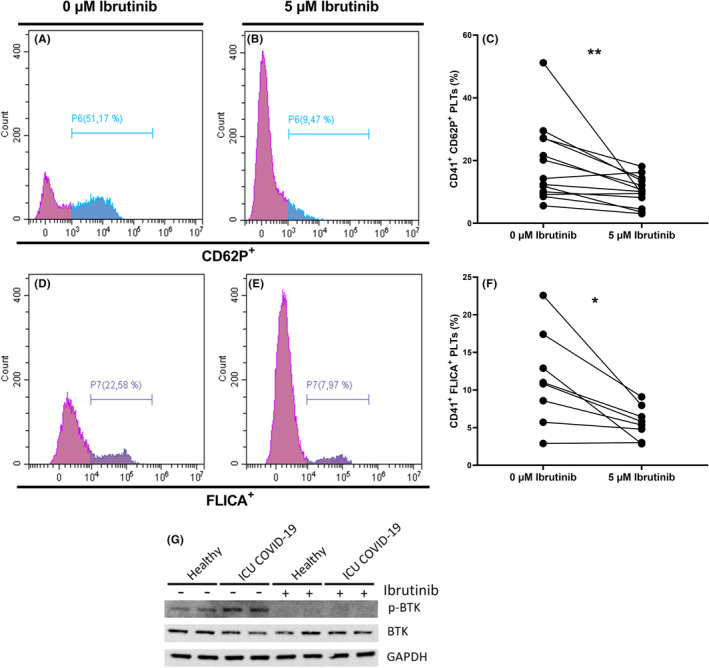
Ibrutinib inhibits platelet and caspase‐1 activation induced by plasma of ICU patients with COVID‐19. (A–F) Healthy platelets were preincubated or not with 5 μM of ibrutinib during 10 min, then incubated with plasma of ICU patients with COVID‐19 during 1 h. (G) Healthy platelets were preincubated or not with 5 μM of ibrutinib during 10 min and thereafter with plasma of healthy subjects or ICU patients with COVID‐19 during 1 h. (A–C) P‐selectin expression and (D–F) caspase‐1 activations were measured by flow cytometry. Platelets were distinguished by specific binding of anti‐CD41 and characteristic forward and side scattering. Expressions of P‐selectin (A,B) and caspase‐1 (D,E) on CD41+ platelets without or with ibrutinib 5 μM were then analyzed on specific histograms. Differences in the expression of P‐selectin (CD62P) and caspase‐1 (FLICA) on healthy platelets incubated with ICU COVID‐19 plasma without or with ibrutinib 5 μM are illustrated in graphs C and F respectively. (G) Representative western blot of p‐BTK, BTK, and GAPDH proteins. p value was calculated with (F) paired t test or (C) Wilcoxon test. *p < 0.05. **p < 0.01. Bruton tyrosine kinase; COVID‐19, coronavirus disease 2019; FLICA, fluorescent labeled inhibitor of caspase‐1; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; ICU, intensive care unit; p‐BTK, phosphorylated Bruton tyrosine kinase; PLT, platelet
3.7. Cytokine concentration
We measured the levels of different cytokines: IL‐2, IL‐12 p70, IFNγ, TNFα, IL‐6, and IL‐1β. Non‐ICU patients had increased levels of all cytokines compared with healthy controls. Surprisingly, non‐ICU patients exhibited also increased levels of the investigated cytokines when compared to ICU patients (Figure S1). No difference was observed between ICU patients and healthy controls.
4. DISCUSSION
In the present report, we showed an increase in platelet activation in patients with COVID‐19 that is more pronounced in those exhibiting the worst clinical conditions. We also documented for the first‐time activation of caspase‐1 and BTK. Moreover, we demonstrated in vitro that plasma of ICU patients with COVID‐19 is able to activate platelets from healthy subjects and increase caspase‐1 activation and BTK phosphorylation. These effects were reversed by the use of ibrutinib. Interestingly, we observed that ICU patients with COVID‐19 exhibit low levels of cytokines induced by helper T lymphocytes (Th1) compared to non‐ICU patients.
Thrombotic complications are common among severely ill patients with COVID‐19. 28 , 29 , 30 , 31 In our cohort, we observed that ICU patients with COVID‐19 have significantly higher levels of D‐dimer, and prothrombin time is significantly longer compared to non‐ICU patients as previously described. 2 , 4 In other viral infections, platelets are also known to be responsible for these hemostasis abnormalities. 26 Patients admitted to ICU for septic shock typically exhibit thrombocytopenia associated with higher morbidity and mortality. 32 But this thrombocytopenia seems to be moderate in SARS‐CoV‐2 infection. 2
Several studies described a direct relationship between thrombocytopenia and the severity of the disease in patients with COVID‐19. 9 In contrast, few patients of our series were thrombocytopenic. In addition, we did not detect difference in platelet count between ICU and non‐ICU groups in contrast with previous studies. 9 This discrepancy could be related to the ethnic origin of the patients included in our study, most of them being Afro‐Caribbean and knowing that ethnic variation of thrombotic risk has been previously reported. 33
Several studies described the platelet activation in COVID‐19, showing their degranulation, the release of platelet extracellular vesicles, the production of proinflammatory cytokines and the platelet‐leukocyte aggregates formation. 11 , 12 , 34 , 35 Our study confirmed the platelet activation in COVID‐19 through the increase of P‐selectin surface expression. 11 We further extend these results by showing that P‐selectin expression varies according to the disease severity since the highest expression was observed in ICU patients. It is well known that platelets can be activated during blood collection. To avoid this pitfall, we collected the blood of all the patients and healthy controls according to the same protocol, namely, with a 21‐gauge needle, and we treated the platelets within an hour after sample collection. We also evidenced positive correlations between the P‐selectin expression and expression of two coagulation markers, namely, D‐dimers and prothrombin time. Several mechanisms and intracellular pathways are able to induce P‐selectin expression and the implication of platelets in the inflammation state of many infectious diseases is now well documented. 34 , 36 , 37 , 38 , 39 , 40 It has been suggested that this activation could be the result of the stimulation of the mitogen‐activated protein kinase pathway by the binding of SARS‐CoV‐2 spike protein to angiotensin‐converting enzyme 2, 41 while an in vitro study suggested that immune complexes containing recombinant SARS‐CoV‐2 spike protein and anti‐spike IgG enhanced platelet‐mediated thrombosis on von Willebrand factor. 42 In our study, we show that platelet activation is associated with caspase‐1 activation and BTK phosphorylation. Caspase‐1 activation is known to be induced by the NLRP3 inflammasome in platelets in several infectious diseases. 26 , 43 Our results suggest a role of platelet activation in the severity degree of the disease and the procoagulant state of the patients probably mediated by NLRP3 inflammasome. However, in the present report, we did not show a direct link between the activation of caspase‐1 and that of NLRP3. Therefore, additional studies are required to determine if the NLRP3 inflammasome is directly responsible for caspase‐1 activation in platelets from patients with COVID‐19. In addition, it would be interesting to explore the platelet‐signaling pathways potentially involved upstream of BTK and caspase‐1, using specific inhibitors. Among the pathways potentially involved, we can mention the way of GPVI, GPIb or TLRs, since platelets express some TLRs. 44
Furthermore, several studies have also suggested a potential role of the NLRP3 inflammasome during the COVID‐19 cytokine storm. 45 , 46 , 47 , 48 A heterogeneous NLRP3 inflammasome signature in circulating monocytes and granulocytes, depending on the COVID‐19 severity, has been recently described. 49 Surprisingly, we did not detect an association between cytokine levels and disease severity. While the severity of the disease and admission to the ICU are associated with an exacerbated immune stimulation leading to a cytokine storm, 2 this association is not always observed. In agreement with previous reports, we observed that most severe patients from our cohort exhibited lower Th1 cytokine levels like IL‐12, IFNγ, and TNFα. An explanation for these discrepancies remained unsolved but could be related to the time course of the disease at which the samples were obtained. 50 , 51 Blood collections were performed as soon as possible after hospital admission (generally within 24 h), but median duration between symptoms onset and hospital admission was more than 1 week, so we cannot exclude a decrease of cytokine levels, explaining differences with other works. This result also suggests that observed platelet activation and platelet caspase‐1 activation are not or little involved in this cytokine storm. But additional experiments would be needed to test this hypothesis on a more homogeneous patient population (on the timing of symptom development). Furthermore, we showed that platelets could be activated in vitro by plasma obtained from patients with severe COVID‐19, in agreement with the study of Hottz et al. 11 We extend this finding by showing that this activation process also involved caspase‐1 and BTK. While the plasmatic triggering factors have not been studied in the present study and remained elusive, it is worth noting that Althaus et al. 52 showed that COVID‐19 serum induces platelet apoptosis and that this effect is triggered by IgG through the FcγRIIA pathway. Further studies are warranted to identify these factors and the involved signaling pathways.
Nevertheless, we also showed that ibrutinib was able to strongly reduce the in vitro activation of platelets induced by the plasma of patients with COVID‐19. It is tempting to speculate that this reduction of platelet activation is related by its impact on BTK phosphorylation, but it has been shown that this compound may counteract platelet activation through FcγRIIA stimulation. 53 It is worth noting that patients with B‐cell malignancies treated by BTK inhibitors who developed COVID‐19 seem to be protected from severe illness. 54 , 55 Altogether, these results strongly suggested that BTK could be a promising therapeutic target.
Several limitations of this report could be noticed such as the limited number of patients included as well as the cross‐sectional design of the study. Patient inclusion was conducted from the second wave of the COVID‐19 pandemic, implying that all patients from our cohort were infected by the same initial variant.
Another limitation is that ibrutinib was used at 5 μM, which is a high dose known to also inhibit Src kinases in platelets, 56 , 57 , 58 , 59 and Src kinases are essential for platelet activation and act upstream of BTK in the GPVI‐signaling pathway. 60 Further experiments are needed to learn the precise involvement of Src kinase inhibition in the observed ibrutinib effect.
Despite these limitations, we have been able to evidence platelet activation, at least partly mediated by plasmatic factors, associated with COVID‐19 severity and to document the inhibitory effect of ibrutinib on platelet activation in vitro.
In summary, our study shows caspase‐1 activation in the platelets of patients with COVID‐19 that is related to disease severity. Moreover, we confirmed the potential therapeutic interest of ibrutinib to reduce platelet activation in COVID‐19. Our study shows another target pathway for BTK inhibitors and reinforces the possibility of using this therapeutic molecule in the treatment of COVID‐19.
AUTHOR CONTRIBUTIONS
V.B. designed the research; L.C., V.B., and F.M. performed the experiments; F.M., B.C., and P.M.R. recruited patients; P.H. performed and supervised the Luminex experiments; L.C., B.T., M.B., Y.G., M.R., and V.B. analyzed the results; L.C. and V.B. made the figures; L.C., Y.C., C.L.V.K., M.R., and V.B. wrote the manuscript; M.R. and V.B. obtained ethical approval.
FUNDING INFORMATION
This work was supported by the Universitary Hospital of Guadeloupe (Grant/Award Nos. IDRCB: 2020–401,908‐31 and CNRIPH: 19–11–26‐67,312) and Janssen‐Cilag.
RELATIONSHIP DISCLOSURE
L.C., F.M., P.H., B.C., P.M.R., Y.G., B.T., Y.C., C.L.V.K., and M.R. declare no conflicts of interest. M.d.B. is scientific manager at Pharmafield, on mission for Janssen. She presented the research project to Janssen V.B. received grant support from Janssen‐Cilag during the conduct of this study.
Supporting information
Figure S1
ACKNOWLEDGMENTS
The authors thank Karubiotec for the use of their P2 laboratory. This work was supported by a grant of V.B. from the University Hospital of Guadeloupe and another grant from Janssen‐Cilag. The authors thank also Ms Annick Bourguignon for the rereading of the manuscript.
Claude L, Martino F, Hermand P, et al. Platelet caspase‐1 and Bruton tyrosine kinase activation in patients with COVID‐19 is associated with disease severity and reversed in vitro by ibrutinib. Res Pract Thromb Haemost. 2022;6:e12811. doi: 10.1002/rth2.12811
Handling Editor: Prof. Yotis Senis
Contributor Information
Frédéric Martino, @fonitram.
Véronique Baccini, Email: veronique.baccini@chu-guadeloupe.fr.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe COVID‐19: interleukin‐6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents. 2020;55(5):105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thachil J, Tang N, Gando S, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID‐19. J Thromb Haemost. 2020;18(5):1023‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Y, Sun W, Guo Y, et al. Association between platelet parameters and mortality in coronavirus disease 2019: retrospective cohort study. Platelets. 2020;31(4):490‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y, Zeng X, Jiao Y, et al. Mechanisms involved in the development of thrombocytopenia in patients with COVID‐19. Thromb Res. 2020;193:110‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guan WJ, Ni ZY, Hu Y, et al. China medical treatment expert group for COVID‐19. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang X, Yang Q, Wang Y, et al. Thrombocytopenia and its association with mortality in patients with COVID‐19. J Thromb Haemost. 2020;18(6):1469‐1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID‐19) infections: a meta‐analysis. Clin Chim Acta. 2020;506:145‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henry BM, de Oliveira MHS, Benoit S, Plebani M, Lippi G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID‐19): a meta‐analysis. Clin Chem Lab Med. 2020;58(7):1021‐1028. [DOI] [PubMed] [Google Scholar]
- 11. Hottz ED, Azevedo‐Quintanilha IG, Palhinha L, et al. Platelet activation and platelet‐monocyte aggregate formation trigger tissue factor expression in patients with severe COVID‐19. Blood. 2020;136(11):1330‐1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manne BK, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID‐19. Blood. 2020;136(11):1317‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Connors JM, Levy JH. COVID‐19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033‐2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boilard E, Paré G, Rousseau M, et al. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood. 2014;123(18):2854‐2863. [DOI] [PubMed] [Google Scholar]
- 15. Campbell RA, Schwertz H, Hottz ED, et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood. 2019;133(19):2013‐2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chao CH, Wu WC, Lai YC, et al. Dengue virus nonstructural protein 1 activates platelets via toll‐like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. 2019;15(4):e1007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koupenova M, Corkrey HA, Vitseva O, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10(1):1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2‐activated platelets enhance dengue virus‐induced lethality via CLEC5A/TLR2. Nat Commun. 2019;10(1):2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34‐45. [DOI] [PubMed] [Google Scholar]
- 20. Garraud O, Cognasse F. Platelet toll‐like receptor expression: the link between « danger » ligands and inflammation. Inflamm Allergy Drug Targets. 2010;9(5):322‐333. [DOI] [PubMed] [Google Scholar]
- 21. Quek LS, Bolen J, Watson SP. A role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137‐1140. [DOI] [PubMed] [Google Scholar]
- 22. Oda A, Ikeda Y, Ochs HD, et al. Rapid tyrosine phosphorylation and activation of Bruton's tyrosine/Tec kinases in platelets induced by collagen binding or CD32 cross‐linking. Blood. 2000;95(5):1663‐1670. [PubMed] [Google Scholar]
- 23. Liu J, Fitzgerald ME, Berndt MC, Jackson CW, Gartner TK. Bruton tyrosine kinase is essential for botrocetin/VWF‐induced signaling and GPIb‐dependent thrombus formation in vivo. Blood. 2006;108(8):2596‐2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nicolson PLR, Nock SH, Hinds J, et al. Low‐dose Btk inhibitors selectively block platelet activation by CLEC‐2. Haematologica. 2021;106(1):208‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murthy P, Durco F, Miller‐Ocuin JL, et al. The NLRP3 inflammasome and bruton's tyrosine kinase in platelets co‐regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem Biophys Res Commun. 2017;483(1):230‐236. [DOI] [PubMed] [Google Scholar]
- 26. Hottz ED, Lopes JF, Freitas C, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3‐inflammasome activation. Blood. 2013;122(20):3405‐3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ozen M, Yilmaz A, Cakmak V, et al. D‐Dimer as a potential biomarker for disease severity in COVID‐19. Am J Emerg Med. 2021;40:55‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Llitjos JF, Leclerc M, Chochois C, et al. High incidence of venous thromboembolic events in anticoagulated severe COVID‐19 patients. J Thromb Haemost. 2020;18(7):1743‐1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beyrouti R, Adams ME, Benjamin L, et al. Characteristics of ischaemic stroke associated with COVID‐19. J Neurol Neurosurg Psychiatry. 2020;91(8):889‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klok FA, Kruip MJHA, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID‐19. Thromb Res. 2020;191:145‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lodigiani C, Iapichino G, Carenzo L, et al. Venous and arterial thromboembolic complications in COVID‐19 patients admitted to an academic hospital in Milan, Italy. Thromb Res. 2020;191:9‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akca S, Haji‐Michael P, de Mendonça A, Suter P, Levi M, Vincent JL. Time course of platelet counts in critically ill patients. Crit Care Med. 2002;30(4):753‐756. [DOI] [PubMed] [Google Scholar]
- 33. Fogarty H, Townsend L, Ni Cheallaigh C, et al. More on COVID‐19 coagulopathy in Caucasian patients. Br J Haematol. 2020;189(6):1060‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lanza GA, Barone L, Scalone G, et al. Inflammation‐related effects of adjuvant influenza A vaccination on platelet activation and cardiac autonomic function. J Intern Med. 2011;269(1):118‐125. [DOI] [PubMed] [Google Scholar]
- 35. Wool GD, Miller JL. The impact of COVID‐19 disease on platelets and coagulation. Pathobiology. 2021;88(1):15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hottz ED, Quirino‐Teixeira AC, Valls‐de‐Souza R, Zimmerman GA, Bozza FA, Bozza PT. Platelet function in HIV plus dengue coinfection associates with reduced inflammation and milder dengue illness. Sci Rep. 2019;9(1):7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Damien P, Cognasse F, Lucht F, et al. Highly active antiretroviral therapy alters inflammation linked to platelet cytokines in HIV‐1‐infected patients. J Infect Dis. 2013;208(5):868‐870. [DOI] [PubMed] [Google Scholar]
- 38. Kelesidis T, Papakonstantinou V, Detopoulou P, et al. The role of platelet‐activating factor in chronic inflammation, immune activation, and comorbidities associated with HIV infection. AIDS Rev. 2015;17(4):191‐201. [PMC free article] [PubMed] [Google Scholar]
- 39. Jin L, Ying ZH, Yu CH, Zhang HH, Yu WY, Wu XN. Isofraxidin ameliorated influenza viral inflammation in rodents via inhibiting platelet aggregation. Int Immunopharmacol. 2020;84:106521. [DOI] [PubMed] [Google Scholar]
- 40. Lê VB, Schneider JG, Boergeling Y, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med. 2015;191(7):804‐819. [DOI] [PubMed] [Google Scholar]
- 41. Zhang S, Liu Y, Wang X, et al. SARS‐CoV‐2 binds platelet ACE2 to enhance thrombosis in COVID‐19. J Hematol Oncol. 2020;13(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen G, Wu D, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620‐2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cornelius DC, Baik CH, Travis OK, et al. NLRP3 inflammasome activation in platelets in response to sepsis. Physiol Rep. 2019;7(9):e14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of toll‐like receptor molecules on human platelets. Immunol Cell Biol. 2005;83(2):196‐198. [DOI] [PubMed] [Google Scholar]
- 45. Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Merad M, Martin JC. Author Correction: Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(7):448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parisi V, Leosco D. Precision medicine in COVID‐19: IL‐1β a potential target. JACC Basic Transl Sci. 2020;5(5):543‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodrigues TS, de Sá KSG, Ishimoto AY, et al. Inflammasomes are activated in response to SARS‐CoV‐2 infection and are associated with COVID‐19 severity in patients. J Exp Med. 2021;218(3):e20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Courjon J, Dufies O, Robert A, et al. Heterogeneous NLRP3 inflammasome signature in circulating myeloid cells as a biomarker of COVID‐19 severity. Blood Adv. 2021;5(5):1523‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dayarathna S, Jeewandara C, Gomes L, et al. Similarities and differences between the « cytokine storms » in acute dengue and COVID‐19. Sci Rep. 2020;10(1):19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gadotti AC, de Castro Deus M, Telles JP, et al. IFN‐γ is an independent risk factor associated with mortality in patients with moderate and severe COVID‐19 infection. Virus Res. 2020;289:198171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Althaus K, Marini I, Zlamal J, et al. Antibody‐induced procoagulant platelets in severe COVID‐19 infection. Blood. 2021;137(8):1061‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bye AP, Hoepel W, Mitchell JL, et al. Aberrant glycosylation of anti‐SARS‐CoV‐2 IgG is a pro‐thrombotic stimulus for platelets. Blood. 2021;138(16):1481‐1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Treon SP, Castillo J, Skarbnik AP, et al. The BTK‐inhibitor ibrutinib may protect against pulmonary injury in COVID‐19 infected patients. Blood. 2020;135(21):1912‐1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roschewski M, Lionakis MS, Sharman JP, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID‐19. Sci Immunol. 2020;5(48):eabd0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Levade M, David E, Garcia C, et al. Ibrutinib treatment affects collagen and von Willebrand factor‐dependent platelet functions. Blood. 2014;124(26):3991‐3995. [DOI] [PubMed] [Google Scholar]
- 57. Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen‐mediated but not ADP‐mediated platelet aggregation. Leukemia. 2015;29(4):783‐787. [DOI] [PubMed] [Google Scholar]
- 58. Bye AP, Unsworth AJ, Vaiyapuri S, Stainer AR, Fry MJ, Gibbins JM. Ibrutinib inhibits platelet integrin αIIbβ3 outside‐in signaling and thrombus stability but not adhesion to collagen. Arterioscler Thromb Vasc Biol. 2015;35(11):2326‐2335. [DOI] [PubMed] [Google Scholar]
- 59. Rigg RA, Aslan JE, Healy LD, et al. Oral administration of Bruton's tyrosine kinase inhibitors impairs GPVI‐mediated platelet function. Am J Physiol Cell Physiol. 2016;310(5):C373‐C380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gratacap MP, Martin V, Valéra MC, et al. The new tyrosine‐kinase inhibitor and anticancer drug dasatinib reversibly affects platelet activation in vitro and in vivo. Blood. 2009;114(9):1884‐1892. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.