

Abstract
Drug development is a lengthy and challenging process that can be accelerated at early stages by new mathematical approaches and modern computers. To address this important issue, we are developing new mathematical solutions for the detection and characterization of protein binding sites that are important for new drug development. In this review, we present algorithms based on graph theory combined with molecular dynamics simulations that we have developed for studying biological target proteins to provide important data for optimizing the early stages of new drug development. A particular focus is the development of new protein binding site prediction algorithms (ProBiS) and new web tools for modeling pharmaceutically interesting molecules—ProBiS Tools (algorithm, database, web server), which have evolved into a full-fledged graphical tool for studying proteins in the proteome. ProBiS differs from other structural algorithms in that it can align proteins with different folds without prior knowledge of the binding sites. It allows detection of similar binding sites and can predict molecular ligands of various types of pharmaceutical interest that could be advanced to drugs to treat a disease, based on the entire Protein Data Bank (PDB) and AlphaFold database, including proteins not yet in the PDB. All ProBiS Tools are freely available to the academic community at http://insilab.org and https://probis.nih.gov.
Keywords: Structural proteome, Protein binding sites, Prediction, ProBiS
Introduction
In developing new drugs and vaccines, the pharmaceutical industry is increasingly turning to molecular modeling, a field in science with potential to shorten the drug discovery process, which studies the properties of molecules by recreating them as models on computers (Martinez-Mayorga et al. 2020). In this paper, we will describe our newly developed molecular modeling tools that enable studying of protein binding sites, which are the targets of most drugs, and enable the prediction of their biochemical functions, and ligands that could be potentially used as drugs. The research questions addressed by these tools are important for the entire pharmaceutical industry, since the rational development of new drugs is only possible if the properties of protein binding sites and their ligands are known and well characterized.
In recent decades, researchers have made discoveries that have revolutionized our understanding of cell structure and function, most notably the fact that proteins are capable of mutual communication and adapt their functions to current conditions in the cell (Phizicky and Fields 1995; Jones and Thornton 1996). Proteins have been found to form protein complexes by binding temporarily or permanently to each other or to other ligands. The binding occurs at a site on the protein surface called a binding site. Interactions between proteins are crucial for the functioning of biological systems, as they influence the function of proteins and ensure their self-regulation.
Protein binding sites can form complexes with small molecules, such as receptor ligands and enzyme substrates, or with large molecules, such as nucleic acids, peptides, and other proteins, the nature of the binding being determined by the specific physicochemical properties of their surfaces (Fig. 1). The prerequisite for two proteins to interact is a complementary pattern of interactions on their surfaces or binding sites, so that, the proteins are attracted to each other (Weiner et al. 1982). In modeling protein interactions, the surface plays an important role, while the interior of the proteins plays a lesser role, since only the surface amino acids contribute to the attractive bonds with which the protein binds to its partners (Schmitt et al. 2002).
Fig. 1.

Binding site and different possible ligands on a protein
The binding sites for small molecules, nucleic acid proteins, ions, and certain water molecules change slowly during evolution (Abrusán and Marsh 2018) and are conserved in the structures of related proteins found in the Protein Data Bank (PDB) (Berman et al. 2003; Kinjo et al. 2017). If we know the structure of a protein but not its binding sites, we can find binding sites on that protein by comparing it to the approximately 190,000 known structures in the PDB; similarities found between the structure studied and those from the database generally coincide with the binding sites on those proteins, and if the function of similar proteins in the database is known, the comparison also allows us to predict the function of the proteins studied (Konc et al. 2013).
Many computational tools have been reported for binding site analysis and prediction (Kinoshita et al. 2002; Kinoshita and Nakamura 2005; Salentin et al. 2015; Jakubec et al. 2022), some of which are based on clique-finding algorithms (Ren et al. 2010; Chartier and Najmanovich 2015), and conservation of hot-spot structure may be insufficient for detection (Cukuroglu et al. 2012; Chen et al. 2012). Phylogenetic protein sequences have been used to detect the conservation of surface residue sequences (Glaser et al. 2003). Other traditional approaches take advantage of the fact that the 3D structure is evolutionarily more conserved than the residue sequence. The function of a protein can be determined by finding at least one structurally similar protein whose function is already known in the PDB. These methods compare the overall 3D shape of proteins or protein folds.
We have developed protein binding site tools (ProBiS) consisting of web servers, databases, and protein and ligand binding site prediction algorithms, all based on a graph theoretic algorithm, a fast and improved maximum clique algorithm that we developed in 2007 (Fig. 2) (Konc and Janezic 2007). The ProBiS Tools have been independently validated and are widely used in pharmaceutical research (Ehrt et al. 2016, 2018; Fu et al. 2016; Vankayala et al. 2017; Ramatenki et al. 2017; Bancroft et al. 2019). One such application is the development of novel biological drugs, where a combination of ProBiS structure comparison and molecular dynamics simulations was used to reduce unwanted side effects of an antibody-based drug, ipilimumab, without compromising its stability or increasing its immunogenicity (Lešnik et al. 2020). The ProBiS algorithm compares local physicochemical and geometric properties of protein surface structures to identify common amino acid motifs independent of protein folding (Konc and Janežič 2010). This algorithm is unique because it does not require the binding site on the protein to be known in advance, but instead compares the entire surface of the protein in question to the surfaces of other proteins and identifies similar binding sites based on the detectable local surface similarities. An extension of this algorithm, the ProBiS-ligands web server (Konc and Janežič 2014), predicts the interactions and positions of ligands with a given protein based on the detection of similarities within protein binding sites in the PDB. Ligands that bind to similar binding sites identified by this algorithm are transferred to the query protein binding site by rotating and translating their coordinates based on the superposition of binding sites, with each group of ligands of the same type representing a predicted binding site.
Fig. 2.
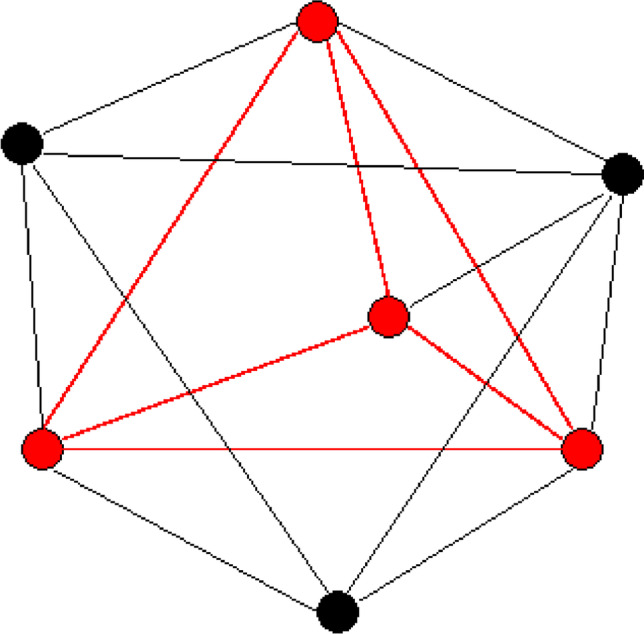
A maximum clique problem. Maximum clique (red) is the largest fully connected subgraph within a graph
In the following sections, we provide an overview of the development of ProBiS Tools and present examples of their capabilities.
What is ProBiS?
The ProBiS algorithm is computer software that allows the prediction of binding sites and the corresponding ligands for a given protein structure (Konc and Janežič 2010). It was originally developed in 2010 and has since been expanded into several ProBiS web servers and databases, all under the name ProBiS Tools that:
enable rapid determination of binding sites for the entire PDB
are, to the best of our knowledge, currently the only tools that can accurately determine the type of ligand for a predicted binding site
are currently the only ones that allow the determination of binding sites and ligands for AlphaFold proteins not yet included in the PDB (Varadi et al. 2022).
What is maximum clique algorithm?
A maximum clique problem is an NP-hard problem for which there is most likely no polynomial solution. A maximum clique algorithm finds the largest fully connected subgraph (a clique) in an undirected graph, i.e., the one with the most vertices. We have developed an algorithm for finding a maximum clique in an undirected graph that is up to 100 times faster than the best comparable algorithm (Konc and Janezic 2007; Depolli et al. 2013; Reba et al. 2022).
Protein graphs
Proteins can be represented as protein graphs (Konc and Janežič 2007, 2010; Depolli et al. 2013). In protein graphs, the vertices have spatial coordinates, and they are located at the geometric centers of the functional groups of the amino acids of the protein surface. The vertices are labeled with five different colors corresponding to the five physicochemical properties, i.e., acceptor, donor, π-π-stacking, aliphatic, and acceptor–donor, of the protein surface amino acids at the resolution of the functional groups. Two vertices ui and uj of a protein graph G are adjacent, i.e., an edge (ui, uj) ∈ E(G) exists between them if the distance (ui, uj) is less than 15 Å.
How can a maximum clique detect the local similarity of two proteins?
A pair of protein graphs can be compared by finding a maximum clique, i.e., the clique with the most vertices, in their product graph, where the maximum clique represents the superposition that aligns the most vertices of the compared protein graphs (Konc and Janežič 2007, 2010; Depolli et al. 2013). The protein product graph of two protein graphs G1 and G2 is defined by the set of vertices V(G1, G2) = V(G1) × V(G2). Each vertex of the protein product graph (ui, vi) consists of two subvertices: a subvertex from the first protein graph (ui ∈ G1) and a subvertex from the second protein graph (vi ∈ G2). In general, a protein product graph has x × y vertices if the respective protein graphs have x and y vertices; however, we reduce its size by considering as product graph vertices only those where the two subvertices have identical colors, i.e., identical physicochemical properties, and similar neighborhoods. We connect two protein product graph vertices (ui, vi) and (uj, vj), where (ui, uj) ∈ E(G1) and (vi, vj) ∈ E(G2), by inserting an edge between them if |distance(ui, uj) − distance(vi, vj)| is less than 0.5 Å, which means that the distances between the respective first and second subvertices in both protein graphs must be nearly equal. A maximum clique in the protein product graph constructed in this way represents the largest similarity between the two compared protein graphs in terms of physicochemical and geometric properties and allows us to identify pairs of similar binding sites and other similar surface regions in proteins independent of their protein folds.
ProBiS Tools development
We have developed new methodological solutions for the prediction and study of protein binding sites and their ligands based on graph theoretical approaches combined with molecular simulations (Fig. 3). These are:
Fig. 3.

Timeline of ProBiS Tools development
MaxCliqueDyn algorithm
We have developed a new algorithm for finding a maximum clique in an undirected graph (http://insilab.org/maxclique), in which we have improved approximate coloring algorithm that is used by the maximum clique algorithm to provide bounds to the size of the maximum clique (Konc and Janezic 2007). We then extended this algorithm to include dynamically varying bounds to adapt the maximum clique search to the type of input graph (Depolli et al. 2013; Reba et al. 2022). We show that by applying tighter, more computationally expensive upper bounds on a fraction of the search space, it is possible to reduce the time to find the maximum clique. This resulting algorithm is significantly faster (between 10 × and 100 ×) than comparable algorithms.
ProBiS algorithm
The ProBiS algorithm (http://insilab.org/probis-algorithm) enables local structural matching of entire protein surface structures against a large database of protein structures in a reasonable amount of time (Konc and Janežič 2007, 2010). The comparison includes geometry and physicochemical properties and is performed at the amino acid functional group level. The algorithm compares the query protein to each of the database proteins, using the maximum clique algorithm (Konc and Janezic 2007; Depolli et al. 2013), which allows it efficiently to detect the largest similar subgraphs of compared protein graphs. For each pairwise comparison of the query protein with a database protein, the algorithm generates multiple local alignments of the surface regions found in both proteins; no attempt is made to align the proteins globally, and similar folding is not a requirement for a relationship between the two proteins. Because no assumptions are made about the localization of binding sites prior to comparison, ProBiS can discover new binding sites and suggest ligands that might host these binding sites. Due to the high computational cost, the comparison of such a number of proteins on a local level and in such a short time has not been possible before, even with high-performance computers.
ProBiS-ligands web server
The ProBiS-ligands web server (http://probis.cmm.ki.si) predicts the binding of ligands to a protein structure (Konc and Janežič 2014). Given a protein structure or binding site, ProBiS-ligands first identify template proteins in the PDB that have similar binding sites. Based on the superimpositions of the query protein and the similar binding sites found, the server then transfers the ligand structures from these sites to the query protein. Such ligand prediction supports many activities, such as drug repurposing (Štular et al. 2016). In addition to identifying protein ligands, the ProBiS-ligands web server can also be used to accurately identify structurally similar binding sites in protein structures or structural evolutionary conservation values.
ProBiS-CHARMMing web server
Unlike the ProBiS web servers, ProBiS-CHARMMing (https://probis.nih.gov) is hosted at the National Institutes of Health, Bethesda, MD, USA (Konc et al. 2015). ProBiS-CHARMMing provides all the features of the ProBiS web servers, plus molecular modeling capabilities, and allows minimization of predicted ligands and their binding sites and calculation of their interaction energies. This is achieved by integrating ProBiS with the CHARMMing web server at https://charmming.org (Miller et al. 2008; Brooks et al. 2009). The main strength of the ProBiS CHARMMing web server is that it is able to remove steric clashes between predicted ligands and proteins, which can be the cause of unrealistic models, and also remove the lack of energy-based scores to assess the strength of ligand binding. The web interface also facilitates the creation of CHARMM-friendly protein–ligand systems, including CHARMM input scripts for further modeling. The server can be used to predict energy-minimized holo protein structures, that is, protein–protein, protein-small molecule, and protein-ion complexes with unliganded (apo) protein structures as queries, and provides an interactive environment where users can explore the predicted protein–ligand complexes and calculate and compare their energy properties.
GenProBiS
The GenProBiS web server (http://genprobis.insilab.org) links sequence variants to protein structures and also to protein–protein, protein-nucleic acid, protein-compound, and protein-metal ion binding sites (Konc et al. 2017). This server enables intuitive visual exploration of extensive mapped variants, such as human cancer-associated somatic missense mutations and nonsynonymous single nucleotide polymorphisms from 21 species, within predicted binding site regions for approximately 80,000 PDB protein structures. It also enables the discovery of potentially deleterious sequence variants and the development of new hypotheses for drug discovery, e.g., to explain the sensitivity of a particular drug to a specific mutation in a protein binding site.
ProBiS H2O
We have developed the ProBiS H2O plugin for PyMOL (http://insilab.org/probis-h2o) that allows rapid identification of conserved water ligands in a protein structure or protein binding site using experimental protein structures from the PDB or a set of custom protein structures available to the user (Jukic et al. 2017). Identifying conserved water sites in protein structures is a challenging task that has applications in molecular docking and protein stability prediction. Using a protein structure, binding site, or single water molecule as a query, ProBiS H2O collects similar proteins from the PDB and performs local or binding site-specific superimpositions of the query structure with similar proteins using the ProBiS algorithm. It collects the experimental water molecules from similar proteins and transfers them to the query protein. The transferred water molecules are clustered according to their mutual proximity, identifying discrete sites in the query protein with high water conservation.
ProBiS H2O MD
The ProBiS H2O MD plugin (http://insilab.org/probis-h2o-md) is an extension of the ProBiS H2O approach and allows the identification of conserved waters as ligands from molecular dynamics trajectories of proteins in water (Jukič et al. 2020). It uses snapshots of a protein in water from a MD trajectory to identify conserved water sites and allows visualization of the identified conserved water sites on a protein.
ProBiS-Dock database
This is a web server and interactive web repository of small ligand–protein binding sites (http://probis-dock-database.insilab.org) for drug design of more than 1.4 million small ligand–protein binding sites in the PDB, which allows these binding sites to be ranked according to their druggability (Konc et al. 2021). A new druggability score is used to measure the suitability of a binding site for drug development. It is defined as the extent to which the binding site is currently used in drug development, as reflected by the proportion of PDB structures of this and similar binding sites bound with ligands with druglike properties. The druglike nature of a ligand is measured by the molecular complexity of the ligand, which takes into account the elemental composition and the number of rings in the compound’s structure. This helps screen out binding sites that bind to small molecules with simple structures that could bind nonspecifically to many proteins and favors binding sites that bind to molecules that are similar to most existing drugs. Another unique feature of the database is the division of ligand binding site into compound (substrate-competitive) and cofactor (cofactor-competitive), which may be particularly suitable for drug design, where typically inhibitors against a substrate or against a cofactor are developed.
ProBiS-Dock algorithm
ProBiS-Dock (http://insilab.org/probisdock) is a hybrid multitemplate homology algorithm for flexible docking enabled by protein binding site comparison (Konc et al. 2022). It is a small molecule docking (and inverse docking) approach based on predicted binding sites that enables flexible docking of small ligands to flexible protein binding sites. The ProBiS-Dock algorithm can be used in drug development for new drug candidate discovery, drug repositioning, and off-target effects. It complements the ProBiS-Dock Database in the sense that its input, the prepared binding sites, can be obtained from that database. The algorithm treats small molecules and proteins as fully flexible entities and allows conformational changes in both after ligand binding. A new scoring function is described that consists of a binding site-specific scoring function (ProBiS-Score) and a general statistical scoring function. This allows the scoring function to adapt to each protein binding site in the PDB. ProBiS-Dock enables rapid docking of small molecules to proteins and has been successfully validated in silico against standard benchmarks. It enables the search for new active ligands by leveraging existing knowledge in the PDB. The potential of the software for drug discovery has been confirmed in vitro by the discovery of new inhibitors of human indoleamine 2,3-dioxygenase 1, an enzyme that is an attractive target for cancer therapy (Dolšak et al. 2021).
ProBiS-Fold web server
This web server and database (http://probis-fold.insilab.org) enables annotation of human structures from the AlphaFold database (Varadi et al. 2022) with no corresponding structure in the PDB to discover new druggable binding sites (Konc and Janežič 2022). It contains predictions of binding sites and their corresponding ligands from the whole human structural proteome (Fig. 4). The predicted binding sites are divided into protein, peptide, nucleic acid, small molecule, further subdivided into compound (for substrate/agonist competitive ligands) and cofactor (for cofactors and cofactor-competitive ligands) binding sites, conserved water, metal ion, and glycan-binding sites according to the type of ligand they bind. In contrast to our previous approach, peptide ligand binding sites are detected separately from protein binding sites because peptides are an important new class of drugs that are distinct from proteins. For ion and water ligands, only biologically relevant metal ions and conserved water molecules are considered. Conserved water molecules are those found in more than 10 PDB structures bound to a similar motif and have a high conservation score greater than 0.6. Biologically relevant metal ions are those found in more than 10 PDB structures at the same location. A total of 149,960 binding sites were predicted for the entire human structural proteome. Importantly, binding sites were identified on protein structures for small molecules that do not have a corresponding structure in the PDB; 573 of these binding sites are highly druggable and 921 other sites are druggable as judged by our druggability score. These represent a novel pool of binding sites for previously unknown protein structures that could enter pharmaceutical pipelines. ProBiS-Fold is an extension of the ProBiS-ligands (Konc and Janežič 2014) and the ProBiS-CHARMMing web interface (Konc et al. 2015) for prediction and optimization of ligands in protein binding sites, as well on a recent addition, the GenProBiS web server (Konc et al. 2017) and in particular, it is an extension of the ProBiS-Dock Database (Konc et al. 2021) with protein models from the recently developed AlphaFold database (Jumper et al. 2021; Varadi et al. 2022) which provides open access to protein structure predictions for the human proteome and 20 other key organisms (DeepMind, Google, https://alphafold.ebi.ac.uk) is thus opening up completely new possibilities for drug research on virtually the entire human proteome as well as on proteomes of other species. The ProBiS-Fold web server enables the characterization of binding sites for novel protein targets and greatly increases the number of potential protein targets that could be used in drug discovery.
Fig. 4.

ProBiS-Fold web server as an extension of ProBiS-Dock Database with AlphaFold DB
Using ProBiS-Fold, we can predict binding sites for protein structures predicted by AlphaFold, which may or may not already have a structure in the PDB. The predicted binding sites are classified by the type of ligand they bind, and the server also allows construction of complexes of the protein with predicted ligands. An example of the output is shown in Fig. 5, in which angiotensin-converting enzyme 2 was used as a query and for which ProBiS-Fold predicted the ligand of the spike protein of SARS-CoV-2 and a corresponding binding site.
Fig. 5.

ProBiS-Fold web server results page shows a 3D view of a predicted binding site (blue surface) for the SARS-CoV-2 spike protein (gray cartoon) on a human angiotensin-converting enzyme 2 (ACE2) protein model from the AlphaFold database (model confidence-colored cartoon). Binding site residues on ACE2 are CPK-colored sticks. The list of predicted ligands for this binding site is below the viewer. Links to all the different predicted binding sites (protein, compound, cofactor, glycan, metal ion, and peptide) for the ACE2 protein are on the left
Conclusions
We have developed ProBiS Tools for protein binding site detection and ligand prediction and characterization. The newly developed ProBiS-Fold web server is the latest addition to the suite of tools. It annotates the AlphaFold human protein structure database of more than 24,000 predicted protein structures with ligand binding sites and sites for post-translational modifications, and 3D structures of ligands that bind to these sites, using a structure-based, comparative approach, and for the first time makes it possible to examine structures in the AlphaFold Database for which there is no corresponding structure in the PDB and to predict in detail where the binding sites are located, to which ligands they bind, and whether the binding sites are suitable for drug development. It can show the reliability of the AlphaFold structure, especially at the binding sites. As a world first, the binding sites are categorized into protein, peptide, nucleic acid, small molecule (substrate and cofactor competitive), metal ion, conserved water, and glycan types, depending on which ligands they bind. All of our past, present, and future web servers and tools are freely available to academic users at http://insilab.org and at https://probis.nih.gov.
Acknowledgements
We gratefully acknowledge the support of NVIDIA Corporation with the donation of Tesla GPUs.
Author contribution
J.K. and D.J. conceptualized the project and performed all aspects of the research process and wrote the entire manuscript. Both authors read and approved the final version of the manuscript.
Funding
This work was supported by the Slovenian Research Agency project grants N1-0142, L7-8269, N1-0209, J1-1715 and J1-9186 and a bilateral grant BI-JP/16–18-002.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Code availability
Not applicable.
Declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Footnotes
This review article is in honor of Prof. Haruki Nakamura’s 70th birthday.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abrusán G, Marsh JA. Ligand binding site structure influences the evolution of protein complex function and topology. Cell Rep. 2018;22:3265–3276. doi: 10.1016/j.celrep.2018.02.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft AJ, Levy CW, Jowitt TA, et al. The major secreted protein of the whipworm parasite tethers to matrix and inhibits interleukin-13 function. Nat Commun. 2019;10:2344. doi: 10.1038/s41467-019-09996-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Mol Biol. 2003;10:980–980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, Mackerell AD, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier M, Najmanovich R. Detection of binding site molecular interaction field similarities. J Chem Inf Model. 2015;55:1600–1615. doi: 10.1021/acs.jcim.5b00333. [DOI] [PubMed] [Google Scholar]
- Chen YC, Wright JD, Lim C. DR_bind: a web server for predicting DNA-binding residues from the protein structure based on electrostatics, evolution and geometry. Nucleic Acids Res. 2012;40:W249–W256. doi: 10.1093/nar/gks481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukuroglu E, Gursoy A, Keskin O. HotRegion: a database of predicted hot spot clusters. Nucleic Acids Res. 2012;40:D829–D833. doi: 10.1093/nar/gkr929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depolli M, Konc J, Rozman K, et al. Exact parallel maximum clique algorithm for general and protein graphs. J Chem Inf Model. 2013;53:2217–2228. doi: 10.1021/ci4002525. [DOI] [PubMed] [Google Scholar]
- Dolšak A, Bratkovič T, Mlinarič L, et al. Novel selective IDO1 inhibitors with isoxazolo[5,4-d]pyrimidin-4(5H)-one scaffold. Pharmaceuticals. 2021;14:265. doi: 10.3390/ph14030265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt C, Brinkjost T, Koch O. Impact of binding site comparisons on medicinal chemistry and rational molecular design. J Med Chem. 2016;59:4121–4151. doi: 10.1021/acs.jmedchem.6b00078. [DOI] [PubMed] [Google Scholar]
- Ehrt C, Brinkjost T, Koch O. A benchmark driven guide to binding site comparison: an exhaustive evaluation using tailor-made data sets (ProSPECCTs) PLoS Comput Biol. 2018;14:e1006483. doi: 10.1371/journal.pcbi.1006483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Zhang G, Liu R, et al. Mechanistic study of human glucose transport mediated by GLUT1. J Chem Inf Model. 2016;56:517–526. doi: 10.1021/acs.jcim.5b00597. [DOI] [PubMed] [Google Scholar]
- Glaser F, Pupko T, Paz I, et al. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- Jakubec D, Skoda P, Krivak R, et al. PrankWeb 3: accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Res. 2022;50:W593–W597. doi: 10.1093/nar/gkac389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukic M, Konc J, Gobec S, Janezic D. Identification of conserved water sites in protein structures for drug design. J Chem Inf Model. 2017;57:3094–3103. doi: 10.1021/acs.jcim.7b00443. [DOI] [PubMed] [Google Scholar]
- Jukič M, Konc J, Janežič D, Bren U. ProBiS H2O MD approach for identification of conserved water sites in protein structures for drug design. ACS Med Chem Lett. 2020;11:877–882. doi: 10.1021/acsmedchemlett.9b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinjo AR, Bekker G-J, Suzuki H, et al. Protein Data Bank Japan (PDBj): updated user interfaces, resource description framework, analysis tools for large structures. Nucleic Acids Res. 2017;45:D282–D288. doi: 10.1093/nar/gkw962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita K, Nakamura H. Identification of the ligand binding sites on the molecular surface of proteins. Protein Sci. 2005;14:711–718. doi: 10.1110/ps.041080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita K, Furui J, Nakamura H. Identification of protein functions from a molecular surface database, eF-site. J Struct Func Genom. 2002;2:9–22. doi: 10.1023/A:1011318527094. [DOI] [PubMed] [Google Scholar]
- Konc J, Janezic D. An improved branch and bound algorithm for the maximum clique problem. MATCH Commun Math Comput Chem. 2007;58:569–590. [Google Scholar]
- Konc J, Janežič D. Protein−protein binding-sites prediction by protein surface structure conservation. J Chem Inf Model. 2007;47:940–944. doi: 10.1021/ci6005257. [DOI] [PubMed] [Google Scholar]
- Konc J, Janežič D. ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics. 2010;26:1160–1168. doi: 10.1093/bioinformatics/btq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J, Janežič D. ProBiS-ligands: a web server for prediction of ligands by examination of protein binding sites. Nucleic Acids Res. 2014;42:W215–W220. doi: 10.1093/nar/gku460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J, Janežič D. ProBiS-Fold approach for annotation of human structures from the AlphaFold Database with no corresponding structure in the PDB to discover new druggable binding sites. J Chem Inf Model. 2022 doi: 10.1021/acs.jcim.2c00947. [DOI] [PubMed] [Google Scholar]
- Konc J, Hodošček M, Ogrizek M, et al. Structure-based function prediction of uncharacterized protein using binding sites comparison. PLOS Comput Biol. 2013;9:e1003341. doi: 10.1371/journal.pcbi.1003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J, Miller BT, Štular T, et al. ProBiS-CHARMMing: web interface for prediction and optimization of ligands in protein binding sites. J Chem Inf Model. 2015;55:2308–2314. doi: 10.1021/acs.jcim.5b00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J, Skrlj B, Erzen N, et al. GenProBiS: web server for mapping of sequence variants to protein binding sites. Nucleic Acids Res. 2017;45:W253–W259. doi: 10.1093/nar/gkx420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J, Lešnik S, Škrlj B, Janežič D. ProBiS-Dock Database: a web server and interactive web repository of small ligand–protein binding sites for drug design. J Chem Inf Model. 2021;61:4097–4107. doi: 10.1021/acs.jcim.1c00454. [DOI] [PubMed] [Google Scholar]
- Konc J, Lešnik S, Škrlj B, et al. ProBiS-Dock: a hybrid multitemplate homology flexible docking algorithm enabled by protein binding site comparison. J Chem Inf Model. 2022;62:1573–1584. doi: 10.1021/acs.jcim.1c01176. [DOI] [PubMed] [Google Scholar]
- Lešnik S, Hodošček M, Podobnik B, Konc J. Loop grafting between similar local environments for fc-silent antibodies. J Chem Inf Model. 2020;60:5475–5486. doi: 10.1021/acs.jcim.9b01198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Mayorga K, Madariaga-Mazon A, Medina-Franco JL, Maggiora G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin Drug Discov. 2020;15:293–306. doi: 10.1080/17460441.2020.1696307. [DOI] [PubMed] [Google Scholar]
- Miller BT, Singh RP, Klauda JB, et al. CHARMMing: a new, flexible web portal for CHARMM. J Chem Inf Model. 2008;48:1920–1929. doi: 10.1021/ci800133b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Fields S. Protein-protein interactions: methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramatenki V, Dumpati R, Vadija R, et al. Identification of new lead molecules against UBE2NL enzyme for cancer therapy. Appl Biochem Biotechnol. 2017;182:1497–1517. doi: 10.1007/s12010-017-2414-7. [DOI] [PubMed] [Google Scholar]
- Reba K, Guid M, Rozman K, et al. Exact maximum clique algorithm for different graph types using machine learning. Mathematics. 2022;10:97. doi: 10.3390/math10010097. [DOI] [Google Scholar]
- Ren J, Xie L, Li WW, Bourne PE. SMAP-WS: a parallel web service for structural proteome-wide ligand-binding site comparison. Nucleic Acids Res. 2010;38:W441–W444. doi: 10.1093/nar/gkq400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin S, Schreiber S, Haupt VJ, et al. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S, Kuhn D, Klebe G. A new method to detect related function among proteins independent of sequence and fold homology. J Mol Biol. 2002;323:387–406. doi: 10.1016/S0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- Štular T, Lešnik S, Rožman K, et al. Discovery of Mycobacterium tuberculosis InhA inhibitors by binding sites comparison and ligands prediction. J Med Chem. 2016;59:11069–11078. doi: 10.1021/acs.jmedchem.6b01277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankayala SL, Kearns FL, Baker BJ, et al. Elucidating a chemical defense mechanism of Antarctic sponges: a computational study. J Mol Graph Model. 2017;71:104–115. doi: 10.1016/j.jmgm.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:D439–D444. doi: 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner PK, Langridge R, Blaney JM, et al. Electrostatic potential molecular surfaces. Proc Natl Acad Sci USA. 1982;79:3754–3758. doi: 10.1073/pnas.79.12.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Not applicable.