

Abstract
Hepatic adeno-associated viral (AAV) gene transfer has the potential to cure the X-linked bleeding disorder hemophilia A. However, declining therapeutic coagulation factor VIII (FVIII) expression has plagued clinical trials. To assess the mechanistic underpinnings of this loss of FVIII expression, we developed a hemophilia A mouse model that shares key features observed in clinical trials. Following liver-directed AAV8 gene transfer in the presence of rapamycin, initial FVIII protein expression declines over time in the absence of antibody formation. Surprisingly, loss of FVIII protein production occurs despite persistence of transgene and mRNA, suggesting a translational shutdown rather than a loss of transduced hepatocytes. Some of the animals develop ER stress, which may be linked to hepatic inflammatory cytokine expression. FVIII protein expression is preserved by interleukin-15/interleukin-15 receptor blockade, which suppresses CD8+ T and natural killer cell responses. Interestingly, mice with initial FVIII levels >100% of normal had diminishing expression while still under immune suppression. Taken together, our findings of interanimal variability of the response, and the ability of the immune system to shut down transgene expression without utilizing cytolytic or antibody-mediated mechanisms, illustrate the challenges associated with FVIII gene transfer. Our protocols based upon cytokine blockade should help to maintain efficient FVIII expression.
Keywords: adeno-associated virus, hemophilia A, factor VIII, liver, immune response, rapamycin, interleukin-15, cellular stress
Graphical abstract
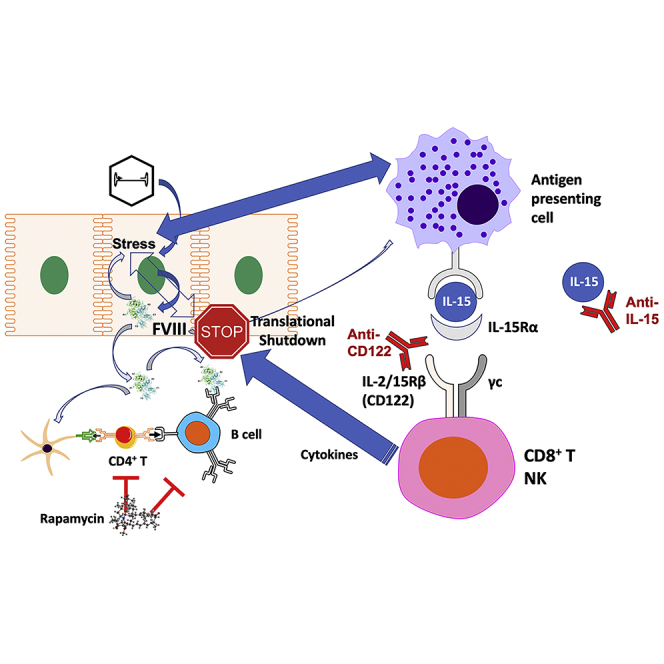
Clinical adeno-associated viral gene therapy for hemophilia A is hampered by variability and decline of factor VIII expression. Butterfield et al. use a mouse model to demonstrate that multiple factors contribute to a shutdown of protein expression in transduced hepatocytes that can be limited by rapamycin combined with interleukin-15 blockade.
Introduction
Gene therapy for the X-linked bleeding disorder hemophilia is currently being evaluated in multiple phase I/II and III clinical trials.1,2,3,4,5,6,7,8,9,10 These utilize adeno-associated viral (AAV) vectors with capsids that provide tropism for the liver upon intravenous infusion. Transduction of hepatocytes results in expression of coagulation factor VIII (FVIII, in the treatment of hemophilia A) or factor IX (FIX, to treat hemophilia B). Despite a large number of therapeutic options and competing technologies in today’s treatment of hemophilia, gene therapy has the distinctive advantage of potentially resulting in multi-year and perhaps even life-long therapy after a single drug administration. AAV vectors offer efficient in vivo gene transfer and limited innate immunity compared with other viral vectors. Gene therapy trials are currently performed primarily in adult patients with severe disease (<1% coagulation activity) who have not formed “inhibitors” (anti-drug antibodies) in conventional protein replacement therapy. Sustained correction of coagulation into or near the normal range for at least 3 years has been achieved in hemophilia B patients, which is in part facilitated by use of a hyperactivated variant of FIX that allows for reduced vector doses.4 Compared with other routes of vector administration, hepatic gene transfer has an increased likelihood to result in immune tolerance to the transgene product.11,12
Gene therapy for hemophilia A has been more challenging. FVIII is a considerably larger molecule that serves as a co-factor for the FIX enzyme in the coagulation cascade. While the coding sequence for full-length FVIII exceeds the packaging capacity of AAV vectors, B-domain-deleted (BDD) FVIII can be incorporated when combined with a small hepatocyte-specific promoter. Codon optimization is typically performed, and high vector doses (>1013 vector genomes [vg]/kg) are given to achieve levels approaching the normal range (which is 50%–150% of average coagulation activity in hemostatically normal individuals).4,13 Through this strategy, normal FVIII levels were reached in several patients in a trial that utilized doses of 6 × 1013 vg/kg of an AAV5 vector.7 These levels were sustained in the first year despite prolonged elevations in liver enzyme levels, prompting the use of steroid drugs (an approach originally developed to counter CD8+ T cell responses against the viral capsid). Between the second and fifth year after gene transfer, FVIII levels unexpectedly gradually declined toward the lower end of the therapeutic range.5,14 While the lack of bleeding episodes during this time period underscores the potent therapeutic effect of the gene therapy, it is concerning that levels were not sustained. Given alternative treatment options such as weekly to monthly subcutaneous injections of a bispecific antibody and the emergence of factor products with extended half-lives, gene therapy for patients in developed countries may only be desirable if levels near the normal range are sustained.1 Moreover, the reasons for the loss of expression remain unclear.15,16,17,18,19 Neither inhibitor formation nor CD8+ T cell response against capsid or FVIII was detected in peripheral blood. Other clinical trials have encountered similar problems of initially high FVIII expression levels declining.2,10
While the normal site of FIX expression is hepatocytes, FVIII is typically made by endothelial cells.20 Steady-state circulating levels are thought to be mostly derived from liver sinusoidal endothelial cells. Although FVIII circulates at considerably lower levels than FIX (200 versus 5,000 ng/mL), its inefficient production and secretion pose a challenge. Furthermore, high levels of FVIII expression tend to induce ER stress related to inefficient secretion, resulting in induction of the unfolded protein response (UPR), cellular toxicities, and aggregation upon formation of amyloid structures.21,22,23,24,25
Thus far, there has not been an animal model showing a gradual decline in FVIII expression following hepatic gene transfer in the absence of antibody formation, making preclinical studies of this phenomenon difficult. Here, we report gradual loss of FVIII expression following hepatic AAV8-FVIII gene transfer combined with rapamycin administration in a strain of hemophilia A mice. A 2-month regimen of rapamycin suppressed T follicular helper (Tfh) cell responses and prevented antibody formation against FVIII and viral capsid. Nonetheless, hepatic and systemic FVIII expression subsequently declined and often became undetectable by 16 weeks. Interleukin-15 (IL-15) blockade (using antibodies against IL-15 or its receptor) partially rescued FVIII protein production. Further experiments revealed a loss of FVIII protein expression in the liver that occurred between weeks 8 and 14 with minimal changes in transcript levels, suggesting a translation shutdown.
Results
Prophylactic immune modulation prevents antibody formation against human FVIII
We previously reported that an AAV8 vector expressing codon-optimized human F8 sequence (BDD) from a hepatocyte-specific promoter improved systemic human FVIII (hFVIII) activity by 5- to 10-fold compared with the native coding sequence and induced immune tolerance to FVIII in hemophilia A mice.26 After intravenous (i.v.) administration of 1 × 1011 vg into mice of the most widely used hemophilia A strain (C57BL6/129 F8e16−/−), we again observed sustained hFVIII expression in circulation (n = 8, Figure S1A). In contrast, hemophilia A mice with the same F8 mutation on BALB/c background (BALB/c-HA) failed to show hFVIII activity (<0.4% of normal) and developed inhibitors (n = 18, Figure S1B), prompting us to explore immune modulation in this model.
The first iteration of our immune-tolerance regimen included the mammalian target of rapamycin (mTOR) inhibitor rapamycin, the cytokine FMS-like tyrosine kinase 3 ligand (Flt3L), and hFVIII protein (Figure S2A), a protocol that we previously found to promote FoxP3+ regulatory T cells (Tregs) and to prevent inhibitor formation in hFVIII protein therapy.27 Within 4 weeks of transduction with AAV8-hFVIII, BALB/c-HA control mice (n = 7) again all formed inhibitors against FVIII (Figure S2C). Antibody formation was delayed by several weeks in seven of eight mice that received the immunomodulating regimen (Figures S2C and S2D). One of eight mice did not form antibodies and retained hFVIII activity (Figure S2B), suggesting that the tolerance protocol could be effective but required further optimization.
In a subsequent experiment, we extended the duration of immune modulation to 10 weeks after gene transfer (Figure S3A) and doubled the vector dose. We also eliminated hFVIII protein from the treatment regimen so that immune tolerance could be induced by hepatocyte-derived hFVIII. We again found that control mice without immune suppression developed inhibitors (Figure S3C). Encouragingly, none of the immune-modulation-treated mice formed antibodies against hFVIII (Figures S3C–S3E). Thus, rapamycin as the only immune-modulatory reagent was sufficient to reliably prevent antibody formation. Regardless of whether Flt3L was included, two of six mice retained hFVIII activity for at least 4 weeks after immune modulation was stopped (Figure S3B).
Next, we wanted to determine if the immune-modulation protocol could be shortened. Thus, we administered immune modulation for 2, 4, 6, or 8 weeks following AAV8-hFVIII (Figure S4A). Flt3L was not included, since it did not provide a clear advantage over rapamycin alone. Rapamycin was administered at 6 mg/kg twice per week. Except for 8 weeks of rapamycin, each group formed antibodies against hFVIII to varying degree by 12–16 weeks after gene transfer (Figures S4C and S4D). Eight weeks of rapamycin also prevented formation of antibodies against AAV capsid (Figure S4E). Disappointingly, by 16 weeks after gene transfer, hFVIII activity was only detectable in three of six mice that received 8 weeks of rapamycin treatment (Figure S4B), and only at levels 1%–2% of normal. To distinguish whether this was a general property of the mouse strain or related to the hFVIII transgene, we transduced BALB/c-HA mice with 2 × 1011 vg of an AAV8 vector expressing a different clotting factor (human FIX [hFIX], n = 7). This resulted in sustained hFIX expression at levels ∼10-fold of normal (400 times higher than normal hFVIII levels), without a need for immune suppression and accompanied by minimal changes in alanine aminotransferase (ALT) levels (Figure S5). This strongly suggested that the instability of expression is due to the hFVIII transgene. Although 8 weeks of rapamycin prevented antibody formation against hFVIII, the regimen still failed to preserve long-term hFVIII expression.
Rapamycin combined with IL-15 blockade preserves hFVIII expression
Given that FVIII activity had been lost over time despite lack of antibody formation, we hypothesized that cellular immune responses had occurred. Therefore, we sought to prevent induction of cellular immune responses using an approach that is clinically feasible but more targeted than broad suppressors of lymphocyte proliferation. Recent publications documented the critical role of the cytokine IL-15 for maintenance of CD8+ memory T cells (Tmem) and natural killer (NK) cells.28,29,30,31,32 Combining rapamycin with anti-IL-15 for the 8 weeks following hepatic gene transfer (Figure 1A) resulted in preservation of FVIII activity in seven of nine BALB/c-HA mice through week 16 (Figure 1B), with four of nine expressing in the mild hemophilia range (5%–50% FVIII activity). In stark contrast, by 16 weeks, seven of eight mice treated with rapamycin alone had undetectable hFVIII activity. Mice treated with rapamycin or the rapamycin/IL-15 combination lacked antibodies against FVIII or viral capsid (Figures 1C, 1D and S6B). IL-15 blockade by itself (i.e., without rapamycin) did not prevent antibody formation against hFVIII but somewhat reduced antibodies against capsid (Figure S7; these differences are likely because antibodies against capsid are immunoglobulin G2a (IgG2a), reflecting T helper cell 1 (Th1) immunity, while antibodies against hFVIII are mostly IgG1, indicating differences in cytokines produced by Th subsets in the response to the virus versus hFVIII). However, the addition of anti-IL-15 to the rapamycin regimen helped to sustain hFVIII expression.
Figure 1.

Sustained systemic hFVIII expression after transient rapamycin administration combined with anti-IL-15 therapy
(A) Experimental outline. Hemophilia A mice (BALB/c F8e16−/−) received hepatic gene transfer with AAV8-hFVIII vector (codon-optimized) at a dose of 2 × 1011 vg/mouse (n = 8–10 per experimental group). Animals were additionally treated twice per week with rapamycin (Rapa) or rapamycin combined with anti-IL-15 (Rapa + Anti-IL-15) for the first 8 weeks after gene transfer, starting the day of vector administration. Control mice received vector but no immune modulation (AAV only). (B) FVIII activity in plasma as a function of time after gene transfer (averages ± SEM). Mice received a second vector administration at week 17 (AAV8-hFIX, 2 × 1011 vg/mouse). (C) Inhibitory antibody titers (BU/mL) against FVIII at week 16. (D) IgG1 antibody titers against hFVIII at week 16. Significance was determined by one-way ANOVA for each time point and corrected for multiple comparisons using the Tukey post hoc test (B) and multiple unpaired t tests, corrected for multiple comparisons using the Holm-Šídák post hoc test (C and D). ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
AAV8 readministration is possible in mice treated with rapamycin or rapamycin/anti-IL-15
Since repeated rapamycin treatment abrogated antibody formation against AAV8, we tested whether vector readministration was possible. A subset of mice initially transduced with AAV8-hFVIII was injected with an identical dose of AAV8-hFIX (expressing hFIX) 17 weeks later (Figure S6A). We chose a different transgene to obtain a clean readout for efficacy of readministration. No immune suppression was applied at the time of this second vector administration. Three weeks after AAV8 vector readministration, previously immune-suppressed mice formed antibodies against AAV8 capsid at levels comparable with those in naive control mice (with no prior vector administration or immune modulation) (Figure S6C), indicating that the immune system regained functionality (based on our prior experience, immune competence is recovered by ∼1 month after rapamycin treatment in mice).27 Mice that initially received AAV8-hFVIII without immune modulation had increased anti-AAV8 titers after the second vector injection to approximately twice that of the other groups. Following injection with AAV8-hFIX, the groups that had received AAV8-hFVIII and rapamycin or rapamycin/anti-IL-15 combination expressed hFIX at average levels of 38%–63% of those in naive control mice (Figure S6D). In contrast, no hFIX expression was found in mice that had received the first vector without immune suppression.
IL-15 blockade via targeting of CD122 is effective in preserving hFVIII expression and has broad anti-inflammatory effects
We repeated the comparison of rapamycin and rapamycin/anti-IL-15 treatments in a larger number of animals (Figure 2A). This time, we achieved higher initial FVIII activities of 40%–60% on average. Among rapamycin-only-treated mice (n = 21), 38% entirely lost FVIII activity by week 16. In the rapamycin/anti-IL-15 treatment group (n = 18), 17% lost activity, resulting in significantly higher average FVIII activity by week 16 (Figure 2C). With these higher starting levels, there was more variability in the rapamycin-only-treated animals, and thus, as compared with results in Figure 1, not all lost expression completely. However, the rapamycin/anti-IL-15 group yielded very similar endpoints in both experiments.
Figure 2.

Effect of alternative approaches to IL-15 blockade and of CD8+ T cell depletion on hFVIII expression
(A) Experimental outline. Hemophilia A mice (BALB/c F8e16−/−) received hepatic gene transfer with AAV8-hFVIII vector at a dose of 2 × 1011 vg/mouse. Animals were additionally treated twice per week with rapamycin (Rapa, n = 21) or rapamycin combined with anti-IL-15 (Rapa + Anti-IL-15, n = 18) or with anti-CD122 (Rapa + Anti-CD122, n = 9) for the first 8 weeks after gene transfer, or received no immune modulation (AAV only, n = 6). Other animals were treated with rapamycin for the first 8 weeks after gene transfer followed by anti-CD8 administration (twice a week) for 8 more weeks (Rapa + Anti-CD8, n = 8). (B) Inhibitory antibody titers (BU/mL) against FVIII at the end of the experiment (week 16; averages ± SEM). (C) hFVIII activity in plasma as a function of time after gene transfer (averages ± SEM). (D) Vector copy numbers per cell in the liver at week 16 (averages ± SEM). (E) Liver F8 transgene mRNA levels (normalized to endogenous β-actin mRNA levels) relative to average of “AAV only”-treated mice (averages ± SEM; week 16). (F–I) F8 mRNA levels in the liver versus FVIII activity in plasma for the indicated treatment groups. Symbols represent individual animals. (J–N) F8 mRNA levels versus vector copy numbers in the liver for the indicated treatment groups. Group sizes for (D–N), n = 8–9/experimental group and n = 6 for “AAV only”-treated mice. Significance was determined by one-way ANOVA for each time point and corrected for multiple comparisons using the Tukey post hoc test (B–E) and Pearson correlation coefficient with two-tailed p values and simple linear regression line (F–N). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
While there is currently no clinical equivalent to anti-IL-15, monoclonal antibodies (mAbs) against the IL-15 receptor are in clinical development. Recent progress led to the ability to target CD122, the β chain of the IL-15 receptor that is shared with the IL-2 receptor, without disruption of Treg function. A murine version of such an anti-CD122 mAb (ChMBC7) was successfully used in murine models of autoimmune diseases.28,29 A limited amount of ChMBC7 was available to us, which we tested in combination with rapamycin in our model (Figure 2A). Following rapamycin/anti-CD122 treatment, only one of nine mice entirely lost activity, so that the percentage of mice retaining FVIII activity was further improved to 89% (Figure 2C). FVIII activities were on average higher compared with rapamycin/anti-IL-15-treated group and significantly higher than for the rapamycin-only group at 16 weeks. As expected for this protocol, none of the mice that had received immune suppression developed inhibitors against hFVIII (Figure 2B). As a positive control for inhibitor formation, a group of mice receiving gene transfer without immune suppression was also included (n = 6). In summary, mAb therapy directed against the CD122 subunit of IL-15 receptor is a promising tool to preserve hFVIII activity.
To further interrogate the role of CD8+ T cells in the loss of expression in the absence of IL-15 blockade, we transduced BALB/c-HA mice with AAV8-hFVIII and treated them for 8 weeks with rapamycin only. At this time point, i.e., when immune suppression was stopped, we administered a depleting anti-CD8 mAb twice per week for the remainder of the experiment (Figure 2A). As a result, nearly all animals (7 of 8, 88%) retained detectable FVIII activity by week 16, which was, however, on average ∼4-fold lower than in the rapamycin/anti-CD122-treated group (Figure 2C).
To study the effect of anti-CD122, we examined NK cells, CD8+ effector and memory T cells (CD8+ Teff and Tmem, respectively), and Kupffer cells (KCs) in the liver at the end of the experiment (week 16, n = 8–9/experimental group). Most striking were the effects of rapamycin/anti-CD122 on hepatic NK cells (Figures 3A–3F and S8). Despite withdrawal of immune-modulation treatments 8 weeks earlier, NK cells remained nearly fully ablated, and their expression of NKG2D (an activating cell surface receptor) was also markedly reduced (Figures 3A and 3B). Moreover, the small population of remaining NK cells was almost entirely composed of less mature subsets (CD11blow), whereas the more mature subsets (CD11bhigh) made up approximately half of the hepatic NK cells in the other groups (Figures 3C–3F). Rapamycin/anti-IL-15-treated mice had modestly increased NK cell frequencies (Figure 3A). While mice treated with rapamycin/anti-IL-15 had moderately decreased CD8+ Teff cells compared with naive or rapamycin-treated mice, CD8+ Teff cells were most potently suppressed by rapamycin/anti-CD122 (Figure 3G); no lasting effect was seen on CD8+ Tmem cells in any of the treatment groups (Figure 3H). Compared with baseline, both rapamycin and rapamycin/anti-IL-15 treatment increased expression of NKG2D in CD8+ Teff and Tmem cells, whereas NKG2D was moderately decreased from baseline with rapamycin/anti-CD122 treatment (Figures 3I and 3J). In KCs, expression of CD206 (a marker for M2/anti-inflammatory macrophage polarization) was increased in mice treated with either rapamycin/anti-IL-15 or rapamycin/anti-CD122 (Figures 3L and S9). Additionally, we found that expression of inducible nitric oxide synthase (iNOS, a pro-inflammatory marker) in mice receiving immune modulation was lowest in rapamycin/anti-CD122 treated mice (Figure 3K). In conclusion, loss of hFVIII expression after rapamycin treatment is only partially mediated by CD8+ T cells; thus, IL-15-dependent cellular responses need to be more broadly targeted, e.g., through anti-CD122 treatment.
Figure 3.

Late effects of rapamycin treatment and IL-15 blockade on hepatic immune cells
Hemophilia A mice (BALB/c F8e16−/−) received hepatic gene transfer with AAV8-hFVIII vector at a dose of 2 × 1011 vg/mouse. Animals were additionally treated twice per week with rapamycin (Rapa) or rapamycin combined with anti-IL-15 (Rapa + Anti-IL-15) or with anti-CD122 (Rapa + Anti-CD122) for the first 8 weeks after gene transfer, or received no immune modulation (AAV only). Livers were collected weeks 16 after gene transfer. (A) Percent NK cells of intrahepatic lymphocytes. (B) Mean fluorescence intensity (MFI) of NKG2D on intrahepatic NK cells. (C) Percent NK1 of intrahepatic NK cells. (D) Percent NK2 of intrahepatic NK cells. (E) Percent NK3 of intrahepatic NK cells. (F) Percent NK4 of intrahepatic NK cells. (G) Percent Teff cells of intrahepatic CD8+ T cells. (H) Percent Tmem cells of intrahepatic CD8+ T cells. (I) MFI of NKG2D on intrahepatic CD8+ Teff cells. (J) MFI of NKG2D on intrahepatic CD8+ Tmem cells. (K) MFI of iNOS stain in KCs. (L) MFI of CD206 stain on KCs. Immunophenotypes used to define cell types and their activation status is provided in Table S1. Data are average ± SEM (n = 8–9/experimental group). Significance was determined by one-way ANOVA and corrected for multiple comparisons using the Tukey post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Next, we administered AAV8-hFVIII and immune-tolerance regimens to additional BALB/c-HA mice for 3 weeks to examine lymphocyte populations while still under immune suppression (Figure 4A). After 3 weeks of rapamycin/anti-CD122 treatment, hepatic NK cells were nearly fully depleted, and those remaining were mostly immature (Figures 4E–4I and S10). Rapamycin/anti-IL-15 decreased hepatic NK cells as well, but to a much lesser extent. All groups receiving immune suppression had reduced hepatic CD8+ Teff cells, while rapamycin combined with either anti-IL-15 or anti-CD122 decreased hepatic CD8+ resident memory T cells (CD8+ Trm) similarly (Figures 4B and 4D). Rapamycin/anti-CD122 combination had a marginally stronger effect on CD8+ effector memory T cells (CD8+ Tem) (Figure 4C) compared with the other rapamycin-based regimens. Splenic T follicular helper (Tfh) and T follicular regulatory (Tfr) cells were markedly reduced at 3 weeks by all the rapamycin-based regimens (Figures 4J, 4K, and S11). We found that all the rapamycin-based regimens also depleted immature B cells (resulting in increased frequency of follicular B cells), and most importantly, ablated germinal center (GC) B cells, which were markedly increased in mice that received vector without immune suppression (Figures 4L, 4N, 4O, and S12). Unexpectedly, marginal zone (MZ) B cells were significantly increased over the other treatment groups with rapamycin/anti-IL-15 (Figure 4M).
Figure 4.

Early effects of rapamycin treatment and IL-15 blockade on hepatic immune cells
(A) Experimental outline. Hemophilia A mice (BALB/c F8e16−/−) received hepatic gene transfer with AAV8-hFVIII vector at a dose of 2 × 1011 vg/mouse. Animals were additionally treated twice per week with Rapa, Rapa + Anti-IL-15, Rapa + Anti-CD122, or received no immune modulation (AAV only). Livers and spleens were collected 3 weeks after gene transfer. (B) Percent Teff cells of intrahepatic CD8+ T cells. (C) Percent Tem cells of intrahepatic CD8+ T cells. (D) Percent Trm cells of intrahepatic CD8+ T cells. (E) Percent NK cells of intrahepatic lymphocytes. (F) Percent NK1 of intrahepatic NK cells. (G) Percent NK2 of intrahepatic NK cells. (H) Percent NK3 of intrahepatic NK cells. (I) Percent NK4 of intrahepatic NK cells. (J) Percent Tfh cells of CD4+ T cells in spleen. (K) Percent Tfr cells of CD4+ T cells in spleen. (L) Percent immature B of total B220+ cells in spleen. (M) Percent marginal zone B cells of mature B cells in spleen. (N) Percent follicular B cells of mature B cells in spleen. (O) Percent germinal center B cells of mature B cells in spleen. Immunophenotypes used to define cell types and their activation status are provided in Table S1. Data are average ± SEM (n = 4–5/experimental group). Significance was determined by one-way ANOVA and corrected for multiple comparisons using the Tukey post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Circulating hFVIII activity levels correlate with hepatic hFVIII expression and not with transcript levels
To assess whether loss or persistence of systemic hFVIII expression correlated with hepatic expression, we harvested livers at the end of the experiment (week 16, n = 8–9/experimental group, n = 6 for group treated with only AAV). Mice treated with rapamycin or rapamycin/anti-IL-15 that had circulating FVIII activity also showed hepatic expression when analyzed by immunostaining for hFVIII, while those that lost systemic hFVIII expression also lacked hFVIII expression in hepatocytes (compare, for instance, the animals shown in Figure 5E with those in Figures 5F, 5G, and 5H, and animals in Figure 5I with those in 5J). Furthermore, four of five examined livers from AAV8-hFVIII transduced animals that did not receive immune suppression showed no hFVIII protein-expressing hepatocytes (see examples in Figures 5B and 5C), despite this treatment group showing the highest gene copy numbers and mRNA levels. The one animal in this group with hepatic hFVIII expression at week 16 additionally showed substantial CD8+ T cell infiltration (Figures 5D and S13). Some of the rapamycin-treated animals had modest CD8+ T cell infiltrates (Figures 5E and 5F), whereas few CD8+ T cells were observed in rapamycin/anti-IL-15-treated and none in rapamycin/anti-CD122- or rapamycin/anti-CD8-treated mice (Figures 5H–5N). FVIII-expressing hepatocytes appeared in clusters, typically around blood vessels. Surprisingly, AAV-hFVIII transduced livers of animals that had not received immune suppression (and thus had no circulating hFVIII due to inhibitor formation, Figures 2B and 2C) showed 2- to 2.5-fold higher vector copy numbers and levels of transgene transcript than the rapamycin-treated groups, which all had similar vector copy numbers (Figures 2D, 2E, and S13). Thus, the improved systemic hFVIII expression observed upon IL-15 blockade did not correlate with changes in transcription or maintenance of vector genomes. All rapamycin-treated groups of mice showed nearly identical average vector copy and F8 mRNA levels, except that anti-CD8 treatment very modestly increased vector copy numbers (but not transcription). Similarly, there was no correlation between FVIII activity and transcript levels when individual mice were analyzed within each treatment group (see low r and high p values in Figures 2F–2I). Vector copy numbers and mRNA levels, however, correlated well (Figures 2J, 2L, and 2N), except for mice with CD8 T cell depletion or anti-CD122 treatment (Figures 2K and 2M). In summary, blocking IL-15 cytokine signaling preserved hFVIII protein expression in hepatocytes and markedly reduced CD8+ T cell responses, but had no effect on transcription of mRNA or gene copy numbers.
Figure 5.

Hepatic FVIII expression 16 weeks after AAV8-hFVIII gene transfer
Hemophilia A mice (BALB/c F8e16−/−) received hepatic gene transfer with AAV8-hFVIII vector at a dose of 2 × 1011 vg/mouse. Shown are immunofluorescent stains of liver sections for hFVIII (green) and CD8 (red). Blue stain is DAPI. White asterisks indicate CD8+ T cells. (A) Example of naive mouse (no gene transfer, negative control). (B–N) Examples of stains for mice of the indicated treatment groups. Percentages are plasma FVIII activity levels at week 16. Original magnification, 200×.
To better understand the changes in hepatic hFVIII expression over time, we transduced another cohort of BALB/c-HA mice. Half of the animals were treated with the rapamycin regimen, which was extended for the duration of the experiment (14 weeks, Figure 6A). Expression levels at week 4 were nearly identical to those in the earlier experiment (Figure 6B versus Figure 2B), and rapamycin-treated mice lacked antibody formation against hFVIII (data not shown). FVIII activity in mice that were continuously immune suppressed were generally stable, with most ranging from 20% to 61% of normal at week 14 (Figure 6E). However, two animals that had initial levels of >100% of normal showed a marked decline, in one case from 225% at week 4 to 6% by week 14. Interestingly, this animal had the second-highest gene copy number of the group and intermediate F8 transcript levels by week 14 but showed very few hFVIII-expressing hepatocytes (Figure S15). Regardless of whether immune suppression was applied or not, systemic ALT levels were significantly elevated compared with mice that did not receive gene transfer (reaching ∼2.5-fold increase by week 14, Figure S16), while aspartate aminotransferase (AST) levels were not increased (data not shown). In conclusion, hFVIII levels <100% of normal may be stable under conditions of continued immune suppression while higher levels are not.
Figure 6.

Time course of vector persistence and FVIII protein and mRNA expression after AAV8-hFVIII gene transfer in the presence or absence of immune suppression
(A) Experimental outline. Hemophilia A mice (BALB/c F8e16−/−) received hepatic gene transfer with AAV8-hFVIII vector at a dose of 2 × 1011 vg/mouse (n = 7–8 per experimental group). Animals were additionally treated with rapamycin twice per week for 14 weeks (throughout the experiment) or received no immune modulation (AAV only). Plasma was collected at weeks 4, 8, 12, and 14. Liver tissues were collected at weeks 4, 8, and 14. Naive mice served as negative control for gene transfer. (B) Plasma FVIII activity levels as a function of time after gene transfer in the rapamycin-treated mice were measured by chromogenic assay (averages ± SEM). (C) Gene copy number was measured by qPCR of DNA extracted from liver tissue (averages ± SEM; ∗p < 0.05; ∗∗p < 0.001). (D) F8 transgene-derived and endogenous β-actin (reference gene) mRNA transcripts were measured by RT-qPCR of RNA extracted from liver tissue and fold change was calculated by the 2–ΔΔCt method (averages ± SEM; ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.0001). Note that copy numbers and mRNA for the “AAV only” group at 16 weeks (denoted by an asterisk) were from a previous experiment (see Figure 2) and are shown here again for comparison (run side by side). (E) Plasma FVIII activity levels as a function of time after gene transfer. Each dot represents an individual rapamycin-treated mouse. (F–Q) Representative immunofluorescence images of livers showing hFVIII protein expression and localization of CD8+ T cells (indicated by white asterisks) from the AAV-only or Rapa groups at weeks 4, 8, and 14. Percentages shown at the left-bottom corner of each image are hFVIII activities of the respective animal at the indicated time points. (R–T) Representative images of single molecular fluorescent in situ hybridization (smFISH) for AAV vector genome and F8 transgene mRNA in mouse liver tissue sections. Zoomed-in images are high magnifications of the region surrounded by dotted lines in the merged panel images and highlight the distribution of AAV vector genome and hF8 mRNA in the nucleus. Scale bars, 50 μm. (U) Quantitation of percentage of cells that are positive for fluorescence signals corresponding to AAV vector genome, F8 mRNA, or both AAV vector genome and F8 mRNA as determined by analysis of smFISH images.
To track hepatic transduction over time, livers were harvested 4, 8, and 14 weeks after gene transfer (n = 7–8 per time point and group). Again, vector copy numbers were consistently somewhat higher at all time points (≤2-fold) in mice that were not immune suppressed (Figure 6C). Furthermore, copy numbers were similar for 4- and 8-week time points. However, these declined ∼5-fold by week 14 (at which time they were 2.5-fold higher compared with week 16 in the earlier experiment shown in Figure 2D). F8 mRNA levels were similar by week 8 for both groups and without a decline between weeks 4 and 8 (Figure 6D). By 14 weeks, there was a mere 2-fold decline in transcript levels despite the aforementioned 5-fold reduction in vector copy numbers, suggesting that transcriptionally inactive vector forms were lost. This interpretation is further supported by improved correlation between vector copy numbers and F8 mRNA levels by week 14 compared with the earlier time points (Figures S17D–S17F). FVIII activity in circulation and transcription again did not correlate at any time point for rapamycin-treated mice (Figures S17A–S17C). When comparing the 14-week time point with week 16 of the prior experiment (Figure 2E; samples from both experiments/time points were run side by side), week 16 transcript levels were drastically lower (Figure 6D), much more so than the difference in vector copy numbers (22-fold versus 2.5-fold), suggesting that transcription may eventually be downregulated. However, transcription of the transgene is minimally affected during the first 3 months after gene transfer.
Consistent with high-dose AAV8 gene transfer to murine liver, both rapamycin-treated and non-immune-suppressed groups showed widespread expression in liver sections at weeks 4 and 8 (n = 3 per time point and group; representative examples are shown in Figures 6F–6H and 6L–6N). However, at week 14 this pattern was only seen for rapamycin-treated animals (Figures 6O and 6P). No CD8+ T cell infiltrates were seen in rapamycin-treated mice. In the absence of immune suppression, mice entirely lost hepatic hFVIII protein expression by week 14 (Figures 6I and 6J), except for a few areas that contained some hFVIII-expressing hepatocytes but also CD8+ T cell infiltrates (Figure 6K). CD8+ T cells were rarely observed at the earlier time points. Single-molecule fluorescence in situ hybridization (smFISH) detected AAV genomes and/or F8 transgene mRNA in 43%–77% of hepatocytes, which was similar at all time points (n = 3–5 per group and time point, Figure 6U). Therefore, the loss of hFVIII protein production in the liver by week 14 in the absence of immune suppression cannot be explained by elimination of transduced cells or loss of transcription, and the decline in gene copy numbers between weeks 8 and 14 represents a loss in copy numbers per cell rather than a reduction in the number of genome-containing cells.
Evidence for cellular stress responses and role of inflammatory cytokines
Both inflammation and expression of hFVIII may be sources of cellular stress. Western blot analysis shows induction of ER stress markers GRP94 and BiP (both key ER chaperones) and UPR proapoptotic factor C/EBP homologous protein (CHOP) in livers of a subset of animals by 14 weeks after AAV8-FVIII transduction (Figures 7A–7C and S18). Among the rapamycin-treated animals, the same four mice (marked #1 to #4 in Figures 7A–7C) had highest levels of all three markers in identical order for each marker. Interestingly, these animals had FVIII activity levels of 20%–57% of normal and thus were not the highest expressors. The two animals with the most elevated stress marker levels (#1 and #2) also had the most elevated levels of phosphorylated eukaryotic translation initiation factor 2α (p-eIF2α) (Figure 7D), which is known to promote translational shutdown. These mice had intermediate FVIII activity levels at week 14 of 20%–30% of normal. Among non-immune-suppressed animals at week 14, the same four mice (#5 to #8) with highest GRP94 also had the highest BiP levels (Figures 7A and 7B), with one showing elevated CHOP (Figure 7C) and three having higher p-eIF2α levels than the other animals in that group (Figure 7D). Again, a subset of animals showed elevation in multiple markers, providing consistent evidence for cellular stress responses.
Figure 7.

Induction of cellular stress in AAV8-hFVIII transduced livers of hemophilia A mice
Quantification of immunoblots for (A) GRP94, (B) BiP, (C) CHOP, and (D) phospho-eIF2α levels in mouse livers at the indicated time points after gene transfer normalized to β-actin control and relative to average levels in naive animals. Mice (7–9 mice/group and time point) received gene transfer only (AAV) or were additionally treated with rapamycin (rapa) as outlined in Figure 6. Bar graphs show average values ± SD. Circles are data for individual mice.
In the earlier 16-week experiment (Figure 2), we determined CHOP mRNA expression. Mice treated with rapamycin/anti-IL-15 had variable transcript levels, which were not seen for the other experimental groups (Figure S19). The animal with the highest CHOP mRNA levels had FVIII activity levels of ∼30% of normal for up to 12 weeks, which subsequently declined to undetectable by week 16, further corroborated by FVIII expression in liver tissue sections (Figure 5J). This mouse also had the lowest vector copy number in its group at week 16; therefore, it is possible that cellular stress induced loss of transduced hepatocytes in this animal.
Cellular stress, translational shutdown, and inflammation may be interlinked.33,34,35,36,37,38 Hence, we decided to compare phosphorylation of eIF2α (as determined by western blot of liver protein extracts) and hepatic interferon-γ (IFN-γ) mRNA levels between two experimental groups that differed in their long-term FVIII protein expression: mice treated with rapamycin or with rapamycin/anti-IL-15 combination. Compared with naive control mice (which received no vector or immune suppression), both groups had elevated p-eIF2α (p = 0.0111 for rapamycin and p = 0.0591 for rapamycin/anti-IL-15) and IFN-γ transcripts (p = 0.2265 for rapamycin and p = 0.0105 for rapamycin/anti-IL-15) (Figures 8A, 8B, and S20). Elevated p-eIF2α levels correlated with IFN-γ transcript levels in mice treated with rapamycin but not rapamycin/anti-IL15 (Figures 8C and 8D). FVIII activity levels tended to inversely correlate with increased phospho-eIF2α in animals treated with rapamycin but not rapamycin/anti-IL-15 (Figures 8E and 8F). In contrast, IFN-γ expression was undetectable in livers of rapamycin/anti-CD122 treated mice (data not shown). In conclusion, cellular stress and inflammation are interlinked, a connection that may be broken by anti-CD122 treatment.
Figure 8.

Correlation between phosphorylated eIF2α and IFN-γ expression in livers of AAV8-hFVIII transduced hemophilia A mice
Hemophilia A mice (BALB/c F8e16−/−) received hepatic gene transfer with a vector dose of 2 × 1011 vg/mouse. Animals were additionally treated twice per week with rapamycin (Rapa) or rapamycin combined with anti-IL-15 (Rapa + Anti-IL-15). Livers were collected 16 weeks after gene transfer. (A) Phosphorylated eIF2α levels (p-eIF2α, relative to total eIF2α) as quantitated by western blot on liver protein extracts at week 16 for Rapa and Rapa + Anti-IL-15 groups and for the naive group (no gene transfer or immune modulation). Data are averages ± SEM. (B) IFN-γ transcript levels (normalized to β-actin mRNA levels) extracted from liver tissue. (C–F) IFN-γ transcript levels versus p-eIF2α levels. Group sizes: n = 9–13 per experimental group. Significance was determined by comparing treatment groups with the naive control group by one-way ANOVA and corrected for multiple comparisons using Dunnett’s post hoc test (A, B) and Pearson’s correlation coefficient with two-tailed p values and simple linear regression line (C–F). ∗p < 0.05.
Discussion
A recent phase I/II gene therapy trial utilizing AAV5 vector to express hFVIII in patients with hemophilia A achieved curative levels (i.e., in the normal range) that, however, were not sustained for reasons that are unclear.15,16,17,18 Median levels fell from initial ∼60% (by chromogenic assay) to ∼8% over a period of 5 years.14 Patients neither formed inhibitors against hFVIII nor had a detectable T cell response against hFVIII or capsid in peripheral blood.5 The subsequent phase III trial has been plagued by substantial interpatient variability,39 prompting repeated calls for basic science studies on the biology of hepatic AAV-hFVIII gene transfer. Several other trials are based on this approach, which relies on high-dose i.v. administration of a hepatotropic vector expressing BDD-hFVIII (since full-length hFVIII sequence exceeds the packaging capacity of AAV vectors). The vectors in these trials differ in their choice of capsid and in their expression cassettes (although all use short hepatocyte-specific promoters).4 Elevated liver enzyme levels were observed for several weeks to months in the first year after gene transfer in the initial trial, suggesting damage to hepatocytes and prompting a prolonged course of steroid drugs, a strategy that was originally employed to counter CD8+ T cell responses against the viral capsid.7,40,41 Initial reports from competing trials again point to the difficulty of maintaining high levels of FVIII, which in some instances already declined within the first year. One trial reported expression of FVIII at levels that stabilized at 10%–20% of normal and have been sustained for at least 3 years.2 Thus far, preclinical studies on the stability of FVIII expression had not been possible because of a lack of an animal model (FVIII expression was lost only if the animals developed inhibitors). Here, we introduce a murine model that shares some of the characteristics seen in clinical trials, allowing us to experimentally interrogate instability and variability of FVIII expression. Furthermore, we identify IL-15 signaling-dependent shutdown of protein production in the liver as a major contributor for systemic loss of expression, which can be targeted by newly developed mAb therapy.
FVIII synthesis is highly inefficient unless codon-optimized sequences are used, with the goal of increasing protein translation. However, hFVIII is also inefficiently secreted and tends to aggregate, so that high levels of expression—such as those achieved through codon optimization—tend to cause ER stress in cells, which in turn may trigger the UPR.21,23,24,25,42 We previously showed that expression in AAV-transduced hepatocytes can induce ER stress in murine livers, as indicated by upregulation of the chaperone BiP and the pro-apoptotic transcription factor CHOP.25 Others recently documented that limiting expression per cell, e.g., by enhancer/promoter choice, reduces the potential for ER stress induction in mice.24 In our experience, ER stress induction and its consequences are mouse strain dependent. While our construct includes the apolipoprotein E (ApoE) hepatocyte control region that has been found by others to contribute to induction of ER stress, we previously found, using the identical vector, that this cellular stress resolves over time without causing a loss of FVIII expression in C57BL/6 mice.25 In the BALB/c strain, we find that very high initial expression (>100% of normal) triggers a substantial decline even under conditions of immune suppression, suggesting that this loss of expression is not immune mediated. Among our various experiments, seven mice had initial levels of >100%, none of which were sustained. These results are reminiscent of clinical trial results reported by George et al., albeit with a more compressed time frame.2 Further experiments are needed to determine whether UPR induced early after gene transfer or a different mechanism triggers this decline. Regardless of immune suppression, approximately half of the transduced mice developed ER stress in the liver over time, which was still detectable in non-immune-suppressed mice after they had lost expression and thus may have peaked earlier in these animals (between weeks 8 and 14).
Independent of immune suppression, vector copy numbers were substantially reduced between weeks 8 and 14, with minimal effect on F8 transcription and without reduction in percentage of transduced cells (see smFISH results, Figures 6R–6U). Others also report elimination of unstable genomes.43 Interestingly, non-immune-suppressed mice lost protein expression in the liver between 8 and 14 weeks. Multiple more lines of evidence (including the lack of correlation between transcript levels and hFVIII production, the partial rescue of protein production but not transcription by IL-15 blockade, and the correlation between hepatic protein and systemic expression) point to an immune-mediated translational shutdown mechanism. Alternatively, UPR can also direct translational reprogramming. Moreover, ER stress activates innate immune pathways. For instance, stress-induced protein kinase RNA-like ER kinase (PERK)-mediated phosphorylation of eIF2α activates nuclear factor κB by suppressing translation of its inhibitor IκBα, leading to inflammatory cytokine expression. Conversely, inflammatory cytokines can induce ER stress and UPR.37,38,44 It is therefore possible that, reminiscent of destruction of pancreatic islet cells in type 1 diabetes, cellular immune responses and ER stress are interconnected in the loss of hFVIII expression in hepatocytes.33,34,35,36 While the mechanism of translational shutdown requires further study, we provide first evidence for increased phosphorylation of eIF2α, which is known to direct translational shutdown, inhibit ER-stress-induced cell death, and thus promote survival of stressed cells. Both cellular stress and viral infection can lead to eIF2α phosphorylation through the kinases PERK or RNA-activated protein kinase R (PKR), which in turn can enhance inflammatory immune responses. In its extreme form, UPR may induce apoptosis through upregulation of CHOP, which, however, was not typically observed in our study. Follow-up studies should address whether the shutdown is specific to hFVIII or whether more global changes in translation occur. Such investigations may include spatial patterns of gene expression in the liver, relationship to ER stress, and the possibility of reversing the shutdown.
Numerous studies have demonstrated immune-tolerance induction to the transgene product by hepatic AAV gene transfer, which, however, does not always occur.45,46,47 For example, inhibitor formation against hFVIII is typically observed in non-human primate studies.48 Rapamycin has proved to be an effective drug to delete Teff and promote immune regulation.49,50,51 Moreover, rapamycin suppresses humoral immunity by inhibition of GC formation,52 as shown here through the absence of Tfh, Tfr, and GC B cell responses. Thus, repeated administration of rapamycin for 8 weeks effectively prevented antibody formation not only against FVIII but also against AAV8 capsid, allowing for a second administration of AAV8. Whether this strategy can be used to boost FVIII levels over the long term will likely depend on the ability to avoid immune and stress responses to hFVIII. While others found rapamycin to enhance AAV transduction of liver (due to enhanced autophagy),53 we consistently observed a modest (∼2-fold) decrease in gene transfer and mRNA expression, which may be related to differential effects of the degree of mTOR inhibition depending on rapamycin dose (which was 2-fold higher in our study).
A concern with using rapamycin is the escape of CD8+ Tmem cells.54,55 There is also increasing evidence showing that the liver contains NK cells with memory phenotype.56,57 IL-15 stimulates proliferation of CD8+ Tmem cells and is required for their maintenance as well as that of NK cells. Current development of clinical candidates for IL-15 blockade is focused on mAbs that block CD122, the β chain of the IL-15 receptor. This approach was historically complicated by the fact that this subunit is shared with the receptor for IL-2, thus potentially inhibiting Tregs. A recently developed anti-CD122 mAb (ChMBC7) circumvents this problem. ChMBC7 is a chimeric mouse IgG2a/κ that blocks IL-15 (trans-presented by CD215) from inducing STAT5 signaling and IL-2 interaction with dimeric CD122/CD132 but not binding of IL-2 to the trimeric/high-affinity IL-2 receptor.28,32 Thus, Treg function is minimally affected. Furthermore, ChMBC7 contains two amino acid substitutions in the heavy chain (L234A/L235A) that hamper its ability to induce effector functions through Fc receptors without significantly reducing binding to FcRn (which maintains its half-life in circulation). Others showed that administration of ChMBC7 twice per week for 7 weeks effectively suppressed development of type 1 diabetes in NOD mice.29 Two weeks after treatment, CD8+ T cells (particularly CD8+ Tmem cells) in the pancreatic islet were distinctly reduced, and NK cells were nearly fully ablated. ChMBC7 also showed strong efficacy in a model of vitiligo, an autoimmune disease that causes skin lesions.28 A human version of this mAb is a candidate for clinical trial testing.
Although our study establishes a role for CD8+ T cells in the immune response against AAV8-FVIII transduced liver, these are clearly not solely responsible for the loss of expression, which also cannot merely be explained by killing/elimination of hepatocytes. Rather, loss of expression is primarily the result of a translational shutdown, which is in part dependent on IL-15 signaling and appears orchestrated by a complex interaction between innate and adaptive immune responses. Interestingly, IFN-γ expression at week 16 showed a strong correlation with phosphorylation of eIF2α in animals treated with rapamycin but not rapamycin/anti-IL-15, consistent with improved hFVIII expression in the latter group. Thus, IL-15 blockade disrupted this connection between IFN-γ expression and translational reprogramming but ultimately may have merely delayed the shutdown of expression, as rapamycin/anti-IL-15-treated mice showed enhanced inflammatory gene expression by week 16. Anti-CD122 treatment had a more potent and durable effect on NK cells and IFN-γ expression and also limited pro-inflammatory gene expression in KCs as well as frequencies of CD8+ Teff and Tem cells even at late time points. These features likely explain the improved hFVIII expression at week 16. How anti-CD122 can resolve this response remains to be determined. In the experiments performed here, IL-15 blockade on average preserved ∼25%–50% of initial FVIII levels. Furthermore, FVIII protein expression was restricted to clusters of hepatocytes rather than the widespread and more uniform expression pattern seen at early time points, and there was no improvement of vector copy numbers or transcription. Thus, the approach has both advantages and limitations. Nonetheless, anti-CD122 treatment created an anti-inflammatory environment in the liver that eliminated CD8+ T cell infiltrates and memory cells, reduced NK cell numbers and activation, and reduced KC activation, thus also modulating antigen-presenting cells. Elimination of CD8+ T cell infiltrates (through anti-CD8 or anti-CD122 treatment) caused a lack of correlation between vector copy numbers and F8 transcript levels, so that mice with low F8 transgene copies had transcription similar to that of those with higher levels of gene transfer. Thus, CD8+ T cells also have a modulatory effect on transcription of the F8 transgene.
While declining hFVIII expression upon hepatic AAV gene transfer has now been observed in several clinical trials, the timing and kinetics vary substantially between trials and between patients enrolled in the same trial. Our study in hemophilia A mice underscores this potential for variability. Decline of expression may be triggered by very high initial levels of expression or by a cellular immune response that involves CD8+ T cells and likely also innate immune cells, while cellular stress may also contribute and may be interlinked to the immune response. Ultimately, these responses result in loss of hFVIII protein production (likely reflecting a translational shutdown) that can be partially prevented by blocking IL-15 cytokine signaling (Figure S21 and Table S1). Thus, cellular immune responses against AAV-transduced tissues are not necessarily destructive but instead are able to promote shutdown of transgene expression. Liver biopsies from AAV5-hFVIII transduced patients support the conclusion that vector genomes can persist at a high level in hepatocytes despite lack of F8 transgene expression.58
As already mentioned, results from different clinical trials range from delayed and gradual decline in FVIII levels to more rapid decline to early peak followed by stable plateau levels, albeit there is also interpatient variability within trials. Such differences may be attributed to differences in expression levels and kinetics as a function of capsid and dose and the varying use and duration of immune suppressants. Difficulty of maintaining high FVIII levels (within or above the normal range), however, appears to be a common theme. These outcomes are likely related to the consequences of inefficient folding of FVIII and of interactions with the immune system. Our study highlights the complex biology of hFVIII expression in hepatocytes. The model we established will allow for investigations into the loss of initially high levels of hFVIII expression over time and into the inflammatory and cellular stress mechanisms of translational shutdown, and into potential roles of pathogen- and damage-associated molecular patterns.
Materials and methods
Animal studies
Male hemophilia A mice (6–8 weeks of age) with targeted deletion of F8 exon 16 were used for all studies.26,59,60 These mice were either on C57BL6/129 (C57BL6/19 F8e16−/−, JAX) or BALB/c (BALB/c F8e16−/−, backcrossed to BALB/c for >10 generations). Mice received AAV vectors and immune-modulatory drugs through tail vein injections. Rapamycin was given at doses of 4–6 mg/kg, 2–3/week.50 Flt3L was given together with rapamycin at a dose of 80 μg/kg.27 Recombinant human B-domain deleted FVIII (Xyntha, Pfizer) was injected at 0.3 IU/mouse.27 Monoclonal rat anti-IL-15 (clone AIO.3; BioXCell, Lebanon, NH) or anti-CD122 (clone ChMBC7, a chimeric mouse IgG2a/κ with two amino acid substitutions in the heavy chain [L234A/L235A]; JN Biosciences) was given at 4 mg/kg twice per week along with rapamycin.28 Blood was collected from the retro-orbital venous plexus using non-heparinized microcapillary tubes followed by transfer into tubes prefilled with one-tenth volume of sodium citrate (3.8% [w/v]). Animals were housed and treated at the University of Florida and at Indiana University under Institutional Animal Care and Use Committee-approved protocols.
Analyses of plasma samples
FVIII activity was determined by chromogenic assay (Chromogenix Coatest SP4 Factor VIII kit; Diapharma) using a 7-point standard curve made with reconstituted normal pooled plasma (Trinicheck Level 1, Stago) diluted in 4% BSA to give a range of 1%–150% of normal FVIII activity. Bethesda assays to measure inhibitor titers (Nijmegen modification; 1 Bethesda unit/mL equals 50% inhibition of coagulation), ELISA to measure hFVIII- and capsid-specific IgG1 and IgG2a titers, as well as ELISA for hFIX antigen levels, were as published.60,61,62 ALT and AST levels in murine plasma were measured with the alanine transaminase activity colorimetric assay kit from Abcam (Waltham, MA).
AAV vectors
ApoE/hAAT-FVIII vector expressing codon-optimized human BDD-FVIII from hepatocyte-specific promoter was as previously described.25,26 ApoE/hAAT-hFIX vector expressing human FIX was also as published.63,64 Vector genomes were packaged into AAV8 capsid by triple transfection of HEK-293 cells and purified by iodixanol gradient as published by others.65 Titers were determined by quantitative PCR (qPCR) using transgene-specific probes.
Flow cytometry
Mouse tissue was mashed through a 70-μm nylon mesh filter strainer with buffer (0.5% BSA and 2 mM EDTA in PBS) into single-cell suspension. Immune cells of interest were then separated with magnetic beads by using either MACS (Miltenyi Biotec) or EasySep (STEMCELL Technologies) isolation kits. To collect KCs and hepatic NK and CD8+ T cells, liver tissue was passed through 200-μm nylon filter strainers and resuspended in 50 mL of RPMI-1640 medium containing GlutaMAX-1, 25 mM HEPES, and 10% fetal calf serum (FCS) (pH 7.4). The single-cell suspension was then centrifuged at 60 × g for 1 min at room temperature, and the supernatant was transferred to a new tube and centrifuged at 480 × g for 8 min at room temperature. The pellet was resuspended in 10 mL of 37% Percoll in Hanks’ balanced salt solution and centrifuged at 850 × g for 30 min at room temperature. The pellet was resuspended in 2 mL of RBC lysis buffer at room temperature for 5 min, then 1 mL of FCS was added and the mixture was centrifuged at 480 × g for 8 min at 8°C. The pellet was resuspended in buffer solution (0.5% BSA and 2 mM EDTA in PBS) and enriched for KCs by positive selection using the F4/80 MicroBead Kit (Miltenyi Biotec). The F4/80+ fraction was used for analysis of KCs and the F4/80− fraction was used for analysis of NK and CD8+ T cells. Immunostaining for flow cytometry was performed using standard protocols. Non-specific binding of immunoglobulin to Fc receptors was blocked with 0.5 μg/μL anti-mouse CD16/32 (clone S17011E; BioLegend). Dead cells were labeled with PBS-diluted amine-reactive fluorescent viability dye using the Zombie Aqua Fixable Viability Kit (BioLegend). Flow cytometry was performed on either LSR-II instrumentation (BD Biosciences) or an Attune NxT Flow Cytometer (Thermo Fisher Scientific). Flow-cytometry data were analyzed with FCS Express 6 or 7 software (De Novo Software). Antibodies are listed and gating strategies provided in Tables S2 and S3, and Figures S8–S12.
Immunohistochemistry
Liver tissue was frozen in optimal cutting temperature compound, cryosectioned, and stained with antibodies as published.66 Tissues were stained with IHC-plus polyclonal sheep anti-human FVIII IgG (LS-B2979; LSBio, Seattle, WA) and rat anti-mouse CD8α IgG2a (ab25478; Abcam, Cambridge, UK) for 30 min in a humidified chamber at room temperature. After being washed with 1× PBS for 2 min, slides were incubated with cross-adsorbed donkey anti-sheep IgG Alexa Fluor 488 (A-11015; Thermo Fisher, Rockford, IL) and pre-adsorbed donkey anti-rat IgG Alexa Fluor 647 (ab150155; Abcam) for secondary antibodies (10 μg/mL). Slides were mounted with a coverslip using ProLong Gold antifade mounting medium with DAPI (Thermo Fisher). Images were taken and stitched together into a single image with an Axio Observer 7 and Zen Blue 2.6 Software (Carl Zeiss, Germany).
In situ hybridization
smFISH was performed as described by others.67 Formalin-fixed paraffin-embedded cross-sections were prepared with a thickness of 10 μm from mouse livers. Slides were baked for 1 h at 60°C for deparaffinization, followed by dehydration twice with xylene for 5 min and 100% ethanol for 2 min at room temperature, then air-dried for 5 min at 60°C. DNA and RNA in situ hybridization was performed using the RNAscope Multiplex Fluorescent V2 kit (Advanced Cell Diagnostics, Newark, CA), according to the manufacturer’s instructions. Air-dried slides were treated with hydrogen peroxide for 10 min at room temperature and washed four times with ddH2O, followed by incubation with target-retrieval reagent for 30 min at 99°C. After another wash with ddH2O for 15 s at room temperature, slides were incubated with 100% ethanol for 3 min at room temperature, air-dried, and treated with protease plus for 30 min at 40°C. Slides were washed twice with ddH2O and hybridized with RNAscope probes for AAV genome and hF8 mRNA, and DNA and RNA signals were detected using a TSA Cyanine 3 Plus and Fluorescein Plus Evaluation kit (PerkinElmer, Waltham, MA) according to the manufacturer’s protocol. All slides were counterstained with DAPI and mounted with a coverslip using ProLong Gold antifade mounting medium (Thermo Fisher). Images were collected with an Axio Observer 7 or a LSM800 confocal microscope (Carl Zeiss).
Western blots
Liver tissues from mice were homogenized in RIPA buffer (9806; Cell Signaling Technology, Danvers, MA) containing cOmplete Mini EDTA-free Protease Inhibitor Cocktail tablets (Roche, Basel, Switzerland) and PhosSTOP Phosphatase Inhibitor Cocktail tablets (Roche) according to manufacturer’s protocol. Fifty micrograms of protein lysates was resolved by electrophoresis on a 4%–20% SDS-polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA), and blotted onto a polyvinylidine fluoride membrane. Membranes were incubated with antibodies to phospho-eIF2α (Ser51; Cell Signaling), eIF2α (L57A5; Cell Signaling), GRP94 (9G10; Enzo Life Sciences, Farmingdale, NY), BiP (3183; Cell Signaling), CHOP (L63F7; Cell Signaling), and β-Actin (13E5; Cell Signaling). LI-COR Biosciences (Lincoln, NE) anti-rabbit or anti-mouse (1:20,000) secondary antibodies were used for detection. Blots were visualized and analyzed using an Odyssey infrared scanner and Image Studio software version 3.1 (LI-COR Biosciences).
Vector copy numbers and mRNA levels
AAV copies per liver cell were determined by qPCR. DNA from tissue samples was isolated using a DNeasy Blood & Tissue Kit (Qiagen), according to the manufacturer’s instructions. qPCR was performed on 100 ng of DNA in duplicate with SsoAdvanced Universal Probes Supermix protocol (Bio-Rad) using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The amplification protocol was 95°C for 2 min followed by 39 cycles of 95°C for 5 s and 60°C for 30 s. F8 transgene-specific primers for “hFVIII-mid” were FWD 5′-CTA CTA CGA GGA CAG CTA TG-3′ and REV 5′-GAT GGT GTC GTC GTA ATC-3′, and probe was 5′-6-FAM-TCA CCC GGA CCA CCC TGC AGT CC-Iowa Black FQ-3′ (Figures S14A and S14C). F8 transgene-specific primers for “hFVIII-start” were FWD 5′-CTT CAG GCA CCA CCA CTG-3′ and REV 5′-AAG AAG CAG GTA GAC AGC TC-3′, and probe was 5′-Cy5.5-TGC GGC CTC GAC GGT ATC GAT GCC ACC A-Iowa Black RQ-Sp-3′ (Figures S14B and S14C). To detect β-actin gene copies, primers FWD 5′-GCC ATG GAT GAC GAT ATC-3′ and REV 5′-CAC ATA GGA GTC CTT CTG AC-3′ were used, along with probe 5′-Cy5.5-CGG CGA TGC TCC CCG GGC TG-Iowa Black RQ-Sp-3′. Standard curves were generated on each plate for F8 and β-actin by sequential dilution of a 215 bp gBlock that included the target region for β-actin, and a 222 bp gBlock that included the target region for hFVIII, both synthesized by Integrated DNA Technologies. Samples were run in duplicate, and assays were multiplexed to simultaneously detect transgene and β-actin.
Levels of hepatic mRNA transcript of F8 transgene and endogenous genes were determined by reverse transcription qPCR (RT-qPCR). Total RNA was isolated from tissue samples using an RNeasy Plus Mini Kit (Qiagen). RT and qPCR was performed using Reliance One-Step Multiplex RT-qPCR Supermix (Bio-Rad) and a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Each RT-qPCR reaction was multiplexed to combine an assay that targeted β-actin as the reference gene, with a second assay targeting F8 or different cellular gene. Primers to detect mRNA transcripts for the hFVIII transgene and β-actin were as described above. CHOP transcript was detected using Bio-Rad Assay ID qMmuCIP0030753. Fold gene expression was calculated by the ΔΔCt method using the following formulas: ΔCt = Ct(target gene) − Ct(reference gene); ΔΔCt = ΔCt − ΔCt(average of mice that received AAV only); fold gene expression = 2(−ΔΔCt).
Statistical analyses
Statistical analysis was performed using GraphPad Prism version 8.1.1 (GraphPad Software). Multiple unpaired t tests were used when comparing two groups, corrected for multiple comparisons with the Holm-Šídák post hoc test. One-way ANOVA was used at each time point when comparing more than two groups, corrected for multiple comparisons with the Tukey post hoc test. A simple linear regression line was drawn, and Pearson correlation coefficient with two-tailed p values was calculated for each correlation graph. All graphed data were analyzed together unless otherwise specified, and all p values presented have been adjusted for multiple comparisons.
Acknowledgments
The authors thank Drs. J. Yun Tso and Naoya Tsurushita of JN Biosciences for the kind gift of ChMBC7 monoclonal antibody against murine CD122. This work was supported by NIH grants R01HL131093 to C.T., Y.P.d.J., and R.W.H.; by P01HL160472 to Y.P.d.J., R.J.K., and R.W.H.; by R01AI51390, U54HL142012, Indiana Collaborative Initiative for Talent Enrichment (INCITE) funds (provided by Lilly Endowment), and the Riley Children's Foundation to R.W.H. The authors thank the members of the Indiana University Melvin and Bren Simon Cancer Center Flow Cytometry Resource Facility (FCRF) for their outstanding technical support. The Indiana University Melvin and Bren Simon Comprehensive Cancer Center Flow Cytometry Resource Facility is funded in part by NIH, National Cancer Institute grant P30CA082709, and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant U54DK106846 (Cooperative Center of Excellence in Hematology). The FCRF is further supported in part by NIH instrumentation grant 1S10D012270. N.L. was supported by NIH grant T32AI060519. Finally, the authors thank Advanced Cell Diagnostics for design of probes for smFISH and for sharing protocols.
Author contributions
J.S.S.B., K.Y., T.B.B., F.S., S.R.P.K., X.L., S.A., A.R.P., A.T., C.A.R., N.L., and J.R. performed experiments. J.S.S.B., K.Y., T.B.B., M.B., Y.P.d.J., C.T., R.J.K., and R.W.H. designed, analyzed, and interpreted experiments. J.S.S.B., K.Y., M.B., Y.P.d.J., C.T., R.J.K., and R.W.H. wrote the manuscript. R.W.H. supervised the study.
Declaration of interests
R.W.H. is serving on the scientific advisory board of the Regeneron Pharmaceuticals – Intellia Therapeutics collaboration on gene editing for hemophilia B, has served on a Biomarin roundtable on clinical gene therapy for hemophilia A, and has received funding from Spark Therapeutics for preclinical gene therapy studies.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.07.005.
Supplemental information
References
- 1.Butterfield J.S.S., Hege K.M., Herzog R.W., Kaczmarek R. A molecular revolution in the treatment of hemophilia. Mol. Ther. 2020;28:997–1015. doi: 10.1016/j.ymthe.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.George L.A., Monahan P.E., Eyster M.E., Sullivan S.K., Ragni M.V., Croteau S.E., Rasko J.E.J., Recht M., Samelson-Jones B.J., MacDougall A., et al. Multiyear factor VIII expression after AAV gene transfer for hemophilia A. N. Engl. J. Med. 2021;385:1961–1973. doi: 10.1056/NEJMoa2104205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konkle B.A., Walsh C., Escobar M.A., Josephson N.C., Young G., von Drygalski A., McPhee S.W.J., Samulski R.J., Bilic I., De La Rosa M., et al. BAX 335 hemophilia B gene therapy clinical trial results - potential impact of CpG sequences on gene expression. Blood. 2021;137:763–774. doi: 10.1182/blood.2019004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendell J.R., Al-Zaidy S.A., Rodino-Klapac L.R., Goodspeed K., Gray S.J., Kay C.N., Boye S.L., Boye S.E., George L.A., Salabarria S., et al. Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 2020;29:464–488. doi: 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 6.Pipe S.W., Ferrante F., Reis M., Wiegmann S., Lange C., Braun M., Michaels L.A. First-in-human gene therapy study of AAVhu37 capsid vector technology in severe hemophilia A -BAY 2599023 has broad patient eligibility and stable and sustained long-term expression of FVIII. Blood. 2020;136:44–45. doi: 10.1182/blood-2020-139803. [DOI] [Google Scholar]
- 7.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 8.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J., et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Von Drygalski A., Giermasz A., Castaman G., Key N.S., Lattimore S., Leebeek F.W.G., Miesbach W., Recht M., Long A., Gut R., et al. Etranacogene dezaparvovec (AMT-061 phase 2b): normal/near normal FIX activity and bleed cessation in hemophilia B. Blood Adv. 2019;3:3241–3247. doi: 10.1182/bloodadvances.2019000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leavitt A.D., Konkle B.A., Stine K., Visweshwar N., Harrington T.J., Adam Giermasz A., Arkin S., Fang A., Plonski F., Smith L., et al. Updated Follow-up of the Alta Study, a phase 1/2 study of giroctocogene fitelparvovec (SB-525) gene therapy in adults with severe hemophilia A. Blood. 2020;136:12. doi: 10.1182/blood-2020-137648. [DOI] [Google Scholar]
- 11.Shirley J.L., de Jong Y.P., Terhorst C., Herzog R.W. Immune responses to viral gene therapy vectors. Mol. Ther. 2020;28:709–722. doi: 10.1016/j.ymthe.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verdera H.C., Kuranda K., Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrin G.Q., Herzog R.W., Markusic D.M. Update on clinical gene therapy for hemophilia. Blood. 2019;133:407–414. doi: 10.1182/blood-2018-07-820720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasi K.J., Laffan M., Rangarajan S., Robinson T.M., Mitchell N., Lester W., Symington E., Madan B., Yang X., Kim B., et al. Persistence of haemostatic response following gene therapy with valoctocogene roxaparvovec in severe haemophilia A. Haemophilia. 2021;27:947–956. doi: 10.1111/hae.14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaczmarek R., Pierce G.F., Noone D., O'Mahony B., Page D., Skinner M.W. Eliminating Panglossian thinking in development of AAV therapeutics. Mol. Ther. 2021;29:3325–3327. doi: 10.1016/j.ymthe.2021.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierce G.F. Gene therapy for hemophilia: are expectations matching reality? Mol. Ther. 2020;28:2097–2098. doi: 10.1016/j.ymthe.2020.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierce G.F., Kaczmarek R., Noone D., O'Mahony B., Page D., Skinner M.W. Gene therapy to cure haemophilia: is robust scientific inquiry the missing factor? Haemophilia. 2020;26:931–933. doi: 10.1111/hae.14131. [DOI] [PubMed] [Google Scholar]
- 18.Sheridan C. A reprieve from hemophilia A, but for how long? Nat. Biotechnol. 2020;38:1107–1109. doi: 10.1038/s41587-020-0693-y. [DOI] [PubMed] [Google Scholar]
- 19.Muhuri M., Levy D.I., Schulz M., McCarty D., Gao G. Durability of transgene expression after rAAV gene therapy. Mol. Ther. 2022;30:1364–1380. doi: 10.1016/j.ymthe.2022.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fahs S.A., Hille M.T., Shi Q., Weiler H., Montgomery R.R. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. 2014;123:3706–3713. doi: 10.1182/blood-2014-02-555151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poothong J., Pottekat A., Siirin M., Campos A.R., Paton A.W., Paton J.C., Lagunas-Acosta J., Chen Z., Swift M., Volkmann N., et al. Factor VIII exhibits chaperone-dependent and glucose-regulated reversible amyloid formation in the endoplasmic reticulum. Blood. 2020;135:1899–1911. doi: 10.1182/blood.2019002867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hetz C., Zhang K., Kaufman R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020;21:421–438. doi: 10.1038/s41580-020-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malhotra J.D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S.W., Kaufman R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong S., Handyside B., Sihn C.R., Liu S., Zhang L., Xie L., Murphy R., Galicia N., Yates B., Minto W.C., et al. Induction of ER stress by an AAV5 BDD FVIII construct is dependent on the strength of the hepatic-specific promoter. Mol. Ther. Methods Clin. Dev. 2020;18:620–630. doi: 10.1016/j.omtm.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zolotukhin I., Markusic D.M., Palaschak B., Hoffman B.E., Srikanthan M.A., Herzog R.W. Potential for cellular stress response to hepatic factor VIII expression from AAV vector. Mol. Ther. Methods Clin. Dev. 2016;3:16063. doi: 10.1038/mtm.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sack B.K., Merchant S., Markusic D.M., Nathwani A.C., Davidoff A.M., Byrne B.J., Herzog R.W. Transient B cell depletion or improved transgene expression by codon optimization promote tolerance to factor VIII in gene therapy. PLoS One. 2012;7:e37671. doi: 10.1371/journal.pone.0037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biswas M., Sarkar D., Kumar S.R.P., Nayak S., Rogers G.L., Markusic D.M., Liao G., Terhorst C., Herzog R.W. Synergy between rapamycin and FLT3 ligand enhances plasmacytoid dendritic cell-dependent induction of CD4+CD25+FoxP3+ Treg. Blood. 2015;125:2937–2947. doi: 10.1182/blood-2014-09-599266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richmond J.M., Strassner J.P., Zapata L., Jr., Garg M., Riding R.L., Refat M.A., Fan X., Azzolino V., Tovar-Garza A., Tsurushita N., et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aam7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan X., Dong Y., Tsurushita N., Tso J.Y., Fu W. CD122 blockade restores immunological tolerance in autoimmune type 1 diabetes via multiple mechanisms. JCI Insight. 2018;3 doi: 10.1172/jci.insight.96600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathews D.V., Dong Y., Higginbotham L.B., Kim S.C., Breeden C.P., Stobert E.A., Jenkins J., Tso J.Y., Larsen C.P., Adams A.B. CD122 signaling in CD8+ memory T cells drives costimulation-independent rejection. J. Clin. Invest. 2018;128:4557–4572. doi: 10.1172/JCI95914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mackay L.K., Rahimpour A., Ma J.Z., Collins N., Stock A.T., Hafon M.L., Vega-Ramos J., Lauzurica P., Mueller S.N., Stefanovic T., et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 32.Yokoyama S., Watanabe N., Sato N., Perera P.Y., Filkoski L., Tanaka T., Miyasaka M., Waldmann T.A., Hiroi T., Perera L.P. Antibody-mediated blockade of IL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc. Natl. Acad. Sci. USA. 2009;106:15849–15854. doi: 10.1073/pnas.0908834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark A.L., Urano F. Endoplasmic reticulum stress in beta cells and autoimmune diabetes. Curr. Opin. Immunol. 2016;43:60–66. doi: 10.1016/j.coi.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engin F. ER stress and development of type 1 diabetes. J. Investig. Med. 2016;64:2–6. doi: 10.1097/JIM.0000000000000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marre M.L., Profozich J.L., Coneybeer J.T., Geng X., Bertera S., Ford M.J., Trucco M., Piganelli J.D. Inherent ER stress in pancreatic islet beta cells causes self-recognition by autoreactive T cells in type 1 diabetes. J. Autoimmun. 2016;72:33–46. doi: 10.1016/j.jaut.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Back S.H., Scheuner D., Han J., Song B., Ribick M., Wang J., Gildersleeve R.D., Pennathur S., Kaufman R.J. Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in β cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grootjans J., Kaser A., Kaufman R.J., Blumberg R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016;16:469–484. doi: 10.1038/nri.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang K., Kaufman R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozelo M.C., Mahlangu J., Pasi K.J., Giermasz A., Leavitt A.D., Laffan M., Symington E., Quon D.V., Wang J.D., Peerlinck K., et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N. Engl. J. Med. 2022;386:1013–1025. doi: 10.1056/NEJMoa2113708. [DOI] [PubMed] [Google Scholar]
- 40.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C., et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lange A.M., Altynova E.S., Nguyen G.N., Sabatino D.E. Overexpression of factor VIII after AAV delivery is transiently associated with cellular stress in hemophilia A mice. Mol. Ther. Methods Clin. Dev. 2016;3:16064. doi: 10.1038/mtm.2016.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sihn C.R., Handyside B., Liu S., Zhang L., Murphy R., Yates B., Xie L., Torres R., Russell C.B., O'Neill C.A., et al. Molecular analysis of AAV5-hFVIII-SQ vector-genome-processing kinetics in transduced mouse and nonhuman primate livers. Mol. Ther. Methods Clin. Dev. 2022;24:142–153. doi: 10.1016/j.omtm.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D.T., Back S.H., Kaufman R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 45.Keeler G.D., Markusic D.M., Hoffman B.E. Liver induced transgene tolerance with AAV vectors. Cell Immunol. 2019;342:103728. doi: 10.1016/j.cellimm.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sack B.K., Herzog R.W., Terhorst C., Markusic D.M. Development of gene transfer for induction of antigen-specific tolerance. Mol. Ther. Methods Clin. Dev. 2014;1:14013. doi: 10.1038/mtm.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S.R.P., Xie J., Hu S., Ko J., Huang Q., Brown H.C., Srivastava A., Markusic D.M., Doering C.B., Spencer H.T., et al. Coagulation factor IX gene transfer to non-human primates using engineered AAV3 capsid and hepatic optimized expression cassette. Mol. Ther. Methods Clin. Dev. 2021;23:98–107. doi: 10.1016/j.omtm.2021.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elkouby L., Armour S.M., Toso R., DiPietro M., Davidson R.J., Nguyen G.N., Willet M., Kutza S., Silverberg J., Frick J., et al. Preclinical assessment of an optimized AAV-FVIII vector in mice and non-human primates for the treatment of hemophilia A. Mol. Ther. Methods Clin. Dev. 2022;24:20–29. doi: 10.1016/j.omtm.2021.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kishimoto T.K. Development of ImmTOR tolerogenic nanoparticles for the mitigation of anti-drug antibodies. Front. Immunol. 2020;11:969. doi: 10.3389/fimmu.2020.00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nayak S., Cao O., Hoffman B.E., Cooper M., Zhou S., Atkinson M.A., Herzog R.W. Prophylactic immune tolerance induced by changing the ratio of antigen-specific effector to regulatory T cells. J. Thromb. Haemost. 2009;7:1523–1532. doi: 10.1111/j.1538-7836.2009.03548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meliani A., Boisgerault F., Hardet R., Marmier S., Collaud F., Ronzitti G., Leborgne C., Costa Verdera H., Simon Sola M., Charles S., et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018;9:4098. doi: 10.1038/s41467-018-06621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye L., Lee J., Xu L., Mohammed A.U.R., Li W., Hale J.S., Tan W.G., Wu T., Davis C.W., Ahmed R., Araki K. mTOR promotes antiviral humoral immunity by differentially regulating CD4 helper T cell and B cell responses. J. Virol. 2017;91 doi: 10.1128/JVI.01653-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hosel M., Huber A., Bohlen S., Lucifora J., Ronzitti G., Puzzo F., Boisgerault F., Hacker U.T., Kwanten W.J., Kloting N., et al. Autophagy determines efficiency of liver-directed gene therapy with adeno-associated viral vectors. Hepatology. 2017;66:252–265. doi: 10.1002/hep.29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Araki K., Turner A.P., Shaffer V.O., Gangappa S., Keller S.A., Bachmann M.F., Larsen C.P., Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Araki K., Youngblood B., Ahmed R. The role of mTOR in memory CD8 T-cell differentiation. Immunol. Rev. 2010;235:234–243. doi: 10.1111/j.0105-2896.2010.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang X., Chen Y., Peng H., Tian Z. Memory NK cells: why do they reside in the liver? Cell Mol. Immunol. 2013;10:196–201. doi: 10.1038/cmi.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paust S., Gill H.S., Wang B.Z., Flynn M.P., Moseman E.A., Senman B., Szczepanik M., Telenti A., Askenase P.W., Compans R.W., von Andrian U.H. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 2010;11:1127–1135. doi: 10.1038/ni.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fong S., Yates B., Sihn C.R., Mattis A.N., Mitchell N., Liu S., Russell C.B., Kim B., Lawal A., Rangarajan S., et al. Interindividual variability in transgene mRNA and protein production following adeno-associated virus gene therapy for hemophilia A. Nat. Med. 2022;28:789–797. doi: 10.1038/s41591-022-01751-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bi L., Lawler A.M., Antonarakis S.E., High K.A., Gearhart J.D., Kazazian H.H., Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat. Genet. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 60.Sherman A., Su J., Lin S., Wang X., Herzog R.W., Daniell H. Suppression of inhibitor formation against FVIII in a murine model of hemophilia A by oral delivery of antigens bioencapsulated in plant cells. Blood. 2014;124:1659–1668. doi: 10.1182/blood-2013-10-528737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bertolini T.B., Shirley J.L., Zolotukhin I., Li X., Kaisho T., Xiao W., Kumar S.R.P., Herzog R.W. Effect of CpG depletion of vector genome on CD8(+) T cell responses in AAV gene therapy. Front. Immunol. 2021;12:672449. doi: 10.3389/fimmu.2021.672449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shirley J.L., Keeler G.D., Sherman A., Zolotukhin I., Markusic D.M., Hoffman B.E., Morel L.M., Wallet M.A., Terhorst C., Herzog R.W. Type I IFN sensing by cDCs and CD4(+) T cell help are both requisite for cross-priming of AAV capsid-specific CD8(+) T cells. Mol. Ther. 2020;28:758–770. doi: 10.1016/j.ymthe.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J.E., Ozelo M.C., Hoots K., Blatt P., Konkle B., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 64.Mingozzi F., Liu Y.L., Dobrzynski E., Kaufhold A., Liu J.H., Wang Y., Arruda V.R., High K.A., Herzog R.W. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J. Clin. Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crosson S.M., Dib P., Smith J.K., Zolotukhin S. Helper-free production of laboratory grade AAV and purification by iodixanol density gradient centrifugation. Mol. Ther. Methods Clin. Dev. 2018;10:1–7. doi: 10.1016/j.omtm.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rogers G.L., Hoffman B.E. Optimal immunofluorescent staining for human factor IX and infiltrating T cells following gene therapy for hemophilia B. J. Genet. Syndr. Gene Ther. 2012;S1 doi: 10.4172/2157-7412.s1-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kusmartseva I., Wu W., Syed F., Van Der Heide V., Jorgensen M., Joseph P., Tang X., Candelario-Jalil E., Yang C., Nick H., et al. Expression of SARS-CoV-2 entry factors in the pancreas of normal organ donors and individuals with COVID-19. Cell Metab. 2020;32:1041–1051.e6. doi: 10.1016/j.cmet.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.