

Abstract
Background:
MRL/MpJ-Tnfrsf6lpr (MRL/lpr) mice, a murine model of systemic lupus erythematosus (SLE), have defective expression of Fas, substantially reducing signaling for apoptosis via this mechanism. However, it is known that MRL/lpr mice have increased spontaneous apoptosis of leukocytes. These conflicting observations have stimulated interest in apoptosis in this SLE model. MRL/lpr mice overproduce nitric oxide (NO) as autoimmune disease progresses. In vitro administration of NO may induce or decrease apoptosis depending on the cell type. Therefore, we hypothesized that NO induces MRL/lpr spleen lymphocyte apoptosis independent of Fas receptor engagement.
Methods:
Percentages of apoptotic spleen lymphocytes from MRL/lpr and BALB/cJ mice were determined ex vivo after in vivo treatment with NG-monomethyl-l-arginine (NMMA), a nitric oxide synthase (NOS) inhibitor. After culture in varying concentrations of a slow-acting NO donor, the following were determined in spleen lymphocytes: (1) levels of apoptosis, (2) the effect of phorbol myristate acid (PMA) on levels of NO-induced apoptosis, and (3) protein kinase C (PKC) activity.
Results:
Spleen lymphocytes from MRL/lpr mice with active disease had increased levels of ex vivo apoptosis when compared with BALB/cJ controls. This increase was reduced by pharmacologic inhibition of NOS in MRL/lpr but not in BALB/cJ mice. Exogenous administration of NO in vitro reduced PKC activity and induced apoptosis in MRL/lpr spleen lymphocytes, an effect that could be reduced via coadministration of PMA in vitro.
Conclusion:
These results suggest that NO plays a role in spleen lymphocyte apoptosis in MRL/lpr mice, possibly via inhibition of PKC, despite a Fas defect.
Keywords: apoptosis, lupus, nitric oxide
MRL/MpJ-Tnfrsf6lpr (MRL/lpr) mice spontaneously develop a lupus-like disease with immune complex glomerulonephritis, arthritis, and anti-double-stranded deoxyribonucleic acid (DNA) antibody production. These mice have a mutation in the Fas or Tnfrsf6 gene, which leads to reduced signaling for apoptosis.1 Much emphasis has therefore been placed on the role of dysregulated apoptosis in the progression of autoimmune disease in these mice.2,3 Despite a deficit in Fas, MRL/lpr mice have displayed increased apoptosis of peripheral lymphocytes.4 Our laboratory has shown that MRL/lpr mice overproduce nitric oxide (NO) and that blocking NO production inhibits disease progression.5,6 Therefore, we hypothesized that increased levels of NO induce apoptosis in these mice via an alternative mechanism to Fas receptor engagement.
NO is a soluble inter- and intracellular messenger formed by the deamination of arginine by nitric oxide synthase (NOS) to form NO and l-citrulline.7 The inducible form of NOS (iNOS) produces large amounts of NO and can be found in macrophages, neutrophils, lymphocytes, Kupffer’s cells, and mast cells. NO is known to have vasodilatory, antitumor, antimicrobial, and inflammatory properties. A growing body of evidence indicates that NO plays a role in autoimmunity.5,6,8–11 The mechanism by which NO is pathogenic in autoimmune syndromes is not clear, however.
Several studies implicate NO as a regulator of apoptosis in humans and mice. NO induces apoptosis in murine macrophages, CD4+CD8+ thymocytes, mastocytoma cells, and pancreatic islet cells.12–17 On the other hand, low concentrations of NO (1–100 μM) can reduce the percentage of apoptotic murine B cells via increased expression of BCL2.18 In humans, NO increases apoptosis among HL-60 promyelocytic leukemia and U937 monocytic cell lines,19 whereas it reduces apoptosis in B lymphocytes a eosinophils.20,21 The role of NO in regulating apoptosis of circulating immune cells in lupus is unknown.
In this study, we investigated the role of NO in regulating MRL/lpr spleen lymphocyte apoptosis by blocking the production of NO with oral NG-monomethyl-l-arginine (NMMA) in MRL/lpr mice with active disease. With no treatment, spleen lymphocytes from diseased MRL/lpr mice had enhanced levels of apoptosis when compared with BALB/cJ controls. NMMA treatment reduced the percentage of apoptotic MRL/lpr spleen 1ymphocytes, particularly B cells, in vivo, whereas the spleen lymphocytes of BALB/cJ controls were not affected by NMMA treatment. These results were confirmed in vitro by exogenous administration of NO via a stable NO donor (Deta NONOate, Alexis Biochemicals, San Diego, CA), which enhanced apoptosis of spleen lymphocytes. Furthermore, exogenous NO inhibited protein kinase C (PKC) activity, and a PKC agonist reduced the level of NO-induced apoptosis. These data offer NO-mediated signaling as a potential mechanism for the seemingly contradictory observation that apoptosis is increased in MRL/lpr lymphocytes despite a homozygous Fas defect.
METHODS
Mice and Sample Collection
Female MRL/lpr and BALB/cJ mice were purchased from Jackson Laboratory (Bar Harbor, ME) at 6 weeks of age and housed under specific pathogen-free conditions in the animal research facility at the Ralph H. Johnson Veterans Affairs Medical Center. Mice were serologically tested and confirmed negative for common murine pathogens on a random basis. Mice from both in vivo treatment groups were fed a low-nitrate and nitrite (NOx), arginine-free diet (Zeigler Brothers, Gardners, PA) throughout the 2-week treatment period to eliminate dietary NOx as a contaminating source of serum NOx. Arginine was eliminated from the diet to prevent competitive inhibition of NMMA absorption because NMMA is an analogue of arginine. Retrobulbar blood collection for serum NOx determinations was performed after anesthesia but prior to sacrifice. Mice used for the in vitro studies were fed a regular diet and sacrificed via cervical dislocation after anesthesia. Untreated MRL/lpr and BALB/cJ mice were used for analysis of spontaneous apoptosis of spleen lymphocytes in vivo. All MRL/lpr mice used for in vivo experiments were 17 weeks old or older because MRL/lpr mice begin overproducing NO at approximately 12 weeks of age.5
In Vivo Treatment
Treated mice were given 50 mM NMMA (Cyclopss Biochemical Corporation, Salt Lake City, UT) ad libitum in their drinking water, whereas controls received distilled water.6,22 Eight mice in each group were given these treatments daily for 2 weeks prior to sacrifice and analysis of spleen lymphocytes. Two separate experiments containing both treatments were performed for the in vivo studies.
Culture of Spleen Lymphocytes
In vitro studies were performed with BALB/cJ and MRL/lpr mice as described above. Mice were sacrificed, and spleens were disrupted by gently compressing and shearing spleens between frosted slide ends using a rotary motion. Red cells were lysed by suspension in a lysis buffer (17 mM Tris HCl pH 7.5 and 140 mM NH4Cl, Sigma, St. Louis, MO) for 2 minutes. Spleen lymphocytes were then washed twice in Dulbecco’s Modified Eagle medium (DMEM) without phenol red, containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco BRL, Grand Island, NY). Cells were resuspended in medium at 2.5 × 106 cells/mL, and 0.2 mL of the suspension was added to each 6 mm well. Deta NONOate was prepared to 0.12 M in 0.01 M NaOH within 30 minutes of culture and diluted to 10× solutions in DMEM just prior to culture to prevent release of NO prior to dilution in medium.23 Sterile Deta NONOate (final concentration: 0, 100, 250, 500, or 1,000 μM) was added to each well. Cells remained in culture for 13 to 20 hours. The results of five separate experiments were combined for MRL/lpr mice, and the results of two experiments were combined for BALB/cJ mice. To determine the effects of phorbol myristate acetate (PMA) on NO-induced apoptosis, MRL/lpr spleen lymphocytes were also cultured in three separate experiments with or without additional PMA (10 ng/mL). PMA or control solvent was added simultaneously with the Deta NONOate as above.
NOx Determinations
All serum samples were collected, frozen at −20°C, and thawed at the same time followed by filtration with a 10,000 nominal molecular weight limit filter (Millipore Corporation, Bedford, MA) to remove protein prior to analysis. Some of the supernatant samples were diluted 1:10 to keep NOx concentrations within the linear detection range of the assay (0–125 μM). Known concentrations of nitrate and nitrite were serially diluted to create a standard curve. Serum and supernatant NOx were determined in duplicate using lie Greiss reagent after reduction of nitrate to nitrite with a modified nitrate reductase reaction using the following final concentrations of reagents: 2 μM reduced nicotinamide adenine dinucleotide phosphate, 1 mM glucose-6-phosphate, 0.3 U/mL glucose-6-phosphate dehydrogenase, 0.1 U/mL nitrate reductase (all Boehringer Mannheim, now Roche Diagnostics Corp., Indianapolis, IN), and 0.22 M Tris HCI, pH 7.5.24
Analysis of Spleen Lymphocyte Apoptosis
Fresh and cultured cells were transferred to a “V bottom” 96-well plate at 2.5 × 105 cells per well. Cells were washed in phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA) (Sigma, St. Louis, MO) and pelleted. To resuspend pelleted cells, 0.2 μg of phycoerythrin (PE)-conjugated anti-CD4, -CD8, or -immunoglobulin M (IgM) antibodies (Promega, Madison, WI) in 50 μL of 1% BSA in PBS was added to wells. The plates were incubated on ice in the dark for 30 minutes.25 The cells were pelleted, washed in PBS, and suspended in 100 μL of binding buffer. Ten microliters each of fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide (PI) from an apoptosis detection kit (R & D Systems, Minneapolis, MN) were added to each well, and the plates were incubated at room temperature for 15 minutes in the dark. Cells were transferred to 12 × 75 mm tubes through a 40 μ filter (Falcon, #2235), placed on ice in a dark container, and analyzed by flow cytometry (Becton Dickinson FACS Vantage, 488 nm excitation wavelength). FITC-conjugated annexin V was detected by fluorescence detector 1 (FL1) using a band filter (530 ± 30 nm). PE-conjugated cell surface markers were detected by fluorescence detector 2 (FL2) using a band filter (585 ± 42 nm). PI staining was detected on fluorescence detector 3 (FL3) using a long pass filter (> 670 nm). Ruptured cells stained for either annexin V, PE cell surface stain, or PI alone were used for positive controls to adjust the compensation and photomultiplier tube voltage prior to analysis and gating after analysis. PE-conjugated isotype controls were used to set gates for analysis. Flow cytometry data were analyzed using the CELLQuest program (Becton Dickinson, San Jose, CA). Viable cells (membranes intact) were first gated on as fluorescence channel 3 (FL3) (PI) negative, middle- and upper-range forward scatter cells. To determine overall spleen lymphocyte apoptosis, viable FL3-negative gated cells were analyzed by quadrant on a dot plot of FL1 versus FL2, whereby FL1-positive gated cells were apoptotic. FL2-positive cells were CD4+, CD8+, or IgM+ depending on which stain was used. In a specific sample, live, nonapoptotic cells were quantitated by determining the percentage of viable annexin V− cells in each subset.26
PI DNA Staining for Analysis of Apoptosis
Spleen lymphocytes (5 × 105) from each sample were fixed in 70% ethanol for 1 to 7 days at −20°C. After fixation, fixed nuclei were transferred to a V bottom 96-well plate, washed twice in PBS, pelleted, resuspended in 100 μL of ribonuclease A (20 μg/mL [R6513, Sigma]), and incubated for 30 minutes in the dark at 37°C. Ten microliters of 110 μg/mL PI in PBS was added to the suspension, and cells were incubated at room temperature for 10 minutes in the dark. Cells were analyzed by flow cytometry using the same equipment and software as with the annexin V apoptosis assay. Pulse processing was used for this assay, and cells were gated using FL2 width versus FL2 amplitude plots to exclude doublets and cell fragments. Gated cells were analyzed using an FL2 histogram. Apoptotic cells appeared as a sub-G1 peak.27
Confirmation of Annexin V Assay for Apoptosis
Spleen lymphocytes from several experiments were analyzed using both the annexin V technique and PI nuclear staining techniques. Annexin V and PI assay results of samples treated at the same time correlated well (r = .88, p = .022; data not shown), so combined annexin V-FITC and cell surface marker-PE staining was used in subsequent experiments to analyze simultaneously for apoptosis and cellular subsets.
Proliferation Assay
Spleen lymphocytes were cultured for 2 hours prior to addition of varying concentrations of Deta NONOate as described above. After adding 2 μCi of [3H]thymidine to the wells, cells were cultured for 24 hours at 37°C in 5% carbon dioxide. Cells were lysed with distilled water and ethanol, and lysates were extracted with a cell harvester (Skatron Instruments, Lier, Norway). After addition of scintillation fluid, counts were read on a Beckman LS 6500 scintillation counter (Beckman Coulter, Inc., Fullerton, CA).The results of two separate experiments with triplicate readings for each well were averaged after normalizing the data to the percentage of counts per treatment relative to control wells.
PKC Activity of Cell Lysates
MRL/lpr spleen lymphocytes were cultured in 0, 250, and 1,000 μM Deta NONOate overnight. Cells were harvested and analyzed for PKC assay using a Calbiochem assay (#539484, Alexis Biochemicals) according to the manufacturer’s instructions. The results of three different experiments performed in triplicate were averaged after the data were normalized to the percentage of activity relative to control wells.
Statistical Analysis
All statistical calculations were performed using SigmaStat 2.03 software (SPSS Inc., Chicago, IL). All values for the percentage of apoptotic cells were reported as a percentage of controls, thus allowing for a comparison of treatment effect between experiments. All comparisons of data between individual in vivo and in vitro treatment groups were made by Wilcoxon rank sum analysis or Student’s t-test depending on the distribution of the data. Simple linear regression analyses were used to correlate serum or supernatant NOx with the percentage of apoptotic cells ex vivo. The same analysis was used to compare annexin V and PI assay results for apoptosis. Two-way repeated-measures analysis of variance was used to determine the effect of a treatment at several concentrations of Deta NONOate on cells cultured simultaneously in vitro. The results were reported as mean ± standard error and were considered significant if p values were <.05. Percentages of CD4−/CD8−/IgM− annexin V+ intact cells were determined by subtracting the sum of the percentages of CD4+, CD8+, and IgM+ cells from averages of all annexin V+ cells.
RESULTS
In Vivo Apoptosis of MRL/lpr Spleen Lymphocytes Was Greater than That of BALB/cJ Spleen Lymphocytes
Splenocytes from MRL/lpr mice (that were old enough to overproduce NO) and BALB/cJ mice were isolated and analyzed for apoptosis using annexin V and PI staining and flow cytometry. MRL/lpr spleen lymphocytes had greater levels of spontaneous in vivo apoptosis than BALB/cJ controls (Figure 1).
FIGURE 1.
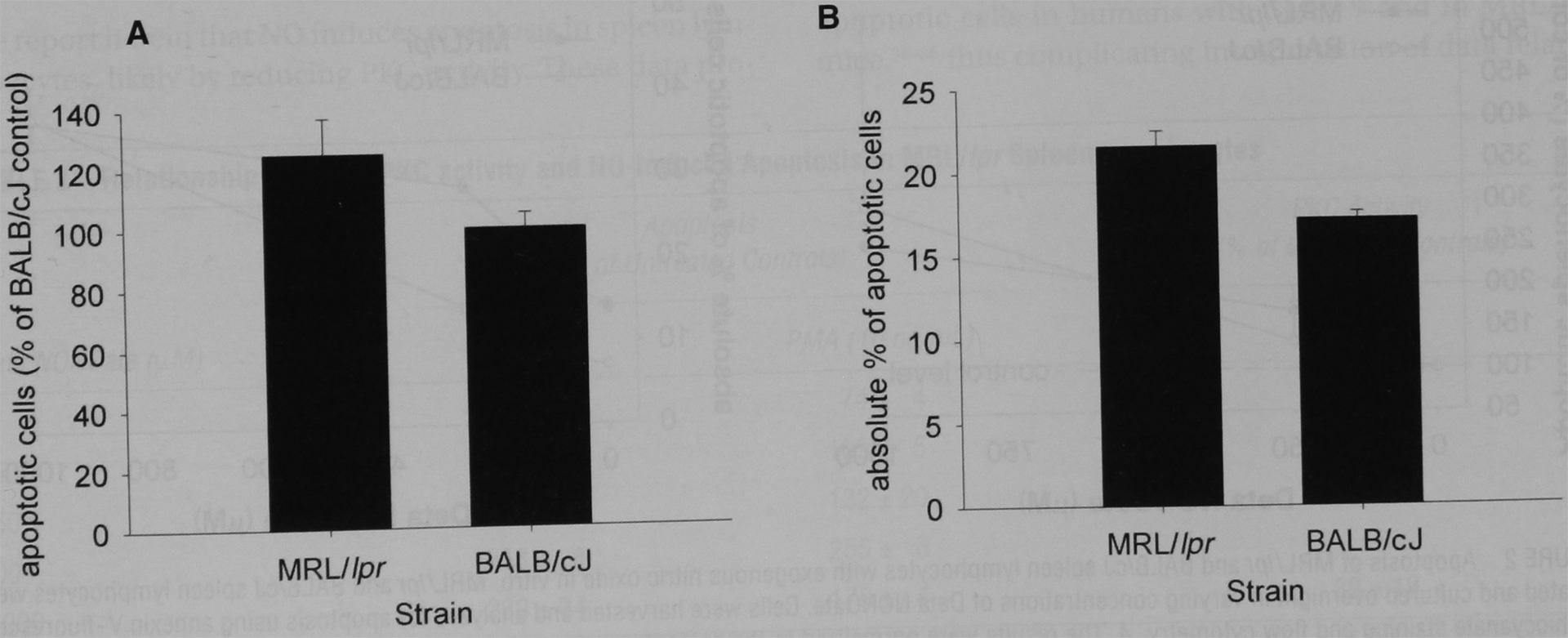
Apoptosis of MRL/lpr and BALB/cJ isolated spleen cells in vivo. Spleen lymphocytes from MRL/lpr mice (that were old enough to overproduce nitric oxide) and BALB/cJ mice were isolated and analyzed for apoptosis using annexin V and propidium iodide staining and flow cytometry. A, Apoptosis of MRL/lpr spleen lymphocytes was reported as a percentage of that for BALB/cJ spleen lymphocytes. The results were an average of several experiments. Apoptosis of MRL/lpr spleen lymphocytes was greater than that seen in BALB/cJ mice (p < .05). B, The results of a single, representative experiment, reported as the percentage of all intact cells that are apoptotic.
NMMA Significantly Reduced MRL/lpr and Not BALB/cJ Spleen Lymphocyte Apoptosis In Vivo
MRL/lpr mice with active disease and BALB/cJ mice were given NMMA or distilled water for 2 weeks. Spleen lymphocytes were then isolated and analyzed for apoptosis. NMMA significantly reduced spleen lymphocyte apoptosis in MRL/lpr but not BALB/cJ spleen lymphocytes (Table 1). Spleen lymphocyte subset analysis of apoptotic MRL/lpr spleen lymphocytes was performed using flow cytometry. NMMA significantly reduced apoptosis of only IgM+ cells in MRL/lpr cells and did not significantly affect apoptosis of BALB/cJ subsets (see table 1). The percentage of live, intact cells in each subset was not significantly affected by NMMA treatment (data not shown). Sera NOx levels were determined at the time of spleen lymphocyte isolation tor MRL/lpr mice in both treatment groups. Serum NOx levels correlated significantly with IgM+ spleen lymphocyte apoptosis (r = .60, p = .015).
TABLE 1.
Oral Administration of the NOS Inhibitor NMMA Inhibited Apoptosis of Spleen Lymphocytes from MRL/lpr but not BALB/cJ Control Mice
| MRL/lpr | Apoptosis (% of Control Treatment) |
p Value | Representative Apoptosis (Absolute % Of Leukocytes) |
||
|---|---|---|---|---|---|
| Distilled Water (n=8) |
NMMA (n = 9) |
Distilled Water | NMMA | ||
| All spleen lymphocytes | 100 ± 7 | 67 ± 6 | .003 | 32.9 ± 3.6 | 26.3 ± 1.4 |
| Cell surface marker | |||||
| IgM+ | 100 ±18 | 36 ± 7 | .005 | 7.8 ± 1.6 | 4.1 ± 0.4 |
| CD8+ | 100 ±21 | 62 ±12 | .1 | 1.9 ± 0.3 | 1.4 ± 0.3 |
| CD4+ | 100 ± 9 | 106 ± 14 | .7 | 4.0 ± 0.5 | 5.0 ± 0.8 |
| IgM−CD4−CD8− | 100 ± 20 | 89 ± 9 | .6 | 19.2 ± 3.3 | 16.6 ± 1.9 |
| BALB/cJ | Distilled Water (n=5) |
NMMA (n = 5) |
p Value | Distilled Water | NMMA |
| All spleen lymphocytes | 100 ± 20 | 108 ± 22 | .36 | 15.3 ± 3.1 | 16.7 ± 3.4 |
| Cell surface marker | |||||
| IgM+ | 100 ± 0.1 | 119 ± 0.1 | .3 | 6.6 ± 0.2 | 6.2 ± 0.5 |
| CD8+ | 100 ± 0.2 | 112 ± 0.1 | .7 | 2.0 ± 0.5 | 2.0 ± 0.2 |
| CD4+ | 100 ± 0.1 | 95 ± 0.1 | .5 | 5.0 ± 0.7 | 6.0 ± 0.6 |
| IgM−CD4−CD8− | 100 ± 27 | 150 ± 14 | .3 | 1.8 ± 0.3 | 2.2 ± 0.5 |
NMMA = NG-monomethyl-l-arginine; NOS = nitric oxide synthase.
On the left side of the table, data are from MRL/lpr mice with high inducible nitric oxide activity and BALB/cJ mice treated for 2 weeks with either distilled water or NMMA over separate experiments. Spleen lymphocytes were isolated and analyzed for annexin V staining for apoptosis and cell surface markers by three-color flow cytometry. The results were reported as a percentage of apoptotic cells ± standard error relative to the percentage of apoptotic spleen lymphocytes in distilled water-treated control mice (so that data could be normalized across several experiments). On the right side of the table, representative single experiments are reported for BALB/cJ and MRL/lpr mice to display the exact percentages of intact cells in each subset staining with annexin V.
Exogenous NO Induces Apoptosis in MRL/lpr and BALB/cJ Spleen Lymphocytes In Vitro
MLR/lpr and BALB/cJ spleen lymphocytes were isolated and cultured with varying concentrations of Deta NONOate (a slow-releasing NO donor) Tor 12 to 18 hours overnight. Spleen lymphocytes were then analyzed for apoptosis using annexin V and cell surface markers. Deta NONOate significantly increased apoptosis of both MRL/lpr and BALB/cJ spleen lymphocytes in a similar dose-dependent fashion (Figure 2). When subset analysis of apoptosis was performed, Deta NONOate induced apoptosis in all subsets, with statistical significance achieved for CD4+ and CD8+ cells in isolated MRL/lpr spleen cells (Figure 3). Subset analysis was not performed for BALB/cJ isolated spleen cells.
FIGURE 2.
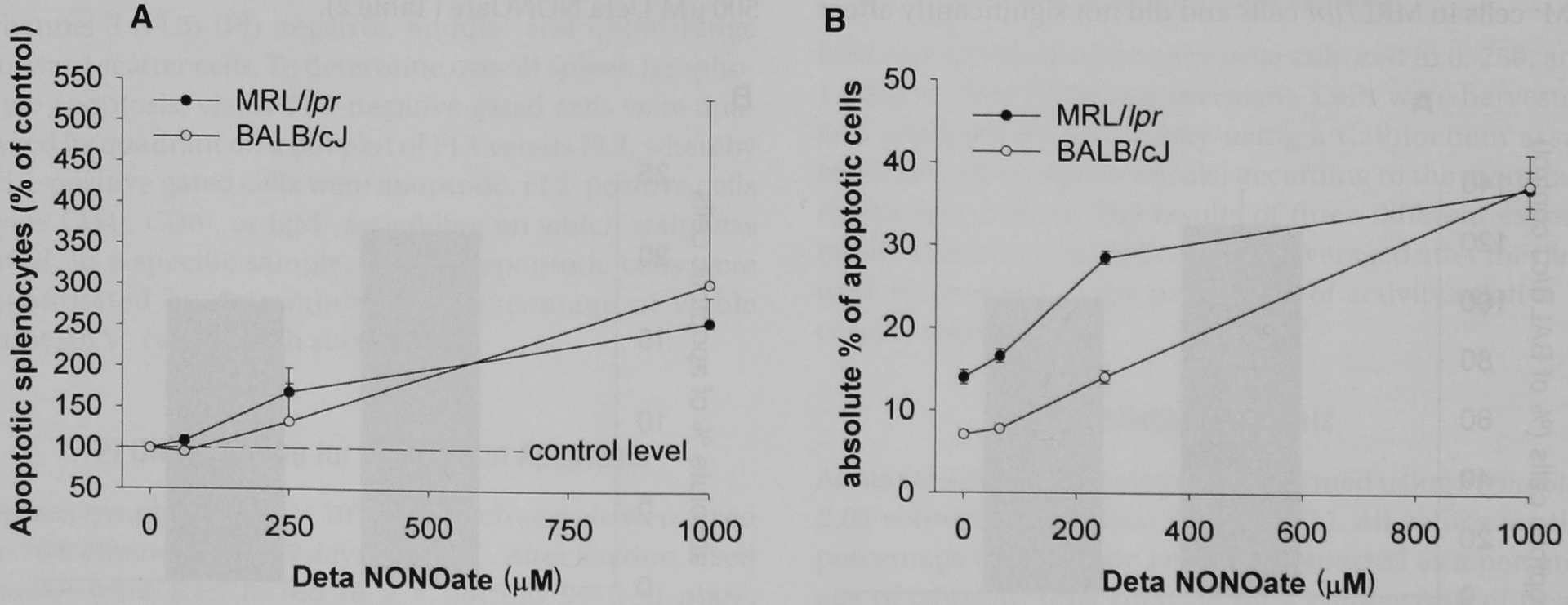
Apoptosis of MRL/lpr and BALB/cJ spleen lymphocytes with exogenous nitric oxide in vitro. MRL/lpr and BALB/cJ spleen lymphocytes were isolated and cultured overnight in varying concentrations of Deta NONOate. Cells were harvested and analyzed for apoptosis using annexin V—fluorescein isothiocyanate staining and flow cytometry. A, The results were normalized to the percentage of apoptotic cells relative to control wells (not exposed to Deta NONOate) and reported as the average ± standard error of five experiments for MRL/lpr mice and two experiments for BALB/cJ mice MRL/lpr spleen lymphocyte apoptosis was represented with closed circles, whereas BALB/cJ apoptosis was represented with open circles. The results were reported as a percentage of control conditions (no added Deta NONOate). For MRL/lpr spleen lymphocytes, p < .001 for treatment effect. For BALB/cJ spleen lymphocytes, treatment effect was not statistically significant across two experiments. B, Representative single experiment from data reported in A The results were reported as the percentage of intact cells that were apoptotic.
FIGURE 3.
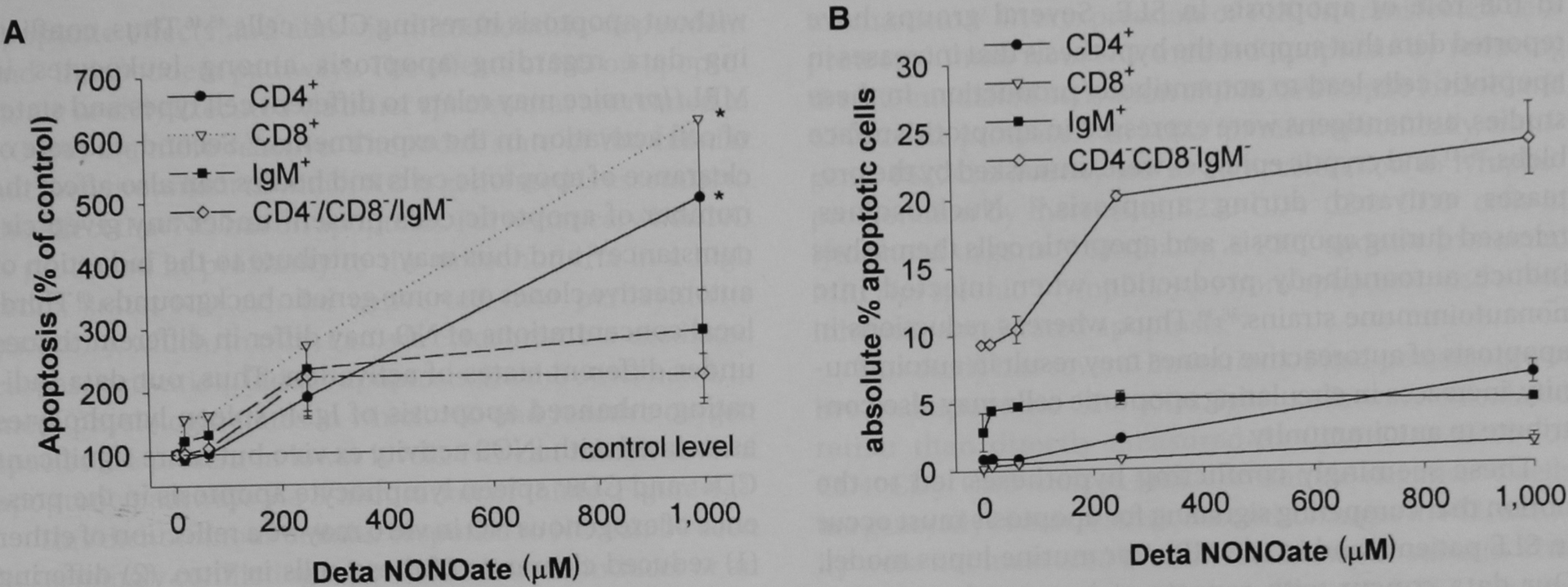
Apoptosis of MRL/lpr spleen lymphocyte subsets with exogenous nitric oxide in vitro. MRL/lpr spleen lymphocytes were isolated and cultured overnight in varying concentrations of Deta NONOate. Cells were harvested and analyzed for apoptosis using flow cytometry with annexin V–fluorescein isothiocyanate and phycoerythrin cell surface marker (CD4+, CD8+, and IgM+) staining. A, The results were normalized to the percentage of apoptotic cells relative to control wells (not exposed to Deta NONOate) ± standard error and were calculated from the average of five experiments. Apoptosis of subsets was reported as follows: CD4+ cells as closed circles, CD8+ cells as open inverted triangles, IgM+ cells as closed squares, and CD4−CD8−IgM− cells as open diamonds. Deta NONOate induced significant apoptosis among CD4+, CD8+, and CD4−CD8−IgM− cells (asterisk signifies p < .05). B, The results of a representative single experiment reported as the percentage of apoptotic intact cells in each subset. Subset analysis was not performed for BALB/cJ spleen lymphocytes.
PKC Agonist PMA Inhibits NO-Induced Apoptosis of MRL/lpr Spleen Lymphocytes
MRL/lpr spleen lymphocytes were isolated and cultured overnight in varying concentrations of Deta NONOate with or without the PKC agonist PMA (10 ng/mL) added simultaneously. PMA reduced NO-induced apoptosis of MRL/lpr spleen lymphocytes except in those cultured with 500 μM Deta NONOate (Table 2).
TABLE 2.
Relationship between PKC activity and NO-Induced Apoptosis in MRL/lpr Spleen Lymphocytes
| Deta NONOate (μM) |
Apoptosis (% of Untreated Controls) |
PKC Activity (% of Untreated Controls) |
|
|---|---|---|---|
| No PMA | PMA (10 ng/mL) | No PMA | |
| 0 | 100 ± 4 | 74 ± 4 | 100 ± 0 |
| 100 | 154 ± 13 | 109 ± 5 | — |
| 250 | 191 ± 18 | 132 ± 20 | 76 ± 27 |
| 500 | 253 ± 8 | 255 ± 6 | — |
| 1,000 | 233 ± 34 | 179 ± 4 | 39 ± 18 |
NO = nitric oxide; PKC = protein kinase C; PMA = phorbol myristate acid.
MRL/lpr spleen lymphocytes were cultured overnight in varying concentrations of Deta NONOate (0–1,000 μM) with or without PMA (10 ng/mL). Cells were analyzed by flow cytometry for the percentage of apoptotic cells present. The results reported are a percentage (± standard error) of the control wells (no additive) and were the average of triplicate readings from three different experiments. Cells were also analyzed for PKC activity in two separate experiments.
Deta NONOate Inhibits PKC in MRL/lpr Spleen Lymphocytes
MRL/lpr spleen lymphocytes were isolated and cultured overnight in 0, 250, and 1,000 μM Deta NONOate. Cells were harvested after culture and analyzed for PKC activity. Deta NONOate reduced PKC activity in a dose-dependent fashion with a statistically significant effect at 1,000 μM Deta NONOate (see Table 2; p = .04).
DISCUSSION
We report herein that NO induces apoptosis in spleen lymphocytes, likely by reducing PKC activity. These data provide insight into the conflicting literature regarding the role of apoptosis in this murine model of systemic lupus erythematosus (SLE) and highlight the importance of multiple competing pathways in apoptosis signaling. Initial interest in apoptosis of immune cells in this model arose from the discovery that the lymphoproliferative or lpr phenotype arose from defective Fas signaling.28 This discovery led to the hypothesis that reduced signaling for apoptosis in these mice not only led to lymphoproliferation but contributed to autoimmunity by reducing programmed deletion of autoreactive B and T cells.28,29 However, several laboratories have reported increased numbers of circulating apoptotic cells in humans with SLE30–35 and in MRL/lpr mice,36–38 thus complicating interpretation of data related to the role of apoptosis in SLE. Several groups have reported data that support the hypothesis that increases in apoptotic cells lead to autoantibody production. In these studies, autoantigens were expressed in apoptotic surface blebs,39–41 and cryptic epitopes were unmasked by the proteases activated during apoptosis.42 Nucleosomes, released during apoptosis, and apoptotic cells themselves induce autoantibody production when injected into nonautoimmune strains.36–39 Thus, whereas reductions in apoptosis of autoreactive clones may result in autoimmunity, increases in circulating apoptotic cells may also contribute to autoimmunity.
These seemingly conflicting hypotheses led to the notion that competing signaling for apoptosis must occur in SLE patients and in the MRL/lpr murine lupus model. Our data concur with reports of increased apoptosis among thymocytes from MRL/lpr mice when compared with those from BALB/cJ mice.43 Others, however, have reported reduced apoptosis of (1) CD4+ cells from MRL/lpr mice during receptor engagement3 and (2) in spleen lymphocytes and thymocytes when studied by labeling of end-nicked DNA.44 There are three possible explanations for conflicting data regarding apoptosis in immune cells in these mice. First, pro- and antiapoptotic signaling varies according to cell type and state of activation. For example, CTLA4 cross-linking in lpr/lpr mice led to apoptosis in stimulated CD4+ cells but resulted in reduced proliferation without apoptosis in resting CD4+ cells.45–46 Thus, conflicting data regarding apoptosis among leukocytes in MRL/lpr mice may relate to different cell types and states of cell activation in the experiments.47 Second, the rate of clearance of apoptotic cells and bodies can also affect the number of apoptotic cells present under any given circumstance48 and thus may contribute to the induction of autoreactive clones on some genetic backgrounds.49 Third, local concentrations of NO may differ in different tissues under different states of activation. Thus, our data indicating enhanced apoptosis of IgM+ spleen lymphocytes associated with iNOS activity ex vivo but more significant CD4+ and CD8+ spleen lymphocyte apoptosis in the presence of exogenous NO in vitro may be a reflection of either (1) reduced clearance of these cells in vitro, (2) differing states of cell activation in vivo versus in vitro, or (3) differing local concentrations of NO in vivo compared with uniform exposure in vitro.50 The fact that BALB/cJ and MRL/lpr spleen lymphocytes undergo apoptosis in a similar fashion in response to exogenous NO in vitro (see Figure 1) suggests that the stimulus for excessive apoptosis of MRL/lpr spleen lymphocytes is not intrinsic to these cells but is instead due to the overproduction of NO or increased oxidative stress at the site of NO production (discussed below) observed in these mice.5
NO, either as produced endogenously or when added to culture medium via NO donors, has both pro- and antiapoptotic effects and acts via mitochondria-dependent and -independent pathways. The effects of NO on apoptosis are usually executed via other species that are related to NO but are more reactive. The redox state of the cellular microenvironment in which NO is generated is essential to the effect that NO has on apoptosis. For instance, when NO is produced in proximity to the mitochondria in a high redox state, NO can be converted into peroxynitrite (ONOO−), which, in turn, can, via cytochrome-c–mediated caspase activation, induce apoptosis. On the other hand, in more physiologic states in which NO and reactive oxygen species are present in lower levels, NO can inhibit several apoptotic pathways via S-nitrosation of thiols (Figure 4).51
FIGURE 4.
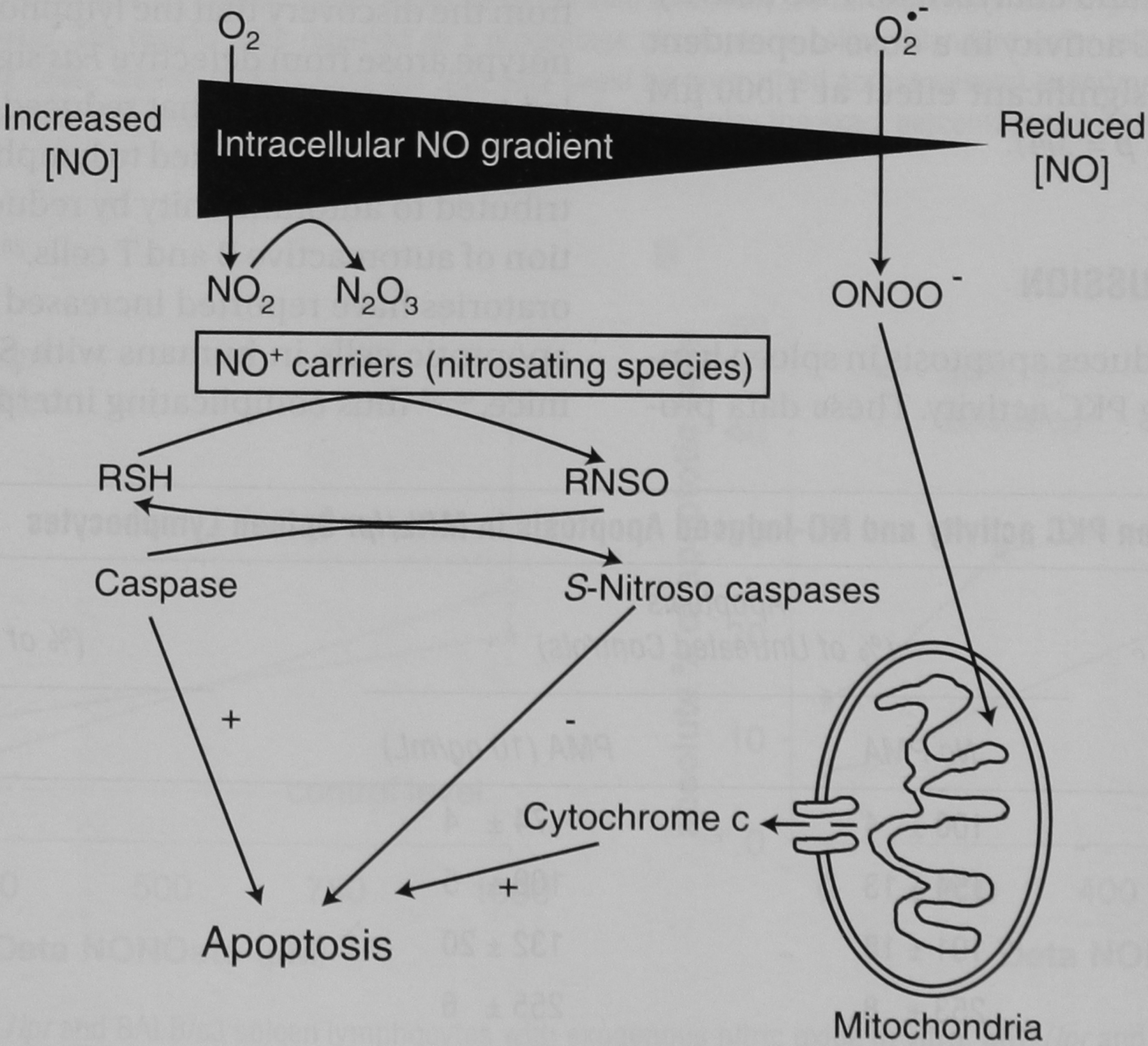
Effect of intracellular nitric oxide (NO) and oxygen gradients on the formation of NO and NO-related species: subsequent effect on mitochondria- and non–mitochondria-dependent apoptosis. The fate of intracellular NO is highly dependent on the relative concentrations of NO and reactive oxygen species. This simplified diagram demonstrates that NO, when produced in the presence of superoxide (O2−), forms peroxynitrite (ONOO−) which stimulates cytochrome-c release from mitochondria. This release can result in apoptosis of the cell. On the other hand, when NO is produced under more physiologic circumstances, it can S-nitrosate caspases and inhibit apoptosis. Adapted from Boyd CS and Cadenas E.51 RNSO = S-nitrosated thiols; RSH = reduced thiol species.
This dichotomous cellular response to NO can be seen in seemingly conflicting reports of NO as an inducer or an inhibitor of apoptosis in different cell types or under different culture conditions. For instance, in thymocytes, tyrosine nitration by reactive nitrogen species appears to be an important step in NO-induced apoptosis.52 When exposed to methylprednisolone and etoposide, rat thymocytes signal for apoptosis by expression of iNOS in mitochondrial membranes and subsequent NO production.53 Induction of iNOS in these settings appeared to serve as an intracellular or autocrine signal or apoptosis.54 Exogenous NO has also been demonstrated to induce apoptosis in thymocytes,52,55 dendritic cells,56 and monocyte and macrophage cell lines in vitro.57 In a more physiologic setting, dendritic cells in the thymus express iNOS in response to self-antigens and may play a role in inducing tolerance by inducing thymocyte apoptosis in an autocrine fashion.58
In contrast to our observations in MRL/lpr lgM+ cells, Mannick and colleagues reported a distinct inhibitory action of exogenous and endogenous NO production in human B lymphocyte apoptosis.21 The same group later reported that NO inhibits Fas-mediated apoptosis in human leukocytic cell lines by S-nitrosation of proapoptotic caspases.59,60 The proapoptotic effect of NO in our hands versus the antiapoptotic effect in the hands of Mannick and colleagues could be explained by differing culture conditions leading to differences in an exogenous or endogenous redox state. However, Mozart and colleagues cultured two different B-cell lines with the same concentration of an exogenous NO donor. Whereas one clone underwent cell cycle arrest and apoptosis, the other did not.61 These results suggest that there is a clone-dependent susceptibility to NO-mediated apoptosis that is consistent with the notion that the effects of NO on apoptosis are highly dependent on reactive oxygen and nitrogen states in the local cellular microenvironment. Such differences may explain the divergent response to NO between human B-cell clones and isolated IgM+ murine spleen cells.
Several laboratories have investigated the role of PKC in NO-mediated signaling for apoptosis. NO induced apoptosis in RAW 264.7 macrophages via a PKC-mediated mechanism.62 Overexpression of PKC in transfected cells protected them from NO-induced apoptosis by reducing the NO-mediated activation of JNK/SAPK, p38 kinase, and CPP32-like protease.63 In MRL/lpr mice specifically, transgenic PKCK2α activity led to an acceleration in lymphoproliferation by increasing B220+CD4−CD8−CD3+ cells in lymphoid organs.64 Similarly, a PKC agonist protected MRL/lpr splenic T lymphocytes from apoptosis, and a PKC inhibitor accelerated apoptosis.65
This study has several limitations. First, percentages of live and apoptotic CD4−CD8−IgM− cells were calculated rather than directly measured as CD4−CD8−B220+ or CD4−CD8−CD3+ cells as MRL/lpr “double-negative” T cells are generally characterized. Second, changes in PKC activity were not measured in individual cellular subsets. This opens the possibility that changes in overall PKC activity may have occurred owing to changes in the composition of remaining live cells. However, in vitro treatment of MRL/lpr spleen cells failed to significantly affect the percentage of live cell subsets (data not shown). There are multiple mechanisms of inducing apoptosis by NO-related species that were not addressed in this report. Among these are effects on cytochrome-c release, caspase activity, and tyrosine kinase activity.51 Therefore, any data regarding PKC-mediated mechanisms of apoptosis induction should be interpreted with caution.
This is the first description of NO-mediated apoptosis in the MRL/lpr model of SLE. This contradicts the notion that this model exhibits a phenotype of universal defective apoptotic signaling. However, it does offer a possible mechanism by which targeted signaling for apoptosis of autoreactive clones may be defective during induction of tolerance yet increased in a nonspecific manner in the periphery during inflammatory states such as infection or autoimmune syndromes. Further work regarding the effects of NO on leukocytes of humans with SLE is ongoing.
ACKNOWLEDGMENTS
Special thanks go to the Medical University of South Carolina Flow Cytometry Facility under the direction of Makio Ogawa, MD, PhD, and Haiqun Zeng, MD, without whom this study would not have been possible.
Supported by a University Research Committee grant from the Medical University of South Carolina, Office of Research and Sponsored Programs; a Career Development Award from the Medical Research Service, Ralph H. Johnson Veterans Affairs Medical Center; National Institute of Arthritis and Musculoskeletal and Skin Diseases grant numbers AR45476 and AR002193; and an Arthritis Foundation Arthritis Investigator Award.
REFERENCES
- 1.Mariani SM, Matiba B, Armandola EA, Krammer PH. The APO-1/Fas (CD95) receptor is expressed in homozygous MRL/lpr mice. Eur J Immunol 1994;24:3119–23. [DOI] [PubMed] [Google Scholar]
- 2.Singh AK. Lupus in the Fas lane? J R Coll Phys Lond 1995;29:475–8. [PMC free article] [PubMed] [Google Scholar]
- 3.Bossu P, Singer GG, Andres P, et al. Mature CD4+ T lymphocytes from MRL/lpr mice are resistant to receptor-mediated tolerance and apoptosis. J Immunol 1993;151.7233–9. [PubMed] [Google Scholar]
- 4.Van Houten N, Budd RC. Accelerated programmed cell death of MRL-lpr/lpr T lymphocytes. J Immunol 1992;149:2513–7. [PubMed] [Google Scholar]
- 5.Weinberg JB, Granger DL, Pisetsky DS, et al. The role of nitric oxide in the pathogenesis of spontaneous murine autoimmune disease: increased nitric oxide production and nitric oxide synthase expression in MRL-lpr/lpr mice, and reduction of spontaneous glomerulonephritis and arthritis by orally administered NG-monomethyl-l-arginine. J Exp Med 1994;179:651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oates JC, Ruiz R, Alexander A, et al. Effect of late modulation of nitric oxide production on murine lupus. Clin Immunol Immunopathol 1997;83:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowenstein CJ, Dinerman JL, Snyder SH. Nitric oxide: a physiologic messenger. Ann Intern Med 1994;120:227–37. [DOI] [PubMed] [Google Scholar]
- 8.Mills CD, Stevens RB, Sutherland DE. Nitric oxide in autoimmunity and cell transplantation. Transplant Proc 1994;26:3348. [PubMed] [Google Scholar]
- 9.Farrell AJ, Blake DR, Palmer RM, Moncada S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann Rheum Dis 1992;51:1219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McInnes IB, Leung BP, Field M, et al. Production of nitric oxide in the synovial membrane of rheumatoid and osteoarthritis patients. J Exp Med 1996;184:1519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.St Clair EW, Wilkinson WE, Lang T, et al. Increased expression of blood mononuclear cell nitric oxide synthase type 2 in rheumatoid arthritis patients. J Exp Med 1996;184:1173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimaoka M, Iida T, Ohara A, et al. NOC, a nitric-oxide–releasing compound, induces dose dependent apoptosis in macrophages. Biochem Biophys Res Commun 1995;209:519–26. [DOI] [PubMed] [Google Scholar]
- 13.Kitajima I, Kawahara K, Nakajima T, et al. Nitric oxide–mediated apoptosis in murine mastocytoma. Biochem Biophys Res Commun 1994;204:244–51. [DOI] [PubMed] [Google Scholar]
- 14.Cui S, Reichner JS, Mateo RB, Albina JE. Activated murine macrophages induce apoptosis in tumor cells through nitric oxide–dependent or -independent mechanisms. Cancer Res 1994;54:2462–7. [PubMed] [Google Scholar]
- 15.Ankarcrona M, Dypbukt JM, Brune B, Nicotera P. Interleukin-1 beta–induced nitric oxide production activates apoptosis in pancreatic RINm5F cells. Exp Ceil Res 1994;213:172–7. [DOI] [PubMed] [Google Scholar]
- 16.Albina JE, Cui S, Mateo RB, Reichner JS. Nitric oxide–mediated apoptosis in murine peritoneal macrophages. J Immunol 1993;150:5080–5. [PubMed] [Google Scholar]
- 17.Fehsel K, Kroncke KD, Meyer KL, et al. Nitric oxide induces apoptosis in mouse thymocytes. J Immunol 1995;155:2858–65. [PubMed] [Google Scholar]
- 18.Genaro AM, Hortelano S, Alvarez A, et al. Splenic B lymphocyte programmed cell death is prevented by nitric oxide release through mechanisms involving sustained Bcl-2 levels. J Clin Invest 1995;95:1884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin KT, Xue JY, Nomen M, et al. Peroxynitrite-induced apoptosis in HL-60 cells. J Biol Chem 1995;270:16487–90. [DOI] [PubMed] [Google Scholar]
- 20.Beauvais F, Michel L, Dubertret L. The nitric oxide donors, azide and hydroxylamine, inhibit the programmed cell death of cytokine-deprived human eosinophils. FEBS Lett 1995;361:229–32. [DOI] [PubMed] [Google Scholar]
- 21.Mannick JB, Asano K, Izumi K, et al. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus reactivation. Cell 1994;79:1137–46. [DOI] [PubMed] [Google Scholar]
- 22.Reilly CM, Farrelly LW, Viti D, et al. Modulation of renal disease in MRL/lpr mice by pharmacologic inhibition of inducible nitric oxide synthase. Kidney Int 2002;61:839–46. [DOI] [PubMed] [Google Scholar]
- 23.Keefer LK, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol 1996;268:281–93. [DOI] [PubMed] [Google Scholar]
- 24.Granger DL, Taintor RR, Boockvar KS, Hibbs JB Jr. Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol 1996;268:142–51. [DOI] [PubMed] [Google Scholar]
- 25.Koopman G, Reutelingsperger CR, Kuijten GA, et al. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994;84:1415–20. [PubMed] [Google Scholar]
- 26.Vermes I, Haanen C, Steffens–Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labeled annexin V. J Immunol Methods 1995;184:39–51. [DOI] [PubMed] [Google Scholar]
- 27.Darzynkiewicz Z, Bruno S, Del Bino G, et al. Features of apoptotic cells measured by flow cytometry. Cytometry 1992;13:795–808. [DOI] [PubMed] [Google Scholar]
- 28.Watson ML, Rao JK, Gilkeson GS, et al. Genetic analysis of MRL-lpr mice: relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J Exp Med 1992;176:1645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musette P, Pannetier C, Gachelin G, Kourilsky P. The expansion of a CD4+ T cell population bearing a distinctive beta chain in MRL lpr/lpr mice suggests a role for the fas protein in peripheral T cell selection. Eur J Immunol 1994;24:2761–6. [DOI] [PubMed] [Google Scholar]
- 30.Emlen W, Niebur J, Kadera R. Accelerated in vitro apoptosis of lymphocytes from patients with systemic lupus erythematosus. J Immunol 1994;152:3685–92. [PubMed] [Google Scholar]
- 31.Richardson BC, Yung RL, Johnson KJ, et al. Monocyte apoptosis in patients with active lupus. Arthritis Rheum 1996;39:1432–4. [DOI] [PubMed] [Google Scholar]
- 32.Courtney PA, Crockard AD, Williamson K, et al. Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann Rheum Dis 1999;58:309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perniok A, Wedekind F, Herrmann M, et al. High levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosus. Lupus 1998;7:113–8. [DOI] [PubMed] [Google Scholar]
- 34.Chan EY, Ko SC, Lau CS. Increased rate of apoptosis and decreased expression of bcl-2 protein in peripheral blood lymphocytes from patients with active systemic lupus erythematosus. Asian Pac J Allergy Immunol 1997;15:3–7. [PubMed] [Google Scholar]
- 35.Lorenz HM, Grunke M, Hieronymus T, et al. In vitro apoptosis and expression of apoptosis-related molecules in lymphocytes from patients with systemic lupus erythematosus and other autoimmune diseases. Arthritis Rheum 1997;40:306–17. [DOI] [PubMed] [Google Scholar]
- 36.Kanai Y, Kyuwa S, Miura K, Kurosawa Y. Induction and natural occurrence of serum nucleosomal DNA in autoimmune MRL/lpr/lpr mice: its relation to apoptosis in the thymus. Immunol Lett 1995;46:207–14. [DOI] [PubMed] [Google Scholar]
- 37.Giudice ED, Ciaramella A, Balestro N, et al. Neutrophil apoptosis in autoimmune Fas-defective MRL lpr/lpr mice. Eur Cytokine Netw 2001;12:510–7. [PubMed] [Google Scholar]
- 38.Licht R, van Bruggen MC, Oppers-Walgreen B, et al. Plasma levels of nucleosomes and nucleosome-autoantibody complexes in murine lupus: effects of disease progression and lipopolysaccharide administration. Arthritis Rheum 2001;44:1320–30. [DOI] [PubMed] [Google Scholar]
- 39.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med 1998;188:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang B, Dong X, Yuan Z, et al. SSA/Ro antigen expressed on membrane of UVB-induced apoptotic keratinocytes is pathogenic but not detectable in supernatant of cell culture. Chin Med J (Engl) 1999;112:512–5. [PubMed] [Google Scholar]
- 41.Casciola–Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 1994;179:1317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosen A, Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ 1999;6:6–12. [DOI] [PubMed] [Google Scholar]
- 43.Takeoka Y, Taguchi N, Shultz L, et al. Apoptosis and the thymic microenvironment in murine lupus. J Autoimmun 1999;13:325–34. [DOI] [PubMed] [Google Scholar]
- 44.Ravirajan CT, Sarraf CE, Anilkumar TV, et al. An analysis of apoptosis in lymphoid organs and lupus disease in murine systemic lupus erythematosus (SLE). Clin Exp Immunol 1996;105:306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noel PJ, Boise LH, Thompson CB. Regulation of T cell activation by CD28 and CTLA4. Adv Exp Med Biol 1996;406:209–17. [DOI] [PubMed] [Google Scholar]
- 46.Gribben JG, Freeman GJ, Boussiotis VA, et al. CTLA4 mediates antigen-specific apoptosis of human T cells. Proc Natl Acad Sci U S A 1995;92:811–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lucas M, Sanchez-Margalet V. Protein kinase C involvement in apoptosis. Gen Pharmacol 1995;26:881–7. [DOI] [PubMed] [Google Scholar]
- 48.Potter PK, Cortes-Hernandez J, Quartier P, et al. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol 2003;170:3223–32. [DOI] [PubMed] [Google Scholar]
- 49.Mitchell DA, Pickering MC, Warren J, et al. C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J Immunol 2002;168:2538–43. [DOI] [PubMed] [Google Scholar]
- 50.Laderach D, Bach JF, Koutouzov S. Nucleosomes inhibit phagocytosis of apoptotic thymocytes by peritoneal macrophages from MRL+/+ lupus-prone mice. J Leukoc Biol 1998;64:774–80. [DOI] [PubMed] [Google Scholar]
- 51.Boyd CS, Cadenas E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol Chem 2002;383:411–23. [DOI] [PubMed] [Google Scholar]
- 52.Moulian N, Truffault F, Gaudry-Talarmain YM, et al. In vivo and in vitro apoptosis of human thymocytes are associated with nitrotyrosine formation. Blood 2001;97:3521–30. [DOI] [PubMed] [Google Scholar]
- 53.Bustamante J, Bersier G, Romero M, et al. Nitric oxide production and mitochondrial dysfunction during rat thymocyte apoptosis. Arch Biochem Biophys 2000;376:239–47. [DOI] [PubMed] [Google Scholar]
- 54.Bustamante J, Bersier G, Badin RA, et al. Sequential NO production by mitochondria and endoplasmic reticulum during induced apoptosis. Nitric Oxide 2002;6:333–41. [DOI] [PubMed] [Google Scholar]
- 55.Tai XG, Toyo-oka K, Yamamoto N, et al. Expression of an inducible type of nitric oxide (NO) synthase in the thymus and involvement of NO in deletion of TCR-stimulated double-positive thymocytes. J Immunol 1997;158:4696–703. [PubMed] [Google Scholar]
- 56.Lu L, Bonham CA, Chambers FG, et al. Induction of nitric oxide synthase in mouse dendritic cells by IFN-gamma, endotoxin, and interaction with allogeneic T cells: nitric oxide production is associated with dendritic cell apoptosis. J Immunol 1996;157:3577–86. [PubMed] [Google Scholar]
- 57.Ramirez R, Carracedo J, Castedo M, et al. CD69-induced monocyte apoptosis involves multiple nonredundant signaling pathways. Cell Immunol 1996;172:192–9. [DOI] [PubMed] [Google Scholar]
- 58.Aiello S, Noris M, Piccinini G, et al. Thymic dendritic cells express inducible nitric oxide synthase and generate nitric oxide in response to self- and alloantigens. J Immunol 2000;164:4649–58. [DOI] [PubMed] [Google Scholar]
- 59.Mannick JB, Miao XQ, Stamler JS. Nitric oxide inhibits Fas-induced apoptosis. J Biol Chem 1997;272:24125–8. [DOI] [PubMed] [Google Scholar]
- 60.Mannick JB, Hausladen A, Liu L, et al. Fas-induced caspase denitrosylation. Science 1999;284:651–4. [DOI] [PubMed] [Google Scholar]
- 61.Mozart M, Scuderi R, Celsing F, Aguilar-Santelises M. Nitric oxide induces apoptosis in NALM-6, a leukaemia cell line with low cyclin E protein levels. Cell Prolif 2001;34:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Messmer UK, Lapetina EG, Brune B. Nitric oxide–induced apoptosis in RAW 264.7 macrophages is antagonized by protein kinase C- and protein kinase A–activating compounds. Mol Pharmacol 1995;47:757–65. [PubMed] [Google Scholar]
- 63.Jun CD, Oh CD, Kwak HJ, et al. Overexpression of protein kinase C isoforms protects RAW 264.7 macrophages from nitric oxide–induced apoptosis: involvement of c-Jun N-terminal kinase/stress-activated protein kinase, p38 kinase, and CPP-32 protease pathways. J Immunol 1999;162:3395–401. [PubMed] [Google Scholar]
- 64.Rifkin IR, Channavajhala PL, Kiefer HL, et al. Acceleration of lpr lymphoproliferative and autoimmune disease by transgenic protein kinase CK2 alpha. J Immunol 1998;161:5164–70. [PubMed] [Google Scholar]
- 65.Ohkusu K, Isobe K, Hidaka H, Nakashima I. Elucidation of the protein kinase C–dependent apoptosis pathway in distinct subsets of T lymphocytes in MRL-lpr/lpr mice. Eur J Immunol 1995;25:3180–6. [DOI] [PubMed] [Google Scholar]