

Abstract
Traumatic brain injury (TBI) is associated with chronic psychiatric complications and increased risk for development of neurodegenerative pathology. Aged individuals account for most TBI-related hospitalizations and deaths. Nonetheless, neurobiological mechanisms that underlie worsened functional outcomes after TBI in the elderly remain unclear. Therefore, this study aimed to identify pathways that govern differential responses to TBI with age. Here, adult (2 months of age) and aged (16–18 months of age) male C57BL/6 mice were subjected to diffuse brain injury (midline fluid percussion), and cognition, gliosis, and neuroinflammation were determined 7 or 30 d postinjury (dpi). Cognitive impairment was evident 7 dpi, independent of age. There was enhanced morphologic restructuring of microglia and astrocytes 7 dpi in the cortex and hippocampus of aged mice compared with adults. Transcriptional analysis revealed robust age-dependent amplification of cytokine/chemokine, complement, innate immune, and interferon-associated inflammatory gene expression in the cortex 7 dpi. Ingenuity pathway analysis of the transcriptional data showed that type I interferon (IFN) signaling was significantly enhanced in the aged brain after TBI compared with adults. Age prolonged inflammatory signaling and microgliosis 30 dpi with an increased presence of rod microglia. Based on these results, a STING (stimulator of interferon genes) agonist, DMXAA, was used to determine whether augmenting IFN signaling worsened cortical inflammation and gliosis after TBI. DMXAA-treated Adult-TBI mice showed comparable expression of myriad genes that were overexpressed in the cortex of Aged-TBI mice, including Irf7, Clec7a, Cxcl10, and Ccl5. Overall, diffuse TBI promoted amplified IFN signaling in aged mice, resulting in extended inflammation and gliosis.
SIGNIFICANCE STATEMENT Elderly individuals are at higher risk of complications following traumatic brain injury (TBI). Individuals >70 years old have the highest rates of TBI-related hospitalization, neurodegenerative pathology, and death. Although inflammation has been linked with poor outcomes in aging, the specific biological pathways driving worsened outcomes after TBI in aging remain undefined. In this study, we identify amplified interferon-associated inflammation and gliosis in aged mice following TBI that was associated with persistent inflammatory gene expression and microglial morphologic diversity 30 dpi. STING (stimulator of interferon genes) agonist DMXAA was used to demonstrate a causal link between augmented interferon signaling and worsened neuroinflammation after TBI. Therefore, interferon signaling may represent a therapeutic target to reduce inflammation-associated complications following TBI.
Keywords: aging, interferon, microglia, neuroinflammation, STING, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is associated with chronic psychiatric complications and increased risk for the development of neurodegenerative pathologies (Gardner et al., 2015; Ramos-Cejudo et al., 2018; Stein et al., 2019). Elderly individuals (>70 years old) are especially vulnerable to fall-related TBI and have the highest rates of TBI-related hospitalizations and deaths per year (https://www.cdc.gov/traumaticbraininjury/index.html). Furthermore, these individuals are more likely to experience new or worsened cognitive symptoms and may even develop neurodegenerative pathology associated with Alzheimer's disease and Parkinson's disease (Gardner et al., 2018; Ramos-Cejudo et al., 2018). Although age at time of injury is a well known predictor of clinical outcomes, the neurobiological mechanisms that underlie impaired functional recovery after TBI in aged individuals remain unclear.
In the aged brain, there are many transcriptional changes in both microglia and astrocytes that affect glial dynamics. For instance, heightened microglial reactivity (Fenn et al., 2014; Muccigrosso et al., 2016) coupled with astrocyte immunosenescence with aging (O'Neil et al., 2022) may negatively influence outcome after TBI. Several studies of TBI (diffuse and focal) in aged rodents have highlighted age-associated pathology and impaired functional recovery. For instance, fluid percussion injury (FPI) caused greater learning deficits in the Morris water maze task and somatomotor deficits in aged rats compared with adult rats 7 d postinjury (dpi; Hamm et al., 1992). Furthermore, controlled cortical impact (CCI; a focal injury model) in aged mice enhanced microglial reactive phenotypes in the cortex, hippocampus (HPC), and thalamus (Kumar et al., 2013). In other studies, amplified microglial response in the aged brain following TBI augmented the production of nitric oxide (NO) and reactive oxygen species (ROS; Kumar et al., 2016; Ritzel et al., 2019), activation of complement pathway with synapse loss (Krukowski et al., 2018), and recruitment of peripheral CCR2+ monocytes (Morganti et al., 2016). Moreover, there was progressive astrocyte reactivity in aged compared with adult cortex and hippocampus after focal TBI (Early et al., 2020). All of these factors aggravate secondary injury and hamper recovery. Though these studies implicate inflammatory processes in the aging brain after TBI (focal injury), the specific biological pathways governing this dysregulated response are less well understood.
Recent single-cell RNA sequencing of the cortex 7 d after diffuse TBI showed unique clusters of trauma-associated microglia that were characterized by expression of type I interferon (IFN) genes. Genes associated with IFN signaling were elevated 7 dpi, concurrent with an increase in microglia showing primed profiles (Witcher et al., 2021). Moreover, these TBI-associated microglia were involved in dendrite remodeling, suppression of neuronal homeostasis, and cognitive impairment (Witcher et al., 2021). At 7 and 30 dpi, preinjury PLX5622-mediated microglial elimination prevented inflammatory and IFN-associated gene expression, neuronal synaptic damage, and, ultimately, cognitive impairment (Witcher et al., 2021). Furthermore, microglia priming and immune reactivity following TBI were also reduced with delayed forced turnover of microglia starting 7 dpi (Bray et al., 2022). Another recent study provided evidence of increased expression of IFN gene transcripts in microglia and astrocytes 7 d after FPI (Todd et al., 2021). Furthermore, IFNβ signaling within the first 24 h after CCI (focal injury) was critical to the promotion of downstream inflammation, tissue pathology, and functional deficits (Barrett et al., 2020; Fritsch et al., 2022). Another study showed enhancement of type I IFN mediators Irf7, pSTAT1, and cGMP–cAMP synthase (cGAS) 24 h after CCI in aged mice (Barrett et al., 2021). Collectively, these data indicate type I IFN signaling may be key for the development of chronic inflammation after TBI.
We aimed to expand these studies by investigating the pathways that underlie differential responses to diffuse TBI with age. Here, we provide novel evidence that amplified interferon signaling promotes aggravated and prolonged microglia-mediated inflammation and gliosis in aged mice after diffuse TBI.
Materials and Methods
Mice.
Adult (2 months of age) and aged (16–18 months of age) male C57BL/6 mice were purchased from The Jackson Laboratory. Mice were group housed under a 12 h light/dark cycle with ad libitum access to food and water. All procedures were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and the Public Health Service Policy on Human Care and Use of Laboratory Animals. The Guide for the Care and Use of Laboratory Animals was approved by The Ohio State University (OSU) Institutional Laboratory Animal Care and Use Committee.
Midline fluid percussion injury.
Mice received a midline diffuse TBI using an FPI apparatus (Custom Design & Fabrication) as previously described (Fenn et al., 2014, 2015; Rowe et al., 2016; Witcher et al., 2018). Briefly, mice were anesthetized in an isoflurane chamber at 4–5% with a flow rate of 0.8 L/min. After the surgical site was shaved, mice were secured to the stereotaxic apparatus (catalog #51731, Stoelting) and maintained under anesthesia with a mask attachment (catalog #51609 M, Stoelting). The surgical site was then sterilized with alternating applications of iodine and 70% ethanol. Mice received a 3 mm craniectomy between the landmark sutures bregma and λ, and a rigid Luer Loc needle hub was secured over the craniectomy site. Following this procedure, mice were moved to a heated (37°C) recovery cage and monitored until fully conscious (upright, responsive, walking). After recovery, mice were briefly reanesthetized in an isoflurane chamber at 5% (flow rate, 0.8 L/min) for 5 min. The Luer Loc hub was filled with saline, and the hub was then attached to the injury device. Once a positive toe-pinch response was elicited (∼30 s), a 10 m/s pulse of saline (1.2 atmospheres; 670–720 mV) was imposed on the dura (Kelley et al., 2007; Fenn et al., 2014; Lifshitz et al., 2016; Rowe et al., 2016). Immediately after injury, the hub was removed, dural integrity was confirmed, and surgical staples were used to close the incision. Mice were then placed on a heated pad and the time to self-right was recorded. Righting time is a relative index of injury severity and can be used as exclusion criteria (Witcher et al., 2018, 2021; Bray et al., 2022). Because control (naive) mice are not exposed to the surgical and midline FPI (mFPI) procedures, righting time is not a relevant measure. The average righting times for adult and aged C57BL/6 mice were within the moderate TBI range (200–560 s), and mice with a compromised dura were excluded from the study.
Postoperative/injury care.
Mice with TBI were monitored for 1 h postinjury, then allowed to recover overnight in a heated recovery cage with accessible food and hydrogel. The next day, mice were returned to the vivarium. To avoid confounding variables that modulate inflammation, no analgesics were provided in these studies. Mice were weighed and checked for signs of lethargy (lack of movement) and infection (redness and pus around the incision site) daily throughout the experiments (7 or 30 d). Removal criteria included a loss of 20% of baseline body weight, sustained lethargy, paralysis, or surgical site infection. In the current study, no TBI-injured mice met this exclusion criteria. Wound clips (7 mm) used to close the incision site were removed between 10 and 14 dpi.
Novel object recognition and location memory.
To determine cognitive impairment following TBI, novel object recognition (NOR) and novel object location (NOL) memory tasks were performed as previously described (Witcher et al., 2021; Bray et al., 2022). In brief, mice underwent the following four 10 min trials, with 24 h intertrial intervals: habituation (no objects), acquisition (two objects), recognition testing (two objects, with one new object), and location testing (two objects, with one in new location). Behavioral chambers and objects were cleaned with 70% ethanol between trials. Total time of exploration [(TTE) = timenovel + timefamiliar], percentage of time spent with novel (timenovel/TTE × 100), and discrimination index [(DI) = (timenovel-timefamiliar)/TTE] were calculated for both NOR and NOL memory trials (n = 8–10/experimental group; Denninger et al., 2018). All trials were recorded and analyzed by an investigator blinded to groups.
Iba-1 and GFAP immunofluorescence.
Iba-1 and GFAP were labeled as previously described (Witcher et al., 2018). In brief, mice were transcardially perfused with ice-cold PBS, pH 7.4, followed by 4% formaldehyde. Brains were postfixed (24 h), dehydrated in 30% sucrose (48 h), snap frozen using dry ice-cooled isopentane, and cryosectioned at 30 µm. Cortical and hippocampal regions were identified using the Allen Mouse Brain Atlas (Allen Institute). Coronal sections were washed in PBS, blocked for 1 h (0.1% Triton X-100, 1% BSA (bovine serum albumin), and 5% NDS (normal donkey serum)) and incubated with primary antibodies [1:1000; rabbit anti-Iba-1, catalog #019–19471, Wako (RRID:AB_880202); 1:500; goat anti-GFAP, catalog #ab53554, Abcam (RRID:AB_880202)] overnight. Sections were then washed and incubated for 2 h with fluorochrome-conjugated secondary antibodies (1:500; donkey anti-rabbit Alexa Fluor 647, catalog #A31573, Thermo Fisher Scientific; 1:500; donkey anti-goat Alexa Fluor 488, catalog #A11055, Thermo Fisher Scientific), mounted onto glass slides, and coverslipped with DAPI Fluoromount (catalog #00–4959-52, Thermo Fisher Scientific). All tissue underwent the following additional protocol to quench autofluorescence (common in aged tissue) before coverslipping with DAPI: slides were washed in 0.1% Sudan Black B (Sigma-Aldrich) in 70% ethanol solution for 2 min, rinsed in 70% ethanol for 2 s, and finally washed in PBS for 5 min.
Microscopy and image analysis.
Fluorescent labeling within the cortex and hippocampus was visualized and imaged using an EVOS FL Auto 2 Imaging System (Thermo Fisher Scientific). Imaging parameters were consistent throughout each experiment. To determine the percentage of area of GFAP+ and Iba-1+ labeling, single-channel images were converted to 8 bit TIFF format and constant thresholds were used to quantify positively labeled pixels (ImageJ). Values from five to eight images per mouse were averaged, and these values were used to calculate group averages and variance for each injury and/or age group (n = 5–6/experimental group). Investigators remained blinded to experimental groups throughout image analyses. Rod morphology of Iba-1+ cells was quantified using the morphologic definition provided as previously described (Ziebell et al., 2012; Taylor et al., 2014). In brief, microglia were considered rod shaped if they possessed elongated cell bodies and processes that aligned perpendicularly to the surface of the cortex. In select figures, insets were thresholded in PowerPoint to generate black and white images.
RNA isolation and quantitative PCR.
RNA was isolated from microdissected coronal tissue (Tri-Reagent, catalog #T9424, Sigma-Aldrich) and used in quantitative PCR (qPCR) as described previously (Witcher et al., 2018). Briefly, RNA was normalized by concentration, then reverse transcribed to cDNA (HiCap RT PCR Kit, catalog #43–688-14, Thermo Fisher Scientific). Taq-Man Gene Expression Assay (Thermo Fisher Scientific) was used to perform quantitative real-time qPCR to amplify the following cDNA genes of interest: Stat1, Irf7, Ifi27Ia, Tnf, Il1b, Tlr2, C1qa, Ccl2, Ccl12, C3, Ccl5, Cxcl10, Cxcl16, Clec7a, Tyrobp, H2-Eb1, and Atf3. Target genes and reference cDNA (Gapdh) were amplified simultaneously using an oligonucleotide probe with 5′ fluorescent reporter dye (FAM) and 3′ nonfluorescent quencher (NFQ). The following probes were used: Stat1 (Mm01257286_m1), Irf7 (Mm00516791_g1), Ifi27Ia, (Mm00835449_g1), Tnf (Mm00443258_m1), Il1b (Mm00434228_m1), Tlr2 (Mm0044234_m1), C1qa (Mm00432142_m1), Ccl2 (Mm00441242_m1), Ccl12 (Mm01617100_m1), C3 (Mm01232779_m1), Ccl5 (Mm01302427_m1), Cxcl10 (Mm00445235_m1), Cxcl16 (Mm00469712_m1), Clec7a (Mm01183349_m1), Tyrobp (Mm00449152), H2-eb1 (Mm00439221_m1), Atf3 (Mm00476033_m1), and Gapdh (Mm99999915_g1). When Taq DNA polymerase synthesizes a new strand and reaches the TaqMan probe, the FAM is cleaved from the NFQ and increases the fluorescent intensity proportional to the amount of amplicon synthesized. Fluorescence was determined using a QuantStudio 5 Real-Time PCR system (Thermo Fisher Scientific). Data were analyzed by the comparative threshold cycle method (ΔΔCT), and results are expressed as the fold-change from the control group (n = 6/experimental group).
RNA isolation and NanoString gene expression analysis.
RNA was isolated from microdissected coronal tissue (Tri-Reagent, catalog #T9424, Sigma-Aldrich). Cortical RNA integrity was confirmed by BioAnalyzer (Agilent). Gene expression was quantified using a NanoString nCounter Neuropathology panel (with 30 added genes, 760 genes total; Witcher et al., 2021) by the OSU Comprehensive Cancer Center Genomics Shared Resource facility (The Ohio State University, Columbus, OH). Data were normalized and differential expression testing was performed using DESeq2 in R (Love et al., 2014). In brief, EstimateSizeFactors in DeSeq2 was used for normalization to housekeeping genes (Aars and Ccdc127). The DeSeq design parameter included the following two variables: one controlling for NanoString cartridge and a second for experimental group (four levels: Adult-Control, Adult-TBI, Aged-Control, Aged-TBI; n = 3/experimental group). Results tables were generated for pairwise comparisons of interest (e.g., Aged-TBI vs Aged-Control, Aged-TBI vs Adult-TBI). To account for multiple comparisons, Benjamini–Hochberg correction was used. Adjusted p-values (p-adj) below a false discovery rate of 5% (i.e., p-adj < 0.05) were considered differentially expressed. Heatmaps were generated using pheatmap in R Studio with scale = “row,” which centers and scales values by gene such that colors reflect z scores rather than absolute values. Statistically significant genes (p-adj < 0.05) were subsequently used for ingenuity pathway analysis (IPA; Qiagen). Gene names and fold changes were submitted to compare expression patterns in our dataset to the IPA database. IPA results for canonical pathways and master regulators with p < 0.05 were considered significant.
DMXAA administration.
DMXAA is a murine stimulator of interferons (STING) agonist. To determine interactions between diffuse brain injury and DMXAA, adult male C57BL/6 mice were injected intraperitoneally with vehicle or DMXAA (25 mg/kg in 7.5% sodium bicarbonate; catalog #14617, Cayman Chemical) 1 h before injury and then every 24 h up to 6 dpi (n = 4–7/experimental group).
Experimental design and statistical analyses.
The number of mice per experiment (including the number of images analyzed) is included in the Materials and Methods for each experiment. Statistical analysis using DESeq2 for NanoString is described above. GraphPad Prism software was used for ANOVA of histologic, behavioral, and biochemical data. Two-way ANOVA was used to determine the main effects and interactions between factors. Bonferroni's test for multiple comparisons was used for post hoc analysis when significant main effects and/or interactions were determined. The resulting post hoc significance (p < 0.05) in the Bonferroni's test is denoted in the figures using lower case letters (a, b, c, or d). Groups with a different letter (e.g., a, b, c, or d) are significantly different from each other (p < 0.05). Groups sharing the same letter or letters are not different. All data are expressed as the group mean ± SEM.
Results
Cognitive impairment in adult and aged mice 7 dpi
Age at the time of injury is an important factor that influences functional recovery after TBI in humans (Marquez de la Plata et al., 2008; Brickler et al., 2017). In adult mice, diffuse brain injury has been shown to cause persistent neuroinflammation mediated by microglia and associated with reduced neuronal plasticity and connectivity (Witcher et al., 2018, 2021). In the aged brain, there are several transcriptional changes in both microglia and astrocytes that may affect glial dynamics (O'Neil et al., 2022), especially in the context of brain injury. Therefore, the purpose of this study was to identify signaling pathways that govern the response to brain injury in the aged brain. To address this aim, adult (2 months) and aged (18 months) male C57BL/6 mice were subjected to a diffuse brain injury induced by mFPI. NOR and NOL memory and indices of inflammation and pathology were assessed at 7 dpi (Fig. 1A). Time to self-right was determined in mice immediately after mFPI-mediated TBI as previously described (Lifshitz et al., 2007). As expected, aged mice had a longer average time to self-right compared with adult mice (Fig. 1B; p < 0.0197).
Figure 1.

Cognitive impairment in adult and aged mice 7 dpi. A, Adult (2 months of age) and aged (18 months of age) male C57BL/6 mice were subjected to midline fluid percussion injury (TBI) or left as uninjured CONs, and NOR/NOL memory was assessed (n = 6) 6 and 7 dpi. B, Self-righting time (in seconds) was determined immediately after TBI (n = 39). C–E, For NOR at 6 dpi, the total time spent exploring the objects (C), the percentage of time spent investigating the novel object (D), and the discrimination index for the novel object were determined (E). F–H, For NOL at 7 dpi, the total time spent exploring the objects (F), the percentage of time spent investigating the object in a novel location (G), and the discrimination index for the object in a novel location (H) were determined. Bars represent the mean ± SEM, and individual data points are provided. Means with different letters (e.g., a, b, c) indicate significant post hoc differences between groups (p < 0.05). Groups with the same letter or letters are not significantly different.
Next, cortical and hippocampal function were assessed using the NOR and NOL memory tasks (Antunes and Biala, 2012; Denninger et al., 2018; Witcher et al., 2021). These tests were assessed in the same mice on sequential days with NOR testing 6 dpi and NOL testing 7 dpi (Fig. 1A). There were no significant differences in the total time spent exploring the objects among the four experimental groups during either the NOR or NOL memory tasks (Fig. 1C,F). NOR at 6 dpi (Fig. 1D) was influenced by age (F(1,31) = 29.0, p < 0.0001) and TBI (F(1,31) = 41.3, p < 0.0001), and tended to be influenced by both age and injury (interaction: F(1,31) = 3.0, p = 0.09). Post hoc analysis indicated that Aged-Control, Adult-TBI, and Aged-TBI mice investigated the novel object for less time (percentage of total time spent exploring objects) than Adult-Control mice (p < 0.05, for each). Similar results were obtained in the NOL memory task. NOL at 7 dpi (Fig. 1G) was influenced by both age (F(1,31) = 14.50, p = 0.0006) and injury (F(1,31) = 33.6, p < 0.0001). Post hoc analysis showed that Aged-Control, Adult-TBI, and Aged-TBI mice spent less time (percentage of total time spent exploring objects) with the object in a novel location compared with the Adult-Controls 7 dpi (p < 0.05, for each). Discrimination index measurements for NOR and NOL indicated that Aged-Control, Adult-TBI, and Aged-TBI mice tended to spend more time with the familiar object than Adult-Controls (Fig. 1E,J; p < 0.05, for each). Overall, cognitive impairment was evident in adult and aged mice 7 dpi.
Amplified morphologic restructuring of astrocytes and microglia in aged mice 7 dpi
Next, the labeling of astrocyte (GFAP+) and microglia (Iba-1+) was assessed in the cortex and hippocampus 7 dpi in adult and aged mice. Representative images from the somatosensory cortex (SS-Ctx) show that there was increased GFAP+ labeling of astrocytes in both adult and aged mice after TBI (Fig. 2A). Thresholded black and white images (from insets) highlight differences between adult and aged mice in morphologic restructuring of astrocytes in the cortex 7 dpi (Fig. 2B). GFAP+ labeling in the cortex (Fig. 2C) was increased by TBI (F(1,19) = 143.6, p < 0.0001), and this increase was age dependent (interaction: F(1,19) = 10.7, p = 0.0041). Post hoc analysis confirmed that Aged-TBI mice had the highest level of GFAP+ labeling (percentage of area) in the cortex compared with all other groups, including Adult-TBI mice (Fig. 2C; p < 0.05). Similar effects of TBI and age were evident in the HPC (Fig. 2D). Thresholded black and white representative images from the HPC show that there was increased GFAP+ labeling of astrocytes in aged mice 7 dpi (Fig. 2E). In the HPC (Fig. 2F), GFAP+ labeling was increased by TBI (F(1,19) = 8.1, p = 0.0103), and this increase was influenced by age (interaction: F(1,19) = 8.3, p = 0.0097). Post hoc analysis confirmed that Aged-TBI mice had the highest level of GFAP+ labeling (percentage of area) in the HPC compared with all other groups (Fig. 2F; p < 0.05). Overall, TBI-associated astrogliosis in the somatosensory cortex and hippocampus was augmented in aged mice compared with adults 7 dpi.
Figure 2.
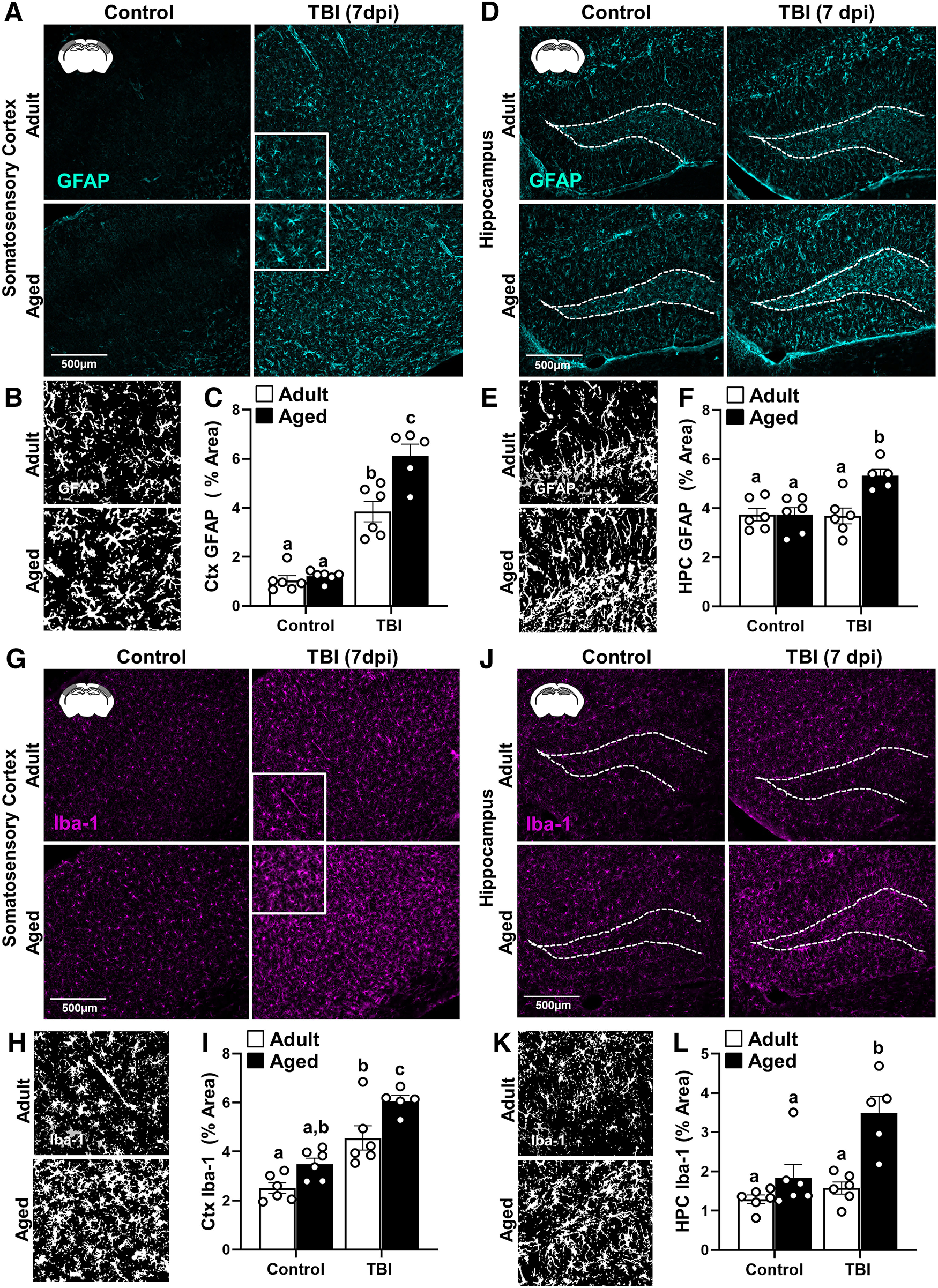
Amplified morphologic restructuring of astrocytes and microglia in aged mice after TBI. Adult (2 months of age) and aged (18 months of age) male C57BL/6 mice were subjected to midline fluid percussion injury (TBI) or left as uninjured CONs. GFAP+ and Iba-1+ labeling was determined in the SS-Ctx and HPC of adult and aged mice 7 dpi. A, Representative images of GFAP+ labeling in the SS-Ctx. B, Thresholded black and white images of GFAP+ labeling in the SS-Ctx of adult and aged mice 7 dpi. C, Percentage of the area of GFAP+ labeling in the SS-Ctx. D, Representative images of GFAP+ labeling in the HPC. E, Thresholded black and white images of GFAP+ labeling in the HPC of adult and aged mice 7 dpi. F, Percentage of the area of GFAP+ labeling in the HPC. G, Representative images of Iba-1+ labeling in the SS-Ctx. H, Thresholded black and white images of Iba-1+ labeling in the SS-Ctx of adult and aged mice 7 dpi. I, Percentage of the area of Iba-1+ labeling in the SS-Ctx. J, Representative images of Iba-1+ labeling in the HPC. K, Thresholded black and white images of Iba-1+ labeling in the HPC of adult and aged mice 7 dpi. L, Percentage of the area of Iba-1+ labeling in the HPC. Bars represent the mean ± SEM, and individual data points are provided. Means with different letters (e.g., a, b, c) indicate significant post hoc differences between groups (p < 0.05). Groups with the same letter or letters are not significantly different.
For microglia, representative images from the SS-Ctx show that there was increased Iba-1+ labeling of microglia in both Adult-TBI and Aged-TBI mice 7 dpi (Fig. 2G). Thresholded black and white images (from insets) highlight differences between experimental groups in morphologic restructuring of microglia in the cortex 7 dpi (Fig. 2H). The percentage of the area labeling Iba-1+ microglia in the cortex (Fig. 2I) was increased by TBI (F(1,19) = 50.0, p < 0.0001) and by age (F(1,19) = 14.5, p = 0.0012). Post hoc analysis revealed that Aged-TBI mice had the highest level of Iba-1+ labeling in the somatosensory cortex compared with all other groups (p < 0.05). There was a similar effect of age on microglial morphologic restructuring at 7 dpi in the HPC. Representative images from the HPC show that there was increased Iba-1+ labeling of microglia in both adult and aged mice after TBI (Fig. 2J). Thresholded black and white images highlight the structural differences between adult and aged microglia in the HPC at 7 dpi (Fig. 2K). Iba-1+ labeling in the HPC (Fig. 2L) was increased by TBI (F(1,19) = 19.7, p = 0.0003), and this increase was influenced by age (interaction: F(1,19) = 6.2, p < 0.0219). Aged-TBI mice had the highest level of Iba-1+ labeling (percentage of area) in the HPC compared with all other groups (p < 0.05). Overall, microglial morphologic restructuring in the cortex and hippocampus was augmented by age 7 dpi.
Inflammatory gene expression in adult and aged cortex 7 dpi
To gain further insight into the influence of age and TBI on brain inflammation, cortical mRNA was assessed using a NanoString nCounter Neuropathology panel (Fig. 3A; 760 genes total). First, the effect of age in the cortex is highlighted in the comparison between the Adult-Control and Aged-Control mice (Aged-Control vs Adult-Control). Figure 3B shows a volcano plot (genes displayed by –log10 p value vs log2 fold change) of differentially expressed genes (DEGs) between Adult-Control and Aged-Control mice (p-adj < 0.05). A total of 15 genes were differentially expressed with age (1 increased, 14 decreased). Red dots are DEGs increased with age (i.e., Pvalb), and blue dots are DEGs decreased with age (e.g., Calb1, Chl1, Stx1a, and Hras). These 15 DEGs were used in IPA, and significant master regulators are listed (Fig. 3C). In Aged-Control mice, P2RY11, calcineurin A, CYP, clathrin, and Mek master regulators were inhibited compared with Adult-Control mice. In addition, MARK2, ZMPSTE24, and MMS22L were all activated in Aged-Control mice compared with Adult-Control mice.
Figure 3.
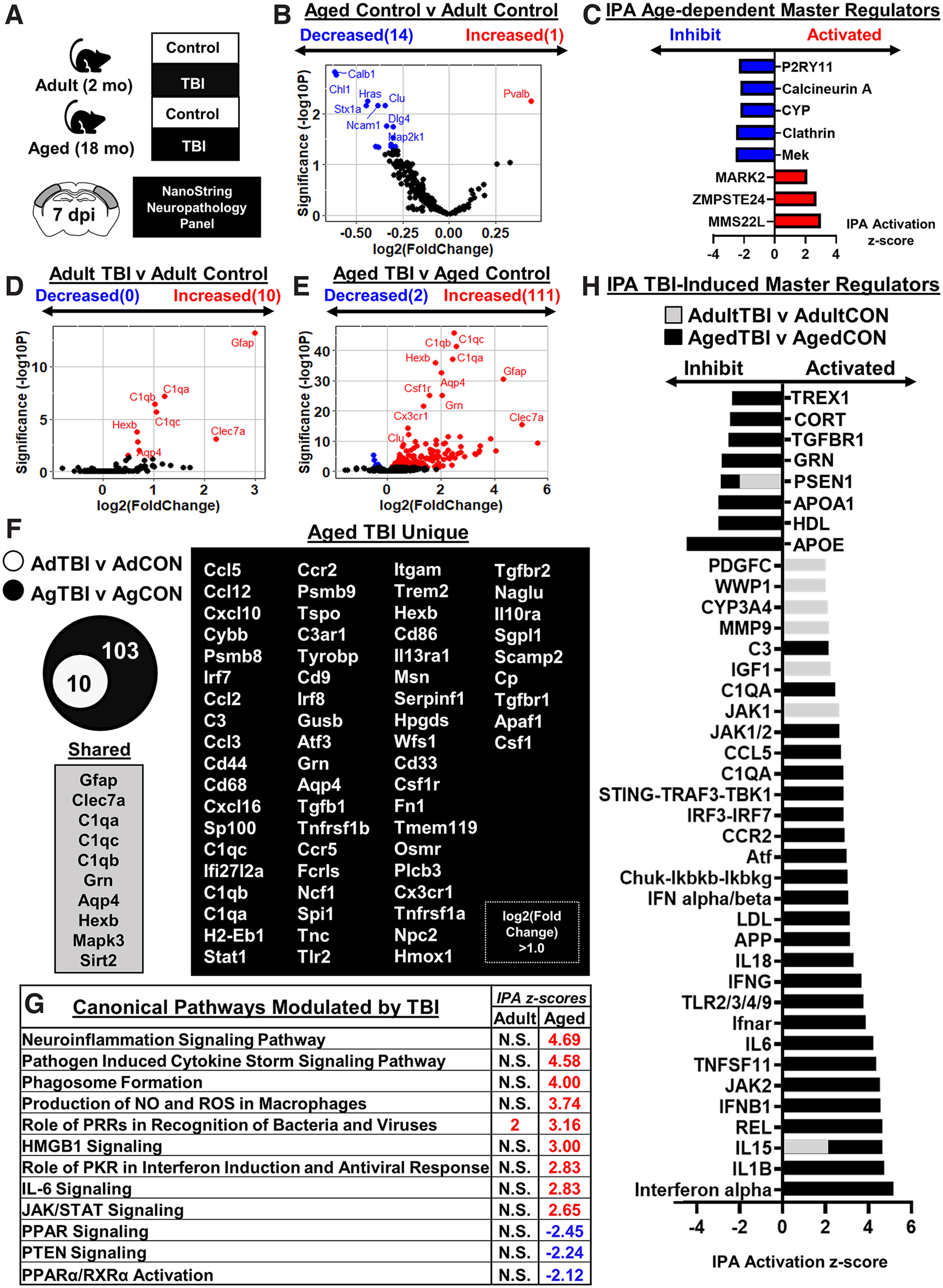
Inflammatory gene expression in adult and aged cortex 7 dpi. A, Adult (2 months of age) and aged (18 months of age) male C57BL/6 mice were subjected to midline fluid percussion injury (TBI) or left as uninjured CONs. Cortical tissue was microdissected and flash frozen at 7 dpi. RNA was collected, and mRNA copy number was determined using a NanoString nCounter Neuropathology panel (n = 3). B, Volcano plot of significantly differentially expressed genes (p < 0.05) compared between Aged-Control and Adult-Control mice. Genes in red were increased with age, and genes in blue were decreased with age. C, IPA of canonical pathways compared between Aged-Control and Adult-Control mice. Positive z score indicates activated and negative z score indicates inhibited in Aged-Control versus Adult-Control mice. D, E, Volcano plots of significantly differentially expressed genes (p < 0.05) compared between Adult-TBI and Adult-Control mice (42 genes; D) and Aged-TBI and Aged-Control mice (242 genes; E). Genes in red were increased, and genes in blue were decreased by TBI in each age group. F, Venn diagram of significantly differentially expressed genes in the Adult-TBI versus Adult-Control and Aged-TBI versus Aged-Control comparison. Genes were grouped as follows: (1) unique to Adult-TBI (6 genes), (2) shared between TBI groups (36 genes), and (3) unique to Aged-TBI (206 genes). G, IPA of significant canonical pathways (z score) activated or inhibited by TBI in adult and aged mice. N.S. denotes IPA canonical pathways that are not significantly activated. H, IPA of significant master regulators (z score) activated or inhibited by TBI for both adult mice (gray bars) and aged mice (black bars). Adult (gray) bars were overlapped on top of the aged (black) bars.
Next, the effect of TBI on cortical gene expression in adult mice and in aged mice 7 dpi is highlighted (Fig. 3D–H). Adult-TBI versus Adult-Control and Aged-TBI versus Aged-Control comparisons were made. Figure 3D shows a volcano plot (–log10 p value vs log2 fold change) of significant DEGs between Adult-TBI and Adult-Control mice. There were 10 DEGs with TBI in adult mice (all increased). Red dots are DEGs increased with TBI (e.g., Gfap, Clec7a, C1qa/b/c, Hexb, Aqp4). Figure 3E shows a volcano plot (–log10 p value vs log2 fold change) of significant DEGs between Aged-TBI and Aged-Control mice. There were 113 DEGs with TBI in aged mice (111 increased, 2 decreased). Red dots are DEGs increased with TBI (e.g., Gfap, Clec7a, C1qa/b/c, Grn, etc.), and blue dots are DEGs decreased with TBI (i.e., Scn1a, Pvalb). The Venn diagram (Fig. 3F) shows genes that were (1) expressed in Adult-TBI (10) mice, all shared with Aged-TBI; and (2) genes that were uniquely expressed in Aged-TBI (103 genes total, genes with greater than onefold change shown).
IPA canonical pathways activated or inhibited by TBI in aged and adult mice are shown (Fig. 3G). IPA shows activation of the role of Pattern Recognition Receptors (PRRs) in recognition of bacteria and viruses in both age groups after TBI. Pathways uniquely activated by TBI in the aged brain include neuroinflammation signaling pathway, pathogen induced cytokine storm signaling pathway, phagosome formation, production of NO and ROS in macrophages, HMGB1 signaling, role of protein kinase RNA (PKR) in interferon induction and antiviral response, IL-6 signaling, and Jak/Stat signaling. Activation of peroxisome proliferator-activated receptor (PPAR), PTEN, and PPARa/retinoid X receptor a (RXRa) was decreased selectively in aged mice 7 dpi. Consistent with these pathways, IPA (Fig. 3H) showed that Aged-TBI mice had more activated master regulators compared with adults, including complement (C1QA, C3), cytokine/chemokine (IL-1B, IL-15, IL-6, IL-18, CCR2, CCL5), Nuclear factor-κB (NF-κB)-associated (REL, TNFRSF11, Chuk-Ikbkb-Ikbkg), and interferon-associated [IFN-α, IFNB1, JAK2, Ifnar, IFNG, IFN-α/β, interferon regulatory transcription factor 3 (IRF3)-IRF7, STING-TRAF3-TBK1, and JAK1/2]. Expression of several anti-inflammatory mediators, including APOE (apolipoprotein E), TGFBR1 (transforming growth factor receptor 1), and CORT (corticosterone), were further inhibited in Aged-TBI mice rather than in Adult-TBI mice. Adult-TBI mice shared the activation of IL-15 and inhibition of PSEN1 with Aged-TBI, though not to the same degree. Unique to Adult-TBI mice was the activation of MMP9, CYP3A4, WWP1, and PDGFC (platelet-derived growth factor C). Collectively, neuropathology-associated gene expression after TBI was strongly influenced by age.
Exacerbated cortical inflammation in aged mice 7 dpi
The final comparison of the cortical RNA 7 dpi was between Aged-TBI and Adult-TBI mice. Figure 4A shows a volcano plot (–log10 p value vs log2 fold change) of significant DEGs between Aged-TBI and Adult-TBI mice. There were 63 DEGs between Aged-TBI and Adult-TBI mice (61 increased, 2 decreased). Red dots are DEGs increased by age with TBI (e.g., Cxcl10, Stat1, Psmb8, Ccl5, Cxcl16), and blue dots are DEGs decreased by age with TBI (i.e., Camk4, Cx3cl1). The heatmap highlights genes in cell-specific categories based on predicted cell expression from the Barres Lab Brain-Seq dataset (www.brainrnaseq.org). These genes were differentially expressed between Aged-TBI and Adult-TBI mice (Fig. 4B, normalized counts in supplemental data). The majority of DEGs in all cell types were most highly expressed in the Aged-TBI group compared with all other experimental groups (Fig. 4B). The Venn diagram (Fig. 4C) shows genes that were (1) TBI dependent, not enhanced with age (1 gene); (2) TBI dependent in both groups and enhanced with age (9 genes); and (3) genes that were uniquely enhanced in Aged-TBI mice (54 genes). Of the genes amplified by age with TBI, those with the highest fold changes are shown in Figure 4D. These include genes associated with chemokines (Cxcl10, Ccl12, Ccl5, Ccl2, Cxcl16, Ccr2), complement components (C3), interferons (Irf7, Stat1, Ifi27I2a, Sp100), innate immunity (Psmb8, Cybb, Clec7a, Ncf1), antigen presentation (H2-Eb1), and injury (Cd44, Tspo). The majority of DEGs with the top fold changes were expressed in microglia (www.brainrnaseq.org). Overall, there was exaggerated inflammation and neuropathological gene expression that was associated with increased microglial gene expression in aged mice 7 dpi compared with adult mice.
Figure 4.

Exacerbated cortical inflammation in aged mice 7 dpi. Adult (2 months of age) and aged (18 months of age) male C57BL/6 mice were subjected to midline fluid percussion injury (TBI) or left as uninjured CONs, and mRNA copy number was determined using a NanoString nCounter Neuropathology panel (n = 3), as in Figure 3. A, Volcano plot of significantly differentially expressed genes (p < 0.05) compared between Aged-TBI and Adult-TBI mice. Genes in red were increased by age and genes in blue were decreased by age with TBI. B, Heatmap of genes that were significantly enhanced by age compared between Aged-TBI and Adult-TBI mice grouped by predicted cell expression (Barres Lab Brain-Seq). C, Venn diagram of significantly differentially expressed genes in Adult-TBI versus Adult-Control and Aged-TBI versus Adult-TBI. Genes were grouped as follows: (1) TBI dependent, not influenced by age (13 genes); and (2) TBI dependent, age enhanced (29 genes), and TBI dependent, uniquely enhanced with age (121 genes). D, Top genes (by fold change) with functional annotation that were amplified in Aged-TBI compared with Adult-TBI mice. Positive z score indicates increase by age, and negative z score indicates decrease by age with the TBI compared with the Adult-TBI group.
Amplified cortical inflammation in aged mice associated with interferon signaling 7 dpi
IPA was used to assess significant canonical pathways and master regulators modulated by age within TBI. Significant pathways activated or inhibited by age with TBI are shown (Fig. 5A). The canonical pathways enhanced with age at 7 dpi notably included neuroinflammation signaling, pathogen induced cytokine storm signaling, phagosome formation, acute phase response, and wound healing signaling, all of which indicate an amplified injury-associated inflammatory response in aged mice 7 dpi. Furthermore, activation of pathways associated with PKR in interferon induction and antiviral response, cross talk between dendritic cells (DCs) and natural killer (NK) cells, DC maturation, role of PRRs in recognition of bacteria and viruses, and tumor microenvironment pathway were also enhanced with age at 7 dpi. There was evidence of metabolic differences in the groups with activation of production of NO and ROS in macrophages and insulin secretion signaling pathway. Other inflammatory pathways had enhanced activation, including IL-13, IL-8, and TREM1 (triggering receptor expressed on myeloid cells 1) signaling. Consistent with earlier comparisons, IPA-generated master regulators revealed that interferon-driven inflammation was robustly enhanced with age 7 dpi. For example, Aged-TBI mice had enhanced activation of IFN-α, Ifnar, IFN type I receptor, MAVS, IFNG, IFNB1, IFN-α/β, IRF3-IRF7, STING-TRAF3-TBK1, IRF7, IRF1, STING1, Stat2, and Stat1 dimer (Fig. 5B). Overall, many of the significant DEGs between Aged-TBI and Adult-TBI mice at 7 dpi were driven by amplified IFN signaling after TBI.
Figure 5.

Amplified cortical inflammation in aged mice that was associated with interferon signaling 7 dpi. Adult (2 months of age) and aged (18 months of age) male C57BL/6 mice were subjected to midline fluid percussion injury (TBI) or left as uninjured CONs. mRNA copy number was determined using a NanoString nCounter Neuropathology panel (n = 3), as in Figure 3. A, IPA of significant canonical pathways (z score) activated or inhibited by TBI in Aged-TBI mice compared with Adult-TBI mice. B, IPA of significant master regulators (z score) amplified by age in TBI mice.
Prolonged cortical astrocyte and microglia morphologic restructuring and amplified cortical inflammation in aged mice 30 dpi
To determine the chronic effects of diffuse TBI, astrocyte (GFAP+) and microglia (Iba-1+) labeling was assessed in the cortex 30 dpi in adult and aged mice. Adult (2 months of age) and aged (18 months of age) male C57BL/6 mice were subjected to midline fluid percussion injury (TBI) or left as uninjured controls (CONs). Representative images from the SS-Ctx show that there was increased GFAP+ labeling of astrocytes in both adult and aged mice 7 dpi (Fig. 6A). Thresholded black and white images (from insets) highlight differences in morphologic restructuring of astrocytes in the cortex 30 dpi between adult and aged mice (Fig. 6B). There was a main effect of TBI on GFAP+ labeling in the cortex (Fig. 6C; F(1,14) = 9.7, p = 0.0076). The increased GFAP+ labeling in the cortex 30 dpi, however, was not influenced by age.
Figure 6.
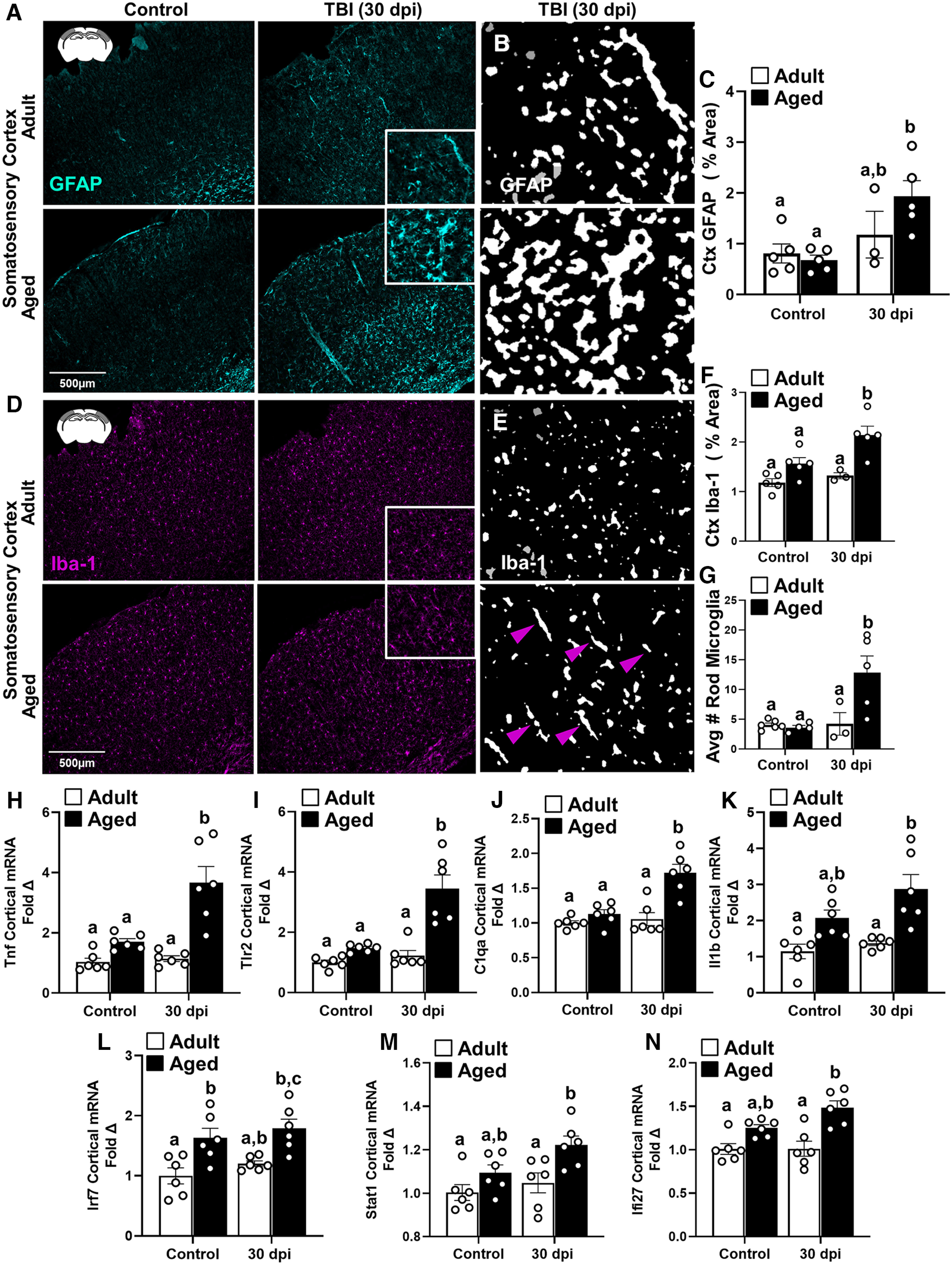
Prolonged cortical astrocyte and microglia morphology differences and amplified cortical inflammatory gene expression in aged mice 30 dpi. Adult (2 months of age) and aged (18 months of age) male C57BL/6 mice were subjected to midline fluid percussion injury (TBI) or left as uninjured CONs, and, at 30 dpi, GFAP+ and Iba-1+ labeling was determined in the SS-Ctx of adult and aged mice. A, Representative images of GFAP+ labeling in the SS-Ctx. B, Thresholded black and white images of GFAP+ labeling in the SS-Ctx of adult and aged mice 30 dpi. C, Percentage of the area of GFAP+ labeling in the SS-Ctx. D, Representative images of Iba-1+ labeling in the SS-Ctx. E, Thresholded black and white images of Iba-1+ labeling in the SS-Ctx of adult and aged mice 30 dpi. F, Percentage of the area of Iba-1+ labeling in the SS-Ctx. G, Quantification of the average number of Iba-1+ rod microglia in SS-Ctx 30 dpi. In a parallel experiment, RNA was isolated from the cortex of adult and aged mice 30 dpi. H–N, Relative IFN/inflammatory-related gene expression was determined by qPCR (n = 6): Tnf (H), Tlr2 (I), C1qa (J), Il-1b (K), Irf7 (L), Stat1 (M), and Ifi27 (N). Bars represent the mean ± SEM, and individual data points are provided. Means with different letters (e.g., a, b, c) indicate significant post hoc differences between groups (p < 0.05). Groups with the same letter or letters are not significantly different from each other.
For microglia, representative images from SS-Ctx are shown (Fig. 6D), and thresholded black and white cortical images (Fig. 6D, insets) highlight differences in morphologic restructuring of microglia in the cortex 30 dpi between adult and aged mice (Fig. 6E). Morphologic restructuring of microglia (Fig. 6F; percentage of the area of Iba-1+ labeling) was increased by TBI (F(1,14) = 7.0, p = 0.0196) and age (F(1,14) = 19.6, p = 0.0006). Post hoc analysis confirmed that Aged-TBI mice had the highest percentage of the area of Iba-1+ labeling 30 dpi compared with all other groups (p < 0.05). In addition, the average number of Iba-1+ rod-shaped microglia at 30 dpi (Fig. 6E, arrows in insets) was increased by TBI and age (interaction: F(1,14) = 6.7, p = 0.0218; Fig. 6G). Post hoc analysis confirmed that Aged-TBI mice had the highest average number of Iba-1+ rod microglia compared with all other groups (p < 0.05). Overall, there was elevated microgliosis and structural diversity of microglia in the Aged-TBI mice 30 dpi compared with adults.
Next, relative expression of inflammation-related genes (7 total) were determined in the cortex 30 dpi. Overall, the expression of these genes was influenced by age and TBI. For example, Il1b (Fig. 6K) and Stat1 (Fig. 6M) were enhanced by age (F(1,20) = 22.8, p = 0.0001; F(1,20) = 11.0, p = 0.0035) and by injury (F(1,20) = 4.080, p = 0.057; F(1,20) = 4.6, p = 0.0448). Age alone enhanced Irf7 (Fig. 6L; F(1,20) = 21.2, p = 0.0002) and Ifi27Ia (Fig. 6N; F(1,20) = 27.4, p < 0.0001). Relative gene expression of Tnf, Tlr2, and C1qa was increased by TBI and amplified by age 30 dpi [Fig. 6H–J; interactions: Tnf (F(1,20) = 10.5, p = 0.0041), Tlr2 (F(1,20) = 12.3, p = 0.0022), C1qa (F(1,20) = 10.4, p = 0.0043)]. Post hoc analysis confirmed that expression of these genes was highest in Aged-TBI mice 30 dpi compared with all other groups (p < 0.05). Collectively, these data indicate that amplified NF-κB-mediated pattern recognition receptor, and complement-associated inflammation in aged mice persisted 30 dpi.
STING agonist DMXAA amplified cortical gliosis and inflammatory gene expression in adult mice 7 dpi
We interpret the NanoString mRNA data to indicate that there was amplified activation of type I IFN signaling in aged mice compared with adult mice 7 dpi. Therefore, DMXAA, a STING agonist, was used to determine whether enhancing IFN signaling in adult mice with TBI would augment gliosis and neuropathological gene expression in the cortex. Here, adult (2 months) male C57BL/6 mice were administered vehicle or DMXAA (25 mg/kg) intraperitoneally 1 h before midline fluid percussion injury (TBI or control) and then daily for 6 d (Fig. 7A). Gliosis and cortical mRNA expression were determined 7 dpi (Fig. 7A). First, time to self-right was determined immediately after mFPI-mediated TBI. Mice injected with the STING agonist (1 h before injury) had longer self-righting times after TBI compared with vehicle controls (Fig. 7B; p = 0.023).
Figure 7.
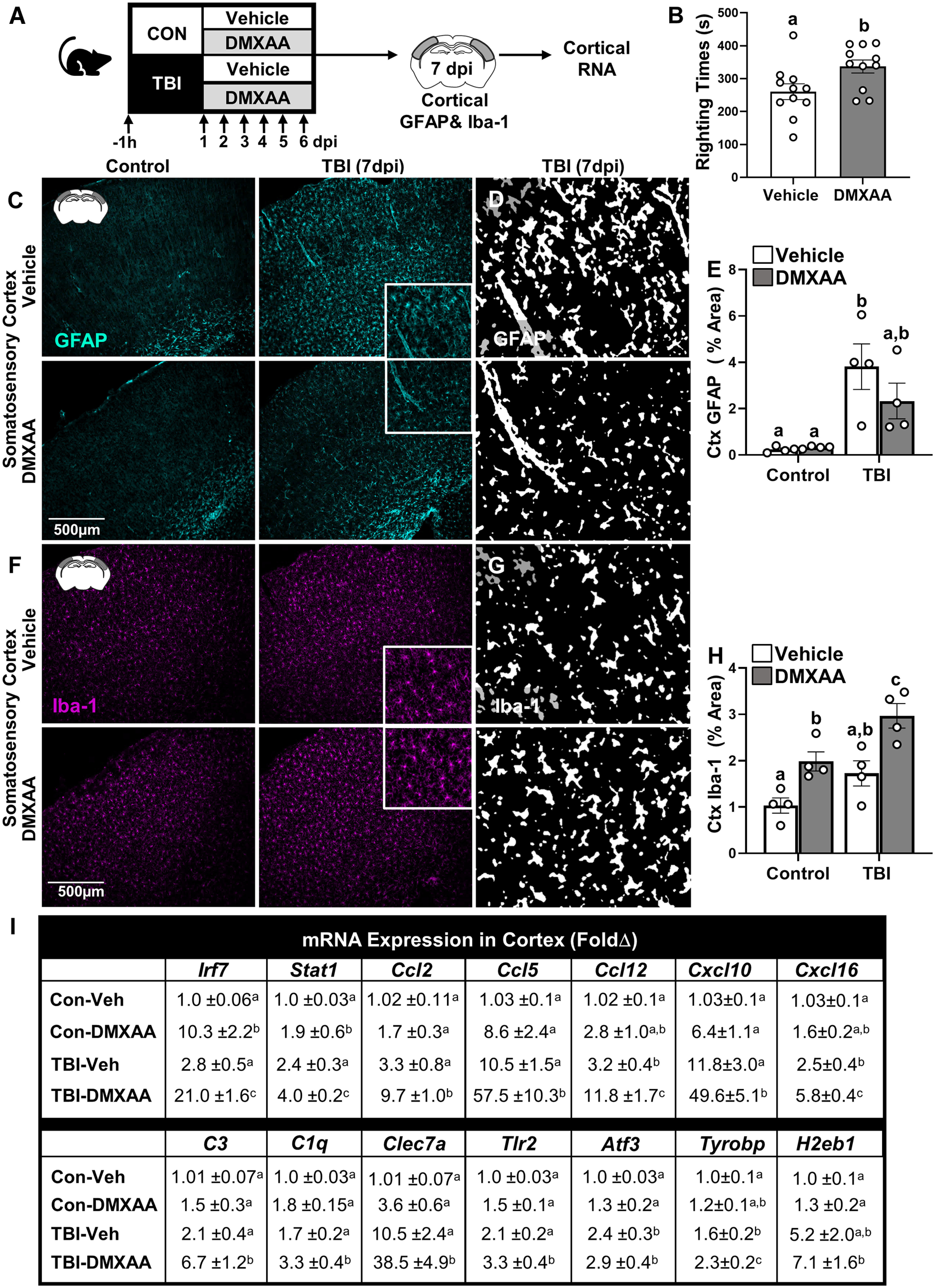
STING agonist DMXAA amplified cortical gliosis and inflammatory gene expression in adult mice 7 dpi. A, Adult (2 months of age) male C57BL/6 mice were administered vehicle or DMXAA (25 mg/kg) intraperitoneally 1 h before midline fluid percussion injury (TBI) and then again daily up to 6 dpi in control and TBI mice. B, Self-righting time (in seconds) was determined immediately after injury. At 7 dpi, mice were perfused, and the brain was fixed, flash frozen, and sectioned. GFAP+ and Iba-1+ labeling was determined in the SS-Ctx and HPC 7 dpi. C, Representative images of GFAP+ cells in the SS-Ctx. D, Thresholded black and white images of GFAP+ labeling in the SS-Ctx of adult and aged mice 7 dpi. E, Percentage of the area of GFAP+ labeling in the SS-Ctx. F, Representative images of Iba-1+ cells in the SS-Ctx. G, Thresholded black and white images of Iba-1+ labeling in the SS-Ctx of adult and aged mice 7 dpi. H, Percentage of the area of Iba-1+ labeling in the SS-Ctx. In a parallel experiment, adult mice were treated as above, and RNA was isolated from the cortex 7 dpi. Relative gene expression was determined by qPCR (n = 6). I, Genes associated with IFNs (Cxcl10, Irf7, Stat1, H2eb1), chemokines (Ccl2, Ccl5, Ccl12, Cxcl16), complement (C3, C1qa), and innate immune (Tlr2, Clec7a, Tyrobp) responses were determined in the cortex 7 dpi. Values represent the mean ± SEM. Means with different letters (e.g., a, b, c) indicate significant post hoc differences between groups (p < 0.05). Groups with the same letter or letters are not significantly different from each other.
Next, gliosis was assessed 7 dpi. Representative images from the SS-Ctx show that there was increased GFAP+ labeling of astrocytes 7 dpi in both vehicle-treated and DMXAA-treated mice (Fig. 7C). Thresholded black and white images (from insets) highlight differences between vehicle-treated and DMXAA-treated astrocyte morphologic restructuring in the cortex 7 dpi (Fig. 7D). GFAP+ labeling (percentage of area) in the cortex was increased by TBI (F(1,12) = 19.9, p = 0.0008), but this increase was DMXAA independent (Fig. 7E). For microglia morphologic restructuring, representative images from the SS-Ctx show that there was increased Iba-1+ labeling of microglia in both vehicle-treated and DMXAA-treated mice 7 dpi (Fig. 7F). Thresholded black and white images (Fig. 7G, insets) highlight differences in microglia morphologic restructuring between vehicle-treated and DMXAA-treated mice in the cortex 7 dpi (Fig. 7G). Iba-1+ labeling (percentage of area) of microglia in the cortex (Fig. 7H) was increased by TBI (F(1,12) = 13.3, p = 0.0034) and by DMXAA (F(1,12) = 22.7, p = 0.0005). Post hoc analysis confirmed that TBI-DMXAA mice had the highest level of Iba-1+ labeling (percentage of area) in the SS-Ctx compared with all other groups (p < 0.05). Overall, DMXAA treatment concurrent with TBI did not affect astrocyte structure but did produce the highest degree of microgliosis in the cortex 7 dpi.
Neuroinflammatory status 7 dpi was assessed in the cortex by determining the relative expression of several inflammatory genes. Fourteen genes were selected based on the results of the NanoString nCounter Neuropathology analysis (Fig. 4D). These TBI-associated genes were enhanced as a function of age and include IFN-associated (Irf7, Stat1), complement (C3, C1qa), chemokine (Ccl2, Ccl12, Ccl5, Cxcl10, Cxcl16), pattern recognition receptors (Clec7a, Tlr2), injury associated (Atf3, Tyrobp), and antigen presentation effector (H2-Eb1). Figure 7I shows the average (±SEM) fold changes of these genes for each of the experimental groups. There was a significant effect of TBI on the expression of all 14 genes (F(1,15), F = 17.71, p < 0.05, for all). DMXAA administration amplified the expression of Irf7, Stat1, Tlr2, Ccl2, Ccl12, C3, Ccl5, Cxcl10, Cxcl16, and Clec7a (interactions: F(1,15), F = 3.417, all p < 0.05). DMXAA also enhanced the expression of Tyrobp, C1qa, H2-Eb1, and Atf3. Post hoc analysis confirmed that DMXAA-treated mice had enhanced expression of inflammatory genes compared with vehicle-treated mice 7 dpi. Collectively, the stimulation of IFN signaling after TBI with DMXAA administration exacerbated microgliosis and inflammation in the cortex 7 dpi.
Discussion
This study investigated mechanisms that drive impaired recovery with age after diffuse TBI. We show that exaggerated cortical inflammation and gliosis in the aged brain after injury was associated with IFN signaling. Furthermore, augmenting IFN signaling using a STING agonist, DMXAA, concomitant with TBI enhanced cortical inflammation and microgliosis in adult mice. Overall, amplified IFN signaling in the aged brain after diffuse TBI was associated with prolonged cortical inflammation and gliosis.
One point for discussion is that, contrary to our expectation, cognitive deficits were not worse in aged mice after TBI. Deficits in NOR/NOL memory performance 7 dpi were independent of age. Aged-Control mice had memory impairments, however, that may represent a “floor effect” and obscured “worsened” cognitive impairment in Aged-TBI mice. Nonetheless, exacerbated cognitive impairment (spatial memory) was reported in aged rats 7 dpi (Hamm et al., 1992). Collectively, both age groups had cognitive impairment 7 dpi, and more sensitive testing may reveal worsened cognitive impairment in Aged-TBI mice.
A relevant finding of this study is that aged mice had enhanced gliosis 7 dpi. There was an enhanced percentage of the area of GFAP+ astrocytes in both cortex and hippocampus of aged mice after TBI. This increased astrogliosis in the cortex of Aged-TBI mice corroborates CCI studies showing progressive astrogliosis in the cortex and hippocampus of aged mice (Early et al., 2020). Another study reported increased reactive phenotypes of microglia in aged mice 24 h after CCI (Kumar et al., 2013). We extended these data by showing an enhanced percentage of the area of Iba-1+ microglia in aged mice 7 and 30 d after diffuse TBI. Based on previous work, a higher percentage of the area of Iba-1+ microglia represents morphologic restructuring, including hypertrophy of processes and increased soma size (Norden et al., 2016). Additionally, there were more rod-shaped microglia in the cortex. Rod-shaped microglia were detected 7 dpi in both adult and aged mice (data not shown), but only aged mice had rod-shaped microglia at 30 dpi. We previously reported that rod-shaped microglia in the cortex of adult mice 7 dpi aligned with the apical dendrites of damaged neurons (Witcher et al., 2018). Moreover, rod-shaped microglia are present in neurodegenerative diseases, including Alzheimer's disease, chronic traumatic encephalopathy, and multiple sclerosis (Wierzba-Bobrowicz et al., 2002; Bachstetter et al., 2017; Madathil et al., 2018; van Wageningen et al., 2019). Therefore, the detection of rod-shaped microglia in aged mice 30 dpi may reflect persistent neuropathology. The enhanced microglial morphologic restructuring and corresponding increased inflammatory gene expression (Tnf, Tlr2, and C1qa) is interpreted to indicate prolonged microglial activation in the aged brain after TBI. Overall, amplified gliosis after TBI in aged mice was accompanied by persistent inflammation and rod-shaped microglia 30 dpi.
Several novel findings from the RNA analysis highlight the influence of age on cortical inflammation 7 dpi. For instance, there were age-dependent increases in gene expression of complement, chemokine, interferon, and innate immunity-related transcripts. Consistent with these data, there was activation of several master regulators in aged mice after TBI, including complement, cytokine/chemokine, NF-κB mediated, and interferon associated. Previously, focal brain injury in aged mice caused the overactivation of complement components, which was associated with synapse loss and cognitive impairment (Krukowski et al., 2018). Enhanced complement activation in the aged brain after TBI may mediate synapse degradation (Stevens et al., 2007) and astrocyte reactivity (Asano et al., 2020). In terms of the enhanced chemokine signaling, significant recruitment of peripheral leukocytes was undetected in adult mice after diffuse TBI (Witcher et al., 2018). Nonetheless, recruitment of CCR2+ macrophages was linked to amplified inflammation and memory deficits in aged mice after focal TBI (Morganti et al., 2016). Thus, it is plausible that the recruitment of leukocytes in the aged brain (or meninges) after diffuse TBI contributes to enhanced inflammation. We did not, however, address this experimentally. Overall, the activation of numerous neuroinflammatory pathways was more pronounced in aged mice after TBI.
Aged-TBI mice also had a mRNA profile consistent with increased production of NO and ROS in macrophages, including the expression of Cybb, Irf8, Mapk3, Ncf1, Spi1, Tlr2, and Tnfrsf1b. These reactive mediators play a significant role in secondary injury following TBI and further promote cellular damage (Khatri et al., 2018; Ma et al., 2018). Other studies using focal injury showed that Aged-TBI mice had higher levels of NOX2 and ROS production after CCI that corresponded with worsened behavioral recovery (Ritzel et al., 2019). Thus, interventions aimed at reducing NO and ROS may decrease inflammation and functional deficits after TBI in aging.
mRNA analysis also showed several pathways that were suppressed after TBI in aged mice but not in adults. PPARs and RXRa are key transcription factors that inhibit NF-κB and IFN signaling (Zhao et al., 2011; Ma et al., 2014) and serve as a therapeutic target for TBI-related pathology (Yi et al., 2008; Qi et al., 2010; Thal et al., 2011; Deng et al., 2020). Furthermore, PTEN, a tumor suppressor, improved neuronal survival after TBI (Kitagishi and Matsuda, 2013; Goh et al., 2014). There was also further suppression of anti-inflammatory master regulators IL-10, TGFBR1, CORT, and APOE. Overall, there was enhanced suppression of key regulatory and neuroprotective pathways in the aged brain after TBI.
A key finding of this study was that IFN (type I and II) signaling pathways were prominently enhanced in aged mice after TBI compared with adults. Myriad central mediators of IFN signaling (IFNAR1, IFN-α/β/γ, IRFs 1/3/7, STING1, and Stat1) were enhanced in the Aged-TBI mice compared with Adult-TBI mice. Moreover, the activation of IFN-γ, which is produced primarily by T cells and NK cells, may reflect enhanced infiltration of leukocytes in the brain or meninges (Louveau et al., 2015). Additionally, several reports indicate that type I IFN signaling after both diffuse and focal TBI is critical to the transition from acute to chronic inflammation in adult mice (Abdullah et al., 2018; Barrett et al., 2020; Witcher et al., 2021; Fritsch et al., 2022). Our current data complement work showing enhanced IFN signaling mediators (Irf7, pSTAT1, and cGAS) 24 h after CCI in aged mice (Barrett et al., 2021). While it is unclear how the IFN pathway is activated in aged mice after TBI, a recent study of CCI in adult mice reported that the activation of cGAS–STING signaling was dependent on mitochondrial DNA (Fritsch et al., 2022). The current study confirms that IFN signaling is associated with worsened outcomes following diffuse TBI in aged mice.
Building on studies that correlate TBI and IFN signaling, our study demonstrated a causative relationship between augmented type I IFN signaling and amplified microgliosis and cortical inflammation following TBI. In this study, repeated intraperitoneal injections of the STING agonist DMXAA increased type I IFN signaling in the brain, resulting in exaggerated microglia restructuring in the cortex and HPC 7 dpi. DMXAA also amplified expression of myriad inflammatory genes in the cortex of adult mice after TBI. Thus, enhanced type I IFN signaling post-TBI caused enhanced microglia activation in the cortex 7 dpi. One caveat is that DMXAA was administered peripherally, which may have contributed to the longer self-righting times and neuroinflammation detected. Studies in stroke and cancer models report that DMXAA penetrates into the CNS (Yung et al., 2014; Kundu et al., 2022). Notably, DMXAA alone increased IFN signaling in the adult brain with increased expression of Irf7, Cxcl10, Cxcl16, Stat1, and Clec7a. These findings indicate that there is increased IFN signaling in the brain (direct or indirect) after intraperitoneal administration of DMXAA. Nonetheless, only DMXAA-treated TBI mice had amplified inflammatory gene expression and microgliosis in the cortex. Overall, STING augmentation coupled with TBI promoted significant exacerbation of cortical inflammation with amplified expression of Irf7, Stat1, Tlr2, Ccl2,5,12, C3, Cxcl10 and Cxcl16, and Clec7a. These genes markedly overlapped with the genes that were age enhanced after TBI.
Our study provides compelling evidence that amplified IFN signaling in aging is associated with enhanced cortical inflammation and reactive microgliosis. While there was amplified astrogliosis in the cortex of aged mice 7 dpi, this was not recapitulated in DMXAA_treated Adult-TBI mice. This finding is interpreted to indicate that astrogliosis in aged mice after TBI is age dependent, but not type I IFN dependent. There are many age-dependent differences in the transcriptional profile of astrocytes (Norden et al., 2016; Palmer and Ousman, 2018; O'Neil et al., 2022) that likely account for the enhanced astrogliosis after TBI. For instance, astrocytes are activated by danger-associated molecular patterns (HMGB1, ROS, and NO) after injury, which do not require IFN signaling (Burda et al., 2016). Collectively, augmenting type I IFN signaling in adult mice concomitant with TBI paralleled the amplified response to TBI in aged mice.
In summary, age is an important factor in determining the extent of inflammation and gliosis after TBI. Here, we provide novel data showing exaggerated and persistent neuroinflammation in aged mice after diffuse TBI. Moreover, our data indicate type I IFNs as critical mediators of amplified microgliosis and persistent inflammation in aged mice following diffuse TBI. Future studies will use IFN antagonists in aged mice with TBI to determine the extent to which this intervention reduces amplified and prolonged inflammation. Therefore, attenuating IFN signaling postinjury may be a promising therapeutic target to ameliorate persistent neuroinflammation, especially in the aged.
Footnotes
This research was supported by National Institute of Health Grants R01-AG-051902 and R01-NS-118037 to J.P.G. L.M.W., J.M.P., and A.C.D. were supported by The Ohio State University (OSU) Distinguished University Fellowships. S.M.O. was supported by an OSU Dean's Distinguished University Fellowship. C.E.B. was supported by the OSU College of Medicine Samuel J. Roessler Scholarship. Z.M.T. was supported by National Institute of Neurological Disorders and Stroke (NINDS) Neuroimmunology Training Grant T32-NS-105864. In addition, this work was supported by NINDS P30 Core Grant P30-NS-045758 to the Center for Brain and Spinal Cord Repair. We thank Dr. Paolo Fadda at The Ohio State University Comprehensive Cancer Center Genomics Shared Resource (under Grant P30-CA-016058), and the Chronic Brain Injury Program, Discovery Themes Initiative at The Ohio State University.
The authors declare no competing financial interests.
References
- Abdullah A, Zhang M, Frugier T, Bedoui S, Taylor JM, Crack PJ (2018) STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J Neuroinflammation 15:323. 10.1186/s12974-018-1354-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes M, Biala G (2012) The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process 13:93–110. 10.1007/s10339-011-0430-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Hayashi Y, Iwata K, Okada-Ogawa A, Hitomi S, Shibuta I, Imamura Y, Shinoda M (2020) Microglia-astrocyte communication via C1q contributes to orofacial neuropathic pain associated with infraorbital nerve injury. Int J Mol Sci 21:6834. 10.3390/ijms21186834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Ighodaro ET, Hassoun Y, Aldeiri D, Neltner JH, Patel E, Abner EL, Nelson PT (2017) Rod-shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol Aging 52:98–105. 10.1016/j.neurobiolaging.2016.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JP, Henry RJ, Ann Shirey K, Doran SJ, Makarevich OD, Ritzel RR, Meadows VA, Vogel SN, Faden AI, Stoica BA, Loane DJ (2020) Interferon-β plays a detrimental role in experimental traumatic brain injury by enhancing neuroinflammation that drives chronic neurodegeneration. J Neurosci 40:2357–2370. 10.1523/JNEUROSCI.2516-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JP, Knoblach SM, Bhattacharya S, Gordish-Dressman H, Stoica BA, Loane DJ (2021) Traumatic brain injury induces cGAS activation and type I interferon signaling in aged mice. Front Immunol 12:710608. 10.3389/fimmu.2021.710608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray CE, Witcher KG, Adekunle-Adegbite D, Ouvina M, Witzel M, Hans E, Tapp ZM, Packer J, Goodman E, Zhao F, Chunchai T, O'Neil S, Chattipakorn SC, Sheridan J, Kokiko-Cochran ON, Askwith C, Godbout JP (2022) Chronic cortical inflammation, cognitive impairment, and immune reactivity associated with diffuse brain injury are ameliorated by forced turnover of microglia. J Neurosci 42:4215–4228. 10.1523/JNEUROSCI.1910-21.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickler T, Morton P, Hazy A, Theus MH (2017) Age-dependent responses following traumatic brain injury. In: Traumatic brain injury: pathobiology, advanced diagnostics and acute management (Gorbunov NV, Long JB, eds), pp 21–38. London: IntechOpen. [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV (2016) Astrocyte roles in traumatic brain injury. Exp Neurol 275:305–315. 10.1016/j.expneurol.2015.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Jiang X, Deng X, Chen H, Xu J, Zhang Z, Liu G, Yong Z, Yuan C, Sun X, Wang C (2020) Pioglitazone ameliorates neuronal damage after traumatic brain injury via the PPARγ/NF-κB/IL-6 signaling pathway. Genes Dis 7:253–265. 10.1016/j.gendis.2019.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denninger JK, Smith BM, Kirby ED (2018) Novel object recognition and object location behavioral testing in mice on a budget. J Vis Exp (141):e58593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early AN, Gorman AA, Van Eldik LJ, Bachstetter AD, Morganti JM (2020) Effects of advanced age upon astrocyte-specific responses to acute traumatic brain injury in mice. J Neuroinflammation 17:115. 10.1186/s12974-020-01800-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP (2014) Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 76:575–584. 10.1016/j.biopsych.2013.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Skendelas JP, Moussa DN, Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP (2015) Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J Neurotrauma 32:127–138. 10.1089/neu.2014.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch LE, Ju J, Gudenschwager Basso EK, Soliman E, Paul S, Chen J, Kaloss AM, Kowalski EA, Tuhy TC, Somaiya RD, Wang X, Allen IC, Theus MH, Pickrell AM (2022) Type I interferon response is mediated by NLRX1-cGAS-STING signaling in brain injury. Front Mol Neurosci 15:852243. 10.3389/fnmol.2022.852243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K (2015) Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol 77:987–995. 10.1002/ana.24396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, Yaffe K (2018) Mild TBI and risk of Parkinson disease: a Chronic Effects of Neurotrauma Consortium Study. Neurology 90:e1771–e1779. 10.1212/WNL.0000000000005522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh CP, Putz U, Howitt J, Low LH, Gunnersen J, Bye N, Morganti-Kossmann C, Tan SS (2014) Nuclear trafficking of Pten after brain injury leads to neuron survival not death. Exp Neurol 252:37–46. 10.1016/j.expneurol.2013.11.017 [DOI] [PubMed] [Google Scholar]
- Hamm RJ, White-Gbadebo DM, Lyeth BG, Jenkins LW, Hayes RL (1992) The effect of age on motor and cognitive deficits after traumatic brain injury in rats. Neurosurgery 31:1072–1077. 10.1227/00006123-199212000-00013 [DOI] [PubMed] [Google Scholar]
- Kelley BJ, Lifshitz J, Povlishock JT (2007) Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol 66:989–1001. 10.1097/NEN.0b013e3181588245 [DOI] [PubMed] [Google Scholar]
- Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK (2018) Oxidative stress: major threat in traumatic brain injury. CNS Neurol Disord Drug Targets 17:689–695. 10.2174/1871527317666180627120501 [DOI] [PubMed] [Google Scholar]
- Kitagishi Y, Matsuda S (2013) Diets involved in PPAR and PI3K/AKT/PTEN pathway may contribute to neuroprotection in a traumatic brain injury. Alzheimers Res Ther 5:42. 10.1186/alzrt208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukowski K, Chou A, Feng X, Tiret B, Paladini MS, Riparip LK, Chaumeil MM, Lemere C, Rosi S (2018) Traumatic brain injury in aged mice induces chronic microglia activation, synapse loss, and complement-dependent memory deficits. Int J Mol Sci 19:3753. 10.3390/ijms19123753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ (2013) Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging 34:1397–1411. 10.1016/j.neurobiolaging.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ (2016) NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun 58:291–309. 10.1016/j.bbi.2016.07.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu N, Kumar A, Corona C, Chen Y, Seth S, Karuppagounder S, Ratan R (2022) A STING agonist preconditions against ischaemic stroke via an adaptive antiviral type 1 interferon response. Brain Commun 4:fcac133. 10.1093/braincomms/fcac133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz J, Kelley BJ, Povlishock JT (2007) Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J Neuropathol Exp Neurol 66:218–229. 10.1097/01.jnen.0000248558.75950.4d [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Rowe RK, Griffiths DR, Evilsizor MN, Thomas TC, Adelson PD, McIntosh TK (2016) Clinical relevance of midline fluid percussion brain injury: acute deficits, chronic morbidities and the utility of biomarkers. Brain Inj 30:1293–1301. 10.1080/02699052.2016.1193628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. 10.1038/nature14432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madathil SK, Wilfred BS, Urankar SE, Yang W, Leung LY, Gilsdorf JS, Shear DAE (2018) Microglial activation following closed-head concussive injury is dominated by pro-inflammatory M-1 type. Front Neurol 9:964. 10.3389/fneur.2018.00964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Liu S-Y, Razani B, Arora N, Li B, Kagechika H, Tontonoz P, Núñez V, Ricote M, Cheng G (2014) Retinoid X receptor α attenuates host antiviral response by suppressing type I interferon. Nat Commun 5:5494. 10.1038/ncomms6494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MW, Wang J, Dhandapani KM, Wang R, Brann DW (2018) NADPH oxidases in traumatic brain injury - promising therapeutic targets? Redox Biol 16:285–293. 10.1016/j.redox.2018.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez de la Plata CD, Hart T, Hammond FM, Frol AB, Hudak A, Harper CR, O'Neil-Pirozzi TM, Whyte J, Carlile M, Diaz-Arrastia R (2008) Impact of age on long-term recovery from traumatic brain injury. Arch Phys Med Rehabil 89:896–903. 10.1016/j.apmr.2007.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti JM, Riparip L-K, Chou A, Liu S, Gupta N, Rosi S (2016) Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. J Neuroinflammation 13:80. 10.1186/s12974-016-0547-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccigrosso MM, Ford J, Benner B, Moussa D, Burnsides C, Fenn AM, Popovich PG, Lifshitz J, Walker FR, Eiferman DS, Godbout JP (2016) Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun 54:95–109. 10.1016/j.bbi.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden DM, Trojanowski PJ, Walker FR, Godbout JP (2016) Insensitivity of astrocytes to interleukin 10 signaling following peripheral immune challenge results in prolonged microglial activation in the aged brain. Neurobiol Aging 44:22–41. 10.1016/j.neurobiolaging.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil SM, Hans EE, Jiang S, Wangler LM, Godbout JP (2022) Astrocyte immunosenescence and deficits in interleukin 10 signaling in the aged brain disrupt the regulation of microglia following innate immune activation. Glia 70:913–934. 10.1002/glia.24147 [DOI] [PubMed] [Google Scholar]
- Palmer AL, Ousman SS (2018) Astrocytes and aging. Front Aging Neurosci 10:337. 10.3389/fnagi.2018.00337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Jacob A, Wang P, Wu R (2010) Peroxisome proliferator activated receptor-y and traumatic brain injury. Int J Clin Exp Med 3:283–292. [PMC free article] [PubMed] [Google Scholar]
- Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, Fossati S (2018) Traumatic brain injury and Alzheimer's disease: the cerebrovascular link. EBioMedicine 28:21–30. 10.1016/j.ebiom.2018.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, Doran SJ, Glaser EP, Meadows VE, Faden AI, Stoica BA, Loane DJ (2019) Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging 77:194–206. 10.1016/j.neurobiolaging.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RK, Griffiths DR, Lifshitz J (2016) Midline (central) fluid percussion model of traumatic brain injury. Methods Mol Biol 1462:211–230. 10.1007/978-1-4939-3816-2_13 [DOI] [PubMed] [Google Scholar]
- Stein MB, et al. (2019) Risk of posttraumatic stress disorder and major depression in civilian patients after mild traumatic brain injury: a TRACK-TBI study. JAMA Psychiatry 76:249–258. 10.1001/jamapsychiatry.2018.4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Taylor SE, Morganti-Kossmann C, Lifshitz J, Ziebell JM (2014) Rod microglia: a morphological definition. PLoS One 9:e97096. 10.1371/journal.pone.0097096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal SC, Heinemann M, Luh C, Pieter D, Werner D, Engelhard K (2011) Pioglitazone reduces secondary brain damage after experimental brain trauma by PPAR-y-independent mechanisms. J Neurotrauma 28:983–993. 10.1089/neu.2010.1685 [DOI] [PubMed] [Google Scholar]
- Todd BP, Chimenti MS, Luo Z, Ferguson PJ, Bassuk AG, Newell EA (2021) Traumatic brain injury results in unique microglial and astrocyte transcriptomes enriched for type I interferon response. J Neuroinflammation 18:151. 10.1186/s12974-021-02197-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wageningen TA, Vlaar E, Kooij G, Jongenelen C, AM, Geurts JJG, van Dam AM (2019) Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol Commun 7:206. 10.1186/s40478-019-0850-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzba-Bobrowicz T, Gwiazda E, Kosno-Kruszewska E, Lewandowska E, Lechowicz W, Bertrand E, Szpak GM, Schmidt-Sidor B (2002) Morphological analysis of active microglia–rod and ramified microglia in human brains affected by some neurological diseases (SSPE, Alzheimer's disease and Wilson's disease. Folia Neuropathol 40:125–131. [PubMed] [Google Scholar]
- Witcher KG, Bray CE, Dziabis JE, McKim DB, Benner BN, Rowe RK, Kokiko-Cochran ON, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP (2018) Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia 66:2719–2736. 10.1002/glia.23523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher KG, Bray CE, Chunchai T, Zhao F, O'Neil SM, Gordillo AJ, Campbell WA, McKim DB, Liu X, Dziabis JE, Quan N, Eiferman DS, Fischer AJ, Kokiko-Cochran ON, Askwith C, Godbout JP (2021) Traumatic brain injury causes chronic cortical inflammation and neuronal dysfunction mediated by microglia. J Neurosci 41:1597–1616. 10.1523/JNEUROSCI.2469-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Park S, Brooks N, Lang BT, Vemuganti R (2008) PPARγ agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res 1244:164–172. 10.1016/j.brainres.2008.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung R, Seyfoddin V, Guise C, Tijono S, McGregor A, Connor B, Ching LM (2014) Efficacy against subcutaneous or intracranial murine GL261 gliomas in relation to the concentration of the vascular-disrupting agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), in the brain and plasma. Cancer Chemother Pharmacol 73:639–649. 10.1007/s00280-014-2395-y [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang L, Zhang M, Wang P, Zhang L, Yuan C, Qi J, Qiao Y, Kuo PC, Gao C (2011) Peroxisome proliferator-activated receptor gamma negatively regulates IFN-beta production in Toll-like receptor (TLR) 3- and TLR4-stimulated macrophages by preventing interferon regulatory factor 3 binding to the IFN-beta promoter. J Biol Chem 286:5519–5528. 10.1074/jbc.M110.149823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J (2012) Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation 9:247. 10.1186/1742-2094-9-247 [DOI] [PMC free article] [PubMed] [Google Scholar]