

Abstract
Objectives
Peripheral neuropathy is a relevant dose‐limiting adverse event that can affect up to 90% of oncologic patients with colorectal cancer receiving oxaliplatin treatment. The severity of neurotoxicity often leads to dose reduction or even premature cessation of chemotherapy. Unfortunately, the limited knowledge about the molecular mechanisms related to oxaliplatin neurotoxicity leads to a lack of effective treatments to prevent the development of this clinical condition. In this context, the present work aimed to determine the exact molecular mechanisms involved in the development of oxaliplatin neurotoxicity in a murine model to try to find new therapeutical targets.
Methods
By single‐cell RNA sequencing (scRNA‐seq), we studied the transcriptomic profile of sensory neurons and satellite glial cells (SGC) of the Dorsal Root Ganglia (DRG) from a well‐characterized mouse model of oxaliplatin neurotoxicity.
Results
Analysis of scRNA‐seq data pointed to modulation of inflammatory processes in response to oxaliplatin treatment. In this line, we observed increased levels of NF‐kB p65 protein, pro‐inflammatory cytokines, and immune cell infiltration in DRGs and peripheral nerves of oxaliplatin‐treated mice, which was accompanied by mechanical allodynia and decrease in sensory nerve amplitudes.
Interpretation
Our data show that, in addition to the well‐described DNA damage, oxaliplatin neurotoxicity is related to an exacerbated pro‐inflammatory response in DRG and peripheral nerves, and open new insights in the development of anti‐inflammatory strategies as a treatment for preventing peripheral neuropathy induced by oxaliplatin.
Introduction
Oxaliplatin is a third‐generation platinum‐based cytostatic agent used as a first‐line treatment against colorectal, gastric, and pancreatic cancer. 1 Similar to other platinum‐based drugs, the main mechanism reported for oxaliplatin anti‐tumor activity is its binding to the DNA template, thereby forming platinum‐DNA adducts, leading to a replication failure of cancer cells and their death by apoptosis. 2 Although oxaliplatin has proven to be very effective in the treatment of gastrointestinal cancers, the development of peripheral neuropathy (a dose‐limiting side effect of oxaliplatin) limits its clinical use and has a direct impact on patient's survival and quality of life due to its long‐lasting nature. 3 , 4
Oxaliplatin neurotoxicity is characterized by an acute and transient syndrome mostly related to cold hyperesthesia, appearing within hours or days following oxaliplatin infusion in more than 90% of patients. Moreover, a progressive and long‐lasting neuropathy linked to the continuous treatment with oxaliplatin also appears in a high percentage (73%) of oncologic patients treated with this agent; being markedly symptomatic in about half of them 5 The acute syndrome has been attributed to ion channel dysregulation due to calcium and magnesium chelation by oxalate, a by‐product of oxaliplatin biotransformation. 6 On the other hand, chronic neuropathy has been classically related to the binding of oxaliplatin to the nuclear and mitochondrial DNA of sensory neurons located in the dorsal root ganglia (DRG), resulting in increased oxidative stress, myotoxicity, and their death by apoptosis. 6 Moreover, the severity of the acute syndrome has been linked to the risk of developing chronic neuropathy. 5 , 7 Despite all the knowledge accumulated during the last decades in the pathophysiological mechanisms related to oxaliplatin neurotoxicity, 5 , 6 , 7 there is a current lack of effective treatment for this clinical condition ‐ likely due to the lack of knowledge of the full mechanisms involved. In fact, most of the works focused on targeting peripheral neuropathy induced by oxaliplatin are hypothesis‐driven from the known actions of platinum drugs in neoplastic cells and are not based on the changes that take place in sensory neurons of the DRG which are the main target of oxaliplatin in the peripheral nervous system.
In this study, we aimed to determine the most relevant mechanisms related to the establishment of oxaliplatin neurotoxicity to find new therapeutic targets. With a view to achieving this aim, we investigated the changes in gene expression of DRG sensory neurons and satellite glial cells (SGCs) by single‐cell RNA‐sequencing (scRNA‐seq) in a well‐characterized murine model of peripheral neuropathy induced by oxaliplatin.
Materials and Methods
Animal model
10‐week‐old female BALB/cAnNCrl (BALB/c) mice (Janvier, 19–22 g on arrival at the housing room) were used for the studies. Animals were housed in a limited‐access animal facility with ad libitum access to water and food. Artificial lighting provided a 24‐h cycle of 12 h light/12 h dark (light 8 am–8 pm). Oxaliplatin (Selleckchem, Cat#12224) was freshly prepared on the day of its use by dissolving it in a 5% glucose solution to a concentration of 1 mg/mL. A total of 35 mice were intravenously (i.v.) injected with 5 mg/kg oxaliplatin twice a week for 8 weeks reaching a total cumulated dose (TCD) of 80 mg/kg (Oxaliplatin group, modified from Marmiroli et al. 2017 8 in basis of own preliminary study results). As a control, 15 mice received an i.v. injection of 5% glucose solution twice a week for 8 weeks. Animals were randomly assigned to experimental groups. All the procedures involving mice were approved by the Ethics Committee of the Universitat Autònoma de Barcelona (CEEA) and followed ARRIVE guidelines.
Nerve conduction studies
To assess compound muscle action potential (CMAP) and sensory nerve action potential (SNAP), the sciatic nerve was stimulated percutaneously through a pair of needle electrodes placed at the sciatic notch. After stimulation, CMAP was recorded by electrodes placed at the plantar muscle, whereas SNAP was recorded from the fourth toe near the digital nerve. Compound nerve action potential (CNAP) was also recorded by placing a pair of recording needle electrodes at the base of the tail and a couple of stimulating needle electrodes at 5 cm distally to the active recording points, elucidating the orthodromic sensory conductions (for more details, see Bruna et al. 2020 9 ). Pentobarbital (50 mg/kg i.p.) was used to anesthetize the mice during the tests and mice body temperature was maintained by means of a thermostated heating pad.
Algesimetry tests
Mechanical allodynia was tested using electronic von Frey device (Bioseb). On testing days, mice were placed in individual clear plastic cages on a wire mesh screen with a flat‐surfaced plastic platform with holes through which von Frey hairs were inserted and applied to the plantar surface of the paw. The combined mean percent withdrawal of three applications to the left foot was calculated for each animal, and group means were calculated.
A plantar test was used to assess heat‐evoked hyperalgesia using a Plantar algesimeter (Ugo Basile). Tests were performed at baseline and then every 2 weeks after starting the oxaliplatin administration and during all the induction and coasting‐effect times. Mice were individually placed into the transparent chambers situated in a farmed glass panel. To perform the test, the infrared emitter/detector was placed underneath the center of the mice paw and time (seconds) until paw withdrawal was measured. Three applications to the left foot were assessed for each animal, and group means were calculated. We set the infrared intensity (IR) at 50 units.
Cold hyperalgesia was assessed with a Cold plate test (Bioseb). Test was conducted at baseline and every 2 weeks during all the induction and coasting‐effect time. On testing days, mice were individually placed in the testing apparatus, a metal plate surrounded by a transparent metiplax cylinder. The metal plate was cooled at −4°C and cold hyperalgesia was assessed by measuring the withdrawal threshold time (seconds). Withdrawal was assessed as the first hind paw lift. Lifting for normal locomotion was excluded. Four tests were assessed for each mouse and group means were calculated. 10 min interval between each measurement was established. To prevent tissue damage, a cut‐off time of 60 seconds was adopted.
All the tests were conducted at baseline and either every 2 weeks during all the induction and coasting‐effect times. Mice underwent training sessions 3 days prior to the first day of testing. Before every session, mice were acclimatized for 15 min to the experimenter, the room in which the behavioral experiments took place and the transparent chamber used for von Frey testing.
Single‐cell sorting of DRG cells
At 8 weeks of the study, all DRGs from three control and three oxaliplatin‐treated mice were removed and cleaned from blood, connective tissue, and root debris. Then, DRGs were enzymatically dissociated in Ca2+ and Mg2+‐free Hank's medium (Sigma) with 10x trypsin (Sigma), 1 μg/mL collagenase A (Sigma), and 1 μg/mL DNAse (Roche) for 30 min at 37°C and with constant agitation. DMEM medium (Sigma) supplemented with 10% hiFBS (Sigma) was added to stop the enzymatic reaction, and DRGs were exposed to mechanical dissociation with a glass pipette. After complete dissociation, cells were filtered in a 70 μm cell strainer and centrifuged at 500 rcg for 5 min to remove enzymes. Cells were incubated for 1 h at 4°C and with constant agitation with primary antibodies rabbit anti‐TrkA (1:200, Abcam), goat anti‐TrkB (1:200, R&D systems), and goat anti‐TrkC (1:200, R&D Systems), diluted in 200 μL incubation medium (Neurobasal‐A (NB‐A, Gibco) supplemented with 6 mg/mL glucose, 2 mmol/L Glutamine (Sigma) and 5% hiFBS). After incubation time, collector tubes were completely filled with washing medium (Gey's Balanced medium (Sigma) supplemented with 6 mg/mL glucose and 5% hiFBS) and centrifuged three times at 500 rcg for 5 min at 4°C. Samples were then incubated for 45 min at room temperature in dark conditions and with constant agitation with secondary antibodies Alexa 488‐conjugated anti‐goat (1:200, Invitrogen) and Alexa 488‐conjugated anti‐rabbit (1:200, Invitrogen) diluted in 200 μL incubation medium. After incubation time, collector tubes were filled up with washing medium and centrifuged at 500 rcg for 5 min at 4°C. This step was repeated a total of three times to remove the secondary antibody excess. On the same day, single‐cell sorting of DRG cells was carried out using a FacsAria Fusion sorter (Beckton Dickinson), equipped with the ACDU (Automatic Cell Deposition Unit) option. Just before the sorting, cells were labeled with propidium iodide (PI, excitation at 561 nm – emission at 610/20 nm) to exclude permeabilized cells. Alexa488 was excited with a blue (488 nm) laser, and fluorescence collected at 530/30 nm. An unstained control was used to place the gate for the positive events. PI‐/TRK+ single‐cells were sorted into 96‐well plates containing RNase Inhibitor solution (Life technologies). After sorting, plates were centrifuged for 1 min at 4°C and stored at −80°C until their analysis.
RNA‐isolation and library construction (Smart‐seq2)
scRNA‐seq libraries were prepared using the NEBNext® Single Cell/Low Input cDNA Synthesis & Amplification Module (New England Biolabs) following the manufacturer's protocol. cDNA was amplified with 20 PCR cycles and validated in a Bioanalyzer High Sensitivity DNA Kit (Agilent), and between 2 and 5 ng of cDNA was then used to prepare the libraries which were also validated using a Bioanalyzer High Sensitivity DNA Kit. Equimolar proportions of each library were mixed, and the subsequent pool was quantified by qPCR and sequenced in a NextSeq High Output 2 × 75 run (Illumina).
Cell type clustering and transcriptome analysis
Reads were mapped using STAR software against the mouse reference GRCm38 and the Gencode annotation vM23. Thereafter, counts were obtained with HTSeq software and those results were assessed using MultiQC software. The total counts and percentage of mitochondrial (%mit) counts regarding all the counts per cells were calculated as a quality control measure. Cells that have more than 3 MADs (median absolute deviation) in some of these parameters were removed from the analysis as they were considered low‐quality cells. After that, normalization by deconvolution was applied: pool‐based size factors (pool counts from many cells to increase the size of the counts for accurate size factor estimation) were “deconvoluted” into cell‐based factors for normalization of each cell's expression profile. For dimensionality reduction, principal component analysis (PCA) was performed after scaling and centering the data. After that, a total of eight corrected clusters were obtained using t‐distributed stochastic neighborhood embedding (t‐SNE) graph‐based method. The determination of cluster identity (cell type) was performed by SingleR comparing expression to published datasets (http://mousebrain.org/ atlas as reference ‐Linnarsson lab Mouse Brain Atlas, 10 RRID:SCR_016999). Differentially expressed genes (DEGs) between cells coming from control and oxaliplatin‐treated animals were determined using the “SCDE” method. Gene Ontology (GO) enrichment analysis for biological processes was conducted by topGO. 11 Enrichment Analysis for Gene Ontology. R package version (2.42.0) on DEGs with p < 0.01 in both neuronal and SGC populations.
Western blot analysis
All DRGs from lumbar to cervical segments were dissected from cold saline (NaCl 0.9%) perfused mice and immediately snap freeze in liquid nitrogen. DRGs were then homogenized in RIPA lysis buffer adding 10 μL/mL of Protease Inhibitor cocktail (Sigma) and PhosSTOP phosphatase inhibitor cocktail (Roche). Protein was quantified by BCA protein assay (Thermo Scientific) following the manufactured protocol. 15–30 μg of protein of each sample were loaded into SDS‐polyacrylamide gels. The transfer was made 1.5 h at room temperature with a constant voltage of 90 V. Membranes were then blocked with 5% non‐fat dried milk (NFDM) or 5% Bovine Serum Albumin (BSA) in 0.1% Tween‐20 in tri‐buffered saline (TBS) for 1 h, and then incubated overnight at 4°C and in constant agitation with primary antibodies: phospho‐histo H2AX (Ser139) (γH2AX, 1:1000, Cell signaling), p21 (1:1000, Abcam), NF‐kB p65 (1:1000, Cell Signaling), calreticulin (1:1000, Thermo Fisher), Hsp70 (1:10000, Abcam), Hsp90 (1:1000, Abcam), eIF2α (1:1000, Cell Signalling), phosphor‐eIF2a (Ser51) (p‐eIF2α, 1:1000, Cell Signalling), actin (1:1000, Sigma), and GAPDH (1:1000, Millipore). After incubation with the appropriate horseradish peroxidase‐conjugated IgG, specific signals were determined using the enhanced chemiluminescence method with the clarity western ECL substrate (BioRad). Western blots were then analyzed using the Lane and band plug‐in from the Image Lab software (BioRad). Data were normalized first by the loading control (GAPDH or Actin) and afterward by the mean of the control samples.
Multiplex analysis for cytokine profile
Sciatic nerves and DRGs were rapidly harvested from mice perfused intracardially with cold saline, snap‐frozen in liquid nitrogen and kept at −80°C. The two sciatic nerves for each mouse were pooled for protein tissue extraction. Similarly, all the DRGs from lumbar to cervical segments were pooled for protein tissue extraction. For tissue processing, sciatic nerves and DRGs were manually homogenized in HEPES buffer (HEPES 6.25 mmol/L, IGEPAL 2%, PMSF 1 mmol/L, EGTA 1 mmol/L (pH 8), EDTA 16.9 mmol/L (pH 8), MgCl2 5 mmol/L) adding 10 μL/mL of protease inhibitory cocktail (Sigma) and PhosSTOP phosphatase inhibitor cocktail (Roche). Protein concentration was set to 3.3 μg/μL in DRG samples and 3 μg/μL in sciatic nerve samples. The protein levels of different cytokines were then determined and analyzed using a custom‐designed Milliplex Cytokine/Chemokine Magnetic Bead Panel on a MAGPIX system (EMD Millipore) in accordance with the manufacturer's protocol. Standard curves were generated using the specific standards supplied by the manufacturer. Data were normalized by the total protein concentration and afterward by the mean of the control samples.
Intraepidermal nerve fiber density (IENFD)
Plantar pads were fixed in 4% PFA in PBS for 1.5 h at 4°C, mounted in OCT and cryosections of 60 μm thickness were obtained. Sections were immunostained with anti‐protein gene product 9.5 (PGP9.5, 1:500, Cederlane) or anti‐calcitonin gene‐related peptide (CGRP, 1:500, Abcam) antibodies. The number of nerve fibers present in the epidermis of the paw pads was quantified in an epifluorescence microscope (Olympus BX51) using an appropriate filter. Five sections from each mouse were used to quantify the mean number and density of nerve fibers present in the epidermis of the paw pads.
Histological evaluation of myelinated axons
A segment of the sciatic nerve at mid‐thigh and the distal part of the tibial nerve were fixed in glutaraldehyde‐PFA (3%:3%) in PB 0.1 mol/L. The samples were then post‐fixed with 2% osmium tetroxide (Sigma) for 2 h, dehydrated in graded concentrations of ethanol and embedded in Eponate 12™ resin (Ted Pella Inc). Semithin sections 0.5 μm thick were stained with toluidine blue. To estimate the number of myelinated fibers in the sciatic and tibial nerves, axons were counted in images taken on a light microscope (Olympus BX40) attached to a digital camera (Olympus DP73) at 100x final magnification using Image J software (NIH, Bethesda, MA). At least 30% of the nerve cross‐section area was analyzed. Myelinated axons were manually counted, and total area of the field used was measured. The whole area of sciatic and tibial nerves was measured in 40x final magnification images using Image J software (NIH, Bethesda, MA). Finally, the total number of myelinated axons per nerve was estimated.
Immunofluorescence of DRG slices
DRGs were dissected, fixed in 4% PFA for 1.5 h at 4°C and mounted in OCT (Tissue Tek). 20 μm thick cryosections were obtained and incubated overnight at 4°C with primary antibodies against calreticulin (1:200, Thermo Fisher), Iba1 (1:500, FUJIFILM Wako Shibayagi), KLK5 (1:200, Abcam), and β‐III‐tubulin (1:500, Biolegend). Slices were then incubated with secondary antibodies overnight at 4°C. To detect signal, some antibodies were amplified by Biotin‐Streptavidin (calreticulin) or TSA method (KLK5) with the TSA Biotin Systems (Perkin Elmer, Cat#NEL700A001KT) following the manufactured protocol. Finally, samples were mounted with DAPI Fluoromount‐G media (Southern Biotech, Cat#0100–20). Using appropriate filters, images were taken with a confocal microscope (LSM 700 Axio Observer, Carl Zeiss) at 20× magnification chosen by systematic random sampling of squares. The same threshold of detection and binarization was applied to all the images. The number of positive neuronal nuclei for KLK5 or latexin proteins was quantified. Percentage of positive nuclei was calculated with respect to the total number of neurons. For calreticulin analysis, the percentage of neurons with surface expression of calreticulin was calculated. For KLK5 analysis, the percentage of neurons with nuclear expression of KLK5 was calculated. In both cases, among 150–250 neurons were quantified for each animal. For Iba1 analysis, the integrated density of Iba1 was measured in random sampling squares. β‐III‐tubulin antibody was used to label sensory neurons. All the editing and analysis of images were performed using the Image J software.
Statistical analysis
Statistical analysis of NCS and algesimetry tests were performed by a repeated measure (RM) two‐way ANOVA test. To compare one variable between multiple groups, a one‐way ANOVA or two‐way ANOVA test were used. The Bonferroni post hoc test was applied for multiple comparisons when needed. For more details on the specific statistical test used for each comparison and the exact value of n, please refer to the corresponding figure legend. GraphPad Prism v8.4.0 software was used for statistical inference analysis and graphically represents the data, which is expressed as group mean ± SD. The dots plotted within figures represent individual data points. Differences among groups or time points were considered significant at p < 0.05.
Results
Characterization of oxaliplatin neurotoxicity in the mouse
To explore the molecular mechanisms involved in oxaliplatin neurotoxicity, we first aimed to develop a mouse model of oxaliplatin neurotoxicity that reproduces the features observed in patients. 5 In order to do this, we injected oxaliplatin intravenously (i.v.) into adult mice for 8 weeks until reaching a total cumulated dose (TCD) of 80 mg/kg at the end of the administration time (Fig. 1A). Functional and behavioral tests to determine the severity of neuropathy were performed during the induction time and up to 2 weeks after oxaliplatin discontinuation to assess the so‐called “coasting‐effect,” a phenomenon seen in oxaliplatin‐treated patients that consists of the worsening of the neuropathy after chemotherapy discontinuation. 12 In the developed model, oxaliplatin treatment induced a pure sensory neuropathy in mice, seen by a progressive decrease in the amplitudes of SNAP and CNAP, statistically significant since 8 weeks, but without alterations in CMAP (Fig. 1B–D). After treatment discontinuation, the impairments in sensory amplitudes remained stable, and no coasting effect was observed (Fig. 1B and C). The absolute values of CMAP, SNAP, and CNAP are specified in Table S1. No signs of demyelination were observed in oxaliplatin‐treated mice as motor, sensory, and compound nerve latencies and nerve conduction velocities (NCV) were not affected along the treatment (Table S1).
Figure 1.
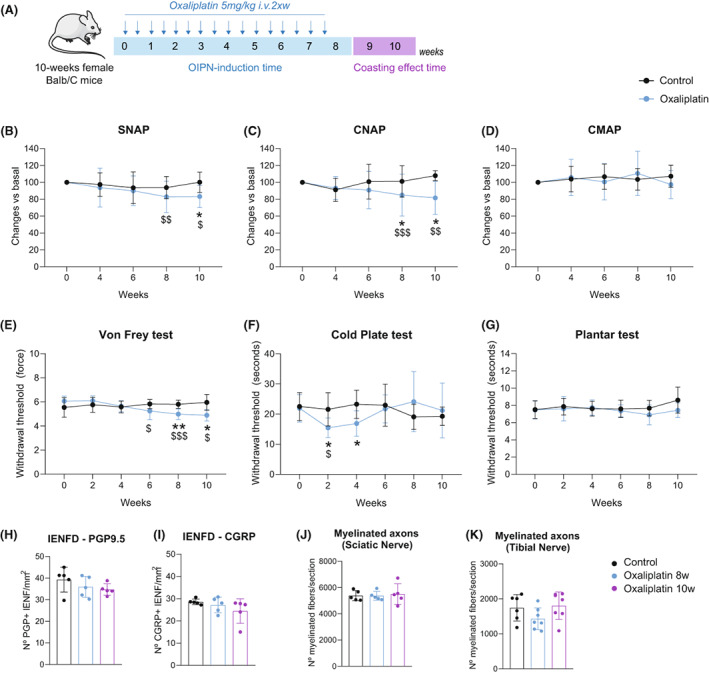
Electrophysiological, behavioral, and morphological characterization of the OIPN mouse model. (A) Oxaliplatin was administered i.v. twice a week (2 × w) for 8 weeks (Induction time). After this time, animals were further evaluated to assess the coasting effect for 2 additional weeks (coasting‐effect time). (B–D) Nerve conduction studies (NCS): SNAP (B), CNAP (C), and CMAP (D) recordings. Data are expressed as change vs basal. n = 15 Control mice and 35 Oxaliplatin‐treated mice. (E) Von Frey test of control and oxaliplatin‐treated mice. Withdrawal threshold (force, G) to mechanical stimuli is represented. n = 9 Control mice and 19 Oxaliplatin‐treated mice. (F) Cold plate test of control and oxaliplatin‐treated mice. Time (seconds) of first hind paw lift to −4°C noxious stimuli is represented. n = 10 Control mice and 13 Oxaliplatin‐treated mice. (G) Plantar test of control and oxaliplatin‐treated mice. Time (seconds) of paw lift to heat stimuli is represented. n = 8 Control mice and 10 Oxaliplatin‐treated mice. (H, I) Quantification of the number of PGP+ (H), and CGRP+ (I) IENF. n = 5 mice/group. (J, K) Quantification of the number of myelinated axons in the sciatic (J) and the tibial (K) nerves of control and oxaliplatin‐treated mice at 8 and 10 weeks. n = 4–5 mice/group in sciatic nerve; n = 7 mice/group in tibial nerve. *p < 0.05 **p < 0.01 vs Control;$ p < 0.05$$ p > 0.01$$$ p < 0.001 veruss Basal. 2‐Way RM ANOVA with Bonferroni post hoc test was used for the analysis of NCS and algesimetry tests. One‐way ANOVA with Bonferroni post hoc test was used for the analysis of histological studies. All data are represented as group mean ± SD.
In the Von Frey test, oxaliplatin‐treated animals had significantly reduced withdrawal thresholds to mechanical stimulation applied to the plantar paw ‐ indicative of mechanical allodynia. It started at 6 weeks of the study and remained until the end of the experiment (Fig. 1E). In the cold plate test, oxaliplatin treatment induced an early decrease in the time of first paw flick at 2 weeks and 4 weeks of treatment (Fig. 1F). However, at weeks 6, 8, and 10, animals treated with oxaliplatin had a slightly increased withdrawal time to the cold stimuli (Fig. 1F). The plantar hot test did not show differences between control and oxaliplatin‐treated mice at any time point evaluated (Fig. 1G).
At the morphological level, there was no marked evidence of degeneration of peripheral nerve fibers after oxaliplatin treatment, since both IENFD and number of myelinated axons in the sciatic and tibial nerves were not significantly different from control mice (Fig. 1H–K).
Identification of DRG cell populations by sc‐RNAseq
To reveal the changes in gene expression profile of sensory neurons and SGC after oxaliplatin treatment, a scRNA‐seq of previously isolated cells from the DRG of control and oxaliplatin‐treated mice was performed (Fig. 2A). The time point chosen for the gene expression analysis was 8 weeks, as it was the earlier time point in which reductions of SNAP and CNAP were observed.
Figure 2.
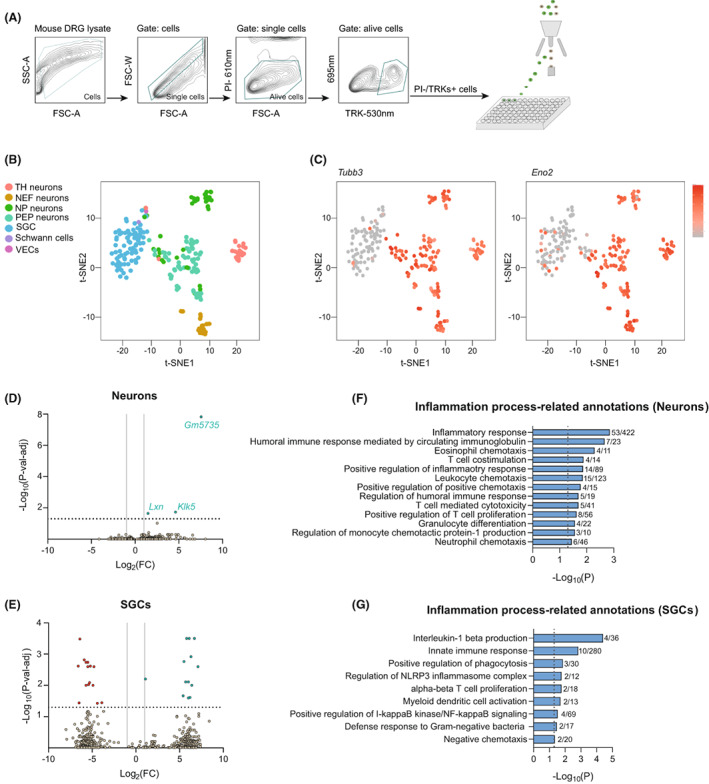
scRNA‐seq analysis of DRG cell population after oxaliplatin treatment. (A) Single‐cell sorting strategy used to isolate DRG cells. Only cells that were negative for propidium iodide (PI) and positive for TRKs labeling were selected and sorted into the 96‐well plates. All other events were discarded. (B) t‐SNE plot showing the distribution of different cell populations obtained in the analysis. (C) Neuronal populations overexpress the specific neuronal markers Tubb3 (left) and Eno2 (right). Color key represents normalized gene expression with the highest expression marked red and the lowest marked gray. (D, E) Volcano plots of DEGs (p < 0.05) between control and oxaliplatin‐treated mice in sensory neurons (D) and SGCs (E). Each point represents a single DEG. The negative log of p‐val‐adj (base 10) is plotted on the Y‐axis, and the log of the FC (base 2) is plotted on the X‐axis. Only DEGs above the dashed line have a p‐val‐adj <0.05. Green genes are up‐regulated and red genes are down‐regulated in oxaliplatin‐treated mice. Vertical lines indicate Log2(FC) of 1 or −1. Horizontal line indicates the point in which p‐val‐adj <0.05 (−log10(p‐val‐adj) = 1.3). (F, G) Inflammation process‐related GO annotations enriched in neurons (F) and SGCs (G) of oxaliplatin‐treated mice. Vertical lines indicate the point in which p < 0.05. The numbers right of the bars correspond to the “number of genes associated with the given GO term and found in the oxaliplatin vs control comparison”/“number of genes associated with the given GO term in the reference genome”.
After single‐cell sorting of DRG cells, only the ones meeting the quality control parameters (refer to methods for more specification about quality control parameters) were used for the comparative analysis. It corresponded to 113 cells from control mice and 136 cells from oxaliplatin‐treated mice. These cells were classified into seven distinct cell populations, which corresponded to Peptidergic neurons (PEP), Non‐Peptidergic neurons (NP), TH neurons, Neurofilament neurons (NEF), SGCs, Schwann cells, and Vascular Endothelial cells (VEC) (Fig. 2B). The top 20 genes that better differentiate each cellular population from the other ones can be found in File S1. The neuronal identity of NEF, PEP, NP, and TH populations is seen by the over‐expression of Tubb3 and Eno2 neuronal markers in those subsets 13 (Fig. 2C). To increase the number of neurons to be compared between experimental conditions (Oxaliplatin vs Control), we grouped the different neuronal clusters in one single population called “Neurons”. With that strategy, we increased the number of cells to be compared between groups and so the chances to find DEGs. At the end, the total number of cells used for the comparison analysis corresponded to 97 Neurons and 39 SGCs coming from oxaliplatin‐treated mice, and 68 Neurons and 45 SGCs coming from control mice. VECs and Schwann cells were not compared between experimental groups. The average number of reads per cell obtained corresponded to 1.57 × 106.
When comparing the gene expression profile in the neuronal population, we found a total of 561 DEGs with p < 0.05 between control and oxaliplatin‐treated mice. From those genes, only three have a p‐adj‐val < 0.05. These genes corresponded to Klk5, Lxn, and the unprocessed pseudogene Gm573 (Fig. 2D; File S2). Regarding SGCs, we found a total of 516 DEGs with p < 0.05. From those, only 28 genes had a p‐adj‐val < 0.05 (Fig. 2E; File S2). GO analysis on DEGs with p < 0.01 revealed an enrichment of biological processes related to the global function “inflammation process” in both neuronal and SGC populations of oxaliplatin‐treated mice (Fig. 2F and G; File S3).
Inflammatory response as a mechanism of oxaliplatin neurotoxicity
The protein product of the Klk5 gene is a member of the kallikrein (KLK) protein family, serine proteases known to be involved in a diverse range of physiological and pathophysiological processes. 14 We did not find increased levels of KLK5 protein in DRGs of oxaliplatin‐treated mice by western blot when compared with control animals (Fig. S1A). However, we did find an increased number of neuronal nuclei positive for KLK5 protein in a 66.6% of animals treated with the platinum drug and used for the scRNA‐seq (Fig. S1B). As proteases, kallikreins activate protease‐activated receptors (PARs), which in turn regulate different signaling pathways and physiological functions. Among them is the pathway of mitogen‐activated protein kinases that links PARs with NF‐kB pathways modulating pro‐inflammatory responses, 15 , 16 hyperexcitability of nociceptors and sensitization of afferent neurons to mechanical stimuli. 17 We observed that animals treated with oxaliplatin had increased levels of NF‐kB p65 protein in their DRGs at 8 weeks (Fig. 3A). It has been reported that activation of PARs favors the migration of inflammatory cells, vascular permeability, and leukocyte extravasation, contributing to the development of an acute inflammatory environment with increased levels of pro‐inflammatory cytokines. 18 Consistent with that, oxaliplatin‐treated mice had increased levels of different pro‐inflammatory cytokines in DRGs and sciatic nerves at 8 weeks of the study (Fig. 3B). These cytokines have been previously related to the recruitment of immune cells, 19 and DRGs of animals treated with oxaliplatin showed increased immunoreactivity against Iba1 at 8 weeks, compatible with infiltration of myeloid cells (Fig. 3C). All these changes were restored at 10 weeks of the study. As no signs of nerve degeneration were seen at the histological level in our model (Fig. 1H–K), the pro‐inflammatory response reported above was attributed to an intrinsic response of DRG cells after oxaliplatin treatment and not a consequence of axonal degeneration. A higher number of infiltrated cells was seen in semithin sections of the sciatic nerve in oxaliplatin‐treated mice compared to controls at 8 weeks (Fig. S2). However, these cells had no features that suggested active phagocytosis that characterize the foamy macrophages found during Wallerian degeneration 20 ; supporting the idea that no peripheral degeneration was taking place in our model.
Figure 3.
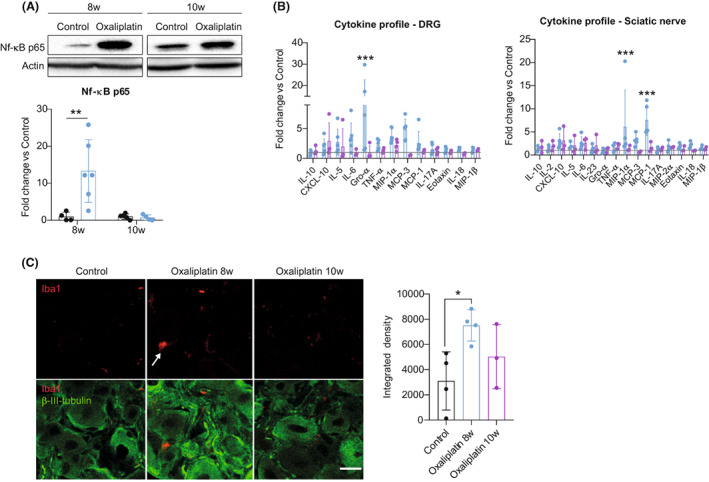
Oxaliplatin treatment induces a pro‐inflammatory response to mice peripheral nervous system. (A) Representative western blots of NF‐kB p65 protein levels in DRGs of control and oxaliplatin‐treated mice at 8 and 10 weeks of the study. Graph below shows the corresponding quantification. n = 4–6 mice/group. Two‐way ANOVA with Bonferroni post hoc test. (B) Bar graphs showing the fold change of cytokine protein levels in DRGs (left) and sciatic nerves (right) of oxaliplatin‐treated mice at 8 and 10 weeks. Horizontal lines indicate fold increase equal 1 corresponding to control animals. n = 3–6 mice/group. Two‐way ANOVA with Bonferroni post hoc test. (C) Representative images of Iba1 staining (red) in the DRG of control and oxaliplatin‐treated mice at 8 and 10 weeks. β‐III‐tubulin (green) was used to label neurons. Scale bar: 20 μm. Graph in the right shows the quantification of Iba1 intensity (Integrated density). n = 3–4 mice/group. One‐way ANOVA with Bonferroni post hoc test. *p < 0.05 vs control **p < 0.01 versus control ***p < 0.001 versus control. All data are represented as group mean ± SD.
On the other hand, oxaliplatin has been involved in the activation of the so‐called Immunogenic Cell Death (ICD) in front of tumor cells, 21 and some cytokines up‐regulated in our model participate in the establishment of this response. 19 To check whether ICD plays a role in oxaliplatin neurotoxicity, we checked different hallmarks of this response, 22 including the protein levels of calreticulin, the Heat Shock Protein (Hsp) 70, Hsp90, and the phosphorylated eIF2α protein at Ser51 (p‐eIF2α). Levels of these ICD‐related proteins were not different between the control and oxaliplatin‐treated mice at any time point evaluated (Fig. 4A). Moreover, when checking the translocation of calreticulin protein to the cell surface of DRG neurons ‐ another classical marker of ICD ‐ we found no changes between experimental groups (Fig. 4B). This data suggested that ICD was not activated in sensory neurons in response to oxaliplatin treatment.
Figure 4.
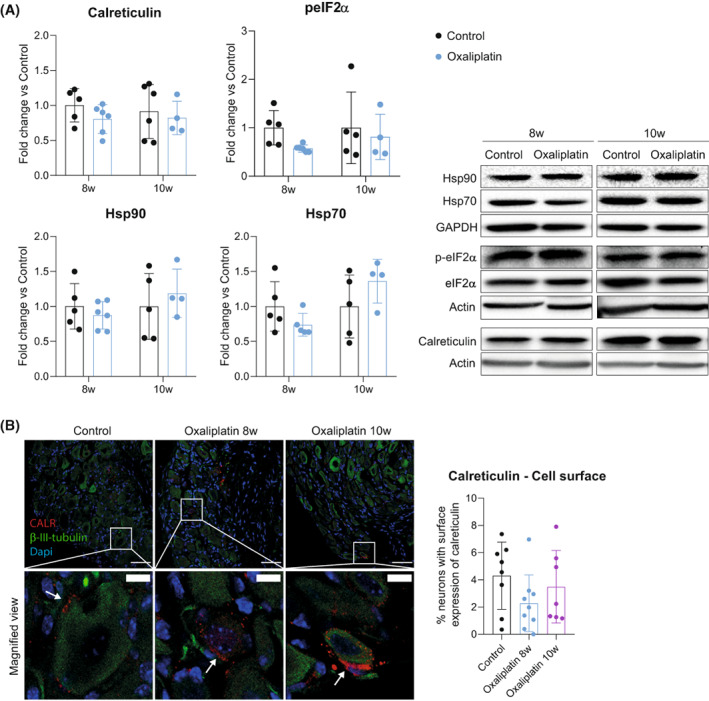
ICD is not activated in the DRG as a response to oxaliplatin treatment. (A) Left: quantification of the protein levels of ICD hallmarks (calreticulin, peIF2a, Hsp90, and Hsp70) in DRGs of control and oxaliplatin‐treated mice at 8 and 10 weeks. Right: representative western blots of each protein analyzed. n = 4–6 mice/group. Two‐way ANOVA with Bonferroni post hoc test. Data are expressed as fold change vs the average of the control group for each time point (group mean ± SD). (B) Representative images of calreticulin staining (CALR, red) in DRG of control and oxaliplatin‐treated mice at 8 and 10 weeks of the study. β‐III‐tubulin (green) was used to label neurons and nuclei were counterstained with Dapi (blue). White boxes are amplified in the images below (magnified view) for a better visualization of calreticulin staining. White arrows indicate calreticulin staining in the cell surface. Scale bar: 100 μm; Scale bar of magnified views: 20 μm. Graph in the right shows the quantification of the percentage of neurons that express calreticulin in their cell surface. n = 7–9 mice/group. One‐way ANOVA with Bonferroni post hoc test. All data are represented as group mean ± SD.
DNA damage is rapidly repaired after discontinuation of oxaliplatin treatment
The main mechanism reported for platinum drugs neurotoxicity, including oxaliplatin, is the death of sensory neurons by apoptosis due to DNA damage. 6 In the scRNA‐seq analysis, some DEGs (p < 0.05) of the neuronal population were related to the DNA damage response (DDR), including Mif, Rbbp8, Xrcc1, Spred1, and Mpm1 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 (File S2). In line with that, the levels of γH2AX protein, a fast responder in front of DNA damage 34 were increased in DRG of animals treated with oxaliplatin compared to controls at 8 weeks. This increase was restored at 10 weeks, that is, after discontinuing the treatment (Fig. 5). Previous results showed that DNA damage induced by cisplatin, another platinum drug, activates a senescence phenotype to DRG sensory neurons. 35 , 36 Considering that γH2AX expression has been related to the establishment of senescence, 34 we checked for p21 protein levels; a key inductor of senescence in general 34 , 37 and in the cisplatin model previously reported by our group. 35 Although p21 protein levels tend to increase in DRG of oxaliplatin‐treated mice at 8 weeks, it did not reach one of significance. Moreover, p21 protein levels were normalized at 10 weeks (Fig. 5).
Figure 5.
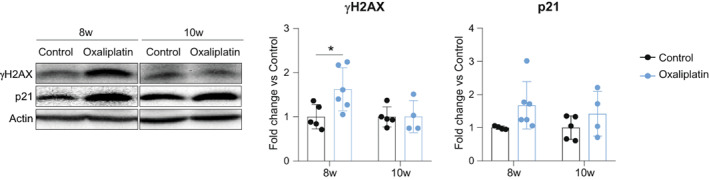
Oxaliplatin treatment induces a transient DNA damage in mice DRGs. Left: representative western blots of γH2AX and p21 protein levels in DRGs of control and oxaliplatin‐treated mice at 8 and 10 weeks of the study. Right: graphs with the corresponding quantification. n = 4–5 mice/group. Two‐way ANOVA with Bonferroni post hoc test. *p < 0.05. Data are represented as group mean ± SD.
Discussion
The findings of this study indicate that oxaliplatin administration causes a deleterious impact on the conduction of sensory nerve fibers and a reduction in mechanical and cold withdrawal thresholds to mice – all together with a transient DNA damage and inflammatory response in peripheral nerves and DRGs. Supporting these findings, we found increased levels of pro‐inflammatory cytokines, NF‐kB p65 protein, and Iba1 myeloid cells in DRG after oxaliplatin treatment. Moreover, an increase in the levels of pro‐inflammatory cytokines and mononuclear infiltrating cells were observed in peripheral nerves of oxaliplatin‐treated mice. These changes are endorsed by the results of the scRNA‐seq, in which many GO annotations related to the general biological process “inflammatory process” appeared enriched in the oxaliplatin‐treated mice.
More specifically, the Klk5 gene appeared up‐regulated in sensory neurons of oxaliplatin‐treated mice. As previously mentioned, the KLK‐PAR‐NFkB axis favors the migration of inflammatory cells to tissue, increases the levels of pro‐inflammatory cytokines and sensitizes peripheral nociceptors which results in the establishment of pain‐like behaviors. 14 , 15 , 16 , 17 Therefore, up‐regulation of Klk5 in DRG neurons may orchestrate the pro‐inflammatory response observed in oxaliplatin‐treated animals, which in turn could lead to the mechanical allodynia seen by the Von Frey test in our model. Although we did not find increased levels of KLK5 protein in DRGs of oxaliplatin‐treated mice, about 40% of animals had increased number of neurons with KLK5 protein translocation to the nucleus. KLKs are mainly known as secreted proteins with extracellular functions, and the contribution of their nuclear localization to the pathogenesis of human diseases has not been studied. Nevertheless, it has been reported that KLK4, another member of the kallikrein family, is predominantly a nuclear protein acting as a transcriptomic factor in prostate cancer. 38 , 39 Considering that data, other KLKs could have nuclear roles in some pathological conditions.
In addition to Klk5, other DEGs obtained by the scRNA‐seq endorse the pro‐inflammatory response seen in the peripheral nervous system of mice treated with oxaliplatin. Lxn, the other DEG found in the neuronal population, has been associated with inflammatory responses like acute pancreatitis and inflammatory lung disease. 40 In fact, it is highly expressed in inflammatory mast cells and is induced by stimulation of mouse macrophages. 41 , 42 Thus, Lxn up‐regulation in sensory neurons after oxaliplatin treatment could favor the inflammatory response observed in our animal model. On the other hand, the Kyat3 gene, which appeared down‐regulated in SGCs of oxaliplatin‐treated mice, encodes for an enzyme participating in the kynurenine pathway ‐ the major metabolic route of tryptophan metabolism. It has been reported that cytokines can affect the expression of various kynurenine enzymes, thus orchestrating metabolic changes that can influence immune cell responses and vice versa. 43 Several studies have demonstrated that altered kynurenine metabolism plays an important role in the pathogenesis of several neurodegenerative diseases. 44 In that sense, Kyat3 down‐regulation in SGCs could favor the accumulation of excitotoxic metabolites and pro‐inflammatory cytokines contributing to oxaliplatin neurotoxicity. Moreover, it highlights the importance of studying SGCs as active players in the induction of peripheral neuropathies. In fact, it is reported that increased reactivity of other glial cells (i.e., astrocytes and microglia) in the spinal cord is involved in oxaliplatin‐induced neuropathic pain, 45 , 46 thus supporting the relevance of glial cells in the neurotoxic effects of oxaliplatin.
In addition to an inflammatory response, oxaliplatin treatment also induced DNA damage to DRG neurons, seen by increased expression of γH2AX protein at 8 weeks of the study. However, the levels of this protein were completely restored after 2 weeks of treatment discontinuation, indicating that the DNA damage was rapidly repaired. Indeed, results obtained from the scRNA‐seq show that some genes related to the DDR were modulated toward a DNA damage repair. 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 γH2AX has been related to the cellular senescence response, 34 and previous results from our laboratory showed that the permanent cisplatin‐induced DNA damage activates a senescence phenotype in DRG sensory neurons. 35 The fast DNA repair after oxaliplatin treatment cessation could avoid the development of a full and durable senescent phenotype. In fact, permanent activation of DDR is necessary for both the initiation and the maintenance of senescent phenotypes. 47 Whether the lesions in the DNA template induced by oxaliplatin can be more easily repaired than those induced by cisplatin it is unknown. However, it has been reported that the presence of a DACH group in the oxaliplatin compound, which makes it different from the rest of platinum drugs, results in the formation of platinum‐DNA adducts that are structurally different from those induced by cisplatin. 48 , 49 The failure to establish a senescence response after oxaliplatin treatment can also be related to the intensity of the genotoxic damage induced. Despite our mouse model of oxaliplatin neurotoxicity encompassing both acute and chronic neuropathy features observed in patients, the TCD of oxaliplatin would correspond to 236.48 mg/m2 in the human equivalent. 50 , 51 At this dose, the phenotypic acute syndrome in patients is fully established, but the chronic features start to appear in patients 52 a fact that matches with the mild severity of NCS alterations in our mice model.
Overall, the results obtained from this study indicate that oxaliplatin treatment induces a mild neuropathy in mice characterized by a pro‐inflammatory response in DRG and peripheral nerves. Interestingly, at this stage of low‐intensity damage, the pro‐inflammatory response is attenuated after treatment discontinuation. Thus, it may be an early and key step toward a progressive and long‐lasting neuropathy. Moreover, it may also be implicated with some of the features related to the acute syndrome. In that scenario, anti‐inflammatory‐based strategies emerge as promising therapies for peripheral neuropathy induced by oxaliplatin, at least during the early phases to prevent further neuropathic events.
Conflict of Interest
The authors declare no competing interests.
Author Contributions
EU and JB designed the study. AC and JB conducted the experiments. AC and LME acquired the data. MT, NB, and FC conducted the scRNA‐seq data analysis. AC, ATE, LME, XN, VY, EU, and JB analyzed and interpreted the data. AC, LME, XN, VY, EU, and JB wrote the manuscript. All authors have read and approved the final version.
Supporting information
Figure S1. KLK5 protein levels in the DRG of oxaliplatin‐treated mice. A representative blots of KLK5 protein levels in control and oxaliplatin‐treated mice at 8 and 10 weeks of the study. Graph below shows the quantification of protein levels of KLK5 at each time point. n = 4–6 mice per group. Two‐way ANOVA with Bonferroni post hoc for multiple comparisons. Data are expressed as fold change vs the average of the control group for each time point (group mean ± SD). B Left: Representative images of KLK5 staining (red) in the DRG of control and oxaliplatin‐treated mice at 8 weeks. b‐III‐tubulin (green) was used to stain the neuronal population and nuclei were counterstained with Dapi (blue). White box is magnified for a better visualization of KLK5+ neuronal nuclei. White arrows indicate KLK+ neuronal nuclei. Scale bar: 100 μm; Scale bar of magnified view: 50 μm. Graph in the right shows the quantification of the percentage of DRG neuronal nuclei immunoreactive against KLK5 protein in both experimental groups. n = 7–9 mice per group. One‐way ANOVA with Bonferroni post hoc test for multiple comparisons. Data are represented as group mean ± SD.
Figure S2. Infiltrated cells in sciatic nerves of oxaliplatin‐treated mice. Representative image of a sciatic nerve cross‐section of a mouse treated with oxaliplatin at 8 weeks of the study. Infiltrated cell is highlighted with a red box and amplified for a better visualization. Scale bar: 20 μm. Scale bar of magnified view: 5 μm. Red asterisk indicates a myelinated axon. Graph in the right shows the quantification of the number of infiltrated cells in the sciatic nerve of control and oxaliplatin‐treated mice at 8 and 10 weeks. n = 4–6 mice per group. One‐way ANOVA with Bonferroni post hoc test for multiple comparisons. *p < 0.05 versus Control. Data are represented as group mean ± SD.
Table S1. Absolute values of nerve amplitudes, latencies, and conduction velocities recorded in control and oxaliplatin‐treated mice at the different time points of the study. CMAP: Compound muscle action potential; SNAP: Sensory nerve action potential; CNAP: Compound nerve action potential; MNCV: Motor nerve conduction velocity; SNCV: Sensory nerve conduction velocity; CNVC: Compound nerve conduction velocity; AV: Group Average; SD: Standard Deviation. n = 15 control mice; n = 35 oxaliplatin‐treated mice.
File S1. Top20 genes of each cellular population identified. Top20 genes that better differentiate NEF (Tab2), TH (Tab3), NP (Tab4), PEP (Tab5), and SGC (Tab6) populations from the rest. Columns correspond to: gene name (A), rank of the gene, where the set of genes with Top ≤X is the union of the top X genes (ranked by p‐value) from each pairwise comparison involving each cluster analyzed comparison (B), p‐value (C), adjusted p‐value by the FDR method (D), summary effect size for each gene and cluster, defined as the effect size from the pairwise comparison with the min.prop‐smallest p‐value (E), fold change of the specific gene in a given population when comparing to other cellular populations (F‐I).
File S2. List of DEGs for Oxaliplatin versus Control experimental conditions in Neurons (Tab2) and SGCs (Tab3). Columns correspond to: gene name (A), gene type (B), log2 fold expression difference value, reported as maximum likelihood estimate (mle, C), 95% lower bound (lb), and upper bound (ub) of the log2 fold expression difference value (D, E), conservative estimate (ce) of the log2 fold expression difference value (upper or lower bound, whichever is closer to 0; or 0 if the 95% confidence interval includes 0) (F), statistical significance of the expression differences, expressed as a signed Z score (G), Z score adjusted for multiple hypothesis testing (cZ, H), adjusted‐p‐value obtained from cZ (p‐val‐adj, I).
File S3. List of GO terms (Biological process) enriched in Oxaliplatin vs Control comparison in Neurons (Tab2) and SGCs (Tab3). Columns correspond to: ID of the GO process (A), name of the GO process (B), Number of genes associated with a given GO term in the reference genome (Mus musculus, C), number of genes associated with a given GO term and found in the Oxaliplatin versus Control comparison (D), number of genes expected to be found randomly (E), statistical significance of the expression differences detected by fisher, weight method (F).
Acknowledgments
The author's research was supported by funds from CIBERNED and TERCEL networks to XN, and by a PI210018 grant to JB from the Instituto de Salud Carlos III of Spain, co‐funded by European Union (ERDF/ESF, “Investing in your future”). JB has received support from grant number SLT008/18/00028 from the Department of Health of the Government of Catalonia, CERCA Program. AC was recipient of a predoctoral fellowship from the Secretaria d'Universitats i Recerca of the Catalan Government and the European Social Fund (FI fellowship).
Funding Information The author's research was supported by funds from CIBERNED and TERCEL networks to XN, and by a PI210018 grant to JB from the Instituto de Salud Carlos III of Spain, co‐funded by European Union (ERDF/ESF, “Investing in your future”). JB has received support from grant number SLT008/18/00028 from the Department of Health of the Government of Catalonia, CERCA Program. AC was recipient of a predoctoral fellowship from the Secretaria d'Universitats i Recerca of the Catalan Government and the European Social Fund (FI fellowship). FC was funded by grants RTI2018‐096824‐B‐C22 and PID2021‐125106OB‐C32 from MCIN/AEI/10.13039/501100011033/ and FEDER Una manera de hacer Europa.
Funding Statement
This work was funded by Agència de Gestió d'Ajuts Universitaris i de Recerca grant FI‐DGR; Instituto de Salud Carlos III of Spain grant PI210018; the Department of Health of the Government of Catalonia, CERCA Program grant SLT008/18/00028; CIBERNED ; TERCEL; European Union ; European Social Fund ; MCIN/AEI grant RTI2018‐096824‐B‐C22; FEDER Una manera de hacer Europa grant PID2021‐125106OB‐C32.
References
- 1. Grothey A, Goldberg RM. A review of oxaliplatin and its clinical use in colorectal cancer. Expert Opin Pharmacother. 2004;5(10):2159‐2170. [DOI] [PubMed] [Google Scholar]
- 2. Hato SV, Khong A, de Vries IJ, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum‐based chemotherapeutics. Clin Cancer Res. 2014;20(11):2831‐2837. [DOI] [PubMed] [Google Scholar]
- 3. Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy‐induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol. 2021;82(1):51‐77. [DOI] [PubMed] [Google Scholar]
- 4. Mols F, Beijers T, Lemmens V, van den Hurk CJ, Vreugdenhil G, van de Poll‐Franse LV. Chemotherapy‐induced neuropathy and its association with quality of life among 2‐ to 11‐year colorectal cancer survivors: results from the population‐based PROFILES registry. J Clin Oncol. 2013;31(21):2699‐2707. [DOI] [PubMed] [Google Scholar]
- 5. Park SB, Lin CS, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Oxaliplatin‐induced neurotoxicity: changes in axonal excitability precede development of neuropathy. Brain. 2009;132(Pt 10):2712‐2723. [DOI] [PubMed] [Google Scholar]
- 6. Calls A, Carozzi V, Navarro X, Monza L, Bruna J. Pathogenesis of platinum‐induced peripheral neurotoxicity: insights from preclinical studies. Exp Neurol. 2020;325:113141. [DOI] [PubMed] [Google Scholar]
- 7. Velasco R, Bruna J. Oxaliplatin neurotoxicity. Curr Colorectal Cancer Rep. 2014;10(3):303‐312. [Google Scholar]
- 8. Marmiroli P, Riva B, Pozzi E, et al. Susceptibility of different mouse strains to oxaliplatin peripheral neurotoxicity: phenotypic and genotypic insights. PloS One. 2017;12(10):e0186250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruna J, Alberti P, Calls‐Cobos A, Caillaud M, Damaj MI, Navarro X. Methods for in vivo studies in rodents of chemotherapy induced peripheral neuropathy. Exp Neurol. 2020;325:113154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexa A, Rahnenfuhrer J. topGO: enrichment analysis for gene ontology. R package version 2.48.0. 2020.
- 11. Zeisel A, Hochgerner H, Lönnerberg P, et al. Molecular architecture of the mouse nervous system. Cell. 2018;174(4):999‐1014.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brouwers EE, Huitema AD, Boogerd W, Beijnen JH, Schellens JH. Persistent neuropathy after treatment with cisplatin and oxaliplatin. Acta Oncol. 2009;48(6):832‐841. [DOI] [PubMed] [Google Scholar]
- 13. Usoskin D, Furlan A, Islam S, et al. Unbiased classification of sensory neuron types by large‐scale single‐cell RNA sequencing. Nat Neurosci. 2015;18:145‐153. [DOI] [PubMed] [Google Scholar]
- 14. Oikonomopoulou K, Hansen KK, Saifeddine M, et al. Kallikrein‐mediated cell signalling: targeting proteinase‐activated receptors (PARs). Biol Chem. 2006;387:817‐824. [DOI] [PubMed] [Google Scholar]
- 15. Clements JA, Willemsen NM, Myers SA, Dong Y. The tissue kallikrein family of serine proteases: functional roles in human disease and potential as clinical biomarkers. Cri Rev Clin Lab Sci. 2004;41(3):265‐312. [DOI] [PubMed] [Google Scholar]
- 16. Rothmeier AS, Ruf W. Protease‐activated receptor 2 signaling in inflammation. Semin Immunopathol. 2012;34:133‐149. [DOI] [PubMed] [Google Scholar]
- 17. Jimenez‐Vargas NN, Pattison LA, Zhao P, et al. Protease‐activated receptor‐2 in endosomes signals persistent pain of irritable bowel syndrome. Proc Natl Acad Sci USA. 2018;115(31):E7438‐E7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mella C, Figueroa CD, Otth C, Ehrenfeld P. Involvement of kallikrein‐related peptidases in nervous system disorders. Front Cellular Neurosci. 2020;14:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Showalter A, Limaye A, Oyer JL, et al. Cytokines in immunogenic cell death: applications for cancer immunotherapy. Cytokine. 2017;97:123‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Udina E, Voda J, Gold BG, Navarro X. Comparative dose‐dependence study of FK506 on transected mouse sciatic nerve repaired by allograft or xenograft. J Peripher Nerv Syst. 2003;8(3):145‐154. [DOI] [PubMed] [Google Scholar]
- 21. Rébé C, Demontoux L, Pilot T, Ghiringhell F. Platinum derivatives effects on anticancer immune response. Biomolecules. 2019;10(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galluzzi L, Vitale I, Warren S, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8(1):e000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003;22(22):6068‐6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurki S, Peltonen K, Latonen L, et al. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2‐mediated degradation. Cancer Cell. 2004;5(5):465‐475. [DOI] [PubMed] [Google Scholar]
- 25. Quennet V, Beucher A, Barton O, Takeda S, Löbrich M. CtIP and MRN promote non‐homologous end‐joining of etoposide‐induced DNA double‐strand breaks in G1. Nucleic Acids Res. 2011;39(6):2144‐2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Makharashvili N, Tubbs AT, Yang SH, et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double‐strand break end resection. Mol Cell. 2014;54(6):1022‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. London RE. The structural basis of XRCC1‐mediated DNA repair. DNA Repair. 2015;2015(30):90‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pasmant E, Gilbert‐Dussardier B, Petit A, et al. SPRED1, a RAS MAPK pathway inhibitor that causes Legius syndrome, is a tumour suppressor downregulated in paediatric acute myeloblastic leukaemia. Oncogene. 2015;34(5):631‐638. [DOI] [PubMed] [Google Scholar]
- 29. Zhang R, Zhang Y, Lu X, et al. SPRED1 is downregulated and a prognostic biomarker in adult acute myeloid leukemia. Front Oncol. 2020;10:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia W, Zhang F, Xie C, Jiang M, Hou M. Macrophage migration inhibitory factor confers resistance to senescence through CD74‐dependent AMPK‐FOXO3a signaling in mesenchymal stem cells. Stem Cell Res Ther. 2015;6(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xia W, Hou M. Macrophage migration inhibitory factor rescues mesenchymal stem cells from doxorubicin‐induced senescence though the PI3K‐Akt signaling pathway. Int J Mol Med. 2018;41(2):1127‐1137. [DOI] [PubMed] [Google Scholar]
- 32. Yamakuchi M, Hashiguchi T. Endothelial cell aging: how miRNAs contribute? J Clin Med. 2018;7(7):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dominic A, Banerjee P, Hamilton DJ, Le N, Abe JI. Time‐dependent replicative senescence vs. disturbed flow‐induced pre‐mature aging in atherosclerosis. Redox Biol. 2020;37:101614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21‐dependent senescence‐like phenotype driven by a DNA damage response. Aging Cell. 2012;11(6):996‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Calls A, Torres‐Espin A, Navarro X, Yuste VJ, Udina E, Bruna J. Cisplatin‐induced peripheral neuropathy is associated with neuronal senescence‐like response. Neuro Oncol. 2021;23(1):88‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Acklin S, Zhang M, Du W, et al. Depletion of senescent‐like neuronal cells alleviates cisplatin‐induced peripheral neuropathy in mice. Sci Rep. 2020;10:14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Georgakilas AG, Martin OA, Bonner WM. p21: a two‐faced genome Guardian. Trends Mol Med. 2017;23(4):310‐319. [DOI] [PubMed] [Google Scholar]
- 38. Klokk TI, Kilander A, Xi Z, et al. Kallikrein 4 is a proliferative factor that is overexpressed in prostate cancer. Cancer Res. 2007;67(11):5221‐5230. [DOI] [PubMed] [Google Scholar]
- 39. Xi Z, Klokk TI, Korkmaz K, et al. Kallikrein 4 is a predominantly nuclear protein and is overexpressed in prostate cancer. Cancer Res. 2004;64(7):2365‐2370. [DOI] [PubMed] [Google Scholar]
- 40. Ji B, Chen XQ, Misek DE, et al. Pancreatic gene expression during the initiation of acute pancreatitis: identification of EGR‐1 as a key regulator. Physiol Genomics. 2003;14(1):59‐72. [DOI] [PubMed] [Google Scholar]
- 41. Uratani Y, Takiguchi‐Hayashi K, Miyasaka N, Sato M, Jin M, Arimatsu Y. Latexin, a carboxypeptidase a inhibitor, is expressed in rat peritoneal mast cells and is associated with granular structures distinct from secretory granules and lysosomes. Biochem J. 2000;346(Pt 3):817‐826. [PMC free article] [PubMed] [Google Scholar]
- 42. Aagaard A, Listwan P, Cowieson N, et al. An inflammatory role for the mammalian carboxypeptidase inhibitor latexin: relationship to cystatins and the tumor suppressor TIG1. Structure. 2005;13(2):309‐317. [DOI] [PubMed] [Google Scholar]
- 43. Baumgartner R, Forteza MJ, Ketelhuth D. The interplay between cytokines and the kynurenine pathway in inflammation and atherosclerosis. Cytokine. 2019;122:154148. [DOI] [PubMed] [Google Scholar]
- 44. Tan L, Yu JT, Tan L. The kynurenine pathway in neurodegenerative diseases: mechanistic and therapeutic considerations. J Neurol Sci. 2012;323(1‐2):1‐8. [DOI] [PubMed] [Google Scholar]
- 45. Lee JH, Kim W. The role of satellite glial cells, astrocytes, and microglia in oxaliplatin‐induced neuropathic pain. Biomedicines. 2020;8(9):324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Di Cesare ML, Pacini A, Micheli L, Tani A, Zanardelli M, Ghelardini C. Glial role in oxaliplatin‐induced neuropathic pain. Exp Neurol. 2014;261:22‐33. [DOI] [PubMed] [Google Scholar]
- 47. Rodier F, Coppé JP, Patil CK, et al. Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chaney SG, Campbell SL, Bassett E, Wu Y. Recognition and processing of cisplatin‐ and oxaliplatin‐DNA adducts. Crit Rev Oncol Hematol. 2005;53(1):3‐11. [DOI] [PubMed] [Google Scholar]
- 49. Woynarowski JM, Faivre S, Herzig MC, et al. Oxaliplatin‐induced damage of cellular DNA. Mol Pharmacol. 2020;58(5):920‐927. [DOI] [PubMed] [Google Scholar]
- 50. Reagan‐Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659‐661. [DOI] [PubMed] [Google Scholar]
- 51. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7(2):27‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bruna J, Videl S, Argyriou AA, et al. Efficacy of a novel Sigma‐1 receptor antagonist for oxaliplatin‐induced neuropathy: a randomized, double‐blind, placebo‐controlled phase IIa clinical trial. Neurotherapeutics. 2018;15:178‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. KLK5 protein levels in the DRG of oxaliplatin‐treated mice. A representative blots of KLK5 protein levels in control and oxaliplatin‐treated mice at 8 and 10 weeks of the study. Graph below shows the quantification of protein levels of KLK5 at each time point. n = 4–6 mice per group. Two‐way ANOVA with Bonferroni post hoc for multiple comparisons. Data are expressed as fold change vs the average of the control group for each time point (group mean ± SD). B Left: Representative images of KLK5 staining (red) in the DRG of control and oxaliplatin‐treated mice at 8 weeks. b‐III‐tubulin (green) was used to stain the neuronal population and nuclei were counterstained with Dapi (blue). White box is magnified for a better visualization of KLK5+ neuronal nuclei. White arrows indicate KLK+ neuronal nuclei. Scale bar: 100 μm; Scale bar of magnified view: 50 μm. Graph in the right shows the quantification of the percentage of DRG neuronal nuclei immunoreactive against KLK5 protein in both experimental groups. n = 7–9 mice per group. One‐way ANOVA with Bonferroni post hoc test for multiple comparisons. Data are represented as group mean ± SD.
Figure S2. Infiltrated cells in sciatic nerves of oxaliplatin‐treated mice. Representative image of a sciatic nerve cross‐section of a mouse treated with oxaliplatin at 8 weeks of the study. Infiltrated cell is highlighted with a red box and amplified for a better visualization. Scale bar: 20 μm. Scale bar of magnified view: 5 μm. Red asterisk indicates a myelinated axon. Graph in the right shows the quantification of the number of infiltrated cells in the sciatic nerve of control and oxaliplatin‐treated mice at 8 and 10 weeks. n = 4–6 mice per group. One‐way ANOVA with Bonferroni post hoc test for multiple comparisons. *p < 0.05 versus Control. Data are represented as group mean ± SD.
Table S1. Absolute values of nerve amplitudes, latencies, and conduction velocities recorded in control and oxaliplatin‐treated mice at the different time points of the study. CMAP: Compound muscle action potential; SNAP: Sensory nerve action potential; CNAP: Compound nerve action potential; MNCV: Motor nerve conduction velocity; SNCV: Sensory nerve conduction velocity; CNVC: Compound nerve conduction velocity; AV: Group Average; SD: Standard Deviation. n = 15 control mice; n = 35 oxaliplatin‐treated mice.
File S1. Top20 genes of each cellular population identified. Top20 genes that better differentiate NEF (Tab2), TH (Tab3), NP (Tab4), PEP (Tab5), and SGC (Tab6) populations from the rest. Columns correspond to: gene name (A), rank of the gene, where the set of genes with Top ≤X is the union of the top X genes (ranked by p‐value) from each pairwise comparison involving each cluster analyzed comparison (B), p‐value (C), adjusted p‐value by the FDR method (D), summary effect size for each gene and cluster, defined as the effect size from the pairwise comparison with the min.prop‐smallest p‐value (E), fold change of the specific gene in a given population when comparing to other cellular populations (F‐I).
File S2. List of DEGs for Oxaliplatin versus Control experimental conditions in Neurons (Tab2) and SGCs (Tab3). Columns correspond to: gene name (A), gene type (B), log2 fold expression difference value, reported as maximum likelihood estimate (mle, C), 95% lower bound (lb), and upper bound (ub) of the log2 fold expression difference value (D, E), conservative estimate (ce) of the log2 fold expression difference value (upper or lower bound, whichever is closer to 0; or 0 if the 95% confidence interval includes 0) (F), statistical significance of the expression differences, expressed as a signed Z score (G), Z score adjusted for multiple hypothesis testing (cZ, H), adjusted‐p‐value obtained from cZ (p‐val‐adj, I).
File S3. List of GO terms (Biological process) enriched in Oxaliplatin vs Control comparison in Neurons (Tab2) and SGCs (Tab3). Columns correspond to: ID of the GO process (A), name of the GO process (B), Number of genes associated with a given GO term in the reference genome (Mus musculus, C), number of genes associated with a given GO term and found in the Oxaliplatin versus Control comparison (D), number of genes expected to be found randomly (E), statistical significance of the expression differences detected by fisher, weight method (F).