

Abstract
The pathogenesis of acute kidney injury (AKI) is still not fully understood, and effective interventions are lacking. Here, we explored whether methyltransferase 3 (METTL3) was involved in the progression of AKI via regulation of cell death. We reported that PT(proximal tubule)-METTL3-knockout (KO) noticeably suppressed ischemic-induced AKI via inhibition of renal cell apoptosis. Furthermore, we also found that the expression of mmu-long non-coding RNA (lncRNA) 121686 was upregulated in antimycin-treated Boston University mouse proximal tubule (BUMPT) cells and a mouse ischemia-reperfusion (I/R)-induced AKI model. Functionally, mmu-lncRNA 121686 could promote I/R-induced mouse renal cell apoptosis. Mechanistically, mmu-lncRNA 121686 acted as a competing endogenous RNA (ceRNA) to prevent microRNA miR-328-5p-mediated downregulation of high-temperature requirement factor A 3 (Htra3). PT-mmu-lncRNA 121686-KO mice significantly ameliorated the ischemic-induced AKI via the miR-328-5p/HtrA3 axis. In addition, hsa-lncRNA 520657, homologous with lncRNA 121686, sponged miR-328-5p and upregulated Htra3 to promote I/R-induced human renal cell apoptosis. Interestingly, we found that mmu-lncRNA 121686/hsa-lncRNA 520657 upregulation were dependent on METTL3 via N6-methyladenosine (m6A) modification. The mmu-lncRNA 121686/miR-328-5p or hsa-lncRNA 520657/miR-328-5p /HtrA3 axis was induced in vitro by METTL3 overexpression; in contrast, this effect was attenuated by METTL3 small interfering RNA (siRNA). Furthermore, we found that PT-METTL3-KO or METTL3 siRNA significantly suppressed ischemic, septic, and vancomycin-induced AKI via downregulation of the mmu-lncRNA 121686/miR-328-5p/HtrA3 axis. Taken together, our data indicate that the METTL3/mmu-lncRNA 121686/hsa-lncRNA 520657/miR-328-5p/HtrA3 axis potentially acts as a therapeutic target for AKI.
Keywords: mmu_lncRNA 121686, hsa_lncRNA 520657, METTL3, miR-328-5pHtrA3, AKI
Graphical abstract
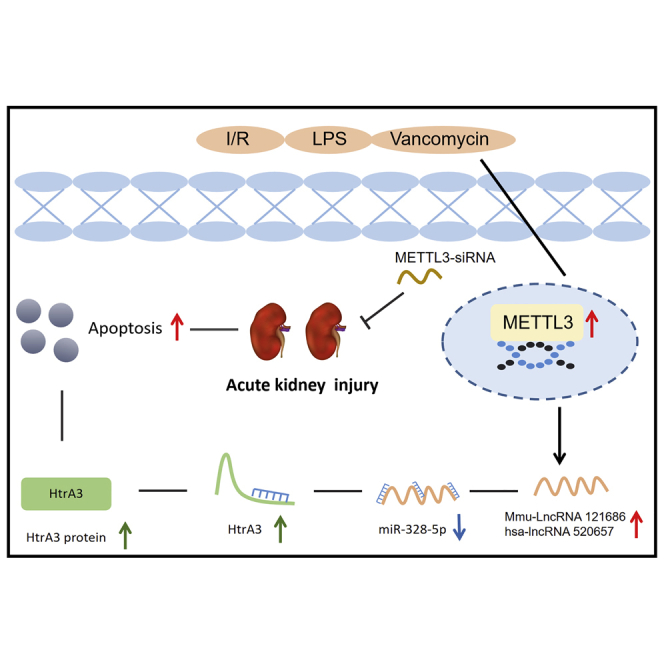
Zhang et al. revealed that the METTL3/mmu-lncRNA 121686 or hsa-lncRNA 520657/miR-328-5p/HtrA3 axis is responsible for the progression of I/R-, CLP-, and VAN-induced AKI. Moreover, METTL3 siRNA is potential therapy way for the AKI.
Introduction
Acute kidney injury (AKI) is characterized by a rapid decline in renal function caused by ischemia-reperfusion (I/R), sepsis, and various nephrotoxins and has been associated with high morbidity and mortality.1 It is estimated that about 2 million people die of AKI every year, according to a global survey.2,3 Apoptosis of proximal tubular cells has been known to play a pivotal role in the progression of AKI.4 However, although great efforts have been made in this area, the underlying mechanism of proximal tubular cell death in AKI remains largely unknown.
Long non-coding RNA (lncRNA), a linear non-coding RNA with a length over 200 nt, participates in multiple cell processes.5 Generally, lncRNAs can modulate the activity of microRNAs (miRNAs) to control target gene expression by acting as a competing endogenous RNA (ceRNA).6 A recent study has shown that lncRNAs are involved in the development of AKI.7 For example, lncRNA nuclear-enriched abundant transcript 1 (NEAT1), lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), and lncRNA TCONS_00016233 were associated with lipopolysaccharide (LPS)-induced septic AKI.8,9 Besides, renal cell apoptosis in ischemic AKI could be induced by several lncRNAs such as lncRNA GAS5, lncRNA NEAT1, lncRNA-PRINS, LINC00520, and lncRNA SNHG14.10,11,12,13,14 On the contrary, lncRNAs RNA H19, TUG1, and MEG3 had an anti-apoptotic role in ischemic injury.15,16,17 Additionally, the regulatory mechanism of lncRNAs in AKI remains to be clarified.
N6-Methyladenosine (m6A) RNA methylation is a major RNA modification in eukaryotic cells. Fat mass and obesity-associated protein (FTO) is one of the m6A “erasers” that mediate demethylation of m6A sites.18 A recent study has reported that FTO could prevent cisplatin-kidney injury.19 In contrast, methyltransferase 3 (METTL3) is an m6A “writer” that modifies adenosine. Interestingly, a previous study has reported that METTL3 also exerts a renoprotective role in colistin-induced nephrotoxicity via regulation of miR-873-5p.20,21,22 However, it remains unknown whether METTL3 can regulate AKI induced by other factors.
In this study, we discovered that mmu-lncRNA 121686 was upregulated in ischemic AKI models both in vitro and in vivo. Moreover, mmu-lncRNA 121686 promoted I/R-induced mouse renal cell apoptosis via targeting miR-328-5p and upregulating high-temperature requirement factor A 3 (HtrA3). In addition, hsa-lncRNA 520657, homologous with lncRNA 121686, drives I/R-induced human renal cell apoptosis via the miR-328-5p/HtrA3 axis. Interestingly, we found that the mmu-lncRNA 121686/miR-328-5p or hsa-lncRNA 520657/miR-328-5p/HtrA3 axis was positively and directly regulated by METTL3 in vitro. Furthermore, we demonstrated that PT-METTL3-KO or METTL3 siRNA significantly attenuated ischemic, septic, and vancomycin (VAN)-induced AKI via downregulation of the mmu-lncRNA 121686/hsa-lncRNA520657/miR-328-5p/HtrA3 axis.
Results
METTL3 is induced by I/R treatment both in vitro and in vivo
In this study, METTL3 was induced at 2 h after ischemia, but its expression and localization remained largely unknown. Quantitative RT-PCR and immunoblot analysis demonstrated that the expression levels of METTL3 in Boston University mouse proximal tubule (BUMPT) cells were gradually increased at 0 h after reperfusion, reached the peak at 2 h after reperfusion, and then declined at 4 h after reperfusion (Figure 1A, 1D, and 1G), which was further supported by the immunofluorescence staining results (Figures 1J and 1K). In the cortex and medulla of ischemic AKI mice, the mRNA and protein expression levels of METTL3 were also gradually increased at 24 h after reperfusion and achieved the peak at 48 h (Figures 1B, 1C, 1E, 1F, 1H, and 1I), which was confirmed by the immunofluorescence results (Figures 1L and 1M). Altogether, these data indicated that I/R injury could induce the expression of METTL3 both in vitro and in vivo.
Figure 1.

METTL3 was induced by I/R injury in BUMPT cells and C57/BL6 mice
BUMPT cells were treated with I/R (2/0, 2, and 4 h). C57/BL6 mice were subjected to bilateral renal ischemia for 28 min and reperfusion for 24 and 48 h. (A–C) qRT-PCR analysis of METTL3 expression in BUMPT cells, and the cortex and medulla of kidney. (D–F) The immunoblot analysis of METTL3 expression in BUMPT cells, and the cortex and medulla of kidney. (G–I) Analysis of the gray scale image between METTL3 and β-tubulin. (J) Immunofluorescence staining of METTL3 in BUMPT cells at indicated time points. Scale bar: 20 μM. (K) Quantification of METTL3 staining. (L and M) Immunohistochemical staining of METTL3 in kidney treated with or without I/R condition. Original magnification ×400. Scale bar: 100 μM. Data are expressed as means ± SD. (n = 6). ∗p < 0.05 versus sham or saline group.
I/R-, cecal ligation and puncture (CLP)-, and VAN-induced AKIs are attenuated in PT-METTL3-KO mice
Male PT-METTL3-wild-type (WT) littermates and PT-METTL3-knockout (KO) mice were treated with or without I/R (28 min/48 h), VAN (days 7), and CLP (18 h). PT-METTL3-KO mice significantly ameliorated I/R-, CLP-, and VAN-induced elevation in blood urine nitrogen and creatinine (Figure 2A–2F), as well as tubular damage (Figures 2G, 2H, 2K, 2L, 2O, and 2P) and tubular cell apoptosis (Figures 2I, 2J, 2M, 2N, 2Q, and 2R). Altogether, these data indicated that METTL3 could mediate I/R-, VAN-, and CLP-induced AKIs in mice.
Figure 2.

IR-, VAN-, and CLP-induced renal injuries and tubular cell apoptosis were ameliorated in PT-METTL3-KO mice
The bilateral renal arteries of PT-METTL3-KO and PT-METTL3-WT littermate mice were clamped for 28 min and then underwent reperfusion for 48 h. The PT-METTL3-KO and PT-METTL3-WT littermate mice were injected intraperitoneally with VAN at a dose of 600 mg/kg for 7 consecutive days. For the CLP model, the cecum of PT-METTL3-KO and PT-METTL3-WT littermate mice were ligated and punctured for 18 h. (A–C) BUN (blood urea nitrogen). (D–F) Serum creatinine. (G, K, and O) H&E staining. (I, M, and Q) TUNEL staining. (H, L, and P) Tubular damage score. (J, N, and R) The number of TUNEL-positive cells. Original magnification ×400. Scale bar, 100 μM. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus sham or saline group. #p < 0.05 versus PT-METTL3-WT with IR, VAN, or CLP group.
I/R induces the expression of mmu-lncRNA 121686/hsa-lncRNA520657 in BUMPT and HK-2 cells
The expression profiles of all lncRNAs in ischemic AKI were examined. Firstly, C57BL/6 mice were treated with I/R (2/48 h), and total RNA was extracted from the kidney cortex tissue for lncRNA chip assay. The heatmap of lncRNAs is shown in Figure 3A. Significantly, the upregulated expression (fold change >7) of lncRNAs was observed in the I/R group compared with the sham group (Figure 3B). Among them, the upregulation of mmu-lncRNA 121686(Ensembl: ENSMUST00000121686) was 7.99-fold, which was further confirmed by qRT-PCR assay in a mouse ischemic AKI model (Figure 3C). To determine the expression of mmu-lncRNA 121686 in BUMPT cells under I/R conditions, the cells were treated with ATP/glucose depletion and calcium ionophore for 2 h and then reperfusion at 0–4 h. qRT-PCR analysis revealed that the expression of mmu-lncRNA 121686 was gradually increased at 0 h after reperfusion, reached the peak at 2 h after reperfusion, and then declined at 4 h after reperfusion (Figure 3D). The immunoblot results demonstrated that the expression trend of cleaved caspase-3 was consistent with that of mmu-lncRNA 121686 (Figures 3E and 3F). Furthermore, hsa-lncRNA 520657(Ensembl: ENST00000520657) was found in the Homo database (https://www.ncbi.nlm.nih.gov/) and had homology with the mmu-lncRNA 121686 sequence (Figure 3G). qRT-PCR analysis and immunoblot results also showed that the expression of mmu-lncRNA 520657 was correlated with the expression of cleaved caspase-3 (Figures 3H–3J). Both the expression of mmu-lncRNA 121686 and hsa-lncRNA 520657 are distributed in the cytoplasm and nucleus of BUMPT and HK-2 cells via qRT-PCR analysis (Figures 3K–3L), which was further confirmed by the results of fluorescence in situ hybridization (FISH) assay (Figures 3M and 3N). Collectively, these findings indicated that the expression of mmu-lncRNA 121686 and hsa-lncRNA 520657 was induced by AKI model.
Figure 3.

The expression of mmu_lncRNA 121686 and hsa_lncRNA 520657 were induced by ischemic injury in BUMPT and HK-2 cells
The bilateral renal arteries of male C57BL/6 mice were clamped for 28 min and then underwent reperfusion for 24 or 48 h. BUMPT cells were treated with I/R (2/0, 2, and 4 h). (A) The heatmap. (B) Upregulation of lncRNAs (fold changes >7). (C) qRT-PCR analysis of mmu_lncRNA 121686 expression in mice with I/R operation. (D) qRT-PCR analysis of mmu_lncRNA 121686 expression at 0, 2, and 4 h reperfusion after 2 h ATP depletion. (E) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, and β-tubulin in BUMPT cells. (F) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, and β-tubulin in BUMPT cells. (G) Comparison of the sequence of mmu _lncRNA 121686 and hsa_lncRNA 520657. (H) qRT-PCR analysis of hsa_LncRNA 520657 expression at 0, 2, and 4 h reperfusion after 2 h ATP depletion. (I) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, and β-tubulin in HK-2 cells. (J) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, and β-tubulin in HK-2 cells. (K) qRT-PCR analysis of the expression of mmu_lncRNA 121686 in nucleus and cytoplasm. (L) qRT-PCR analysis of the expression of hsa_lncRNA520657 in nucleus and cytoplasm. (M and N) FISH probe detection of the localization of mmu_lncRNA 121686 and hsa_lncRNA 520657 in BUMPT and HK-2 cells. U6 and 18S were used as control for nucleus and cytoplasm, respectively. Scale bar: 20 μM. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus control group.
mmu-lncRNA 121686 and hsa-lncRNA 520657 mediate I/R-induced BUMPT and HK-2 cell apoptosis
Next, the deletion of mmu-lncRNA 121686 or hsa-lncRNA 520657 in BUMPT or HK-2 cells was established, respectively. Briefly, we designed two guide RNAs (gRNAs) targeting both ends of lncRNA 121686 and 520657 sequences and then embedded them into pSpCas9(BB)-2A-Puro (PX459) vectors. The vectors were transfected into BUMPT and HK-2 cells, respectively, and then treated with the puromycin at 1 μg/mL for selection of the positive cells (Figure 4A). The flow cytometry (FCM) analysis demonstrated that knocking down mmu-lncRNA 121686 and hsa-lncRNA 520657 noticeably decreased I/R-induced BUMPT and HK-2 cell apoptosis (Figures 4B–4D), which was further confirmed by the immunoblotting of cleaved capase-3 (Figures 4E–4H). These data illustrated that mmu-lncRNA 121686 and hsa-lncRNA 520657 were involved in I/R-induced BUMPT and HK-2 cell apoptosis.
Figure 4.

mmu_lncRNA 121686 mediated I/R-induced BUMPT cell apoptosis while hsa_lncRNA 520657 mediated I/R-induced HK-2 cells apoptosis
BUMPT and HK-2 cells were transfected with hsa_lncRNA 520657 sgRNA or mmu_lncRNA 121686 sgRNA and then were subjected to I/R (2/2 h). (A) Schema of Cas9 KO mmu_lncRNA 121686 and has_lncRNA 520657 (B and C) Flow cytometry analysis of BUMPT and HK-2 cells apoptosis. (D) Representative apoptosis rate of BUMPT cells and HK-2 cells. (E) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, and β-tubulin in BUMPT cells. (F) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, and β-tubulin in BUMPT cells. (G) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, and β-tubulin in HK-2 cells. (H) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, and β-tubulin in HK-2 cells. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus scramble with saline group. #p < 0.05 versus scramble with I/R group.
mmu-lncRNA 121686 and his-lncRNA 520657 directly binds to miR-328-5p
Generally, lncRNAs can act as ceRNAs that compete for binding to shared miRNAs. We found that both mmu-lncRNA 121686 and hsa-lncRNA 520657 contained the complementary sequences of miR-328-5p via a target-prediction analysis software (Figures 5A and 5C). The luciferase reporter assay illustrated that a miR-328-5p mimic significantly decreased the luciferase activity of mmu-lncRNA 121686-WT and hsa-lncRNA 520657-WT but not mmu-lncRNA 121686-mutant (Mut) and hsa-lncRNA-Mut (Figures 5B and 5D). The FISH results showed that miR-328-5p with mmu-lncRNA 121686 or hsa-lncRNA 520657 co-localized in the cytoplasm of BUMPT cells or HK-2 cells in in vitro ischemic AKI model, respectively (Figures 5E and 5F). Further, qRT-PCR analysis demonstrated that the expression of miR-328-5p was increased by KO of mmu-lncRNA 121686 and hsa-lncRNA 520657 under basic and ischemic injury conditions; on the contrary, it was suppressed by transfection of mmu-lncRNA 121686 and hsa-lncRNA 520657 (Figures 5G–5J). These data also found that mmu-lncRNA 121686 and hsa-lncRNA 520657 could directly sponge to the miR-328-5p.
Figure 5.

hsa_lncRNA 520657 and mmu_lncRNA 121686 directly bind to miR-328-5p
(A and C) The complementary and mutated sequences of mmu_lncRNA 121686, hsa_lncRNA 520657, and miR-328-5p. (B) The detection of relative luciferase activity after BUMPT cells co-transfected with miR-328-5p plus mmu_lncRNA 121686-WT or -Mut plasmid. (D) The detection of relative luciferase activity after HK-2 cells co-transfected with miR-328-5p plus lncRNA 520657-WT or -Mut plasmid. (E) The FISH probe detection of co-localization of mmu_lncRNA 121686 and miR-328-5p in BUMPT cells. (F) The FISH probe detection of co-localization of hsa_lncRNA 520657 and miR-328-5p in HK-2 cells. (G and H) BUMPT cells were transfected with hsa_lncRNA 520657 sgRNA or plasmid and then treated with I/R (2 /2 h). The expression of miR-328-5p was examined by qRT-PCR. (I and J) BUMPT cells were transfected with mmu_lncRNA 121686 sgRNA or plasmid and then were treated with I/R (2/2 h). The expression of miR-328-5p was examined by qRT-PCR. Scale bar: 20 μM. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus scramble or control plasmid with saline group. #p < 0.05 versus scramble or control plasmid with IR group.
Overexpression of miR-328-5p attenuated I/R-induced BUMPT and HK-2 cell apoptosis
We explore the role of miR-328-5p in I/R-induced apoptosis in BUMPT and HK-2 cells. miR-328-5p mimic was transfected into BUMPT and HK-2 cells and then subjected to the I/R (2/2 h) treatment. The qRT-PCR analysis demonstrated the expression of miR-328-5p was significantly increased after transfection of the miR-328-5p mimic in HK-2 and BUMPT cells, respectively (Figure 6A). The FCM analysis showed that the miR-328-5p mimic significantly suppressed the I/R-induced HK-2 and BUMPT cell apoptosis (Figures 6B–6D), which was confirmed by immunoblot analysis of cleaved caspase-3 in BUMPT and HK-2 cells (Figures 6E–6H).
Figure 6.

I/R-induced HK-2 and BUMPT cell apoptosis was attenuated by the overexpression of miR-328-5p
BUMPT and HK-2 cells were transfected with miR-328-5p mimic or scramble and then subjected to I/R (2/2 h). (A) qRT-PCR analysis of the expression of miR-328-5p. (B and C) Flow cytometry analysis of BUMPT and HK-2 cell apoptosis. (D) Representative apoptosis rate of BUMPT and HK-2 cells. (E) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, and β-tubulin in BUMPT cells. (F) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, and β-tubulin in BUMPT cells. (G) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, and β-tubulin in HK-2 cells. (H) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, and β-tubulin in HK-2 cells. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus scramble group. #p < 0.05 versus I/R with scramble group.
HtrA3 is a target gene of miR-328-5p
To explore the mechanism of the anti-apoptotic role of miR-328-5p, we predicted the target gene of miR-328-5p. As shown in Figures 7A and 7C, miR-328-5p also binds to the sequence of 3′ UTR of HtrA3. The luciferase reporter analysis showed that miR-328-5p suppressed the luciferase activity of the HtrA3-WT-3’ UTR but not the HtrA3-Mut-3’ UTR (Figures 7B and 7D). To confirm the above finding, the miR-328-5p mimic was transfected into BUMPT and HK-2 cells and treated with I/R. The RT-PCR analysis showed that the miR-328-5p mimic significantly suppressed the expression of HtrA3 under basic and I/R treatment conditions (Figures 7E and 7H), which was further confirmed by the immunoblot analysis of HtrA3 (Figures 7F, 7G, 7I, and 7J). Taken altogether, these data suggest that HtrA3 is a direct target gene of miR-328-5p.
Figure 7.

HtrA3 is the target gene of miR-328-5p
(A and C) The complementary and mutated sequences of HtrA3 and miR-328-5p. (B and D) The luciferase activities after co-transfection with HtrA3-WT or HtrA3-Mut plus with or without miR-328-5p. (E) qRT-PCR analysis of HtrA3 expression after transfection with miR-328-3p or scramble under I/R status in BUMPT cells. (F) Immunoblots analysis of the expression of HtrA3 and β-tubulin in BUMPT cells. (G) Grayscale analysis of western blot bands of HtrA3 and β-tubulin in BUMPT cells. (H) qRT-PCR analysis of HtrA3 expression after transfection with miR-328-3p or scramble under I/R status in HK-2 cells. (I) Immunoblots analysis of the expression of HtrA3 and β-tubulin in HK-2 cells. (J) Grayscale analysis of western blot bands of HtrA3 and β-tubulin in HK-2 cells. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus HtrA3-Mut or scramble with saline group. #p < 0.05 versus IR with scramble group.
HtrA3 mediates I/R-induced BUMPT cell apoptosis
HtrA3 was involved in the regulation of cell proliferation, differentiation, and death in various cancers, including gastric, lung, and ovarian cancers.23 However, the role of HtrA3 in I/R-induced renal cell apoptosis has not been fully understood. We detected whether HtrA3 was induced by I/R in in HK-2 cells. The immunoblot results verified that the expression level of HtrA3 was gradually increased at 0 h after reperfusion, reached the peak at 2 h after reperfusion, and then declined at 4 h after reperfusion (Figures 8A and 8B). To confirm the role of HtrA3, we decided to KO HtrA3 in BUMPT and HK-2 cells. The single-guide RNA (sgRNA) targeted the sequence of HtrA3, embedded them into pSpCas9(BB)-2A-Puro (PX459) vectors, and then transfected into BUMPT and HK-2 cells followed by treatment with the puromycin at 1 μg/mL for selection of the positive cells (Figure 8C). The FCM analysis and immunoblot results showed that KO HtrA3 attenuated the I/R-induced BUMPT and HK-2 cell apoptosis and activation of caspase-3 (Figures 8D–8K). These data indicated that HtrA3 could promote I/R-induced BUMPT and HK-2 cell apoptosis.
Figure 8.

HtrA3 mediates I/R-induced BUMPT and HK-2 cell apoptosis
BUMPT and HK-2 cells were transfected with HtrA3 sgRNA, plasmid, or scramble and then were subjected to I/R (2/2 h). (A) Immunoblots analysis of HtrA3 expression at indicated time points after reperfusion. (B) Grayscale analysis of western blot bands. (C) Schema of Cas9 KO HtrA3. (D) Flow cytometry analysis of BUMPT cell apoptosis. (E) Representative apoptosis rate of BUMPT cells. (F) Immunoblots analysis of HtrA3, caspase 3, cleaved caspase-3, and β-tubulin. (G) Grayscale analysis of western blot bands of HtrA3, caspase 3, cleaved caspase-3, and β-tubulin. (H) Flow cytometry analysis of HK-2 cell apoptosis. (I) Representative apoptosis rate of HK-2 cells. (J) Immunoblots analysis of HtrA3, caspase-3, cleaved caspase-3, and β-tubulin. (K) Grayscale analysis of western blot bands of HtrA3, caspase-3, cleaved caspase-3, and β-tubulin in HK-2 cells. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus scramble with saline group. #p < 0.05 versus IR with sgRNA group.
miR-328-5p inhibitor reverses the protective role of KO of mmu-lncRNA 121686 and hsa-lncRNA 520657 during I/R-induced BUMPT and HK-2 cell apoptosis
To clarify whether mmu-lncRNA 121686 and hsa-lncRNA 520657 can mediate I/R-induced BUMPT and HK-2 cell apoptosis by sponging miR-328-5p, the miR-328-5p inhibitor was transfected into the BUMPT and HK-2 cells with deletion of mmu-lncRNA 121686 and hsa-lncRNA 520657. qRT-PCR analysis demonstrated that I/R suppressed the expression of miR-328-5p that was reversed by the KO of mmu-lncRNA 121686 or hsa-lncRNA 520657; however, this effect was diminished by the transfection of the miR-328-5p inhibitor (Figures 9A and 9F). Deletion of mmu-lncRNA 121686 and hsa-lncRNA 520657 significantly suppressed I/R-induced BUMPT and HK-2 cell apoptosis and decreased the expression levels of HtrA3 and cleaved caspase-3, which could be reversed by the miR-328-5p inhibitor (Figures 9B–9E and 9G–9J). These findings demonstrated that mmu-lncRNA 121686 and hsa-lncRNA 520657 also mediate I/R-induced BUMPT and HK-2 cell apoptosis by sponging miR-328-5p.
Figure 9.

KO of mmu_lncRNA 121686 and hsa_lncRNA 520657 attenuated I/R-induced apoptosis, and this effect was reversed by miR-328-5p inhibitor
The KO cell lines of mmu_lncRNA 121686 and hsa_lncRNA 520657 were transfected with miR-328-5p inhibitor and then subjected to I/R treatment. (A) qRT-PCR analysis of the expression of mmu-miR-328-5p. (B) Flow cytometry analysis of BUMPT apoptosis. (C) The apoptosis rate of BUMPT cells. (D) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, HtrA3, and β-tubulin in BUMPT cells. (E) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, HtrA3, and β-tubulin in BUMPT cells. (F) qRT-PCR analysis of the expression of hsa-miR-328-5p. (G) Flow cytometry analysis of HK-2 apoptosis. (H) The apoptosis rate of HK-2 cells. (I) Immunoblots analysis of the expression of cleaved caspase-3, caspase-3, HtrA3, and β-tubulin in HK-2 cells. (J) Grayscale analysis of western blot bands of cleaved caspase-3, caspase-3, HtrA3, and β-tubulin in HK-2 cells. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus scramble with saline group. #p < 0.05 versus I/R with scramble group. ▵p < 0.05 versus IR group with mmu_lncRNA 121686 or hsa_lncRNA 520657 KO group.
I/R-induced AKI is attenuated by PT-mmu-lncRNA 121686-KO mice
In order to verify whether mmu-lncRNA 121686 also plays the same role in vivo, we established PT-mmu-lncRNA 121686-KO mice. The littermates of male PT-mmu-lncRNA 121686-WT and PT-mmu-lncRNA 121686-KO mice were subjected to I/R (28 min/48 h). PT-mmu-lncRNA 121686-KO noticeably attenuated I/R-induced elevation in blood urine nitrogen and creatinine (Figure 10A and 10B), as well as tubular damage (Figures 10C and 10E) and tubular cell apoptosis (Figures 10D and 10F). The qRT-PCR analysis showed that the expression of mmu-lncRNA 121686 was decreased by PT-mmu-lncRNA 121686-KO under basic and I/R treatment conditions (Figure 10G). However, I/R-induced suppression of miR-328-5p was reversed by PT-mmu-lncRNA 121686-KO (Figure 10H). The immunoblot results demonstrated that PT-mmu-lncRNA 1212686-KO mice significantly attenuated I/R-induced expression of cleaved caspase-3 and HtrA3 (Figures 10I and 10J). Altogether, these data indicated that mmu-lncRNA 1212686 could mediate I/R-induced AKI in mice.
Figure 10.

mmu_lncRNA 121686 mediates I/R-induced apoptosis via miR-328-5p/HtrA3 axis
The bilateral renal arteries of PT-mmu_lncRNA 121686-WT and PT-mmu_LncRNA 121686-KO littermate mice were clamped for 28 min and then underwent reperfusion for 48 h. (A) BUN level. (B) Creatinine level. (C) H&E staining. (D) TUNEL staining. (E) Tubular damage score. (F) TUNEL-positive cells. (G) qRT-PCR analysis of the expression of mmu_lncRNA 121686. (H) qRT-PCR analysis of the expression of miRNA-328-5p. (I) Immunoblots analysis of HtrA3, caspase-3, cleaved caspase-3, and β-tubulin. (J) Grayscale analysis of western blot bands of HtrA3, caspase-3, cleaved caspase-3, and β-tubulin. Data are expressed as means ± SD (n = 6). ∗p < 0.05 versus PT-mmu_LncRNA 121686-WT with sham group. #p < 0.05 versus PT-mmu_LncRNA 121686-WT with I/R group.
mmu-lncRNA 121686 and hsa-lncRNA 520657 contain m6A sites that interact with METTL3
To explore upstream regulation mechanisms of mmu-lncRNA 121686 and hsa-lncRNA 520657, we focused on m6A RNA methylation, a predominant RNA modification that occurs in eukaryotic cells. As shown in Figures S3A, S3B, S4A, and S4B, we found that 3 and 2 out of 6 m6A sites in mmu-lncRNA 121686 and hsa-lncRNA 520657 sequences had very high scores through m6A-modification site prediction, respectively (http://www.dcuilab.cn/sramp). We hypothesized that m6A METTL3 could bind to the m6A sites of mmu-lncRNA 121686 and hsa-lncRNA 520657. The RNA immunoprecipitation (RIP) assay was used for experimental validation. Briefly, the RNA samples extracted from control, and I/R-exposed BUMPT and HK-2 cells were segmented into 100-bp fragments and then pulled down using anti-METTL3 antibody. qRT-PCR analysis verified that METTL3 could directly bind to 3 predicted m6A sites of mmu-lncRNA 121686 while binding to 2 predicted m6A sites of hsa-lncRNA 520657 (Figures S3C and S4C). Next, the WT and Mut luciferase reporter vectors of these 3 predicted m6A sites in BUMPT cells and 2 predicted m6A sites in HK-2 cells were co-transfected with or without METTL3 plasmid. The dual-luciferase reporter assay demonstrated that METTL3 significantly suppressed the luciferase activity of WT plasmids with m6A sites but not that of mutated m6A site plasmids (Figures S3D–S3I and S4D–S4G). To detect the stability of mmu-lncRNA 121686 and hsa-lncRNA 520657, scramble or METTL3-siRNA was transfected into BUMPT and HK-2 cells followed by actinomycin D at 5 μg/mL. The results indicated that knockdown METTL3 could suppress the stability of mmu-lncRNA 121686 and hsa-lncRNA 520657 at 2 and 3 h (Figures S3J and S4H). These data suggest METTL3 can interact with mmu-lncRNA 121686 and hsa-lncRNA 520657 m6A sites.
METTL3 mediates I/R-induced BUMPT and HK-2 cell apoptosis
Next, we hypothesized that METTL3 could promote I/R-induced apoptosis in BUMPT or HK-2 cell via the mmu-lncRNA 121686 or hsa-lncRNA 520657/miR-328-5p/HtrA3 axis, respectively. As shown in Figures S5A–S5G and S6A–S6F, METTL3 siRNA significantly suppressed I/R-induced BUMPT cell and HK-2 apoptosis, downregulated mmu-lncRNA 121686 and hsa-lncRNA 520657 expression, and decreased the levels of METTL3, cleaved caspase-3, and HtrA3. However, I/R-induced suppression of miR-328-5p was reversed by METTL3 siRNA (Figures S5A–S5G and S6A–S6F). On the contrary, these changes induced by I/R were further enhanced by the overexpression of METTL3 (Figures S5G–S5L and S6H–S6N). Collectively, these data reveal that METTL3 can promote I/R-induced BUMPT cell apoptosis.
The effect of overexpression of METTL3 aggravated I/R-induced AKI was diminished by PT-mmu-lncRNA 121686-KO mice
To verify whether METTLE3 drives the development of AKI via regulating lncRNA 121686, the littermates of PT-mmu-lncRNA 121686-WT and PT-mmu-lncRNA 121686-KO mice were subjected to I (28 min)/R(48 h) treatment with or without tail vein injection of METTL3 vector or METTL3 plasmid. We found that overexpression of METTL3 aggravated the I/R-induced decline of renal function, tubular damage, and renal cell apoptosis in PT-mmu-lncRNA 1212686 mice; by contrast, this effect was diminished in PT-mmu-lncRNA 121686-KO mice (Figures S7A–S7F). These data further support that METTL3 promoted the progression of AKI via mmu-lncRNA 121686.
IR-, VAN-, and CLP-induced AKIs are also ameliorated in PT-METTL3-KO mice via inhibition of mmu-lncRNA 121686/miR-328-5p/HtrA3 axis
Although we have illustrated the regulatory role of METTL3 in I/R-induced AKI, its roles in other types of AKIs, such as septic AKI and nephrotoxic AKI, remain unclear. Therefore, in the subsequent experiments, male PT-METTL3-WT littermates and PT-METTL3-KO mice were subjected to I(28minutes)/R(48 h), injection with VAN at a dose of 600 mg/kg for 7 days intraperitoneally, or performed with moderate CLP for 18 h as described previously.24,25 qRT-PCR analysis revealed that in I/R-, VAN-, and CLP-induced upregulation of mmu-lncRNA 121686, downregulation of miR-328-5p was noticeably reversed in PT-METTL3-KO mice (Figures S8A–S8F). The immunoblot results indicated that I/R-, VAN-, and CLP-induced overexpression of METTL3, HtrA3, and cleaved caspase-3 were significantly attenuated in PT-METTL3-KO mice (Figures S8J–S8L). Altogether, these data suggest that the METTL3/mmu-lncRNA 121686/miR-328-5p/HtrA3 axis not only promotes I/R-induced AKI but also mediates AKI induced by VAN and CLP.
METTL3 siRNA ameliorates I/R-, VAN-, and CLP-induced AKIs through suppression of mmu-lncRNA 121686/miR-328-5p/HtrA3 axis
To evaluate the therapeutic potential of METTL3 siRNA for AKIs, C57BL/6 mice were injected with 15 mg/kg METTL3 siRNA through tail vein before exposure to I/R, VAN, and CLP. The results showed that METTL3 siRNA significantly ameliorated I/R-, VAN-, and CLP-induced renal function damage, tubular morphological change, and tubular cell apoptosis (Figures S9, S10, and S11A–S11F). In addition, METTL3 siRNA reversed the upregulation of mmu-lncRNA 121686, METTL3, HtrA3, cleaved caspase-3 and downregulation of miR-328-5p caused by I/R-, VAN-, and CLP models (Figures S9, S10, and S11G–S11K). Altogether, these findings imply that METTL3 siRNA attenuates I/R-, VAN-, and CLP-induced AKIs via suppression of the mmu-lncRNA 121686/miR-328-5p/HtrA3 axis.
Discussion
The role of METTL3 in AKI remains controversial. In the present study, we reported that METTL3 promoted the progression of AKI. Furthermore, lncRNAs can be classified according to their distinct positional relationships with the coding gene, which in turn affect their functions.5 We provided evidence of the regulatory and functional roles of mmu-lncRNA 121686 in AKIs both in vitro and in vivo. mmu-lncRNA 121686 was upregulated in antimycin-treated BUPMT cells and a mouse I/R-induced AKI model. Mechanically, mmu-lncRNA 121686 sponged miR-328-5p to increase HtrA3 expression and induce BUMPT cell apoptosis. Interestingly, hsa-lncRNA 520657, a homolog with mmu-lncRNA 121686, also mediated HK-2 cell apoptosis via the miR-328-5p/HtrA3 axis. In addition, mmu-lncRNA 121686 and hsa-lncRNA 520657 could be regulated by METTL3. Collectively, our findings indicate that the METTL3/mmu-lncRNA 121686/hsa-lncRNA 520657/miR-328-5p/HtrA3 axis plays a pivotal role in the progression of AKIs.
Several lncRNAs have been reported to be associated with kidney injury and repair.7 In the present study, we also demonstrated that 19 lncRNAs were upregulated in the kidney cortex of ischemic mouse at 48 h (fold change>7). Among these, the upregulation of mmu-lncRNA 121686 was the most prominent in ischemic AKI mice, which was further confirmed in antimycin-treated BUMPT cells (Figure 3). hsa-lncRNA 520657 has a homology with mmu-lncRNA 121686. It was also found that mmu-lncRNA 121686 and hsa-lncRNA 520657 could mediate I/R-induced BUMPT and HK-2 cell apoptosis (Figure 4). The above findings initially suggested that hsa-lncRNA 520657 might mediate the progression of patients with AKI; however, these effects needed to be confirmed in kidney from patients with AKI in a future study. In light of our earlier findings that TCONS_00016233 promoted septic AKI by acting as a molecular sponge of the anti-apoptotic miR-22-3p,26 we postulated that mmu-lncRNA 121686 or hsa-lncRNA 520657 also inhibited the expression of an anti-apoptotic miRNA. This hypothesis was initially verified by identifying miR-328-5p as the target miRNA, and the co-localization between miR-328-5p and mmu-lncRNA 121686 or hsa-lncRNA 520657 was confirmed by dual-luciferase reporter assays (Figure 5). In addition, we found that miR-328-5p was not only negatively regulated by mmu-lncRNA 121686 in vitro and vivo under normal and ischemic injury conditions but also negatively regulated by hsa-lncRNA 520657 in HK-2 cells with or without ischemic injury (Figures 4 and 5). Furthermore, the anti-apoptotic roles of mmu-lncRNA 121686 and hsa-lncRNA 520657 were reversed by miR-328-5p inhibitor (Figure 9).
Recent research has provided evidence that many miRNAs take part in different phenotypes of AKI by inducing renal cell apoptosis.7 Although miR-328-5p is the target miRNAs of mmu-lncRNA 121686, while miR-328-5p is one of the target miRNAs of hsa-lncRNA 520657, the role and regulation mechanism of miR-328-5p in AKIs remain unclear. In this study, we reported that miR-328-5p had an anti-apoptotic role, and this was supported by the evidence that the miR-328-5p mimic suppressed BUMPT and HK-2 cell apoptosis during antimycin treatment (Figure 6). Moreover, we found that HtrA3 was a direct target of miR-328-5p, which was supported by the following evidence. Firstly, miRNA target prediction analysis indicated that miR-328-5p could bind to the 3’ UTR of HtrA3 (Figure 7). Secondly, dual-luciferase reporter assays showed that miR-328-5p suppressed the luciferase activity of HtrA3-WT but not HtrA3-MUT (Figure 7). HtrA3 is a highly conserved serine protease that belongs to the Htra family. Recent studies of different pathological conditions, such as lung cancer, pancreatic cancer, and neurodegenerative diseases, provided evidence that HtrA3 could promote cell death.23,27,28,29,30 In addition, a study reported that HtrA3 suppressed the hypoxia-reoxygenation-induced inflammation response.31 However, whether HtrA3 can induce renal cell apoptosis has not been discovered yet during ischemic injury. The results demonstrated that the deletion of HtrA3 attenuated BUMPT and HK-2 cell apoptosis during ATP-depletion injury (Figure 8), thereby supporting its pro-apoptotic role.
lncRNAs share the similar structure of mRNAs, and over 160 kinds of RNA epigenetic modifications, including the predominant RNA m6A methylation, exist in eukaryotic cells.32 The m6A-modification site prediction showed that 3 and 2 out of 6 m6A sites in mmu-lncRNA 121686 and hsa-lncRNA 520657 sequences were found to have high m6A scores, respectively. In addition, METTL3 was the most well-known methyltransferase of m6A RNA methylation.33,34,35,36,37,38 Hence, we assumed that METTL3 could bind to these 3 or 2 m6A sites with high scores. This is supported by the following facts. Firstly, RIP was performed to test this hypothesis, and the RT-PCR results clearly demonstrated that METTL3 directly bound to the m6A sites of mmu-lncRNA 121686 and hsa-lncRNA 520657 (Figures S3A–S3C and S4A–S4C). Secondly, the dual-luciferase reporter assay further verified that METTL3 significantly suppressed the luciferase activity of the WT plasmids of mmu-lncRNA 121686 and hsa-lncRNA 520657 m6A sites but not mutated m6A site plasmids (Figures S3D–S3I and S4D–S4G). In addition, METTL3 could enhance the stability of mmu-lncRNA 121686 and hsa-lncRNA 520657(Figures S3J and S4H). Overexpression of METTL3 enhanced I/R-induced ischemic injury was diminished by PT-mmu-lncRNA 121686-KO (Figure S7). Therefore, METTL3 was considered an upstream molecule of mmu-lncRNA 121686 and hsa-lncRNA 520657.
Furthermore, the roles of METTL3 in nephrotoxicity AKI remain controversial. A previous study reported that METTL3 prevented colistin-induced AKI;22 by contrast, it enhanced cisplatin-induced HK-2 cell apoptosis.39 The above-mentioned data suggested that the role of METTL3 in apoptosis was associated with the stimulation factors. To clarify the function of it, VAN-induced nephrotoxicity AKI was used in current study. The data suggested that PT-METTL3-KO or METTL3 siRNA significantly alleviated the VAN-induced AKI (Figure2B, 2E, 2K–2N, S8B, S8E, S8I, S8J, and S10), which is consistent with cisplatin-induced AKI. In addition, we investigated the roles of METTL3 in I/R- and CLP-induced AKI. Firstly, we found that I/R induced the expression of METTL3 both in vitro and in vivo (Figure 1). Secondly, our data further demonstrated that I/R induced BUMPT cell apoptosis via the mmu-lncRNA 121686/miR-328-5p/HtrA3 signaling pathway, and this axis was enhanced and attenuated by METTL3 overexpression and knockdown, respectively (Figure S5 and S6). Thirdly, I/R- and CLP-induced AKIs were significantly attenuated in PT-METTL3-KO mice via the mmu-lncRNA 121686/miR-328-5p/HtrA3 axis (Figures S8A, S8C, S8D, S8F–S8H, S8K, and S8L). Finally, METTL3 siRNA also noticeably suppressed I/R- and CLP-induced AKI by downregulating mmu-lncRNA 121686/miR-328-5p/HtrA3 (Figures S9–S11).
In conclusion, our data reveal that the regulatory pathway METTL3/mmu-lncRNA 121686/miR-328-5p/HtrA3 is responsible for the progression of I/R-, CLP-, and VAN-induced AKIs. Moreover, METTL3/hsa-lncRNA 520657/miR-328-5p/HtrA3 is responsible for human renal tubular cell apoptosis during the in vitro ischemic AKI model. The data also suggest that METTL3 is a promising therapeutic target for AKI.
Materials and methods
Antibodies and reagents
Anti-METTL3 (ab195352) were purchased from Abcam (Cambridge Science Park, Cambridge, UK). Anti-Caspase3 (9662) and cleaved capase-3 (9664) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-METTL3 and anti-β-tubulin antibodies were purchased from Proteintech (Rosemont, IL, USA). All secondary antibodies were obtained from Affinity (Affinity). The calcium ionophore (MCE, HY-N6687) was purchased from MCE company (Shanghai, China). Antimycin A (ab141904) was obtained from Abcam (Cambridge Science Park, Cambridge, UK).
Establishment of PT-METTL3-KO and PT-mmu-lncRNA 121686-KO mouse models
To substantiate the role of tubular METTL3 and mmu-lncRNA 121686 in AKIs, proximal tubule-specific METTL3 and mmu-lncRNA 121686 KO mouse models were established. The detailed procedures are described in Figures S1 and 2A. Specifically, male floxed mice harboring METTL3 alleles (METTL3f/f XY) and mmu-lncRNA 121686 alleles (mmu-lncRNA 121686 f/f XY) were crossed with female phosphoenolpyruvate carboxy kinase-cAMP response element (PEPCK-Cre) transgenic mice (METTL3+/+ or mmu-lncRNA 121686 XcreXcre) to produce the first generation. Later, the heterozygous female offspring (METTL3 or mmu-lncRNA 121686 f/+XcreX) were crossed with METTL3 or mmu-lncRNA 121686 f/fXcreY males to create littermate mice with proximal tubule-specific METTL3 or mmu-lncRNA 121686 WT (PT-METTL3 or mmu-lncRNA 121686-WT) and PT-METTL3 or mmu-lncRNA 121686-KO (METTL3 or mmu-lncRNA 121686f/fXcreY). Genotyping of mice was identified using 3 sets of PCR: (1) the 290- or 366-bp DNA fragment METTL3 or mmu-lncRNA 121686 floxed allele amplification (Figure S1B, lanes 5 and 6, or S2B, lanes 3 and 6), respectively; (2) deficiency of the 234- or 302-bp DNA fragment WT allele amplification (Figure S1B, lanes 1 and 2, or S2B, lanes 1 and 5), respectively; and (3) the 370-bp DNA fragment of Cre gene amplification (Figure S1B, lanes 2, 3, 5, and 6, or S2B, lanes 3, 4, and 6). Then, PT-METTL3 or mmu-lncRNA 121686-WT and PT-METTL3 or mmu-lncRNA 121686-KO littermate mice were subjected to I/R (28 min/48 h) treatment. Quantitative immunoblot analysis demonstrated that the expression of METTL3 was downregulated in the kidney cortical tissues of PT-METTL3-KO mice compared with PT-METTL3-WT mice under normal and ischemic injury conditions, which was further confirmed by the immunofluorescence staining results (Figures S1C–S1F). In addition, the qRT-PCR analysis demonstrated that the expression of METTL3 was downregulated in the kidney cortical tissues of PT-mmu-lncRNA 121686-KO mice compared with PT-mmu-lncRNA 121686-WT mice under normal and ischemic injury conditions (Figure S2C). These data revealed that tubular METTL3 and mmu-lncRNA 121686 were successfully deleted in PT-METTL3-KO mice.
The mouse AKI models
C57BL/6J mice were purchased from Hunan SJA Laboratory Animal Company (Hunan, China). The METTL3 (flox/flox) mice (Cyagen) and lncRNA 121686 (flox/flox) mice (Shanghai Model Organisms Center) were crossed with PEPCK-Cre mice (provided by Volker Haase), and then mice were produced with the proximal tubule-specific METTL3 deletion according to a previous description.40 Male mice (8–10 weeks) were subjected to ischemia, CLP, and VAN nephrotoxic AKIs. For ischemic AKI, the bilateral renal artery was continuously clipped for 28 min followed by reperfusion for 24 or 48 h. The body temperature of mice was maintained at approximately 37°C ± 0.5°C throughout the surgery. For CLP-induced AKI, the cecum was exposed and tightly ligated 1.5 cm from the tip with 4–0 silk thread, which was then followed by puncture for 18 h.25 For VAN-induced AKI, mice were intraperitoneally injected with a single dose of VAN at a dose of 600 mg/kg for 7 consecutive days as described previously.24,41 In addition, the C57BL/6 mice were injected with METTL3-siRNA (at a dose of 15 mg/kg) via the tail vein twice a week, and saline was used as an injection control. The C57BL/6 mice were injected with METTL3 plasmid (at a dose of 25 μg) via the tail vein once every 3 weeks, and the same vector was used as an injection. All animal experiments followed the guiding principles approved by the Animal Care Ethics Committee of Second Xiangya Hospital, People’s Republic of China (no. 2018065). Mice were housed in a 12 h light/dark environment and had free access to a standard rodent diet and clean water.
Cell culture and in vitro ischemia reperfusion model
BUMPT cells were cultured with DMEM (Gibco, 11965092) with an added 10% FBS (Gibco, 10100147) and 1% penicillin-streptomycin (Gibco, 15140163) at 37°C in 5% CO2. HK-2 cells were cultured with DMEM/F12 (Gibco, 11320033) with an added 10% FBS (Gibco, 10100147) and 1% penicillin-streptomycin (Gibco, 15140163) at 37°C in 5% CO2. For the ischemic cell model, BUMPT and HK-2 cells were treated with 10 μM antimycin A (an inhibitor of mitochondrial complex III, Abcam, ab141904) and 1.5 μM calcium ionophore (MCE, HY-N6687) in Hanks’ balanced salt solution (Hyclone, SH30030.02) for 2 h as previously described.42 The DMEM medium was replaced with HBSS at 0–4 h after reperfusion. Several plasmids or siRNA or inhibitor were created and then transfected into BUMPT cells by Lipofection 2000. The plasmids of lncRNA 121686 and METTL3 were obtained from HonorGene(Changsha,Hu nan, China). METTL3 siRNA (mouse): 5’- TCGGACACGTGGAGCTCTA-3’; METTL3 siRNA (human): 5’- CTGCAAGTATGTTCACTATGA-3’; HtrA3 siRNA: 5’-GCAGTGGAATATATGTTCA-3’. The culture medium was replaced with DMEM at 8 h after transfection.
Non-homologous end joining (NHEJ)-based CRISPR KO
To induce gene silence, corresponding sgRNAs of mmu_LncRNA 121686, hsa-lncRNA 520657, and HtrA3 were designed and constructed into the pSpCas9(BB)-2A-Puro (PX459) vectors by Han Biotechnology (Changsha, Hunan Province, China), respectively. Their plasmids were transfected into BUMPT and HK-2 cells using Lipofection 2000 (Invitrogen) for 6 h and then were replaced with normal medium for 48 h. The puromycin at 1 μg/mL was added into cell culture medium to select positive clones. HtrA3 (mouse) gRNA:GCTCCAGCACTGCCTCCGGC; HtrA3 (human) gRNA:CCGCGCGCTGCGACGTGTC; has_lncRNA 520657 gRNA1 AAAAAGGATTGAGCAAAGAC; gRNA2 GTTGCGCCCCCA GCAGCGAG; mmu_lncRNA 121686 gRNA1 GCTGCAGCCAGACTCCTAGC; gRNA2 CGG CTTATCTTCTGGGCTGC.
FISH
The fluorescent probes of LncRNA 121686 were designed and purchased from Ruibo (Guangzhou, China), the fluorescent probes of mmu_LncRNA 121686 and miR-328-5p were purchased from GenePharma (Shanghai, China), and then they were performed according to the instructions of the in situ hybridization kit (C10910, Ruibo). The probes of U6, 18S, Lnc121686, and miR-328-5p were added separately to the confocal dishes and the prepared frozen sections. Probe hybridization was incubated overnight at 37°C, and then we washed the dishes with 2×SSC and used DAPI for nuclear staining. The confocal microscope was used to observe the staining.
FCM
BUMPT and HK-2 cells following different treatment were collected by using trypsin without EDTA and washed three times with PBS, then operated following the protocol of the FITC Annexin V Apoptosis Detection Kit (BD, 556547). Briefly, we performed cell staining for 1 h before the FCM test according to the instructions. Cells were added with FITC for 15 min and then PI for 5 min at room temperature, and finally tested by the FCM.
Immunofluorescence
For immunofluorescence staining, the cells were incubated with specific primary antibody (METTL3 1:200) overnight at 4°C followed by a secondary fluorescent antibody in the dark for 1 h at 37°C. The nuclear marker DAPI was added for 3–5 min. The cells were observed using fluorescent microscopy.
Relative qPCR
Total RNA was extracted from BUMPT cells, HK-2 cells, or kidney tissue using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and then reverse transcripted into first-strand cDNA using the Prime Script RT Reagent kit and gDNA Eraser (TaKaRa, RR037A) according to previous descriptions.43,44,45,46 In addition, for the cell cytoplasm and nucleus RNA separation, fractionation and purification of BUMPT cells were carried according to the Cytoplasmic & Nuclear RNA Purification Kit (Norgen, #21000, 37400) instruction. Briefly, BUMPT cells were lysed with Lysis Buffer J before centrifugation, and then the RNAs were extracted and separated into parts of cytoplasmic and nuclear. The separated RNAs were then purified for the further detection. Real-time qPCR was carried with Bio-Rad (Hercules, CA, USA) IQ SYBR green supermix with Opticon (MJ Research, Waltham, MA, USA) following the manufacturer’s instructions. The primers used were as follow: mmu_LncRNA 121686: 5’-GTGTCGGAACTCACCTGTGG-3’ (forward) and 5’-CTTGCTTGGGAGGCTCATCG-3’ (reverse); METTL3: 5’-CACGCTGCCTCCGATGTTGATC-3’(forward) and 5’-ATGGACTGTTCCTTGGCT GTTGTG-3’ (reverse); HtrA3: 5’-TCTGAGTTCCAAAACAAGCATG-3’ (forward) and 5’-TCCTCTCTGAGAAGGTGAATTG-3’ (reverse); GAPDH: 5’-CGTGCCGCCTGGAGAAACC -3’ (forward) and 5’- TGGAAGA GTGGGAGTTGCTGTTG-3’ (reverse). U6 primers were described in a previous report.43 The relative quantification was performed by determining ΔΔCt values.
The staining of H&E, TUNEL, and immunochemistry and immunoblotting
Renal tissue was embedded in paraffin and then cut into sections for various types of staining according to a previous description.47 Histology was assessed by H&E staining in accordance with the criteria of score renal tubular injury as described previously.47,48 The renal cell apoptosis was evaluated using TUNEL staining. The quantitative indicator of apoptosis was used with the proportion (%) of TUNEL-positive cells in 10 to 20 microscopic fields per tissue section. For immunochemistry staining, tissue sections were incubated with specific primary antibody (METTL3 1:200) overnight at 4°C and then incubated with secondary antibody for 30 min at 37°C and reacted with DAB for 5 to 10 min. Protein lysates from BUMPT cells or kidneys were harvested and then centrifuged to collect the supernatant containing the proteins. The supernatant was subjected to SDS-PAGE and then transferred to a PVDF membrane. The membranes were then incubated with primary antibody (METTL3 1:1,000, caspase-3 1:1,000, cleaved caspase-3 1:1,000, HtrA3 1:1,000, β-tubulin 1:2,000) overnight at 4°C followed by a secondary antibody for 1 h at room temperature.
Luciferase reporter assays
The luciferase reporter analysis was performed as previously described.43 The miRNA activity was evaluated by inserting luciferase gene (luc2) into pmirGLO double-luciferase miRNA target expression vector. Luciferase vectors containing responsive elements of HtrA3-3’ UTR (WT HtrA3) or mmu_lncRNA 121686 (WT lncRNA 121686) interacting with miR-328-5p and Mut plasmids of HtrA3 (Mut HtrA3) or mmu_lncRNA 121686 (Mut lncRNA 121686) lacking responsive elements were used, respectively. PGMLR-TK luciferase reporter was used as control vector. All plasmids were constructed by Tsingke Biotechnology (Beijing, China). In short, the plasmids of WT mmu_lncRNA 121686, WT HtrA3, Mut mmu_lncRNA 121686, Mut HtrA3, and miR-328-5p mimics were transfected into BUMPT and HK-2 cells for 6 h. Then, the luciferase activity was measured with a Dual-Luciferase Reporter Assay System kit (Promega, E1910) and normalized according to mmu_lncRNA 121686-Mut or hsa_lncRNA 520657-Mut activity.
RNA stability
HK-2 and BUMPT cells were transfected with scramble and METTL3 siRNA, and then we added actinomycin D (Sigma, SBR00013) at a dose of 5 μg/mL. The total RNA was extracted using Trizol and then was detected by qRT-PCR for the expression of mmu_lncRNA 121686 and hsa_lncRNA 520657 at different time points.
Statistical analyses
The two-groups comparison was done with two-tailed Student’s t tests. The multiple-group comparison was done with one-way or two-way ANOVA. The Kruskal-Walls test was used for the data with non-normal distributions. GraphPad software 8.0 was used to analyze the data. Quantitative data were expressed as mean ± SD. p <0.05 was considered statistically significant.
Acknowledgments
The study was supported in part by a grant from National Natural Science Foundation of China (81870475, 81570646, 82171088, and 81770692); the Changsha Science and Technology Bureau project (kq2001039); the Key Project of Hunan Provincial Science and Technology innovation (2020SK1014); the Department of Science and Technology of the Hunan Province Project of International Cooperation and Exchanges (2020WK2009), the Hunan Key Research and Development Program(2021SK2034); and the Natural Science Foundation of Hunan Province(2021JJ30935) Fundamental Research Funds for the Central Universities of Central South University (2020zzts282 and 2021zzts0362). This work was also supported by the Hunan Provincial Innovation Foundation for Postgraduate (CX20200290 and CX20210364) and the China Hunan Provincial Science and Technology Department (2021SK4004). Cardiovascular Multidisciplinary Integration Research Fund (2021-N-15).
Author contributions
D.Z. conceived and designed the experiments; J.P., Y.X., and H.L. carried out the experiments; X.L.,X.L, J.Z., X.T., Z.H., and Z.P. analyzed the data; H.Z., Y.L., X.X., J.C., and Y.Y. contributed reagents/materials/analysis tools; D.Z. wrote the main manuscript text, but all authors reviewed the manuscript.
Declaration of interest
The authors have declared that no conflict of interest exists.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.07.014.
Supplemental information
References
- 1.Ronco C., Bellomo R., Kellum J.A. Acute kidney injury. Lancet. 2019;394:1949–1964. doi: 10.1016/s0140-6736(19)32563-2. [DOI] [PubMed] [Google Scholar]
- 2.Gammelager H., Christiansen C.F., Johansen M.B., Tønnesen E., Jespersen B., Sørensen H.T. One-year mortality among Danish intensive care patients with acute kidney injury: a cohort study. Crit. Care. 2012;16:R124. doi: 10.1186/cc11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masewu A., Makulo J.R., Lepira F., Amisi E.B., Sumaili E.K., Bukabau J., Mokoli V., Longo A., Nlandu Y., Engole Y., et al. Acute kidney injury is a powerful independent predictor of mortality in critically ill patients: a multicenter prospective cohort study from Kinshasa, the Democratic Republic of Congo. BMC Nephrol. 2016;17:118. doi: 10.1186/s12882-016-0333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linkermann A., Chen G., Dong G., Kunzendorf U., Krautwald S., Dong Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014;25:2689–2701. doi: 10.1681/asn.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopp F., Mendell J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomson D.W., Dinger M.E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z., Wang Y., Shu S., Cai J., Tang C., Dong Z. Non-coding RNAs in kidney injury and repair. Am. J. Physiol. Cell Physiol. 2019;317:C177–C188. doi: 10.1152/ajpcell.00048.2019. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y., Qiu J., Chen B., Lin Y., Chen Y., Xie G., Qiu J., Tong H., Jiang D. Long non-coding RNA NEAT1 plays an important role in sepsis-induced acute kidney injury by targeting miR-204 and modulating the NF-κB pathway. Int. Immunopharmacol. 2018;59:252–260. doi: 10.1016/j.intimp.2018.03.023. [DOI] [PubMed] [Google Scholar]
- 9.Ding Y., Guo F., Zhu T., Li J., Gu D., Jiang W., Lu Y., Zhou D. Mechanism of long non-coding RNA MALAT1 in lipopolysaccharide-induced acute kidney injury is mediated by the miR-146a/NF-κB signaling pathway. Int. J. Mol. Med. 2018;41:446–454. doi: 10.3892/ijmm.2017.3232. [DOI] [PubMed] [Google Scholar]
- 10.Geng X., Song N., Zhao S., Xu J., Liu Y., Fang Y., Liang M., Xu X., Ding X. LncRNA GAS5 promotes apoptosis as a competing endogenous RNA for miR-21 via thrombospondin 1 in ischemic AKI. Cell Death Discov. 2020;6:19. doi: 10.1038/s41420-020-0253-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang X., Li D., Shen W., Shen X., Liu Y. LncRNA NEAT1 promotes hypoxia-induced renal tubular epithelial apoptosis through downregulating miR-27a-3p. J. Cell. Biochem. 2019;120:16273–16282. doi: 10.1002/jcb.28909. [DOI] [PubMed] [Google Scholar]
- 12.Yu T.M., Palanisamy K., Sun K.T., Day Y.J., Shu K.H., Wang I.K., Shyu W.C., Chen P., Chen Y.L., Li C.Y. RANTES mediates kidney ischemia reperfusion injury through a possible role of HIF-1α and LncRNA PRINS. Sci. Rep. 2016;6:18424. doi: 10.1038/srep18424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian X., Ji Y., Liang Y., Zhang J., Guan L., Wang C. LINC00520 targeting miR-27b-3p regulates OSMR expression level to promote acute kidney injury development through the PI3K/AKT signaling pathway. J. Cell. Physiol. 2019;234:14221–14233. doi: 10.1002/jcp.28118. [DOI] [PubMed] [Google Scholar]
- 14.Xue Q., Yang L., Wang H., Han S. Silence of long noncoding RNA SNHG14 alleviates ischemia/reperfusion-induced acute kidney injury by regulating miR-124-3p/MMP2 Axis. Biomed. Res. Int. 2021;2021:8884438. doi: 10.1155/2021/8884438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haddad G., Kölling M., Wegmann U.A., Dettling A., Seeger H., Schmitt R., Soerensen-Zender I., Haller H., Kistler A.D., Dueck A., et al. Renal AAV2-mediated overexpression of long non-coding RNA H19 attenuates ischemic acute kidney injury through sponging of microRNA-30a-5p. J. Am. Soc. Nephrol. 2021;32:323–341. doi: 10.1681/ASN.2020060775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L., Xu J.Y., Tan H.B. LncRNA TUG1 regulates the development of ischemia-reperfusion mediated acute kidney injury through miR-494-3p/E-cadherin axis. J. Inflamm. 2021;18:12. doi: 10.1186/s12950-021-00278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu D., Liu Y., Zheng X., Liu N. c-MYC-induced long noncoding RNA MEG3 aggravates kidney ischemia-reperfusion injury through activating mitophagy by upregulation of RTKN to trigger the Wnt/β-catenin pathway. Cell Death Dis. 2021;12:191. doi: 10.1038/s41419-021-03466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng X., Su R., Stanford S., Chen J. Critical enzymatic functions of FTO in obesity and cancer. Front. Endocrinol. 2018;9:396. doi: 10.3389/fendo.2018.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taranenko L.D., Medvedenko A.F., Sinepupov N.A., Bondarev V.I., Belonenko G.A. [Diagnosis and treatment of acute pancreatitis] Klin. Med. 1985;63:102–106. [PubMed] [Google Scholar]
- 20.Frye M., Harada B.T., Behm M., He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–1349. doi: 10.1126/science.aau1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer K.D., Jaffrey S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J., Ishfaq M., Xu L., Xia C., Chen C., Li J. METTL3/m(6)A/miRNA-873-5p attenuated oxidative stress and apoptosis in colistin-induced kidney injury by modulating keap1/Nrf2 pathway. Front. Pharmacol. 2019;10:517. doi: 10.3389/fphar.2019.00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bowden M.A., Drummond A.E., Fuller P.J., Salamonsen L.A., Findlay J.K., Nie G. High-temperature requirement factor A3 (Htra3): a novel serine protease and its potential role in ovarian function and ovarian cancers. Mol. Cell. Endocrinol. 2010;327:13–18. doi: 10.1016/j.mce.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Xu L., Li X., Zhang F., Wu L., Dong Z., Zhang D. EGFR drives the progression of AKI to CKD through HIPK2 overexpression. Theranostics. 2019;9:2712–2726. doi: 10.7150/thno.31424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Y., Liu B., Pan J., Liu J., Li X., Li H., Qiu S., Xiang X., Zheng P., Chen J., et al. MBD2 mediates septic AKI through activation of PKCη/p38MAPK and the ERK1/2 Axis. Mol. Ther. Nucleic Acids. 2021;23:76–88. doi: 10.1016/j.omtn.2020.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang P., Yi L., Qu S., Dai J., Li X., Liu B., Li H., Ai K., Zheng P., Qiu S., et al. The biomarker TCONS_00016233 drives septic AKI by targeting the miR-22-3p/AIFM1 signaling Axis. Mol. Ther. Nucleic Acids. 2020;19:1027–1042. doi: 10.1016/j.omtn.2019.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J., Feng M., Liu D., Liu H., Shi M., Zhang J., Qu J. Antagonism between HTRA3 and TGFβ1 contributes to metastasis in non-small cell lung cancer. Cancer Res. 2019;79:2853–2864. doi: 10.1158/0008-5472.Can-18-2507. [DOI] [PubMed] [Google Scholar]
- 28.Wenta T., Rychlowski M., Jurewicz E., Jarzab M., Zurawa-Janicka D., Filipek A., Lipinska B. The HtrA3 protease promotes drug-induced death of lung cancer cells by cleavage of the X-linked inhibitor of apoptosis protein (XIAP) Febs j. 2019;286:4579–4596. doi: 10.1111/febs.14977. [DOI] [PubMed] [Google Scholar]
- 29.Li Y., Gong L., Qi R., Sun Q., Xia X., He H., Ren J., Zhu O., Zhuo D. Paeoniflorin suppresses pancreatic cancer cell growth by upregulating HTRA3 expression. Drug Des. Devel. Ther. 2017;11:2481–2491. doi: 10.2147/dddt.S134518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skorko-Glonek J., Zurawa-Janicka D., Koper T., Jarzab M., Figaj D., Glaza P., Lipinska B. HtrA protease family as therapeutic targets. Curr. Pharm. Des. 2013;19:977–1009. doi: 10.2174/1381612811319060003. [DOI] [PubMed] [Google Scholar]
- 31.Gong Q., Shen Z.M., Sheng Z., Jiang S., Ge S.L. Hsa-miR-494-3p attenuates gene HtrA3 transcription to increase inflammatory response in hypoxia/reoxygenation HK2 Cells. Sci. Rep. 2021;11:1665. doi: 10.1038/s41598-021-81113-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coker H., Wei G., Brockdorff N. m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 2019;1862:310–318. doi: 10.1016/j.bbagrm.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Shi H., Wei J., He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell. 2019;74:640–650. doi: 10.1016/j.molcel.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H., Hu X., Huang M., Liu J., Gu Y., Ma L., Zhou Q., Cao X. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat. Commun. 2019;10:1898. doi: 10.1038/s41467-019-09903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y., Xie L., Wang M., Xiong Q., Guo Y., Liang Y., Li J., Sheng R., Deng P., Wang Y., et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 2018;9:4772. doi: 10.1038/s41467-018-06898-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y., Gao M., Zhu F., Li X., Yang Y., Yan Q., Jia L., Xie L., Chen Z. METTL3 is essential for postnatal development of brown adipose tissue and energy expenditure in mice. Nat. Commun. 2020;11:1648. doi: 10.1038/s41467-020-15488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W., Shao F., Yang X., Wang J., Zhu R., Yang Y., Zhao G., Guo D., Sun Y., Wang J., et al. METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N(6)-methyladenosine-dependent YTHDF binding. Nat. Commun. 2021;12:3803. doi: 10.1038/s41467-021-23501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dorn L.E., Lasman L., Chen J., Xu X., Hund T.J., Medvedovic M., Hanna J.H., van Berlo J.H., Accornero F. The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. 2019;139:533–545. doi: 10.1161/circulationaha.118.036146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou P., Wu M., Ye C., Xu Q., Wang L. Meclofenamic acid promotes cisplatin-induced acute kidney injury by inhibiting fat mass and obesity-associated protein-mediated m(6)A abrogation in RNA. J. Biol. Chem. 2019;294:16908–16917. doi: 10.1074/jbc.RA119.011009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X., Pan J., Li H., Li G., Liu X., Liu B., He Z., Peng Z., Zhang H., Li Y., et al. DsbA-L mediated renal tubulointerstitial fibrosis in UUO mice. Nat. Commun. 2020;11:4467. doi: 10.1038/s41467-020-18304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X., Pan J., Li H., Li X., Fang F., Wu D., Zhou Y., Zheng P., Xiong L., Zhang D. Atg7 mediates renal tubular cell apoptosis in vancomycin nephrotoxicity through activation of PKC-δ. Faseb j. 2019;33:4513–4524. doi: 10.1096/fj.201801515R. [DOI] [PubMed] [Google Scholar]
- 42.Li X., Pan J., Li H., Li G., Liu B., Tang X., Liu X., He Z., Peng Z., Zhang H., et al. DsbA-L interacts with VDAC1 in mitochondrion-mediated tubular cell apoptosis and contributes to the progression of acute kidney disease. EBioMedicine. 2022;76:103859. doi: 10.1016/j.ebiom.2022.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ge Y., Wang J., Wu D., Zhou Y., Qiu S., Chen J., Zhu X., Xiang X., Li H., Zhang D. lncRNA NR_038323 suppresses renal fibrosis in diabetic nephropathy by targeting the miR-324-3p/DUSP1 Axis. Mol. Ther. Nucleic Acids. 2019;17:741–753. doi: 10.1016/j.omtn.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi L., Ai K., Li H., Qiu S., Li Y., Wang Y., Li X., Zheng P., Chen J., Wu D., et al. CircRNA_30032 promotes renal fibrosis in UUO model mice via miRNA-96-5p/HBEGF/KRAS axis. Aging (Albany NY) 2021;13:12780–12799. doi: 10.18632/aging.202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ai K., Li X., Zhang P., Pan J., Li H., He Z., Zhang H., Yi L., Kang Y., Wang Y., et al. Genetic or siRNA inhibition of MBD2 attenuates the UUO- and I/R-induced renal fibrosis via downregulation of EGR1. Mol. Ther. Nucleic Acids. 2022;28:77–86. doi: 10.1016/j.omtn.2022.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ai K., Pan J., Zhang P., Li H., He Z., Zhang H., Li X., Li Y., Yi L., Kang Y., et al. Methyl-CpG-binding domain protein 2 contributes to renal fibrosis through promoting polarized M1 macrophages. Cell Death Dis. 2022;13:125. doi: 10.1038/s41419-022-04577-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang D., Liu Y., Wei Q., Huo Y., Liu K., Liu F., Dong Z. Tubular p53 regulates multiple genes to mediate AKI. J. Am. Soc. Nephrol. 2014;25:2278–2289. doi: 10.1681/ASN.2013080902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J., Pan J., Li H., Long J., Fang F., Chen J., Zhu X., Xiang X., Zhang D. lncRNA ZEB1-AS1 was suppressed by p53 for renal fibrosis in diabetic nephropathy. Mol. Ther. Nucleic Acids. 2018;12:741–750. doi: 10.1016/j.omtn.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.