

Abstract
Two vaccine formulations previously shown to induce protective immunity in mice and prevention of long-term necrosis in guinea pigs were tested as potential immunotherapeutic vaccines in mice earlier infected by aerosol with Mycobacterium tuberculosis. Neither vaccine had any effect on the course of the infection in the lungs, but both reduced the bacterial load in the spleen. Similarly, inoculation with Mycobacterium bovis BCG had no effect whatsoever and, if given more than once, appeared to induce an increasingly severe pyogranulomatous response in the lungs of these mice.
The current vaccine against tuberculosis, Mycobacterium bovis BCG, is cheap to produce and safe to administer, but it is far from effective, especially when the individual reaches the teenage years and above (15, 16). This lack of protective efficacy, now observed in several controlled clinical trials, has resulted in considerable effort being directed toward the construction of a new generation of tuberculosis vaccines (9–11).
Not only will this process be difficult, despite the innovative approaches that have already emerged, but the question remains as to when a new vaccine should be given and how its efficacy should then be monitored. The most obvious approach would be to vaccinate early in life, prior to any exposure to or infection with Mycobacterium tuberculosis, and then to monitor individuals for many decades to see if they then contract disease. Here the unanswered question is whether the vaccine should replace BCG, be given in tandem, or used to boost the immunity induced by the BCG vaccine.
There is an alternative approach recently suggested in which the vaccine is given as a postexposure or immunotherapeutic vaccine after the individual has been infected with M. tuberculosis (4). The idea here is that this type of vaccine not only will amplify the ongoing response but also may have other benefits such as overcoming aberrant responses to other “environmental” mycobacteria which may compromise the capacity of the individual to respond adequately to pulmonary infection.
In the current study, we examined the possibility that two newly developed subunit and DNA vaccines, recently shown to be effective in the mouse and guinea pig models against pulmonary tuberculosis (1, 5), might have some effect if given as an immunotherapeutic vaccine. The results of the study show, however, that neither these materials, nor BCG itself, modulated the course of an aerosol infection with M. tuberculosis. Interestingly, however, both nonliving vaccines reduced bacterial counts in the spleen, either directly or by reducing dissemination from the lungs. Finally, in mice given additional inoculations with BCG, an increasingly severe pyogranulomatous response was seen in the lungs as the tuberculosis infection progressed.
Specific-pathogen-free female C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, Mass.). Mice 6 to 8 weeks in age were kept in ABL-3 biohazard facilities throughout the study and maintained with sterile water, bedding, and mouse chow. M. tuberculosis Erdman was originally obtained from the Trudeau Mycobacteria Collection, Saranac Lake, N.Y. Bacteria were grown in Proskauer-Beck liquid medium containing 0.05% Tween 80 to mid-log phase and frozen in aliquots at −70°C until needed. Animals were infected by the aerosol route with a low dose of bacteria; briefly, the nebulizer compartment of an airborne infection apparatus (Glas-Col, Terre Haute, Ind.) was filled with 5 ml of a suspension containing 107 bacteria to allow uptake of 50 to 100 viable bacteria per lung during a 30-min exposure. The numbers of viable bacteria in the lung, liver, and spleen were determined at various time points by plating serial dilutions of whole-organ homogenates onto nutrient Middlebrook 7H11 agar (Life Technologies, Gaithersburg, Md.) and counting bacterial colonies after 20 to 30 days of incubation at 37°C in humidified air. The data are expressed as log10 values of the mean numbers of bacteria recovered per organ (n = 4 animals).
Vaccination was administered twice, 50 and 73 days after infection; control mice were given 200 μl of saline subcutaneously. BCG (strain Pasteur) was given in a dose of 106 subcutaneously in 200 μl of sterile saline. Mid-log-phase culture filtrate proteins (CFP) from M. tuberculosis were prepared and purified as described previously (14) and then emulsified in MPL-TeoA adjuvant (Ribi Immunochem, Hamilton, Mont.) supplemented with 100 μg of recombinant interleukin 2 (IL-2) (Chiron Corp., Emeryville, Calif.). DNA vaccines consisting of the control plasmid vector V1Jns or V1Jns containing the gene encoding the secreted form of M. tuberculosis antigen 85A (Ag85A) protein (Ag85-DNA) were kindly provided by Merck Research Laboratories (West Point, Pa.). Mice were given 50 μg of plasmid DNA in saline by injection into a hind quadriceps muscle.
Lungs were harvested, fixed in formal saline, and then sectioned and stained with hematoxylin and eosin. Slides were read by a veterinary pathologist who lacked prior knowledge of the treatment groups.
As shown in Fig. 1, the infection grew progressively in the lungs for approximately 30 days before entering into a chronic stage of disease in which the bacterial load did not fluctuate appreciably. The various vaccines and appropriate control inoculations were given on days 50 and 73, and potential changes in bacterial load were assayed on days 95 and 150. No differences were observed in lung loads at either time point, in any of the groups (Table 1). Interestingly, despite no apparent effects in the lungs, mice receiving the Ag85-DNA vaccine or the CFP-based vaccine both showed evidence of statistically significant reductions in splenic bacterial loads which were not seen in vaccine vehicle controls.
FIG. 1.

Course of infection in the lungs of control mice. Data are shown as mean values (n = 4) ± standard errors of the means. Arrows indicate administration of immunotherapeutic vaccines to test groups.
TABLE 1.
Changes in bacterial load
| Day | Postexposure vaccine | Value for organ:
|
P | |||
|---|---|---|---|---|---|---|
| Lung
|
Spleen
|
|||||
| CFU (mean log10) | SEM | CFU (mean log10) | SEM | |||
| 95 | Saline control | 5.2 | 0.2 | 4.2 | 0.5 | |
| BCG | 5.4 | 0.4 | 3.9 | 0.1 | ||
| DNA vector | 5.2 | 0.3 | 4.5 | 0.4 | ||
| DNA-Ag85 | 5.3 | 0.2 | 3.0 | 0.4 | <0.01 | |
| MPL-CFP | 5.3 | 0.4 | 4.7 | 0.6 | ||
| MPL–CFP–IL-2 | 5.4 | 0.3 | 3.4 | 0.1 | 0.059 | |
| MPL | 4.9 | 0.3 | 4.2 | 0.3 | ||
| 150 | Saline control | 5.1 | 0.1 | 4.8 | 0.3 | |
| BCG | 5.1 | 0.5 | 4.4 | 0.5 | ||
| DNA vector | 5.0 | 0.3 | 4.1 | 0.3 | ||
| DNA-Ag85 | 5.0 | 0.1 | 3.8 | 0.4 | <0.02 | |
| MPL-CFP | 5.2 | 0.4 | 4.5 | 0.1 | ||
| MPL–CFP–IL-2 | 5.5 | 0.3 | 3.8 | 0.4 | <0.01 | |
| MPL | 5.2 | 0.2 | 4.1 | 0.3 | ||
No histologic changes were seen in the lungs of mice given the experimental vaccines, with all animals exhibiting a typical multifocal lymphocytic granulomatous response after exposure to the challenge infection. In mice receiving BCG given after the tuberculosis infection, some adverse pathology was noted, and so in a second experiment, the BCG vaccine was given up to three times at 15-day intervals after challenge. In this experiment, the control mice again exhibited expected pathology as reported above (Fig. 2), but animals given BCG after the tuberculosis infection developed increasingly severe pyogranulomatous responses (lesions containing large numbers of neutrophils) with the degree of severity seeming to mirror the number of BCG inoculations. Such abscesses are known to have the potential to be fatal (3).
FIG. 2.
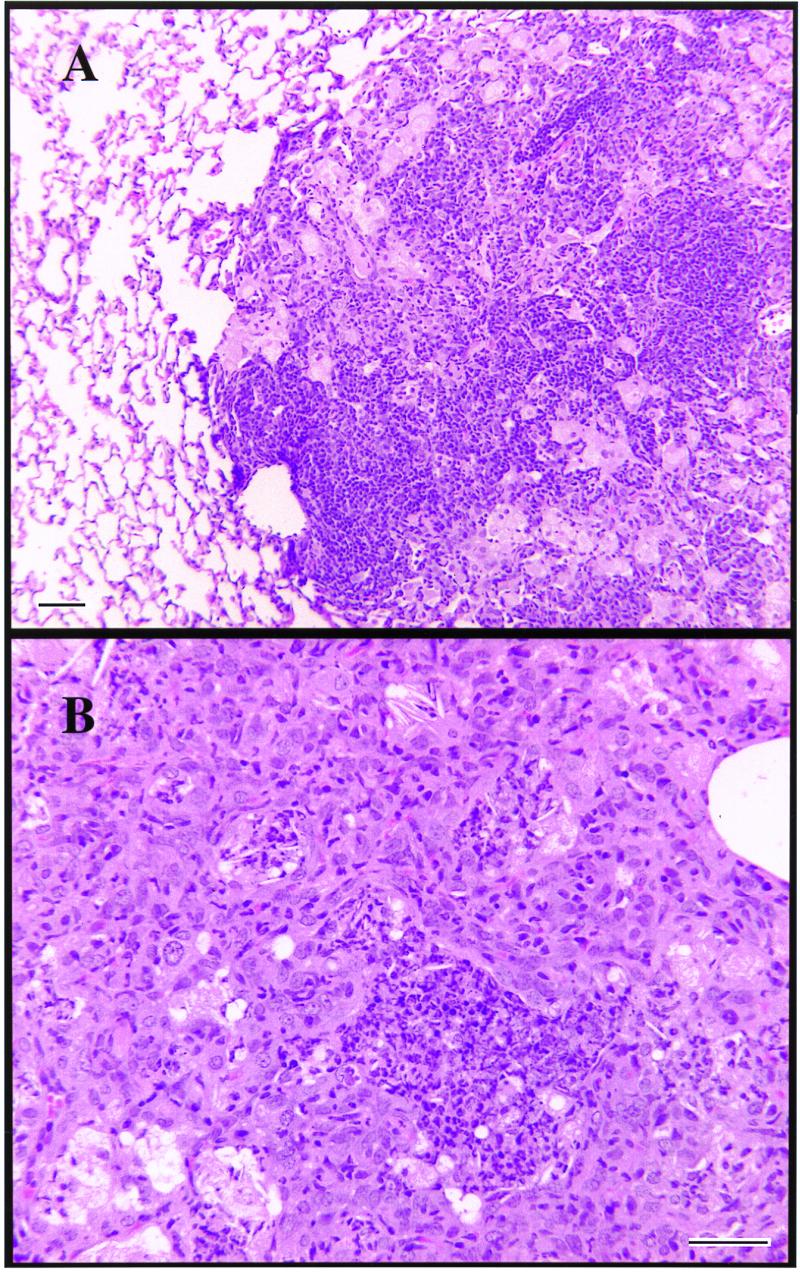
(A) Large lymphocytic granuloma in the lungs of infected control mice (day 95). (B) Pyogranulomatous cellular infiltration creating abscess-like lesion in lungs of mice given three separate injections of BCG postaerosol (day 120). Bars, 100 μm.
The results of this study show that immunotherapeutic inoculation of mice with vaccine formulations that have previously been shown to have a protective effect when given prior to challenge did not have any apparent effect on the course of the infection in the lungs. Interestingly, however, mice given the DNA-based vaccine and the CFP-based vaccine during the tuberculosis infection exhibited reductions in the bacterial load seen in the spleen. This is hard to interpret, since it could be due to a prevention of bacterial dissemination from the lungs to the spleen or could indicate better expression of immunity in the spleen itself, curtailing bacterial growth, or a combination of both.
The immunotherapeutic vaccination was given at a time when the animal was entering into a phase of chronic disease in the lungs, when the granulomatous response was well underway (13). The failure of T cells potentially boosted by the vaccines could simply be due to the fact that they could not focus on the granuloma structures, which by this time were relatively large, or alternatively could have happened because the bacteria themselves shut down metabolically and entered a latent stage in which relevant antigens such as Ag85 were not presented. Currently, we favor the first hypothesis, simply because bacteria in this chronic stage are still susceptible to isoniazid in vivo (8), indicating that mycolic acid biosynthesis, in which the Ag85 mycolyl transferase plays a key role (2), is continuing.
The observation that BCG given after challenge not only was ineffective but also seemed to trigger detrimental pathologic changes in the lungs was completely unexpected. The effect was marginal if BCG was given once but became more prolonged and severe if the vaccine was given twice or three times (at 15-day intervals). At this time, we can offer only speculations on this event. The first would be that BCG is boosting some type of immunity induced by the tuberculosis infection that causes direct local tissue damage, which would in turn cause the attraction of polymorphonuclear phagocytes. A second, perhaps more subtle, speculation is that the boosting response induced by the presence of BCG down-regulates or subverts the ordered generation of the granuloma. In this regard, we have seen a similar type of pathology developing in mice lacking γδ T cells (3) and have hypothesized (12) that such cells are involved in granuloma development with the minimum of tissue damage. Other mechanisms also could be subverted, such those mediated by IL-10 and transforming growth factor β in the down-regulation of acquired immunity (7, 17).
It has been suggested previously that BCG be used as a vector to carry M. tuberculosis genes no longer present in BCG or to express mammalian cytokines designed to boost immunity or used as an auxotrophic vaccine (9). While these are all promising avenues, the results here suggest caution in the use of BCG as an immunotherapeutic vaccine candidate. In fact, to date, only one vaccine candidate, a DNA vaccine derived from the hsp60 heat shock protein of Mycobacterium leprae, has been shown to have an immunotherapeutic vaccine effect (6). This effect is spectacular, with a thousandfold reduction in bacterial counts in both the spleen and lungs and complete prevention of reactivation of bacteria in a Cornell model system.
Acknowledgments
This work was supported by NIH grant AI-44072 and NIH contract AI-75320 and by a generous grant from Merck, Inc.
We thank Terry Ulrich and Marty Giedlin for providing additional vaccine reagents.
REFERENCES
- 1.Baldwin S L, D'Souza C, Roberts A D, Kelly B P, Frank A A, Lui M A, Ulmer J B, Huygen K, McMurray D M, Orme I M. Evaluation of new vaccines in the mouse and guinea pig model of tuberculosis. Infect Immun. 1998;66:2951–2959. doi: 10.1128/iai.66.6.2951-2959.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belisle J T, Vissa V D, Sievert T, Takayama K, Brennan P J, Besra G S. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276:1420–1422. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 3.D'Souza C D, Cooper A M, Frank A A, Mazzaccaro R J, Bloom B R, Orme I M. An anti-inflammatory role for gamma delta T lymphocytes in acquired immunity to Mycobacterium tuberculosis. J Immunol. 1997;158:1217–1221. [PubMed] [Google Scholar]
- 4.Fine P E. Vaccines, genes and trials. Novartis Found Symp. 1998;217:57–69. doi: 10.1002/0470846526.ch5. [DOI] [PubMed] [Google Scholar]
- 5.Huygen K, Content J, Denis O, Montgomery D L, Yawman A M, Deck R R, DeWitt C M, Orme I M, Baldwin S, D'Souza C, Drowart A, Lozes E, Vandenbussche P, Van Vooren J P, Liu M A, Ulmer J B. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine. Nat Med. 1996;2:893–898. doi: 10.1038/nm0896-893. [DOI] [PubMed] [Google Scholar]
- 6.Lowrie D B, Tascon R E, Bonato V L, Lima V M, Faccioli L H, Stavropoulos E, Colston M J, Hewinson R G, Moelling K, Silva C L. Therapy of tuberculosis in mice by DNA vaccination. Nature. 1999;400:269–271. doi: 10.1038/22326. [DOI] [PubMed] [Google Scholar]
- 7.Murray P J, Wang L, Onufryk C, Tepper R I, Young R A. T cell-derived IL-10 antagonizes macrophage function in mycobacterial infection. J Immunol. 1997;158:315–321. [PubMed] [Google Scholar]
- 8.Orme I M. Antimycobacterial activity in vivo of LM427 (rifabutin) Am Rev Respir Dis. 1988;138:1254–1257. doi: 10.1164/ajrccm/138.5.1254. [DOI] [PubMed] [Google Scholar]
- 9.Orme I M. New vaccines against tuberculosis. The status of current research. Infect Dis Clin N Am. 1999;13:169–185. doi: 10.1016/s0891-5520(05)70049-0. , vii–viii. [DOI] [PubMed] [Google Scholar]
- 10.Orme I M. Progress in the development of new vaccines against tuberculosis. Int J Tuberc Lung Dis. 1997;1:95–100. [PubMed] [Google Scholar]
- 11.Orme I M. Vaccination against tuberculosis: recent progress. Adv Vet Med. 1999;41:135–143. doi: 10.1016/s0065-3519(99)80013-5. [DOI] [PubMed] [Google Scholar]
- 12.Orme I M, Cooper A M. Cytokine/chemokine cascades in immunity to tuberculosis. Immunol Today. 1999;20:307–312. doi: 10.1016/s0167-5699(98)01438-8. [DOI] [PubMed] [Google Scholar]
- 13.Rhoades E R, Frank A A, Orme I M. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:57–66. doi: 10.1016/s0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- 14.Roberts A D, Sonnenberg M G, Ordway D J, Furney S K, Brennan P J, Belisle J T, Orme I M. Characteristics of protective immunity engendered by vaccination of mice with purified culture filtrate protein antigens of Mycobacterium tuberculosis. Immunology. 1995;85:502–508. [PMC free article] [PubMed] [Google Scholar]
- 15.Rodrigues L C, Smith P G. Tuberculosis in developing countries and methods for its control. Trans R Soc Trop Med Hyg. 1990;84:739–744. doi: 10.1016/0035-9203(90)90172-b. [DOI] [PubMed] [Google Scholar]
- 16.Sterne J A, Rodrigues L C, Guedes I N. Does the efficacy of BCG decline with time since vaccination? Int J Tuberc Lung Dis. 1998;2:200–207. [PubMed] [Google Scholar]
- 17.Toossi Z, Ellner J J. The role of TGF beta in the pathogenesis of human tuberculosis. Clin Immunol Immunopathol. 1998;87:107–114. doi: 10.1006/clin.1998.4528. [DOI] [PubMed] [Google Scholar]