

Abstract
Lysine demethylase 5A (KDM5A) is a histone demethylase frequently involved in cancer progression. This research aimed to explore the function of KDM5A in prostate adenocarcinoma (PRAD) and the molecular mechanism. KDM5A was highly expressed in collected PRAD tissues and acquired PRAD cells. High KDM5A expression was correlated with reduced survival and poor prognosis of patients with PRAD. Knockdown of KDM5A suppressed the proliferation, colony formation, migration, and invasiveness of PRAD cells and reduced angiogenesis ability of endothelial cells. Downstream molecules implicated in KDM5A mediation were predicted using integrated bioinformatic analyses. KDM5A enhanced ETS proto-oncogene 1 (ETS1) expression through demethylation of H3K4me2 at its promoter. ETS1 suppressed the transcription activity of miR-330-3p, and either further ETS1 overexpression or miR-330-3p inhibition blocked the functions of KDM5A knockdown in PRAD. miR-330-3p targeted coatomer protein complex subunit β2 (COPB2) mRNA. Downregulation of miR-330-3p restored the expression of COPB2 and activated the PI3K/AKT pathway in PRAD. The results in vitro were reproduced in vivo where KDM5A downregulation suppressed the growth and metastasis of xenograft tumors in nude mice. In conclusion, this study demonstrated that KDM5A promoted PRAD by suppressing miR-330-3p and activating the COPB2/PI3K/AKT axis in an ETS1-dependent manner.
Keywords: Lysine demethylase 5A; ETS proto-oncogene 1; miR-330-3p, COPB2; PI3K/AKT signaling pathway
Introduction
Prostate cancer (PCa), with an estimated 1,276,106 new diagnoses and 358,989 deaths worldwide in 2018, represents the second most frequent neoplastic disease and the fifth leading cause of cancer mortality in men (Bray et al. 2018). Prostate adenocarcinoma (PRAD) accounts for over 99% of all PCa cases (Shen and Abate-Shen 2010; Torre et al. 2015). A timely diagnosis is crucial for the survival of patients. The 5-year survival rate of PCa patients with localized disease was reported to be approximately 100%, but that of patients with distant metastases was decreased to approximately 31% (Ward and Purysko 2020). The application of prostate-specific antigen has improved the opportunity of an early diagnosis of PRAD; however, approximately 40% of patients developed drug resistance, metastasis, and distant seeding (Foroozan et al. 2017). Identifying molecular markers for cancer diagnosis and treatment for advanced diseases is yet of great importance.
Histone modification is a crucial mechanism that mediates cellular processes (Kouzarides 2007). The lysine demethylase 5 (KDM5) family of histone demethylases (HDMs) specifically remove mono-, di-, or tri-methylation from lysine 4 in histone H3 (H3K4me1/2/3) in histone proteins and leads to transcriptional activation or suppression (Petronikolou et al. 2020; Janevska et al. 2018). KDM5A, also named RBP2 or JARID1A, is a JmjC domain-containing demethylase for H3K4me2/3 (Blair et al. 2011). H3K4me3 usually indicates transcriptional activation while H3K4me2 can serve as a repressive epigenetic mark (Liu et al. 2019; Voichek et al. 2018). KDM5A affects gene expression and exert multiple functions by regulating epigenome, transcription factors and synthesis of translational machinery (Kirtana et al. 2020). It mediates mitochondrial metabolism, differentiation and development, and its aberrant expression is frequently linked to cancer progression (Cui et al. 2020; Petronikolou et al. 2020). For instance, KDM5A played oncogenic roles in lung cancer (Oser et al. 2019) and pancreatic cancer (Cui et al. 2020); however, it showed played anti-tumorigenic properties in glioma (Dai et al. 2018). Increased abundance of KDM5B (Xiang et al. 2007) and KDM5C (Hong et al. 2019) while loss of KDM5D (Komura et al. 2018; Li et al. 2016) have reportedly been correlated with the androgen receptor signaling and the progression of PCa. High expression of KDM5A has been detected in PCa tissues (Vieira et al. 2014), but its roles and epigenetic regulations in PCa remain to be elucidated. Therefore, the specific function of KDM5A in PCa attracted our attention.
The ETS family of transcription factors which play crucial roles in development, differentiation, proliferation and apoptosis, and tissue remodeling, are frequently involved in tumorigenesis (Kar and Gutierrez-Hartmann 2013). ETS1 governs a wide array of pro-tumorigenic effects such as invasiveness, epithelial-mesenchymal transition (EMT), drug resistance, and angiogenesis by regulating the well-known targets such as matrix metalloproteinases (MMPs) and vascular endothelial growth factor receptors (VEGFRs) (Nagarajan et al. 2010; Dittmer 2015). In agreement with this, ETS1 has been reported to promote EMT in PCa (Rodgers et al. 2019). But the exact roles and interacted molecules of ETS1 in PCa remain largely unknown. The integrated bioinformatic analyses in this work predicted ETS1 as a KDM5A-related gene and suggested KDM5A and H3K4me2 have significant binding peaks with ETS1 promoter. We therefore postulated that KDM5A might demethylate H3K4me2 at ETS1 promoter to induce its expression.
Although most studies focused on the ETS1 regulated transcriptome, a small number of studies also reported that ETS1 can regulate miRNAome (Harris et al. 2010; Taylor et al. 2016). MicroRNAs (miRNAs) are the mostly studied non-coding RNAs that are heavily dysregulated in diseases, including cancer and play versatile functions due to their potent regulation on thousands of mRNAs (Adams et al. 2014). There is no exception for PCa where miRNAs represent a class of promising tools for assessment of disease course and prognosis (Ghafouri-Fard et al. 2020). In this study, ETS1 was predicted to own a putative binding site with the promoter region of miR-330-3p. miR-330-3p has been reported with both oncogenic (Shen et al. 2019) and tumor-suppressing (Huang et al. 2020) functions in human cancers, with its role in PRAD unknown yet. Taken together, we surmised that there might be a KDM5A/ETS1/miR-330-3p axis in PRAD. Altered expression of these molecules was introduced in cells for both in vitro and in vivo experiments to examine their interactions and functions in PRAD.
Materials and methods
Collection of clinical samples
Sixty patients with PRAD treated at Affiliated Hospital of Jiangnan University from February 2013 to May 2014 were included in this study. All patients were first diagnosed as PRAD according to digital rectal examination, serum prostate-specific antigen examination, transrectal ultrasound, pelvic magnetic resonance imaging, and the prostate biopsy. All recruited patients had complete clinical information without other malignancies or a history of anti-cancer treatment. Major clinical characteristics of patients are shown in Table 1. PRAD tissues and the adjacent tissues (control) were collected during surgery and instantly stored at – 80 °C. After surgery, a 5-year follow-up study was performed at a six-month interval to monitor the prognosis of patients. This research was ratified by the Ethics Committee of Affiliated Hospital of Jiangnan University and performed in line with the Declaration of Helsinki. Written informed consent was obtained from each eligible respondent.
Table 1.
Correlations between KDM5A expression with the clinical characteristics of patients with PRAD
| Characteristics | N = 60 | KDM5A expression | p value | ||
|---|---|---|---|---|---|
| Low (n = 31) | High (n = 29) | ||||
| Age (years) | < 65 | 25 | 14 | 11 | 0.609 |
| ≥ 65 | 35 | 17 | 18 | ||
| PSA (ng/mL) | ≥ 20 | 33 | 14 | 19 | 0.1287 |
| < 20 | 27 | 17 | 10 | ||
| Bone metastasis | Positive | 29 | 9 | 20 | 0.0041** |
| Negative | 31 | 22 | 9 | ||
| Gleason score | ≥ 7 | 39 | 16 | 23 | 0.032* |
| < 7 | 21 | 15 | 6 | ||
| Lymph-node metastasis | Positive | 32 | 12 | 20 | 0.023* |
| Negative | 28 | 19 | 9 | ||
| Tumor stage | T2 | 24 | 17 | 7 | 0.0193* |
| T3 + T4 | 36 | 14 | 22 | ||
Clinical characteristics of patients were analyzed using the Fisher's exact test; *p < 0.05; ** p < 0.01; KDM5A lysine demethylase 5A, PRAD prostate adenocarcinoma, PSA prostate-specific antigen
Cell transfection
A normal human prostatic epithelial cell line RWPE1 (CRL-11609), and PRAD cell lines DU145 (HTB-81), PC-3 (CRL-1435), VCaP (CRL-2876), and C4-2 (CRL-3314) were acquired from ATCC (Manassas, VA, USA). Cells were cultured in 10% FBS-contained DMEM (Gibco Company (Grand Island, NY, USA) at 37 °C in air enriched with 5% CO2.
Short-hairpin RNAs (shRNAs) of KDM5A (sh-KDM5A 1, 2, 3#), sh-negative control (NC), pcDNA-ETS1, pcDNA empty vector, miR-330-3p mimic/inhibitor, and NC mimic/inhibitor were provided by GenePharma Co., Ltd. (Shanghai, China). A Lipofectamine 2000 kit (Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA) was utilized for cell transfection according to the manufacturer’s instructions. After 48 h, the cells were harvested for the subsequent experiments. The sequences of shRNA Oligos are as follows: sh-KDM5A 1#: 5ʹ- CACCGCCTCCATTTGCCTGTGAAGTCGAAACTTCACAGGCAAATGGAGGC-3ʹ; sh-KDM5A 2#: 5ʹ-CACCGCAAGATTGTTGCCAGCAAAGCGAACTTTGCTGGCAACAATCTTG C-3ʹ; KDM5A 3#; 5ʹ- CACCGGATGAACATTCTGCCGAAGACGAATCTTCGGCAGAATGTTCATCC -3ʹ.
A PI3K/AKT-specific agonist Recilisib (CAS No. 334969–03-8) was procured from MedChemExpress (Monmouth Junction, NJ, USA). Cells were treated with Recilisib at 50 μM for 24 h to activate the PI3K/AKT pathway (Kang et al. 2013; Li et al. 2021). Cells treated with an equal volume of DMSO was set to control.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA from tissues and cells was isolated using the TRIzol reagent (Invitrogen). The cDNA was synthetized using a one-step PrimeScript® miRNA cDNA Synthesis kit (Takara Holdings Inc., Kyoto, Japan) or a PrimeScript™ RT reagent kit (Takara). Thereafter, real-time qPCR was conducted using a TB Green® Premix Ex Taq™ II kit (Takara) and the fluorescence quantitation PCR kit (MX3000p; Stratagene, La Jolla, CA, USA). Relative gene expression was quantified by the 2−ΔΔCt method. The primers are listed in Table 2. GAPDH and U6 were used as internal loadings for mRNA and miRNA, respectively.
Table 2.
Primer sequences for RT-qPCR
| Gene | Primer sequence (5'–3') |
|---|---|
| KDM5A | F: GCTAAGGTCTGCCTACAGGCAA |
| R: CCACTTTAGCGGTCCATTCTCG | |
| ETS1 | F: GAGTCAACCCAGCCTATCCAGA |
| R: GAGCGTCTGATAGGACTCTGTG | |
| miR-330-3p | F: TCTCTGGGCCTGTGTC |
| R: GAACATGTCTGCGTATCTC | |
| COPB2 | F: GCTTTGGGCTATGATGAAGGGAG |
| R: GCTGGACTTCTGAATGCTTGGC | |
| JPH1 | F: CTGTCACCTGATTTCTACCAACC |
| R: GGAGACTCCTTTGGTGTAGGTG | |
| BCL11B | F: CTCCCTTTGGATGCCAGTGTCA |
| R: GGCTCCAGGTAGATGCGGAAG | |
| KPNA1 | F: TTCCAAAAGCCCAGAGCAACAGC |
| R: CCACTACTCCTGGTGTGCTGAT | |
| TGFBR3 | F: TGGAGTCTCCTCTGAATGGCTG |
| R: CCATTATCACCTGACTCCAGATC | |
| ATL3 | F: TGGACTCCAAGGAGGAATGGCA |
| R: GAGAAAGCAGGTGACATCGGAG | |
| PCMT1 | F: GAAGTGATGCTGGCTACAGACC |
| R: ATGTGTGGAGCACTGATTGTTGC | |
| MAP3K2 | F: TACACCCGTCAGATTCTGGAGG |
| R: ATGGTCTGAAGCCGTTTGCTGG | |
| ATF2 | F: GGTAGCGGATTGGTTAGGACTC |
| R: TGCTCTTCTCCGACGACCACTT | |
| SMAD2 | F: GGGTTTTGAAGCCGTCTATCAGC |
| R: CCAACCACTGTAGAGGTCCATTC | |
| MMP1 | F: ATGAAGCAGCCCAGATGTGGAG |
| R: TGGTCCACATCTGCTCTTGGCA | |
| MMP2 | F: AGCGAGTGGATGCCGCCTTTAA |
| R: CATTCCAGGCATCTGCGATGAG | |
| MMP9 | F: GCCACTACTGTGCCTTTGAGTC |
| R: CCCTCAGAGAATCGCCAGTACT | |
| VEGFR1 | F: CCTGCAAGATTCAGGCACCTATG |
| R: GTTTCGCAGGAGGTATGGTGCT | |
| VEGFR2 | F: GGAACCTCACTATCCGCAGAGT |
| R: CCAAGTTCGTCTTTTCCTGGGC | |
| GAPDH | F: GTCTCCTCTGACTTCAACAGCG |
| R: ACCACCCTGTTGCTGTAGCCAA | |
| U6 | F: ACGCTTCACGAATTTGCGTGTC |
| R: GCTTCGGCAGCACATATACTAAAAT |
RT-qPCR reverse transcription quantitative polymerase chain reaction, KDM5A lysine demethylase 5A, ETS1 ETS proto-oncogene 1, miR-330-3p microRNA-330-3p, COPB2 COPI coat complex subunit β 2, JPH1 junctophilin 1, BCL11B BAF chromatin remodeling complex subunit, KPNA1 karyopherin subunit alpha 1, TGFBR3 transforming growth factor beta receptor 3, ATL3 atlastin GTPase 3, protein-L-isoaspartate (D-aspartate) O-methyltransferase, MAP3K2 mitogen-activated protein kinase kinase kinase 2, MMP matrix metalloproteinase, VEGFR vascular endothelial growth factor receptor, GAPDH glyceraldehyde-3-phosphate dehydrogenase, F forward, R reverse
Immunohistochemistry (IHC)
The collected tumor tissues were fixed in 10% formalin, embedded in paraffin, cut into 5-μm sections, and placed on glass slides. The tissue slides were soaked in peroxidase solution to block the activity of endogenous peroxidase, and then treated with 5% bovine serum albumin to block non-specific binding. Thereafter, the sections were co-cultured with anti-KDM5A (ab217292, Abcam Inc., Cambridge, MA, USA), anti-p-AKT (#4060, Cell Signaling Technologies (CST), Beverly, MA, USA), anti-coatomer protein complex subunit β2 (COPB2, ab192924, Abcam), anti-ETS1 (14,069, CST), anti-Ki67 (ab217292, Abcam), anti-VEGFA (ab1316, Abcam) and anti-CCND1 (ab16663, Abcam) at 4 °C overnight, and then with anti-immunoglobulin G (IgG, Abcam) at 23 °C for 2 h. Diaminobenzidine was used for color development, and the nuclei were counter-stained by hematoxylin. After that, the tissue slides were sealed by neutral balsam. The staining was observed under an optical microscope (Nikon Eclipse NI, Tokyo, Japan). The number of positively stained cells was calculated using Image J (NIH).
Cell counting kit-8 (CCK-8) method
A CCK-8 kit (Beyotime Biotechnology Co. Ltd., Shanghai, China) was used to examine the proliferation ability of cells. In short, transfected cells were incubated in 96-well plates at 1,000 cells per well. At the 0, 24, 48 and 72 h of incubation, respectively, 10 μL CCK-8 reagent was loaded into the wells, followed by another 4 h of warm incubation. After that, the optical density at 450 nm was read by a microplate reader (EL340, Bio-Tek Instruments, Hopkinton, MA, USA).
Colony formation assay
Transfected DU145 and PC-3 cells were cultured in 6-well plates at 500 cells per well. The medium was refreshed every 3 d. After a 14-d incubation, the cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet for 5 min. The number of cell colonies (≥ 50 cells) was counted under a microscope (Nikon, Tokyo, Minato City, Japan).
5-ethynyl-2’-deoxyuridine (EdU) labeling assay
A Cell-Light EdU DNA replication kit (RiboBio Co., Ltd., Guangdong, China) was used to measure the DNA replication of cells. In brief, transfected cells were cultured in 96-well plates at 2 × 104 cells per well. Each well was filled with 50 μM EdU solution for 3 h of incubation. After that, the cells were fixed and stained with Apollo solution, and the nuclei were stained by 4', 6-diamidino-2-phenylindole (DAPI; Beyotime). The labeling was observed and photographed under an inverted microscope (Olympus Optical Co., Ltd, Tokyo, Japan). The number of EdU-positive (red) and DAPI-positive (blue) cells in five random fields was counted.
Flow cytometry
An Annexin V-fluorescein isothiocyanate (FITC) cell apoptosis detection kit (Thermo Fisher Scientific) was utilized to examine cell apoptosis according to the kit’s instructions. Transfected cells (5 × 104 cells) were centrifuged at 1000 g for 5 min to discard the supernatant. The cells were resuspended in Annexin V-FITC binding buffer and incubated with propidium iodide at 20–25 °C for 10–20 min. The number of apoptotic cells was detected by a flow cytometer (FACS Calibur, BD, Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed by the Flow J software.
Scratch test
A scratch assay was conducted to examine the migration ability of cells. When the cell confluence reached 90%, a sterile 20-μL pipette tip was used to produce scratches on monolayer cells. The cell debris was washed away with D-hanks solution (Solarbio Science & Technology Co., Ltd., Beijing, China), and the remaining cells were cultured in serum-free medium (Gibco) for 48 h. The width of scratch at 0 h and 48 h was photographed under a microscope (Eclipse E600, Nikon Instruments Inc., Tokyo, Japan).
Transwell assay
Transwell chambers (Corning Incorporated, Corning, NY) were used to determine the invasiveness of cells. After transfection, 2 × 105 cells were resuspended in 200 μL serum-free medium and loaded in the apical chambers pre-coated with Matrigel (BD Biosciences). The basolateral chambers were filled with 600 μL 10% FBS-contained DMEM. After 24 h of incubation, the non-invaded cells were discarded using cotton swabs, whereas the invading cells were fixed and stained with 0.1% crystal violet for 15 min. The number of invasive cells in five random fields was counted under the inverted microscope (Nikon).
Tube formation assay
Human umbilical vein endothelial cells (HUVECs) were acquired from ATCC and cultured in Matrigel-coated 24-well plates. HUVECs were incubated at 37 °C for 30 min for polymerization and cultured in different PARD-sourced conditioned medium (CM). After 6 h of incubation, the number of tubes formed by HUVECs was counted under the inverted microscope.
Western blot analysis
Total protein from cells was extracted using the radio-immunoprecipitation assay (RIPA) buffer reagent. The concentration of protein was examined using a bicinchoninic acid kit (Thermo Fisher Scientific). Next, an equal amount of protein sample was separated by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and loaded onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Thereafter, the protein samples were blocked in 5% non-fat milk and incubated with the primary antibodies at 4 °C overnight, followed by further incubation with secondary antibodies at 25 °C for 2 h. The antibodies are listed in Table 3. The protein bands were developed by the Amersham ECL prime western blotting detection reagent (Cytiva, Danaher, Eugene, OR, USA). Relative protein expression was evaluated by Image J software.
Table 3.
Antibodies used for western blot analysis
| Antibodies | Dilution | Item number | Producer |
|---|---|---|---|
| Primary antibodies | |||
| Bax | 1:1000 | ab32503 | Abcam |
| Bcl-2 | 1:1000 | ab32124 | Abcam |
| E-cadherin | 1:1,0000 | ab40772 | Abcam |
| Snail | 1:1000 | ab216347 | Abcam |
| H3K4me2 | 1:500 | ab32356 | Abcam |
| GAPDH | 1:1,0000 | ab181603 | Abcam |
| COPB2 | 1:2000 | ab192924 | Abcam |
| ETS1 | 1:1000 | 14,069 | CST |
| PI3K | 1:1000 | 4257 | CST |
| p-PI3k (Y607) | 1:1000 | ab182651 | Abcam |
| AKT | 1:1000 | 4685 | CST |
| p-AKT (Ser473) | 1:2000 | 4060 | CST |
| KDM5A | 1:5000 | ab194286 | Abcam |
| KDM5B | 1:1000 | ab181089 | Abcam |
| KDM5C | 1:2000 | ab259913 | Abcam |
| KDM5D | 1:1000 | #78,995 | CST |
| Secondary antibodies | |||
| Goat anti-rabbit IgG | 1:2000 | ab205718 | Abcam |
| Goat anti-mouse IgG | 1:5000 | ab205719 | Abcam |
Bax Bcl-2-associated X, Bcl-2 B-cell lymphoma-2, GAPDH glyceraldehyde-3-phosphate dehydrogenase, COPB2 COPI coat complex subunit β 2, ETS1 ETS proto-oncogene 1, PI3K phosphatidyl inositol 3-kinase, AKT protein kinase B, IgG Immunoglobulin G, Abcam Abcam Inc., Cambridge, MA, USA, CST Cell Signaling Technology, Beverly, MA, USA
Chromatin immunoprecipitation (ChIP)-qPCR
PRAD cells were cross-linked in 1% formaldehyde at 37 °C for 10 min, terminated by 0.125 mol/L glycine, and lysed in SDS lysis. The lysates were ultrasonicated using Bioruptor Pico (Diagenode s.a., Seraing, Belgium) to obtain ~ 1000 bp fragments. After centrifugation, the supernatant was diluted in ChIP buffer and cleared with 80 µL protein A/G magnetic beads at 4 °C for 4 h. Thereafter, the lysates were co-cultured with anti-KDM5A (ab194286, Abcam Inc., Cambridge, MA, USA), anti-H3K4me2 (ab32356, Abcam), anti-ETS1 (#14,069, Cell Signaling Technologies, Beverly, MA, USA), or control IgG (sc-2025, Santa Cruz, CA, USA) overnight at 4 °C. The immune complexes were collected with magnetic beads, eluted, and de-crosslinked at 65 °C overnight. After detachment in RNase (Thermo Fisher Scientific) at 37 °C and in proteinase K (Thermo Fisher Scientific) at 45 °C, the immunoprecipitated DNA was extracted and quantified by real time-qPCR.
Examination of activity of HDMs
The protein component from nuclei was isolated using the PARIS™ kit (Invitrogen) and quantified using a Pierce™ BCA kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The HDM activity was examined using a Histone Demethylase (H3K4) Activity Quantification Assay kit (ab113455, Abcam). In brief, each sample well and control well was loaded with 50 μL diluted HDM substrate. Each sample well was further added with 28 μL HDM assay buffer and 2 μL nuclear protein (4–20 μg). Each control well was added with 30 μL HDM assay buffer. Blank wells were set by adding with 30 μL HDM assay only. After incubation at 37 °C for 60 min, each well was added with diluted capture antibodies, followed by shaking incubation at 22–25 °C for 60 min. Thereafter, each well was added with diluted detection antibodies for 25–30 min of incubation. After that, fluorescence development reagents were added and for 2–5 min of incubation in the dark. The fluorescence was examined and red using a fluorescence microplate reader at Ex/Em = 530/590 nm. The HDM activity was calculated as follows: HDM activity (RFU/h/μg) = [(Control RFU – Blank RFU) – (Sample RFU – Blank RFU)]/[Reaction time (h)x protein amount added (μg)].
Dual-luciferase reporter gene assay
To verify the binding between ETS1 and the promoter of miR-330-3p, the miR-330-3p promoter sequence (chr19:45,639,088–45,640,087) containing the putative binding site with ETS1 was inserted into the pGL3 (R2.2)-basic vector (E6441, Promega) to construct luciferase reporter vectors, which was named Promoter. To validate the binding relationship between miR-330-3p and the 3ʹuntranslated region (3ʹ-UTR; located at 2821 bp ~ 3275 bp) of COPB2 mRNA (NM_004766.3), the wild-type (WT) COPB2 sequence containing the putative binding site with miR-330-3p (located at 2892 bp ~ 2899 bp of COBP2 mRNA) was inserted into the pGL3 promoter vector (E1761, Promega) to construct COPB2-WT luciferase vector. The mutant-type (MT) COPB2 3ʹ -UTR sequence (Fig. 3f), was designed and inserted into the pGL3 promoter vector to construct COPB2-MT luciferase vector. The Promoter vector was co-transfected with pcDNA-ETS1 or the pcDNA empty vector into 293 T cells (ATCC) using the Lipofectamine 2000. The COPB2-WT and COPB2-MT vectors were co-transfected with miR-330-3p mimic or mimic control into 293 T cells. Each group cells were transfected with pRL Renilla luciferase control reporter vectors (E2231, Promega) for endogenous loading. After 48 h, the activities of irefly and Renilla luciferase in cells were determined using a luciferase reporter detection system (E1910, Promega), and the firefly luciferase activity was normalized to Renilla luciferase activity.
Fig. 3.

KDM5A enhances ETS1 expression through demethylation modification of H3K4me2. A KDM5A-related genes in PRAD predicted in UALCAN; B pathway enrichment analysis of the KDM5A-related genes; C a PPI network of proteins enriched in the androgen receptor signaling pathway; D mRNA expression of ATF2, SMAD2, and ETS1 in PRAD cells with KDM5A knockdown determined by RT-qPCR; E, F, binding peaks between KDM5A (E) or H3K4me2 (F) and the ETS1 promoter predicted in the ChIP-seq Cistrome Data Browser; G mRNA expression of ETS1 in collected PRAD tissues and adjacent normal tissues examined by RT-qPCR (n = 60); H correlation between KDM5A and ETS1 expression in PRAD tissues; I mRNA expression of ETS1 in DU145 and PC-3 cells and in RWPE1 cells determined by RT-qPCR; J protein levels of KDM5A, ETS1, H3K4me2, KDM5B, KDM5C, and KDM5D in DU145 and PC-3 cells after KDM5A silencing determined by western blot analysis; K activity of H3K4-specific HDMs in the nuclear extracts of PRAD cells; L ETS1 promoter fragments enriched by anti-KDM5A and anti-H3K4me2 after sh-KDM5A transfection determined by ChIP assay. Data were collected from three independent experiments and expressed as mean ± SEM. Differences were analyzed by paired t test (G), one-way ANOVA (I, K, and L), or two-way ANOVA (D and J); correlation between KDM5A and ETS1 (H) was analyzed by Pearson’s correlation analysis, r = 0.749, p < 0.01; **p < 0.01 compared to adjacent tissues; #p < 0.05 compared to RWPE1; &p < 0.05 compared to sh-NC; @p < 0.05, @@p < 0.01 compared to IgG
RNA-binding protein immunoprecipitation (RIP) assay
An EZ-Magna RIP kit was utilized in accordance with the manufacturer’s instructions. In brief, magnetic beads were conjugated with human anti-Ago2 (Millipore) and the incubated with the DU145 or PC-3 cell lysates in RIPA lysis buffer. IgG (Millipore) was used as a control. The RNA was purified using Proteinase K, and the expression of miR-330-3p and COPB2 mRNA was determined by RT-qPCR.
RNA pull-down assay
A total of 50 nM Bio-labeled miR-330-3p or the control Bio-NC (RiboBio Co., Ltd, Guangzhou, Guangdong, China) was transfected with PRAD cells for 48 h. After that, RNase-free BSA and yeast tRNA were used to pre-block M-280 streptavidin magnetic beads, and the cells were lysed in cell lysis buffer. The lysis buffer was collected and centrifuged at 500 g at 4 °C for 10 min to collect the supernatant, which was then incubated with the magnetic beads at 4 °C for 3 h. The beads coated with Bio-labeled RNA was collected and washed. The RNA was resuspended and isolated by TRIzol again, and the abundance of COPB2 fragments enriched by miR-330-3p was examined by RT-qPCR.
Growth and metastasis of xenograft tumors in vivo
Male BALB/c mice (4–6 weeks old) were procured from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and used for tumor growth and metastasis assays in vivo. In the tumor growth assay, DU145 cells stably transfected with sh-KDM5A and the control sh-NC, or transfected with sh-KDM5A + miR-330-3p inhibitor and the control sh-KDM5A + NC inhibitor were subcutaneously injected into the mice at the dorsal side (2 × 105 cells per mouse, n = 6 in each group). After that, the volume (V) of tumors was determined weekly since the second week of injection: V (mm3) = a2 × b/2, where ‘a’ refers to the width and ‘b’ refers to the length of the tumors. Four weeks later, the mice were euthanized through intraperitoneal injection of pentobarbital sodium (150 mg/kg), and the xenograft tumors were weighed and collected for IHC.
In the tumor metastasis assay, stably transfected DU145 cells were injected into the mice through tail veins (2 × 105 cells per mouse; n = 6 in each group). These mice were euthanized on the 45th day (d), and the liver and lung tissues were collected for hematoxylin and eosin (HE) staining to observe the number of metastatic nodules. All animal experiments were ratified by the Ethics Committee of Affiliated Hospital of Jiangnan University and adhered to the Guide for the Care and Use of Laboratory Animals (NIH, Bethesda, Maryland, USA). Significant efforts were made to minimize the suffering of animals.
Statistical analysis
Data were analyzed using Prism 8.0 (GraphPad, La Jolla, CA, USA). All data were collected from three independent experiments and presented as the mean ± standard error of mean (SEM). Differences between two groups were analyzed by the t test, and differences among multiple groups were evaluated by the one- or two-way analysis of variance (ANOVA) followed by Tukey’s multiple test. The 5-year survival rate of patients was analyzed by Log-rank test. Enumeration data were analyzed by Fisher's exact test. The correlation between variables was evaluated by Pearson's correlation analysis. *p < 0.05 represents statistical significance.
Results
High KDM5A expression indicates unfavorable prognosis of patients with PRAD
According to the RT-qPCR results, high KDM5A mRNA level was detected in PRAD tumor tissues compared to the paracancerous tissues (control) (Fig. 1a). A similar trend was validated by the IHC assay that the positive staining of KDM5A was increased in PRAD tumor tissues compared to the adjacent tissues (Fig. 1b). According to the mean value of KDM5A mRNA expression (cutoff value = 3.25), the patients with PRAD were divided into high KDM5A expression group (n = 29) and low KDM5A expression group (n = 31). The 5-year follow-up results suggested that patients with higher KDM5A expression had a lower 5-year survival rate than those with low KDM5A expression (Fig. 1c). The relevance of KDM5A expression to the clinical characteristics of patients was analyzed. It was found that high KDM5A expression was linked to increased bone and lymph node metastases, elevated Gleason score, and advanced clinical stage in patients with PRAD (Table 1).
Fig. 1.

KDM5A abundancy indicates unfavorable prognosis of PRAD patients. A mRNA expression of KDM5A in PRAD tumor tissues and the adjacent tissues evaluated by RT-qPCR (n = 60); B protein level of KDM5A in the PRAD tumor tissues and the adjacent tissues examined by IHC; C relevance of the KDM5A expression to the 5-year survival rate or PRAD patients. Data were collected from three independent experiments and expressed as mean ± SEM. Differences were analyzed by the paired t test (A and B), *p < 0.05, **p < 0.01. Survival rate of patients was analyzed by Log-rank test (C); #p < 0.05
Knockdown of KDM5A suppresses the activity of PRAD cells in vitro
The KDM5A expression in PRAD cell lines (DU145, VCaP, PC-3, and C4-2) and in normal RWPE1 cells was then examined. RT-qPCR results confirmed increased expression of KDM5A in the PRAD cell lines compared to RWPE1 cells (Fig. 2a). Among the PRAD cells, DU-145 and PC-3 cells with the highest KDM5A expression were selected for the subsequent experiments. Thereafter, three shRNAs (sh-KDM5A 1, 2, 3#) were transfected into DU-145 and PC-3 cells for KDM5A knockdown, and the successful transfection was confirmed by RT-qPCR (Fig. 2b). Specifically, sh-KDM5A 1# and sh-KDM5A 2# presenting prominent interfering efficacy were used in the subsequent KDM5A knockdown experiments.
Fig. 2.

Knockdown of KDM5A suppresses the malignancy of PRAD cells in vitro. A mRNA expression of KDM5A in PRAD cell lines (DU145, VCaP, PC-3 and C4-2) and in normal RWPE1 cells examined by RT-qPCR; B transfection efficacy of sh-KDM5A 1, 2, 3# in PRAD cells detected by RT-qPCR; C proliferation ability of DU145 and PC-3 cells detected by CCK-8 assay; D colony formation of DU145 and PC-3 cells examined by colony formation assay; E DNA replication ability of cells detected by EdU labeling assay; F apoptosis rate of DU145 and PC-3 cells determined by flow cytometry; G, migration ability of DU145 and PC-3cells measured by scratch test; H invasiveness of DU145 and PC-3 determined by Transwell assay; I angiogenesis ability of HUVECs in different CM detected by tube formation assay; J protein levels of apoptosis related factors (Bax and Bcl-2) and EMT-related factors (E-cadherin and Snail) in DU145 and PC-3 after sh-KDM5A transfection examined by western blot analysis. Data were collected from three independent experiments and expressed as mean ± SEM. Differences were analyzed by one-way ANOVA (A, B, C, D, E, F, G, H, I and J) or two-way ANOVA (C); #p < 0.05 compared to RWPE1; *p < 0.05 compared to sh-NC
Thereafter, the CCK-8 method results indicated that the proliferation ability of cells was obviously weakened by sh-KDM5A 1# or sh-KDM5A 2# compared to the sh-NC (Fig. 2c). Similarly, the colony formation assay found that the number of colonies formed by DU-145 and PC-3 cells was decreased upon KDM5A inhibition (Fig. 2d). The EdU labeling assay suggested that the DNA replication ability of DU-145 and PC-3 cells was significantly reduced by sh-KDM5A (Fig. 2e). For cell apoptosis, the flow cytometry suggested that knockdown of KDM5A increased the apoptosis rate in both DU-145 and PC-3 cells (Fig. 2f).
The aggressiveness of cells was examined as well. The scratch test showed that the wound healing rate, namely the migratory ability of cells, was significantly decreased after KDM5A knockdown (Fig. 2g). The Transwell assay showed that the number of cells invaded to the lower membranes in a given time was significantly reduced after KDM5A inhibition (Fig. 2h). Moreover, the tube formation assay suggested that the angiogenesis ability of HUVECs was decreased when they were cultured in a condition of KDM5A knockdown (Fig. 2i). We further examined the expression of apoptosis-related factors Bax (pro-apoptotic) and Bcl-2 (anti-apoptotic), and the expression of EMT marker proteins (E-cadherin and Snail). The western blot assay results showed that downregulation of KDM5A led to an increase in the expression of Bax and E-cadherin while a decline in the expression of Bcl-2 and Snail (Fig. 2j), indicating that sh-KDM5A promoted cell apoptosis whereas reduced EMT of cells. In the subsequent experiments, sh-KDM5A 1# that showed the best interfering efficacy was used for KDM5A knockdown experiments.
KDM5A enhances ETS1 expression through histone demethylation of H3K4me2
To examine the molecules downstream of KDM5A, we first predicted the KDM5A-related genes in PRAD in the UALCAN system (http://ualcan.path.uab.edu/index.html) (Fig. 3a). The top 1,000 genes with the highest correlation coefficient were used for pathway enrichment analysis in Metascape (https://metascape.org/gp/index.html#/main/step1) (Fig. 3b). The androgen receptor signaling pathway was found to have a strong correlation with PRAD. Proteins enriched in this signaling pathway were collected, based on which a protein–protein interaction (PPI) network was established (Fig. 3c) in the STRING system (https://www.string-db.org/cgi/input?sessionId=bvC52xOMp58u&input_page_show_search=on). Seven proteins whose node degree over 6 were defined as HUB proteins, including RB1, AR, CREBBP, EP300, SP1, CREB1, and MAPK1. Thereafter, all proteins were scored according to their correlations with the 7 HUB proteins. An interaction with one HUB protein was scored 1, and the score for each protein ranged 0 – 7. Among the proteins, three proteins had the highest interaction score (4): ATF2, SMAD2, and ETS1. The expression of three candidate genes after KDM5A knockdown was examined by RT-qPCR. Compared to sh-NC, sh-KDM5A led to a significant decline in the expression of ETS1, whereas it had no significant effect on the expression of ATF2 and SMAD2 (Fig. 3d). In addition, according to data in the ChIP-seq Cistrome Data Browser (http://cistrome.org/db/#/) and the UCSC (https://genome.ucsc.edu/index.html) system, KDM5A (Fig. 3e) and the transcriptional suppressive marker H3K4me2 (Fig. 3f) were suggested to have significant binding peaks with ETS1 promoter. We therefore surmised that KDM5A might regulate histone demethylation of H3K4me2 at ETS1 promoter to activate transcription.
To validate this, we explored the expression of ETS1 in clinically collected tissues. RT-qPCR detected high expression of ETS1 in PRAD tissues compared to the normal control tissues (Fig. 3g), which showed a significant positive correlation with KDM5A (Fig. 3h). Similar results were observed in cells. Compared to RWPE1 cells, high expression of ETS1 was detected in DU145 and PC-3 cells (Fig. 3i).
Thereafter, to explore whether the regulation of KDM5A on ETS1 expression was achieved through demethylation of H3K4me2, we detected the protein levels of, KDM5A, ETS1, H3K4me2, and other H3K4me2 demethylases KDM5B, KDM5C, and KDM5D in cells by western blot assay. Compared to sh-NC, sh-KDM5A significantly suppressed the protein levels of KDM5A and ETS1 and elevated the level of H3K4me2, while it had little impact on the protein levels of KDM5B, KDM5C, and KDM5D (Fig. 3j), indicating that the increase in H3K4me2 was resultant mainly form knockdown of KDM5A only. Moreover, the Histone Demethylase (H3K4) activity kit suggested that the activity of H3K4-specific HDMs in the nuclear extracts of PRAD cells was significantly decreased after KDM5A knockdown (Fig. 3k). Next, the ChIP assay was conducted to verify whether H3K4me2 is the site mediated by KDM5A at ETS1 promoter (Fig. 3l). Compared to the control IgG, an enrichment of ETS1 promoter fragments was found in the complexes formed by anti-KDM5A and anti-H3K4me2. After KDM5A knockdown, the ETS1 promoter fragments enriched by anti-KDM5A were reduced whereas those by anti-H3K4me2 were increased.
Overexpression of ETS1 blocks the function of sh-KDM5A in PRAD cells
To confirm the interaction between KDM5A and ETS1 in PRAD progression, the sh-KDM5A-transfected DU145 and PC-3 cells were further transfected with pcDNA-ETS1 or the control pcDNA. The successful upregulation of ETS1 was detected by RT-qPCR (Fig. 4a). Under this condition, we found that the proliferation ability of DU145 and PC-3 cells, initially suppressed by sh-KDM5A, was recovered after ETS1 upregulation (Fig. 4b). Likewise, the colony formation and EdU labeling assays suggested that the colony formation and DNA replication abilities of PRAD cells were increased after ETS1 overexpression (Fig. 4c-d). In addition, the flow cytometry results suggested that upregulation of ETS1 reduced the apoptosis of DU145 and PC-3 cells (Fig. 4e). Moreover, we observed that the migratory (Fig. 4f) and invasive (Fig. 4g) abilities were significantly enhanced. Likewise, cultivation in an ETS1-overexpressing condition restored the angiogenesis ability of HUVECs (Fig. 4h).
Fig. 4.

Overexpression of ETS1 blocks the function of sh-KDM5A on PRAD cells. A transfection efficacy of pcDNA-ETS1 in PRAD cells examined by RT-qPCR; B proliferation ability of DU145 and PC-3 cells examined by CCK-8 assay; C colony formation of DU145 and PC-3 cells detected by colony formation assay; D DNA replication ability of cells measured by EdU labeling assay; E apoptosis rate of DU145 and PC-3 cells detected by flow cytometry; F migration ability of DU145 and PC-3 cells measured by scratch test; G invasiveness of DU145 and PC-3 evaluated by the Transwell assay; H angiogenesis ability of HUVECs in different CM detected by tube formation assay. Data were collected from three independent experiments and expressed as mean ± SEM. Differences were analyzed by one-way ANOVA (A, C, D, E, F, G and H) or two-way ANOVA (B); *p < 0.05 compared to sh-KDM5A + pcDNA
ETS1 suppresses transcription of miR-330-3p
As mentioned above, ETS is related to invasiveness, EMT, drug resistance and neo-angiogenesis in carcinoma, and the well-known targets of ETS1 include MMPs and VEGFRs (Dittmer 2015). Thereafter, we observed that ETS1 overexpression significantly elevated the levels of MMPs (MMP1/2/9) and VEGFR1/2 in PRAD cells (Fig. 5a). This might partially explain how ETS1 led to an increase in the invasiveness and dissemination of PRAD cells but could not explain how the proliferation of cells was affected by ETS1. Although most studies focused on the ETS1 regulated transcriptome, a small number of studies also reported that ETS1 can regulate miRNAome (Harris et al. 2010; Taylor et al. 2016). However, in PRAD, the miRNAome of ETS1 has not been investigated. Therefore, we examined the candidate target miRNAs of ETS1 in the hTFtarget system (http://bioinfo.life.hust.edu.cn/hTFtarget/#!/). After that, the pathway enrichment analyses were performed based on these candidate miRNAs (Fig. 5b). Among the four pathways with the highest degree of enrichment (GO:0,035,195; ko05206; GO:0,043,535 and WP1545), a total of 17 miRNAs were found to be intersected (Fig. 5c). Among them, miR-330-3p has been suggested to serve as a potential tumor suppressor in PRAD (Li et al. 2020b). Importantly, data in ChIP-seq Cistrome Data Browser suggested that ETS1 has significant binding buffer with the miR-330-3p promoter (Fig. 5d).
Fig. 5.

ETS1 suppresses transcription of miR-330-3p. A mRNA expression of MMPs (MMP1/2/9) and VEGFR1/2 in PRAD cells determined by RT-qPCR; B pathway enrichment analyses based on ETS1 target mRNAs predicted from the hTFtarget system; C common miRNAs intersected in the four pathways with the highest degree of enrichment; D binding between ETS1 and miR-330-3p promoter analyzed in ChIP-seq Cistrome Data Browser; E miR-330-3p as a target miRNA of ETS1 predicted in hTFtarget; F conservative binding site of ETS1; G putative binding sites between ETS1 and miR-330-3p promoter; H miR-330-3p expression in PRAD tumor tissues and the adjacent tissues measured by RT-qPCR (n = 60); I a negative correlation between miR-330-3p and ETS1 in the PRAD tumor tissues; J miR-330-3p expression in DU145, PC-3 and RWPE1 cells examined RT-qPCR; K miR-330-3p expression in DU145 and PC-3 cells after sh-KDM5A or pcDNA-ETS1 transfection quantified by RT-qPCR; L the sequence of luciferase reporter vector Promoter containing the binding site of miR-330-3p promoter; M binding relationship between ETS1 and miR-330-3p promoter validated through luciferase assay; N enrichment of miR-330-3p by anti-ETS1 examined by ChIP assay. Data were collected from three independent experiments and expressed as mean ± SEM. Differences were analyzed by paired t test (G), one-way ANOVA (I, J, L and M); correlation between ETS1 and miR-330-3p expression (I) was analyzed by Pearson’s correlation analysis, r = -0.712, p < 0.01; **p < 0.01 compared to adjacent tissues; #p < 0.05 compared to RWPE1; &p < 0.05 compared to sh-NC; @p < 0.05 compared to sh-KDM5A + pcDNA or pcDNA; $p < 0.05 compared to Promoter + pcDNA; ^^p < 0.01 compared to anti-IgG
The miR-330-3p was suggested as a candidate downstream target of ETS1 in hTFtarget (Fig. 5e). Thereafter, we obtained the conservative binding sequence of the transcription factor ETS1 from Jaspar (http://jaspar.genereg.net/) (Fig. 5f). According to the sequence, the corresponding DNA promoter sequences that might bind to ETS1 were predicted on another bioinformatic system UCSC (https://genome.ucsc.edu/index.html), and the miR-330-3p promoter sequence was suggested to own a binding relationship with ETS1 (Fig. 5g). Next, we examined the expression of miR-330-3p in the collected PRAD tissues and the adjacent normal tissues using RT-qPCR, and poor expression of miR-330-3p was detected in PRAD tumor tissues (Fig. 5h), which showed a negative correlation with ETS1 (Fig. 5i). Likewise, poor expression of miR-330-3p was detected in DU145 and PC-3 cells compared to that in RWPE1 cells (Fig. 5j).
We further examined the expression of miR-330-3p in DU145 and PC-3 cells after sh-KDM5A or pcDNA-ETS1 transfection. The RT-qPCR results showed that compared to sh-NC, sh-KDM5A upregulated miR-330-3p expression, whereas further transfection of pcDNA-ETS1 significantly reduced the miR-330-3p expression in cells (Fig. 5k). Subsequently, the binding site between ETS1 and miR-330-3p promoter with the highest predictive score was used to construct luciferase reporter vector (Promoter) (Fig. 5l). The Promoter was co-transfected with pcDNA or pcDNA-ETS1 into 293 T cells. After 48 h, we observed that compared to pcDNA empty vector, pcDNA-ETS1 led to a significant decline in the luciferase activity of the Promoter in cells (Fig. 5m). The binding between ETS1 and miR-330-3p promoter was further validated through a ChIP assay. Compared to IgG, anti-ETS1 enriched the promoter fragments of miR-330-3p after immunoprecipitation reaction at 4 °C overnight. (Fig. 5n). Collectively, these results confirmed that ETS1 bound to the promoter region of miR-330-3p to suppress its transcription.
miR-330-3p targets COPB2
miRNAs exert their versatile roles by binding to a multitude number of mRNAs. Thereafter, we predicted the candidate target mRNAs of miR-330-3p using six bioinformatic systems including StarBase, TargetScan (http://www.targetscan.org/vert_72/), miRDB (http://mirdb.org/), miRDIP (http://ophid.utoronto.ca/mirDIP/), miRWalk (http://mirwalk.umm.uni-heidelberg.de/) and miRTarBase (http://mirtarbase.cuhk.edu.cn/php/search.php), and a total of 8 mRNAs were found to be intersected (Fig. 6a). After that, miR-330-3p mimic and NC mimic were transfected into PRAD cells, and then the expression of miR-330-3p and the candidate target genes was examined by RT-qPCR. Compared to NC mimic, miR-330-3p mimic significantly elevated the expression of miR-330-3p whereas reduced the mRNA expression of COPB2, but it did not affect the mRNA expression of the rest seven candidate genes (Fig. 6b). We next analyzed the correlation of COPB2 with ETS1 and KDM5A in patients with PRAD in the StarBase Pan-Cancer system. It was found that COPB2 showed a significant positive correlation with ETS1 and KDM5A (Fig. 6c). After that, high COPB2 expression was detected in PRAD tumor tissues compared to the adjacent tissues (Fig. 6d), which was negatively correlated with miR-330-3p but positively correlated with ETS1 and KDM5A expression (Fig. 6e).
Fig. 6.

miR-330-3p targets COPB2. A candidate target mRNAs of miR-330-3p predicted using six bioinformatic systems; B expression of miR-330-3p and COPB2 in DU145 and PC-3 cells after miR-330-3p mimic transfection determined by RT-qPCR; C correlations between each COPB2 with ETS1 and KDM5A in PRAD analyzed on StarBase; D COPB2 expression in the collected PRAD tumor tissues and the adjacent tissues evaluated by RT-qPCR (n = 60); E correlations between COPB2 expression and miR-330-3p, ETS1 and KDM5A expression in the collected PRAD tumor tissues; F sequences of COPB2-WT/COPB2-MT vectors for luciferase assay; G binding relationship between miR-330-3p and COPB2 in cells examined by a dual luciferase reporter gene assay; H enrichment of miR-330-3p and COPB2 fragments by anti-Ago2 in RIP assay; I enrichment of COPB2 fragments by Bio-miR-330-3p examined by RNA pull-down assay. Data were collected from three independent experiments and expressed as mean ± SEM. Differences were analyzed by paired t test (D) or two-way ANOVA (B, G and H); correlations between variables (E) were analyzed by Pearson’s correlation analysis (COPB2 vs. miR-330-3p: r = -0.616, p < 0.001; COPB2 vs. KDM5A; r = 0.418, p < 0.01; COPB2 vs. ETS1: r = 0.509, p < 0.001); **p < 0.01 compared to adjacent tissues; #p < 0.05 NC mimic; &&p < 0.01 compared to anti-IgG; @@p < 0.01 compared to Bio-NC
Subsequently, the putative binding site between COPB2 and miR-330-3p was obtained from StarBase, and the COPB2-WT/COPB2-MT vectors were constructed (Fig. 6f) for luciferase assays. These vectors were co-transfected with miR-330-3p mimic or NC mimic into 293 T cells. Compared to NC mimic, miR-330-3p mimic significantly reduced the luciferase activity of COPB2-WT in cells (Fig. 6g). The subsequent RIP assay further suggested that compared to IgG, anti-Ago2 enriched abundant miR-330-3p and COPB2 fragments in the complexes (Fig. 6h). In addition, the RNA pull-down assay suggested that COPB2 mRNA was significantly pulled down by Bio-miR-330-3p compared to Bio-NC (Fig. 6i).
Knockdown of miR-330-3p reactivates the COPB2/PI3K/AKT axis and blocks the inhibitory function of sh-KDM5A
Downregulation of COPB2 has been reported to reduce the phosphorylation of AKT in gastric cancer (An et al. 2019). The PI3K/AKT has been reported as an important signaling pathway whose activation is involved in PRAD pathogenesis as well (Singh et al. 2019). Here, we further transfected miR-330-3p inhibitor or the control NC inhibitor into PRAD cells after sh-KDM5A transfection, and the successful miR-330-3p inhibition by miR-330-3p inhibitor was confirmed by RT-qPCR (Fig. 7a). Thereafter, the protein level of COPB2 and the activation of PI3K/AKT in cells were examined by western blot analysis. Compared to sh-NC, sh-KDM5A significantly suppressed the protein level of COPB2 as well as the phosphorylation of PI3K and AKT in DU145 and PC-3 cells. However, compared to NC inhibitor, the miR-330-3p inhibitor restored the COPB2 protein and the phosphorylation of PI3K/AKT (Fig. 7b). After that, a PI3K/AKT-specific agonist Recilisib was used to treat the sh-KDM5A-transfected cells for 24 h. Compared to DMSO treatment, Recilisib increased phosphorylation of PI3K/AKT, but the PI3K/AKT activation did not affect the protein levels of KDM5A and COPB2 (Fig. 7c), indicating that induction of PI3K/AKT by KDM5A is unidirectional.
Fig. 7.

Knockdown of miR-330-3p activates the COPB2/PI3K/AKT axis and blocks the inhibitory function of sh-KDM5A. A transfection efficacy of miR-330-3p in DU145 and PC-3 cells examined by RT-qPCR; B protein level of COPB2 and the phosphorylation of PI3K and AKT in DU145 and PC-3 cells after sh-KDM5A and miR-330-3p inhibitor transfections examined by western blot analysis; C protein levels of KDM5A and COPB2 and phosphorylation of PI3K and AKT in DU145 and PC-3 cells after Recilisib or DMSO treatment determined by western blot analysis; D proliferation ability of DU145 and PC-3 cells examined by the CCK-8 assay; E colony formation of DU145 and PC-3 cells detected by the colony formation assay; F DNA replication ability of cells measured by the EdU labeling assay; G apoptosis rate of DU145 and PC-3 cells detected by flow cytometry; H migration ability of DU145 and PC-3cells measured by the scratch test; I invasiveness of DU145 and PC-3 evaluated by the Transwell assay; J angiogenesis ability of HUVECs in different CM detected by tube formation assay. Data were collected from three independent experiments and expressed as mean ± SEM. Differences were analyzed by two-way ANOVA (A–J); #p < 0.05 compared to sh-NC; *p < 0.05 compared to sh-KDM5A + NC inhibitor or sh-KDM5A + DMSO
Whether the miR-330-3p inhibitor-induced COPB2 restoration and PI3K/AKT reactivation affects the behaviors of cells was further examined. Compared to the sh-KDM5A + NC inhibitor group, the proliferation and colony formation abilities of DU145 and PC-3 cells in the sh-KDM5A + miR-330-3p inhibitor group were recovered (Fig. 7d-7e). Also, downregulation of miR-330-3p restored the DNA replication ability of the PRAD cells (Fig. 7f). The flow cytometry results suggested that miR-330-3p inhibition reduced the apoptosis rate in cells (Fig. 7g), and the subsequent scratch test and Transwell assay results indicated that the migration (Fig. 7h) and invasiveness (Fig. 7i) of DU145 and PC-3 were restored in the setting of miR-330-3p inhibition. As expected, cultivation in the CM of PRAD cells transfected with miR-330-3p inhibitor rescued the angiogenesis ability of HUVECs (Fig. 7j).
miR-330-3p inhibition rescues malignant growth and metastasis of xenograft tumors suppressed by sh-KDM5A in mice
To further examine the functions of the molecules above in tumor growth and metastasis in vivo, DU145 cells stably transfected with sh-NC, sh-KDM5A, sh-KDM5A + NC inhibitor and sh-KDM5A + miR-330-3p inhibitor were injected into mice (to reduce the usage of animals, only one representative cell line was used for animal experiments).
For tumor growth, the cells were administrated into nude mice through subcutaneous injection. After that, the volume of the xenograft tumor in nude mice was determined weekly. The growth rate of tumors in mice was significantly suppressed by sh-KDM5A (compared to sh-NC) but then enhanced by miR-330-3p inhibitor (compared to NC inhibitor) (Fig. 8a). Four weeks later, the tumor tissues were collected and weighed. Likewise, transfection of sh-KDM5A in DU145 cells limited the weight of xenograft tumors, but further transfection of miR-330-3p inhibitor led to an increase in the tumor weight (Fig. 8b). The tumor tissues were collected for IHC. It was found that the staining intensity of ETS1, the proliferation marker KI67, the angiogenesis marker VEGFA, p-AKT, and the PI3K/AKT downstream factor CCND1 in the tissues were suppressed by sh-KDM5A. Importantly, downregulation of miR-330-3p rescued the levels of KI67, VEGFA, and CCND1 and the phosphorylation of AKT (Fig. 8c). However, inhibition of miR-330-3p has little effect on the expression of ETS1 in tumor tissues.
Fig. 8.

Knockdown of KDM5A blocks the inhibition of ETS1 on miR-330-3p and suppresses the COPB2/AKT axis to limit the tumor growth in vivo. A weekly change of the xenograft tumors in nude mice; B weight of the xenograft tumors on week 4; C protein levels of ETS1, KI67, VEGFA and CCND1, and phosphorylation of AKT in the collected tumor tissues examined by IHC; D, E number of metastatic nodules in mouse lung and liver tissues examined by HE staining in the setting of caudal vein injection of DU145 cells. Data were collected from three independent experiments and expressed as the mean ± SEM. Differences were analyzed by one-way ANOVA (B, D and E) or two-way ANOVA (A and C); *p < 0.05 compared to sh-NC; #p < 0.05 compared to sh-KDM5A + NC inhibitor
For tumor metastasis, the cells were injected into mice through caudal veins. On the 45th d, the mice were euthanized to collect the liver and lung tissues. The subsequent HE staining showed that sh-KDM5A significantly reduced the number of metastatic nodules in mouse lung and liver tissues. Again, further administration of miR-330-3p-inhibitor increased the number of metastatic nodules in both tissues (Fig. 8d-e).
Discussion
Globally speaking, the incidence and mortality rates of PCa have declined in the past decades, particularly in the high-income countries (Culp et al. 2020). However, given the overall large coverage and high morbidity, exploring effective biomarkers for cancer diagnosis, prognosis, and even treatment remains a core issue for PCa management. In this study, we reported that KDM5A triggered the growth and aggressiveness of PRAD cells by suppressing miR-330-3p expression and activating the COPB2/PI3K/AKT axis in an ETS1-dependent manner.
The oncogenic role of KDM5A has been well-recognized (Kirtana et al. 2020; Oser et al. 2019; Ren et al. 2020). Selective inhibition of KDM5A has shown significant anti-cancer activity in several malignancies, such as breast cancer (Yang et al. 2019a, 2018) and acute myeloid leukemia (Shokri et al. 2018). Interestingly, targeting KDM5A had more significant anti-leukemic effects compared to targeting KDM5B (Shokri et al. 2018). Amplification of KDM5A has been observed in PCa as well (Petronikolou et al. 2020; Vieira et al. 2014). Here, we validated that high expression of KDM5A in patients with PRAD was relevant to reduced survival time, increased lymph node metastasis and advanced clinical stage of the patients. A similar trend was reported in a recent report by Du et al. that high expression of KDM5A was correlated with poor prognosis in patients (Du et al. 2020). We also found that downregulation of KDM5A significantly reduced proliferation, resistance to death, and migration, invasiveness and metastasis of the PRAD cells both in vitro and in vivo. Collectively, our results demonstrated that KDM5A may serve as a prognostic marker and a therapeutic target for PRAD.
The integrated bioinformatic analyses in this paper identified ETS1 as an important target of KDM5A, and binding peaks of KDM5A and H3K4me2 were predicted in ETS1 promoter. A significant positive correlation between KDM5A and ETS1 was confirmed in PRAD tissues. ETS1 has been found to be activated by KDM3A through epigenetic regulation (Sechler et al. 2017; Sobral et al. 2020). Importantly, we observed that KDM5A knockdown in cells led to an increase in H3K4me2 expression whereas a decline in ETS1 expression, indicating that KDM5A enhances ETS1 expression through the demethylation of H3K4me2. Deregulation of ETS1 has been frequently observed in human malignancies and is associated with tumor extracellular matrix degradation and metastasis (Itoh et al. 2010; Luo et al. 2020; Yalim-Camci et al. 2019). Targeting ETS1 by a miRNA has been demonstrated to suppress proliferation and metastasis of PCa cells (Xu et al. 2017). The subsequent rescue experiments in this study showed that the malignant behaviors of cancer cells such as proliferation and invasiveness weakened upon KDM5A silencing were rescued after ETS1 overexpression, indicating that upregulation of ETS1 was, at least partially, accountable for the oncogenic roles of KDM5A in PRAD.
Although ETS1 have been largely studied as a target of miRNAs involved in tumorigenesis (Chou et al. 2018; Xu et al. 2017; Yang et al. 2019b), it can also mediate the transcription activity of miRNAs by serving as a transcription factor involving both direct and indirect mechanisms (Kern et al. 2012; Taylor et al. 2016). ETS1 has been observed as a transcriptional activator for some oncogenes (Kfir-Elirachman et al. 2018; Nazir et al. 2019), though, it can also inhibit the transcription of its targets. For instance, ETS1 has been witnessed as a negative regulator of the IL-10 gene (Lee et al. 2012). The ETs factor Pea3 has been demonstrated as consistent repressor of miR-21 transcription (Kern et al. 2012). In addition, ETS1 deficiency or knockdown has been associated with increased expression of miR-192 (Kato et al. 2013). Our bioinformatics analyses and subsequent luciferase and ChIP assays validated miR-330-3p as a target of ETS1. miR-330-3p has been revealed as a tumor suppressor in several cancers, including gastric cancer (Ma et al. 2020; Wang et al. 2018), liver cancer (Jin et al. 2019), and PCa as well (Li et al. 2020a, b). On the other hand, miR-330-3p has been reported as an oncogene in several cases (Chen et al. 2019; Xiong et al. 2019). This functional discrepancy might be owing to the difference in target genes in different diseases. We identified miR-330-3p was poorly expressed in the collected PRAD tissues and acquired cancer cell lines. Further downregulation of miR-330-3p in PRAD cells after sh-KDM5A transfection restored the proliferation and invasiveness of cells both in vivo and in vitro.
Thereafter, prediction via several bioinformatics tools suggested COPB2 mRNA as a target mRNA of miR-330-3p in PRAD. COPB2 has been increasingly recognized as an oncogene by inducing cell proliferation and metastasis, while suppressing apoptosis in multiple human cancers (An et al. 2019; Bhandari et al. 2019; Wang et al. 2020). Importantly, our previous report preliminarily found that COPB2 was upregulated in PCa and its silencing suppressed cancer cell proliferation and cell cycle progression, and promoted cell apoptosis (Mi et al. 2016). Importantly, COPB2 has been reported as a positive regulator of AKT phosphorylation (An et al. 2019). The PI3K/AKT signaling pathway is one of the most common pathways activated in multiple cancers by governing nutrient metabolism, cell survival, migration, and angiogenesis (Chen et al. 2016; Yan and Huang 2019). Targeting this signaling pathway has also been recommended as a promising option for the management of PCa (Toren and Zoubeidi 2014). Importantly, we confirmed that silencing of KDM5A reduced the protein level of COPB2 and activation of the PI3K/AKT signaling pathway in PRAD cells and in the xenograft tumors from nude mice. KDM5B has to be essential for the hyperactivation of PI3K/AKT signaling in the tumorigenesis of PCa by binding to the promoter of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (Li et al. 2020a). KDM5C suppressed transcription of methyltransferase-like 14 via H3K4me3 to activate the PI3K/AKT and the progression of colorectal cancer (Chen et al. 2020). Intriguingly, inhibition of AKT has been reported to lead to a reduction in the expression level of KDM5 proteins, especially KDM5B (Khan et al. 2019). However, in this work, treatment with Recilisib, the PI3K/AKT agonist, did not affect the expression levels of KDM5A in cells, suggesting that the induction of PI3K/AKT by KDM5A is unidirectional.
In conclusion, this study confirmed the oncogenic role of KDM5A in PRAD by suppressing miR-330-3p expression and activating the COPB2/PI3K/AKT axis through epigenetic activation of ETS1 (Fig. 9). These findings may offer novel insights into the management of PRAD. In the current work, KDM5A and miR-330-3p were selected as two major molecules involved in the functional network. With the aim of reducing animal usage, functional loss experiments in animals were only performed on KDM5A and miR-330-3p. This might be a limitation of the research, though, we included expression of ETS1, COPB2 and the phosphorylation of AKT in tumor tissues to validate the interaction among all major molecules in vivo. In addition, the exact mechanism by which COPB2 regulates the PI3K/AKT needs further explanation. We would like to focus on these issues in our future experiments.
Fig. 9.
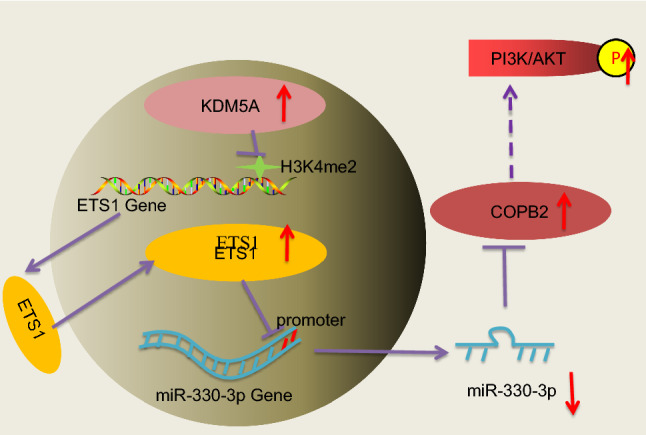
A diagram for the molecular mechanism. In PRAD cells, highly expressed KDM5A increases ETS1 expression through demethylating H3K4me2 at its promoter. ETS1 suppresses miR-330-3p to restore the level of COPB2 mRNA and activate the PI3K/AKT signaling pathway, which augments proliferation, migration, invasiveness and angiogenesis in PRAD
Acknowledgements
This work was supported by National Natural Science Foundation (No. 81802576, 81902565, 81372316), Wuxi City Medical Young Talent (No. QNRC043), Wuxi Commission of Health and Family Planning (No. T202102, T202024, J202012, Z202011, CXTDPY2021003), the Science and Technology Development Fund of Wuxi (No. WX18IIAN024, N20202021), Jiangnan University Wuxi School of Medicine (No. 1286010242190070), Wuxi “Taihu Talent Program”-High-end Talent in Medical and Healthentalent plan of Taihu Lake in Wuxi (Double Hundred Medical Youth Professionals Program) from Health Committee of Wuxi (No. BJ2020061), clinical trial of Affiliated Hospital of Jiangnan University (No. LCYJ202227). The authors are thankful for the guidance of Professor Yongquan Chen.
Authors’ contributions
YYM, LFZ, CYS and YYF conceived the design of the research and carried out most of the experiments. JS, JW, DJY, XWQ, HYW, SW, GWX and LJZ obtained, analyzed and interpreted the data. YYM, LFZ, CYS and YYF wrote and revised the manuscript. SW and GWX prepared the fgures and provides financial support for all experiments. All authors read and approved the fnal manuscript. We confirmed that no third parties were used in either the writing of the paper or the generation of data.
Data availability
The datasets used and analyzed in the current study are available from the corresponding author upon request.
Declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This research was ratified by the Ethics Committee of Affiliated Hospital of Jiangnan University and performed in line with the Declaration of Helsinki. Written informed consent was obtained from each eligible respondent. All animal experiments were ratified by the Ethics Committee of Affiliated Hospital of Jiangnan University and conducted in line with the Guide for the Care and Use of Laboratory Animals issued by the National Institutes of Health (NIH, Bethesda, Maryland, USA). Significant efforts were made to minimize the suffering of animals.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yuanyuan Mi, Lifeng Zhang, Xiaowei Qi have contributed equally to this work.
Contributor Information
Sheng Wu, Email: shengW_7@163.com.
Lijie Zhu, Email: zhulijie_09@163.com.
References
- Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol. 2014;24:R762–776. doi: 10.1016/j.cub.2014.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An C, Li H, Zhang X, Wang J, Qiang Y, Ye X, Li Q, Guan Q, Zhou Y. Silencing of COPB2 inhibits the proliferation of gastric cancer cells and induces apoptosis via suppression of the RTK signaling pathway. Int J Oncol. 2019;54:1195–1208. doi: 10.3892/ijo.2019.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari A, Zheng C, Sindan N, Sindan N, Quan R, Xia E, Thapa Y, Tamang D, Wang O, Ye X, Huang D. COPB2 is up-regulated in breast cancer and plays a vital role in the metastasis via N-cadherin and Vimentin. J Cell Mol Med. 2019;23:5235–5245. doi: 10.1111/jcmm.14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair LP, Cao J, Zou MR, Sayegh J, Yan Q. Epigenetic Regulation by Lysine Demethylase 5 (KDM5) enzymes in cancer. Cancers (basel) 2011;3:1383–1404. doi: 10.3390/cancers3011383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhou L, Wu X, Li R, Wen J, Sha J, Wen X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front Biosci (landmark Ed) 2016;21:1084–1091. doi: 10.2741/4443. [DOI] [PubMed] [Google Scholar]
- Chen T, Yang Z, Liu C, Wang L, Yang J, Chen L, Li W. Circ_0078767 suppresses non-small-cell lung cancer by protecting RASSF1A expression via sponging miR-330-3p. Cell Prolif. 2019;52:e12548. doi: 10.1111/cpr.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu M, Xu X, Zeng K, Liu X, Pan B, Li C, Sun L, Qin J, Xu T, He B, Pan Y, Sun H, Wang S. METTL14-mediated N6-methyladenosine modification of SOX4 mRNA inhibits tumor metastasis in colorectal cancer. Mol Cancer. 2020;19:106. doi: 10.1186/s12943-020-01220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou NH, Lo YH, Wang KC, Kang CH, Tsai CY, Tsai KW. MiR-193a-5p and -3p play a distinct role in gastric cancer: miR-193a-3p suppresses gastric cancer cell growth by targeting ETS1 and CCND1. Anticancer Res. 2018;38:3309–3318. doi: 10.21873/anticanres.12596. [DOI] [PubMed] [Google Scholar]
- Cui J, Quan M, Xie D, Gao Y, Guha S, Fallon MB, Chen J, Xie K. A novel KDM5A/MPC-1 signaling pathway promotes pancreatic cancer progression via redirecting mitochondrial pyruvate metabolism. Oncogene. 2020;39:1140–1151. doi: 10.1038/s41388-019-1051-8. [DOI] [PubMed] [Google Scholar]
- Culp MB, Soerjomataram I, Efstathiou JA, Bray F, Jemal A. Recent global patterns in prostate cancer incidence and mortality rates. Eur Urol. 2020;77:38–52. doi: 10.1016/j.eururo.2019.08.005. [DOI] [PubMed] [Google Scholar]
- Dai B, Huang H, Guan F, Zhu G, Xiao Z, Mao B, Su H, Hu Z. Histone demethylase KDM5A inhibits glioma cells migration and invasion by down regulating ZEB1. Biomed Pharmacother. 2018;99:72–80. doi: 10.1016/j.biopha.2018.01.020. [DOI] [PubMed] [Google Scholar]
- Dittmer J. The role of the transcription factor Ets1 in carcinoma. Semin Cancer Biol. 2015;35:20–38. doi: 10.1016/j.semcancer.2015.09.010. [DOI] [PubMed] [Google Scholar]
- Du C, Lv C, Feng Y, Yu S. Activation of the KDM5A/miRNA-495/YTHDF2/m6A-MOB3B axis facilitates prostate cancer progression. J Exp Clin Cancer Res. 2020;39:223. doi: 10.1186/s13046-020-01735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foroozan M, Roudi R, Abolhasani M, Gheytanchi E, Mehrazma M. Clinical significance of endothelial cell marker CD34 and mast cell marker CD117 in prostate adenocarcinoma. Pathol Res Pract. 2017;213:612–618. doi: 10.1016/j.prp.2017.04.027. [DOI] [PubMed] [Google Scholar]
- Ghafouri-Fard S, Shoorei H, Taheri M. Role of microRNAs in the development, prognosis and therapeutic response of patients with prostate cancer. Gene. 2020;759:144995. doi: 10.1016/j.gene.2020.144995. [DOI] [PubMed] [Google Scholar]
- Harris TA, Yamakuchi M, Kondo M, Oettgen P, Lowenstein CJ. Ets-1 and Ets-2 regulate the expression of microRNA-126 in endothelial cells. Arterioscler Thromb Vasc Biol. 2010;30:1990–1997. doi: 10.1161/ATVBAHA.110.211706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Wu G, Xiang ZD, Xu CD, Huang SS, Li C, Shi L, Wu DL. KDM5C is transcriptionally regulated by BRD4 and promotes castration-resistance prostate cancer cell proliferation by repressing PTEN. Biomed Pharmacother. 2019;114:108793. doi: 10.1016/j.biopha.2019.108793. [DOI] [PubMed] [Google Scholar]
- Huang Y, Sun H, Ma X, Zeng Y, Pan Y, Yu D, Liu Z, Xiang Y. HLA-F-AS1/miR-330-3p/PFN1 axis promotes colorectal cancer progression. Life Sci. 2020;254:117180. doi: 10.1016/j.lfs.2019.117180. [DOI] [PubMed] [Google Scholar]
- Itoh T, Takeda S, Akao Y. MicroRNA-208 modulates BMP-2-stimulated mouse preosteoblast differentiation by directly targeting V-ets erythroblastosis virus E26 oncogene homolog 1. J Biol Chem. 2010;285:27745–27752. doi: 10.1074/jbc.M110.105080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janevska S, Guldener U, Sulyok M, Tudzynski B, Studt L. Set1 and Kdm5 are antagonists for H3K4 methylation and regulators of the major conidiation-specific transcription factor gene ABA1 in Fusarium fujikuroi. Environ Microbiol. 2018;20:3343–3362. doi: 10.1111/1462-2920.14339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Jia B, Tan L, Liu Y. miR-330-3p suppresses liver cancer cell migration by targeting MAP2K1. Oncol Lett. 2019;18:314–320. doi: 10.3892/ol.2019.10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang AD, Cosenza SC, Bonagura M, Manair M, Reddy MV, Reddy EP. ON01210.Na (Ex-RAD(R)) mitigates radiation damage through activation of the AKT pathway. PLoS ONE. 2013 doi: 10.1371/journal.pone.0058355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar A, Gutierrez-Hartmann A. Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Crit Rev Biochem Mol Biol. 2013;48:522–543. doi: 10.3109/10409238.2013.838202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Dang V, Wang M, Park JT, Deshpande S, Kadam S, Mardiros A, Zhan Y, Oettgen P, Putta S, Yuan H, Lanting L, Natarajan R. TGF-beta induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci Signal. 2013 doi: 10.1126/scisignal.2003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern HB, Niemeyer BF, Parrish JK, Kerr CA, Yaghi NK, Prescott JD, Gutierrez-Hartmann A, Jedlicka P. Control of MicroRNA-21 expression in colorectal cancer cells by oncogenic epidermal growth factor/Ras signaling and Ets transcription factors. DNA Cell Biol. 2012;31:1403–1411. doi: 10.1089/dna.2011.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfir-Elirachman K, Ortenberg R, Vizel B, Besser MJ, Barshack I, Schachter J, Nemlich Y, Markel G. Regulation of CEACAM1 protein expression by the transcription factor ETS-1 in BRAF-mutant human metastatic melanoma cells. Neoplasia. 2018;20:401–409. doi: 10.1016/j.neo.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MI, Hamid A, Rath S, Ateeq B, Khan Q, Siddiqui IA, Adhami VM, Choudhry H, Zamzami MA, Mukhtar H. AKT inhibition modulates H3K4 demethylase levels in PTEN-null prostate cancer. Mol Cancer Ther. 2019;18:356–363. doi: 10.1158/1535-7163.MCT-18-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirtana R, Manna S, Patra SK. Molecular mechanisms of KDM5A in cellular functions: Facets during development and disease. Exp Cell Res. 2020;396:112314. doi: 10.1016/j.yexcr.2020.112314. [DOI] [PubMed] [Google Scholar]
- Komura K, Yoshikawa Y, Shimamura T, Chakraborty G, Gerke TA, Hinohara K, Chadalavada K, Jeong SH, Armenia J, Du SY, Mazzu YZ, Taniguchi K, Ibuki N, Meyer CA, Nanjangud GJ, Inamoto T, Lee GM, Mucci LA, Azuma H, Sweeney CJ, Kantoff PW. ATR inhibition controls aggressive prostate tumors deficient in Y-linked histone demethylase KDM5D. J Clin Invest. 2018;128:2979–2995. doi: 10.1172/JCI96769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lee CG, Kwon HK, Sahoo A, Hwang W, So JS, Hwang JS, Chae CS, Kim GC, Kim JE, So HS, Hwang ES, Grenningloh R, Ho IC, Im SH. Interaction of Ets-1 with HDAC1 represses IL-10 expression in Th1 cells. J Immunol. 2012;188:2244–2253. doi: 10.4049/jimmunol.1101614. [DOI] [PubMed] [Google Scholar]
- Li N, Dhar SS, Chen TY, Kan PY, Wei Y, Kim JH, Chan CH, Lin HK, Hung MC, Lee MG. JARID1D is a suppressor and prognostic marker of prostate cancer invasion and metastasis. Cancer Res. 2016;76:831–843. doi: 10.1158/0008-5472.CAN-15-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Kanagasabai T, Lu W, Zou MR, Zhang SM, Celada SI, Izban MG, Liu Q, Lu T, Ballard BR, Zhou X, Adunyah SE, Matusik RJ, Yan Q, Chen Z. KDM5B is essential for the hyperactivation of PI3K/AKT signaling in prostate tumorigenesis. Cancer Res. 2020;80:4633–4643. doi: 10.1158/0008-5472.CAN-20-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wang W, Zhang M, Sun W, Shi W, Li F. Circular RNA circ-0016068 promotes the growth, migration, and invasion of prostate cancer cells by regulating the miR-330-3p/BMI-1 axis as a competing endogenous RNA. Front Cell Dev Biol. 2020;8:827. doi: 10.3389/fcell.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li H, Ma Z, Lei Y. The expression of kappa-opioid receptor promotes the migration of breast cancer cells in vitro. BMC Anesthesiol. 2021;21:210. doi: 10.1186/s12871-021-01429-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu K, Yin L, Yu Y, Qi J, Shen WH, Zhu J, Zhang Y, Dong A. H3K4me2 functions as a repressive epigenetic mark in plants. Epigenetics Chromatin. 2019;12:40. doi: 10.1186/s13072-019-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Liu F, Yan C, Qu W, Zhu L, Guo Z, Zhou F, Zhang W. Long non-coding RNA CASC19 sponges microRNA-532 and promotes oncogenicity of clear cell renal cell carcinoma by increasing ETS1 expression. Cancer Manag Res. 2020;12:2195–2207. doi: 10.2147/CMAR.S242472. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ma B, Ma J, Yang Y, He X, Pan X, Wang Z, Qian Y. Effects of miR-330-3p on invasion, migration and EMT of gastric cancer cells by targeting PRRX1-mediated Wnt/beta-catenin signaling pathway. Onco Targets Ther. 2020;13:3411–3423. doi: 10.2147/OTT.S238665. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mi Y, Yu M, Zhang L, Sun C, Wei B, Ding W, Zhu Y, Tang J, Xia G, Zhu L. COPB2 is upregulated in prostate cancer and regulates PC-3 cell proliferation, cell cycle, and apoptosis. Arch Med Res. 2016;47:411–418. doi: 10.1016/j.arcmed.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Nagarajan P, Chin SS, Wang D, Liu S, Sinha S, Garrett-Sinha LA. Ets1 blocks terminal differentiation of keratinocytes and induces expression of matrix metalloproteases and innate immune mediators. J Cell Sci. 2010;123:3566–3575. doi: 10.1242/jcs.062240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazir SU, Kumar R, Singh A, Khan A, Tanwar P, Tripathi R, Mehrotra R, Hussain S. Breast cancer invasion and progression by MMP-9 through Ets-1 transcription factor. Gene. 2019;711:143952. doi: 10.1016/j.gene.2019.143952. [DOI] [PubMed] [Google Scholar]
- Oser MG, Sabet AH, Gao W, Chakraborty AA, Schinzel AC, Jennings RB, Fonseca R, Bonal DM, Booker MA, Flaifel A, Novak JS, Christensen CL, Zhang H, Herbert ZT, Tolstorukov MY, Buss EJ, Wong KK, Bronson RT, Nguyen QD, Signoretti S, Kaelin WG., Jr The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes Dev. 2019;33:1718–1738. doi: 10.1101/gad.328336.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronikolou N, Longbotham JE, Fujimori DG. Extended recognition of the histone H3 tail by histone demethylase KDM5A. Biochemistry. 2020;59:647–651. doi: 10.1021/acs.biochem.9b01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren F, Shrestha C, Shi H, Sun F, Zhang M, Cao Y, Li G. Targeting of KDM5A by miR-421 in human ovarian cancer suppresses the progression of ovarian cancer cells. Onco Targets Ther. 2020;13:9419–9428. doi: 10.2147/OTT.S266211. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rodgers JJ, McClure R, Epis MR, Cohen RJ, Leedman PJ, Harvey JM, Australian Prostate Cancer B, Thomas MA, Bentel JM. ETS1 induces transforming growth factor beta signaling and promotes epithelial-to-mesenchymal transition in prostate cancer cells. J Cell Biochem. 2019;120:848–860. doi: 10.1002/jcb.27446. [DOI] [PubMed] [Google Scholar]
- Sechler M, Parrish JK, Birks DK, Jedlicka P. The histone demethylase KDM3A, and its downstream target MCAM, promote Ewing Sarcoma cell migration and metastasis. Oncogene. 2017;36:4150–4160. doi: 10.1038/onc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Yi S, Huang L, Li S, Bai F, Lei S, Breitzig M, Czachor A, Sun H, Zheng Q, Wang F. miR-330-3p promotes lung cancer cells invasion, migration, and metastasis by directly targeting hSOD2b. Biotechnol Appl Biochem. 2019;66:21–32. doi: 10.1002/bab.1691. [DOI] [PubMed] [Google Scholar]
- Shokri G, Doudi S, Fathi-Roudsari M, Kouhkan F, Sanati MH. Targeting histone demethylases KDM5A and KDM5B in AML cancer cells: A comparative view. Leuk Res. 2018;68:105–111. doi: 10.1016/j.leukres.2018.02.003. [DOI] [PubMed] [Google Scholar]
- Singh SK, Apata T, Gordetsky JB, Singh R. Docetaxel combined with thymoquinone induces apoptosis in prostate cancer cells via inhibition of the PI3K/AKT signaling pathway. Cancers (basel) 2019 doi: 10.3390/cancers11091390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobral LM, Hicks HM, Parrish JK, McCann TS, Hsieh J, Goodspeed A, Costello JC, Black JC, Jedlicka P. KDM3A/Ets1 epigenetic axis contributes to PAX3/FOXO1-driven and independent disease-promoting gene expression in fusion-positive Rhabdomyosarcoma. Mol Oncol. 2020;14:2471–2486. doi: 10.1002/1878-0261.12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MA, Wappett M, Delpuech O, Brown H, Chresta CM. Enhanced MAPK signaling drives ETS1-mediated induction of miR-29b leading to downregulation of TET1 and changes in epigenetic modifications in a subset of lung SCC. Oncogene. 2016;35:4345–4357. doi: 10.1038/onc.2015.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toren P, Zoubeidi A. Targeting the PI3K/Akt pathway in prostate cancer: challenges and opportunities (review) Int J Oncol. 2014;45:1793–1801. doi: 10.3892/ijo.2014.2601. [DOI] [PubMed] [Google Scholar]
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Vieira FQ, Costa-Pinheiro P, Ramalho-Carvalho J, Pereira A, Menezes FD, Antunes L, Carneiro I, Oliveira J, Henrique R, Jeronimo C. Deregulated expression of selected histone methylases and demethylases in prostate carcinoma. Endocr Relat Cancer. 2014;21:51–61. doi: 10.1530/ERC-13-0375. [DOI] [PubMed] [Google Scholar]
- Voichek Y, Mittelman K, Gordon Y, Bar-Ziv R, Lifshitz Smit D, Shenhav R, Barkai N. Epigenetic control of expression homeostasis during replication is stabilized by the replication checkpoint. Mol Cell. 2018 doi: 10.1016/j.molcel.2018.05.015. [DOI] [PubMed] [Google Scholar]
- Wang Z, Qu H, Gong W, Liu A. Up-regulation and tumor-promoting role of SPHK1 were attenuated by miR-330-3p in gastric cancer. IUBMB Life. 2018;70:1164–1176. doi: 10.1002/iub.1934. [DOI] [PubMed] [Google Scholar]
- Wang X, Shi J, Niu Z, Wang J, Zhang W. MiR-216a-3p regulates the proliferation, apoptosis, migration, and invasion of lung cancer cells via targeting COPB2. Biosci Biotechnol Biochem. 2020;84:2014–2027. doi: 10.1080/09168451.2020.1783197. [DOI] [PubMed] [Google Scholar]
- Ward RD, Purysko AS. Magnetic resonance imaging of prostate adenocarcinoma: detection and staging. Top Magn Reson Imaging. 2020;29:17–30. doi: 10.1097/RMR.0000000000000226. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A. 2007;104:19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Shi Q, Yang X, Wang W, Tao J. LINC00052 functions as a tumor suppressor through negatively modulating miR-330-3p in pancreatic cancer. J Cell Physiol. 2019;234:15619–15626. doi: 10.1002/jcp.28209. [DOI] [PubMed] [Google Scholar]
- Xu S, Ge J, Zhang Z, Zhou W. MiR-129 inhibits cell proliferation and metastasis by targeting ETS1 via PI3K/AKT/mTOR pathway in prostate cancer. Biomed Pharmacother. 2017;96:634–641. doi: 10.1016/j.biopha.2017.10.037. [DOI] [PubMed] [Google Scholar]
- Yalim-Camci I, Balcik-Ercin P, Cetin M, Odabas G, Tokay N, Sayan AE, Yagci T. ETS1 is coexpressed with ZEB2 and mediates ZEB2-induced epithelial-mesenchymal transition in human tumors. Mol Carcinog. 2019;58:1068–1081. doi: 10.1002/mc.22994. [DOI] [PubMed] [Google Scholar]
- Yan Y, Huang H. Interplay Among PI3K/AKT, PTEN/FOXO and AR Signaling in Prostate Cancer. Adv Exp Med Biol. 2019;1210:319–331. doi: 10.1007/978-3-030-32656-2_14. [DOI] [PubMed] [Google Scholar]
- Yang GJ, Wang W, Mok SWF, Wu C, Law BYK, Miao XM, Wu KJ, Zhong HJ, Wong CY, Wong VKW, Ma DL, Leung CH. Selective Inhibition of Lysine-Specific Demethylase 5A (KDM5A) using a Rhodium(III) complex for triple-negative breast cancer therapy. Angew Chem Int Ed Engl. 2018;57:13091–13095. doi: 10.1002/anie.201807305. [DOI] [PubMed] [Google Scholar]
- Yang GJ, Ko CN, Zhong HJ, Leung CH, Ma DL. Structure-based discovery of a selective KDM5A inhibitor that exhibits anti-cancer activity via inducing cell cycle arrest and senescence in breast cancer cell lines. Cancers (basel) 2019 doi: 10.3390/cancers11010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YB, Tan H, Wang Q. MiRNA-300 suppresses proliferation, migration and invasion of non-small cell lung cancer via targeting ETS1. Eur Rev Med Pharmacol Sci. 2019;23:10827–10834. doi: 10.26355/eurrev_201912_19786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analyzed in the current study are available from the corresponding author upon request.