

Abstract
In this paper, a reinforcement learning model is proposed that can maximize the predicted binding affinity between a generated molecule and target proteins. The model used to generate molecules in the proposed model was the Stacked Conditional Variation AutoEncoder (Stack-CVAE), which acts as an agent in reinforcement learning so that the resulting chemical formulas have the desired chemical properties and show high binding affinity with specific target proteins. We generated 1000 chemical formulas using the chemical properties of sorafenib and the three target kinases of sorafenib. Then, we confirmed that Stack-CVAE generates more of the valid and unique chemical compounds that have the desired chemical properties and predicted binding affinity better than other generative models. More detailed analysis for 100 of the top scoring molecules show that they are novel ones not found in existing chemical databases. Moreover, they reveal significantly higher predicted binding affinity score for Raf kinases than for other kinases. Furthermore, they are highly druggable and synthesizable.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13321-022-00666-9.
Keywords: De novo drug design, Reinforcement learning, Conditional Variational AutoEencoder, Sorafenib, Raf kinases
Background
The goal of drug discovery is to identify novel molecules with desired chemical or pharmacological properties. However, the search space for identification of such molecules is vast and discontinuous, which makes drug discovery difficult and costly [1]. To address this problem, many computational methods have been studied [2] and Computer-Aided Drug Discovery (CADD), such as Virtual Screening and Structure/Ligand-Based Drug Design, have been applied for drug discovery [3–5].
Recently, various deep generative models have been proposed and used to solve a variety of problems in drug discovery [6]. Many deep learning-based drug discovery studies express the chemical structure using the Simplified Molecular Input Line Entry System (SMILES) [7]. Models for generating SMILES expressions of chemicals include Recurrent Neural Network (RNN)-based models [8–10], Generative Adversarial Network (GAN)-based models [11], and AutoEncoder (AE)-based models [12–15] and their combinations.
The RNNs and their variations are powerful generative models, especially in natural language processing. Because SMILES is a string of characters, it is also effective for SMILES generation. However, while RNN is suitable for a training set, it may have limited ability to generate novel chemicals that are significantly different from the training set [10]. For this reason, RNNs and their variations are often used as the generators of GAN, or decoders and encoders of AE. For example, Guimaraes et al. [16] proposed a sequence-based GAN framework named objective-reinforced generative adversarial network (ORGAN). In this model, a CNN model was used as the discriminator to classify tests, and an RNN model with LSTM units was used as the generator.
GANs often give better results than other generation methods. However, a GAN has two main problems. First, a GAN is hard to converge and is unstable. For successful training of GAN-based models, a relatively large amount of computing and human effort is required. Another problem is the mode collapse problem. Generators of GAN-based models are trained to deceive the discriminator and cannot capture multimode distributions of real data [17].
Compared to GAN, an AE and its variations are relatively easy to train. For drug design, the encoder and decoder of AEs and their variations such as VAE (Variational AutoEncoder) and CVAE (Conditional Variational AutoEncoder) are often implemented using RNNs and their variations. A SMILE string can be generated by putting latent vectors made by the encoder into a decoder and using a predictor to measure the chemical properties [8]. A new molecule with the desired properties can be created by attaching conditions to the input data and latent vectors [9].
Meanwhile, reinforcement learning can be used to fine-tune pre-trained models [12–14, 18–20].In ReLeaSE [13], the policy to select a behavior is a generation model that is implemented using a stack-augmented RNN (Stack-RNN). The reward to learn policies is measured through the properties of the generated chemicals, and the generated model is retrained to have the desired chemical properties. PaccManRL [14] applied reinforcement learning to create an effective anticancer drug structure for given transcriptomic profiles. PaccManRL is trained to maximize a reward that is calculated by a drug sensitivity prediction model called PaccMann [15]. Olivecrona et al. [20] proposed a reinforcement learning model to generate drugs similar to existing drugs. This model measures the similarity between a specific drug and SMILES produced by the model and is trained to maximize the similarity. Liu et al. [18] introduced an RNN model with an exploration strategy. This model was trained with a reward that is created by a pre-trained predictor to predict whether the molecules generated are active or not.
The proposed method exploits not only the chemical properties of molecules but also the binding affinity between molecules and target proteins, that is, to where molecules should or should not bind. For a generative model, we designed Stack-CVAE, which applies Stack-RNN [19] to CVAE. We can input the desired chemical properties such as molecular weight, LogP, and the topological polar surface area (TPSA) [21] into Stack-CVAE. Then reinforcement learning is used to maximize or minimize the binding affinity calculated using DeepPurpose [22], and to maximize RAscore [23] to increase the synthesizability of the generated drugs.
In this study, we aimed to generate structures of a targeted anticancer drug of which indications and chemical properties are equivalent to or better than those of sorafenib. To achieve this goal, we first pre-trained Stack-CVAE using 1.5 million chemicals in the ChEMBL database [24] with the molecular weight, LogP, and TPSA of sorafenib. Then reinforcement learning was performed to maximize the RAscore and binding affinity score to sorafenib target Raf kinases (A-Raf, B-Raf, and C-Raf). Reinforcement learning was also used to minimize the binding affinity to sorafenib non-target kinases (ERK-1, MEK-1, EGFR, HER-2, IGFR-1, c-met, PKB, PKA, CDK1, PKCα, PKC γ, and pim-1) [25].
We generated 1000 chemical formulas and confirmed that Stack-CVAE generates more of the valid and unique chemical compounds that have the desired chemical properties and predicted binding affinity than other generative models do. For more detailed analysis, we selected 100 of the top scoring molecules from among 1000 molecules. The top 100 molecules are novel ones not found in existing chemical databases and have significantly higher binding affinity for Raf kinases. Their Druglikeness (DL), TPSA, and calculated LogP (cLogP) are similar to those of pre-existing drugs, and in silico ADME and toxicity profiles show that they are druggable. Furthermore, the synthetic accessibility (SA) score was comparable to those of approved drugs, which shows that they are synthesizable.
Methods
Conditional variational autoencoder with Stack augmented RNN (Stack-CVAE)
We designed a novel generative model, Stack-CVAE, that combines stack-RNN with CVAE to generate structural expressions in SMILES. Stack-CVAE is based on CVAE, which produces substances similar to, but not the same as, the substances used for training. The objective function of CVAE is as follows:
| 1 |
In formula (1), and approximate the probability distributions of an encoder and a decoder, respectively. The term is the Kullback–Leibler divergence, and and indicate input data and latent spaces, respectively. Here, the term indicates a condition vector that is associated with encoding and decoding.
The biggest difference between Stack-CVAE and CVAE is that CVAE uses regular RNN, LSTM, or GRU, and Stack-CVAE uses stack-augmented RNN (Stack-RNN). Stack-RNN is an augmented recurrent network with structured and growing memory. Stack-RNN has three operations. The POP operation deletes an element of a stack; the PUSH operation adds a new element to the top of a stack; and the NO-OP operation does not do anything. One of three operations is selected at each time step by a three-dimensional variable , which is calculated using a hidden variable as in formula (2).
| 2 |
In formula (2), is a matrix ( is the size of the hidden layer) and is a Softmax function. We denote by the probability of the PUSH operation, by the probability of the POP operation and by the probability of the NO-OP operation. The stack is stored in a vector with size p at time t, and p is not fixed in order to increase the capacity of the model. The top element is stored in position 0 with value of . The PUSH operation adds a new element to the position 0 as in formula (3).
| 3 |
where is matrix. If is equal to 1, the top element is popped and all the other elements are moved up one position. If is 1, all elements are moved down one position and the new element is added to the top of the stack. If is 1, the stack is not changed. Similarly, for an element stored at a depth > 0 in the stack, elements in the previous stack are stored in the current stack as in the formula (4).
| 4 |
An element of a stack is propagated to the next hidden layer, which is calculated by formula (5).
| 5 |
In formula (5), is matrix, is a top element of the stack of time point t-1, and is the size of an element. Because Stack-RNN is an RNN of which a cell has a stack structure, it has strength to learn longer and more complex data. Stack memory increases the probability of generating valid SMILES because RNN without a stack structure cannot learn ring structure or brackets. Furthermore, RNN without stack structure tends to generate SMILES that are similar to the training SMILES [16]. Because Stack-CVAE uses Stack-RNN as its encoder, the proportion of valid and unique SMILES of Stack-CVAE can be higher than that of CVAE of which the encoder is an RNN, GRU, or LSTM. We used GRU as a decoder. The model is depicted in Fig. 1.
Fig. 1.

Training and generation steps: Training steps of encoder and decoder of stack-CVAE are shown in a and b, respectively. Generation of SMILES by a decoder of stack-CVAE is shown in c
First, the '<' character, which indicates the start of a SMILES, is put in front of the SMILES string, and the '>' character, which indicates the end of a SMILES, is put at the end of a SMILES string. When input SMILES is put into the generation model, an embedding vector is generated and combined with a pre-produced condition vector to generate an input matrix. The condition vector is made by calculating the molecular weight, LogP, and TPSA of input the SMILES using RDkit [26]. The input matrix generated is input to the encoder of the Stack-CVAE and converted into a latent vector. The process for generating a latent vector is illustrated in Fig. 1a. The encoder of Stack-CVAE used a 3-layer Stack-RNN with 512 hidden nodes, a stack-width of size 50, a stack-depth of size 10, and a 3-layer GRU with 512 hidden nodes as a decoder. The cross entropy was used as the cost function of the reconstruct error, and a linear neural network was used for each output of the decoder cell.
A generated latent vector is combined with the condition vector and input to the decoder. Finally, the output of the decoder generates a distribution of the probability that each token will be selected through Softmax and the decoder is trained to predict a token correctly. This process is illustrated in Fig. 1b.
After training, a new molecule can be generated using the generator, that is, the decoder of stack-CVAE. When the start token '<' is entered, the decoder generates a new token. A generated token is re-entered into a decoder and this process is repeated until a decoder generates an end token '>'. This process is illustrated in Fig. 1c.
Reinforcement learning for drug properties
In the pre-training stage, the Stack-CVAE model learns the rules of SMILES given conditions such as molecular weight, LogP, and TPSA. Then, reinforcement learning is applied to the pre-trained Stack-RNN model to generate a new chemical compound with the desired properties. In reinforcement learning, an agent determines a behavior according to a policy that specifies the probability of taking an action in a state. An environment then rewards an agent, and an agent is trained based on rewards. Reinforcement learning is performed to maximize accumulated rewards returned by an environment.
In the proposed model, an agent corresponds to a decoder of stack-CVAE, and a policy is a probability distribution of a decoder of stack-CVAE to generate tokens. An action and a state of an agent mean creating one token and a generated token, respectively. When all actions are completed, a chemical formula in the form of SMILES is generated, and the reward is measured by calculating the RAscore and binding affinity of the SMILES generated.
More specifically, a state of a policy means the current token. If a current token is combined with the latent vector z generated using a random value from a normal distribution with mean = 0 and sd = 1, and put into a decoder of stack-CVAE, then the probability distribution for the next token given the latent vector z is calculated. The next token is sampled according to the probability distribution, and this process is repeated until the ending character '>' is generated. Until then, the reward is zero, and an environment calculates reward based on two predicted properties of the generated SMILES.
The first property is the predicted Retrosynthetic Accessibility score (RAscore), which is calculated by a synthesis planning tool, AiZynthFinder [27]. RAscore is a coefficient indicating the synthesis possibility and has the value 1 if synthesis is possible, and 0 if not. The reward by RAscore is reward1 and is calculated by formula (6).
| 6 |
The second property is the binding affinity of the produced SMILES to the target proteins. The binding affinity was calculated using DeepPurpose. For multiple target proteins, binding affinities were averaged. We had two groups of target proteins, group A and group D. Group A included target proteins that should bind to the generated SMILES, and group D included target proteins that should not bind to the generated SMILES. The averaged binding affinity for group A and D is affA and affD, respectively. The reward by the binding affinity to group A is reward2 and is calculated by formula (7).
| 7 |
Reward by binding affinity to group D is reward3, and is 6, if affD < 5.5 and 1, otherwise. The entire reward is a sum of reward1, reward2, and reward3.
A decoder of stack-CVAE is trained to increase the reward and decrease loss, which is calculated by formula (8).
| 8 |
In formula (8), means the length of the generated SMILES string, and is the probability that the th token appears. Discount reward is reward multiplied by a discount rate, which we set to 0.1. The overall reinforcement learning process is illustrated in Fig. 2.
Fig. 2.
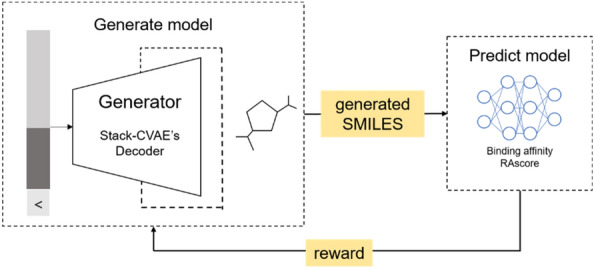
Pipeline of the reinforcement learning model for novel molecule generation
Results and discussion
Model performance
The Stack-CVAE model was trained with 1,499,939 SMILES data collected from the ChEMBL database. The length of the SMILES strings was limited to < 100, and molecular weight was limited to between 150 and 500. After pre-training, we generated 10,000 SMILES strings and found 6,463 SMILES strings among them that were valid.
In this study, we generated SMILES that show not only specific binding to Raf kinases, but also have properties that are equivalent to or better than those of sorafenib, a Raf kinase inhibitor. Sorafenib is a multikinase inhibitor and is known to inhibit Raf as well as other kinases [25, 28–31]. Recent studies show that sorafenib has inhibitory activity on more than 99 kinases at clinically relevant concentrations [32, 33]. This property may not only reduce the therapeutic efficacy of sorafenib, but could also cause toxicity due to increased drug use. Therefore, it is crucial to develop therapeutic drugs that can only target Raf kinases (i.e., on-target) rather than non-Raf kinases (i.e., off-target). To this end, we used a Stack-CVAE model to make Raf kinase-specific SMILES. We used three Raf kinases (A-Raf, B-Raf, and C-Raf) as sorafenib targets and ten non-Raf kinases (ERK-1, MEK-1, EGFR, HER-2, IGFR-1, c-met, PKB, PKA, CDK1, PKCα, PKCγ, and pim-1) as non-sorafenib targets.
Because Stack-CVAE is based on CVAE and Stack-RNN, we performed reinforcement learning and compared results using pre-trained Stack-CVAE, CVAE, and Stack-RNN as a policy. We also used SSVAE [34] and cRNN [35] as a policy for a comparison test. Figure 3 shows that loss and reward of three generation models decreases and increases, respectively, as training epochs increase.
Fig. 3.

Loss and reward graphs of different generative models: Loss and Rewards graphs of stack-CVAE (a), CVAE (b), SSVAE(c), stack-RNN (d) and cRNN (e)
After reinforcement learning with 500 epochs, we generated 1000 SMILES strings for Stack-CVAE, CVAE, and Stack-RNN. The results are summarized in Table 1, which shows that, although Stack-RNN has higher probability of valid molecules, Stack-CVAE has much higher probability of valid and unique molecules than other algorithms do. Moreover, we can see that Stack-RNN generates molecules that are the same as the training molecules.
Table 1.
Proportion of valid and unique molecules
| % of valid SMILES | % of valid and unique SMILES | % of valid and unique SMILES not in training data | |
|---|---|---|---|
| Stack-CVAE (%) | 98.1 | 81.1 | 81.1 |
| CVAE (%) | 98.1 | 39.9 | 39.9 |
| SSVAE (%) | 98.8 | 16.6 | 16.6 |
| Stack-RNN (%) | 100 | 1.5 | 1.2 |
| cRNN (%) | 99.7 | 25.2 | 25.2 |
Stack-CVAE can generate a greater number of valid and diverse molecules. However, diverse molecules would be meaningless if they do not show the desired properties. To show the proportion of molecules that show desired properties, we calculated the number of SMILES strings of which binding affinity to on-targets and off-targets, RAscore, molecular weight, logP, and TPSA values fell within 10% range of those of sorafenib (among the valid and unique SMILES not in training data for Stack-CVAE, SSVAE, cRNN, CVAE, and StackRNN). Table 2 shows the summarized results. Compared to CVAE, Stack-CVAE has higher probability of generating molecules with the desired properties, except for on-target binding affinity. SSVAE and cRNN show higher probability on RAscore, LogP, Molecular Weight and/or TPSA compared to Stack-CVAE, but the number of molecules is far less than that of Stack-CVAE.
Table 2.
Molecules having desired properties among valid and unique SMILES not in training data
| Stack-CVAE | SSVAE | cRNN | CVAE | Stack-RNN | |
|---|---|---|---|---|---|
| Binding Affinity on on-target proteins | 652 (80.39%) | 133 (80.12%) | 145(57.54%) | 368 (92.23%) | 11 (91.67%) |
| Binding Affinity on off-target proteins | 810 (99.88%) | 139 (83.73%) | 236(93.65%) | 392 (98.25%) | 12 (100%) |
| RAscore | 566 (69.79%) | 158 (95.18%) | 248(98.41%) | 198 (49.62%) | 1 (8.33%) |
| Molecular Weight | 532 (65.60%) | 147 (88.55%) | 198(78.57%) | 134 (33.58%) | 0 (0%) |
| LogP | 623 (76.82%) | 129 (77.71%) | 180(71.43%) | 238 (59.65%) | 0 (0%) |
| TPSA | 594 (73.24%) | 138 (83.13%) | 194(76.98%) | 278 (69.67%) | 0 (0%) |
Quantitative and qualitative analysis of the top 100 scoring molecules
We scored 1000 molecules according to the formula (9).
| 9 |
where mw, logP, and TPSA are 0–1 scaled differences of molecular weights, logP, and TPSA of sorafenib and generated molecules, respectively. The items bindA and bindD are 0–1 scaled values of an average binding affinity between a generated molecule and on-target and off-target proteins, respectively. Molecules with high scores are likely to have more chemical properties similar to those of sorafenib. We selected 100 of the top scoring molecules and performed more detailed analysis. The top 100 molecules are shown in Additional file 1: Fig. S1.
First, we checked the novelty of the top 100 molecules. Figure 4a and b show that the top 100 molecules are dissimilar to sorafenib and 1,585 FDA-approved drugs, respectively. We also checked that the top 100 molecules did not overlap with 9,309 DrugBank [36] drugs and 1,585 FDA-approved drugs within the similarity limit of 90%. All drug data were downloaded from DataWarrior 5.5.0 [37].
Fig. 4.

Novelty of the top 100 molecules: Structures of the top 100 scoring molecules are not similar to that of sorafenib (a) and FDA-approved drugs (b), within a similarity limit of 95%. The top 100 scoring molecules do not overlap DrugBank and FDA-approved drugs within a similarity limit of 90% (see c). All analyses were performed using DataWarrior 5.5.0
Next, we verified whether the top 100 molecules are specific to Raf kinases (Group A) only. Figure 5b shows that, even though sequence and structure homology of the kinase domains are high, the binding of the top 100 molecules is specific to Raf kinases, but not to other kinases (Group B and D). Group B kinases are known to be inhibited by sorafenib. However, the top 100 molecules show higher binding affinity to Raf kinases than that of Group B kinases. This result was further confirmed by structure-based molecular docking analysis, which showed that the top 100 molecules exhibit more favorable interaction with B-RAF (Group A) than with Group B or Group D (Fig. 5c). Taken together, these results indicate that the proposed model can significantly reduce off-target drug interaction.
Fig. 5.

Binding specificity of the top 100 molecules: List of kinase proteins used for study is shown in a. Binding of the top 100 molecules is specific to Raf kinases, but not to other kinases (see b). Binding specificity is confirmed by molecular docking [38, 39] (see c). Box and Violin plots are drawn by BoxPlotR [40] and PlotsOfData [41], respectively
We also found out whether the top 100 molecules are druggable. Figure 6a, b and c show that Druglikeness (DL), topological polar surface area (TPSA), and calculated logP (cLogP) of the top 100 molecules are comparable to those of pre-existing DrugBank and FDA-approved drugs.
Fig. 6.

Comparison of druggability: Distribution of Druglikeness (a), Topological polar surface area (TPSA) (b), and Calculated logP (c) of DrugBank drugs, FDA-approved drugs, and the top 100 molecules. Violin plot drawn by PlotsOfData
We evaluated whether the top 100 molecules have more desirable ADME (Absorption, Distribution, Metabolism, and Excretion) profiles when compared with FDA-approved drugs using ADMETlab2.0 [42]. Among 23 classified models, seven representative models were included in Fig. 7. The overall ADME profile of the top 100 molecules shows similarity to FDA-approved drugs. Specifically, the top 100 molecules have distribution and excretion profiles similar to those of FDA-approved drugs. Furthermore, their evaluation scores for distribution and excretion are excellent (Fig. 7b and d). On the other hand, the evaluation score for absorption and metabolism is moderate and slightly poor, respectively (Fig. 7a and 7c).
Fig. 7.

In silico ADME profiles: Profiles of Absorption (a), Distribution (b), Metabolism (c), and Excretion (d). Violin plot drawn by PlotsOfData
We also performed in silico toxicity profiling analysis. Toxicity data of chemical molecules were obtained from Gadaleta et al. [43]. Figure 8a shows that the chemical acute toxicity of the top 100 molecules was a little higher than non-toxic drugs and FDA-approved drugs, but still very much lower than toxic drugs. Figure 8b shows in silico pharmacological profiling and assessment of the potential interaction for 64 toxicity off-targets, and we can observe that the profiles of the top 100 molecules are similar to non-toxic drugs rather than to toxic drugs. In silico pharmacological profiles were created by ToxProfiler [44]. Figures 6, 7, and 8 all show that the top 100 molecules are highly druggable.
Fig. 8.

In silico toxicity profiles: Acute oral toxicity (median lethal death, LD50) prediction was calculated by Xu et al. [45] (see a). In silico pharmacological profiling and assessment of the potential for off-target interactions of drugs are shown in b. Heatmap and violin plot are drawn by PlotsOfData and ToxProfiler, respectively
Finally, we checked whether the top 100 molecules are synthesizable. Figure 9a shows that the Synthetic Accessibility (SA) scores of the top 100 molecules are slightly higher than those of DrugBank drugs and FDA-approved drugs; nevertheless, the SA scores of the top 100 molecules exist within the acceptable range based upon previous publications [46]. Drugs for comparison were selected according to their molecular weight (300 < MW < 600). Figure 9b shows that 19, 56, and 15 drugs among the top 100 molecules can be synthesized in 2, 3, and 4 steps, respectively, through solved retrosynthetic pathways [47]. Further analysis shows that the top 100 molecules with fewer steps have good RA scores (Fig. 9c). Taken together, these results suggest that 90 out of 100 molecules can be synthesized using chemical reactions.
Fig. 9.

Comparison of synthesizability: Comparison of SA score (a). Feasibility of compound synthesis by solved retrosynthetic routes of top 100 molecules [47] (b). Correlation analysis between retrosynthetic steps and the RA score (c). Violin and box plots were drawn by PlotsOfData and BoxPlotR, respectively
Conclusions
For this study, we proposed a reinforcement learning model that could maximize the predicted binding affinity between a generated molecule and target proteins, while existing methods mainly consider the chemical properties of the created molecules. The agent of the proposed reinforcement learning model is Stack-CVAE, and policy is a probability distribution of the decoder of stack-CVAE. The rewards are measured using the synthesizability of the generated molecules and the binding affinity between the generated molecules and the target proteins, and the model is trained to increase the rewards.
After the model was trained, we generated 1000 chemical formulas using chemical properties and the three Raf target proteins of sorafenib. The proportion of valid and unique chemical compounds of Stack-CVAE is higher than that of other generative models. The proportion of chemical compounds that have the desired chemical properties (such as molecular weight, logP, TPSA, and RAscore) and higher binding affinity of Stack-CVAE is also higher. Further quantitative and qualitative analysis of the top 100 scoring molecules shows that they are novel and have higher binding affinity for Raf proteins than for other proteins. This 100 of the top scoring molecules are also highly druggable and synthesizable.
Supplementary Information
Additional file 1: Figure S1. Top 100 SMILES.
Acknowledgements
Not Applicable
Abbreviations
- AE
AutoEncoder
- CADD
Computer Aided Drug Discovery
- CVAE
Conditional AutoEncoder
- DL
Druglikeness
- TPSA
Topological polar surface
- SA
Synthetic accessibiliry
- DNN
Deep Neural Network
- DTBA
Drug-Target Binding Affinity
- LBDD
Ligand-Based Drug Design
- LSTM
Long Short-Term Memory
- S2SAE
Sequence-To-Sequence Auto-Encoder
- SBDD
Structure-Based Drug Design
- SMILES
Simplified Molecular Input Line Entry System
- Stack-RNN
Stack augmented RNN
- stack-CVAE
Conditional variational autoencoder with Stack augmented RNN
- RAscore
Retrosynthetic accessibility score
- RNN
Recurrent Neural Network
- GAN
Generative Adversarial Network
- GRU
Gated Recurrent Units
- VAE
Variational AutoEncoder
Author contributions
HK, SK, and JA conceived and designed the experiments; HK and SK performed the experiments; HK, SR, and BK analyzed the data; HK, BK, and JA wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT [NRF-2019R1A2C3005212].
Availability of data and materials
https://github.com/HwanheeKim813/stack_CVAE.git, DrugBank, https://www.drugbank.ca/releases/latest, ChEMBL, https://www.ebi.ac.uk/chembl/
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hwanhee Kim, Email: iamhappy@inu.ac.kr.
Soohyun Ko, Email: gerko@genesisego.com.
Byung Ju Kim, Email: byungju@hotmail.com.
Sung Jin Ryu, Email: sjryu2013@gmail.com.
Jaegyoon Ahn, Email: jgahn@inu.ac.kr.
References
- 1.Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–D1395. doi: 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin XX, Li X, Lin XX. A review on applications of computational methods in drug screening and design. Molecules. 2020;25:1–17. doi: 10.3390/molecules25061375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shoichet BK. Virtual screening of chemical libraries. Nature. 2005;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scior T, Bender A, Tresadern G, et al. Recognizing pitfalls in virtual screening: a critical review. J Chem Inf Model. 2012;52:867–881. doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 5.Cheng T, Li Q, Zhou Z, et al. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012;14:133–141. doi: 10.1208/s12248-012-9322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H, Engkvist O, Wang Y, et al. The rise of deep learning in drug discovery. Drug Discov Today. 2018;23:1241–1250. doi: 10.1016/j.drudis.2018.01.039. [DOI] [PubMed] [Google Scholar]
- 7.Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–36. doi: 10.1021/ci00057a005. [DOI] [Google Scholar]
- 8.Gómez-Bombarelli R, Wei JN, Duvenaud D, et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci. 2018;4:268–276. doi: 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim J, Ryu S, Kim JW, Kim WY. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J Cheminform. 2018;10:31. doi: 10.1186/s13321-018-0286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams RJ. Simple statistical gradient-following algorithms for connectionist reinforcement learning. Mach Learn. 1992;8:229–256. doi: 10.1007/bf00992696. [DOI] [Google Scholar]
- 11.Jaques N, Gu S, Bahdanau D, et al (2017) Sequence Tutor: Conservative Fine-Tuning of Sequence Generation Models with KL-control. Proceedings of the 34th International Conference on Machine Learning, PMLR 70 4:1645–1654
- 12.Yu L, Zhang W, Wang J, Yu Y (2017) SeqGAN: Sequence generative adversarial nets with policy gradient. In: 31st AAAI conference on artificial intelligence AAAI, pp 2852–2858
- 13.Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv. 2018;4:aap7885. doi: 10.1126/sciadv.aap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Born J, Manica M, Oskooei A, et al. PaccMann(RL): de novo generation of hit-like anticancer molecules from transcriptomic data via reinforcement learning. iScience. 2021;24:102269. doi: 10.1016/j.isci.2021.102269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cadow J, Born J, Manica M, et al. PaccMann: a web service for interpretable anticancer compound sensitivity prediction. Nucleic Acids Res. 2020;48:W502–W508. doi: 10.1093/nar/gkaa327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guimaraes GL, Sanchez-Lengeling B, Outeiral C, Farias PLC, Aspuru-Guzik A (2017) Objective-reinforced generative adversarial networks (organ) for sequence generation models. arXiv preprint arXiv:1705.10843
- 17.Srivastava A, Valkov L, Russell C, et al (2017) VEEGAN: reducing mode collapse in GANs using implicit variational learning. In: Advances in neural information processing systems 30 (NIPS 2017). Neural Information Processing Systems, pp 3308–3318
- 18.Liu X, Ye K, van Vlijmen HWT, et al. An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: a case for the adenosine A2A receptor. J Cheminform. 2019;11:1–16. doi: 10.1186/s13321-019-0355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joulin A, Mikolov T (2015) Inferring algorithmic patterns with stack-augmented recurrent nets. In: proceedings of the 28th international conference on neural information processing systems, vol 1. MIT Press, Cambridge, MA, USA, pp 190–198
- 20.Olivecrona M, Blaschke T, Engkvist O, Chen H. Molecular de-novo design through deep reinforcement learning. J Cheminform. 2017;9:48. doi: 10.1186/s13321-017-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasanna S, Doerksen RJ. Topological polar surface area: a useful descriptor in 2D-QSAR. Curr Med Chem. 2009;16:21–41. doi: 10.2174/092986709787002817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang K, Fu T, Glass LM, et al. DeepPurpose: a deep learning library for drug-target interaction prediction. Bioinformatics. 2020;36(22-23):5545–5547. doi: 10.1093/bioinformatics/btaa1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thakkar A, Chadimová V, Bjerrum EJ, et al. Retrosynthetic accessibility score (RAscore)-rapid machine learned synthesizability classification from AI driven retrosynthetic planning. Chem Sci. 2021;12:3339–3349. doi: 10.1039/d0sc05401a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bento AP, Gaulton A, Hersey A, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilhelm SM, Carter C, Tang LY, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 26.Landrum G (2013) RDKit: a software suite for cheminformatics, computational chemistry, and predictive modeling. Academic Press, Cambridge
- 27.Genheden S, Thakkar A, Chadimová V, et al. AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J Cheminform. 2020;12:1–9. doi: 10.1186/s13321-020-00472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilhelm S, Carter C, Lynch M, et al. Erratum: Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2007;6:168. doi: 10.1038/nrd2262. [DOI] [PubMed] [Google Scholar]
- 29.Carlomagno F, Anaganti S, Guida T, et al. BAY 43–9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst. 2006;98:326–334. doi: 10.1093/jnci/djj069. [DOI] [PubMed] [Google Scholar]
- 30.Namboodiri HV, Bukhtiyarova M, Ramcharan J, et al. Analysis of imatinib and sorafenib binding to p38α Compared with c-Abl and b-Raf provides structural insights for understanding the selectivity of inhibitors targeting the DFG-out form of protein kinases. Biochemistry. 2010;49:3611–3618. doi: 10.1021/bi100070r. [DOI] [PubMed] [Google Scholar]
- 31.Bergeron P, Koehler MFT, Blackwood EM, et al. Design and development of a series of potent and selective type II inhibitors of CDK8. ACS Med Chem Lett. 2016;7:595–600. doi: 10.1021/acsmedchemlett.6b00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 33.Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 34.Kang S, Cho K. Conditional molecular design with deep generative models. J Chem Inf Model. 2018 doi: 10.1021/acs.jcim.8b00263. [DOI] [PubMed] [Google Scholar]
- 35.Kotsias P-C, Arús-Pous J, Chen H, et al. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat Mach Intell. 2020;2:254–265. doi: 10.1038/s42256-020-0174-5. [DOI] [Google Scholar]
- 36.Law V, Knox C, Djoumbou Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 38.Guedes IA, Barreto AMS, Marinho D, et al. New machine learning and physics-based scoring functions for drug discovery. Sci Rep. 2021;11:3198. doi: 10.1038/s41598-021-82410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guedes IA, Krempser E, Dardenne LE (2017) DockThor 2.0 : a free web server for protein-ligand virtual screening, vol 2013, pp 2013–2014
- 40.Spitzer M, Wildenhain J, Rappsilber J, Tyers M. BoxPlotR: a web tool for generation of box plots. Nat Methods. 2014;11:121–122. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Postma M, Goedhart J. Plotsofdata—a web app for visualizing data together with their summaries. PLoS Biol. 2019;17:1–8. doi: 10.1371/journal.pbio.3000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong G, Wu Z, Yi J, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49:W5–W14. doi: 10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gadaleta D, Vuković K, Toma C, et al. SAR and QSAR modeling of a large collection of LD50 rat acute oral toxicity data. J Cheminform. 2019;11:1–16. doi: 10.1186/s13321-019-0383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.AbdulHameed MDM, Liu R, Schyman P, et al. ToxProfiler: toxicity-target profiler based on chemical similarity. Comput Toxicol. 2021;18:100162. doi: 10.1016/j.comtox.2021.100162. [DOI] [Google Scholar]
- 45.Xu Y, Pei J, Lai L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model. 2017;57:2672–2685. doi: 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- 46.Ertl P, Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform. 2009;1:1–11. doi: 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parrot M, Tajmouati H, Barros Ribeiro da Silva V, et al (2021) Integrating Synthetic Accessibility with AI-based Generative Drug Design. ChemRxiv. doi: 10.26434/chemrxiv-2021-jkhzw This content is a preprint and has not been peer-reviewed. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Top 100 SMILES.
Data Availability Statement
https://github.com/HwanheeKim813/stack_CVAE.git, DrugBank, https://www.drugbank.ca/releases/latest, ChEMBL, https://www.ebi.ac.uk/chembl/