

Abstract
Rat mammary carcinoma susceptibility 5a1 (Mcs5a1), which is concordant to human MCS5A1 breast cancer risk locus, mediates susceptibility by a non-mammary cell-autonomous mechanism associated with T cell differential expression of F-box protein 10 (Fbxo10). Human FBXO10, an evolutionarily conserved ubiquitin ligase gene, was shown to have a potential role in regulating cell death by controlling the degradation of Bcl-2, a key protein involved in apoptosis. Breast cancer susceptibility is controlled by interactions between environmental and genetic factors; therefore, we sought to determine if breast cancer risk-associated environmental chemicals interact with Mcs5a1 variants using luciferase reporter constructs containing 4.2 kb Fbxo10 promoters based on alleles of mammary cancer susceptible Wistar Furth (WF) and resistant Wistar Kyoto (WKY) rat strains. 12-O-Tetradecanoylphorbol-13-acetate (TPA) induced activation of a 4.2 kb WF Fbxo10 promoter region, but lower levels of activation of the homologous WKY Fbxo10 promoter region. Using general and specific protein kinase inhibitors, we identified a protein kinase C (PKC) pathway that mediated TPA activation. We narrowed the possible PKCs to a member of the atypical PKC isoforms, namely PKCμ. We also determined that activator protein 1 (AP1) family member c-Fos mediated TPA activation of the 4.2 kb WF Fbxo10 promoter. TPA was shown to induce endogenous FBXO10 mRNA and FBXO10 protein in Jurkat cells, a human T cell line, with a maximal level of expression from 1.5 to 2.5 h after exposure. These results indicate that FBXO10/Fbxo10 expression is regulated by a PKC-dependent pathway acting through c-Fos, which binds AP1-specific DNA elements in Mcs5a1.
Keywords: breast cancer susceptibility, F-box protein 10, mammary carcinoma susceptibility 5a1, protein kinase C, activator protein 1
INTRODUCTION
Breast cancer is the leading cause of death by cancer in US women age 20–59 years [1]. Risk of developing breast cancer is influenced by genetic and environmental factors. Breast cancer genetic susceptibility consists of high to moderate penetrance genes with rare population frequencies, and low penetrance modifier genes with moderate to high population frequencies. Most low-penetrance susceptibility associated genetic variants identified are located in non-protein coding regions [2].
Susceptibility alleles likely interact with environmental factors to influence modifier gene expression [3,4]. Multiple environmental factors have been associated with either decreased or increased risk of developing breast cancer. For example, Asian populations have a lower risk of developing breast cancer that is partly attributed to higher soy intake compared to Western diets [5]. Dietary calcium and vitamin D have also been associated with lower breast cancer risk [6–8]. Green tea may also confer lower breast cancer risk [9]. With respect to increased risk, pesticides have been associated with a modest elevated risk of breast cancer [10]. Benzo[a]pyrene, a ubiquitous polycyclic aromatic hydrocarbon, induces DNA-damage that may confer a higher chance of developing breast cancer [11]. Bisphenol A has been suspected as a potential breast cancer risk factor [12]. Thus, both genetic and environmental effects on breast cancer risk have been independently documented; however, interactions between genetic and environmental factors involved in breast cancer susceptibility, while plausible, have been difficult to clearly identify. This is likely due to the large sample sizes required to detect interactions between low-penetrance breast cancer susceptibility alleles and environmental factors.
Currently, uncovering interactions between genetic factors and environmental effects is largely dependent on epidemiological approaches [13,14]. Experimental organisms with susceptibility loci concordant to those elucidated in human disease may complement current approaches. Here we developed an in vitro promoter assay system to simplify the study of susceptibility alleles and environment interactions. We focused on the mammary carcinoma susceptibility 5a1 (Mcs5a1) region, a concordant human/rat breast/mammary carcinoma susceptibility locus. We selected an approximately 4.2 kb genomic segment of Mcs5a1 from mammary cancer susceptible Wistar Furth (WF) and resistant Wistar Kyoto (WKY) rat strains. Rat Mcs5a1 is a concordant ortholog of MCS5A1, a non-protein coding low-penetrance human breast cancer susceptibility allele [15]. Rat Mcs5a1 is part of a synthetic quantitative trait locus (QTL) named Mcs5a that acts in a non-mammary cell-autonomous manner and is important for T cell homeostasis [16]. Rat F-box protein 10 (Fbxo10), a ubiquitin ligase gene, is partially located in Mcs5a1, and is differentially expressed between susceptibility and resistance genotypes in thymocytes and T cells [15,17]. FBXO10 is conserved between Caenorhabditis elegans, Drosophila (Sophophora) melanogaster, and mammals [18]. Both FBXO10 and DRE-1, the ortholog of FBXO10 in C. elegans, bind, respectively, to Bcl2 and CED-9 to initiate cell death [19].
12-O-Tetradecanoylphorbol-13-acetate (TPA or phorbol 12-myristate 13-acetate, PMA) is a diester of phorbol and a potent tumor promoter used extensively in carcinogenesis research [20]. A main TPA-mediated pathway is the protein kinase C (PKC) pathway [21,22]. Comprised of a family of serine/threonine kinases, PKCs play important roles in several signal transduction cascades [23]. The PKC family consists of at least ten isozymes classified into three subfamilies based on their second messenger requirements: conventional (or classical), novel, and atypical [24,25]. Conventional (c) PKCs include isoforms α, βI, βII, and γ. These isoforms require Ca2+, diacylglycerol, and a phospholipid such as phosphatidylserine for activation. Novel (n) PKCs are δ, ε, η, and θ isoforms. Members of this group require DAG, but do not require Ca2+ for activation. Atypical (a) PKCs including protein kinase μ, Mζ, and ι/λ isoforms require neither Ca2+ nor DAG for activation [26]. Several general and isozyme specific PKC inhibitors have been developed for both research and clinical use.
Activator protein 1 (AP1) is a transcription factor which is a heterodimeric protein composed of proteins including the c-Fos, c-Jun, and ATF families [27]. Heterodimers and homodimers between AP1 family members determine specific target gene expression by recognizing TRE (TPA-responsive element), CRE (c-AMP response element), MARE (MAF responsive element), or ARE (antioxidant responsive element) as responsive elements [28]. AP1 can be induced by a variety of stimuli, and in turn regulates a number of cellular processes including cell proliferation, transformation, and death [29,30].
In this study, we have utilized an in vitro screen in Jurkat cells, a human T cell line, to identify chemicals that may influence the transcriptional activation of FBXO10, a candidate breast cancer susceptibility gene. We identified TPA as a potent activator of a rat Fbxo10 promoter region within Mcs5a1. We also demonstrated that a PKC mediated AP1 signaling pathway is involved in this response.
MATERIALS AND METHODS
Bioinformatics
A transcription factor database program named TFSEARCH (http://molsun1.cbrc.aist.go.jp/research/db/TFSEARCH.html) was used to search for sites of potential differential DNA binding for 4.2 kb WF and WKY rat Fbxo10 promoters. There are at least two 5′-UTRs for rat Fbxo10. The 4.2 kb WF and WKY rat Fbxo10 promoters we cloned representing a Fbxo10 genomic DNA region located at rat chr5:61 645 816–61 650 035 (The November 2004 rat genome of UCSC browser (Baylor3.4/rn4)) which is orthologous to a human T cell expressed FBXO10 transcript with GenBank accession identifier AF176705.
Chemicals and Reagents
All reagents were either analytical chemistry or molecular biology grade and purchased from commercial vendors (Table S1). Atrazine, benzo[a]pyrene, 17β-estradiol, 2-amino-1-methyl-6-phenylimidazo-[4,5-b] pyridine, and vitamin D3 were purchased from Fisher Scientific (Pittsburgh, PA). Butylated hydroxyanisole, bisphenol A, forskolin, GÖ6976, phorbol 12,13-dibutyrate, t-butylhydroquinone, thapsigargin, and TPA were purchased from Sigma–Aldrich (St. Louis, MO). Dexamethasone was purchased from Tocris (Minneapolis, MN) and recombinant human TGFβ1 was purchased from R&D System (Minneapolis, MN). H-7, H-8, and HA-1004 were purchased from Research Biochemicals International (now affiliated with Sigma–Aldrich), while GÖ6983 was purchased from Calbiochem (Rockland, MA).
Cell Culture
Jurkat cells (ATCC#TIB-152; American Type Culture Collection (ATCC), Manassas, VA) and MOLT-4 (ATCC#CRL-1582; ATCC) were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS) and penicillin/streptomycin solution from Invitrogen in 5% CO2 atmosphere at 37°C. HepG2 cells (ATCC#HB-8065) were cultured in DMEM medium with 10% FBS and penicillin/streptomycin solution from Invitrogen in 5% CO2 atmosphere at 37°C.
Plasmid Construction and Site-Directed Mutagenesis
A rat Fbxo10 (NCBI: AF176705) promoter region of 4.2 kb (rat chr5:61,645,816–61,650,035, UCSC Genome Browser on Rat Nov. 2004 (Baylor 3.4/rn4) Assembly) was cloned from WF and WKY rat spleen genomic DNA using PrimeSTAR® HS DNA Polymerase with GC Buffer (Clontech, Mountain View, CA). The cloned segment of Fbxo10 contains a T cell preferred transcriptional start site cluster in common between humans and rats [31]. Each Fbxo10 reporter was constructed by ligating target DNA into pGL3-enhancer (Promega, Madison, WI) using MluI and XhoI restriction sites. The primers obtained from IDT (Coralville, IA) were: forward: 5′CG ACG CGT AGG GGA GGC ACC GAG AAA ACA AG3′, reverse: 5′CCG CTC GAG AGG CCG GGC AGG GGT TAT CTC3′. For constructing luciferase reporters containing 3.5, 1.8, and 0.7 kb WF and WKY Fbxo10 promoter regions, we adopted the same reverse primer shown above and different forward primers which were, respectively: 5′CGACGCGTCTCCCCAAGCAGCCACTCAAAATG3′, 5′CGACGCGTTGCCCCCACATCCACTACCA3′, and 5′CGACGCGTTCATCCGCACTGAGCCCA3′.
Mutagenesis of a putative AP1 binding site in 4.2 kb WF Fbxo10 promoter (TGACTAA to TGGTTAA) was introduced using Quick-change site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) following the protocol. In brief, 4.2 kb WF Fbxo10 promoter was used as templates for PCR amplification. The sequences of reverse complementary primers designed to introduce a two nucleotide mutation were: GGAGTTTAAACAAATGGTTAATTGAAGGCTGGGATG and CATCCCAGCCTTCAATTAACCATTTGTTTAAACTCC. After PCR amplification, PCR products were treated with DpnI to digest parental DNA template and select for mutation-containing DNA. The DNA containing mutations was then transformed into competent cells for positive clone screening.
All constructs were sequenced using Sanger methods at the University of Louisville Center for Genetics and Molecular Medicine Nucleic Acid Core to confirm correct insertion or mutation of sequences. pRL-TK Renilla vector was purchased from Promega. Expression vectors containing c-Jun and c-Fos were obtained from Truyen Nguyen and Cecil Pickett (Schering-Plough Research Institute, Kenilworth, NJ) and JunB, JunD, Fra1, Fra2 plasmids were from Sunita Agarwal (NIDDK, NIH, Bethesda, MD).
Transient Transfection and Dual Luciferase Activity Assays
TransIT Jurkat Transfection Reagent (Mirus, Madison, WI) was used to transfect 1 μg plasmids into 1 × 106 Jurkat cells according to the manufacturer’s protocol. pRL-TK Renilla luciferase vector (50 ng, Promega) was used as an endogenous control in every transfection. Twenty-four hours after transfection, cells were treated with various compounds as stated for each experiment. After an additional time period (4 h for TPA or 24 h for other compounds), cell lysates were harvested. For luciferase activity assays, cells in 24-well plates were washed three times with cold phosphate-buffered saline. Cell lysates were prepared and luciferase activities measured using a dual luciferase reporter assay system (Promega) and a Biotek Synergy2 plate reader (Biotek, Winooski, VT). Changes in Firefly luciferase activities were calculated after dividing by respective Renilla luciferase activities. Results were based on three replicates per experiment and three independent experiments were performed. Relative luciferase activity was computed by setting control sample luciferase activity in each reporter experiment to one and calculating experimental group luciferase activities accordingly.
Electrophoretic Mobility Shift Assays (EMSA)
Electrophoretic mobility shift assays (EMSA) were performed using Pierce Lightshift Chemiluminescent EMSA Kit according to the manufacturer’s protocol (Pierce, Rockford, IL). Forward and reverse DNA sequence of 25 bp rat SNP36 WF/WKY alleles, rat Mcs5a1 TRE-1 (TRE1), rat Mcs5a1 TRE-2 (TRE2), rat Mcs5a1 TRE-2 mutation (TRE2m), rat Mcs5a1 TRE-3 (TRE3), rat Mcs5a1 TRE-4 (TRE4), and human TRE, respectively, were annealed and labeled with biotin using a biotin 3′ end DNA labeling kit (Pierce). Oligonucleotide sequences are shown in Table S2. Jurkat cells (DMSO or TPA exposed) were collected to obtain nuclear extracts (NE or SNE) as previously described [32]. Labeled oligonucleotides (20 fmol) and 2 μg nuclear proteins were used in EMSA. A 4.5% non-denaturing polyacrylamide gel (4.5% 29:1 acrylamide/bisacrylamide, 0.5 × TBE, 1% glycerol, 0.075% APS, and 0.1% TEMED) was used to resolve protein-DNA complexes in 0.5 × TBE buffer. Gel supershift of protein-DNA complexes was performed by incubating nuclear protein and 2 μg c-Fos antibodies (Santa Cruz) or a negative IgG in EMSA buffer prior to adding biotin-labeled probes.
Quantitative PCR (QPCR) Analysis
Total RNA was extracted from Jurkat cells (1 × 106) using TRI Reagent (Molecular Research Center, Inc. Cincinnati, OH). One μg of total RNA from each sample was reverse transcribed to cDNA using Invitrogen Superscript III reverse transcriptase (Invitrogen). Synthesized cDNA served as template in a 20-μL QPCR reaction. Quantitative PCRs were performed using Taqman probes (Applied Biosystems, Foster city, CA) and Taqman 1000 RXN gold with buffer A (Applied Biosystems) in a StepOnePlus realtime PCR system. Primers obtained from Integrated DNA Technology (Coralville, IA) for FBXO10 were: forward: 5′CTGCCGGCATAGCAGTGAAT3′, reverse: 5′, and probe: 6FAM-CTCATCACAGAAAATGT-MGBNFQ. GAPDH (Part number 402869, Applied Biosystems) was used as an endogenous control. The expression of FBXO10 was adjusted to GAPDH level and relative concentrations of each template were calculated automatically according to a comparative Ct method.
Western Blot Analysis
Whole cell extract was collected using RIPA buffer (Cell Signaling, Danvers, MA). SDS–PAGE (8% or 12%) was used to separate proteins. FBXO10 antibody was obtained from Sigma–Aldrich (QC23691) and used at a 1:2000 dilution. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was purchased from Bethyl (Montgomery, TX) and used at 1:4000. Antirabbit secondary antibody from Cell Signaling was used at a 1:4000 dilution. Chemiluminescence was detected using Pierce Supersignal West Pico Chemiluminescent Substrate (Pierce). Relative protein abundance was calculated based on the density of each western blot band using ImageJ (http://imagej.nih.gov/ij/). FBXO10 expression was adjusted relative to GAPDH levels.
Statistical Analysis
Absolute luciferase activity was calculated by dividing firefly luciferase activity by Renilla luciferase activity. Target transcript level (FBXO10), measured by quantitative PCR (QPCR), was divided by GAPDH transcript level. Values of control samples were set to a mean of one and relative concentrations of each template were calculated automatically according to a comparative Ct method. Results are shown as means ±SDs. Square roots of ratio data were transformed by arcsine function prior to statistical analyses by Student’s t-tests. Analysis of variance (ANOVA) was used when experiments involved multiple comparisons. Comparisons and ANOVA F-tests were considered statistically significant if P ≤ 0.05.
RESULTS
In Vitro Screening of a Rat FBXO10 Promoter Region
Figure 1 is a schematic of the human MCS5A1 5.7 kb chromosome 9 region associated with breast cancer risk and the rat orthologous region that is part of Mcs5a1, a 32 kb segment that is part of the Mcs5a synthetic QTL [15]. The WKY rat Mcs5a allele is associated with mammary cancer resistance and lower Fbxo10 in thymus and splenic T cells compared to the susceptible WF strain [15,17]. Human MCS5A1 breast cancer risk associated polymorphisms are located in and near a CpG island containing a transcription start site for FBXO10 [17]. To study in vitro interactions between genetic and environmental factors, we cloned into independent luciferase reporters mammary carcinoma susceptible WF and resistant WKY alleles from Rattus norvegicus Chr 5: 61 645 816 to 61 650 035, which is contained within rat Mcs5a1. This 4.2 kb rat sequence contains twelve sequence variants between WF and WKY alleles that are within 3.5 kb of a human-rat conserved CpG island containing a T cell preferred Fbxo10/FBXO10 transcription start site cluster. The cloned 4.2 kb of minus strand orientated sequence contained the CpG island (478 bp) plus 3560 bp 5′, and 182 bp 3′ flanking sequences.
Figure 1.

A schematic of human MCS5A1 and the rat genomic region of Mcs5a1 used to make 4.2 kb Fbxo10 luciferase reporter constructs. (A) The human MCS5A1 locus is a 5.7 kb region of human chromosome 9 that contains four breast cancer risk associated polymorphisms (rs7042509, rs6476643, rs10758441, and indel138–9899, marked with black circles). (B) The rat Mcs5a1 locus is a 32 kb region of rat chromosome 5 that contains segments hypothesized to be 5′-Fbxo10 regulatory regions. Relative locations of genetic variants between mammary cancer susceptible WF and resistant WKY alleles are marked with black circles. Asterisks represent potential TRE (predicted AP1 binding sites). The lengths of all reporter constructs are shown and more detailed information regarding these variants is available in Table 1. Human and rat FBXO10/Fbxo10 conserved 5′-UTRs and an intronic segment are shown for reference. The human/rat conserved CpG islands contain FBXO10/Fbxo10 T cell preferred transcription start sites (TSS).
Subsequently, we measured Fbxo10 4.2 kb promoter activity changes in Jurkat cells, a human T cell line, exposed to a variety of potential breast cancer risk environmental modifiers and immune modulators (Table S1). Concentration and incubation times were those reported to be effective for each chemical from the literature. Most agents tested did not have a significant effect on either WF or WKY 4.2 kb Fbxo10 reporter activities. On the other hand, treatment of Jurkat cells with the phorbol ester TPA resulted in differentially increased transcriptional activities of WF compared to WKY 4.2 kb Fbxo10 reporter (P < 0.05). As shown in Figure 2A, addition of 1 μM TPA activated the WF 4.2 kb Fbxo10 promoter fivefold 4-h after TPA-exposure of Jurkat cells, which was more than the WKY 4.2 kb promoter (twofold at 4 and 24 h). This suggests that genetic variation in the 4.2 kb Fbxo10 promoter may contribute to differential TPA-responsiveness. After longer exposure times (up to 24 h), TPA-induced activity of the WF 4.2 kb promoter remained statistically significant, but declined to a level that was statistically insignificant compared to the WKY 4.2 kb promoter (P = 0.0986). The concentration dependence of TPA activation of WF and WKY 4.2 kb Fbxo10 reporters displayed different magnitudes, but similar patterns of response with maximal activation of both at around 100 nM TPA (Figure 2B). Overall, these results indicate that a transient TPA-response of this 4.2 kb Fbxo10 promoter occurs within hours after exposure and the latency and magnitude of the response may be genetically determined.
Figure 2.

In vitro TPA activation of luciferase reporter genes containing a 5′-flanking region of either WF or WKY Fbxo10. TPA activated 4.2 kb WF and WKY Fbxo10 promoters in a time (A) and concentration (B) dependent manner. Jurkat cells were transfected with luciferase reporter genes containing either a WF (lighter bars) or WKY (darker bars) Fbxo10 segment and exposed to TPA for the time and concentrations indicated. Luciferase activities were measured in triplicate and results from triplicate samples are shown as average relative luciferase activity ± SD adjusted to the respective Renilla luciferase activity. Single or double asterisks indicate statistically significant (P ≤ 0.05) relative to DMSO control (no TPA exposure) or WF compared to WKY, respectively.
Like TPA, another phorbol ester family member, phorbol 12, 13-dibutyrate (PDBu) also activated both the WF and WKY 4.2kb Fbxo10 reporter genes (Figure S1). Compared to TPA-exposure, PDBu-exposure did not increase WF reporter activities as much. TPA is known to be a more effective activator of PKC than PDBu, and PDBu is more hydrophilic than TPA. Both attributes, as well as genetic variation, are potential contributors to apparent different effects of TPA and PDBu on 4.2 kb WF promoter activities. Differential activation of both the WF and WKY 4.2 kb Fbxo10 luciferase reporters by TPA was detected in another T cell line, but not in other cell lines examined. For example, we found similar differences in TPA-induced activities of WF and WKY 4.2 kb Fbxo10 promoters in MOLT-4, another T cell line (Figure S2A). In contrast, we did not find TPA induction of Fbxo10 promoters in HepG2 cells, a human liver carcinoma cell line, in which two other TPA responsive reporters (human collagenase TRE luciferase reporter (pCOL-TRE) and rat aldehyde dehydrogenase 3A1 CRE luciferase reporter (pKCF97)) showed significant TPA activation (Figure S2B–D). These results demonstrate that the activation of this 4.2 kb Fbxo10 reporter is apparently regulated in a cell-type specific manner.
PKCμ Is a Potential Candidate Mediating TPA-Activation of Variant Fbxo10 Promoters
We were interested to define the signaling pathways mediating TPA activation of luciferase reporter constructs containing 4.2 kb of the 5′-flanking region of the Fbxo10 gene from the WF and WKY strains. As TPA is a well-known activator of the PKC pathway, inhibitors of multiple protein kinases were tested to determine whether the effect of TPA activation was PKC-dependent (Table S2). As shown in Figure 3A, strong PKC inhibitors such as H-7 and H-8 both antagonized TPA activation at concentrations below 50 μM, whereas weak PKC inhibitor HA-1004 did not inhibit TPA activation at 50 μM. Consistent with this, inhibition by H-7 and H-8 was concentration dependent (Figure 3A), with H-7 being a better inhibitor than H-8. Inhibitor H-7 (5 μM), but not H-8 effectively repressed the 4.2 kb WF Fbxo10 promoter activity more potently than the WKY promoter. This trend is consistent with the fact that H-7 is a stronger PKC inhibitor than H-8 (Table S2). Since Forskolin, a PKA activator, did not activate either 4.2 kb Fbxo10 promoters (Table S1), we concluded that TPA activated WF and WKY 4.2 kb Fbxo10 promoters via a PKC-dependent pathway.
Figure 3.

TPA activation of rat Fbxo10 luciferase reporter is PKC dependent and PKCμ is a potential activating isoform. (A) Jurkat cells were transfected with luciferase reporter plasmids containing either the WF (solid lines) or WKY (dashed lines) 4.2 kb Fbxo10 5′-flanking region. Twenty-four hours after transfection, cells were incubated with 1 μM TPA alone or TPA plus increasing concentrations of H-7 (squares), H-8 (triangles), or HA-1004 (circles) for 4 h. DMSO was used as a control. The results are expressed as average relative luciferase activity ± SD for three independent experiments performed in triplicate. (B) Jurkat cells transfected with WF (gray bars) or WKY (black bars) 4.2 kb Fbxo10 luciferase reporter vectors were incubated with either 1 μM TPA, 1 μM Thapsigargin (TG), TPA + TG, or 0.8 μg/mL 1,2-dioleoyl-sn-glycerol (DAG). (C) Jurkat cells transfected with WF (solid lines) or WKY (dashed lines) 4.2 kb Fbxo10 luciferase reporter were incubated with 1 μM TPA and TPA plus different concentrations of either GÖ6976 (squares) or GÖ6983 (circles) PKC inhibitors as indicated. Asterisks indicate P < 0.05 comparing inhibitor group to DMSO group.
To narrow the list of PKC family members to a specific PKC subfamily based upon the requirement for calcium or 1,2-dioleoyl-sn-glycerol (DAG) for activation, we used a Ca2+-ATPase inhibitor, thapsigargin, and DAG to test whether TPA induced Fbxo10 reporter activity was Ca2+-dependent or DAG dependent. Thapsigargin did not have an effect on promoter basal level or TPA activation of Fbxo10 reporter activities of either construct (Figure 3B). Furthermore, DAG did not induce either the WF or WKY 4.2 kb Fbxo10 reporter activities (Figure 3B). These results ruled out conventional and novel PKC family members and raised the possibility that atypical PKCs were involved in either one or both of the 4.2 kb Fbxo10 variant reporter activities induced by TPA.
A recently identified PKC isoform named PKCμ is involved in several signaling pathways [33]. We sought to determine if PKCμ was involved in TPA induced Fbxo10 luciferase reporter activation using two PKC inhibitors GÖ6976 and GÖ6983, which function, respectively, as a strong PKCμ inhibitor with an IC50 at 20 μM, and a weak PKCμ inhibitor with an IC50 at 20 nM (Table S2). As shown in Figure 3C, GÖ6976 repressed TPA activation of Fbxo10 reporter at 5 nM, whereas GÖ6983 exposure resulted in a more modest repression of TPA activation of Fbxo10 reporters, as a concentration of 50 nM GÖ6983 was required for significant inhibition. In addition, these PKCμ inhibitors had a larger effect on TPA-activation of the WF 4.2 kb Fbxo10 reporter than the respective WKY Fbxo10 reporter construct. These data imply that PKCμ is a potential PKC isoform responsible for differential responses of the mammary cancer susceptible WF and resistant WKY Fbxo10 promoters to TPA.
AP1 Family Member c-Fos Differentially Increased WF and WKY 4.2 kb Fbxo10 Promoter Activities
AP1 is one of the main transcription factors, activated after TPA exposure [34,35]. We co-transfected Jurkat cells with either WF or WKY 4.2 kb Fbxo10 luciferase reporter plasmids along with expression vectors containing either c-Fos or c-Jun. In the absence of added TPA, c-Fos activated both Fbxo10 promoters, whereas c-Jun antagonized c-Fos activation of these Fbxo10 reporters (Figure 4A–C). Because c-Jun suppressed c-Fos activation of Fbxo10 luciferase reporter activities in cells not exposed to TPA, we subsequently determined whether TPA activation of Fbxo10 reporters could also be compromised by coexpression of c-Jun. We found that the WF Fbxo10 reporter in the presence of c-Jun over-expression responded less to TPA than co-transfection with the pcDNA3.1 expression control vector (Figure 4B). However, c-Jun had little or no effect on the TPA activation of the WKY Fbxo10 reporter (P = 0.13). This suggests that c-Jun is effective at either acting to sequester the c-Fos heterodimerization partner or directly competing with c-Fos in the context of the WF 4.2 kb Fbxo10 promoter, but not the WKY variant. We also tested other AP1 family members that included Jun related proteins JunB, JunD, and Fos like proteins Fra1, and Fra2, but none of these activated Fbxo10 luciferase reporter constructs (Figure S3A). Furthermore, JunB and JunD, like c-Jun and NRF2 (Figure 4), antagonized c-Fos activation of Fbxo10 reporters (Figure S3B). Based on these results, it was possible that TPA activated Fbxo10 luciferase promoter constructs through c-Fos and a heterodimerization partner may compete with Jun family members including JunB, JunD, etc. for c-Fos binding. For example, JunD has been shown to be a main determinant in macrophage activity in rats and is higher in WKY rats [36]. We extended our search to NRF2 and c-Maf, which are proteins known to heterodimerize with AP1 proteins. To test this possibility Jurkat cells were co-transfected with variant Fbxo10 reporters and an NRF2 or c-Maf expression vector. Like c-Jun, NRF2 fully inhibited TPA activation of both WF and WKY Fbxo10 reporters in a dose dependent manner (Figure 4D), whereas c-Maf had little or no effect on TPA activation of both WF and WKY Fbxo10 reporters (Figure 4E). We also determined the protein levels of c-Fos, c-Jun, NRF2, and c-Maf in whole cell and nuclear extracts of Jurkat cells with or without TPA stimulation. As shown in Figure S4, c-Fos was increased in nuclear extract of Jurkat cells stimulated with TPA for 4 h compared to Jurkat cells treated with DMSO. Levels of c-Jun were similar between unstimulated and stimulated Jurkat-cell nuclear-extracts; upon TPA treatment, NRF2 disappeared from Jurkat nuclear extracts; and c-Maf was not detected in Jurkat nuclear extracts. The protein expression of c-Fos, c-Jun, NRF2, and c-Maf is reminiscent of their effect on 4.2 kb WF and WKY Fbxo10 reporters (Figure 4A–E). Nevertheless, the potential dimerization partner for c-Fos will be the focus of future studies.
Figure 4.

c-Fos expression activated luciferase reporters containing a 5′-flanking region of either the 4.2 kb WF or WKY Fbxo10 gene in Jurkat cells. (A) Jurkat cells were transfected with 1 μg of luciferase reporter gene containing either the 5′-flanking regions of either the 4.2 kb WF (lighter black) or WKY (dark black) Fbxo10 genes and 50 ng pRL-TK plasmids plus either 1 μg c-Fos, c-Jun, NRF2, or combinations of c-Fos with either c-Jun or NRF2 as indicated. *Statistically significant (P < 0.001) as indicated. (B) The c-Jun (0.2, 0.5, and 1.0 μg) and control plasmids were transfected into Jurkat cells and TPA (1 μM) was added for 4 h. Statistically significant (*P = 0.016, **P = 0.006, and ***P = 0.019 as indicated). (C) The c-Fos (0.2, 0.5, and 1.0 μg) or control vectors were transfected into Jurkat cells; and TPA (1 μM) was added for 4 h. Statistically significant (*P < 0.02, **P = 0.002) as indicated). (D) An NRF2 (0.2, 0.5, and 1.0 μg) or empty vector was transiently transfected into Jurkat cells; TPA (1 μM) was added for 4 h. Statistically significant (*P < 0.02) as indicated). (E) The c-Maf (0.2, 0.5, or 1.0 μg) or control vector was transfected into Jurkat cells, and TPA (1 μM) was added for 4 h. Statistically significant (*P < 0.02) as indicated). (F) Luciferase reporters containing 4.2, 3.5, 1.8, or 0.7 kb WF or WKY rat strain Fbxo10 alleles were transfected into Jurkat cells with pRL-TK as a transfection control. One micromolar TPA was added for 4 h. X-Axis genotype by treatment subgroups are shown below each Fbxo10 segment group (4.2, 3.8, 1.8, and 0.7 kb). Asterisks represent P < 0.05 comparing the respective groups indicated by lines.
We next cloned different lengths of the Fbxo10 5′-UTR flanking sequence of interest into luciferase reporters (0.7, 1.8, 3.5 kb) using the same reverse primer for cloning 4.2 kb Fbxo10 reporters, but different forward primers. The different sequence lengths cloned are shown in Figure 1 and contain the following number of variants between WF and WKY sequence: 4.2 kb—12 variants; 3.5 kb—10 variants; 1.8 kb—7 variants; 0.7 kb—1 SNV. As shown in Figure 4F, we found that of these constructs only the 4.2 kb WF and WKY luciferase reporters were differentially activated by TPA. No significant effect of TPA was seen on 0.7 kb WF or WKY reporters compared to these reporters in cells without TPA. Both 3.5 and 1.8 kb WF and WKY reporters were activated by TPA in a similar manner (basal and inducible expression). This suggested that sequence contained in the 1.8 kb segment may harbor potential AP1 binding sites involved in TPA activation of Fbxo10. However, this sequence is not directly responsible for differential TPA activation of WF and WKY variants. In other words, the integrity of the full-length 4.2 kb Fbxo10 segment was required for differential TPA activation; therefore, additional regulatory elements are likely involved.
A TRE Identified in the 4.2 kb WF and WKY Fbxo10 Promoters May Mediate a TPA Response
We systematically analyzed putative TRE sites in the 4.2 kb WF and WKY Fbxo10 promoters (Table 1). Among the variants previously identified between Mcs5a1 alleles, only SNP36 was predicted to differentially bind to AP1. The AP1 binding score attributed to SNP36 WF was 72.2, whereas the AP1 binding score of SNP36 WKY was 60.8. We performed EMSA using nuclear extract of Jurkat cells (either DMSO or TPA treated) and SNP36 WF/WKY oligonucleotides to determine if AP1 bound to either one. The human collagenase TRE was used as a control. As shown in Figure 5A, an increase in DNA–protein complex for SNP36 WF/WKY and human collagenase TRE oligonucleotides was noted for all ds-oligonucleotides with extracts from TPA-treated Jurkat cells (see solid arrow), but there was an additional DNA–protein complex for the human collagenase ds-oligonucleotide that migrated more slowly (Figure 5A dashed arrow).
Table 1.
Genetic Variants Between WF and WKY Rat 4.2 kb Fbxo10 Promoter Regions
| SNPs, indels (NCBI assay IDs) and AP1 binding sites | Sequence and SNP position of AP1 binding site (relative to TSS of WF rat Fbxo10 promoter) | Dimorphisms between WF and WKY rat alleles | Predicted differentially binding proteins (score) | Constructs containing variants (kb) |
|---|---|---|---|---|
| SNP35 (ss262858665) | TTCCCACCCC T TCAAAGGCCA (−204) | WF = T; WKY = G | E2F; cdxA; TATA; NKX-2.5 | ≤0.7 |
| Indel8 (ss262858666) | TTTTTTTTTTTTTTT (−451 to −439) | WF = Tn=15; WKY ≕ | ≤1.8 | |
| SNP36 (ss262858667) | AACAGGAGCC A CCACCACTTG (−607) | WF = A; WKY = G | AP1 (WF 72.2, WKY 60.8), CREB | ≥0.7 ≤1.8 |
| Indel9 (ss262858668) | CAAA (−861 to −858) | WF = CAAA; WKY ≕ | ≥0.7 ≤1.8 |
|
| Indel10 | AA (−879 to −878) | WF = AA; WKY ≕ | ≥0.7 ≤1.8 |
|
| Indel11 | AAAGA (−917 to −913) | WF = AAAGA; WKY ≕ | ≥0.7 ≤1.8 |
|
| SNP37 (ss262858670) | TAGGGTGCAG A TTGGAGAGAC (−1189) | WF = A; WKY = G | GATA; NF-Y; Oct-1; Evi-1 | ≥0.7 ≤1.8 |
| SNP38 (ss262858671) | CGCTTACAAA C GCTGTTCTTG (−2035) | WF = C; WKY = T | cdxA | ≥1.8 ≤3.5 |
| Indel12 (ss262858672) | TGTGCCAGGC T GCTAAGGGGA (−2175) | WF = T; WKY ≕ | GATA1 | ≥1.8 ≤3.5 |
| Indel13 (ss262858673) | Long nucleotide deletion (−2486 to −2339) | WF = INSERTION; WKY ≕ | ≥1.8 ≤3.5 |
|
| SNP39 (ss262858674) | ACAGTAGGGG A ATTGGGAGGGA (−3365) | WF = A; WKY = G | Tst-1; GATA1; STATx; c-ets; | ≥3.5 ≤4.2 |
| SNP40 (ss262858675) | AAACAAGGCC C CTAAATTAAC (−3779) | WF = C; WKY = T | Stress response elements (STRE) | ≥3.5 ≤4.2 |
| TRE1 61647025–61647031 | TGTCTCA (−795 to −788) | AP1 (83.3) | ≥0.7 ≤1.8 |
|
| TRE2 61647743–61647749 | TGACTAA (−1513 to −1506) | AP1 (89.1) | ≥1.8 ≤3.5 |
|
| TRE3 61647951–61647957 | TGGGTCA (−1721 to −1714) | AP1 (86.6) | ≥1.8 ≤3.5 |
|
| TRE4 61649140–61649146 | −2911 to −2904 | AP1 (87.1) | ≥1.8 ≤3.5 |
SNP, single nucleotide polymorphism; Fbxo10, F-box protein 10; WF, Wistar Furth; WKY, Wistar Kyoto; TRE, TPA response element.
Potential binding proteins were predicted using TFSEARCH. Potential TRE with AP1 binding scores predicted by TFSEARCH are also shown. Genomic locations (Rat genome, Feb. 2009 GRCh37/hg19) are shown. Relative locations of polymorphisms and AP1 binding sites in the constructed promoters are listed. The asterisk indicates the Fbxo10 TRE that was bound by c-Fos and TPA responsive.
Figure 5.
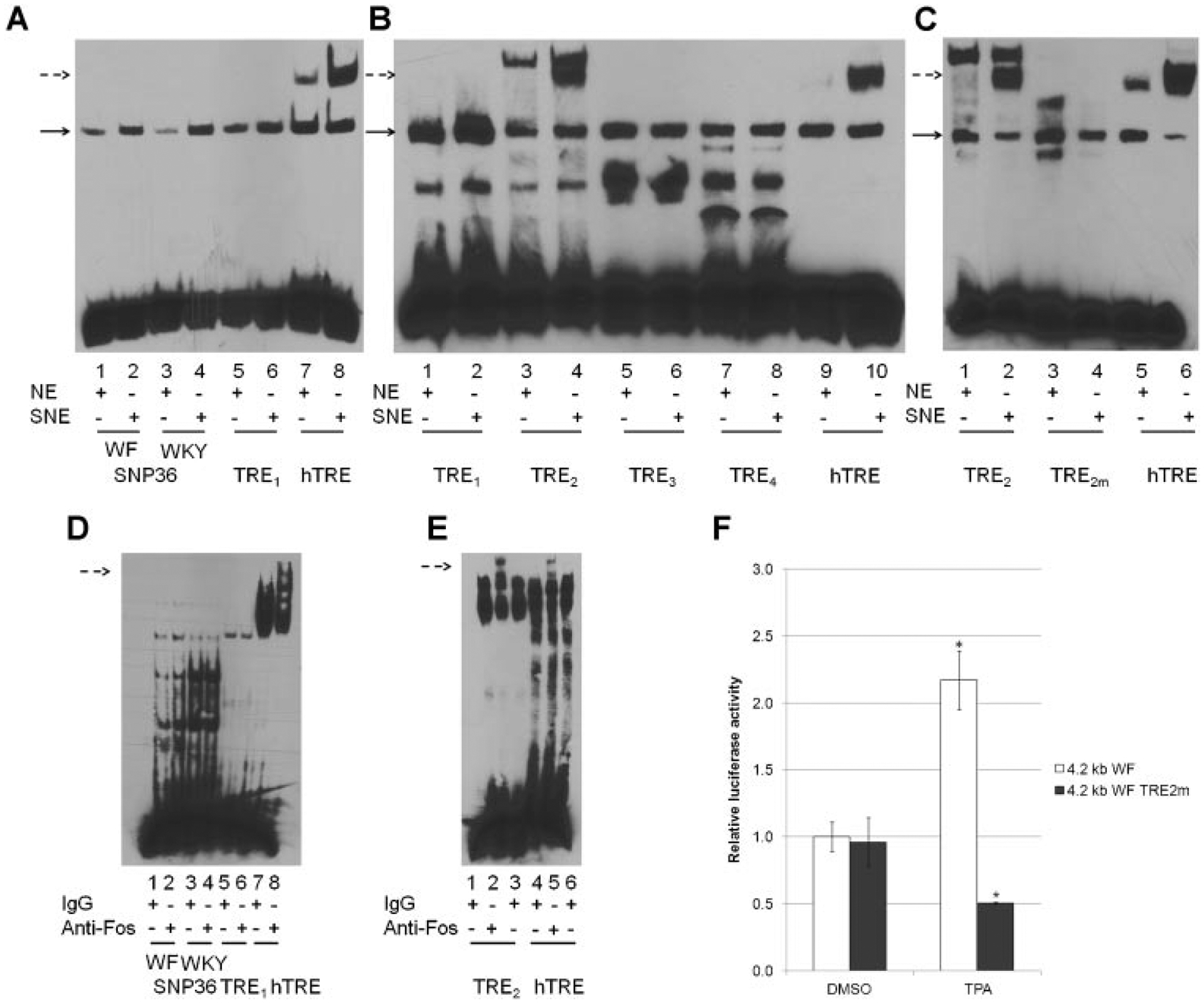
Identification of a TPA responsive element (TRE) in a 4.2 kb regulatory region of Fbxo10. (A) SNP36 WF or WKY, putative rat Mcs5a1 TRE-1 (TRE1) did not show common mobility shifts due to c-Fos antibody, as observed with the human collagenase TRE. EMSA was performed using TPA stimulated Jurkat cell nuclear extract (SNE) and control nuclear extract (NE) as described in methods. DNA–protein complexes that were increased after TPA treatment of Jurkat cells was observed for all SNP36 ds-oligonucleotides, Fbxo10 TRE1, and human collagenase TRE (solid arrow), but an additional slower moving DNA–protein complex was seen with the human TRE ds-oligo (dashed arrow). (B) Among selected rat Mcs5a1 TREs, only Mcs5a1 TRE-2 (TRE2) showed an electrophoretic mobility shift similar to that observed with the human collagenase TRE. The experiment was performed as described in (A). A dashed arrow marks the common shift between TRE2 and human TRE. (C) Oligonucleotides containing TRE2m, a mutation of the core AP1 binding TRE site, resulted in the loss of DNA–protein binding in EMSA. (D) SNP36 WF/WKY and TRE1 failed to show a supershift when anti-c-Fos was added to the reaction mixture, compared to the supershift observed with the human TRE during EMSA. A dashed arrow marks the common supershift between TRE2 and human TRE. (E) Mcs5a1 TRE-2 (TRE2) showed a supershift when anti-c-Fos is added, similar to the supershift observed with the human TRE during EMSA. Supershift was performed as described above. (F) The 4.2 kb WF Fbxo10 promoter harboring TRE2 mutation failed to respond to TPA stimulation. Jurkat cells were transfected with luciferase reporter genes containing either the 4.2 kb WF (white bars) or 4.2 kb WF with TRE2 mutation (gray bars) Fbxo10 5′-flanking region plus pRL-TK plasmids as a control. Cells were exposed to 1 μM TPA for 4 h. Luciferase activities were measured in triplicate, and results from triplicate samples are shown as average relative luciferase activity ± SD adjusted to the respective Renilla luciferase activity *P < 0.05.
In addition to SNP36, we identified using bioinformatics a number of putative AP1 binding sites with higher predicted binding scores (Table 1). One of these, potential TRE1 also formed a complex running at the area indicated by the solid arrow. Further, the apparent density of this DNA–protein complex was increased by nuclear extracts of TPA-treated Jurkat cells (Figure 5A), but did not form a slower migrating band similar to that seen using a human collagenase TRE. These results demonstrated that SNP36 and Fbxo10 potential TRE1 containing oligonucleotides form DNA–protein complexes that are increased in apparent mass by TPA treatment of Jurkat cells. However, they do not form a more slowly migrating complex expected for AP1 proteins, which was seen using human collagenase TRE ds-oligonucleotides.
We selected three additional putative AP1 binding sites in the 4.2 kb WF and WKY Fbxo10 promoters with high predicted binding scores and named them Fbxo10 potential TRE2, TRE3, and TRE4 (Figure 1B). Double-stranded oligonucleotides containing these potential AP1 binding sites were used in EMSA to test for binding to nuclear proteins. Potential TRE1 and human collagenase TRE containing oligonucleotides were used as controls. Only Fbxo10 potential TRE2 displayed a slower migrating complex similar to the human collagenase TRE ds-oligonucleotide (Figure 5B). Nuclear extracts from TPA-activated Jurkat cells did show higher levels of all DNA–protein complexes (solid arrow) suggesting that TPA may increase proteins that bind to all of these potential responsive elements. The more slowly migrating DNA–protein complexes did not form when EMSAs were performed with Fbxo10 potential TRE2 mutant (TRE2m) oligonucleotides (Figure 5C). This indicated that nuclear proteins (c-Fos or other AP1 protein) induced by TPA may uniquely bind to potential TRE2 producing the slowest migrating DNA–protein complex.
Addition of c-Fos antibody to the incubation mixtures containing the various ds-oligonucleotides failed to result in a supershift when SNP36 WF or WKY oligonucleotides were used in EMSA (Figure 5D), whereas a supershift was observed when either the Fbxo10 potential TRE2 or human collagenase TRE were incubated with c-Fos antibodies, but not with non-specific IgG antibodies (Figure 5E). Together, these results ruled out SNP36 variants, and three other predicted TREs as functional c-Fos binding sites in a 4.2 kb WF and WKY Fbxo10 promoter region. Nuclear extracts from c-Fos over-expressing HepG2 cells were also incubated with Fbxo10 potential TREs1–4 and hTRE oligos. Consistent with results from TPA treated Jurkat cells, only TRE2 showed a shift similar to hTRE (Figure S5A). Oligonucleotides containing TRE2m showed a diminished binding to c-Fos compared to Fbxo10 potential TRE2 and hTRE (Figure S5B).
The same two nucleotide mutation of the AP1 binding site sequence in TRE2m was also introduced into the 4.2 kb WF luciferase reporter. We compared luciferase activities of 4.2 kb WF and WF-TRE2m reporters. As shown in Figure 5F, 4.2 kb Fbxo10 WF-TRE2m promoter showed similar activity to the 4.2 kb Fbxo10 WF promoter in Jurkat cells without TPA. However, activity of the 4.2 kb Fbxo10 WF-TRE2m promoter failed to be induced by TPA in Jurkat cells, whereas the 4.2 kb Fbxo10 WF promoter activity was induced by TPA. Results from EMSAs and luciferase reporter assays strongly suggest that Fbxo10 TRE2 is a functional AP1 binding site involved in TPA-induced transcriptional activity of Fbxo10. Our finding that additional putative Fbxo10 predicted TREs interact with induced nuclear proteins in EMSA analysis, but apparently not c-Fos may be indicative of a possible DNA–protein complex that functions to regulate Fbxo10 gene expression by binding AP1 family members not including c-Fos or a heterodimeric partner that is not expected to be c-Jun or other related proteins, such as Maf and NRF2. These possibilities will require further study, considering more complex modes of Fbxo10 gene regulation.
Jurkat Cell FBXO10 mRNA and Protein Levels Increased in Response to TPA
Since rat Fbxo10 promoter driven luciferase activity was increased by TPA, we determined whether TPA had an effect on endogenous FBXO10 mRNA levels in Jurkat cells. First, we treated Jurkat cells with different concentrations of TPA (from 10 nM to 1 μM concentration) for 24 h. No statistically significant changes in FBXO10 mRNA levels were observed at 24 h (Figure 6A). We subsequently treated Jurkat cells with 1 μM, 100 nM, and 10 nM TPA for different times from 0 to 3.5 h. As shown in Figure 6B, TPA exposure rapidly increased FBXO10 levels in a concentration-dependent manner. The highest concentration of TPA (1 μM) increased FBXO10 mRNA levels approximately1.7-fold after 1.5 h compared to control, while 100 nM TPA induced FBXO10 mRNA levels approximately 1.4×. TPA treatment at 10 nM did not effectively increase FBXO10 mRNA levels at 1.5 h. However, after 2.5 h, 10 nM TPA induced an approximately 1.2-fold increase of FBXO10 mRNA. One μM TPA increased FBXO10 protein levels at 2 h, but longer TPA exposure times led to a slight decrease in FBXO10 (Figure 6C and D). These results demonstrate that TPA alters FBXO10 expression at mRNA and protein levels as an early response in Jurkat cells.
Figure 6.

TPA increased FBXO10 mRNA and protein levels in Jurkat cells. (A) TPA (10, 100, and 1000 nM) had no significant effect on FBXO10 mRNA levels 24 h after exposure. Jurkat cells were incubated with different concentrations of TPA indicated for 24 h. Cells were collected for RNA extraction prior to measuring relative FBXO10 mRNA levels by QPCR. (B) The effect of TPA (10, 100, and 1000 nM) on FBXO10 mRNA levels at different time points. *Statistically significant P < 0.05. (C) The effect of TPA (1.0 μM) on FBXO10 protein levels at different time points as indicated. Jurkat cells were treated with 1.0 μM TPA for times indicated and cells were collected for protein extraction and western blot analysis. Relative intensity of the FBXO10 protein was standardized relative to the intensity of GAPDH band using ImageJ.
DISCUSSION
Using luciferase reporter constructs containing a 4.2 kb segment of Fbxo10 gene promoter from either a mammary cancer susceptible WF or resistant WKY rat strain, we screened environmental chemicals associated with breast cancer risk and immune-modulators that might have an effect on FBXO10, a genetic association study nominated breast cancer susceptibility gene. Among chemicals tested, TPA was shown to differentially affect the mammary cancer susceptible-WF Fbxo10 promoter construct by increasing gene expression over fivefold, while the cancer resistant-WKY Fbxo10 promoter construct was increased only twofold. Activation of WF and WKY Fbxo10 promoter containing luciferase reporters occurred in a TPA concentration- and time-dependent manner. Other chemicals tested had no significant effect on the WF or WKY Fbxo10 promoter regions used at the times and concentrations used. Although we used the selected chemicals under conditions that have been shown to be efficacious at increasing expression of other genes, we have not ruled out the possibility that other chemicals may affect WF and WKY Fbxo10 promoters in a time- and concentration-dependent manner. It is also highly possible that considering the entire Mcs5a1 locus would reveal additional environmental exposures capable of interacting with relevant transcriptional regulatory elements.
TPA and other phorbol ester family members were first found to act as tumor promoters via PKC in a mouse skin carcinogenesis model [20–22]. In our study, TPA differentially activated the luciferase reporter genes containing two variant 4.2 kb fragments of Mcs5a1 in Jurkat and MOLT-4 T cell lines, but not in HepG2 cells, a human liver carcinoma cell line. We also showed that endogenous FBXO10 mRNA and protein levels increased in Jurkat cells in response to TPA. It is possible that Fbxo10 plays a role in stress response in rat thymocytes during carcinogenesis and further supports that different levels of Fbxo10 expression between WF and WKY rat thymocytes and T cell populations partly contribute to mammary cancer susceptibility between these two strains. In support of this, Smits et al. reported that conserved rat Mcs5a, a synthetic mammary carcinoma susceptibility QTL that contains Fbxo10, functioned in a non-mammary cell-autonomous fashion through the immune system [37].
Figure 7 is a schematic showing our proposed mechanism of TPA-induced transcription of rat/human Fbxo10/FBXO10. We demonstrated, by using several PKC inhibitors (specifically GÖ6983 and GÖ6976), that PKCμ was a possible candidate signaling protein mediating TPA activation of FBXO10. Also named PKD1, PKCμ is a ubiquitous serine-threonine protein kinase involved in multiple cellular events [38,39]. Other PKCs may also be involved in TPA activation of WF and WKY 4.2 kb Fbxo10 promoters. For example, PKCμ itself can be activated by other PKC family members including PKCε, PKCη, and PKCθ [40,41]. PKCμ has been reported to modulate the MAPK pathway, leading to the activation of c-Fos and other target genes [42]. This supports our conclusion that c-Fos has a role in TPA activation of 4.2 kb cancer susceptible-WF and cancer resistant-WKY Fbxo10 promoters (Figure 7).
Figure 7.

A schematic of TPA-induced PKC-AP1 pathway regulation of 4.2 kb WF and WKY Fbxo10 promoters. Induction of PKCμ activity after TPA exposure results in increased c-Fos and an unidentified heterodimerization partner binding at Fbxo10-TRE (asterisk) in both WF and WKY 4.2 kb Fbxo10 promoters. Either one or both genetic variants marked by factor X? is hypothesized to be responsible for a difference in TPA-responsiveness of the WF and WKY 4.2 kb Fbxo10 promoters observed in this study. Black and white ellipses represent genetic variants between WF and WKY promoter sequences. Transcription factors c-Fos, an unidentified factor X heterodimerization partner, and possible coactivators are represented by squares, circles, and ovals, respectively. Potential differential DNA binding affinity between genetic variants is indicated by different factor X shape sizes.
The AP1 family member c-Fos has an important role in bone remodeling, tooth eruption, and hematopoiesis [43]. Of all AP1 family members tested, only c-Fos had an active role in up-regulation of the luciferase reporters containing 4.2 kb WF and WKY Fbxo10 promoter regions. A heterodimer partner of c-Fos, c-Jun, usually plays an activating role at target genes. However, in our T cell system, c-Jun and NRF2 antagonized both c-Fos and TPA activation of Fbxo10 reporter expression. Other members of the c-Fos and c-Jun family also blunted c-Fos regulation of the gene. Since c-Fos is believed not to form homodimers, these results implicate another yet to be determined c-Fos heterodimerization partner in mediating TPA activation of FBXO10/Fbxo10 (Figure 7) [44].
Previously, we showed that a general transcription factor named LEDGF is involved in the regulation of FBXO10 in response to environmental stress including heat and oxidative stress [45]. LEDGF has been reported to be induced by TPA, which is a known stimulator of H2O2 production, a form of oxidative stress that activates LEDGF [46]. Moreover, LEDGF binding SNP40 lies within 4.2 kb WF/WKY Fbxo10 promoters. Therefore, LEDGF may be responsible for constitutive transcription of FBXO10, while AP1 has a role in increasing transcription of FBXO10 (Figure 7). To this effect there are several predicted AP1-like binding sites in the human promoter region of FBXO10 that will be important to evaluate in future studies.
AP1-related DNA–protein complexes did not bind SNP36 (ss262858667) oligonucleotides containing WF or WKY variants. On the contrary, a highly predicted AP1 binding site with a core binding sequence of TGACTAA was shown to bind c-Fos and mediate TPA activation a of 4.2 kb Fbxo10 promoter region. We have named this TPA-responsive c-Fos binding site Fbxo10 TRE (Figure 7). The two rat strains studied have identical DNA sequences at this site. Considering that 4.2 kb WF and WKY Fbxo10 promoters are different in basal as well as induced promoter activity, it appears that genetic variation may contribute to the TPA responsiveness of these promoters. It is possible that polymorphisms in 4.2 kb WF and WKY Fbxo10 promoters might synergistically modify the TPA responsiveness of the functional Fbxo10 TRE we have identified.
In conclusion, we demonstrated that TPA regulates luciferase reporter genes containing variants of the 5′-flanking region of rat Fbxo10 via PKC and AP1 pathways involving c-Fos and an unidentified heterodimerization partner in Jurkat cells. The significance of this study to breast cancer susceptibility is that many environmental chemicals may activate PKC and c-Fos mediated increases in FBXO10 expression; so that, in addition to genetically determined FBXO10 expression, FBXO10 gene–environment interactions may contribute to breast cancer susceptibility. This knowledge may be useful to research investigating effects of environmental and genetic factors on breast cancer susceptibility, and this information may provide pathways toward prevention of breast cancer.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by National Institutes of Health grants CA137052 and P30ES014443, which supported a Career Development Award and a Pilot Project Grant from a P30 Environmental Health Sciences Core Center, and a Bales Fund Collaborative Matching Grant from the University of Louisville School of Medicine.
Grant sponsor:
National Institutes of Health; Grant numbers: CA137052; P30ES014443; Grant sponsor: P30 Environmental Health Sciences Core Center; Grant sponsor: University of Louisville School of Medicine
Abbreviations
- Mcs5a1
mammary carcinoma susceptibility 5a1 locus
- WF
Wistar Furth
- WKY
Wistar Kyoto
- QTL
quantitative trait locus
- Fbxo10
F-box protein 10
- TPA
12-O-tetradecanoylphorbol-13-acetate
- PKC
protein kinase C
- DAG
1,2-dioleoyl-sn-glycerol
- TRE
TPA response element
- AP1
activator protein 1
- EMSA
electrophoretic mobility shift assay
- QPCR
quantitative polymerase chain reaction
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- PDBu
phorbol 12, 13-dibutyrate
- SNP
single nucleotide polymorphism
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin 2012;62:10–29. [DOI] [PubMed] [Google Scholar]
- 2.Hindorff LA, Gillanders EM, Manolio TA. Genetic architecture of cancer and other complex diseases: lessons learned and future directions. Carcinogenesis 32:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bond GL. Hu W. Bond EE, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004;119:591–602. [DOI] [PubMed] [Google Scholar]
- 4.Meyer KB, Maia AT, O’Reilly M, et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol 2008;6:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu AH, Wan P, Hankin J, et al. Adolescent and adult soy intake and risk of breast cancer in Asian-Americans. Carcinogenesis 2002;23:1491–1496. [DOI] [PubMed] [Google Scholar]
- 6.Lin J, Manson JE, Lee IM, et al. Intakes of calcium and vitamin D and breast cancer risk in women. Arch Intern Med 2007;167: 1050–1059. [DOI] [PubMed] [Google Scholar]
- 7.Garland CF, Gorham ED, Mohr SB, et al. Vitamin D and prevention of breast cancer: pooled analysis. J Steroid Biochem Mol Biol 2007;103:708–711. [DOI] [PubMed] [Google Scholar]
- 8.Lappe JM, Travers-Gustafson D, Davies KM, et al. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr 2007;85:1586–1591. [DOI] [PubMed] [Google Scholar]
- 9.Sun CL, Yuan JM, Koh WP, et al. Green tea, black tea and breast cancer risk: a meta-analysis of epidemiological studies. Carcinogenesis 2006;27:1310–1315. [DOI] [PubMed] [Google Scholar]
- 10.Engel LS, Hill DA, Hoppin JA, et al. Pesticide use and breast cancer risk among farmers’ wives in the agricultural health study. Am J Epidemiol 2005;161:121–135. [DOI] [PubMed] [Google Scholar]
- 11.Xiong P, Bondy ML, Li D, et al. Sensitivity to benzo(a)pyrene diol-epoxide associated with risk of breast cancer in young women and modulation by glutathione S-transferase polymorphisms: A case–control study. Cancer Res 2001;61:8465–8469. [PubMed] [Google Scholar]
- 12.Yang M, Ryu JH, Jeon R, et al. Effects of bisphenol A on breast cancer and its risk factors. Arch Toxicol 2009;83:281–285. [DOI] [PubMed] [Google Scholar]
- 13.Dunning AM, Healey CS, Pharoah PD, et al. A systematic review of genetic polymorphisms and breast cancer risk. Cancer Epidemiol Biomarkers Prev 1999;8:843–854. [PubMed] [Google Scholar]
- 14.Ottman R Gene–environment interaction: Definitions and study designs. Prev Med 1996;25:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuelson DJ, Hesselson SE, Aperavich BA, et al. Rat Mcs5a is a compound quantitative trait locus with orthologous human loci that associate with breast cancer risk. Proc Natl Acad Sci USA 2007;104:6299–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smits B, Sharma D, Samuelson D, et al. The non-protein coding breast cancer susceptibility locus Mcs5a acts in a non-mammary cell-autonomous fashion through the immune system and modulates T-cell homeostasis and functions. Breast Cancer Res 2011;13:R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smits BMG, Traun BD, Devries TL, et al. An insulator loop resides between the synthetically interacting elements of the human/rat conserved breast cancer susceptibility locus MCS5A/Mcs5a. Nucleic Acids Res 2012;40:132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin J, Cardozo T, Lovering RC, et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 2004;18:2573–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiorazzi M, Rui L, Yang Y, et al. Related F-box proteins control cell death in Caenorhabditis elegans and human lymphoma. Proc Natl Acad Sci USA 2013;110:3943–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castagna M, Takai Y, Kaibuchi K, et al. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem 1982;257:7847–7851. [PubMed] [Google Scholar]
- 21.Blumberg PM. Protein kinase C as the receptor for the phorbol ester tumor promoters: Sixth Rhoads memorial award lecture. Cancer Res 1988;48:1–8. [PubMed] [Google Scholar]
- 22.Niedel JE, Kuhn LJ, Vandenbark GR. Phorbol diester receptor copurifies with protein kinase C. Proc Natl Acad Sci USA 1983;80:36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishizuka Y The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984;308:693–698. [DOI] [PubMed] [Google Scholar]
- 24.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J 1998;332:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishizuka Y Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 1995;9:484–496. [PubMed] [Google Scholar]
- 26.Gschwendt M, Dieterich S, Rennecke J, et al. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase cisoenzymes. FEBS Lett 1996;392: 77–80. [DOI] [PubMed] [Google Scholar]
- 27.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol 1997;9:240–246. [DOI] [PubMed] [Google Scholar]
- 28.Eferl R, Wagner EF. AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer 2003;3:859–868. [DOI] [PubMed] [Google Scholar]
- 29.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: Quarrel and harmony among siblings. J Cell Sci 2004;117:5965–5973. [DOI] [PubMed] [Google Scholar]
- 30.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002;4:E131–E136. [DOI] [PubMed] [Google Scholar]
- 31.Smits BM, Traun BD, Devries TL, et al. An insulator loop resides between the synthetically interacting elements of the human/rat conserved breast cancer susceptibility locus MCS5A/Mcs5a. Nucleic Acids Res 2011;40:132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wadman IA, Osada H, Grutz GG, et al. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J 1997;16:3145–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sundram V, Chauhan SC, Jaggi M. Emerging roles of protein kinase D1 in cancer. Mol Cancer Res 2011;9:985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Angel P, Imagawa M, Chiu R, et al. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 1987;49:729–739. [DOI] [PubMed] [Google Scholar]
- 35.Lee W, Mitchell P, Tjian R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 1987;49:741–752. [DOI] [PubMed] [Google Scholar]
- 36.Behmoaras J, Bhangal G, Smith J, et al. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat Genet 2008;40:553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smits BM, Sharma D, Samuelson DJ, et al. The non-protein coding breast cancer susceptibility locus Mcs5a acts in a non-mammary cell-autonomous fashion through the immune system and modulates T-cell homeostasis and functions. Breast Cancer Res 2011;13:R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johannes FJ, Prestle J, Eis S, et al. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem 1994;269:6140–6148. [PubMed] [Google Scholar]
- 39.Valverde AM, Sinnett-Smith J, Van Lint J, et al. Molecular cloning and characterization of protein kinase D: A target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci USA 1994;91:8572–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Lint J, Rykx A, Maeda Y, et al. Protein kinase D: An intracellular traffic regulator on the move. Trends Cell Biol 2002;12:193–200. [DOI] [PubMed] [Google Scholar]
- 41.Rykx A, De Kimpe L, Mikhalap S, et al. Protein kinase D: A family affair. FEBS Lett 2003;546:81–86. [DOI] [PubMed] [Google Scholar]
- 42.Guha S, Tanasanvimon S, Sinnett-Smith J, et al. Role of protein kinase D signaling in pancreatic cancer. Biochem Pharmacol 2010;80:1946–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang ZQ, Ovitt C, Grigoriadis AE, et al. Bone and haematopoietic defects in mice lacking c-fos. Nature 1992; 360:741–745. [DOI] [PubMed] [Google Scholar]
- 44.Porte D, Oertel-Buchheit P, John M, et al. DNA binding and transactivation properties of Fos variants with homo-dimerization capacity. Nucleic Acids Res 1997;25:3026–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu X, Powell DW, Lambring CJ, et al. Human MCS5A1 candidate breast cancer susceptibility gene FBXO10 is induced by cellular stress and correlated with lens epithelium-derived growth factor (LEDGF). Mol Carcinog 2012; doi: 10.1002/mc.21977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kubo E, Fatma N, Sharma P, et al. Transactivation of involucrin, a marker of differentiation in keratinocytes, by lens epithelium-derived growth factor (LEDGF). J Mol Biol 2002;320:1053–1063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.