

Abstract
Small molecule targeting of DNA and RNA sequences has come into focus as a therapeutic strategy for diseases such as myotonic dystrophy type 1 (DM1), a trinucleotide repeat disease characterized by RNA gain-of-function. Herein, we report a novel template-selected, reversible assembly of therapeutic agents in situ via aldehyde-amine condensation. Rationally designed small molecule targeting agents functionalized with either an aldehyde or an amine were synthesized and screened against the target nucleic acid sequence. The assembly of fragments was confirmed by MALDI-MS in the presence of DM1-relevant nucleic acid sequences. The resulting hit combinations of aldehyde and amine inhibited the formation of r(CUG)exp in vitro in a cooperative manner at low micromolar levels and rescued mis-splicing defects in DM1 model cells. This reversible template-selected assembly is a promising approach to achieve cell permeable and multivalent targeting via in situ synthesis and could be applied to other nucleic acid targets.
We developed small molecules that selectively bind to and reversible assemble on the DNA and RNA sequences that are the causative agent of myotonic dystrophy type 1 (DM1). The combination of dialdehyde and diamine inhibited the formation of toxic RNA in vitro and rescued mis-splicing in DM1 model cells.
Keywords: drug design, myotonic dystrophy type 1, nucleic acids, small molecule, transcription inhibition
Graphical Abstract
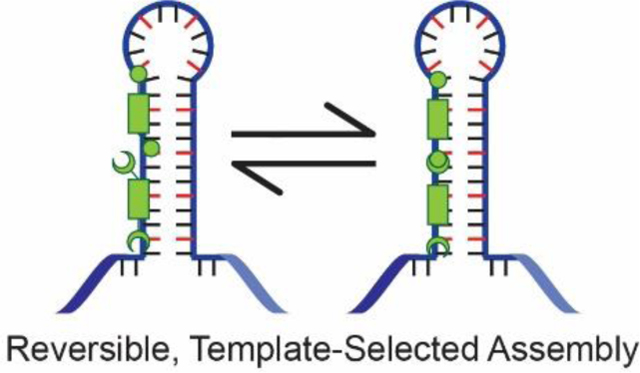
Introduction
Much of the genomic DNA that is transcribed into RNA in the human body is never translated into protein, and thus likely has some other function or benefit to remain in the genome.[1] As a result of recent advances in understanding the roles of both coding and non-coding RNAs in disease, DNA- and RNA-targeting strategies have come into focus for development of therapeutics. Recent drug discovery efforts have enabled selective targeting of DNA and RNA sequences through methods including antisense oligonucleotides,[2] CRISPR/Cas9 genome editing,[3] siRNA[4] and miRNA[5]-based approaches, and small-molecules.[6–9] Several groups have explored the use of these and other RNA-targeting strategies for trinucleotide repeat diseases, in particular myotonic dystrophy type 1 (DM1).[10–16]
DM1 is a neuromuscular disease caused by an expanded d(CTG·CAG)exp repeat sequence in the 3’-untranslated region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19[17] that is bidirectionally transcribed to form RNA with expanded repeats, i.e., r(CUG)exp and r(CAG)exp.[18] Healthy individuals have between 5 and 35 repeats, whereas individuals who manifest symptoms of the disease typically have 80 to more than 2,500 repeats.[19] These expanded repeat transcripts result in a toxic RNA gain-of-function through several mechanisms including splicing mis-regulation[20,21] and the synthesis of toxic homopeptides through repeat-associated non-ATG (RAN) translation (Figure 1).[22] A small molecule or oligomer that targets the parent DNA and inhibits bidirectional transcription would prevent the formation of the toxic RNA and potentially ameliorate DM1 patient symptoms.
Figure 1. Myotonic Dystrophy Type 1 (DM1) Pathogenesis.

d(CTG·CAG)exp is bidirectionally transcribed to form r(CAG)exp and r(CUG)exp. The r(CUG)exp can form hairpin secondary structures that sequester MBNL1 proteins and lead to improper splicing of pre-mRNAs. Both RNA transcripts can undergo RNA translation to yield toxic homopolymeric peptides such as polyglutamine. The use of reactive small molecules to specifically target RNA and DNA and assemble on template could alleviate symptoms of DM1 by preventing bidirectional transcription of d(CTG·CAG)exp or competitively binding the r(CUG)exp hairpin structure to release MBNL1 and prevent RNA translation.
We previously reported that compound 1 selectively binds to r(CUG)exp and shows low toxicity and high cell permeability.[23] A dimeric version of compound 1 proved more effective than other dimeric compounds (e.g., acridine dimers), but had low cell permeability.[24] Both 1 and its dimeric analogue selectively bound the r(CUG)n, but showed no affinity toward d(CTG)n. Although RNA is a suitable target, a potentially more powerful therapeutic strategy would target upstream at the DNA level, thereby inhibiting the formation of the toxic RNA altogether. Thus, we sought to develop bisamidinium-based DNA targeting agents that would be more effective as therapeutic agents while taking advantage of the low cytotoxicity of the groove-binding scaffold.

Given the poor cell permeability exhibited by dimers of 1,[31] we sought a targeting strategy wherein improved affinity and cell permeability could both be achieved by using monomeric units that assembled in situ on the target nucleic acid template. Rideout pioneered the idea of self-assembling therapeutics, where the slightly selective uptake of two agents led to amplified selectivity through localized assembly.[25,26] The 1,3-dipolar cycloaddition reaction, or “click chemistry”,[27] has been employed by Dervan and coworkers to assemble a polyamide-based therapeutic agent in vitro[28] as well as by our group and others for the treatment of DM1.[29–31] In the 1990s, the Lynn group employed imine condensation chemistry to achieve reversible nucleic acid templated backbone ligation that mimics DNA replication.[32–34] The Rayner,[35] Miller,[36] and Liu[37] groups have worked to develop dynamic covalent therapeutic screening approaches via nucleic acid template-assisted synthesis. More recently, Ly and coworkers showed that template-assisted assembly could be achieved on an r(CUG)n template using native chemical ligation.[38] While this work was in progress, the Hargrove group reported a template-guided selection via imine condensation, in which a small library was utilized to identify amplified binders.[39]
Our approach was to functionalize the previously developed selective targeting agents with amine or aldehyde groups that would covalently, but reversibly assemble via imine formation, producing multivalent therapeutic agents in situ (Figure 2). Our approach utilizes two specific binders, whereas previous approaches use one specific binder with general binders or non-binders. The increased complexity of requiring two agents might be offset by the opportunity to amplify selectivity analogous to the Rideout approach (vide supra). In addition, the assembled multivalent structures were expected to have significantly higher affinity for d(CTG)exp and r(CUG)exp relative to their monomeric analogues and exhibit high cell permeability. The reversible covalent, in situ imine condensation is attractive from a therapeutic perspective because the fragments are effectively recyclable. Cellular degradation of the target nucleic acid or dissociation of the assembled product would lead to hydrolysis of the imine bond, thus degrading the oligomer to the original monomers in the absence of template (Figure 2). An additional benefit of applying this assembly approach to the treatment of trinucleotide repeat diseases, such as DM1, is the potential for the oligomers to size-match the repeat length and thereby maximize binding to that sequence. Thus, a personalized treatment could be realized by dosing with just a few monomers that could be used to synthesize an oligomer matching each patient’s repeat length in situ.
Figure 2. Dynamic Covalent Chemistry Template-Selected Assembly Approach.

Cell-permeable monomers reversibly assemble, allowing for template-selected amplification of the potent multivalent targeting agent.
Results and Discussion
Compound Design and Computational Study of Assembly
Based on the success of ligand 1 and its dimeric and oligomeric derivatives,[23,24,40] analogues using this scaffold were designed with benzaldehyde and aniline groups that may condense in situ to form imines. The triazine moieties serve as T-T and U-U mismatch recognition units and the bisamidinium groove-binding linker is designed to bind in the major groove of the RNA helix. In the docked structure of 1, each ligand spans a total of 3 U-U mismatches. Monomers 2 and 3 were designed such that the aldehyde and amine groups would be in close enough proximity on the DNA or RNA to react.
To test the potential for imine formation computationally, ligands 2 and 3 were docked onto the crystal structure of r(CUG)6 (PDB ID: 3gm7)[41] in Molecular Operating Environment (MOE). These ligands were docked individually on the sequence with successive minimizations and the distance between reactive partners (the carbonyl carbon of the aldehyde and the aniline nitrogen) was measured to be 3.95 Å (Figure 3). Of note, a hydrogen bond between the carbonyl oxygen and the aniline nitrogen holds these moieties in close proximity. A ligation was performed manually by linking the two ligands followed by minimization to yield the docked structure shown in Figure 3. Additional images of the docked structures are included in Figure S1. Based on this modeling study, the linker length and distance between reactive groups is sufficient for binding and ligation with each ligand spanning 3 U-U mismatches, consistent with the mode of binding hypothesized for ligand 1.
Figure 3. MOE docking studies for ligands 2 and 3.

(a) Monomers 2 and 3 docked side by side. The fragments were manually ligated, and the structure minimized to form the dimeric product 2+3 on the r(CUG)6 helix shown in (b). Uracil is green (light green are flipped in, dark green flipped out), cytosine is tan and guanine is grey. Hydrogen bonds between ligand and RNA are shown in red. R(CUG)6 PDB ID: 3gm7.[41] The distance between reactive atoms (from carbonyl carbon to aniline nitrogen) was measured to be 3.95 Å (purple dotted line in c.)
Synthesis of Compounds 2 and 3
Compounds 2 and 3 were synthesized according to the methods in Schemes 1 and 2, respectively. In the synthesis of 2, 4-cyano-benzaldehyde (4) was protected with trimethyl orthoformate and the nitrile group was reduced to a primary amine with lithium aluminum hydride. The resulting amine (5) was reacted with 4,6-dicholoro-1,3,5-triazin-2-amine. The disubstituted triazine product was functionalized with 1,4-diaminobutane to yield intermediate 6. Reaction with diethyl terephthalimidate followed by deprotection gave dialdehyde 2.
Scheme 1.

Synthesis of 2. See Supporting Information for detailed methods.
Scheme 2.

Synthesis of 3. See Supporting Information for additional details.
The synthesis of compound 3 began with the successive reaction of 4,6-dicholoro-1,3,5-triazin-2-amine with 4-amino-benzylamine and 1,4-diaminobutane to give triaminotriazine 7. Reaction of 7 with diethyl terephthalimidate gave compound 3. Both 2 and 3 were purified via preparatory HPLC in acetonitrile:water gradient with 0.1% TFA. Detailed methods and characterization data are included in the Supporting Information.
MALDI-MS Analysis of Template-Selected Assembly.
With compound 2 and 3 in hand, we wondered whether the benzaldehyde and aniline groups would react to assemble a dimeric or oligomeric product. Thus, monomers 2 and 3 were mixed in the presence or absence of template, incubated at 37 °C, and reduced with sodium cyanoborohydride. In the presence of both d(CTG)16 and r(CUG)16, MALDI-MS analysis showed peaks corresponding to monomers 2 and 3 in each sample as well as a peak at m/z of 1193.8 consistent with a fragment of dimer 2+3 (Figure 4b-d). The higher mass peak was not observed in the absence of template (Figure 4a). We observed formation of this product selectively on template after as few as 2 min (Figure S7). We were unable to characterize the reaction at a time faster than 2 min due to limitations of mixing the solution, sampling, reducing, and spotting for MALDI-MS. This assembly occurs not only selectively on the template, but also very quickly compared to previous assembly methods that take hours or even days to form product. Notably, any product that may form off-template in the aqueous in situ environment could be hydrolyzed, thus returning to the monomeric form. Therefore, we refer to this assembly as “template-selected.”
Figure 4. MALDI-MS analysis of 2+3 assembly on and off template with reduction by NaCNBH3.

(a) absence of template, (b) d(CTG)16 template, (c) r(CUG)16 template. Buffer: 2 mM each of KCl, MgCl2, CaCl2, and Tri-HCl, pH 7, incubated at 37 oC for 75 min before reduction and analysis. 100 μM compounds, 10 μM d(CTG)16 or r(CUG)16. See procedure S1 for details.
Due to partial reduction of the aldehyde by sodium cyanoborohydride, it was noted that in the MALDI-MS, monomer 2 and the dimeric product both exist in multiple reduction states. For example, monomer 2 could be present as the dialdehyde (M+H = 759.41), aldehyde-alcohol (M+H = 761.42), or diol (M+H = 763.44). The dimeric product could be present as an amine (reduced imine) with aldehyde end group (M+H = 1475.84) or an alcohol end group (M+H = 1477.86).
To determine if larger, oligomeric products could form, the compounds were incubated in the presence or absence of template at 37 °C for 24 h before MALDI-MS analysis. Trimer 2+3+2, tetramer 2+3+2+3, and pentamer 2+3+2+3+2 were observed in the presence of d(CTG)16, r(CUG)16, and r(CUG)90, (Figure 5b–d) but not in the absence of template (Figure 5a). The presence of these longer oligomers supports the potential to use this strategy to synthesize multivalent targeting agents in situ, selected by the nucleic acid template.
Figure 5. High MW MALDI-MS analysis of 2+3 assembly on and off template with reduction by NaCNBH3.

(a) absence of template, (b) d(CTG)16 template, (c) r(CUG)16 template, (d) r(CUG)90 template. Buffer: 2 mM each of KCl, MgCl2, CaCl2, and Tri-HCl, pH 7, incubated at 37 oC for 24 h before reduction with NaCNBH3, and analysis. Compound concentration 100 μM, 10 μM d(CTG)16 and r(CUG)16 or 500 nM r(CUG)90. See Procedure S2 for details.
The MALDI results did raise several questions surrounding which oligomers should be preferentially formed on which templates. For example, templates d(CTG)16 and r(CUG)16 were annealed to form hairpins containing 6 U-U or T-T mismatches with the other 4 U/Ts in the loop. Although this length is suitable for forming dimeric products, it too short to accommodate longer oligomeric products yet trimer, tetramer, and pentamer were observed (Fig. 5). Because these oligomers are self-complementary (e.g., 5’-GGG-(CTG)16)-CCC-3’) we cannot rule out the presence of some duplex that templates the longer oligomers. A more likely scenario is that the cationic ligands allow the formation of aggregates, analogous to polyplexes, and the longer oligomers arise from such aggregates. Another question is why longer oligomeric products were not observed on the longer r(CUG)90 template. The structure of r(CUG)90 is not fully understood, but it is likely a dynamic structure containing multiple hairpins of varied length, rather than one long hairpin.[42–45]
Although it is tempting to compare the absolute and relative intensities of the MALDI oligomer peaks observed using the d(CTG)16 and r(CUG)90 templates (Fig. 5c,d), it is not possible in this study for several reasons. Beyond the MALDI-MS assay not being quantitative, the concentrations of the d(CTG)16 (10 μM) and the r(CUG)90 (0.5 μM) templates used in the assay were different due to the large difference in mismatch content, and we cannot rule out some tight RNA-ligand complexes suppressing the MALDI peak intensities.
Although selective assembly on d(CTG) and r(CUG) templates was observed when comparing to random duplex DNA, there are many other potential nucleic acid targets. To more clearly test the selectivity of the assembly on the DM1-relevant targets d(CTG)16, r(CUG)16, and r(CUG)90, we performed the same MALDI-MS assay with the following control templates: d(CTG)3, d(CTG·CAG)8, d(CAG)16, d(GCC)3, d(CAGG)3, a Spinocerebellar Ataxia Type 10 template containing d(ATTCT)2 random single stranded DNA, r(CAG)90, HIV TAR RNA, and random single stranded RNA (Figure S9). We did not observe any dimeric or oligomeric products on int the presence of any of these control templates. The d(CTG)3 template carries only 3 TT mismatches, and thus should not be long enough to allow both 2 and 3 to bind on the template. Indeed, no assembly was observed on this template. The DNA duplex d(CTG·CAG)8 mimics a healthy repeat length and as expected, no evidence of assembly was observed in the presence of this template. The d(CAG)16 and r(CAG)90 templates are an important control, as they are complimentary to the DM1-relevant d(CTG) and r(CUG) sequences. The other controls tested are mismatch-containing sequences or random templates that could allow for non-specific assembly, but no assembly was observed on any of these templates. Thus, we only observed assembly on the target mismatch-containing d(CTG)16, r(CUG)16, and r(CUG)90 sequences and not on any of the 11 control templates or in the buffer.
Because of its reactive aldehyde groups, we wondered if monomer 2 might react with the nucleic acid template to yield a covalent DNA modification. MALDI-MS was used to screen for modification of d(CTG)3 and a random duplex DNA target by monomer 2. No modification of these DNA sequences was observed (Figure S8), suggesting that interactions are non-covalent.
HPLC Analysis of Template-Selected Assembly
After observing assembly of 2+3 on d(CTG)16 or r(CUG)16 by MALDI-MS, we utilized HPLC to screen for the appearance of new peaks that could be consistent with dimer or oligomer formation. We were particularly interested in better quantifying product formation and determining whether the trimer or longer oligomers would be more prominent in the presence of r(CUG)90, with the longer length of the template potentially leading to longer oligomers being formed.
For HPLC analysis, 2 and 3 were incubated in the presence or absence of template at 37 °C for 24 h before reduction with sodium cyanoborohydride and analysis by HPLC. In the presence of the templates d(CTG)16, r(CUG)16, and r(CUG)90, at least three prominent new peaks appeared (Figure S7). Presumably, these new peaks correspond to the dimeric and oligomeric products observed by MALDI-MS. Attempts to chemically prepare and separate the oligomeric products by reductive amination were not successful so definitive assignments could not be made. The use of LC/MS to identify and quantify the relative amounts of monomer, dimer, and larger oligomers in the solution was inconclusive because of the large amount of fragmentation and the high abundance of multiply charged peaks. Nonetheless, in addition to supporting the formation of a new dimeric and oligomeric product as observed by MALDI-MS, these HPLC studies indicate that an amine product can be characterized from the template-selected assembly of an imine product. As expected, in a control assay with no reductant added, only monomers and no product were observed in the HPLC and only a very small peak consistent with the dimeric imine product was observed by MALDI-MS, suggesting that the lability of imine product when not bound to template (Figure S12).
Isothermal Titration Calorimetry
ITC studies were performed to test the binding affinity of each monomer compared to the mixture of monomers using d(CTG)16 and random duplex DNA as the templates. The calculated Kd value for 2+3 binding d(CTG)16 was 5.6 ± 0.5 μM, whereas the analogous Kd for 2 was measured as 81.6 ± 0.9 μM and Kd > 100 μM observed for 3 (Figure S13a–c). Compound 2, 3, and 2+3 all showed no significant binding to a random duplex DNA sequence (Figure S13d–f). The thermodynamic data for these binding interactions is shown in Table S1. Of additional interest for the treatment of DM1, 2+3 also binds to r(CUG)16 with a calculated Kd value of 4.3 ± 0.5 μM and 2+3 does not bind random ssRNA (Figure S14).
In the ITC experiment there was remaining background heat following saturation, likely caused by a combination of the heat of dilution of N11 as compared to the blank (Figure S15). Of note, the determined stoichiometry, n = 1.85, was calculated based on the concentration of the monomeric units. Because any dimeric or oligomeric product would be formed in situ, an n value of ca. 2 is consistent with either 2 monomers or 1 dimer being bound. A d(CTG)16 hairpin would contain 6 TT mismatches, a length that matches well with that of dimer 2+3.
These binding studies suggest that the transient imine product binds more strongly to a d(CTG)16 target than the monomers themselves and neither the monomers nor 2+3 bind to a random DNA duplex. Further, 2+3 does not bind to random ssRNA, but can bind to r(CUG)16, suggesting its potential as a dual-targeting agent for DM1.
in vitro Transcription Inhibition
Because we are interested in developing a DNA-targeting agent that would prevent the formation of toxic RNA and the associated disease pathobiology, we sought to test if monomers 2 and 3 could inhibit transcription of a DM1-relevant DNA template either independently or cooperatively through in vitro transcription inhibition (Figure 6a). Although compound 1 shows no appreciable inhibition at these concentrations, analogues 2 and 3 both showed a higher level of inhibition, with 2 showing an IC50 value of 59.2 ± 0.5 μM (Figure 6b–c). Because the imine is transient by design, it is difficult to quantify its presence in solution. Thus, to test the mixture of 2+3, equimolar amounts of the compounds were mixed in the treatment such that 20 μM indicates the presence of 20 μM of each monomer, or 20 μM of the dimeric imine that can form in situ. To compare this mixture (2+3) to the inhibition of the monomeric parts, an arithmetic sum of the individual monomer inhibition was calculated. Notably, the calculated IC50 value for the mixture of 2+3 was 20.8 ± 0.6 μM (grey line). For comparison, the arithmetic sum of the independent inhibition of 2 and 3 has a calculated IC50 value of 49.2 μM. This data is consistent with the Kd values observed via ITC.
Figure 6.

in vitro Transcription Inhibition Assay with T7 RNA Polymerase and pSP72 plasmid containing 90 (CTG·CAG) repeats. Results are reported as the average of at least 3 independent experiments are error is reported as standard error of the mean.
As the assembly of the imine presumably occurs in situ, we wondered if providing a 24 h pre-incubation window during which 2 and 3 were incubated with the template before addition of the polymerase would allow for stronger inhibition. As the previous incubation window was 3.5 h, we hypothesized that providing longer incubation would allow for increased template-selected formation of imine and thus stronger inhibition. Indeed, the IC50 for 2+3 with longer pre-incubation of 24 h (black line) dropped to 9.8 ± 0.5 μM. Of note, the combination of 2+3 showed no inhibition in the transcription of a random duplex sequence (Figure S16). Together, these results suggest that the mixture of 2+3 is effective at inhibiting transcription in vitro. This improvement is consistent with the formation of an imine dimer in situ, but the cooperative formation of a noncovalent dimer cannot be ruled out.
A modest improvement was observed here for 2+3 compared to the monomeric units, when considering the almost 1,000-fold improvement over 1 for a groove-binding click dimer.[24] Importantly, the dimers studied here are formed reversibly in situ, compared to the previous approach in which the dimer was synthesized ex vivo and dosed as an assembled unit. Thus, the imine exists in equilibrium with the monomeric components. Assuming the equilibrium favors monomer, the additional binding energy provided by the dimer will come with this energetic penalty. Furthermore, both the spacing and conformation of the linker in this dimeric compound are different from our previously reported click dimer and may not be optimized. These factors alone readily explain why a smaller improvement in activity was observed here. It is important to note that whereas our previously reported groove binders only targeted r(CUG)exp, 2+3 binds both DM1 causative agents, d(CTG)exp and r(CUG)exp.
Insulin Receptor Mis-Splicing Rescue.
The biological activity of 2+3 was further investigated via a cellular mis-splicing assay. Prior to this assay, sulforhodamine B cytotoxicity assays were performed and suggested that the monomers and the combination of 2+3 were not toxic up to a concentration of 200 μM (Figure S17). In DM1 patients sequestration of MBNL1 by r(CUG)exp prevents proper splicing of the insulin receptor (IR) pre-mRNA, leading to insulin resistance due to aberrant splicing regulation.[46] Healthy individuals predominantly express isoform B, with exon 11 included, but DM1 patients predominantly express isoform A, a lower-signaling, non-muscle isoform (Figure 7a).
Figure 7. Insulin Receptor Mis-splicing Assay.

(a) Improper splicing patterns caused by MBNL1 sequestration by toxic RNA products. (b) Combination of ligands 2 and 3 rescued mis-splicing in the insulin receptor minigene in a dose-dependent manner. Results reported as the average of at least 3 independent replicates. Error bars indicate standard error. * indicates p<0.05 compared to negative control.
Detailed methods for this splicing rescue assay can be found in the Supporting Information. Briefly, HeLa cells were transfected with the IR minigene with or without the DT960 minigene, which carries 960 d(CTG⋅CAG) repeats. Cells were treated with varying concentrations of compound ranging from 10 μM to 100 μM and compared to the positive control (IR without DT960, healthy) and the negative control (IR with DT960, diseased). A dose-dependent splicing rescue was observed (Figure 7b), where 65% splicing recovery was achieved at 100 μM. Of note, monomers 2 and 3 on their own did not achieve any significant mis-splicing rescue up to 100 μM (Figure S19). As IR mis-splicing is one of the most difficult to rescue,[47] this strong splicing improvement coupled with the in vitro transcription inhibition suggests that the formation of a transient imine product in situ could be a viable strategy to treat DM1 or other trinucleotide repeat diseases. One potential challenge with utilizing 2 and 3 in cells is that the two compounds both need to reach the target and bind in the proper orientation for assembly. This successful proof-of-concept study with 2+3 could be adapted to treat with a single compound that has an amine end and an aldehyde end. Thus, one could envision the potential of this compound to reversibly form oligomers while overcoming the challenges of colocalization.
Conclusion
In this preliminary study, we reported evidence for a template-selected, reversible covalent coupling of nucleic acid-targeting benzaldehyde 2 and aniline 3. Computational modeling studies suggest that these compounds can be brought into proximity when bound to adjacent sites on a nucleic acid template and can link together via a condensation reaction to reversibly form a more potent targeting agent. Using HPLC and MALDI-MS, monomers 2 and 3 were shown to form an imine dimer 2+3 as well as oligomeric products in the presence of the target oligonucleotides. The therapeutic effects of the combination 2+3 were moderately improved in comparison to the monomers, as the putative imine product bound d(CTG)16 and r(CUG)16 with a low micromolar Kd, inhibited transcription of d(CTG·CAG)90 to form r(CUG)90, and partially rescued IR mis-splicing. We anticipate that this reversible assembly strategy could be broadly applied in drug discovery efforts to reversibly synthesize multivalent targeting agents in situ.
Experimental Section
Computational Methods: Docking of 2+3
The crystal structure of r[(CUG)6]2 was obtained from the Protein Data Bank (code 3gm7)[41] and extended to r[(CUG)7]2 using the Nucleic Acid Builder in Molecular Operating Environment (MOE, v2018.01). Uracil (U) bases (from U-U mismatches denoted 1, 3, 4, and 6) were manually rotated out of the helix to simulate base flipping. Each melamine was docked with 3 hydrogen bonds with the Watson-Crick-Franklin face of the undisturbed U and upon minimization, an additional hydrogen bond was observed with the phosphate backbone of the flipped U base. Bisamidinium units were placed in the major groove of the helix and sequentially minimized. The melamine and bisamidinium units were linked with an alkyl group consisting of four methylene units and minimized, with the melamine units at U-U mismatch 1 and 3 linked to a bisamidinium and those at U-U mismatch 4 and 6 linked, to represent monomers 2 and 3. Upon minimization, the aniline and benzaldehyde functionalities were built onto the internal melamine unit at uracil 3 and 4, respectively. The structure was minimized to give the final docked structure of the monomers. The distance between the carbonyl carbon and aniline nitrogen was measured using USCF Chimera. A manual ligation was performed by linking the carbonyl carbon to the aniline nitrogen atom and by deleting the oxygen atom that would be lost as a water molecule. The docked structure was minimized to yield the final docked structure of the imine dimer. Final docked structures were exported as protein data bank (pdb) files. Images were rendered and hydrogen bonds (H-bonds) were quantified[48] in UCSF Chimera (v1.15.1).[49]
in vitro Transcription Inhibition Assay with T7 RNA Polymerase[50]
This assay was performed as previously reported with minor changes to the compound concentrations and gel running times. Herein, the gels were imaged on a BioRad Gel Doc XR and RNA was quantified based on fluorescence intensity. Bands were quantified with ImageJ by drawing a box of same size around each band, plotting the lanes, drawing a straight line under each curve, and integrating with the wizard tool in ImageJ. Normalized % transcript = treatment / control x 100%. Percent inhibition = 1 – normalized % transcript. Data was plotted in RStudio and fitted to a logistic curve using the nonlinear least function with the SSlogis model. The output logistic equation was used to calculate IC50 values and error is reported from standard error of the coefficients in the fit equation. Curves that did not fit the logistic model were plotted with a linear model for visual comparison.
Insulin Receptor (IR) Mis-splicing Rescue Assay[51]
This assay was performed as previously reported with minor changes to the compound concentrations and gel running times.
Supplementary Material
Acknowledgements
The authors would like to thank the National Institutes of Health (R01 AR069645 to S.C.Z.) and the National Science Foundation through the Graduate Research Fellow Program under Grant No. DGE 1746047 (S.B.K.). We thank Maurice Swanson (University of Florida, Gainesville) for the (CTG)90 plasmid and Nicholas Webster (University of California, San Diego) for the IR minigene plasmid.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Pennisi E, Science 2012, 337, 1159–1161. [DOI] [PubMed] [Google Scholar]
- [2].Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, Pandey SK, Revill M, Rooney C, Buckett LK, Klein SK, Hudson K, Monia BP, Zinda M, Blakey DC, Lyne PD, Macleod AR, Sci. Transl. Med 2017, 9, 1–13. [DOI] [PubMed] [Google Scholar]
- [3].Moreno AM, Fu X, Zhu J, Katrekar D, Shih YRV, Marlett J, Cabotaje J, Tat J, Naughton J, Lisowski L, Varghese S, Zhang K, Mali P, Mol. Ther 2018, 26, 1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chi X, Gatti P, Papoian T, Drug Discov. Today 2017, 22, 823–833. [DOI] [PubMed] [Google Scholar]
- [5].Bahrami A, Aledavood A, Anvari K, Hassanian SM, Maftouh M, Yaghobzade A, Salarzaee O, ShahidSales S, Avan A, J. Cell. Physiol 2018, 233, 774–786. [DOI] [PubMed] [Google Scholar]
- [6].Donlic A, Morgan BS, Xu JL, Liu A, Roble C, Hargrove AE, Angew. Chemie - Int. Ed 2018, 57, 13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu S, Yang Y, Li W, Tian X, Cui H, Zhang Q, Gene 2018, 662, 46–53. [DOI] [PubMed] [Google Scholar]
- [8].Matthes F, Massari S, Bochicchio A, Schorpp K, Schilling J, Weber S, Offermann N, Desantis J, Wanker E, Carloni P, Hadian K, Tabarrini O, Rossetti G, Krauss S, ACS Chem. Neurosci 2018, 9, 1399–1408. [DOI] [PubMed] [Google Scholar]
- [9].Connelly CM, Numata T, Boer RE, Moon MH, Sinniah RS, Barchi JJ, Ferré-D’Amaré AR, Schneekloth JS, Nat. Commun 2019, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gao Z, Cooper TA, Hum. Gene Ther 2013, 24, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rinaldi C, A Wood MJ, Nat. Publ. Gr 2017, DOI 10.1038/nrneurol.2017.148. [DOI] [Google Scholar]
- [12].Provenzano C, Cappella M, Valaperta R, Cardani R, Meola G, Martelli F, Cardinali B, Falcone G, Mol. Ther. - Nucleic Acids 2017, 9, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cerro-Herreros E, Sabater-Arcis M, Fernandez-Costa JM, Moreno N, Perez-Alonso M, Llamusi B, Artero R, Nat. Commun 2018, 9, 2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jenquin JR, Coonrod LA, Silverglate QA, Pellitier NA, Hale MA, Xia G, Nakamori M, Berglund JA, ACS Chem. Biol 2018, 13, 2708–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hsieh WC, Bahal R, Thadke SA, Bhatt K, Sobczak K, Thornton C, Ly DH, Biochemistry 2018, 57, 907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Konieczny P, Selma-Soriano E, Rapisarda AS, Fernandez-Costa JM, Perez-Alonso M, Artero R, Drug Discov. Today 2017, 22, 1740–1748. [DOI] [PubMed] [Google Scholar]
- [17].Mirkin SM, Nature 2007, DOI 10.1038/nature05977. [DOI] [Google Scholar]
- [18].Pearson CE, Nucleic Acids Res. 2002, 30, 4534–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brook DJ, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion J, Hudson T, Sohn R, Zemelman B, Snell RG, Flundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ, Housman DE, Park H, Cell 1992, 68, 799–808. [DOI] [PubMed] [Google Scholar]
- [20].Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA, Hum. Mol. Genet 2006, 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- [21].Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, Kumar A, Ashizawa T, Clark HB, Kimura T, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Gomes-Pereira M, Gourdon G, Sakai N, Nishino S, Foster TC, Ares M, Darnell RB, Swanson MS, Neuron 2012, 75, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MAC, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LPW, Proc. Natl. Acad. Sci 2011, 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wong CH, Nguyen L, Peh J, Luu LM, Sanchez JS, Richardson SL, Tuccinardi T, Tsoi H, Chan WY, Chan HYE, Baranger AM, Hergenrother PJ, Zimmerman SC, J. Am. Chem. Soc 2014, 136, 6355–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Luu LM, Nguyen L, Peng S, Lee JY, Lee HY, Wong CH, Hergenrother PJ, Chan HYE, Zimmerman SC, ChemMedChem 2016, 11, 1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rideout D, Science 1986, 233, 561–563. [DOI] [PubMed] [Google Scholar]
- [26].Rideout D, Calogeropoulou T, Jaworski J, McCarthy M, Biopolymers 1990, 29, 247–262. [DOI] [PubMed] [Google Scholar]
- [27].Huisgen R, Angew. Chem. Int. Ed 1963, 2, 565–632. [Google Scholar]
- [28].Poulin-Kerstein AT, Dervan PB, J. Am. Chem. Soc 2003, 125, 15811–15821. [DOI] [PubMed] [Google Scholar]
- [29].Benhamou RI, Angelbello AJ, Wang ET, Disney MD, Cell Chem. Biol 2020, 27, 223–231.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hagler LD, Krueger SB, Luu LM, Lanzendorf AN, Mitchell NL, Vergara JI, Curet LD, Zimmerman SC, ACS Med. Chem. Lett 2021, 12, 935–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hagler LD, Luu LM, Tonelli M, Lee J, Hayes SM, Bonson SE, Vergara JI, Butcher SE, Zimmerman SC, Biochemistry 2020, 59, 3463–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Goodwin JT, Lynn DG, J. Am. Chem. Soc 1992, 114, 9197–9198. [Google Scholar]
- [33].Zhan Z-YJ, Lynn DG, J. Am. Chem. Soc 1997, 119, 12420–12421. [Google Scholar]
- [34].Li X, Zhan ZYJ, Knipe R, Lynn DG, J. Am. Chem. Soc 2002, 124, 746–747. [DOI] [PubMed] [Google Scholar]
- [35].Bugaut A, Toulmé JJ, Rayner B, Angew. Chemie - Int. Ed 2004, 43, 3144–3147. [DOI] [PubMed] [Google Scholar]
- [36].Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, Miller BL, J. Am. Chem. Soc 2008, 130, 16254–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR, Science 2004, 305, 1601–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bahal R, Manna A, Hsieh WC, Thadke SA, Sureshkumar G, Ly DH, ChemBioChem 2018, 19, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Umuhire Juru A, Cai Z, Jan A, Hargrove AE, Chem. Commun 2020, 56, 3555–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee J, Bai Y, V Chembazhi U, Peng S, Yum K, Luu LM, Hagler LD, Serrano JF, Chan HYE, Kalsotra A, Zimmerman SC, Proc. Natl. Acad. Sci 2019, 116, 8709–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kiliszek A, Kierzek R, Krzyzosiak WJ, Rypniewski W, Nucleic Acids Res. 2009, 37, 4149–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Napierała M, Krzyzosiak WJ, J. Biol. Chem 1997, 272, 31079–31085. [DOI] [PubMed] [Google Scholar]
- [43].Qi X, Zhang F, Su Z, Jiang S, Han D, Ding B, Liu Y, Chiu W, Yin P, Yan H, Nat. Commun 2018, 9, 4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jain A, Vale RD, Nature 2017, 546, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hu T, Morten MJ, Magennis SW, Nat. Commun 2021, 12, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Savkur RS, Philips AV, Cooper TA, Nat. Genet 2001, 29, 40–47. [DOI] [PubMed] [Google Scholar]
- [47].Jog SP, Paul S, Dansithong W, Tring S, Comai L, Reddy S, PLoS One 2012, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mills JEJ, Dean PM, J. Comput. Aided. Mol. Des 1996, 10, 607–622. [DOI] [PubMed] [Google Scholar]
- [49].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- [50].Nguyen L, Luu LM, Peng S, Serrano JF, Chan HYE, Zimmerman SC, J. Am. Chem. Soc 2015, 137, 14180–14189. [DOI] [PubMed] [Google Scholar]
- [51].Jahromi AH, Nguyen L, Fu Y, Miller KA, Baranger AM, Zimmerman SC, ACS Chem. Biol 2013, 8, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.