

Abstract
Hemophilia A gene therapy targets hepatocytes to express B domain deleted (BDD) clotting factor VIII (FVIII) to permit viral encapsidation. Since BDD is prone to misfolding in the endoplasmic reticulum (ER) and ER protein misfolding in hepatocytes followed by high-fat diet (HFD) can cause hepatocellular carcinoma (HCC), we studied how FVIII misfolding impacts HCC development using hepatocyte DNA delivery to express three proteins from the same parental vector: (1) well-folded cytosolic dihydrofolate reductase (DHFR); (2) BDD-FVIII, which is prone to misfolding in the ER; and (3) N6-FVIII, which folds more efficiently than BDD-FVIII. One week after DNA delivery, when FVIII expression was undetectable, mice were fed HFD for 65 weeks. Remarkably, all mice that received BDD-FVIII vector developed liver tumors, whereas only 58% of mice that received N6 and no mice that received DHFR vector developed liver tumors, suggesting that the degree of protein misfolding in the ER increases predisposition to HCC in the context of an HFD and in the absence of viral transduction. Our findings raise concerns of ectopic BDD-FVIII expression in hepatocytes in the clinic, which poses risks independent of viral vector integration. Limited expression per hepatocyte and/or use of proteins that avoid misfolding may enhance safety.
Graphical abstract
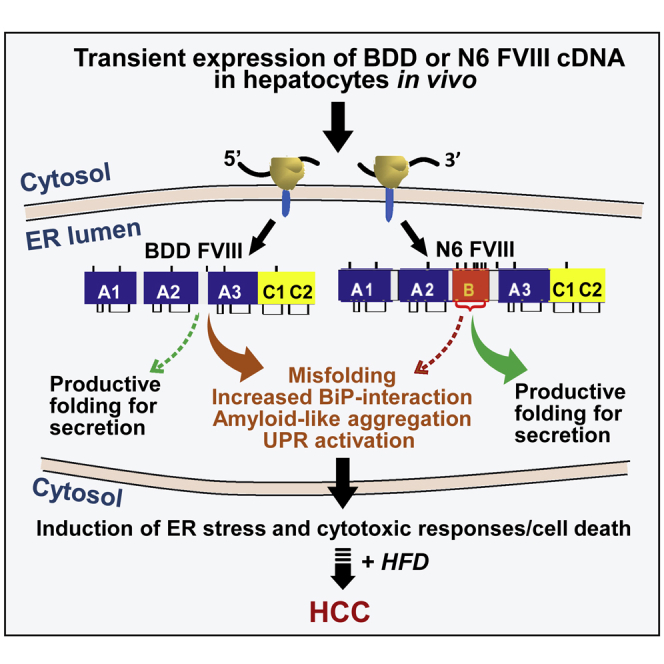
Hemophilia A gene therapy targets hepatocytes to express B domain deleted (BDD) factor VIII, which misfolds and causes stress in the ER. It is shown herein that transient hepatocyte BDD expression with subsequent high-fat diet induces liver tumors in mice, signifying a safety concern for hemophilia A gene therapy.
Introduction
Hemophilia A (HA) is an X chromosome-linked bleeding disorder affecting 24.6 per 100,000 males at birth that results from deficiency of clotting factor VIII (FVIII).1 Protein replacement therapy with recombinant FVIII has significantly reduced morbidity and mortality associated with HA, although concerns remain. First, anti-FVIII inhibitory antibodies develop in ∼25% of patients.2 Second, recombinant FVIII protein bears a high cost with limited availability due to low and variable production from mammalian host cells.3 FVIII secretion from mammalian cells is inefficient, partly due to FVIII protein misfolding, aggregation, and retention in the endoplasmic reticulum (ER).4 Misfolded FVIII activates the unfolded protein response (UPR) to resolve defective protein folding. However, upon chronic protein misfolding in the ER, the UPR shifts to an apoptotic program.
FVIII is a 330-kDa glycoprotein composed of three major domains (A1-A2-B-A3-C1-C2).5 It is primarily produced by liver sinusoidal endothelial cells.6 The amino acid (aa) sequences in the A and C domains exhibit 40% aa identity between species, whereas the ∼900 aa sequences within the large B domain show no homology other than a conserved large number (18) of N-linked oligosaccharides that are also observed in the B domains of FVIII from different species, as well as in the homologous clotting factor V. B domain-deleted FVIII (BDD),7,8 A1-A2-A3-C1-C2 (Figure 1), is functional and similar to Refacto, used for protein replacement therapy for HA in the clinic (i.e., SQ-BDD-FVIII; Pfizer).10,11,12 The only difference between BDD and SQ-BDD is that SQ-BDD contains an extra 14 aa linker (SFSQNPPVLKRHQR) at the junction between the A2 and A3 domains.13,14 Subsequent codon optimization improved SQ-BDD production leading to its use in ongoing HA gene therapy clinical studies.13,14,15,16,17 SQ-BDD is used for gene therapy via adeno-associated viral (AAV) vectors because the size of full-length FVIII is beyond the packaging limit of the vector genome, which is <5 kb.16 However, BDD exhibits similar misfolding as intact FVIII, activates the UPR, and leads to hepatocyte death upon in vivo DNA vector delivery to hepatocytes in mice.9 Besides BDD, we described a partial B domain deletion molecule, herein (N6), that retains an additional 226 aa with 6 N-linked glycosylation sites18 (Figure 1), and is secreted almost 10-fold more efficiently than full-length FVIII or BDD,9 presumably because the 6 N-glycans engage the intracellular lectin chaperone machinery, that was demonstrated to enhance FVIII trafficking from the ER to the Golgi compartment.19,20,21,22,23,24 Thus, N6 is secreted more efficiently and causes less UPR activation than BDD (Figure 1).
Figure 1.

Hepatocyte responses to wtFVIII, BDD, and N6 upon hydrodynamic tail vein injection of vector DNA into mice
Relative FVIII secretion efficiency, activation of the UPR, production of reactive oxygen species (ROS), induction of apoptosis, and the ability for antioxidant butylated hydroxyanisole (BHA) to improve secretion were shown by us previously.9 a1-3 = acidic regions. N-glycosylation sites (N-gly) and disulfide bonds (s-s) are depicted. Red bracket indicates residues from the B domain of wtFVIII that were retained in N6.
Presently, there are numerous ongoing liver-directed gene therapy clinical studies to deliver codon-optimized SQ-BDD via AAV to hepatocytes in men with severe HA. Initial results appeared to be very successful, with correction of FVIII levels into the normal range during the first year.14,15 Unfortunately, elevated liver enzyme levels were observed in the first year, and at a 5-year25 follow-up, FVIII expression levels gradually declined to the lower end of the therapeutic range, starting in the second year.26,27,28,29 While immune response to the AAV vector is known to play an important role in liver pathology and the decline in FVIII expression in the clinical setting,17,30 it is possible that FVIII misfolding and UPR activation impact hepatocyte function, health, and/or survival. Therefore, there is an urgent need to understand mechanisms underlying inefficient FVIII secretion26,27,28 and the long-term pathologic effect of ectopic expression of FVIII in hepatocytes in vivo.
Previously, we demonstrated feeding a high-fat diet (HFD) to mice that transiently over-express liver-specific urokinase-type plasminogen activator (uPA)-transgenic mice, which misfolds in the ER, initiates development of non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma (HCC).31 Thus, it appears that either transient expression of a misfolded protein in the ER that activates the UPR followed by an HFD can initiate HCC development or there are unique properties of uPA that drive HCC progression. These findings led us to test whether transient hepatocyte expression of FVIII molecules with different folding efficiencies in the ER may also promote HCC in mice.
Here we studied the long-term outcome of transient expression of BDD, which aggregates in the cell and activates the UPR,32 and N6 that displays reduced aggregation and UPR activation.9,18 We delivered BDD and N6 expression vectors to murine livers by tail vein hydrodynamic DNA injection. These mice were then fed an HFD for 65 weeks starting at 1 week after vector DNA injection when FVIII expression was undetectable. We observed greater tumor formation in BDD vector-injected mice than N6 vector-injected mice. For the first time these findings support the notion that the degree of ER protein misfolding may be a significant factor in liver disease progression. Our findings point to a potential risk regarding ectopic expression of FVIII in hepatocytes in the context of HA gene therapy.
The potential for hepatic gene transfer to contribute to formation of HCC continues to be widely debated.33,34 For instance, a recent study found that AAV gene transfer to adult murine livers may cause HCC when animals receive an HFD.35 Hence, the combination of low-grade viral vector integration with the diet-induced hepatic steatosis and injury to the liver raises the risk of liver cancer formation. Herein, we found that even transient expression of a protein that is prone to misfolding and induction of ER stress (BDD-FVIII) in hepatocytes of adult mice is a trigger for HCC formation when followed by an HFD diet in the absence of viral delivery. Thus, this fundamentally new concept of protein expression-derived cellular stress predisposing to malignancy when followed by diet-induced inflammation is independent of molecular events caused by viral vectors. Importantly, the risk for HCC formation was reduced when a variant of FVIII was expressed that shows enhanced folding and secretion.
Results
FVIII forms aggregates that resolve at 1-2 h following energy repletion
Stable expression of BDD and N6 FVIII in Chinese hamster ovary (CHO) cells demonstrated that N6 expression was tolerated at an ∼10-fold greater level than BDD.18 We previously characterized full-length FVIII aggregation by filtration of cell lysates through nitrocellulose (NC) membranes, which retain all cell proteins, and cellulose acetate (CA) membranes, which only retain proteins that have β-sheet aggregate structures.32 Lysate proteins retained on membranes were probed with FVIII or β-actin antibodies. Filtration through NC membranes demonstrated that intracellular steady state levels of N6 were ∼5-fold greater than BDD, although secretion of N6 was significantly higher than BDD (Figure 2A, red box). Filtration through CA membranes demonstrated significant aggregation of BDD and >4-fold less aggregation for N6 (ratio of CA/NC), although N6 was expressed at a ∼5-fold greater level than BDD (Figure 2A, blue box). Following energy depletion by treatment with 2-deoxyglucose (2-DG) and sodium azide (NaN3) to inhibit glycolysis and oxidative phosphorylation, respectively, FVIII aggregates accumulated in CHO cells that stably express wild-type FVIII (wtFVIII) (∼500 mU/mL per 106 cells/day, not shown),32 BDD (∼1 U/mL per 106 cells/day), and N6 (∼10 U/mL per 106 cells/day). We previously demonstrated that 2-DG, but not NaN3, was sufficient to induce wtFVIII aggregation.32 It is notable that NaN3 inhibition of oxidative phosphorylation is irreversible. After 2-DG removal and glucose replenishment, BDD and N6 aggregates began to disappear at 1 to 2 h, in a manner that did not require de novo protein synthesis, as addition of the protein synthesis elongation inhibitor cycloheximide (CHX) did not alter aggregate dissolution or significantly reduce secretion of functional FVIII activity (Figure 2A).
Figure 2.

Reversible aggregation of BDD and N6 expressed in CHO cells
(A) FVIII aggregates induced by glucose depletion begin to resolve at 1 h following glucose repletion. CHO cells that express BDD or N6 were treated with either normal media or glucose-free media containing 10 mM 2DG and 20 mM sodium azide (2DG + NaN3). After 2 h, cells were harvested or allowed to recover in complete media for the indicated times, or complete media with 10 μg/mL CHX. Cell lysates were filtered through nitrocellulose (left) or cellulose acetate (CA) (right) membranes and probed with FVIII or β-actin antibodies. Analysis of β-actin is from BDD-expressing CHO cells. Red box = steady state levels of BDD and N6 in untreated cells. Blue box = aggregation of FVIII before and after 2DG + NaN3 treatment. (B) Reversible retention and secretion of active FVIII in CHO cells. 35S-Met/Cys pulse-chase CHO cells were treated in parallel as in (A). CHO cells were pulse-labeled for 20 min and then chased for 20 min with media containing excess unlabeled Met/Cys to complete synthesis of nascent chains (lane 1) before being treated with either normal media (lanes 2–3) or glucose-free media containing 10 mM 2DG and 20 mM NaN3. After 2 h, cells were harvested (lane 4) or allowed to recover in complete media for increasing times (lanes 5–8), or complete media with CHX (lane 9). Lysates and media were collected at indicated time points for FVIII IP and reducing SDS-PAGE. For FVIII aPPT activity assay of media, cells were treated in parallel, but not pulse-labeled. Lanes: 1: Untreated, 20′ chase; 2: Untreated 120′ chase; 3: Untreated 240′ chase; 4–9: 2DG for 120′; 5: 2-DG + 30′ recovery; 6: 2-DG + 60′ recovery; 7: 2DG + 120′ recovery; 8: 2DG + 240′ recovery; 9: 2DG + 240′ recovery + CHX. (C) FVIII activity in the medium via aPTT analysis of BDD and N6 expressed in CHO cells. All results (A–C) were from the same set of experiments performed in parallel.
To identify the destiny of aggregated BDD and N6, we performed 35S-Met/Cys-pulse-chase labeling which confirmed that glucose deprivation retained BDD and N6 within the cell. Importantly, upon glucose repletion, intracellular aggregated BDD and N6 dissolved and were efficiently secreted into the medium (Figure 2A). Analysis of FVIII activity in the conditioned media from cells treated under the same conditions as the pulse-chase assay demonstrated that the secreted FVIII is functional (Figure 2C). Increasing amounts of functional FVIII appeared in the media as early as 1 h following glucose repletion for BDD and at 2 h for N6, which correlated with the rate of disappearance of intracellular aggregated FVIII. Although CHX treatment did not affect the amount of previously metabolically labeled FVIII secreted into the medium following glucose repletion, CHX treatment reduced secreted FVIII activity to 30% to 60% (Figure 2C), presumably due to inhibition of new FVIII synthesis. In sum, these results (Figures 2A–2C) indicate that a major percent of both BDD and N6 form metastable aggregates that can resolve to produce folded, functional, and secreted FVIII.
Ectopic FVIII expression in hepatocytes causes protein aggregation in murine livers
Previously we demonstrated that full-length wtFVIII forms amyloid-like fibrils upon expression in cultured cells.32 Hydrodynamic tail vein injection of vector DNA is an efficient method to express exogenous genes in hepatocytes.18,36 Using this technique, we confirmed expression of exogenous FVIII in hepatocytes of mice injected with BDD or N6 vector DNA induced the UPR, although the stress response to N6 expression was significantly attenuated.9 To determine if the stress response to FVIII expression in hepatocytes is associated with protein aggregation in vivo, we performed thioflavin-S (Thio-S) staining on the liver sections to specifically identify amyloid-like protein aggregates. The livers of BDD vector-injected mice showed significantly more Thio-S positivity than N6 vector-injected mice, evidenced by colocalization of Thio-S staining with FVIII immunostaining (Figure 3). Similar to BDD expression in CHO cells,32 these findings demonstrate that ectopic expression of BDD in hepatocytes in vivo also leads to intracellular accumulation of amyloid-like FVIII aggregates in the liver. As expected, neither FVIII nor Thio-S stains were observed in mice that received the parental vector pMT expressing the cytosolic well-folded enzyme dihydrofolate reductase (DHFR) (Figure 3).
Figure 3.

BDD expression in hepatocytes in vivo causes accumulation of amyloid-like aggregates that colocalize with FVIII in liver sections
Liver sections were prepared from mice injected with the indicated vector DNAs as described9 and stained for FVIII (red) and Thio-S (green).32 Shown are representative images from these analyses. Details of the vector constructs are depicted with plasmid maps on the left-hand side. The parental vector pMT37 (Addgene) contains a transcription unit that is composed of the SV40 origin and enhancer element from the early promoter, the adenovirus major late promoter containing most of the tripartite leader present in adenovirus late mRNAs, a hybrid intron (IVS), the cytosolic dihydrofolate reductase (DHFR)-coding region, and the SV40 early polyadenylation signal (SV40 PolyA). The parental vector efficiently expresses DHFR.37 Human BDD or N6 coding sequences were inserted upstream of the DHFR-coding region at the EcoR1 restriction sites (R1).
BDD misfolds in murine hepatocytes in vivo
To provide mechanistic insight into BDD misfolding and UPR activation, we characterized BDD and N6 interaction with the ER chaperone BiP/GRP78. Misfolded proteins bind BiP, whereas well-folded proteins do not.38,39 Therefore, protein interaction with BiP can be used as a surrogate to measure ER protein misfolding. However, there are no available antibodies that can quantitatively co-immunoprecipitate (IP) BiP-client protein complexes. Hence, we created a genetically modified mouse with a 3x FLAG tag inserted into the endogenous BiP/GRP78/Hspa5 locus (BiP-Flag mice) for quantitative isolation of BiP-client protein complexes using anti- FLAG magnetic beads,40 Tagging BiP with 3x FLAG in this manner did not alter BiP function or BiP expression, which is essential to measure physiological interactions (Figures 4A and 4B40,41). Importantly, hepatocyte-specific BiP-Flag displays the same expression level and is regulated identically as the untagged wild-type BiP (Figure 4A and 4B). To elucidate the folding status of BDD and N6 ectopically expressed in murine hepatocytes, we expressed BDD and N6 in hepatocytes of BiP-Flag homozygous (BiP-Flag+/+) mice by hydrodynamic tail vein delivery of vector DNA and analyzed the interactions between BiP-Flag and BDD or N6 through anti-FLAG co-IP of BDD and N6 (Figure 4C). Consistent with our previous observations,9 hepatic BDD levels in BDD vector-injected mice were significantly higher than those in the N6 vector-injected mice (Figure 4C, lanes 5–10 versus lanes 11–16 and Figure 4D, left panel), probably due to the greater retention of BDD in the ER. Importantly, anti-FLAG IP pulled down a significant portion of BDD from the livers of BDD vector-injected BiP-Flag+/+ mice (25%; Figure 4C, lanes 24–26 versus 8–10 and Figure 4D, right panel), while N6 was pulled down at a significantly lesser level (16%; Figure 4C, lanes 24–26 versus 30–32 versus 14–16 Figure 4D, right panel). These results support the notion that BDD exhibits greater misfolding than N6 in hepatocytes of murine livers, which likely accounts for the different degrees of UPR induction.9
Figure 4.

Epitope-tagging of the endogenous murine BiP/GRP78/Hspa5 locus demonstrates misfolding of BDD in vivo
(A and B) A diagram illustrating location of insertion of 3x FLAG tag to C-terminal region of BiP. (B) Western blot demonstrating similar changes in hepatic levels of endogenous wtBiP (BiP) and BiP-Flag in mice heterozygous for BiP-Flag allele (10 days after activation of BiP-Flag expression in the liver) in response to treatment with tunicamycin for 24 h. (C) Co-IP of BDD (lanes 24–26) and N6 (lanes 30–32) from liver lysates of pMT-BDD or pMT-N6-injected BiP-Flag homozygous mice (BiP-Flag+/+) with anti-FLAG magnetic beads. Hepatic level of BDD and N6 were determined by direct western blot on liver lysates (lanes 5–10 and lanes 11–16 for BDD and N6, respectively). Red asterisk = N6 FVIII bands. (D) Quantification of BDD and N6 bands shown in (C). Data are represented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.001. Each lane in the images for western blots represents an individual mouse, i.e., the number of mice (n) in each group: n = 2 for pMT/Wt and pMT/BiP-Flag; n = 3 for pMT-BDD/Wt, pMT-BDD/BiP-Flag, pMT-N6/Wt, and pMT-N6/BiP-Flag, respectively.
Lentiviral transduction of BDD into human primary hepatocytes activates the UPR
We next asked whether BDD expression can activate the UPR in human hepatocytes. We used mouse-passaged human primary hepatocytes (mpPHHs)42 to investigate the cellular response to ectopic BDD expression. The mpPHHs were transduced in vitro with increasing amounts of a lentiviral vector that expresses a codon-optimized SQ-BDD cDNA13 that is used in all current liver-directed HA gene therapy trials.14,15,17 Western blot analysis demonstrated SQ-BDD expression induced UPR markers BiP, eIF2α phosphorylation, and CHOP, despite its weak signal, at 10 days after infection (Figure 5). Interestingly, the protein levels of two UPR-induced genes in the liver, hepcidin43 and cysteine rich with epidermal growth factor like domains 2 (CRELD2), an oncogene associated with HCC that promotes survival upon ER stress,44,45,46,47 were increased in BDD-expressing mpPHH (Figure 5). These results for the first time show that ectopic BDD expression induces ER stress in human hepatocytes.
Figure 5.

BDD expression induces the UPR in mouse-passaged primary human hepatocytes (mpPHH)
mpPHH were transduced with a lentiviral vector expressing a codon-optimized form of SQ-BDD (Lenti-BDD) at the indicated vector doses and cultured in a hepatocyte-defined medium. The transduced mpPHHs were harvested for western blot analysis 10 days post-transduction. Each lane represents an individual culture well. The numerical numbers represent averaged fold changes after correction with loading control β-actin. Experiment was repeated twice with different batches of mpPHH and similar results were obtained.
HCC development correlates with the degree of ER protein misfolding
Previous findings suggest protein misfolding in the ER can initiate NASH and HCC development.31 Therefore, we tested whether two different human FVIII variants that misfold to different degrees can initiate HCC development. C57BL/6J mice were subjected to hydrodynamic injection of parental vector pMT, pMT-BDD, or pMT-N6 vector DNA.9 After 24 h, plasma levels of N6 FVIII were significantly higher than those for BDD-FVIII (Figures 6A and S1A), consistent with our previous observations.9 After 1 week, when FVIII expression was not detectable (Figures S1B–S1D), mice were fed a 60% HFD for 65 weeks and then analyzed for liver surface tumors. No tumors, neither adenomas nor adenocarcinomas, were observed in mice that received parental vector pMT DNA that expressed the cytosolic DHFR. In contrast, all mice that received pMT-BDD expression vector developed tumors, while reduced tumor formation was observed in mice that received pMT-N6 vector (Figure 6B). All mice in the pMT-BDD group had one liver surface tumor, whereas a couple of tumors were observed in three mice from the pMT-N6 group.
Figure 6.

Transient FVIII expression in hepatocytes causes liver tumors in mice in the context of high-fat diet (HFD) feeding
Wild-type C57BL/6J male mice were injected with pMT vector, pMT-N6 or pMT-BDD DNA vectors by hydrodynamic tail vein injection.
(A) Plasma FVIII levels at 24 h after vector DNA injection. Data shown are mean ± SE (n = 4 for pMT and pMT-BDD, n = 5 for pMT-N6). (B) Incidence of liver tumors in pMT-BDD- or pMT-N6-injected mice after a 65-week-HFD treatment. The difference between the two groups (i.e., pMT-N6, n = 12 and pMT-BDD, n = 10) were tested for statistical significance using the χ2 test (degrees of freedom = 1, 95% confidence intervals). (C) HCC but not adenoma exhibited positivity for glutamine synthase staining. Both tumor tissue (T) and non-tumor tissue (N) are shown in the high-magnification images.
The tumors (both adenomas and carcinomas) in the above mice were evaluated and extensively characterized independently by three clinical pathologists. A detailed summary of this histopathological analysis is provided in Tables S1–S3. First, reticulin staining is intended to demonstrate reticular fibers surrounding tumor cells. Tumors that retain a reticulin network are generally benign or pre-neoplastic, whereas HCC loses the reticulin fiber normal architecture. Second, immunohistochemistry staining of CD34, glutamine synthetase (GS), β-catenin, glypican 3, and CD44 were also performed to differentiate the adenomatoid dysplastic nodule from the HCC lesions. Although no tumors were detected in any of the 13 mice injected with parental vector pMT, we observed that 58% (7 of 12) of mice that received pMT-N6 vector developed liver tumors; in contrast to 100% (10 of 10) of mice that received pMT-BDD vector (Figure 6B, p < 0.02, χ2 test). The findings suggest a higher penetrance of tumors in the presence of BDD misfolded protein. Detailed histopathological assessment of the pMT-BDD-injected mice demonstrated that six of the 10 lesions were adenomas and four carcinomas (Figure 6B), while the pMT-N6-injected mice developed four adenomas and three carcinomas (Figure S2A). CD34 demonstrates neovascularization in HCC, while nontumorous hepatic sinusoids do not stain with CD34.47 The adenomas were overall negative for CD34, whereas the carcinomas displayed a patchy strong CD34-positive staining (Figure S3), further confirming the malignancy.48,49 GS stains positively for zone 3 of the liver parenchyma. Most carcinomas had positive GS staining (Figure 6C), indicative of an early-stage HCC.50 However, a few well-differentiated HCCs and an early evolving HCC, arising from adenomas, were GS negative. As expected, β-catenin immunostaining showed identical results to GS, as they are both frequently overexpressed in early HCC.50 Finally, the adenomas in the BDD cohort were positive for CD44, with the sinusoidal lymphocytes staining while the adenomas in the N6 cohort were negative (Figure S3). Based on these histopathological assessments, the distribution of adenomas and carcinomas appears to be similar in both groups (Figures 6C and S2A). However, BDD mice developed more tumors and thus a more aggressive phenotype. In addition, the staining intensity and cellular distribution of the tumor makers differed considerably in the liver tumors observed in our mice (Table S3), indicating that these tumors were at different stages of development toward HCC. It is important to note that none of the tumors found in these mice expressed BDD or N6, as demonstrated by the analysis of DNA isolated from the liver tumors and their adjacent non-tumor tissues collected at the study endpoint (Table S4), providing evidence supporting the notion that transient expression of misfolded FVIII, over the first week after DNA delivery, in the hepatocyte combined with HFD feeding is sufficient to induce HCC.
Discussion
Hepatic gene transfer of codon-optimized SQ-BDD-FVIII cDNA using AAV vectors has emerged as a promising therapeutic approach for HA.14,15 Substantial improvement in hemostasis was documented at a 5-year follow-up,25 although the approach has encountered two key hurdles: (1) A requirement for very high vector doses to drive FVIII expression in hepatocytes; and (2) in addition to signs of liver damage, transgene expression declined over time.26,27,28,29 Both may be directly linked to FVIII misfolding and retention in the ER. Durability of transgene expression is a major factor for HA gene therapy, as it is safely and effectively treated by prophylaxis with recombinant FVIII as well as FVIII-bypassing molecules such as a bispecific antibody29 raising the safety and efficacy bars for HA gene therapy regimens. The significance of this problem was recently highlighted.26,51,52 Here, we demonstrate that cellular stress resulting from transient expression of FVIII in hepatocytes may contribute to liver tumorigenesis. Encouragingly, we find that a more efficiently secreted FVIII variant can be developed to reduce this risk. While there are differences in the biology of human and murine HCC and AAV gene transfer differs from plasmid vectors, these outcomes warrant further investigations into the potential for gene therapy approaches to cause tumors in patients with HA.
Our previous work identified an FVIII derivative, N6, that is more efficiently secreted and less prone to aggregation than present B-domain deletion molecules used for HA gene therapy.9,18 In addition to the increased secretion efficiency of N6 compared with BDD, N6 elicits reduced cellular toxicity, as measured by UPR activation, apoptosis, levels of reactive oxygen species (ROS), protection by antioxidant butylated hydroxyanisole (BHA) treatment, and HCC (Figure 1).9,18,53,54 Using mice with an FLAG tag inserted into the endogenous BiP/Hspa5 locus for quantitative FLAG IP of BiP-client protein complexes,40 we found that BDD displays greater interaction with BiP compared with N6, i.e., a quarter of intrahepatocellular BDD binds BiP (Figure 3D), indicative of misfolding. The BiP-Flag mouse provides the ability to evaluate the misfolding propensity of FVIII derivatives considered for gene therapy. Ectopic expression of BDD produces a higher level of misfolded FVIII, which can induce a series of cytotoxic responses, including activation of the UPR,9 ultimately leading to initiation of HCC development.
Our findings suggest that characterization of the folding efficiency, host response, and safety in model organisms and non-human primates is essential to ensure safety over the lifetime of an individual, given that HCC takes years to develop in humans. A case of HCC did occur in a hemophilia B patient after hepatic AAV-mediated factor IX gene transfer,55 which was unlikely caused by gene therapy, as it occurred early after gene transfer and the patient had multiple risk factors that predisposed to HCC. While HCC formation was observed in murine studies with AAV vectors using other transgenes,35 this was attributed to insertional mutagenesis and the relevance of this observation to the safety of HA gene therapy remains to be understood. However, our new results show that high levels of FVIII gene expression by itself, even if transient and very likely independent of insertional events, as our studies did not use viral vectors, can induce HCC in the context of an HFD. All murine models of NASH and HCC progression require a hypercaloric diet, presumably to induce an inflammatory environment.56,57 HCC has not been observed in animals treated with AAV-FVIII vectors, but these animals were not subjected an HFD challenge. Interestingly, neonatal AAV gene transfer (expressing other transgenes) led to HCC under a normal dietary regimen but transduction of the same AAV vectors to adult mice did not induce HCC unless they were fed an HFD leading to non-alcoholic fatty liver disease (NAFLD),35 which was associated with increased inflammation and hepatocyte proliferation, consistent with our findings. Nevertheless, it should be considered that our studies do not reflect the course of liver disease in humans, nor the therapeutic approach. For instance, we used DNA delivery in our mouse studies because we previously characterized detailed responses to BDD versus N6.9 It is of great importance to investigate if the amount of FVIII produced by hepatocytes under a setting of AAV gene transfer reaches a threshold required to set off the cascade of molecular events that promotes HCC. Long-term follow-up in patients who have high levels of FVIII expression will be required to answer this question.
Our studies also support the need for careful monitoring of liver function over the time course of gene therapy treatment. We demonstrate the role of diet when expressing a therapeutic protein in the liver that is prone to misfolding and ER stress. While AAV-FVIII gene transfer has not been found to cause the level of hepatocyte apoptosis and liver injury that may occur with DNA vectors, multiple studies have shown induction of ER stress markers in mice in response to AAV-FVIII,58,59,60 and liver injury (albeit of unknown origin) occurred in patients treated with high-dose AAV-FVIII.14,15 We now directly show that expression of SQ-BDD in primary human hepatocytes induces ER stress and the UPR in the dose-dependent manner (Figure 5). Together, our study provides experimental evidence demonstrating the need for rigorous scientific investigation toward the pathophysiological consequences upon AAV-mediated FVIII expression in hepatocytes. HCC is the most common primary liver cancer and the third leading cause of cancer deaths worldwide.61 The incidence of HCC has tripled since 1980 and is the most increasing cause of cancer mortality in the United States.62 While the incidence of viral-related HCC is declining, HCC related to metabolic stress is on the rise, due to the global increase in obesity and the associated metabolic syndrome.63 It is currently estimated that NASH, which is already the leading cause of liver transplants in developed countries, will become the dominant HCC etiology. A quarter of the world’s population suffers from NAFLD characterized by abnormal lipid accumulation in hepatocytes. Approximately 20% of NAFLD patients eventually develop NASH with inflammation, hepatocyte ballooning, Mallory Denk Bodies, and cell death, a subset of which further develop fibrosis, cirrhosis, and HCC.64 Both ER stress and activation of the UPR are documented in many different human diseases,65,66 including NASH and HCC.67,68,69 The prevalence of NASH in the general population may increase the risk for HCC associated with the hepatocyte-targeted gene therapy.
In conclusion, we provide the first evidence that even transient hepatic expression of a protein that is prone to misfolding in the ER to induce the UPR, can trigger HCC formation when followed by a second insult to the liver such as an HFD diet. While these findings in mice using DNA vectors do not necessarily mean that humans treated with viral vectors in current clinical approaches are at increased risk of cancer formation, limited levels of expression of such proteins in hepatocytes and/or use of better secreted variants should be considered in clinical development. Importantly our findings demonstrate that a factor, independent of viral toxicity, can be contributed by protein misfolding in the ER.
Materials and methods
Cell lines
Parental Chinese hamster ovary cells (CHO-K1) and 2 CHO-K1 clones were engineered for constitutive BDD (CHO-BDD) or N6 (CHO-N6) expression were previously described.18
Reagents and standard methods
All reagents and standard methods are specified in supplemental methods (available on the Molecular Therapy website).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 and a two-tailed Student’s t test. Chi-square test was used for comparing of tumor incidence between pMT-BDD and pMT-N6 groups. A p value <0.05 was regarded as statistically significant (p < 0.05 ∗, p < 0.01 ∗∗).
Acknowledgments
This work was supported by NIH grants CA198103 and DK113171 to R.J.K. and in part by the NIDDK-funded San Diego Digestive Diseases Research Center (P30DK 120515). Portions of this work were supported by NIH P01HL160472 (to R.W.H., R.J.K., and Y.P.D.J.). C.L. was supported by NIH training grant T32DK007494. We thank Takeda Pharmaceuticals U.S.A., Inc., for providing us with the Sepharose-coupled and uncoupled monoclonal anti-human FVIII antibodies for this work.
Author contributions
R.J.K. conceived the project, designed, and interpreted experiments, and wrote the manuscript with Z.C. and A.K.L. A.K.L., Z.C., R.D., C.L., L.G., J.M., J.Y., and C.Z. performed various in vivo and in vitro experiments. Z.G., A.L., and P.M. provided expert consultation and supervision on tumor analyses. Y.P.D.J. and R.W.H. interpreted data and edited the manuscript. R.J.K. supported the study.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.10.004.
Supplemental information
References
- 1.Seaman C.D., Xavier F., Ragni M.V. Hemophilia A (factor VIII deficiency) Hematol. Oncol. Clin. North Am. 2021;35:1117–1129. doi: 10.1016/j.hoc.2021.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Meeks S.L., Batsuli G. Hemophilia and inhibitors: current treatment options and potential new therapeutic approaches. Hematol. Am. Soc. Hematol. Educ. Program. 2016;2016:657–662. doi: 10.1182/asheducation-2016.1.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang R., Monroe T., McRogers R., Larson P.J. Manufacturing challenges in the commercial production of recombinant coagulation factor VIII. Haemophilia. 2002;8(Suppl 2):1–5. doi: 10.1046/j.1351-8216.2001.00115.x. [DOI] [PubMed] [Google Scholar]
- 4.Pipe S.W. The promise and challenges of bioengineered recombinant clotting factors. J. Thromb. Haemost. 2005;3:1692–1701. doi: 10.1111/j.1538-7836.2005.01367.x. [DOI] [PubMed] [Google Scholar]
- 5.Pittman D.D., Kaufman R.J. Structure-function relationships of factor VIII elucidated through recombinant DNA technology. Thromb. Haemost. 1989;61:161–165. [PubMed] [Google Scholar]
- 6.Everett L.A., Cleuren A.C.A., Khoriaty R.N., Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123:3697–3705. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toole J.J., Pittman D.D., Orr E.C., Murtha P., Wasley L.C., Kaufman R.J. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc. Natl. Acad. Sci. USA. 1986;83:5939–5942. doi: 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pittman D.D., Alderman E.M., Tomkinson K.N., Wang J.H., Giles A.R., Kaufman R.J. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993;81:2925–2935. [PubMed] [Google Scholar]
- 9.Malhotra J.D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S.W., Kaufman R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lusher J.M., Lee C.A., Kessler C.M., Bedrosian C.L., ReFacto Phase 3 Study Group The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia. 2003;9:38–49. doi: 10.1046/j.1365-2516.2003.00708.x. [DOI] [PubMed] [Google Scholar]
- 11.Lusher J.M., Roth D.A. The safety and efficacy of B-domain deleted recombinant factor VIII concentrates in patients with severe haemophilia A: an update. Haemophilia. 2005;11:292–293. doi: 10.1111/j.1365-2516.2005.01099.x. [DOI] [PubMed] [Google Scholar]
- 12.Di Paola J., Smith M.P., Klamroth R., Mannucci P.M., Kollmer C., Feingold J., Kessler C., Pollmann H., Morfini M., Udata C., et al. ReFacto and Advate: a single-dose, randomized, two-period crossover pharmacokinetics study in subjects with haemophilia A. Haemophilia. 2007;13:124–130. doi: 10.1111/j.1365-2516.2006.01420.x. [DOI] [PubMed] [Google Scholar]
- 13.Ward N.J., Buckley S.M.K., Waddington S.N., Vandendriessche T., Chuah M.K.L., Nathwani A.C., McIntosh J., Tuddenham E.G.D., Kinnon C., Thrasher A.J., McVey J.H. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood. 2011;117:798–807. doi: 10.1182/blood-2010-05-282707. [DOI] [PubMed] [Google Scholar]
- 14.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 15.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 16.Grimm D., Kay M.A. F rom virus evolution to vector revolution: use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr. Gene Ther. 2003;3:281–304. doi: 10.2174/1566523034578285. [DOI] [PubMed] [Google Scholar]
- 17.Arruda V.R., Doshi B.S. Gene therapy for hemophilia: facts and quandaries in the 21st century. Mediterr. J. Hematol. Infect. Dis. 2020;12 doi: 10.4084/MJHID.2020.069. e2020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miao H.Z., Sirachainan N., Palmer L., Kucab P., Cunningham M.A., Kaufman R.J., Pipe S.W. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–3419. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- 19.Nichols W.C., Seligsohn U., Zivelin A., Terry V.H., Hertel C.E., Wheatley M.A., Moussalli M.J., Hauri H.P., Ciavarella N., Kaufman R.J., Ginsburg D. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93:61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 20.Pipe S.W., Morris J.A., Shah J., Kaufman R.J. Differential interaction of coagulation factor VIII and factor V with protein chaperones calnexin and calreticulin. J. Biol. Chem. 1998;273:8537–8544. doi: 10.1074/jbc.273.14.8537. [DOI] [PubMed] [Google Scholar]
- 21.Moussalli M., Pipe S.W., Hauri H.P., Nichols W.C., Ginsburg D., Kaufman R.J. Mannose-dependent endoplasmic reticulum (ER)-Golgi intermediate compartment-53-mediated ER to Golgi trafficking of coagulation factors V and VIII. J. Biol. Chem. 1999;274:32539–32542. doi: 10.1074/jbc.274.46.32539. [DOI] [PubMed] [Google Scholar]
- 22.Cunningham M.A., Pipe S.W., Zhang B., Hauri H.P., Ginsburg D., Kaufman R.J. LMAN1 is a molecular chaperone for the secretion of coagulation factor VIII. J. Thromb. Haemost. 2003;1:2360–2367. doi: 10.1046/j.1538-7836.2003.00415.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B., Cunningham M.A., Nichols W.C., Bernat J.A., Seligsohn U., Pipe S.W., McVey J.H., Schulte-Overberg U., de Bosch N.B., Ruiz-Saez A., et al. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nat. Genet. 2003;34:220–225. doi: 10.1038/ng1153. [DOI] [PubMed] [Google Scholar]
- 24.Zhang B., McGee B., Yamaoka J.S., Guglielmone H., Downes K.A., Minoldo S., Jarchum G., Peyvandi F., de Bosch N.B., Ruiz-Saez A., et al. Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2. Blood. 2006;107:1903–1907. doi: 10.1182/blood-2005-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasi K.J., Laffan M., Rangarajan S., Robinson T.M., Mitchell N., Lester W., Symington E., Madan B., Yang X., Kim B., et al. Persistence of haemostatic response following gene therapy with valoctocogene roxaparvovec in severe haemophilia A. Haemophilia. 2021;27:947–956. doi: 10.1111/hae.14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce G.F. Gene therapy for hemophilia: are expectations matching reality? Mol. Ther. 2020;28:2097–2098. doi: 10.1016/j.ymthe.2020.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierce G.F., Kaczmarek R., Noone D., O'Mahony B., Page D., Skinner M.W. Gene therapy to cure haemophilia: is robust scientific inquiry the missing factor? Haemophilia. 2020;26:931–933. doi: 10.1111/hae.14131. [DOI] [PubMed] [Google Scholar]
- 28.Sheridan C. A reprieve from hemophilia A, but for how long? Nat. Biotechnol. 2020;38:1107–1109. doi: 10.1038/s41587-020-0693-y. [DOI] [PubMed] [Google Scholar]
- 29.Batty P., Lillicrap D. Hemophilia gene therapy: approaching the first licensed product. Hemasphere. 2021;5:e540. doi: 10.1097/HS9.0000000000000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mingozzi F., High K.A. Overcoming the host immune response to adeno-associated virus gene delivery vectors: the race between clearance, tolerance, neutralization, and escape. Annu. Rev. Virol. 2017;4:511–534. doi: 10.1146/annurev-virology-101416-041936. [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa H., Umemura A., Taniguchi K., Font-Burgada J., Dhar D., Ogata H., Zhong Z., Valasek M.A., Seki E., Hidalgo J., et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poothong J., Pottekat A., Siirin M., Campos A.R., Paton A.W., Paton J.C., Lagunas-Acosta J., Chen Z., Swift M., Volkmann N., et al. Factor VIII exhibits chaperone-dependent and glucose-regulated reversible amyloid formation in the endoplasmic reticulum. Blood. 2020;135:1899–1911. doi: 10.1182/blood.2019002867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaczmarek R., Pierce G.F., Noone D., O'Mahony B., Page D., Skinner M.W. Eliminating Panglossian thinking in development of AAV therapeutics. Mol. Ther. 2021;29:3325–3327. doi: 10.1016/j.ymthe.2021.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabatino D.E., McCarty D.M. Topics in AAV integration come front and center at ASGCT AAV Integration Roundtable. Mol. Ther. 2021;29:3319–3320. doi: 10.1016/j.ymthe.2021.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalwadi D.A., Torrens L., Abril-Fornaguera J., Pinyol R., Willoughby C., Posey J., Llovet J.M., Lanciault C., Russell D.W., Grompe M., Naugler W.E. Liver injury increases the incidence of HCC following AAV gene therapy in mice. Mol. Ther. 2021;29:680–690. doi: 10.1016/j.ymthe.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu F., Song Y., Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 37.Kaufman R.J., Wasley L.C., Davies M.V., Wise R.J., Israel D.I., Dorner A.J. Effect of von Willebrand factor coexpression on the synthesis and secretion of factor VIII in Chinese hamster ovary cells. Mol. Cell. Biol. 1989;9:1233–1242. doi: 10.1128/mcb.9.3.1233-1242.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pobre K.F.R., Poet G.J., Hendershot L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J. Biol. Chem. 2019;294:2098–2108. doi: 10.1074/jbc.REV118.002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng D.T., Watowich S.S., Lamb R.A. Analysis in vivo of GRP78-BiP/substrate interactions and their role in induction of the GRP78-BiP gene. Mol. Biol. Cell. 1992;3:143–155. doi: 10.1091/mbc.3.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Y., Chen Z., Jang I., Arvan P., Kaufman R.J. Epitope-tagging of the endogenous murine BiP/GRP78/Hspa5 locus allows direct analysis of the BiP interactome and protein misfolding in vivo. bioRxiv. 2021 doi: 10.1101/2020.01.01.892539. Preprint at. [DOI] [Google Scholar]
- 41.Preissler S., Chambers J.E., Crespillo-Casado A., Avezov E., Miranda E., Perez J., Hendershot L.M., Harding H.P., Ron D. Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. Elife. 2015;4:e08961. doi: 10.7554/eLife.08961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michailidis E., Vercauteren K., Mancio-Silva L., Andrus L., Jahan C., Ricardo-Lax I., Zou C., Kabbani M., Park P., Quirk C., et al. Expansion, in vivo-ex vivo cycling, and genetic manipulation of primary human hepatocytes. Proc. Natl. Acad. Sci. USA. 2020;117:1678–1688. doi: 10.1073/pnas.1919035117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vecchi C., Montosi G., Zhang K., Lamberti I., Duncan S.A., Kaufman R.J., Pietrangelo A. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325:877–880. doi: 10.1126/science.1176639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh-hashi K., Koga H., Ikeda S., Shimada K., Hirata Y., Kiuchi K. CRELD2 is a novel endoplasmic reticulum stress-inducible gene. Biochem. Biophys. Res. Commun. 2009;387:504–510. doi: 10.1016/j.bbrc.2009.07.047. [DOI] [PubMed] [Google Scholar]
- 45.Boyle S.T., Poltavets V., Kular J., Pyne N.T., Sandow J.J., Lewis A.C., Murphy K.J., Kolesnikoff N., Moretti P.A.B., Tea M.N., et al. Publisher Correction: ROCK-mediated selective activation of PERK signalling causes fibroblast reprogramming and tumour progression through a CRELD2-dependent mechanism. Nat. Cell Biol. 2020;22:908. doi: 10.1038/s41556-020-0539-3. [DOI] [PubMed] [Google Scholar]
- 46.Liu G.M., Zeng H.D., Zhang C.Y., Xu J.W. Key genes associated with diabetes mellitus and hepatocellular carcinoma. Pathol. Res. Pract. 2019;215:152510. doi: 10.1016/j.prp.2019.152510. [DOI] [PubMed] [Google Scholar]
- 47.Kern P., Balzer N.R., Blank N., Cygon C., Wunderling K., Bender F., Frolov A., Sowa J.P., Bonaguro L., Ulas T., et al. Creld2 function during unfolded protein response is essential for liver metabolism homeostasis. FASEB J. 2021;35:e21939. doi: 10.1096/fj.202002713RR. [DOI] [PubMed] [Google Scholar]
- 48.Cui S., Hano H., Sakata A., Harada T., Liu T., Takai S., Ushigome S. Enhanced CD34 expression of sinusoid-like vascular endothelial cells in hepatocellular carcinoma. Pathol. Int. 1996;46:751–756. doi: 10.1111/j.1440-1827.1996.tb03544.x. [DOI] [PubMed] [Google Scholar]
- 49.Wasfy R.E., Shams Eldeen A.A. Roles of combined glypican-3 and glutamine synthetase in differential diagnosis of hepatocellular lesions. Asian Pac. J. Cancer Prev. 2015;16:4769–4775. doi: 10.7314/apjcp.2015.16.11.4769. [DOI] [PubMed] [Google Scholar]
- 50.Di Tommaso L., Franchi G., Park Y.N., Fiamengo B., Destro A., Morenghi E., Montorsi M., Torzilli G., Tommasini M., Terracciano L., et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology. 2007;45:725–734. doi: 10.1002/hep.21531. [DOI] [PubMed] [Google Scholar]
- 51.Sidonio R.F., Jr., Pipe S.W., Callaghan M.U., Valentino L.A., Monahan P.E., Croteau S.E. Discussing investigational AAV gene therapy with hemophilia patients: a guide. Blood Rev. 2021;47:100759. doi: 10.1016/j.blre.2020.100759. [DOI] [PubMed] [Google Scholar]
- 52.Kaiser J. How safe is a popular gene therapy vector? Science. 2020;367:131. doi: 10.1126/science.367.6474.131. [DOI] [PubMed] [Google Scholar]
- 53.Rutkowski D.T., Wu J., Back S.H., Callaghan M.U., Ferris S.P., Iqbal J., Clark R., Miao H., Hassler J.R., Fornek J., et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell. 2008;15:829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D.T., Back S.H., Kaufman R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 55.de Jong Y.P., Herzog R.W. Liver gene therapy and hepatocellular carcinoma: a complex web. Mol. Ther. 2021;29:1353–1354. doi: 10.1016/j.ymthe.2021.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Febbraio M.A., Reibe S., Shalapour S., Ooi G.J., Watt M.J., Karin M. Preclinical models for studying NASH-driven HCC: how useful are they? Cell Metab. 2019;29:18–26. doi: 10.1016/j.cmet.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim J.Y., He F., Karin M. From liver fat to cancer: perils of the western diet. Cancers (Basel) 2021;13:1095. doi: 10.3390/cancers13051095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lange A.M., Altynova E.S., Nguyen G.N., Sabatino D.E. Overexpression of factor VIII after AAV delivery is transiently associated with cellular stress in hemophilia A mice. Mol. Ther. Methods Clin. Dev. 2016;3:16064. doi: 10.1038/mtm.2016.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zolotukhin I., Markusic D.M., Palaschak B., Hoffman B.E., Srikanthan M.A., Herzog R.W. Potential for cellular stress response to hepatic factor VIII expression from AAV vector. Mol. Ther. Methods Clin. Dev. 2016;3:16063. doi: 10.1038/mtm.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fong S., Handyside B., Sihn C.R., Liu S., Zhang L., Xie L., Murphy R., Galicia N., Yates B., Minto W.C., et al. Induction of ER stress by an AAV5 BDD FVIII construct is dependent on the strength of the hepatic-specific promoter. Mol. Ther. Methods Clin. Dev. 2020;18:620–630. doi: 10.1016/j.omtm.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 62.White D.L., Thrift A.P., Kanwal F., Davila J., El-Serag H.B. Incidence of hepatocellular carcinoma in all 50 United States, from 2000 through 2012. Gastroenterology. 2017;152:812–820.e5. doi: 10.1053/j.gastro.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahmed O., Liu L., Gayed A., Baadh A., Patel M., Tasse J., Turba U., Arslan B. The changing face of hepatocellular carcinoma: forecasting prevalence of nonalcoholic steatohepatitis and hepatitis C cirrhosis. J. Clin. Exp. Hepatol. 2019;9:50–55. doi: 10.1016/j.jceh.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loomba R., Friedman S.L., Shulman G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537–2564. doi: 10.1016/j.cell.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 65.Wang M., Kaufman R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 66.Wang M., Kaufman R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 67.Malhi H., Kaufman R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lebeaupin C., Vallée D., Hazari Y., Hetz C., Chevet E., Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018;69:927–947. doi: 10.1016/j.jhep.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 69.Rutkowski D.T. Liver function and dysfunction - a unique window into the physiological reach of ER stress and the unfolded protein response. FEBS J. 2019;286:356–378. doi: 10.1111/febs.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.