

Abstract
Recent genome-wide association studies (GWAS) and whole-exome sequencing of neuropsychiatric disorders, especially schizophrenia, have identified a plethora of common and rare disease risk variants/genes. Translating the mounting human genetic discoveries into novel disease biology and more tailored clinical treatments is tied to our ability to causally connect genetic risk variants to molecular and cellular phenotypes. When combined with the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated (Cas) nuclease-mediated genome editing system, human induced pluripotent stem cell (hiPSC)-derived neural cultures (both 2D and 3D organoids) provide a promising tractable cellular model for bridging the gap between genetic findings and disease biology. In this review, we first conceptualize the advances in understanding the disease polygenicity and convergence from the past decade of iPSC modeling of different types of genetic risk factors of neuropsychiatric disorders. We then discuss the major cell types and cellular phenotypes that are most relevant to neuropsychiatric disorders in iPSC modeling. Finally, we critically review the limitations of iPSC modeling of neuropsychiatric disorders and outline the need for implementing and developing novel methods to scale up the number of iPSC lines and disease risk variants in a systematic manner. Sufficiently scaled-up iPSC modeling and a better functional interpretation of genetic risk variants, in combination with cutting-edge CRISPR/Cas9 gene editing and single-cell multi-omics methods, will enable the field to identify the specific and convergent molecular and cellular phenotypes in precision for neuropsychiatric disorders.
Keywords: Neuropsychiatric disorders, Schizophrenia, Genetics, Human induced pluripotent stem cells (hiPSC), Modeling
1. Introduction
The last decade of psychiatric research has witnessed the success of genome-wide association studies (GWAS). Through unbiased genome-wide interrogation of millions of genetic markers, i.e., single nucleotide polymorphisms (SNPs), in large population case/control samples, GWAS of schizophrenia (SZ) and other neuropsychiatric disorders have identified hundreds of risk loci with common genetic risk variants (Psychiatric Genomics Consortium-Schizophrenia, 2011; Psychiatric Genomics Consortium-Schizophrenia, 2014; Mullins et al., 2021; PGC3 et al., 2020; Purcell et al., 2009; Ripke and Concortium, 2013; Shi et al., 2009; Stefansson et al., 2009). Of these, SZ GWAS has the most successes: the recent Psychiatric Genomics Consortium (PGC) (PGC2) reported 145 genome-wide significant SZ risk loci (Psychiatric Genomics Consortium-Schizophrenia, 2014; Pardinas et al., 2018), which were further expanded by the upcoming PGC wave 3 (~270 SZ risk loci) (The Schizophrenia Working Group of the Psychiatric Genomics Consortium et al., 2020). The common disease risk variants implicated by these GWAS often have small population effect sizes (odds ratios, OR < 1.2) (Bassett et al., 2010; Levinson et al., 2011; Marshall et al., 2017; Szatkiewicz et al., 2014), hindering the mechanistic understanding of disease pathophysiology.
These genome-wide studies in large samples also revealed another side of the risk spectrum for neuropsychiatric disorders: rare copy number variants (CNVs, i.e., genomic segments that are duplicated or deleted) of higher penetrance. SZ has the largest number of reproducibly associated CNVs, which include deletions at 1q21.1, 2p16.3 (NRXN1), 3q29, 15q13.3, distal 16p11.2, 22q11.2, and duplications at 7q11.23 and proximal 16p11.2 (Bassett et al., 2010; Levinson et al., 2011; Marshall et al., 2017; Szatkiewicz et al., 2014). These rare and large (usually >100 kb) CNVs often show much larger effect sizes (OR of 2–70) than common SNPs (OR < 1.2) (Bassett et al., 2010; Levinson et al., 2011; Marshall et al., 2017; Szatkiewicz et al., 2014). Besides these rare CNVs, recent large-scale whole-exome sequencing (WES) studies have also unraveled rare protein-coding variants that are associated with SZ with relatively large effect sizes (Singh et al., 2017). By analyzing exome variants in 24,000 SZ patients and 97,000 controls, the SZ Exome Sequencing Meta-Analysis (SCHEMA) Consortium reported ~10 genes with ultra-rare loss-of-function (LoF) mutations (or protein-truncating variants) that collectively reached genome-wide significant association with SZ (Singh et al., 2020). Together with common disease risk variants identified from GWAS, these genetic findings have provided unprecedented opportunities for the neuropsychiatric field to better understand disease biology.
Despite the tremendous progress made in identifying neuropsychiatric risk variants, effective treatments of these disorders remain scarce and largely rely on old drugs. For instance, most antipsychotic drugs for treating positive symptoms of SZ target dopamine D2 receptors (DRD2), a discovery that was made almost half a century ago (Creese et al., 1976; Howes et al., 2012; Seeman and Lee, 1975; Snyder, 1976). Furthermore, their use has been impeded by side effects such as extrapyramidal symptoms and tardive dyskinesia (Nasrallah, 2008). Although atypical antipsychotic drugs such as clozapine and risperidone can improve negative symptoms, and cognitive function with fewer extrapyramidal symptoms by targeting not only DRD2 but also non-dopamine targets such as serotonin and glutamine, such non-specificity of targets may contribute to a number of side effects of concern, e.g., weight gain, glucose dysregulation and dyslipidemia. Thus, translating these genetic findings of neuropsychiatry into novel disease biology and potentially more tailored clinical interventions is highly needed, which requires not only our conceptual understanding of the complexities of polygenic neuropsychiatric disorders but also a comprehensive approach that integrates knowledge from different experimental models.
Human postmortem brain tissues and animal models (Carlson et al., 2011; Dong et al., 2013; Jeong et al., 2006) have provided invaluable insights into plausible disease pathophysiology, but each model has its pros and cons. The postmortem brain is not living tissue and mostly does not capture changes at early neuronal developmental stages (Brennand et al., 2015). Furthermore, postmortem brain study is well-known for confounding factors related to tissue variability and some uncontrollable environmental factors (Lipska et al., 2006), and the postmortem brain is not amenable to genetic modification. Although rodent models can be genetically modified for studying psychiatric disorders, they often do not faithfully recapitulate human pathophysiology and behaviors. Moreover, because regulatory variants are often species-specific (Shen et al., 2012), animal models may not elicit the expected functional impact of human genetic variations (Johnson et al., 2009). Peripheral blood cells and B-cell transformed lymphoblastoid cell lines (LCLs) from psychiatric patients and controls of relatively large numbers have also been used as ex vivo models to reveal disease genetic effects on gene expressions (Arloth et al., 2015; Duan et al., 2018; Kos et al., 2018; Mostafavi et al., 2014). For instance, with LCLs of over 1000 SZ cases and controls, we found that the disease-associated differentially expressed genes upon cellular stimulation by dopamine (DA) were enriched for genes related to immune processes and apoptosis as well as mitochondrial oxidative phosphorylation, and interestingly, were overrepresented by those near genome-wide significant SZ loci and within SZ-associated CNVs (Duan et al., 2018; Kos et al., 2018). Although blood cells and LCLs may be useful in providing mechanistic insight for disease risk factors related to the long-standing immune hypothesis of the neuropsychiatric disorder (Heath and Krupp, 1967; Pouget, 2018; van Mierlo et al., 2020), such a cellular model has its obvious limitations, i.e., further-removed from the brain that is presumably the most relevant tissue for neuropsychiatric disorders.
Compared with postmortem brains or animal models, stem cell-based cellular models provide a promising alternative for studying common and rare genetic risk factors of neuropsychiatric disorders (Panchision, 2016; Wen et al., 2016). Benefiting from the revolutionary induced pluripotent stem cell (iPSC) technology discovered by Dr. Yamanaka (Takahashi et al., 2007; Takahashi and Yamanaka, 2006), somatic cells such as fibroblasts or blood cells from patients or healthy controls can be reprogramed into pluripotent stem cells (i.e., human iPSCs) simply by exogenously expressing some transcription factors (i.e., Yamanaka factors: Oct3/4, Sox2, Klf4, c-Myc) (Takahashi et al., 2007; Takahashi and Yamanaka, 2006). iPSCs can then be re-differentiated into different brain cell types that are relevant to neuropsychiatric disorders. iPSC model enables studying disease-relevant molecular and cellular phenotypic changes in a temporal and cell type-specific manner. More importantly, iPSC models are amenable to genetic modification or epigenomic perturbation, which is important for studying the functional impacts of disease risk variants. When combined with the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated (Cas) nuclease-mediated genome editing system (Cong et al., 2013; Fu et al., 2013; Mali et al., 2013; Sander and Joung, 2014; Shalem et al., 2014; Wang et al., 2013), iPSC-derived neurons represent a powerful cellular model for understanding disease biology underlying neuropsychiatric genetic findings. In light of previous reviews about using iPSC models for studying psychiatric disorders over the years (Das et al., 2020; De Los Angeles et al., 2021; Dolmetsch and Geschwind, 2011; Duan, 2015; Duan et al., 2019; Durak and Tsai, 2014; Fernando et al., 2020; Hoffmann et al., 2019; Jacobs, 2015; Michael Deans and Brennand, 2021; Miller and Kelsoe, 2017; Quadrato et al., 2016; Rajarajan et al., 2020; Soliman et al., 2017; Temme et al., 2016; Wen et al., 2016; Wright et al., 2014; Young-Pearse and Morrow, 2016), here we will focus on conceptualizing the key advances in the field (Fig. 1, Table 1 and Table S1), the validity of the model, interpretation of the results, limitations and new research opportunities.
Fig. 1.
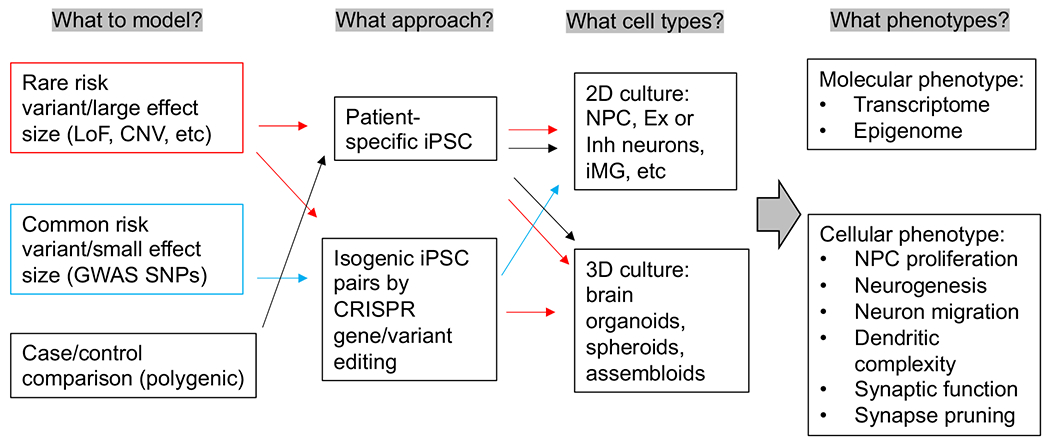
The type of neuropsychiatric risk variants and the disease relevant cellular phenotypes in iPSC modeling. Red, blue or black arrow indicates iPSC modeling for rare variants/CNV, common variants, or polygenic risk factors, respectively. LoF = loss of function; CNV = copy number variants; Ex = excitatory neurons; Inh = inhibitory neurons; iMG = iPSC-derived microglia. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
hiPSC models of neuropsychiatric risk variants (only studies with clearly defined disease risk variants and cellular phenotypes are listed).
| Author/year | Ref | Disease | Risk variant/gene | Perturbation | Sample size | Brain cell type | Cellular phenotypes |
|---|---|---|---|---|---|---|---|
| Urresti 2021 | 34433918 | ASD | 16p11.2 CNV | Comparative | 3 × DEL & 3 × DUP + 3 ctrls | Organoids | Excess of neurons and depletion of NPCs in DELs; Organoid size recapitulates macrocephaly and microcephaly phenotypes observed in the patients. |
| Roth 2020 | 33169669 | ASD | 16p11.2 CNV | Comparative | 10 DEL/3 DUP + 4 ctrls | Cortical neurons | Significant correlation between transcription modules and clinical phenotypes in 16pDS patients. |
| Deshpande 2017 | 29212016 | ASD | 16p11.2 CNV | Comparative | 3 × DEL & 3 × DUP + 4 ctrls | Neurons | Increased soma size and dendrite length in 16p DEL, which were decreased in 16p DUP neurons. Both exhibited reduced synaptic density. |
| Deneault 2019 | 30747104 | ASD | 16p11.2 DEL, NRXN1Δ/+ | Comparative; CRISPR/Cas9 | 14 ASD + 11 ctrls | Glutamatergic neurons | Consistent spontaneous network hyperactivity for CNTN5--deficient or EHMT2/UBE2I-variant. |
| Carbonell 2019 | 31388001 | ASD | ANKS1B | Comparative | 2 cases + 2 ctrls | Neurons | Loss of ANKS1B led to altered synaptogenesis and neurodevelopment. |
| Deneault 2018 | 30392976 | ASD | FF2/FMR2, ANOS1, ASTN2, etc. | CRISPR/Cas9; isogenic | 10 isogenic hiPSC lines | Excitatory neurons | Electrophysiological deficits were distinct for different mutations. Consistent reduction of synaptic activity and reduced sEPSC frequencies. |
| Avazzadeh 2021 | 34525970 | ASD | NRXN1α | Comparative | 3 NRXN1α+/− + 5 ctrls | Cortical neurons | Higher sodium currents and action potential, and accelerated depolarization time. |
| Zaslavsky 2019 | 30911184 | ASD | SHANK2 DEL (R841X) | CRISPR/Cas9 | 2–4 isogenic lines | Cortical neurons | Increased synapse numbers and longer dendrites. Hyperconnectivity and increased sEPSC frequency. |
| Wang 2021 | bioRxiv | ASD | SHANK3 | CRISPR/Cas9; isogenic | 3 mutants + 3 ctrls | Cortico-striatal organoids | Smaller organoids with fewer and smaller neurons. Fewer synapsin1/SHANK3-containing excitatory synaptic puncta and high excitability. |
| Gouder 2019 | 30643170 | ASD | SHANK3 (E809X, Q1243X, G1271Afs*15, L1142Vfs*153) | Comparative | 4 SHANK3-het ASD + 3 ctrls | Pyramidal cortical neurons | Significant reduction in dendritic spine densities and whole spine and spine head volumes. |
| Chiola 2021 | 33558651 | ASD | SHANK3 hemizygosity | CRISPR/Cas9; xenografting | 4 PSC lines + 4 ctrls | Astrocytes & glut neurons | Impaired AMPA-mediated synaptic transmission, dendritic arbors, and spines in the mouse cortex. |
| Fink 2021 | 34538422 | ASD (AS) | 15q11-q13 DUP | Comparative | 4 Dup + 6 ctrls | Excitatory & inhibitory neurons | Increased excitatory synaptic activity, dendritic density, and action potential. Decreased inhibitory synaptic transmission. |
| Utami 2020 | 32653109 | ASD (FXS) | FMR1 | CRISPR/Cas9 | 4 FXS + 3 ctrls (1 isogenic) | GABAergic & glutamatergic | Abnormal FXS neural rosette formation. Overall impairment of electrophysiological network activities. |
| Das Sharma 2020 | 32560741 | ASD (FXS) | FMR1 | CRISPR/Cas9; comparative | 3 FXS + 3 ctrls (1 isogenic) | Cortical neurons | No observed differences in the intrinsic properties of FXS neurons. Shorter and more frequent spontaneous action potential firing in FXS neurons. |
| Achuta 2017 | 27411166 | ASD (FXS) | FMR1 | Comparative | 4 FXS + 3 ctrls | NPCs and neurospheres | Intracellular calcium response to the mGluR agonist DHPG was augmented in FXS NPCs. |
| Lutz 2020 | 32522805 | ASD (PMS) | SHANK3 deficiency | Comparative | 5 PMS + 3 ctrls | Neurons/myotubes | Shortened Z-discs and severe impairment of acetylcholine receptor clustering in PMS myotubes. |
| Roessler 2018 | 30456368 | ASD (PMS) | 22q13.3 DEL | CRISPR/Cas9; comparative | 3 DEL & 2 CRISPR + 1 ctrl | NPCs & cortical neurons | Impairment of neuronal maturation and reduced overall activity in 22q DEL. reduced protein levels of SHANK3, JIP2, DCX, and NeuN. |
| Trujillo 2021 | 33501759 | ASD (RTT) | MECP2 LoF (Q83X, K82 frameshift) | CRISPR/Cas9; drug screening | 2 mutants + 2 ctrls | Neurons & organoids | Nefiracetam and PHA 543613 reversed the cytologic neuropathology in MECP2-KO neurons, increased calcium activity in mosaic neurospheres. |
| Chen 2021 | 32851591 | ASD (RTT) | MECP2 | CRISPR/Cas9 mutagenesis | 2 mutants + 3 isogenic ctrls | Cortical neurons | RTT neurons lack electrophysiological maturation. Spine density is impaired. Synaptic transmission abnormalities were observed in MECP2 KD neurons. |
| Gomes 2020 | 33363173 | ASD (RTT) | MeCP2: R255X(XX), MeCP2:Q83X(XY) | Comparative | 2 MeCP2 RTT + 4 ctrls | Forebrain organoids | Premature development of the deep-cortical layer neurons, low NPCs, and altered calcium dynamics. Impaired interneuron migration in RTT forebrain organoids. |
| Xiang 2020 | 32526163 | ASD (RTT) | BRD4, MECP2 (c.397C > T, C.808C > T, c.916C > T) | CRISPR/Cas9 | 5 RTT + 2 ctrls (1 isogenic) | Interneurons and brain organoids | Abnormal increases in BRD4 binding cause the abnormal transcription of mutant MeCP2 in RTT interneurons.) |
| Nott 2016 | 27428650 | ASD (RTT) | MECP2 (R306C) | CR1SPR/Cas9; comparative | 1 mutant + 1 isogenic ctrl | NPCs | Deficits in HDAC3 and FOXO recruitment and gene expression. CRISPR editing rescued the impaired HDAC3-FOXO-mediated phenotype in NPCs. |
| Williams 2014 | 24419315 | ASD (RTT) | MECP2 | Comparative | 3 RTT + 3 ctrls | Astrocyte & neuron | Mutant astrocytes had adverse effects on the morphology and function of wild-type neurons. |
| Ananiev 2011 | 21966470 | ASD (RTT) | MECP2 (R306C, T158M, R294X, V247X) | CRISPR/Cas9; comparative | 4 MeCP2 + 4 ctrls | Neurons | RTT neurons were smaller than their isogenic controls. |
| Kim 2011 | 21807996 | ASD (RTT) | MECP2 (T158M, Q244X, E235fs, R306C, X487W) | Comparative | 5 RTT + 5 ctrls | NPCs & neurons | Neuronal maturation deficits. |
| Marchetto 2010 | 21074045 | ASD (RTT) | MECP2 (1155del32, R306C, Q244X, T158M) | Comparative | 4 RTT + 5 ctrls | NPCs & neurons | RTT NPCs had normal proliferation. RTT neurons had reduced glutamatergic synapse numbers and abnormal morphologies. |
| Panagiotakos 2019 | 31868578 | ASD (TS) | CACNA1C | Comparative | 3 TS + 3 ctrls | NPCs & neurons | SATB2+ neurons were less abundant while CTIP2+ neurons were more abundant. |
| Paşca 2011 | 22120178 | ASD (TS) | CACNA1C | Comparative | 2 TS + 3 ctrls | NPCs & neurons | Deficient Ca2+ signaling, decreased lower cortical layers, abnormal tyrosine hydroxylase expression, and increased production of norepinephrine and dopamine. |
| Nadadhur 2019 | 30581017 | ASD (TSC) | TSC1 (2249G > A), TSC2 (1563dupA/H522T) | Comparative | 2 TSC + 3 ctrls | Neurons & oligodendrocytes | Increased dendritic branching and network activity. Oligodendrocytes had increased proliferation and decreased maturation. |
| Becker 2020 | 32929080 | ASD, MICPCH | CASK (c. 1296 + 1 G > T), CASK (Xp11.4 DUP) | Comparative | 2 mutants + 2 ctrls | Neurons | Altered presynaptic development and affecting their excitatory/inhibitory balance. |
| Jiang 2019 | 30135510 | BP | TRANK1 (rs9834970T > C, rs906482G > A) | Comparative; drug treatment | 2 BP + 8 ctrls | NPCs & neurons & astrocytes | The NPCs with the risk allele of rs9834970 had lower baseline TRANK1 expression. |
| Yoshimizu 2015 | 25403839 | MDD, BP, SZ | CACNA1C (rs1006737 G > A) | Comparative | 10 BP + 4 SZ + 2 MDD + 8 unaffected | Neurons | L-type VGCC current density, as well as mRNA expression of CACNA1C, were increased. |
| Wen 2014 | 25132547 | MDD, SZ | DISC1 | Comparative | 4 DISC1-het (2 isogenic) + 4 ctrls | NPCs & cortical neurons | Synaptic vesicle release deficits in forebrain neurons. Dysregulated expression of many synaptic genes associated with psychiatric disorders. |
| Sundberg 2021 | 34006844 | SZ | 16p11.2 CNV | CRISPR/Cas9; isogenic | 3 isogenic lines & 1 x 16p11.2 ctrl | Dopaminergic neurons (DA) | 16p DUP showed deficits in neuronal differentiation. 16p DEL had increased soma size and hyperactivity, which can be rescued by RHOA inhibition (Rhosin). |
| de Vrij 2018 | 29302076 | SZ | CSPG4 (c.391G > A [p.A131T]) | Comparative | 3 SZ + 3 related ctrls | OPCs | Aberrant cellular morphology, and myelination potential. |
| Srikanth 2018 | 30410030 | SZ | DISC1, UNC5D | CRISPR/dCas9-VPR for KD | 2 mutants + 2 isogenic ctrls | Neurons | DISC1 mutations did not seem to alter cell fate, presynaptic protein expression, or electrophysiological activity. Reduced neurite outgrowth. |
| Shrode 2019 | 31548722 | SZ | FURIN (rs4702), SNAP91, TSNARE1 | CRISPR/Cas9; CRISPRa/i | 4 CRISPR-edited + 4 ctrls | NPCs, glut & GABAergic | Reduced neurite length and firing rates in rs4702 GG neurons. sEPSCs frequency/amplitude increased in SNAP91 CRISPRa (decreased with CRISPRi). |
| Forrest 2017 | 28803920 | SZ | MIR137 (rs1198588) | CRISPR/Cas9 | 1 isogenic pair (2 clones) | Glutamatergic neurons | The Risk allele is associated with lower MIR137 expression, increased dendritic complexity, and synaptic puncta positive for GluA1 and PSD95. |
| Pak 2021 | 34035170 | SZ | NRXN1 | Patient-specific & TALENs | 3 patients + 3 ctrls; 3 edited | Neurons | A deficit in spontaneous & evoked synaptic events responses, synaptic paired-pulse depression. Impaired synaptic function regardless of genetic background. |
| Zhang 2021 | bioRxiv | SZ | VPS45, AC244033.2, or C1orf54 (rs2027349) | CRISPR/Cas9; isogenic | 2 isogenic pairs (2–3 clones) | Excitatory neurons | Risk allele A is associated with increased dendritic complexity, synaptic puncta density, and hyperactivity; cis-regulated all three genes with synergistic effects. |
| Li 2021c | 34009292 | SZ (DGS) | 22q11.2 DEL | Comparative | 8 × DEL + 6 ctrls | Neurons | Reduced OXPHOS activity and ATP levels. |
| Li 2019 | 31740674 | SZ (DGS) | 22q11.2 DEL, MRPL40 | CRISPR/Cas9; comparative | 4 × DEL & 1 KO + 5 ctrls | Neurons | Reduced levels of mitochondrial-encoded proteins, ATP, and electron transport chain activity. |
| Khan 2020 | 32989314 | SZ (DGS), ASD | 22q11.2 DEL, DGCR8 | CRISPR/Cas9; comparative | 12 × 22q11DS + 11 ctrls | Cerebral cortical organoids | Defects in spontaneous neuronal activity and calcium signaling. DGCR8+/− neurons show similar neuronal dysfunction and behavior to 22q11DS neurons. |
| Flaherty 2019 | 31784728 | SZ (psychotic) | NRXNα | Comparative | 4 NRXN1α+/− + 4 ctrls | Glutamatergic & GABAergic | Aberrant and differentially-expressed NRXN1 isoforms. Deficits in neuronal activity depending on expressed NRXN1α isoforms in a genotype-dependent manner. |
| Li 2021d | 33833053 | SZ, ASD | 16p11.2 CNV, CD47 | Comparative | 11 SZ and ASD lines + 3 ctrls | NPCs & OPCs | CD47 is overexpressed in 16p11.2 DEL contributing to reduced phagocytosis. |
| Johnstone 2018 | 30401811 | SZ, GAD/MDD | 16p13.11 DUP, TSC2 (16:2115634:C/T) | Comparative | 3 × 16p13.11 DUP + 5 ctrls | NPC & organoids | 16p13.11 DUP NPCs had proliferation deficits and cerebral organoids were smaller and had altered radial glial progenitor cell division fates. |
2. What to model: rare vs. common disease risk variants
Translating the mounting human genetic discoveries into novel disease biology and more tailored clinical treatments is tied to our ability to causally connect genetic risk variants to molecular and cellular phenotypes. iPSC model has been used for studying the functional impacts of both rare and common disease risk variants. Depending on the effect size of a modeled variant, either patient-specific iPSC lines or CRISPR-engineered isogenic iPSC lines have been employed. However, because of the polygenic nature of neuropsychiatric disorders, regardless of the variant penetrance or the experimental design, the resulting molecular and cellular phenotypes may be confounded by the individual donor’s genetic background and need to be interpreted cautiously.
2.1. Rare CNVs
There are overwhelmingly more studies of iPSC modeling of rare disease-associated CNVs, of which autism and SZ are the two most commonly modeled disorders. The large effect sizes of these CNVs make them “low-hanging fruits” among other genetic findings, an ideal model for understanding disease biology and interpreting the disease relevance of cellular phenotypes. Of the CNVs with established reproducible associations with SZ and/or autism at 1q21.1, 2p16.3 (NRXN1), 3q29, 7q11.23, 15q13.3, 16p11.2, and 22q11.2 (Bassett et al., 2010; Levinson et al., 2011; Marshall et al., 2017; Szatkiewicz et al., 2014), all except for the SZ-associated 3q29 deletion (Sefik et al., 2020) have at least one iPSC modeling study that reported a cellular phenotype (Adamo et al., 2015; Avazzadeh et al., 2021; Chailangkarn et al., 2016; Chapman et al., 2021; Crockett et al., 2021; Flaherty et al., 2019; Gillentine et al., 2017; Khan et al., 2020; Khattak et al., 2015; Lalli et al., 2016; Li et al., 2021a; Li et al., 2021b; Roth et al., 2020; Sundberg et al., 2021; Toyoshima et al., 2016; Zanella et al., 2019; Zhang et al., 2021b) (Table 1). Because of their large effect sizes, these studies exclusively use patient-specific iPSC lines, i.e., directly derived from patients who carry the CNV, except for one that used CRISPR-Cas9 engineered 16p11.2 duplication (16pdup) and 16p11.2 deletion (16pdel) (Sundberg et al., 2021). The patient-specific CNV carriers are then compared with matched population control lines to determine any meaningful molecular (i.e., often transcriptomic profile) and/or cellular phenotypic characteristics associated with a CNV. Because these CNVs tend to be extremely rare in the population, individual studies often have a very small sample size, ranging from 1 to 15 patient lines. Not surprisingly, the largest study (n = 15 cases) was with 22q11.2 deletion (Khan et al., 2020), the most prevalent CNV with the strongest association with SZ.
Likely due to the small and variable number of CNV carriers in each study, despite the high penetrance of these studied CNVs, iPSC modeling studies frequently yield discordant phenotypes. Even for the same CNV, different studies often test different hypotheses and report discordant molecular and cellular phenotypes. For instance, the largest study of 22q11.2 deletion (Khan et al., 2020) analyzed transcriptomic profiles at different developmental stages of patient-specific iPSC-derived cortical spheroids and identified a deficit gene pathway related to calcium channel activity. Interestingly, although neurons dissociated from the cortical spheroids of 22q11.2 deletion delayed spontaneous hyperactivity, their calcium transmission activity was found significantly impaired upon neuronal depolarization, which can later be reversed by antipsychotic drugs targeting DRD2 (Khan et al., 2020). However, two other studies of patient-specific 22q11.2 deletions reported dysfunction of mitochondria biogenesis and blood-brain barrier (BBB), respectively (Crockett et al., 2021; Li et al., 2021b). For another commonly modeled CNV region, SZ-associated 16pdup and autism-associated 16pdel, one study using patient-specific CNV carriers revealed increased soma size and dendrite length in 16pdel neurons and reduced neuronal size and dendrite length in 16pdup neurons, and interestingly, both 16pdel and 16pdup neurons (excitatory) displayed reduced synaptic density (Deshpande et al., 2017). Although seemingly consistent neuronal hyperactivity was reported for CRISPR-engineered 16pdel, it was only observed in iPSC-derived dopaminergic neurons and no obvious phenotype was observed for 16pdup (Sundberg et al., 2021). Moreover, of the two other studies of patient-specific 16pdel, one reported that the overexpression of CD47 (a “don’t eat me” signal) in both neural progenitor cells (NPCs) and oligodendrocyte progenitor cells (OPCs) of 16pdel carriers may contribute to the reduced phagocytosis and brain overgrowth in autism-associated macrocephaly (Li et al., 2021a), while the other study found substantial transcriptional alterations associated with early neural development without any reported cellular phenotypic changes in 16del carriers (Roth et al., 2020).
Alternatively, such phenotypic discrepancies may be attributed to the fact that these large CNVs span multiple genes. However, even for 2pl6.3 deletion that only involves a single gene, NRXN1, there are still inconsistencies between different studies. A multi-center study of NRXIV1-deletion lines reported a large decrease of spontaneous synaptic events, evoked synaptic responses, and synaptic paired-pulse depression in excitatory neurons, regardless of genetic backgrounds (Pak et al., 2021). While the reduced neuronal activity in NRXIV1-deletion lines was also observed in another study (Flaherty et al., 2019), a most recent study seemed to show larger sodium currents, higher AP amplitude, and accelerated depolarization time, i.e., increased excitability, in cortical neurons carrying NRXN1 deletion (Avazzadeh et al., 2021). Therefore, the discrepancies across iPSC modeling of CNVs were likely due to the intrinsic clinical heterogeneity associated with each CNV and the effects of individual genetic backgrounds. Future iPSC modeling of CNVs with an increased number of patient-specific iPSC lines in combination with CRISPR-engineering of CNVs may help improve the consistency and identify more convergent disease-specific phenotypes.
Because these rare CNVs are usually long (>100 kb) and span multiple genes, it has been a challenge to identify which gene(s) within a CNV region are likely the driver(s) for disease-relevant phenotypes. The challenge is amplified by the possible effects of a CNV on local or distal chromatin architecture and, consequently, the expression of genes outside the CNV region (Franke et al., 2016; Redin et al., 2017). iPSC model in combination with CRISPR/Cas9 gene editing or CRISPRi/CRISPRoff (Kampmann, 2020; Nunez et al., 2021) to knockdown (KD) individual gene expression within a CNV would be an effective approach to solve the problem. The only iPSC modeling study that made such a systematic effort was for 15q13.3 microdeletion by analyzing the transcriptomic similarity of individual gene KD with the entire CNV in day-6 (post neural induction) neurons, which however did not point to any specific gene (Zhang et al., 2021b). The imprecision of mapping the drive gene(s) in this study (Zhang et al., 2021b) may be improved by using more mature neurons (e.g., 4 weeks rather than day-6 neurons). Towards this end, in an independent study, combining with targeted resequencing of CNV genes in an SZ case/control cohort and iPSC modeling of the patient-specific loss-of-function (LoF) mutation of OTUD7A (OTU Deubiquitinase 7A), we found that OUTD7A LoF resulted in reduced dendrite complexities, synaptic protein puncta densities in spines, and impaired electrophysiology (Kozlova et al., 2022), which recapitulates the cellular phenotypes of the CNV modeling in animals (Yin et al., 2018), supporting OUTD7A as a plausible driver gene for 15q13.3 microdeletion.
2.2. Rare protein-coding variants
Most iPSC modeling of rare protein-coding variants, including missense or protein-truncating mutations, is for monogenic-like autism spectrum disorders. Both patient-specific iPSC lines carrying the modeled mutations or isogenic lines with CRISPR-engineered mutations were used. The modeled mutations tend to be extremely rare or de novo that have already been extensively studied in rodents or other model organisms, and iPSC modeling mostly recapitulates the known cellular phenotypes. Overall, the resulted cellular phenotypes in human neurons for these mutations can be classified into two categories: (1) hypofunction such as reduced calcium signaling and activity-dependent dendrite retraction for a missense mutation in the L-type calcium channel Ca(v)1.2 (Krey et al., 2013; Panagiotakos et al., 2019; Pasca et al., 2011), reduced action potentials and peak inward currents for mutations in methyl-CpG-binding protein 2 (MECP2) (Farra et al., 2012); fewer synapses and defects in excitatory synaptic neurotransmission for mutations in postsynaptic SHANK3 (Kathuria et al., 2018; Shcheglovitov et al., 2013) and aberrant dendritic spines for CDKL5 mutations (Ricciardi et al., 2012); (2) hyperfunction such as increased neural progenitor proliferation and organoid overgrowth for protein-truncating LoF mutation in CNTNAP2 (de Jong et al., 2021), accelerated dendritic morphogenesis and enhanced excitatory synaptic strength for LoF variants in SYNGAP1 (Llamosas et al., 2020), and increased dendrite length, complexity, synapse number, and frequency of spontaneous excitatory postsynaptic currents and hyper-connectivity for SHANK2 mutations (Zaslavsky et al., 2019).
Compared to autism risk variants/genes, very few rare risk variants of other neuropsychiatric variants have been modeled in iPSC-derived human neurons. This reflects the different genetic risk architecture of autism spectrum disorders from other psychiatric disorders: autism is more monogenic and has much more established disease-associated protein-coding variants. The most studied rare risk variant is a frame-shift mutation of disrupted in schizophrenia 1 (DISC1) that is co-segregated with major psychiatric disorders in a single family (Millar et al., 2000). Despite the lack of support from the SZ GWAS (PGC2, 2014; PGC3 et al., 2020) or other large-scale exome sequencing projects such as SCHEMA (Singh et al., 2020), the isogenic iPSC lines carrying the DISC1 mutation showed a deficit of synaptic vesicle release and dysregulated expression of genes related to synapses and psychiatric disorders in iPSC-derived forebrain neurons and brain organoids, when the iPSC-derived cells are cultured in a three-dimensional fashion instead of monolayers on a dish, which was also consistent with the results from humanized DISC1 mutant mouse model (Wen et al., 2014; Ye et al., 2017). Similarly lacking strong genetic association evidence, two rare missense mutations Chondroitin Sulfate Proteoglycan 4 (CSPG4) that showed familial segregation with SZ (de Vrij et al., 2019) and a missense mutation, E492K, in NTRK1 that showed familial segregation with bipolar disorder (BP) (Nakajima et al., 2020) were also modeled in iPSC-derived neurons. Given that large exome sequencing projects such as SCHEMA (Singh et al., 2020) start to reproducibly identify rare protein-coding variants associated with SZ and other neuropsychiatric disorders, we anticipate more iPSC modeling of rare protein-coding variants, which will help improve our mechanistic understanding of the contribution of rare protein-coding variants to neuropsychiatric disorders.
2.3. Common variants
Because common GWAS risk variants explain much more disease liability than rare risk variants of high penetrance, it is imperative to tie putative causal GWAS risk SNPs with functionality to understand disease causal mechanisms. iPSC model in combination with CRISPR-based precise SNP allele editing provides a powerful approach to bridge the GWAS findings to novel disease biology. However, modeling common GWAS risk variants is challenging for several reasons:
(1) for most GWAS risk loci, each has many common SNPs equally associated with disease and often spans multiple genes due to linkage disequilibrium (LD), it is difficult to determine which are the likely functional and causal variant/gene to model; (2) most common risk variants are in the noncoding part of the genome and do not change protein sequence, rather regulating gene expression; (3) more importantly, common risk variants have small population effect sizes, which may make it challenging to detect any biological function.
Because of these challenges, modeling common GWAS risk variants often requires prioritization of putatively functional/causal SNPs by integrative computational fine mapping of causal SNP and/or functional genomics interrogation of their putative functionality, e.g., by brain expression quantitative trait locus (eQTL) analysis or chromatin accessibility mapping (Dobrindt et al., 2020; Forrest et al., 2017; Schrode et al., 2019; Zhang et al., 2021a; Zhang et al., 2020). As regulatory variants are often cell-type and developmental stage-specific (Civelek and Lusis, 2014; Nica et al., 2013; Paul et al., 2014), iPSC modeling of common risk variants may need to assay for temporal changes of transcriptional and cellular effects of common variants at different neural differentiation stages. With regard to the feasibility to detect meaningful biological function for common risk variants of small effect sizes, it is arguably proven that simple cellular models like iPSC-derived neurons may reduce the system’s “buffering” to genetic or environmental perturbations compared to the whole organism (Merkle and Eggan, 2013), and common GWAS risk variants can still elicit moderate or even strong effects on molecular/cellular phenotypes (Bauer et al., 2013; Corradin et al., 2014; Kulzer et al., 2014; Miller et al., 2014; Musunuru et al., 2010; Spieler et al., 2014). Furthermore, even assuming a homozygous common GWAS risk variant causes a 20–30% difference of gene expression, the magnitude of the functional impact is not too different from a theoretically 50% reduction of gene expression or protein function resulting from a heterozygous rare LoF mutation of high penetrance. Moreover, it is very likely a subtle expression change may result in an amplified downstream cellular phenotype alteration, for instance, ~ 15% KD of expression of an Alzheimer’s disease GWAS risk gene, PICALM, in astrocytes leads to >50% reduced endocytosis of neuron-derived lipids (Moulton et al., 2021).
We and others have recently successfully studied functional impacts of common GWAS risk variants of SZ in CRISPR-engineered isogenic iPSC-derived neurons. As a proof of concept, we prioritized putatively functional SZ GWAS risk variants through co-localization with open chromatin peaks, and for a leading SZ risk locus spanning M1R137, we showed that the risk allele of common GWAS risk SNP rs1198588 was associated with altered M1R137 promoter chromatin openness, reduced MIR137 expression, and accelerated neuronal maturation (Forrest et al., 2017). More recently, we systematically mapped putatively functional SZ GWAS risk variants that showed differential allelic chromatin accessibility (i.e., allele-specific open chromatin or ASoC) and affect gene expression in iPSC-derived NPCs, glutamatergic neurons, GABAergic neurons, and dopaminergic neurons (Zhang et al., 2020). For the strongest ASoC SNP (rs2027349) associated with SZ at the vacuolar protein sorting 45 homolog (VPS45) locus, we found rs2027349 editing altered the expression of VPS45, lncRNA AC244033.2, and a distal gene, C1orf54, in human neurons. Neurons carrying the risk allele exhibited increased dendritic complexity, synaptic puncta density, and hyperactivity, which were reversed by knocking-down distinct cis-regulated genes (VPS45, AC244033.2, or C1orf54), suggesting a phenotypic contribution from all three genes (Zhang et al., 2021a). Similar to our demonstrated compound non-additive effects from all three genes at the same GWAS locus (Zhang et al., 2021a), another earlier study elegantly showed that common GWAS risk variants/genes, prioritized by brain eQTL mapping, from several different risk loci (FURIN, SNAP91, TSNARE1, and CLCN3), may synergistically affect the expression of genes involved in SZ pathogenesis in iPSC-derived neurons (Schrode et al., 2019). These studies suggest that noncoding GWAS risk variants impact the neurodevelopmental aspect of SZ and show a detectable biological function in iPSC-derived neurons.
Despite the initial success of iPSC modeling of common GWAS risk variants of neuropsychiatric disorders, functional interpretation of the cellular and molecular effects of individual variants in a small sample can be challenging. This is largely due to the small effect sizes of common risk variants and some confounding factors such as variable genetic backgrounds and intrinsic iPSC clonal variation. As a result of purifying selection against deleterious mutations in the population (Cvijovic et al., 2018; Gibson, 2012), the small effect sizes of common risk variants are expected to yield small magnitude of biological effects. In addition, buffering effects from genes in the same biological pathway and/or other allele(s) in the same haplotype background as the risk allele (Gibson, 2012; Hartman et al., 2001) may further complicate the detection and interpretation of any biological effects of common risk variant/gene in iPSC modeling. In this regard, it is noteworthy that, Peng et al. recently showed that the GWAS eQTL variants associated with SZ can have an effect on the expression of some target genes that is inversely correlated with SZ risk (Peng et al., 2021). A systematic and unbiased massive parallel approach will be needed to overcome these limitations to better model common GWAS risk variants (Townsley et al., 2020). Moreover, given that common GWAS risk genes may often act together as part of gene networks, multiplex SNP/gene editing to perturb different genes at the network level will be needed for iPSC modeling of common GWAS risk variants.
2.4. Polygenic risk
Both GWAS of neuropsychiatric disorders (Psychiatric Genomics Consortium-Schizophrenia, 2011; Psychiatric Genomics Consortium-Schizophrenia, 2014; Mullins et al., 2021; PGC3 et al., 2020; Purcell et al., 2009; Ripke and Concortium, 2013; Shi et al., 2009; Stefansson et al., 2009) and large-scale postmortem brain transcriptome studies (e.g., by PsychENCODE) (Fromer et al., 2016; Gandal et al., 2018; Wang et al., 2018) revealed the polygenic nature of neuropsychiatric disorders (Schizophrenia Working Group of the Psychiatric Genomics, 2014; Sullivan et al., 2018). Although modeling individual common GWAS risk variants or rare risk variants can help understand disease mechanisms at individual loci, it is also important to determine the convergent functional effects of polygenic risk on disease-relevant cellular and molecular phenotypes. One of the first iPSC modeling studies of the polygenic risk of neuropsychiatric disorders was with SZ using 4–5 cases compared to matched controls (Brennand et al., 2011). Despite a very small sample, biologically meaningful transcriptomic differences were identified between the SZ case and control iPSC-derived neurons (Brennand et al., 2011). Due to the technical challenge of scaling up the iPSC work, the reported largest case/control sample size in studies aiming to model polygenic effects remains too small, with cortical interneurons of 14 SZ cases and 14 controls (Shao et al., 2019). Likely because of the small sample size, only dysregulated expression of protocadherin genes and protocadherin relevant neuronal phenotypes were identified (Shao et al., 2019).
With small samples that do not reflect the polygenic risk spectrum of neuropsychiatric disorder, reducing the clinical heterogeneity of hiPSC lines by selecting subjects with common clinical manifestations or with rare genetic variants would be critical for drawing meaningful but limited insights (Brennand et al., 2014). Alternatively, because individual polygenic risk score (PRS) often correlates with the severity of disease symptoms or resilience (Hess et al., 2019; Zhang et al., 2019), selecting iPSC lines from donors with extreme PRS may help improve the study power of iPSC modeling of neuropsychiatric disorders. For instance, with iPSC-derived neural progenitors and cortical neurons from 13 SZ individuals with high PRS for SZ, along with 15 neurotypical individuals with low PRS, Page et al. identified neural electrophysiological measures associated with a diagnosis that implicated altered Na+ channel function and GABAergic neurotransmission (Page et al., 2021). However, due to complex yet unclear interactions between rare risk variants and/or non-genetic risk factors (e.g., stress) with individual polygenic risk backgrounds, the results from such study design of comparing patient group of high PRS to control group of low PRS may not reflect the difference between patients of low PRS and healthy controls of high PRS. Regardless, the mechanistic insight on the polygenic risk effects from studies with small sample sizes remains limited. Investigating a sufficient number of samples from each PRS group, affected or healthy, on cellular and genomic/transcriptomic levels, may give us leads to what may be the reliable and valid cellular phenotypes relevant to neuropsychiatric disorders.
With a limited sample size at this time, the iPSC modeling of polygenic risk may benefit from a focused study of the effect of PRS on phenotypic expressivity of rare and highly penetrant risk variants. Patients who carry rare disease risk variants often have an excess burden of common GWAS risk alleles (Tansey et al., 2016). The field has started to understand the interplay between PRS and highly penetrant rare risk variants such as the well-known 22q11 deletion (Cleynen et al., 2020). Some top-ranking genes identified by SCHEMA to have rare but highly penetrant SZ-associated protein-truncating or LoF mutations (Singh et al., 2020) may also be such candidates for exploring PRS effects. For example, for the strongest SZ candidate gene in the SCHEMA study, SETD1A, its LoF mutations are highly penetrant (OR = 20; similar to that of SZ-associated 22q11 deletion) (Singh et al., 2020). However, LoF-mutation-carriers do not always develop SZ rather show other neurodevelopmental phenotypes. Such incomplete penetrance or phenotypic heterogeneity may be explained by individual genetic risk backgrounds: the variable phenotypic expressivity may be modulated by common SZ risk loci, either through additive or synergistic effects, resulting in disease resilience (by low PRS) or vulnerability (by high PRS). iPSC lines with high or low extreme PRS of neuropsychiatric disorders would be very useful for modeling such polygenic risk effects.
In this regard, it is noteworthy that we have recently built a small cohort of iPSC lines with extreme SZ PRS (Dobrindt et al., 2020). These iPSC lines were selected from a few thousand donor lines at the California Institute of Regenerative Medicine (CIRM), representing the extreme PRS compared to the rest. These lines have been characterized for their pluripotency, cell growth, transfection efficiency, neuronal differentiation, and CRISPR-editing efficiencies (Dobrindt et al., 2020). Although a small sample, a cohort of well-characterized iPSC lines is expected to be useful for generating isogenic lines for the functional study of SZ risk variants in the context of high/low PRS backgrounds. However, given the nature of phenotypic heterogeneity and the small population effect size of common variants, even with an isogenic approach the selection of PRS backgrounds in CRISPR-editing needs to be carefully considered. Because of additive effect from risk variants, although unproven, introducing a common risk variant on top of a healthy subject with high SZ PRS may be more likely to yield disease relevant cellular phenotypes, which otherwise may be confounded by effects from non-genetic or rare genetic risk factors on a patient genetic background. Conversely, for modeling rare risk variants of high penetrance, a CRISPR-editing experiment may benefit from “correcting” a risk allele on a patient genetic background in addition to independently introducing a risk allele on a healthy genetic background of high SZ PRS. Nonetheless, CRISPR-editing on different type of genetic backgrounds is imperative for obtaining more reliable and interpretable disease-relevant cellular phenotypes.
3. Which model: 2D vs. 3D neuronal culture systems
Both 2D and 3D neuronal cultures (i.e., organoids) derived from human iPSCs of donors with different genetic backgrounds, or from CRISPR-engineered isogenic lines, are promising tractable cellular models for neuropsychiatric disorders (Duan et al., 2019; Kampmann, 2020; Townsley et al., 2020). Because these neural cultures only recapitulate early neurodevelopment processes, they are most suitable for modeling psychiatric disorders with neurodevelopmental aspects, i.e., autism and SZ. While the 2D culture has been widely used for modeling both common and rare risk variants, 3D cortical or brain organoids have been mainly used to model rare risk variants of large effect sizes.
3.1. 2D culture: monolayer neurons or co-culture system
All major brain cell types (NPCs, glutamatergic, GABAergic, dopaminergic, and cholinergic neurons, oligodendrocyte, astrocyte, and microglia) can now be efficiently differentiated from iPSC (Barretto et al., 2020; Butler Iii et al., 2020; Dobrindt et al., 2020; Douvaras and Fossati, 2015; Giacomelli et al., 2022; McQuade et al., 2018; Yang et al., 2017; Zhang et al., 2020; Zhang et al., 2013). Because of the relatively high efficiency of differentiation and high purity of each iPSC-derived cell type, the 2D culture system has been widely used for studying neuropsychiatric, neurodevelopmental, and neurodegenerative disorders. For instance, forced exogenous expression of NGN2 gives rise to near 100% excitatory neurons (NGN2-iNs) in about 4 weeks (Vierbuchen et al., 2010; Zhang et al., 2013), which makes NGN2-iNs the most commonly used cellular model for studying both common and rare risk factors of neuropsychiatric disorders (Table 1). The relatively homogenous population of differentiated cells also makes it straightforward for transcriptomic analysis by RNA-seq, morphological and electrophysiological analyses. Compared to the postmortem brain, which is well-known to be confounded by tissue variability and environmental factors (Lipska et al., 2006), hiPSC differentiation into neurons can be better controlled, thus making the data more reproducible.
The wide use of 2D culture systems often comes with methodological variations, which may yield different cellular phenotypes that need careful interpretation. For instance, Compared to NGN2-INs directly derived from iPSC, excitatory neurons differentiated from iPSC-derived NPCs represent a slower process but better recapitulates normal neurodevelopmental processes (Wang et al., 2019; Wen et al., 2014). Moreover, on some occasions of modeling neuropsychiatric risk variants, the neural electrophysiological phenotype may be observed in NPC-differentiated excitatory neurons but not in NGN2-INs (personal communication with Dr. Zhiping Pang). By comparing the inhibitory neurons directly derived from iPSC (Yang et al., 2017) and those derived from NPCs (Barretto et al., 2020), we also noted their different electrophysiological characteristics (Dobrindt et al., 2020). Such discrepancies may be due to the variable maturation stages and/or regional identity of the seemingly pure iPSC neurons, a reasoning that was supported by some recent single-cell RNA-seq analysis of the seemingly pure cell populations of NGN2-INs (based on immunofluorescence staining) contain cells of different maturity and even not the expected cortical identify (Wang et al., 2021; Zhang et al., 2021a).
Although 2D culture has the advantage of being relatively homogenous, it is not reminiscent of in vivo neural environment. An improved 2D culture system is the co-culture of excitatory and inhibitory neurons, or at a defined ratio, i.e. 80:20%, that is similar to the neuronal composition of the forebrain (Sahara et al., 2012). Human neurons will then be co-cultured with monolayer glial cells to facilitate maturation and synaptogenesis (Pang et al., 2011; Ullian et al., 2001; Vierbuchen et al., 2010). The cellular phenotypes can be assayed by differentially labeling excitatory and inhibitory neurons in the co-culture and the transcriptomic changes can be interrogated by scRAN-seq. With such a co-culture design, Wang et al. elegantly demonstrated that human knock-in neurons carrying the autism risk variant (R451C) in the NLGN3 gene decreased NLGN3 protein level and enhanced the strength of excitatory synapses without affecting inhibitory synapses (Wang et al., 2021).
3.2. 3D culture: spheroids or organoids
Brain organoids have anatomical structures reminiscent of the developing human brain thus presenting a promising approach for studying early neurodevelopment and for modeling risk factors for neurodevelopmental disorders (Pasca et al., 2015; Paşca, 2019; Quadrato et al., 2016; Rigamonti et al., 2016). Among different types of methods for generating brain organoids or spheroids, cortical spheroids as part of the assembloids have been more commonly used due to their relatively better reproducibility and simplicity (Paşca, 2019). However, for most methods, because of the cellular stress and cell death posed by the lengthy process of organoid development (months), cortical layer expansion and the size of cortical plate of the differentiated brain organoids are usually not reminiscent of the human cortical structure. This limitation can be mitigated by a recently developed method, a sliced neocortical organoid (SNO) system (Qian et al., 2020). This method is based on a previously established protocol using bioreactors (Qian et al., 2018; Qian et al., 2016) in generating cortical organoids, now combined with brain organoid slicing and culturing in vitro, resulting in sustained neurogenesis and radial migration of newborn neurons (Qian et al., 2020). Consequently, SNO forms an expanded cortical plate that establishes distinct upper and deep cortical layers, with diverse subtypes of neurons and astrocytes. The SNO largely resembles the third-trimester embryonic human neocortex (Qian et al., 2020), which will be ideal for studying neurodevelopment and for quantifying the morphological and cellular compositional changes associated with neuropsychiatric risk factors.
Besides the anatomical or morphological resemblance of brain organoids to developing human brains, at a molecular level, what is the validity of the 3D organoid model or how well it can faithfully recapitulate human neurodevelopmental trajectory? To address this question, Gordon et al. performed genome-wide epigenomic and transcriptomic analyses at different stages of cortical organoid differentiation and compared that to human brain development (Gordon et al., 2021). They found the transcriptional profiles of cortical organoids before 250–300 days are more like that of prenatal brains, while the transcriptional patterns of cortical organoids beyond 250–300 days are more like that of postnatal human brains. They also confirmed several known developmental milestones in cortical organoids such as the well-known switches of NMDA receptor subunits in the brain before and after birth (Paoletti et al., 2013), suggesting that cortical organoids, if successfully cultured for a long time, can model not only early neurodevelopment but also mid- to later-fetal stages (Gordon et al., 2021).
Cortical organoids, combined with scRNA-seq of tens of thousands of cells, provide an unprecedented opportunity to dissect the spatial and temporal early neuronal development in a cell-type-specific manner (Khrameeva et al., 2020; Pollen et al., 2019). Despite the single-cell transcriptomic similarities between brain organoids and developing brains (Tasic et al., 2016; Tasic et al., 2018; Zizhen Yao et al., 2020a), the functionality, i.e., electrophysiological properties of different types of neurons in brain organoids remain unexplored. This is largely due to the heterogeneity of different neuronal cells at variable stages of maturity. A promising approach is to generate a cell census map of cortical organoid neuronal subtypes with different functional dimensions by conducting a multimodal analysis of neurons in the 3D cortical organoid system (Personal communication with Dr. Zhiping Pang). To do so, a patch-seq technique (Bakken et al., 2020; Bardy et al., 2016; Cadwell et al., 2016; Cadwell et al., 2017; Chen et al., 2016; Fuzik et al., 2016; Scala et al., 2020; Zizhen Yao et al., 2020b) may be used to collect multimodal profiles, including electrophysiology, morphology, and transcriptomics from the same single cell of 3D organoids. The cellular taxonomy of neurons based on their functional, morphological, and transcriptomics features may be used as a general model to predict the functional identity of any neurons in 3D organoids.
Despite the advantages of using 3D organoids to model neurodevelopment, morphological variability and uneven cellular composition across organoids make the 3D organoid model more suitable for studying rare risk variants of highly penetrance rather than common risk factors of small effect sizes. Cortical organoids have been used to model autism- and SZ-associated 16p11.2 deletion and duplications (Urresti et al., 2021) and SZ-associated 22q11.2 deletion (Khan et al., 2020), illuminating temporal neurodevelopmental deficits in CNV-carriers that can be studied in 2D cell cultures. Using the method to generate cortical spheroids (Paşca, 2019), we also started to model the SETD1A LoF mutation associated with SZ, and the preliminary results showed that SETD1A LoF resulted in precocious neurogenesis (West et al., 2019). There are also attempts to model polygenic effects for SZ (Kathuria et al., 2020a) and BP (Kathuria et al., 2020b), each with cerebral organoids of 8 patents and 8 controls. However, the striking transcriptomic and phenotypic changes in SZ or BP organoids in these studies need to be interpreted cautiously, given the relatively small sample size and the known challenges of analyzing the bulk RNA-seq data and ascertaining the morphological differences in brain organoids.
4. Which cell type: disease relevance and region identify
Human iPSC can be differentiated into different brain cell types. For modeling neuropsychiatric risk variants, the cell type of interest can be informed by the known brain expression profiles and function of the risk gene and in general, by the disease-relevant cell types for each neuropsychiatric disorder.
Disease-relevant cell types and brain region identity for most neuropsychiatric disorders are not well defined. For instance, animal studies, human postmortem brain, and clinical brain imaging studies of SZ implicate almost every part of the brain, leaving the most disease-relevant or vulnerable cell types and their region identities unknown. Recent brain scRNA-seq transcriptomic profiling enables a global view of cell-type-specific gene expression patterns of each cell type/region at single-cell resolution (Habib et al., 2017; Zeisel et al., 2018). By mapping GWAS loci onto each specific brain cell type based on the cellular taxonomy of single-cell gene expression profiles, cortical inhibitory interneurons and excitatory neurons from the cerebral cortex and hippocampus (pyramidal and granule cells) as well as inhibitory medium spiny neurons (in the striatum) were found to be the most genetically vulnerable cell types for SZ (PGC3 et al., 2020; Skene et al., 2018). Across 265 cell types in the mouse central and peripheral nervous systems (Zeisel et al., 2018), glutamatergic (excitatory) neurons in the deep layers of the cortex, amygdala, and hippocampus showed the strongest enrichment for SZ heritability, which was followed by both inhibitory and excitatory neurons from the midbrain, thalamus, and hindbrain (PGC3 et al., 2020). In contrast, major depression disorder (MDD) and neurodegenerative disorders did not show such GWAS risk enrichment in these cell types (PGC3 et al., 2020; Skene et al., 2018). A more systematic analysis of all the most recent neuropsychiatric GWAS datasets with large samples would yield a more informative view of the most relevant brain cell types/regions for each neuropsychiatric disorder.
There has been no comprehensive neuropsychiatric GWAS risk enrichment analysis for iPSC-derived brain cell types to ascertain their disease relevance. Based on the GWAS risk enrichment analysis of open chromatin peaks and allele-specific open chromatin variants in iPSC-derived NPCs, glutamatergic, GABAergic, and dopaminergic neurons, we found all these cell types showed significant enrichment for SZ GWAS risk and to a less extent for BP and MDD (Zhang et al., 2020). Using Bulk RNA-seq data of iPSC-derived NPCs and microglia (iMG), our MAGMA analysis but not LDSC analysis showed enrichment of GWAS risk of SZ and BP in NPCs, while both analyses showed enrichment of GWAS risk of Alzheimer’s disease (AD) in the iMG (Butler Iii et al., 2020). A more systematic analysis of each iPSC-derived pure cell type in both 2D and 3D neural culture systems by scRNA-seq will better inform the genetic relevance of each iPSC-derived cell type to neuropsychiatric disorders.
It is worth noting that the disease-relevant cell type based on genome-wide gene expression and genetic association may not be applicable to some specific set of genes or pathways. For instance, for microRNA-137 (MIR137), a leading neuropsychiatric risk gene and a post-transcriptional master regulator, we conducted a cell type-specific gene set (MIR137 target genes) PRS analysis in both European and Han Chinese SZ samples (Yao et al., 2021). Although the MIR137 target gene set expressed iPSC-derived glutamatergic neurons showed the greatest enrichment of SZ GWAS risk, which was consistent with the notion that glutamatergic neuron is the most disease-relevant cell types for SZ (PGC3 et al., 2020), we also found significant SZ risk enrichment in MIR137 target genes expressed in iPSC-derived NPC or dopaminergic neurons (Yao et al., 2021). Specifically, we found that PRS derived from the predicted MIR137 target genes that are expressed in hiPSC-derived NPCs, GABAergic neurons, dopaminergic neurons, or glutamatergic neurons explains greater SZ risk variance than PRS derived from genes expressed in hiPSCs or other less relevant cell types (Yao et al., 2021). The cell-type-specific enrichment of SZ GWAS risk in different iPSC-derived cell types was further supported by an independent LDSC analysis (Yao et al., 2021).
It is also noteworthy that cell types not enriched for disease GWAS risk may still be important for some specific pathophysiological processes of SZ and other neuropsychiatric disorders. For instance, brain microglia or iPSC-derived microglia are not genetically vulnerable cell types for SZ (Butler Iii et al., 2020; PGC3 et al., 2020); however, dysregulation of synaptic pruning mediated by microglia has been hypothesized to be pathogenic to SZ (Sellgren et al., 2019; Sellgren et al., 2017). Excessive synapse pruning by microglia during adolescence may lead to the reduced synaptic density in the SZ brain, which is correlated with decreased gray matter thickness and reduced overall brain volume (Cannon et al., 2015; Glausier and Lewis, 2013; Lewis and Gonzalez-Burgos, 2008). The role of microglia in dysfunctional synaptic pruning in SZ was also partially supported by SZ GWAS: common SZ risk variants within the complement component 4 (C4) locus are associated with increased neuronal complement deposition and synapse uptake (Sellgren et al., 2019). Some other brain cell types may also play an important role: for example, astrocytes derived from BP patients are functionally less supportive of neuronal excitability and this effect is partially mediated by IL-6, suggesting a potential role of astrocyte-mediated inflammatory signaling in BP.
5. Which phenotype: disease-relevant cellular phenotypes
Disease-relevant specific cellular phenotypes for neuropsychiatric disorders remain largely undefined. Although for some brain cellular phenotypic changes have been observed for certain neuropsychiatric disorders, for instance, the reduced synaptic density in the SZ postmortem brain (Cannon et al., 2015; Glausier and Lewis, 2013; Lewis and Gonzalez-Burgos, 2008), the causal link between genetic risk factors, the observed cellular phenotypes, and clinical features of a neuropsychiatric disorder is lacking. Because of the neurodevelopmental aspects of autism and schizophrenia, compared to other neuropsychiatric disorders, the field has a better understanding of the postulated cellular phenotypes that are associated with neurodevelopmental abnormalities observed from postmortem brain studies and/or clinical brain imaging. The lack of defined disease-relevant cellular phenotypes poses a challenge to the iPSC modeling of neuropsychiatric disorders. On the other hand, well-powered studies of patient-specific iPSC lines, in combination with CRISPR gene editing to engineer specific disease risk variants, will help define disease-relevant cellular phenotypes and establish causal links with genetic risk factors.
Abnormal proliferation of iPSC-derived NPC has been implicated for autism and to a less extent, for SZ. The disease relevance of abnormal NPC proliferation is supported by known clinical features of autism, where either accelerated brain growth (i.e., macrocephaly) (Butler et al., 2005; Chawarska et al., 2011) or reduced brain size (i.e., microcephaly) can be seen in early brain development of some patients (Miles, 2011; van Bon et al., 2016). iPSC modeling microcephaly showed loss of NPCs and premature differentiation (Lancaster et al., 2013), while modeling macrocephaly showed rapid proliferation of NPCs (Marchetto et al., 2017). These opposite clinical phenotypes can be associated with different genetic risk factors, for instance, 16p11.2 duplication is associated with microcephaly while deletion is associated with large head size/macrocephaly (Steinman et al., 2016). However, iPSC modeling of 16p11.2 deletions and duplications did not show significant effects on NPC proliferation, rather recapitulated the opposite effects on neuron size and dendrite length (Deshpande et al., 2017), suggesting NPC proliferation may not be the only cellular phenotype associated with the early brain outgrowth in autism. Abnormal NPC proliferation has also been associated with SZ, but only in iPSC modeling. SZ iPSC-derived NPCs were found to have aberrant migration and increased oxidative stress (Brennand et al., 2015), while our iPSC-derived cortical organoids with SZ-associated SETD1A LoF mutation showed reduced NPC proliferation and precocious neurogenesis at early-developmental stage (West et al., 2019).
Most disease-associated cellular phenotypes are at the neuron level, often involving dendritic length, branches, synaptic puncta density, sodium/potassium/calcium channel activity, electrophysiology properties, and/or neural network activities. Dendrites and spines are the main neuronal structures that receive input from other neurons. For autism, both human postmortem brain and animal studies suggest a reduction of dendrite numbers and spine density (Martínez-Cerdeño, 2017). For schizophrenia, the most consistent findings are the reduced dendritic spine density and dendritic arborization in postmortem brains, and the accelerated adolescent gray matter reduction from brain imaging studies (Moyer et al., 2015). At the level of neuronal function, animal models of autism mutations reported both hyper- and hypoactivity associated with autism-like behaviors (Peça et al., 2011; Sacai et al., 2020; Schmeisser et al., 2012; Tabuchi et al., 2007; Won et al., 2012), while both pharmacology and genetic animal models of schizophrenia converge on hypofunction of glutamatergic synapse despite the reasonable skepticism as to how accurately animal behaviors can be reflective of schizophrenia (Coyle et al., 2020).
However, these cellular phenotypes of autism and SZ postulated from human postmortem brain, brain imaging, and animal studies were not fully recapitulated by the recent iPSC modeling of common or rare risk factors of both disorders (see above section “what to model”). For instance, contrary to the expected reduced dendritic complexities and synaptic function (Duan et al., 2019; Penzes et al., 2011), some iPSC modeling observed the increased dendritic complexities and neuronal hyperexcitability (Blizinsky et al., 2016; Forrest et al., 2017; Schrode et al., 2019; Yi et al., 2016; Zhang et al., 2020). These studied SZ risk variants include both common variation (e.g., a GWAS SNP at M1R137 locus) (Forrest et al., 2017) with small effect sizes and rare risk alleles with high penetrance (e.g., 16p11 duplication or loss-of-function of SHANK3) (Blizinsky et al., 2016; Yi et al., 2016). Our recent modeling of a chromatin accessibility-altering common SZ risk variant rs2027349 at the VPS45 locus in isogenic NGN2-iNs further added another example that risk allele is associated with the increased dendritic complexity, synaptic puncta maturation, and neuronal firing rate (Zhang et al., 2021a). Such seemingly inconsistent cellular phenotypes may be the result of genetic pleiotropy across major psychiatric disorders (Ruderfer et al., 2018). Alternatively, hypo- and hyperfunction for the same disorder may reflect different temporal functional characteristics of neurons at different maturing stages. For instance, for patient-derived human hippocampal neurons carrying a rare autism-associated missense mutation A350V in gene IQSEC2, the immature dentate gyrus (DG) granule neurons are extremely hyperexcitable, while the aged neurons are hypoexcitable (Brant et al., 2021). However, neural maturity may not explain some of the observed increase of dendritic complexities and synaptic puncta in SZ neurons, e.g., the effect of the common risk allele at MIR137 locus (Forrest et al., 2017), which was confirmed by a mouse model of the MIR137 risk allele (i.e., with haploinsufficiency of MIR137) (Cheng et al., 2018). Finally, among other plausible interpretations, the reported both neural hyperfunction (Blizinsky et al., 2016; Forrest et al., 2017, a; Schrode et al., 2019; Yi et al., 2016; Zhang et al., 2020) and hypofunction (Duan et al., 2019; Penzes et al., 2011) in SZ iPSC models provide further support for a neuronal homeostatic model of neuropsychiatric disorders where either excess or inadequate synaptic signaling output may contribute to pathophysiology (Landek-Salgado et al., 2016; Ramocki and Zoghbi, 2008).
As a possible mechanistic link to dendritic and synaptic dysfunction as described above for SZ and autism, the abnormal microglia-mediated synapse pruning may also be considered as a cellular phenotype relevant to neurodevelopmental disorders. For SZ, the hypothesis is that excess synaptic pruning could trigger the disease during the active period of synapse elimination in adolescence; while for autism, human and animal studies imply a deficit of pruning that may lead to excessive synaptic connections (Sakai, 2020). The idea has gained empirical support from iPSC modeling of synaptic pruning in SZ (Sellgren et al., 2019; Sellgren et al., 2017). With an in vitro model of microglia-mediated synapse engulfment, the excessive synaptic pruning in SZ lines was found to result from abnormalities in both microglia-like cells and synaptic structures (Sellgren et al., 2019). However, although the SZ-associated C4 allele is correlated with synapse uptake (Sellgren et al., 2019), a causal relationship between the C4 risk variant and the increased synapse engulfment is lacking. Moreover, because the activated-microglia-conditioned medium has been found to alter metabolism differentially in SZ iPSC-derived cortical interneurons, neural synaptic deficit may play a major role in microglia-mediated synaptic pruning (Park et al., 2020). Therefore, it remains uncertain whether the increased phagocytosis by microglia is a cellular phenotype genetically relevant to SZ.
6. What next: challenges, opportunities, and new research frontier
iPSC modeling of neuropsychiatric risk factors is becoming more popular in the field. However, some limitations of the model and challenges remain to be appreciated: (1) The sample size is relatively small in almost all the iPSC modeling studies, while it is becoming clear that donor genetic background and iPSC clonal variation are common confounding factors. (2) Modeling common risk variants and genes remains challenging simply because functional interpretation and causal inference of noncoding risk variants are not as straightforward as protein-coding variants. (3) Disease risk variants may only manifest their functional effects at a specific cellular state and in a context-specific manner, e.g., neural activation-dependent effects. (4) Nonnuclear DNAs such as mitochondrial DNA (mtDNA) may play a significant role in the etiology of neuropsychiatric disorders.
6.1. Scaling up the iPSC modeling
Confounding factors in iPSC modeling often arise from the line-to-line variation due to the effects of different donor genetic backgrounds, iPSC clonal-to-clone variation originated from cellular reprogramming and/or iPSC cell passaging processes, and technical variations resulted from cell culturing and neuronal differentiation procedures. Compared to 2D culture, 3D organoids show even more variations during the lengthy culture process (months). For instance, although the cell type diversity is quite producible across organoids (Velasco et al., 2019), the cellular composition and subtype specification vary substantially between organoids and across protocols, especially at later stages of cortical organoid maturation (Bhaduri et al., 2020; Velasco et al., 2019). Although line-to-line variation can be mitigated by using CRISPR editing-based isogenic approach to enable the experimental comparison virtually on the same genetic background between isogenic pairs, the phenotypic expressivity can still be influenced by the genetic background of each donor. Furthermore, the iPSC clonal variation often makes transcriptomic data difficult to interpret, even with isogenic design and in a setting with multicenter and cross-lab validation (Pak et al., 2021). As such, when the sample size is small, these potential confounding factors can substantially limit the interpretation of molecular and cellular phenotypes associated with psychiatric risk variants in iPSC modeling.
It is becoming increasingly recognized that the iPSC modeling needs scaling up, both on the number of iPSC lines to be used and the variant numbers to be studied. The current situation in iPSC modeling is somewhat reminiscent of the early stage of the candidate gene association study of neuropsychiatric disorders when a small sample size and a small number of interrogated genetic markers often yield false-positive associations and evidence hanging in the balance (Sanders et al., 2008). A larger sample size can alleviate the experimental variations and produce more rigorous results. However, scaling up iPSC modeling is currently not only associated with higher cost but also needs conceptual and technical innovations on how to delineate the molecular and cellular phenotypes more effectively for a large number of iPSC lines and risk variants/genes (Fig. 2).
Fig. 2.
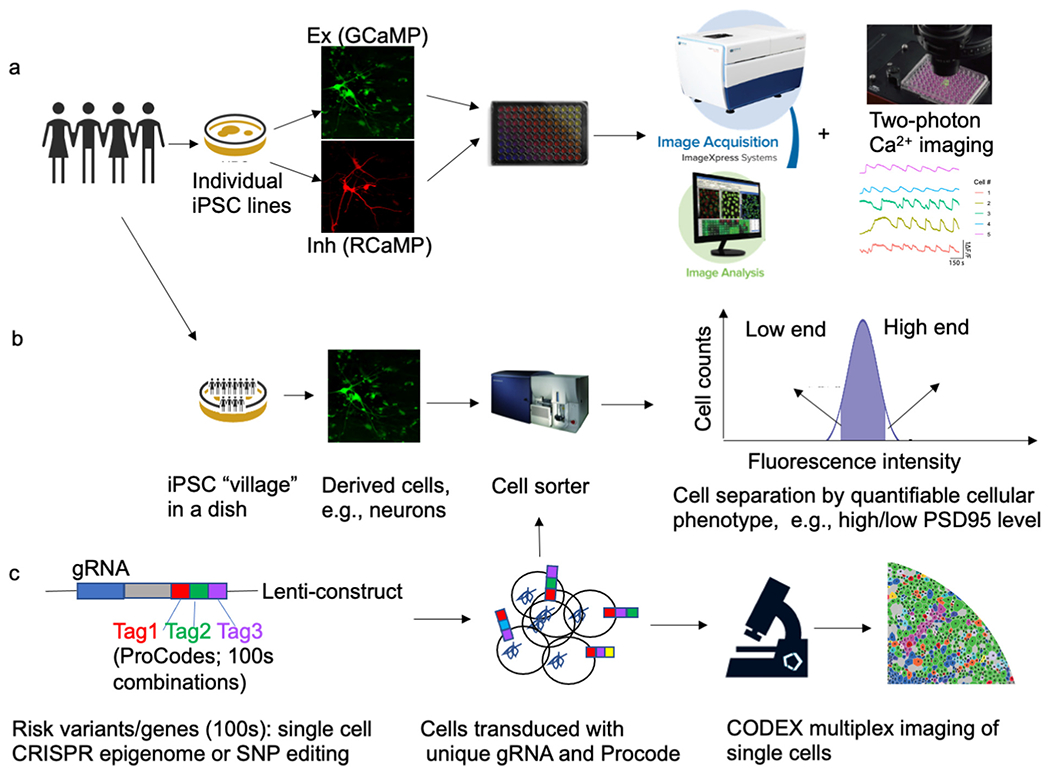
Approaches for scaling up the number of iPSC lines and/or genetic risk variants/genes as well as cellular phenotyping. (a) Individually cultured iPSC lines on multi-well plates assayed by high-content imaging, (b) iPSC lines co-cultured on a dish, i.e., a “cell village” approach, combined with a pooled phenotype screening. (c) Scaling up the number of genetic risk variants for iPSC modeling by employing a ProCode approach combined with CODEX multiplex imaging at single-cell resolution.
To scale up the number of iPSC lines, one straightforward way is to culture individual iPSC lines in separate wells on a multi-well plate, followed by high-content imaging of neuron morphology, synaptic maturation, and electrophysiological properties (Fig. 1)a. With this setting, for each individual line, a co-culture system may be used to differentially label excitatory (e.g., by GCaMP) and inhibitory (e.g., by RCaMP) neurons, which will enable the morphometric analyses of different types of neurons simultaneously. The high-content imaging system (e.g., ImageXpress) can auto-segment the images, which can be analyzed for dendritic complexity and synaptic puncta density using built-in methods or some customized machine learning-based methods such as Intellicount (Fantuzzo et al., 2017). Taking advantage of high-performance Ca2+ sensors (GCaMP and RCaMP) for imaging neuronal activity (Chen et al., 2013; Dana et al., 2019), the cellular electrophysiology property of both types of neurons can also be assayed by high-throughput Ca2+ imaging with a two-photon microscope.
Alternatively, iPSC sample scaling up can be done by an innovative approach “cell village” (Mitchell et al., 2020). With this setting, 10s to 100s iPSC lines can be co-cultured together in a cell culture dish and differentiated into neurons together, followed by cell sorting to groups neurons based on a specific cell surface marker or antigen (i.e., cellular phenotype) and then by Census-seq to associates cellular phenotypes to donors’ genotypes in cell “villages” (Fig. 1)b. Although this approach measures cellular phenotypes in cells from many donors simultaneously, it would be challenging to establish a customized “village” where all the iPSC lines may grow in balance, i.e., without a few overgrowing others. Furthermore, with many co-cultured lines, cell non-autonomous effects may confound the analysis and data interpretation. Nonetheless, “cell village” has been proved to be effective in mapping common genetic variants affecting some cellular phenotypes such as SMN protein levels in spinal muscular atrophy (SMA) (Mitchell et al., 2020).
In a sense, “cell village” (and census-seq) also scales up the variant number, because it assays the effect of many common alleles on a specific phenotype (e.g., SMN protein level) at a cell population level (Mitchell et al., 2020). However, it is still an association-based approach to establish a correlation between genotype and phenotype rather than directly modeling function/causal variant en masse. In a proof-of-concept experiment, Cederquist et al. carried out a multiplex iPSC screening, in which 30 isogenic lines carrying different autism mutations are pooled in a single dish and differentiated into the prefrontal cortex (PFC) lineages to test early developmental hypotheses of autism (Cederquist et al., 2020). With PFC neurogenesis as a cellular phenotype for pooled screening, mutations were sub-grouped into those that enhance or suppress neurogenesis (Cederquist et al., 2020), providing a framework to disentangle genetic heterogeneity associated with autism and identify converging molecular and cellular phenotypes of diverse disease variants. However, the number of modeled variants here remains small, and the way to individually CRISPR-engineer the mutations would not be suitable to further scale up the number of assayed variants.
To substantially increase the number of variants/genes (100s or 1000s) to be modeled, pooled, or multiplex CRISPR/Cas9 SNP/gene editing or epigenome editing will be needed. The editing will be mediated by low-MOI (multiplicity of infection) lentivirus infection to introduce single guide RNAs (sgRNAs) into each cell, followed by molecular and phenotypic screening at a single-cell resolution (F 2c). For modeling LoF mutation, CRISPR/Cas9 editing can be used to systematically create small indels in protein-coding regions through non-homologous end joining (NHEJ) repair of double-strand breaks (DSBs) (Ran et al., 2013), resulting in protein-truncating mutations. Alternatively, LoF mutation can be efficiently generated by introducing premature protein stop codon using CRISPR cytosine base editors (CBE) without creating DSBs (Cuella-Martin et al., 2021; Xu et al., 2021). To mitigate the cell-toxic DSBs and off-target editing of DNAs associated with CRISPR/Cas9 editing (Ran et al., 2013), or the possible off-target RNA editing associated with CBE editing (Cuella-Martin et al., 2021; Xu et al., 2021), CRISPRi (Holtzman and Gersbach, 2018) or its more effective version, CRISPRoff (Nunez et al., 2021) can be used to transcriptionally repress the expression of the sgRNA-targeted genes by rewriting the epigenomic state without changing DNA sequences (i.e., epigenome editing). Compared to modeling LoF mutation, precisely editing many risk SNPs in iPSCs remains challenging due to the low homology-directed repair (HDR) efficiency in CRISPR/Cas9 editing (Ran et al., 2013). However, the rapidly evolving high-efficiency SNP base editing system (CBE or ABE) (Cheng et al., 2021; Cuella-Martin et al., 2021; Hanna et al., 2021; Richter et al., 2020) may enable massive parallel assessment of hundreds or thousands of SNPs in iPSCs.
For the above-described pooled editing of many risk variants/genes, it is essential that sgRNA barcodes individual cells for molecular and cellular phenotyping at single-cell resolution. Molecular phenotyping (e.g., transcriptomic level) is relatively easy because scRNA-seq can distinguish individual cells barcoded by single gRNAs thus allowing to assess SNP/gene editing effect on cis-target gene expression or transcriptomic changes. However, cellular phenotyping can be challenging depending on whether the cellular phenotype can be subject to cell sorting. For a phenotype that can be distinguished by cell sorting, cells that underwent the pooled editing can be sorted into two groups and the effects of genetic variants can be analyzed as described for CRISPRi pooled screening (Nunez et al., 2021). For morphological phenotype such as neuron dendritic branches, a combination of Pro-Code technique to barcode each gRNA/edited cell (Wroblewska et al., 2018) with CODEX multiplex imaging of single cells (Goltsev et al., 2018) may be needed (Fig. 2c). In this setting, each gRNA, and thus each cell infected by the gRNA, will be tagged by a combination of 3 or more genetic barcodes detectable as protein tags (Pro-Codes) (Wroblewska et al., 2018). Then, each pro-code barcoded cell can be imaged sequentially for each protein tag using the CODEX technique that also allows interrogating the immunofluorescence staining of cell-specific markers (e.g., PSD95) as part of the CODEX detection panel (Goltsev et al., 2018). The obtained single-cell imaging data can be used for ascertaining the cellular phenotypic effects of each risk variant/gene in a pooled or multiplexed CRISPR SNP/gene editing.
6.2. Functional and mechanistic interpretation of noncoding risk variants
Functional interpretation of noncoding variants is important, because most GWAS risk variants of neuropsychiatric risk variants, like for other complex disorders, are in the noncoding part of the genome. Furthermore, as whole genome-sequencing data in large samples become available, it is conceivable that some rare noncoding variants may be found associated with disease similarly to those rare coding variants identified by SCHEMA (Singh et al., 2020). However, it is challenging to study the function of common noncoding risk variants in primary human tissues and at early development stages, particularly for brain disorders. Although brain eQTL, chromatin-accessibility QTL (caQTL) and chromatin interaction (Hi-C) data from PsychENCODE (Gandal et al., 2019; Gandal et al., 2018; Li et al., 2018; Rajarajan et al., 2018; Wang et al., 2018) are instrumental for prioritizing functional noncoding risk variants, iPSC-based models can provide additional regulatory dimension at early neurodevelopment stage that may not be captured by brain QTL mapping. Furthermore, because iPSC models are amenable to genetic manipulation, they can provide mechanistic insight on how noncoding risk variants may affect disease risk genes.
eQTL can inform noncoding regulatory variants that are associated with one or more target genes in cis or in trans. The only eQTL mapping study in an iPSC model is for dopaminergic neurons differentiated from 215 iPSC lines, a sizable sample for iPSC study (Jerber et al., 2021). Leveraging the scRNA-seq profiling at a single-neuron resolution, eQTLs were identified for both relatively pure dopaminergic neurons and another intermediate subtype of cells at different differentiation stages. Although iPSC-derived eQTL maps mimic in vivo GTEx brain eQTL maps, about 2366 eQTL were detected only in iPSC models. Of the 1284 eQTL colocalized with known neurological trait risk loci, 46% are not found in the GTEx catalog (Jerber et al., 2021), highlighting the added value of mapping eQTLs in iPSC models. Identifying eQTLs in other brain cell types relevant to major neuropsychiatric disorder, e.g., glutamatergic and GABAergic neurons co-cultured and differentiated from a moderate number of iPSC lines, is warranted.
There has been no available caQTL dataset for iPSC-derived neurons. Chromatin accessibility precedes gene transcription and can help interpret regulatory noncoding variants. Similar to the concept of caQTL, we have recently mapped putatively functional noncoding variants that showed differential allelic chromatin accessibility, as we designated as allele-specific open chromatin (ASoC), in NPC, glutamatergic (including both NPC-derived and NGN2-induced), GABAergic and dopaminergic neurons derived from 20 iPSC lines (Zhang et al., 2021a; Zhang et al., 2020). Because ASoC directly compares the two alleles within an individual, it is expected to be a more sensitive assay of chromatin-regulating variants than traditional QTL analysis using cross-sample variation (i.e., caQTL) (Calderon et al., 2019; Zhang et al., 2020). ASoC SNPs frequently alter expression (Forrest et al., 2017; Zhang et al., 2020) but act upstream of transcription, thus complementing eQTL analysis. Importantly, neuronal ASoC SNPs, compared to open chromatin peak regions, exhibited much stronger enrichment for SZ risk variants (Zhang et al., 2021a; Zhang et al., 2020). The neuronal ASoCs were partially driven by altered transcription factor binding, overrepresented in brain gene enhancers and eQTLs, and frequently associated with distal genes through chromatin contacts. We also identified abundant neuronal open chromatin peaks not detected in brains. Our ASoC analysis in iPSCs highlights ASoC as a functional mechanism of noncoding neuropsychiatric risk variants, providing a powerful framework for identifying disease causal variants and genes.
eQTL mapping is association-based and does not directly inform which is the functional SNP among its LD proxies (R2 >0.8). ASoC may directly point to a functional SNP but still needs orthogonal functional validation as we demonstrated (Zhang et al., 2020). Massive parallel reporter assay (MPRA) provides a powerful approach to systematically test the regulatory potential of tens of thousands of noncoding GWAS risk variants, thus enabling to cross-validate or screen for the putatively functional variants prioritized from QTL mapping and GWAS. MPRA has been used to test GWAS risk variants of neuropsychiatric and neurological disorders in non-neuronal cells (Myint et al., 2019; Myint et al., 2020), where both alleles of each SNP along with the ~ 100-bp flanking sequence at each side are synthesized and cloned next to a minimal promoter, sequence-based barcode, Kozak consensus sequence, and the GFP gene, into a construct for transducing or transfecting mammalian cells. The level of expression of the barcodes in the RNA of both alleles will be compared to determine whether the targeted sequences of both alleles differentially drive gene expression. Among several variations of MAPR such as STARR-seq (Arnold et al., 2013) and capSTARR-seq (Vanhille et al., 2015), a lentivirus-based MPRA approach, as described by Gordon et al. (2020), may be most suitable for systematically efficiently infecting neuronal cells. Despite its limitation of only testing SNP function on a very short surrounding sequence without the intact genomic sequence context, MPRA in iPSC-neurons will be invaluable in providing direct functional evidence for noncoding risk variants of neuropsychiatric disorders.
To gain mechanistic insight on disease GWAS associations, it is imperative to tie the functional noncoding risk variants to their cis- or trans-regulated target genes. While eQTL mapping can help make the connection, chromatin contact mapping (Hi-C) provides direct evidence for physical interaction between a risk variant and its target gene(s). With iPSC-derived NPCs, neurons, and astrocyte-like glial cells as in vitro models of human brain development, Rajarajan et al. mapped genome-wide chromatin contacts, or “three-dimensional genome” (3DG), in different cell types using Hi-C (Rajarajan et al., 2018). It was found that many SZ risk loci showed 3DG connections with genes outside the risk loci, representing long-range distal gene regulation and expanding the putative disease risk genes by 50 to 150% (Rajarajan et al., 2018). Although most 3DG connections involved in these disease risk loci still need orthogonal validation, the concept of long-range regulation was independently confirmed by a recent mapping of cis-regulatory chromatin contacts in neural cells using promoter capture-HiC (Song et al., 2019). Hundreds of thousands of long-range cis-interactions between promoters and distal promoter-interacting regions, many of which are cell type-specific, were identified in iPSC-derived excitatory neurons, lower motor neurons, hippocampal dentate gyrus-like neurons, and in primary astrocytes, enabling to link regulatory elements to their target genes (Song et al., 2019).
In support of the widespread long-distance cis-target genes, our focused study of a SZ risk variant rs2027349 showing strong ASoC near VPS45 found that CRISPR editing of rs2027349 altered the expression of VPS45, lncRNA AC244033.2, and a distal gene, Clorf54, in human neurons. Notably, we found all three cis-target genes of rs2027349 contribute to the cellular phenotype change in a non-additive manner, which was supported by chromatin contacts between the risk variant and all three cis-target genes, including the distal Clorf54 (Zhang et al., 2021a). The compound effects of all three cis-genes in the same GWAS locus (VPS45) (Zhang et al., 2021a) resemble the synergistic transcriptional effects of multiple SZ risk loci that were recently demonstrated in iPSC-derived neurons (Schrode et al., 2019), albeit the lack of 3DG chromatin contacts between those different SZ risk loci (Rajarajan et al., 2018). Thus, iPSC modeling provides mechanistic insight on how noncoding risk variants function and confer disease risk.
6.3. Cellular state- or context-specific effects of risk variants
Context-specific regulatory variants can unmask hidden disease heritability, which has been well demonstrated for immune disorders (Calderon et al., 2019; Farh et al., 2015; Kim-Hellmuth et al., 2017; Lee et al., 2014; Ramos-Rodriguez et al., 2019). Many risk variants of immune disorders are functional only in stimulated cells; for instance, cytokine stimulation of pancreatic β-cell unmasks abundant novel regulatory sequences, and remarkably, only stimulated enhancers are enriched for type 1’ GWAS variants (Ramos-Rodriguez et al., 2019). For neuropsychiatric disorders, the commonly used postmortem brains tissues (e.g., by PsychENCODE) (Amiri et al., 2018; Gandal et al., 2018; Li et al., 2018; Rajarajan et al., 2018), cannot capture the effects of disease variants in specific biological contexts, e.g., developmental stages and responses to stimuli (Calderon et al., 2019; Farh et al., 2015; Kim-Hellmuth et al., 2017; Lee et al., 2014) (Calderon et al., 2019; Farh et al., 2015; Kim-Hellmuth et al., 2017; Lee et al., 2014). In mice, neuronal activity in response to neurotransmitters leads to Ca2+ influx, activating early response genes (ERGs; e.g., FOS) and late response genes (LRGs; e.g., BDNF), regulating dendritic growth, synapse development, and neuronal plasticity (Yap and Greenberg, 2018). Some cytokines (e.g., IL-4, IL-17, and IFN-r) also affect neuronal activity, leading to neurodevelopmental abnormality and behavioral changes in animal models (Filiano et al., 2016; Reed et al., 2020; Ribeiro et al., 2019; Vogelaar et al., 2018). At a molecular level, neural activity in mouse brain induced drastic alterations of OCRs, accompanied by expression changes of 2000–5000 genes within 4–6 h (Fernandez-Albert et al., 2019; Su et al., 2017).
iPSC-derived neurons provide a cellular model suitable for testing neural activity-dependent changes and the effects from disease risk variants. In vitro, a variety of stimuli like membrane-depolarizing levels of potassium chloride (KCl) can induce neuronal activity that mimics the in vivo effects of visual and social experiences, stress, or drugs of abuse in mice (Yap and Greenberg, 2018). With a small sample size (iPSC lines from 4 SZ cases vs. 4 controls), Roussos et al. identified >1000 differentially expressed genes in iPSC-derived neurons in response to KC1 depolarization (Roussos et al., 2016). The robust activity-dependent gene expression may be partially due to activity-dependent secretion of catecholamines—dopamine (DA), norepinephrine (NE), and epinephrine (Epi), which was found elevated in SZ cases (Hook et al., 2014). A recent comprehensive activity-dependent transcriptional and epigenetic profiling of iPSC-derived GABAergic neurons further identified a genome-wide profile of activity-dependent enhancers and promoters (Boulting et al., 2021), and the inducible promoters were found significantly enriched for heritability of ASD, suggesting the sequence variants within activity-inducible promoters of developing human forebrain GABAergic neurons contributes to ASD risk (Boulting et al., 2021).
To systematically identify neuronal activity-dependent functional variants in major neuronal cell types relevant to neuropsychiatric disorders, we have designed an experiment that combined the co-culture of iPSC-derived excitatory and inhibitory neurons and scRNA/ATAC-seq in about 100 iPSC lines (See NIH RePORTER, PI: Duan) (Fig. 3). iPSC lines of different donors in the co-culture can be demultiplexed (Kang et al., 2018) based on their genotypes. Stimulated neurons co-cultured with glial cells will be assayed by scRNA-seq and by scATAC-seq (or by using 10 x Genomics’ Multiome kit), followed by mapping of stimulation-specific eQTLs and ASoC SNPs or caQTLs different time points of neural activation (Fig. 3). The neural activity-dependent regulatory variants will complement our previous ASoC (Zhang et al., 2020) and existing brain eQTL datasets (Amiri et al., 2018; Gandal et al., 2018; Girdhar et al., 2018; Psych et al., 2015; Rhie et al., 2018; Wang et al., 2018), expanding the repertoire of regulatory neuropsychiatric risk variants that may affect chromatin accessibility and gene expression only in activated/stimulated hiPSC neurons.
Fig. 3.
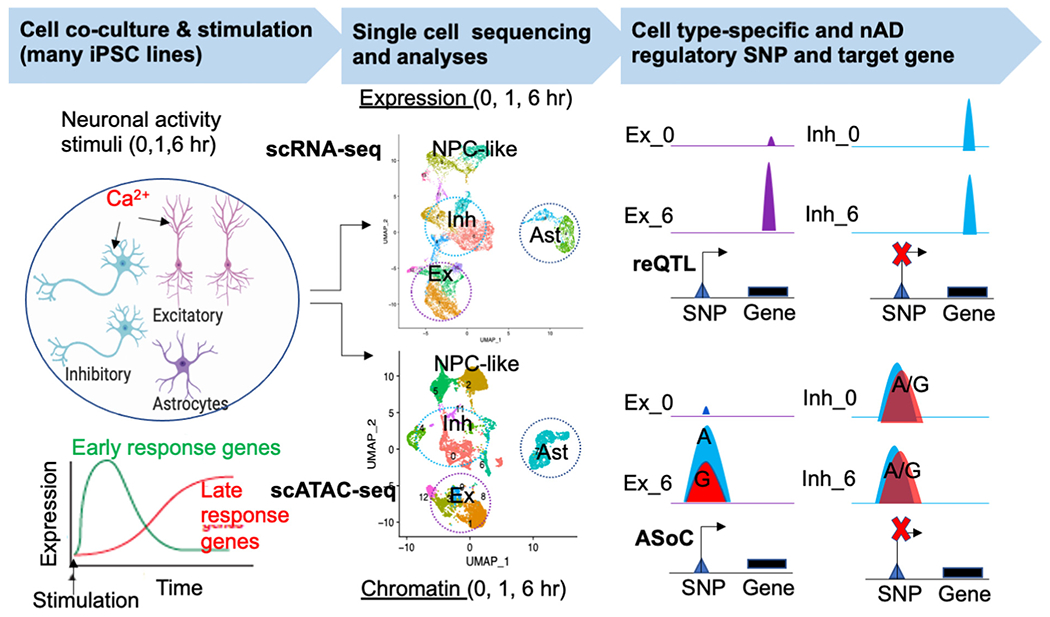
A schematic design of assaying neural activity-dependent functional noncoding SNPs in a co-culture system. The co-cultured excitatory and inhibitory neurons can be stimulated by KCl, followed by scATAC-seq/scRNA-seq to separate Ex and Inh neurons, astrocytes (Ast), and residual NPCs (middle). Cell type-specific regulatory SNPs that affect gene expression (reQTL) and chromatin (ASoC) are identified (right).
6.4. Dysfunctional mitochondria in neuropsychiatric disorders
There is a growing body of evidence that supports the role of mitochondrial dysfunction in neuropsychiatric disorders (Andreazza and Nierenberg, 2018; Ben-Shachar, 2017; Kato et al., 2011; Kim et al., 2019; Morava and Kozicz, 2013; Pei and Wallace, 2018). The reasoning behind the hypothesis was that since the mitochondria are the powerhouse of the cells, and the brain consumes a significant proportion of the bodies energy evident in the abundance of mitochondria in brain cells, individuals whose mitochondria functions suboptimally are more prone to stress-associated pathologies, thus, are more susceptible to neuropsychiatric disorders (Andreazza and Nierenberg, 2018; Kim et al., 2019; Morava and Kozicz, 2013; Pei and Wallace, 2018). Multiple mtDNA deletions have been associated with BD and SZ (Kato, 2011; Mancuso et al., 2008) or depression (Gardner et al., 2003). Some reports also show a higher prevalence of neuropsychiatric disorders in people with mitochondrial diseases compared to patients with other metabolic disorders and their relatives or the general population (Grover et al., 2006; Kato, 2011; Levey et al., 2020; Oexle and Zwirner, 1997). In one mtDNA association study on 50,000 individuals, including cases of eleven different diseases as well as controls, SZ was among the top diseases that had a significant association with certain mtDNA polymorphisms (Hudson et al., 2014). Moreover, alterations in complex I and complex IV in the respiratory chain could be found in SZ patients that had mtDNA deletions in their brain tissues (Whitehurst and Howes, 2022). In addition to evidence from studies on human patients and cells, multiple studies on mice support the theory that mitochondrial dysfunction may be directly involved in the pathology of neuropsychiatric disorders (Hovatta et al., 2010; Kato et al., 2011; Salim et al., 2010).
More recently, the role of mitochondrial dynamics in iPSCs has become of interest (Sercel et al., 2021), and iPSC models of dysfunctional mitochondria in neuropsychiatric disorders are being used to understand the pathogenic aspects that cannot be explained by the nuclear DNA (Zilocchi et al., 2020). Compared to other neuropsychiatric disorders, more focus has been given to studying mitochondrial dysfunction of SZ using iPSC models (Ni and Chung, 2020). iPSC-derived neural cells from SZ patients showed alterations in oxidative phosphorylation (OXPHOS) and ROS levels (Brennand et al., 2015; Li et al., 2019; Li et al., 2021b; Ni et al., 2020), disruption in mitochondrial membrane potential and respiration (Robicsek et al., 2013) (Brennand et al., 2015), and affected biochemical and protein networks in mitochondria in neurons (Li et al., 2019; Ni et al., 2020; Robicsek et al., 2013; Sullivan et al., 2019). One study found that co-culturing iPSC-derived neurons from SZ patients with activated microglia results in disruption in mitochondrial function that was not observed in controls (Park et al., 2020). Specific genetic risk factor of SZ, e.g., 22qdeletion, has also been linked to dysfunctional mitochondria in patient-specific iPSC models (Li et al., 2019). However, it remains to be established whether mitochondria dysfunction is the causal factor or consequence of neuropsychiatric disorders.
Despite the lack of a causal mechanistic link between dysfunctional mitochondria and neuropsychiatric disorders, iPSC modeling may help establish mitochondria as a potential therapeutic target for some neuropsychiatric disorders (Marazziti et al., 2012; Pei and Wallace, 2018). For instance, studies on animals and cellular models found that some current mood-stabilizing drugs and antidepressants, including lithium, valproate, and selective serotonin reuptake inhibitors, are known to affect mitochondrial energy metabolism and antioxidant activity in patients and animal models (Jou et al., 2009; Wu et al., 2013; Zilocchi et al., 2020). Other studies of SZ patients’ iPSC-derived neurons found that stimulating mitochondrial biogenesis by treating cells with bezafibrate and antioxidants (e.g., α-lipoic acid and acetyl-l-carnitine) can reverse mitochondrial deficits and abnormal arborization in SZ patients’ neurons (Li et al., 2021b; Ni et al., 2020; Park et al., 2020). Even some over-the-counter antioxidant supplements, such as mitoquinones, show anxiolytic and neuroprotective effects (Cocheme et al., 2007; Filiou and Sandi, 2019; Nussbaumer et al., 2016; Snow et al., 2010). In this regard, it is noteworthy that treating iPSC-derived neurons of familial Parkinson’s disease (PD) patients with small molecules that target mitochondrial stress can rescue PD-relevant neuronal deficits (Cooper et al., 2012).
Studying mitochondrial dysfunction in iPSCs also has general implications for iPSC modeling of neuropsychiatric risk factors. This is because mtDNA is prone to mutations, which can affect the cell’s survival, health, and pluripotency of iPSCs (Sercel et al., 2021; Tian et al., 2021). To that point, only very few known mtDNA polymorphisms have been studied in hiPSCs (Sercel et al., 2021), which means that controlling cell culture conditions, proliferation, and differentiation in hiPSCs with mtDNA heteroplasmy (and related mutations) is a necessary subject of investigation. Resolving such issues is important for minimizing any mtDNA mutation-caused potential poor reproducibility and functional discrepancies from different iPSC clones in disease modeling. New advances in mtDNA gene editing (Mok et al., 2020) can open the door to better modeling mitochondrial dysfunction in hiPSCs.
7. Conclusion
Tractable and reproducible experimental models are pivotal for bridging the gap between genetic findings and disease biology. For modeling genetic risk factors of neuropsychiatric disorders, because of the inherent limitation on drawing causal inferences about psychiatric disease mechanisms, it is critical to clearly define the research question, carefully select the most appropriate experimental system, and rigorously design a well-powered study (see guidelines in a recent review (Alexander Arguello et al., 2019)). With iPSC-derived 2D and 3D models, studying both common and rare risk variants of neuropsychiatric disorders has provided valuable mechanistic insights on how genetic risk factors contribute to disease risks. However, the exact cellular phenotypes and causal molecular mechanisms for each neuropsychiatric disorder remain poorly understood. Most iPSC modeling studies focus on single variants/genes and use a small number of iPSC lines, which may have contributed to the inconsistencies across studies. To improve the robustness and reproducibility of disease variant modeling, it is critical to implement or develop novel methods to scale up the number of iPSC lines and disease risk variants in a systematic manner. Enough cell lines and variants, in combination with cutting-edge CRISPR/Cas9 gene editing and single-cell multi-omics methods, will enable the field to identify convergent molecular and cellular phenotypes that are relevant to a specific disorder or reflective of cross-disorder genetic pleiotropy. It is equally important to further improve the fidelity of iPSC models, especially the brain organoids, by minimizing the stochastic developmental processes and increasing the spatial and temporal control to better recapitulate the in vivo neurodevelopment and brain function. To account for the functional effects from “missing heritability,” it is imperative to consider cellular state or context-specific regulation, gene x environmental interaction, and mtDNA mutations. Finally, neuropsychiatric disorders are polygenic, or even “omnigenic” where thousands of peripheral genes confer disease liability by perturbing a core set of genes (Boyle et al., 2017), it is thus increasingly clear that most risk genes function as gene networks controlled by master regulators (MRs) such TFs (Doostparast Torshizi et al., 2019). Instead of modeling individual risk variants/genes, network perturbation of MRs and other core genes by multiplex CRISPR/Cas9 editing in iPSCs may be needed to more faithfully recapitulate the molecular and cellular phenotypes that are relevant to neuropsychiatric disorders.
Supplementary Material
Acknowledgement
We thank for Charles R. Walgreen family and for National Institute of Mental Health for supporting the work.
Role of funding source
The work was supported by NIH grants R01MH106575, R01MH116281, R0MH1125528 and R01AG063175.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.schres.2022.04.003.
Declaration of competing interest
Authors declare not conflict of interest.
References
- Adamo A, Atashpaz S, Germain PL, Zanella M, D’Agostino G, Albertin V, Chenoweth J, Micale L, Fusco C, Unger C, Augello B, Palumbo O, Hamilton B, Carella M, Donti E, Pruned G, Selicorni A, Biamino E, Prontera P, McKay R, Merla G, Testa G, 2015. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet 47 (2), 132–141. [DOI] [PubMed] [Google Scholar]
- Alexander Arguello P, Addington A, Borja S, Brady L, Dutka T, Gitik M, Koester S, Meinecke D, Merikangas K, McMahon FJ, Panchision D, Senthil G, Lehner T, 2019. From genetics to biology: advancing mental health research in the genomics ERA. Mol. Psychiatry 24 (11), 1576–1582. [DOI] [PubMed] [Google Scholar]
- Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, Pochareddy S, Shin Y, Safi A, Song L, Zhu Y, Sousa AMM, Psych EC, Gerstein M, Crawford GE, Sestan N, Abyzov A, Vaccarino FM, 2018. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362 (6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreazza AC, Nierenberg AA, 2018. Mitochondrial dysfunction: at the Core of psychiatric Disorders ? Biol. Psychiatry 83 (9), 718–719. [DOI] [PubMed] [Google Scholar]
- Arloth J, Bogdan R, Weber P, Frishman G, Menke A, Wagner KV, Balsevich G, Schmidt MV, Karbalai N, Czamara D, Altmann A, Trumbach D, Wurst W, Mehta D, Uhr M, Klengel T, Erhardt A, Carey CE, Conley ED, , Major Depressive Disorder Working Group of the Psychiatric Genomics C, Ruepp A, Muller-Myhsok B, Hariri AR, Binder EB, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium P.G.C., 2015. Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron 86 (5), 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A, 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339 (6123), 1074–1077. [DOI] [PubMed] [Google Scholar]
- Avazzadeh S, Quinlan LR, Reilly J, McDonagh K, Jalali A, Wang Y, McInerney V, Krawczyk J, Ding Y, Fitzgerald J, O’Sullivan M, Forman EB, Lynch SA, Ennis S, Feerick N, Reilly R, Li W, Shen X, Yang G, Lu Y, Peeters H, Dockery P, O’Brien T, Shen S, Gallagher L, 2021. NRXN1alpha(+/−) is associated with increased excitability in ASD iPSC-derived neurons. BMC Neurosci. 22 (1), 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE, J N, Hu Q, Lake BB, Tian W, Kalmbach BE, Crow M, Hodge Rebecca D., Krienen Fenna M., Sorensen Staci A., Eggermont Jeroen, Yao Zizhen, Aevermann Brian D., Aldridge Andrew I., Bartlett Anna, Bertagnolli Darren, Casper Tamara, Castanon Rosa G., Crichton Kirsten, Daigle Tanya L., Dailey Rachel, Dee Nick, Dembrow Nikolai, Diep Dinh, Ding Song-Lin, Dong Weixiu, Fang Rongxin, Fischer Stephan, Goldman Melissa, Goldy Jeff, Graybuck Lucas T., Herb Brian R., Hou Xiaomeng, Kancherla Jayaram, Kroll Matthew, Lathia Kanan, van Lew Baldur, Li Yang Eric, Liu Christine S., Liu Hanqing, Lucero Jacinta D., Mahurkar Anup, McMillen Delissa, Miller Jeremy A., Moussa Marmar, Nery Joseph R., Nicovich Philip R., Orvis Joshua, Osteen Julia K., Owen Scott, Palmer Carter R., Pham Thanh, Plongthongkum Nongluk, Poirion Olivier, Reed Nora M., Rimorin Christine, Rivkin Angeline, Romanow William J., Sedeño-Cortés Adriana E., Siletti Kimberly, Somasundaram Saroja, Sulc Josef, Tieu Michael, Torkelson Amy, Tung Herman, Wang Xinxin, Xie Fangming, Yanny Anna Marie, Zhang Renee, Ament Seth A., Behrens M.Margarita, Bravo Hector Corrada, Chun Jerold, Dobin Alexander, Gillis Jesse, Hertzano Ronna, Hof Patrick R., Höllt Thomas, Horwitz Gregory D., Keene C.Dirk, Kharchenko Peter V., Ko Andrew L., Lelieveldt Boudewijn P., Luo Chongyuan, Mukamel Eran A., Preiss Sebastian, Regev Aviv, Ren Bing, Scheuermann Richard H., Smith Kimberly, Spai William J., White Owen R., Koch Christof, Hawrylycz Michael, Tasic Bosiljka, Macosko Evan Z., McCarroll Steven A., Ting Jonathan T., Zeng Hongkui, Zhang Kun, Feng Guoping, Ecker Joseph R., Linnarsson Sten, Lein Ed S., 2020. Evolution of Cellular Diversity in Primary Motor Cortex of Human, Marmoset Monkey, and Mouse. biorxiv.org. [Google Scholar]
- Bardy C, van den Hurk M, Kakaradov B, Erwin JA, Jaeger BN, Hernandez RV, Eames T, Paucar AA, Gorris M, Marchand C, Jappelli R, Barron J, Bryant AK, Kellogg M, Lasken RS, Rutten BP, Steinbusch HW, Yeo GW, Gage FH, 2016. Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Mol. Psychiatry 21 (11), 1573–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N, Zhang H, Powell SK, Fernando MB, Zhang S, Flaherty EK, Ho SM, Slesinger PA, Duan J, Brennand KJ, 2020. ASCL1- and DLX2-induced GABAergic neurons from hiPSC-derived NPCs. J. Neurosci. Methods 334, 108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Scherer SW, Brzustowicz LM, 2010. Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. Am. J. Psychiatry 167 (8), 899–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, Sabo PJ, Vierstra J, Voit RA, Yuan GC, Porteus MH, Stamatoyannopoulos JA, Lettre G, Orkin SH, 2013. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342 (6155), 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar D, 2017. Mitochondrial multifaceted dysfunction in schizophrenia; complex I as a possible pathological target. Schizophr. Res 187, 3–10. [DOI] [PubMed] [Google Scholar]
- Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, Pollen AA, Nowakowski TJ, Kriegstein AR, 2020. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578 (7793), 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blizinsky KD, Diaz-Castro B, Forrest MP, Schurmann B, Bach AP, Martin-de-Saavedra MD, Wang L, Csernansky JG, Duan J, Penzes P, 2016. Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc. Natl. Acad. Sci. U. S. A 113 (30), 8520–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting GL, Durresi E, Ataman B, Sherman MA, Mei K, Harmin DA, Carter AC, Hochbaum DR, Granger AJ, Engreitz JM, Hrvatin S, Blanchard MR, Yang MG, Griffith EC, Greenberg ME, 2021. Activity-dependent regulome of human GABAergic neurons reveals new patterns of gene regulation and neurological disease heritability. Nat. Neurosci 24 (3), 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EA, Li YI, Pritchard JK, 2017. An expanded view of complex traits: from polygenic to omnigenic. Cell 169 (7), 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brant B, Stern T, Shekhidem HA, Mizrahi L, Rosh I, Stern Y, Ofer P, Asleh A, Umanah GKE, Jada R, Levy NS, Levy AP, Stern S, 2021. IQSEC2 mutation associated with epilepsy, intellectual disability, and autism results in hyperexcitability of patient-derived neurons and deficient synaptic transmission. Mol. Psychiatry 26 (12), 7498–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I, Abdelrahim M, Matikainen-Ankney B, Chao SH, Mrksich M, Rakic P, Fang G, Zhang B, Yates JR 3rd, Gage FH, 2015. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 20 (3), 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Landek-Salgado MA, Sawa A, 2014. Modeling heterogeneous patients with a clinical diagnosis of schizophrenia with induced pluripotent stem cells. Biol. Psychiatry 75 (12), 936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH, 2011. Modelling schizophrenia using human induced pluripotent stem cells. Nature 473 (7346), 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler Iii RR, Kozlova A, Zhang H, Zhang S, Streit M, Sanders AR, Laudanski K, Pang ZP, Gejman PV, Duan J, 2020. The genetic relevance of human induced pluripotent stem cell-derived microglia to Alzheimer’s disease and major neuropsychiatric disorders. Mol Neuropsychiatry 5 (Suppl. 1), 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C, 2005. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet 42 (4), 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, Reimer J, Shen S, Bethge M, Tolias KF, Sandberg R, Tolias AS, 2016. Electrophysiological, transcriptomic and morphologic profiling of single neurons using patch-seq. Nat. Biotechnol 34 (2), 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell CR, Scala F, Li S, Livrizzi G, Shen S, Sandberg R, Jiang X, Tolias AS, 2017. Multimodal profiling of single-cell morphology, electrophysiology, and gene expression using patch-seq. Nat. Protoc 12 (12), 2531–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon D, Nguyen MLT, Mezger A, Kathiria A, Muller F, Nguyen V, Lescano N, Wu B, Trombetta J, Ribado JV, Knowles DA, Gao Z, Blaeschke F, Parent AV, Burt TD, Anderson MS, Criswell LA, Greenleaf WJ, Marson A, Pritchard JK, 2019. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet 51 (10), 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TG, McEwen S, Addington J, Bearden CE, Cadenhead K, Cornblatt B, Mathalon DH, McGlashan T, Perkins D, Jeffries C, Seidman LJ, Tsuang M, Walker E, Woods SW, Heinssen R, 2015. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol. Psychiatry 77 (2), 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GC, Talbot K, Halene TB, Gandal MJ, Kazi HA, Schlosser L, Phung QH, Gur RE, Arnold SE, Siegel SJ, 2011. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc. Natl. Acad. Sci. U. S. A 108 (43), E962–E970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederquist GY, Tchieu J, Callahan SJ, Ramnarine K, Ryan S, Zhang C, Rittenhouse C, Zeltner N, Chung SY, Zhou T, Chen S, Betel D, White RM, Tomishima M, Studer L, 2020. A multiplex human pluripotent stem cell platform defines molecular and functional subclasses of autism-related genes. Cell Stem Cell 27 (1), 35–49.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX, Brown TT, Marchetto MC, Bardy C, McHenry L, Stefanacci L, Jarvinen A, Searcy YM, DeWitt M, Wong W, Lai P, Ard MC, Hanson KL, Romero S, Jacobs B, Dale AM, Dai L, Korenberg JR, Gage FH, Bellugi U, Halgren E, Semendeferi K, Muotri AR, 2016. A human neurodevelopmental model for Williams syndrome. Nature 536 (7616), 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman G, Alsaqati M, Lunn S, Singh T, Linden SC, Linden DEJ, van den Bree MBM, Ziller M, Owen MJ, Hall J, Harwood AJ, Syed YA, 2021. Using induced pluripotent stem cells to investigate human neuronal phenotypes in 1q21.1 deletion and duplication syndrome. Mol. Psychiatry, 10.1038/S41380-021-01182-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawarska K, Campbell D, Chen L, Shic F, Klin A, Chang J, 2011. Early generalized overgrowth in boys with autism. Arch. Gen. Psychiatry 68 (10), 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS, 2013. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499 (7458), 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang K, Zhou L, Gao X, Wang J, Yao Y, He F, Luo Y, Yu Y, Li S, Cheng L, Sun YE, 2016. Coupled electrophysiological recording and single cell transcriptome analyses revealed molecular mechanisms underlying neuronal maturation. Protein Cell 7 (3), 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Li Y, Qi Q, Xu P, Feng R, Palmer L, Chen J, Wu R, Yee T, Zhang J, Yao Y, Sharma A, Hardison RC, Weiss MJ, Cheng Y, 2021. Single-nucleotide-level mapping of DNA regulatory elements that control fetal hemoglobin expression. Nat. Genet 53 (6), 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wang Z-M, Tan W, Wang X, Li Y, Bai B, Li Y, Zhang S-F, Yan H-L, Chen Z-L, Liu C-M, Mi T-W, Xia S, Zhou Z, Liu A, Tang G-B, Liu C, Dai Z-J, Wang Y-Y, Wang H, Wang X, Kang Y, Lin L, Chen Z, Xie N, Sun Q, Xie W, Peng J, Chen D, Teng Z-Q, Jin P, 2018. Partial loss of psychiatric risk gene Mirl37 in mice causes repetitive behavior and impairs sociability and learning via increased Pde1Oa. Nat. Neurosci 21 (12), 1689–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek M, Lusis AJ, 2014. Systems genetics approaches to understand complex traits. Nat. Rev. Genet 15 (1), 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleynen I, Engchuan W, Hestand MS, Heung T, Holleman AM, Johnston HR, Monfeuga T, McDonald-McGinn DM, Gur RE, Morrow BE, Swillen A, Vorstman JAS, Bearden CE, Chow EWC, van den Bree M, Emanuel BS, Vermeesch JR, Warren ST, Owen MJ, Chopra P, Cutler DJ, Duncan R, Kotlar AV, Mulle JG, Voss AJ, Zwick ME, Diacou A, Golden A, Guo T, Lin JR, Wang T, Zhang Z, Zhao Y, Marshall C, Merico D, Jin A, Lilley B, Salmons HI, Tran O, Holmans P, Pardinas A, Walters JTR, Demaerel W, Boot E, Butcher NJ, Costain GA, Lowther C, Evers R, van Amelsvoort T, van Duin E, Vingerhoets C, Breckpot J, Devriendt K, Vergaelen E, Vogels A, Crowley TB, McGinn DE, Moss EM, Sharkus RJ, Unolt M, Zackai EH, Calkins ME, Gallagher RS, Gur RC, Tang SX, Fritsch R, Ornstein C, Repetto GM, Breetvelt E, Duijff SN, Fiksinski A, Moss H, Niarchou M, Murphy KC, Prasad SE, Daly EM, Gudbrandsen M, Murphy CM, Murphy DG, Buzzanca A, Fabio FD, Digilio MC, Pontillo M, Marino B, Vicari S, Coleman K, Cubells JF, Ousley OY, Carmel M, Gothelf D, Mekori-Domachevsky E, Michaelovsky E, Weinberger R, Weizman A, Kushan L, Jalbrzikowski M, Armando M, Eliez S, Sandini C, Schneider M, Bena FS, Antshel KM, Fremont W, Kates WR, Belzeaux R, Busa T, Philip N, Campbell LE, McCabe KL, Hooper SR, Schoch K, Shashi V, Simon TJ, Tassone F, Arango C, Fraguas D, Garcia-Minaur S, Morey-Canyelles J, Rosell J, Suner DH, Raventos-Simic J, Behavior C, Epstein MP, Williams NM, Bassett AS, International 22q D.S.B., 2020. Genetic contributors to risk of schizophrenia in the presence of a 22q11.2 deletion. Mol Psychiatry 26 (8), 4496–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocheme HM, Kelso GF, James AM, Ross MF, Trnka J, Mahendiran T, Asin-Cayuela J, Blaikie FH, Manas AR, Porteous CM, Adlam VJ, Smith RA, Murphy MP, 2007. Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion 7 (Suppl), S94–S102. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F, 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339 (6121), 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, Romero N, Jiang H, Uitti RJ, Huang Z, Opala G, Scarffe LA, Dawson VL, Klein C, Feng J, Ross OA, Trojanowski JQ, Lee VM, Marder K, Surmeier DJ, Wszolek ZK, Przedborski S, Krainc D, Dawson TM, Isacson O, 2012. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med 4 (141), 141ral90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal Lari R, Lupien M, Markowitz S, Scacheri PC, 2014. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 24 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Ruzicka WB, Balu DT, 2020. Fifty years of research on schizophrenia: the ascendance of the glutamatergic synapse. Am. J. Psychiatry 177 (12), 1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH, 1976. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192 (4238), 481–483. [DOI] [PubMed] [Google Scholar]
- Crockett AM, Ryan SK, Vasquez AH, Canning C, Kanyuch N, Kebir H, Ceja G, Gesualdi J, Zackai E, McDonald-McGinn D, Viaene A, Kapoor R, Benallegue N, Gur R, Anderson SA, Alvarez JI, 2021. Disruption of the blood-brain barrier in 22q11.2 deletion syndrome. Brain 144 (5), 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuella-Martin R, Hayward SB, Fan X, Chen X, Huang J-W, Taglialatela A, Leuzzi G, Zhao J, Rabadan R, Lu C, Shen Y, Ciccia A, 2021. Functional interrogation of DNA damage response variants with base editing screens. Cell 184 (4), 1081–1097 el019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvijovic I, Good BH, Desai MM, 2018. The effect of strong purifying selection on genetic diversity. Genetics 209 (4), 1235–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Sun Y, Mohar B, Hulse BK, Kerlin AM, Hasseman JP, Tsegaye G, Tsang A, Wong A, Patel R, Macklin JJ, Chen Y, Konnerth A, Jayaraman V, Looger LL, Schreiter ER, Svoboda K, Kim DS, 2019. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16 (7), 649–657. [DOI] [PubMed] [Google Scholar]
- Das D, Feuer K, Wahbeh M, Avramopoulos D, 2020. Modeling psychiatric disorder biology with stem cells. Curr. Psychiatry Rep 22 (5), 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong JO, Llapashtica C, Genestine M, Strauss K, Provenzano F, Sun Y, Zhu H, Cortese GP, Brundu F, Brigatti KW, Corneo B, Migliori B, Tomer R, Kushner SA, Kellendonk C, Javitch JA, Xu B, Markx S, 2021. Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder. Nat. Commun 12 (1), 4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Los Angeles A, Fernando MB, Hall NAL, Brennand KJ, Harrison PJ, Maher BJ, Weinberger DR, Tunbridge EM, 2021. Induced pluripotent stem cells in psychiatry: an overview and critical perspective. Biol. Psychiatry 90 (6), 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vrij FM, Bouwkamp CG, Gunhanlar N, Shpak G, Lendemeijer B, Baghdadi M, Gopalakrishna S, Ghazvini M, Li TM, Quadri M, Olgiati S, Breedveld GJ, Coesmans M, Mientjes E, de Wit T, Verheijen FW, Beverloo HB, Cohen D, Kok RM, Bakker PR, Nijburg A, Spijker AT, Hafffnans PMJ, Hoencamp E, Bergink V, Consortium GS, Vorstman JA, Wu T, Olde Loohuis LM, Amin N, Langen CD, Hofman A, Hoogendijk WJ, van Duijn CM, Ikram MA, Vernooij MW, Tiemeier H, Uitterlinden AG, Elgersma Y, Distel B, Gribnau J, White T, Bonifati V, Kushner SA, 2019. Candidate CSPG4 mutations and induced pluripotent stem cell modeling implicate oligodendrocyte progenitor cell dysfunction in familial schizophrenia. Mol. Psychiatry 24 (5), 757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Yadav S, Dao DQ, Wu Z-Y, Hokanson KC, Cahill MK, Wiita AP, Jan Y-N, Ullian EM, Weiss LA, 2017. Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism Spectrum disorder. Cell Rep. 21 (10), 2678–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrindt K, Zhang H, Das D, Abdollahi S, Prorok T, Ghosh S, Weintraub S, Genovese G, Powell SK, Lund A, Akbarian S, Eggan K, McCarroll S, Duan J, Avramopoulos D, Brennand KJ, 2020. Publicly available hiPSC lines with extreme polygenic risk scores for modeling schizophrenia. Complex Psychiatry 6 (3–4), 68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R, Geschwind DH, 2011. The human brain in a dish: the promise of iPSC-derived neurons. Cell 145 (6), 831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Peng J, Guo S, 2013. Stable gene silencing in zebrafish with spatiotemporally targetable RNA interference. Genetics 193 (4), 1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doostparast Torshizi A, Armoskus C, Zhang H, Forrest MP, Zhang S, Souaiaia T, Evgrafov OV, Knowles JA, Duan J, Wang K, 2019. Deconvolution of transcriptional networks identifies TCF4 as a master regulator in schizophrenia. Sci. Adv 5 (9), eaau4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvaras P, Fossati V, 2015. Generation and isolation of oligodendrocyte progenitor cells from human pluripotent stem cells. Nat. Protoc 10 (8), 1143–1154. [DOI] [PubMed] [Google Scholar]
- Duan J, 2015. Path from schizophrenia genomics to biology: gene regulation and perturbation in neurons derived from induced pluripotent stem cells and genome editing. Neurosci. Bull 31 (1), 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Goring HHH, Sanders AR, Moy W, Freda J, Drigalenko EI, Kos M, He D, Gejman PV, Mgs, 2018. Transcriptomic signatures of schizophrenia revealed by dopamine perturbation in an ex vivo model. Transl. Psychiatry 8 (1), 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Sanders AR, Gejman PV, 2019. From schizophrenia genetics to disease biology: harnessing new concepts and technologies. J. Psychiatr. Brain Sci 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durak O, Tsai LH, 2014. Human induced pluripotent stem cells: now open to discovery. Cell Stem Cell 15 (1), 4–6. [DOI] [PubMed] [Google Scholar]
- Fantuzzo JA, Mirabella VR, Hamod AH, Hart RP, Zahn JD, Pang ZP, 2017. Intellicount: high-throughput quantification of fluorescent synaptic protein puncta by machine learning. eNeuro 4 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, Hatan M, Carrasco-Alfonso MJ, Mayer D, Luckey CJ, Patsopoulos NA, De Jager PL, Kuchroo VK, Epstein CB, Daly MJ, Hafler DA, Bernstein BE, 2015. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518 (7539), 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farra N, Zhang WB, Pasceri P, Eubanks JH, Salter MW, Ellis J, 2012. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol. Psychiatry 17 (12), 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Albert J, Lipinski M, Lopez-Cascales MT, Rowley MJ, Martin-Gonzalez AM, Del Blanco B, Corces VG, Barco A, 2019. Immediate and deferred epigenomic signatures of in vivo neuronal activation in mouse hippocampus. Nat. Neurosci 22 (10), 1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando MB, Ahfeldt T, Brennand KJ, 2020. Modeling the complex genetic architectures of brain disease. Nat. Genet 52 (4), 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, Overall CC, Gadani SP, Turner SD, Weng Z, Peerzade SN, Chen H, Lee KS, Scott MM, Beenhakker MP, Litvak V, Kipnis J, 2016. Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature 535 (7612), 425–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiou MD, Sandi C, 2019. Anxiety and brain mitochondria: a bidirectional crosstalk. Trends Neurosci 42 (9), 573–588. [DOI] [PubMed] [Google Scholar]
- Flaherty E, Zhu S, Barretto N, Cheng E, Deans PJM, Fernando MB, Schrode N, Francoeur N, Antoine A, Alganem K, Halpern M, Deikus G, Shah H, Fitzgerald M, Ladran I, Gochman P, Rapoport J, Tsankova NM, McCullumsmith R, Hoffman GE, Sebra R, Fang G, Brennand KJ, 2019. Neuronal impact of patient-specific aberrant NRXN1alpha splicing. Nat. Genet 51 (12), 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Zhang H, Moy W, McGowan H, Leites C, Dionisio LE, Xu Z, Shi J, Sanders AR, Greenleaf WJ, Cowan CA, Pang ZP, Gejman PV, Penzes P, Duan J, 2017. Open chromatin profiling in hiPSC-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell 21 (3), 305–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schopflin R, Kraft K, Kempfer R, Jerkovic I, Chan WL, Spielmann M, Timmermann B, Wittier L, Kurth I, Cambiaso P, Zuffardi O, Houge G, Lambie L, Brancati F, Pombo A, Vingron M, Spitz F, Mundlos S, 2016. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538 (7624), 265–269. [DOI] [PubMed] [Google Scholar]
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, Klei LL, Kramer R, Pinto D, Gumus ZH, Cicek AE, Dang KK, Browne A, Lu C, Xie L, Readhead B, Stahl EA, Xiao J, Parvizi M, Hamamsy T, Fullard JF, Wang YC, Mahajan MC, Derry JM, Dudley JT, Hemby SE, Logsdon BA, Talbot K, Raj T, Bennett DA, De Jager PL, Zhu J, Zhang B, Sullivan PF, Chess A, Purcell SM, Shinobu LA, Mangravite LM, Toyoshiba H, Gur RE, Hahn CG, Lewis DA, Haroutunian V, Peters MA, Lipska BK, Buxbaum JD, Schadt EE, Hirai K, Roeder K, Brennand KJ, Katsanis N, Domenici E, Devlin B, Sklar P, 2016. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci 19(11), 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD, 2013. High-frequency off-target mutagenesis induced by CRISPR-cas nucleases in human cells. Nat. Biotechnol 31 (9), 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuzik J, Zeisel A, Mate Z, Calvigioni D, Yanagawa Y, Szabo G, Linnarsson S, Harkany T, 2016. Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat. Biotechnol 34 (2), 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hard C, Schork AJ, Appadurai V, Bull A, Werge TM, Liu C, White KP, CommonMind Consortium, P.C.i.-B.W.G.S.H, Geschwind DH, 2019. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Focus (Am Psychiatr Publ) 17 (1), 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, Shieh AW, Haney J, Parhami S, Belmont J, Kim M, Moran Losada P, Khan Z, Mleczko J, Xia Y, Dai R, Wang D, Yang YT, Xu M, Fish K, Hof PR, Warrell J, Fitzgerald D, White K, Jaffe AE, Psych EC, Peters MA, Gerstein M, Liu C, Iakoucheva LM, Pinto D, Geschwind DH, 2018. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362 (6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner A, Johansson A, Wibom R, Nennesmo I, von Dobeln U, Hagenfeldt L, Hallstrom T, 2003. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord 76 (1–3), 55–68. [DOI] [PubMed] [Google Scholar]
- Giacomelli E, Vahsen BF, Calder EL, Xu Y, Scaber J, Gray E, Dafinca R, Talbot K, Studer L, 2022. Human stem cell models of neurodegeneration: from basic science of amyotrophic lateral sclerosis to clinical translation. Cell Stem Cell 29 (1), 11–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, 2012. Rare and common variants: twenty arguments. Nat. Rev. Genet 13 (2), 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillentine MA, Yin J, Bajic A, Zhang P, Cummock S, Kim JJ, Schaaf CP, 2017. Functional consequences of CHRNA7 copy-number alterations in induced pluripotent stem cells and neural progenitor cells. Am. J. Hum. Genet 101 (6), 874–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdhar K, Hoffman GE, Jiang Y, Brown L, Kundakovic M, Hauberg ME, Francoeur NJ, Wang YC, Shah H, Kavanagh DH, Zharovsky E, Jacobov R, Wiseman JR, Park R, Johnson JS, Kassim BS, Sloofman L, Mattel E, Weng Z, Sieberts SK, Peters MA, Harris BT, Lipska BK, Sklar P, Roussos P, Akbarian S, 2018. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci 21 (8), 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glausier JR, Lewis DA, 2013. Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, Nolan GP, 2018. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174 (4), 968–981 e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Yoon S-J, Tran SS, Makinson CD, Park JY, Andersen J, Valencia AM, Horvath S, Xiao X, Huguenard JR, Pasca SP, Geschwind DH, 2021. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci 24 (3), 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MG, Inoue F, Martin B, Schubach M, Agarwal V, Whalen S, Feng S, Zhao J, Ashuach T, Ziffra R, Kreimer A, Georgakopoulos-Soares I, Yosef N, Ye CJ, Pollard KS, Shendure J, Kircher M, Ahituv N, 2020. lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements. Nat. Protoc 15 (8), 2387–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover S, Padhy SK, Das CP, Vasishta RK, Sharan P, Chakrabarti S, 2006. Mania as a first presentation in mitochondrial myopathy. Psychiatry Clin. Neurosci 60 (6), 774–775. [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, Weitz DA, Rozenblatt-Rosen O, Zhang F, Regev A, 2017. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14 (10), 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RE, Hegde M, Fagre CR, DeWeirdt PC, Sangree AK, Szegletes Z, Griffith A, Feeley MN, Sanson KR, Baidi Y, Koblan LW, Liu DR, Neal JT, Doench JG, 2021. Massively parallel assessment of human variants with base editor screens. Cell 184 (4), 1064–1080. [DOI] [PubMed] [Google Scholar]
- Hartman J.L.t., Garvik B, Hartwell L, 2001. Principles for the buffering of genetic variation. Science 291 (5506), 1001–1004. [DOI] [PubMed] [Google Scholar]
- Heath RG, Krupp IM, 1967. Schizophrenia as an immunologic disorder. I. Demonstration of antibrain globulins by fluorescent antibody techniques. Arch. Gen. Psychiatry 16 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- Hess JL, Tylee DS, Mattheisen M, Borglum AD, Als TD, Grove J, Werge T, Mortensen PB, Mors O, Nordentoft M, Hougaard DM, Byberg-Grauholm J, Baekvad-Hansen M, Greenwood TA, Tsuang MT, Curtis D, Steinberg S, Sigurdsson E, Stefansson H, Stefansson K, Edenberg HJ, Holmans P, Faraone SV, Glatt SJ, Schizophrenia Working Group of the Psychiatric Genomics C., Lundbeck Foundation Initiative for Integrative Psychiatric R., 2019. A polygenic resilience score moderates the genetic risk for schizophrenia. Mol. Psychiatry 26 (3), 800–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Ziller M, Spengler D, 2019. Progress in iPSC-based modeling of psychiatric disorders. Int. J. Mol. Sci 20 (19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman L, Gersbach CA, 2018. Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet 19, 43–71. [DOI] [PubMed] [Google Scholar]
- Hook V, Brennand KJ, Kim Y, Toneff T, Funkelstein L, Lee KC, Ziegler M, Gage FH, 2014. Human iPSC neurons display activity-dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem Cell Rep. 3 (4), 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatta I, Juhila J, Donner J, 2010. Oxidative stress in anxiety and comorbid disorders. Neurosci. Res 68 (4), 261–275. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S, 2012. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch. Gen. Psychiatry 69 (8), 776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF, 2014. Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet. 10 (5), el004369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs BM, 2015. A dangerous method? The use of induced pluripotent stem cells as a model for schizophrenia. Schizophr. Res 168 (1–2), 563–568. [DOI] [PubMed] [Google Scholar]
- Jeong JY, Einhorn Z, Mercurio S, Lee S, Lau B, Mione M, Wilson SW, Guo S, 2006. Neurogenin1 is a determinant of zebrafish basal forebrain dopaminergic neurons and is regulated by the conserved zinc finger protein Tof/Fezl. Proc. Natl. Acad. Sci. U. S. A 103 (13), 5143–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerber J, Seaton DD, Cuomo ASE, Kumasaka N, Haldane J, Steer J, Patel M, Pearce D, Andersson M, Bonder MJ, Mountjoy E, Ghoussaini M, Lancaster MA, Marioni JC, Merkle FT, Gaffney DJ, Stegle O, HipSci C, 2021. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet 53 (3), 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, Geschwind DH, Mane SM, State MW, Sestan N, 2009. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62 (4), 494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou SH, Chiu NY, Liu CS, 2009. Mitochondrial dysfunction and psychiatric disorders. Chang Gung Med. J 32 (4), 370–379. [PubMed] [Google Scholar]
- Kampmann M, 2020. CRISPR-based functional genomics for neurological disease. Nat. Rev. Neurol 16 (9), 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, Wan E, Wong S, Byrnes L, Lanata CM, Gate RE, Mostafavi S, Marson A, Zaitlen N, Criswell LA, Ye CJ, 2018. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36 (1), 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria A, Lopez-Lengowski K, Jagtap SS, McPhie D, Perils RH, Cohen BM, Karmacharya R, 2020a. Transcriptomic landscape and functional characterization of induced pluripotent stem cell-derived cerebral organoids in schizophrenia. JAMA psychiatry 77 (7), 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria A, Lopez-Lengowski K, Vater M, McPhie D, Cohen BM, Karmacharya R, 2020b. Transcriptome analysis and functional characterization of cerebral organoids in bipolar disorder. Genome Med 12 (1), 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria A, Nowosiad P, Jagasia R, Aigner S, Taylor RD, Andreae LC, Gatford NJF, Lucchesi W, Srivastava DP, Price J, 2018. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 23 (3), 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Nakamura M, Ichiba M, Tomiyasu A, Shimo H, Higuchi I, Ueno S, Sano A, 2011. Mitochondrial DNA deletion mutations in patients with neuropsychiatric symptoms. Neurosci. Res 69 (4), 331–336. [DOI] [PubMed] [Google Scholar]
- Kato T, 2011. Mitochondrial dysfunction and bipolar disorder. Curr. Top. Behav. Neurosci 5, 187–200. [DOI] [PubMed] [Google Scholar]
- Khan TA, Revah O, Gordon A, Yoon SJ, Krawisz AK, Goold C, Sun Y, Kim CH, Tian Y, Li MY, Schaepe JM, Ikeda K, Amin ND, Sakai N, Yazawa M, Kushan L, Nishino S, Porteus MH, Rapoport JL, Bernstein JA, O’Hara R, Bearden CE, Hallmayer JF, Huguenard JR, Geschwind DH, Dolmetsch RE, Pasca SP, 2020. Neuronal defects in a human cellular model of 22q11.2 deletion syndrome. Nat. Med 26 (12), 1888–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattak S, Brimble E, Zhang W, Zaslavsky K, Strong E, Ross PJ, Hendry J, Mital S, Salter MW, Osborne LR, Ellis J, 2015. Human induced pluripotent stem cell derived neurons as a model for Williams-beuren syndrome. Mol. Brain 8 (1), 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrameeva E, Kurochkin I, Han D, Guijarro P, Kanton S, Santel M, Qian Z, Rong S, Mazin P, Sabirov M, Bulat M, Efimova O, Tkachev A, Guo S, Sherwood CC, Camp JG, Paabo S, Treutlein B, Khaitovich P, 2020. Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res. 30 (5), 776–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Vadodaria KC, Lenkei Z, Kato T, Gage FH, Marchetto MC, Santos R, 2019. Mitochondria, metabolism, and redox mechanisms in psychiatric disorders. Antioxid. Redox Signal 31 (4), 275–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Hellmuth S, Bechheim M, Putz B, Mohammadi P, Nedelec Y, Giangreco N, Becker J, Kaiser V, Fricker N, Beier E, Boor P, Castel SE, Nothen MM, Barreiro LB, Pickrell JK, Muller-Myhsok B, Lappalainen T, Schumacher J, Hornung V, 2017. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun 8 (1), 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos MZ, Duan J, Sanders AR, Blondell L, Drigalenko EI, Carless MA, Gejman PV, Goring HHH, Mgs, 2018. Dopamine perturbation of gene co-expression networks reveals differential response in schizophrenia for translational machinery. Transl. Psychiatry 8 (1), 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlova A, Zhang S, Kotlar AV, Jamison B, Zhang H, Shi S, Forrest MP, McDaid J, Cutler DJ, Epstein MP, Zwick ME, Pang ZP, Sanders AR, Warren ST, Gejman PV, Mulle JG, Duan J, 2022. Loss-of-function of OTUD7A in the Schizophrenia-associated 15q13.3 Deletion Impairs Synapse Development and Function in Human Neurons. bioRxiv, 2022.2001.2006.473910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, Dolmetsch RE, 2013. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci 16 (2), 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulzer JR, Stitzel ML, Morken MA, Huyghe JR, Fuchsberger C, Kuusisto J, Laakso M, Boehnke M, Collins FS, Mohlke KL, 2014. A common functional regulatory variant at a type 2 diabetes locus upregulates ARAP1 expression in the pancreatic beta cell. Am. J. Hum. Genet 94 (2), 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli MA, Jang J, Park JH, Wang Y, Guzman E, Zhou H, Audouard M, Bridges D, Tovar KR, Papuc SM, Tutulan-Cunita AC, Huang Y, Budisteanu M, Arghir A, Kosik KS, 2016. Haploinsufficiency of BAZ1B contributes to Williams syndrome through transcriptional dysregulation of neurodevelopmental pathways. Hum. Mol. Genet 25 (7), 1294–1306. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, 2013. Cerebral organoids model human brain development and microcephaly. Nature 501 (7467), 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landek-Salgado MA, Faust TE, Sawa A, 2016. Molecular substrates of schizophrenia: homeostatic signaling to connectivity. Mol. Psychiatry 21 (1), 10–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, Ward LD, Raddassi K, McCabe C, Lee MH, Frohlich IY, Hafler DA, Kellis M, Raychaudhuri S, Zhang F, Stranger BE, Benoist CO, De Jager PL, Regev A, Hacohen N, 2014. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343 (6175), 1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey DF, Gelernter J, Polimanti R, Zhou H, Cheng Z, Aslan M, Quaden R, Concato J, Radhakrishnan K, Bryois J, Sullivan PF, Million Veteran P, Stein MB, 2020. Reproducible genetic risk loci for anxiety: results from approximately 200,000 participants in the million veteran program. Am. J. Psychiatry 177 (3), 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Kendler KS, Freedman R, Dudbridge F, Pe’er I, Hakonarson H, Bergen SE, Fanous AH, Holmans PA, Gejman PV, 2011. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry 168 (3), 302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos G, 2008. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology 33 (1), 141–165. [DOI] [PubMed] [Google Scholar]
- Li J, Brickler T, Banuelos A, Marjon K, Shcherbina A, Banerjee S, Bian J, Narayanan C, Weissman IL, Chetty S, 2021a. Overexpression of CD47 is associated with brain overgrowth and 16p11.2 deletion syndrome. Proc. Natl. Acad. Sci. U. S. A 118 (15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ryan SK, Deboer E, Cook K, Fitzgerald S, Lachman HM, Wallace DC, Goldberg EM, Anderson SA, 2019. Mitochondrial deficits in human iPSC-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl. Psychiatry 9 (1), 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tran OT, Crowley TB, Moore TM, Zackai EH, Emanuel BS, McDonald-McGinn DM, Gur RE, Wallace DC, Anderson SA, 2021b. Association of mitochondrial biogenesis with variable penetrance of schizophrenia. JAMA Psychiatry 78 (8), 911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, Sousa AMM, Werling DM, Kitchen RR, Kang HJ, Pletikos M, Choi J, Muchnik S, Xu X, Wang D, Lorente-Galdos B, Liu S, Giusti-Rodriguez P, Won H, de Leeuw CA, Pardinas AF, BrainSpan C, Psych EC, Psych EDS, Hu M, Jin F, Li Y, Owen MJ, O’Donovan MC, Walters JTR, Posthuma D, Reimers MA, Levitt P, Weinberger DR, Hyde TM, Kleinman JE, Geschwind DH, Hawrylycz MJ, State MW, Sanders SJ, Sullivan PF, Gerstein MB, Lein ES, Knowles JA, Sestan N, 2018. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362 (6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, Kleinman JE, 2006. Critical factors in gene expression in postmortem human brain: focus on studies in schizophrenia. Biol. Psychiatry 60 (6), 650–658. [DOI] [PubMed] [Google Scholar]
- Llamosas N, Arora V, Vij R, Kilinc M, Bijoch L, Rojas C, Reich A, Sridharan B, Willems E, Piper DR, Scampavia L, Spicer TP, Miller CA, Holder JL, Rumbaugh G, 2020. SYNGAP1 controls the maturation of dendrites, synaptic function, and network activity in developing human neurons. J. Neurosci 40 (41), 7980–7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM, 2013. RNA-guided human genome engineering via Cas9. Science 339 (6121), 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso M, Ricci G, Choub A, Filosto M, DiMauro S, Davidzon G, Tessa A, Santorelli FM, Murri L, Siciliano G, 2008. Autosomal dominant psychiatric disorders and mitochondrial DNA multiple deletions: report of a family. J. Affect. Disord 106 (1–2), 173–177. [DOI] [PubMed] [Google Scholar]
- Marazziti D, Baroni S, Picchetti M, Landi P, Silvestri S, Vatteroni E, Catena Dell’Osso M, 2012. Psychiatric disorders and mitochondrial dysfunctions. Eur. Rev. Med. Pharmacol. Sci 16 (2), 270–275. [PubMed] [Google Scholar]
- Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, Beltrao-Braga P, Trujillo CA, Mendes APD, Padmanabhan K, Nunez Y, Ou J, Ghosh H, Wright R, Brennand K, Pierce K, Eichenfield L, Pramparo T, Eyler L, Barnes CC, Courchesne E, Geschwind DH, Gage FH, Wynshaw-Boris A, Muotri AR, 2017. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 22 (6), 820–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, Gujral M, Brandler WM, Malhotra D, Wang Z, Fajarado KVF, Maile MS, Ripke S, Agartz I, Albus M, Alexander M, Amin F, Atkins J, Bacanu SA, Belliveau RA Jr., Bergen SE Jr., Bertalan M Jr., Bevilacqua E Jr., Bigdeli TB Jr., Black DW Jr., Bruggeman R Jr., Buccola NG Jr., Buckner RL Jr., Bulik-Sullivan B Jr., Byerley W Jr., Cahn W Jr., Cai G Jr., Cairns MJ Jr., Campion D Jr., Cantor RM Jr., Carr VJ Jr., Carrera N Jr., Catts SV Jr., Chambert KD Jr., Cheng W Jr., Cloninger CR Jr., Cohen D Jr., Cormican P Jr., Craddock N Jr., Crespo-Facorro B Jr., Crowley JJ Jr., Curtis D Jr., Davidson M Jr., Davis KL Jr., Degenhardt F Jr., Del Favero J Jr., DeLisi LE Jr., Dikeos D Jr., Dinan T Jr., Djurovic S Jr., Donohoe G Jr., Drapeau E Jr., Duan J Jr., Dudbridge F Jr., Eichhammer P Jr., Eriksson J Jr., Escott-Price V Jr., Essioux L Jr., Fanous AH Jr., Farh KH Jr., Farrell MS Jr., Frank J Jr., Franke L Jr., Freedman R Jr., Freimer NB Jr., Friedman JI Jr., Forstner AJ Jr., Fromer M Jr., Genovese G Jr., Georgieva L Jr., Gershon ES Jr., Giegling I Jr., Giusti-Rodriguez P Jr., Godard S Jr., Goldstein JI Jr., Gratten J Jr., de Haan L Jr., Hamshere ML Jr., Hansen M Jr., Hansen T Jr., Haroutunian V Jr., Hartmann AM Jr., Henskens FA Jr., Herms S Jr., Hirschhorn JN Jr., Hoffmann P Jr., Hofman A Jr., Huang H Jr., Ikeda M Jr., Joa I Jr., Kahler AK Jr., Kahn RS Jr., Kalaydjieva L Jr., Karjalainen J Jr., Kavanagh D Jr., Keller MC Jr., Kelly BJ Jr., Kennedy JL Jr., Kim Y Jr., Knowles JA Jr., Konte B Jr., Laurent C Jr., Lee P Jr., Lee SH Jr., Legge SE Jr., Lerer B Jr., Levy DL Jr., Liang KY Jr., Lieberman J Jr., Lonnqvist J Jr., Loughland CM Jr., Magnusson PKE Jr., Maher BS Jr., Maier W Jr., Mallet J Jr., Mattheisen M Jr., Mattingsdal M Jr., McCarley RW Jr., McDonald C Jr., McIntosh AM Jr., Meier S Jr., Meijer CJ Jr., Melle I Jr., Mesholam-Gately RI Jr., Metspalu A Jr., Michie PT Jr., Milani L Jr., Milanova V Jr., Mokrab Y Jr., Morris DW Jr., Muller-Myhsok B Jr., Murphy KC Jr., Murray RM Jr., Myin-Germeys I Jr., Nenadic I Jr., Nertney DA Jr., Nestadt G Jr., Nicodemus KK Jr., Nisenbaum L Jr., Nordin A Jr., O’Callaghan E Jr., O’Dushlaine C Jr., Oh SY Jr., Olincy A Jr., Olsen L Jr., O’Neill FA Jr., Van Os J Jr., Pantelis C Jr., Papadimitriou GN Jr., Parkhomenko E Jr., Pato MT Jr., Paunio T Jr., Psychosis Endophenotypes International, Perkins DO Jr., Pers TH Jr., Pietilainen O Jr., Pimm J Jr., Pocklington AJ Jr., Powell J Jr., Price A Jr., Pulver AE Jr., Purcell SM Jr., Quested D Jr., Rasmussen HB Jr., Reichenberg A Jr., Reimers MA Jr., Richards AL Jr., Roffman JL Jr., Roussos P Jr., Ruderfer DM Jr., Salomaa V Jr., Sanders AR Jr., Savitz A Jr., Schall U Jr., Schulze TG Jr., Schwab SG Jr., Scolnick EM Jr., Scott RJ Jr., Seidman LJ Jr., Shi J Jr., Silverman JM Jr., Smoller JW Jr., Soderman E Jr., Spencer CCA Jr., Stahl EA Jr., Strengman E Jr., Strohmaier J Jr., Stroup TS Jr., Suvisaari J Jr., Svrakic DM Jr., Szatkiewicz JP Jr., Thirumalai S Jr., Tooney PA Jr., Veijola J Jr., Visscher PM Jr., Waddington J Jr., Walsh D Jr., Webb BT Jr., Weiser M Jr., Wildenauer DB Jr., Williams NM Jr., Williams S Jr., Witt SH Jr., Wolen AR Jr., Wormley BK Jr., Wray NR Jr., Wu JQ Jr., Zai CC Jr., Adolfsson R Jr., Andreassen OA Jr., Blackwood DHR Jr., Bramon E Jr., Buxbaum JD Jr., Cichon S Jr., Collier DA Jr., Corvin A Jr., Daly MJ Jr., Darvasi A Jr., Domenici E Jr., Esko T Jr., Gejman PV Jr., Gill M Jr., Gurling H Jr., Hultman CM Jr., Iwata N Jr., Jablensky AV Jr., Jonsson EG Jr., Kendler KS Jr., Kirov G Jr., Knight J Jr., Levinson DF Jr., Li QS Jr., McCarroll SA Jr., McQuillin A Jr., Moran JL Jr., Mowry BJ Jr., Nothen MM Jr., Ophoff RA Jr., Owen MJ Jr., Palotie A Jr., Pato CN Jr., Petryshen TL Jr., Posthuma D Jr., Rietschel M Jr., Riley BP Jr., Rujescu D Jr., Sklar P Jr., St Clair D Jr., Walters JTR Jr., Werge T Jr., Sullivan PF Jr., O’Donovan MC Jr., Scherer SW Jr., Neale BM Jr., Sebat J Jr., Cnv, Schizophrenia Working Groups of the Psychiatric Genomics C, 2017. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 49 (1), 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Cerdeño V, 2017. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol 77 (4), 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, Blurton-Jones M, 2018. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener 13 (1), 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle FT, Eggan K, 2013. Modeling human disease with pluripotent stem cells: from genome association to function. Cell Stem Cell 12 (6), 656–668. [DOI] [PubMed] [Google Scholar]
- Michael Deans PJ, Brennand KJ, 2021. Applying stem cells and CRISPR engineering to uncover the etiology of schizophrenia. Curr. Opin. Neurobiol 69, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles JH, 2011. Autism spectrum disorders—A genetics review. Genetics in Medicine 13 (4), 278–294. [DOI] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, Porteous DJ, 2000. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet 9 (9), 1415–1423. [DOI] [PubMed] [Google Scholar]
- Miller CL, Haas U, Diaz R, Leeper NJ, Kundu RK, Patlolla B, Assimes TL, Kaiser FJ, Perisic L, Hedin U, Maegdefessel L, Schunkert H, Erdmann J, Quertermous T, Sczakiel G, 2014. Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation. PLoS Genet. 10 (3), e1004263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ND, Kelsoe JR, 2017. Unraveling the biology of bipolar disorder using induced pluripotent stem-derived neurons. Bipolar Disord. 19 (7), 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JM, Nemesh J, Ghosh S, Handsaker RE, Mello CJ, Meyer D, Raghunathan K, de Rivera H, Tegtmeyer M, Hawes D, Neumann A, Nehme R, Eggan K, McCarroll SA, 2020. Mapping Genetic Effects on Cellular Phenotypes With “Cell Villages”. bioRxiv, 2020.2006.2029.174383. [Google Scholar]
- Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, Mougous JD, Liu DR, 2020. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583 (7817), 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morava E, Kozicz T, 2013. Mitochondria and the economy of stress (mal)adaptation. Neurosci. Biobehav. Rev 37 (4), 668–680. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Battle A, Zhu X, Potash JB, Weissman MM, Shi J, Beckman K, Haudenschild C, McCormick C, Mei R, Gameroff MJ, Gindes H, Adams P, Goes FS, Mondimore FM, MacKinnon DF, Notes L, Schweizer B, Furman D, Montgomery SB, Urban AE, Koller D, Levinson DF, 2014. Type I interferon signaling genes in recurrent major depression: increased expression detected by whole-blood RNA sequencing. Mol. Psychiatry 19 (12), 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton MJ, Barish S, Ralhan I, Chang J, Goodman LD, Harland JG, Marcogliese PC, Johansson JO, Ioannou MS, Bellen HJ, 2021. Neuronal ROS-induced Glial Lipid Droplet Formation Is Altered by Loss of Alzheimer’s Disease-associated Genes. bioRxiv, 2021.2003.2003.433580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer CE, Shelton MA, Sweet RA, 2015. Dendritic spine alterations in schizophrenia. Neurosci. Lett 601, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, Als TD, Bigdeli TB, Børte S, Bryois J, Charney AW, Drange OK, Gandal MJ, Hagenaars SP, Ikeda M, Kamitaki N, Kim M, Krebs K, Panagiotaropoulou G, Schilder BM, Sloofman LG, Steinberg S, Trubetskoy V, Winsvold BS, Won HH, Abramova L, Adorjan K, Agerbo E, Al Eissa M, Albani D, Alliey-Rodriguez N, Anjorin A, Antilla V, Antoniou A, Awasthi S, Baek JH, Bækvad-Hansen M, Bass N, Bauer M, Beins EC, Bergen SE, Birner A, Bøcker Pedersen C, Bøen E, Boks MP, Bosch R, Brum M, Brumpton BM, Brunkhorst-Kanaan N, Budde M, Bybjerg-Grauholm J, Byerley W, Cairns M, Casas M, Cervantes P, Clarke TK, Cruceanu C, Cuellar-Barboza A, Cunningham J, Curtis D, Czerski PM, Dale AM, Dalkner N, David FS, Degenhardt F, Djurovic S, Dobbyn AL, Douzenis A, Elvsåshagen T, Escott-Price V, Ferrier IN, Fiorentino A, Foroud TM, Forty L, Frank J, Frei O, Freimer NB,Frisén L, Gade K, Garnham J, Gelernter J, Giørtz Pedersen M, Gizer IR, Gordon SD, Gordon-Smith K, Greenwood TA, Grove J, Guzman-Parra J, Ha K, Haraldsson M, Hautzinger M, Heilbronner U, Hellgren D, Herms S, Hoffmann P, Holmans PA, Huckins L, Jamain S, Johnson JS, Kalman JL, Kamatani Y, Kennedy JL, Kittel-Schneider S, Knowles JA, Kogevinas M, Koromina M, Kranz TM, Kranzler HR, Kubo M, Kupka R, Kushner SA, Lavebratt C, Lawrence J, Leber M, Lee HJ, Lee PH, Levy SE, Lewis C, Liao C, Lucae S, Lundberg M, MacIntyre DJ, Magnusson SH, Maier W, Maihofer A, Malaspina D, Maratou E, Martinsson L, Mattheisen M, McCarroll SA, McGregor NW, McGuffin P, McKay JD, Medeiros H, Medland SE, Millischer V, Montgomery GW, Moran JL, Morris DW, Mühleisen TW, O’Brien N, O’Donovan C, Olde Loohuis LM, Oruc L, Papiol S, Pardiñas AF, Perry A, Pfennig A, Porichi E, Potash JB, Quested D, Raj T, Rapaport MH, DePaulo JR, Regeer EJ, Rice JP, Rivas F, Rivera M, Roth J, Roussos P, Ruderfer DM, Sánchez-Mora C, Schulte EC, Senner F, Sharp S, Shilling PD, Sigurdsson E, Sirignano L, Slaney C, Smeland OB, Smith DJ, Sobell JL, Søholm Hansen C, Soler Artigas M, Spijker AT, Stein DJ, Strauss JS, Świątkowska B, Terao C, Thorgeirsson TE, Toma C, Tooney P, Tsermpini EE, Vawter MP, Vedder H, Walters JTR, Witt SH, Xi S, Xu W, Yang JMK, Young AH, Young H, Zandi PP, Zhou H, Zillich L, Adolfsson R, Agartz I, Alda M, Alfredsson L, Babadjanova G, Backlund L, Baune BT, Bellivier F, Bengesser S, Berrettini WH, Blackwood DHR, Boehnke M, Børglum AD, Breen G, Carr VJ, Catts S, Corvin A, Craddock N, Dannlowski U, Dikeos D, Esko T, Etain B, Ferentinos P, Frye M, Fullerton JM, Gawlik M, Gershon ES, Goes FS, Green MJ, Grigoroiu-Serbanescu M, Hauser J, Henskens F, Hillert J, Hong KS, Hougaard DM, Hultman CM, Hveem K, Iwata N, Jablensky AV, Jones I, Jones LA, Kahn RS, Kelsoe JR, Kirov G, Landén M, Leboyer M, Lewis CM, Li QS, Lissowska J, Lochner C, Loughland C, Martin NG, Mathews CA, Mayoral F, McElroy SL, McIntosh AM, McMahon FJ, Melle I, Michie P, Milani L, Mitchell PB, Morken G, Mors O, Mortensen PB, Mowry B, Müller-Myhsok B, Myers RM, Neale BM, Nievergelt CM, Nordentoft M, Nöthen MM, O’Donovan MC, Oedegaard KJ, Olsson T, Owen MJ, Paciga SA, Pantelis C, Pato C, Pato MT, Patrinos GP, Perlis RH, Posthuma D, Ramos-Quiroga JA, Reif A, Reininghaus EZ, Ribasés M, Rietschel M, Ripke S, Rouleau GA, Saito T, Schall U, Schalling M, Schofield PR, Schulze TG, Scott LJ, Scott RJ, Serretti A, Shannon Weickert C, Smoller JW, Stefansson H, Stefansson K, Stordal E, Streit F, Sullivan PF, Turecki G, Vaaler AE, Vieta E, Vincent JB, Waldman ID, Weickert TW, Werge T, Wray NR, Zwart JA, Biernacka JM, Nurnberger JI, Cichon S, Edenberg HJ, Stahl EA, McQuillin A, Di Florio A, Ophoff RA, Andreassen OA, 2021. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet 53 (6), 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, Rader DJ, 2010. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466 (7307), 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myint L, Avramopoulos DG, Goff LA, Hansen KD, 2019. Linear models enable powerful differential activity analysis in massively parallel reporter assays. BMC Genomics 20 (1), 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myint L, Wang R, Boukas L, Hansen KD, Goff LA, Avramopoulos D, 2020. A screen of 1,049 schizophrenia and 30 Alzheimer’s-associated variants for regulatory potential. Am. J. Med. Genet. B Neuropsychiatr. Genet 183 (1), 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Miranda A, Craig DW, Shekhtman T, Kmoch S, Bleyer A, Szelinger S, Kato T, Kelsoe JR, 2020. Ntrk1 mutation co-segregating with bipolar disorder and inherited kidney disease in a multiplex family causes defects in neuronal growth and depression-like behavior in mice. Transl. Psychiatry 10 (1), 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah HA, 2008. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol. Psychiatry 13 (1), 27–35. [DOI] [PubMed] [Google Scholar]
- Ni P, Chung S, 2020. Mitochondrial dysfunction in schizophrenia. Bioessays 42 (6), e1900202. [DOI] [PubMed] [Google Scholar]
- Ni P, Noh H, Park GH, Shao Z, Guan Y, Park JM, Yu S, Park JS, Coyle JT, Weinberger DR, Straub RE, Cohen BM, McPhie DL, Yin C, Huang W, Kim HY, Chung S, 2020. iPSC-derived homogeneous populations of developing schizophrenia cortical interneurons have compromised mitochondrial function. Mol. Psychiatry 25 (11), 2873–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, Halban PA, Dermitzakis ET, 2013. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 23 (9), 1554–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez JK, Chen J, Pommier GC, Cogan JZ, Replogle JM, Adriaens C, Ramadoss GN, Shi Q, Hung KL, Samelson AJ, Pogson AN, Kim JYS, Chung A, Leonetti MD, Chang HY, Kampmann M, Bernstein BE, Hovestadt V, Gilbert LA, Weissman JS, 2021. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184 (9), 2503–2519 e2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaumer M, Asara JM, Teplytska L, Murphy MP, Logan A, Turck CW, Filiou MD, 2016. Selective mitochondrial targeting exerts anxiolytic effects in vivo. Neuropsychopharmacology 41 (7), 1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oexle K, Zwirner A, 1997. Advanced telomere shortening in respiratory chain disorders. Hum. Mol. Genet 6 (6), 905–908. [DOI] [PubMed] [Google Scholar]
- Page SC, Sripathy SR, Farinelli F, Ye Z, Wang Y, Hiler DJ, Pattie EA, Nguyen CV, Tippani M, Moses RL, Chen H-Y, Tran MN, Eagles NJ, Stolz JM, Catallini JL, Soudry OR, Dickinson D, Berman KF, Apud JA, Weinberger DR, Martinowich K, Jaffe AE, Straub RE, Maher BJ, 2021. Electrophysiological Measures From Human iPSC-derived Neurons Are Associated With Schizophrenia Clinical Status and Predict Individual Cognitive Performance. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak C, Danko T, Mirabella VR, Wang J, Liu Y, Vangipuram M, Grieder S, Zhang X, Ward T, Huang YA, Jin K, Dexheimer P, Bardes E, Mitelpunkt A, Ma J, McLachlan M, Moore JC, Qu P, Purmann C, Dage JL, Swanson BJ, Urban AE, Aronow BJ, Pang ZP, Levinson DF, Wernig M, Sudhof TC, 2021. Cross-platform validation of neurotransmitter release impairments in schizophrenia patient-derived NRXN1-mutant neurons. Proc. Natl. Acad. Sci. U. S. A 118 (22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagiotakos G, Haveles C, Arjun A, Petrova R, Rana A, Portmann T, Pasca SP, Palmer TD, Dolmetsch RE, 2019. Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome, elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchision DM, 2016. Concise review: Progress and challenges in using human stem cells for biological and therapeutics discovery: neuropsychiatric disorders. Stem Cells 34 (3), 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, Wernig M, 2011. Induction of human neuronal cells by defined transcription factors. Nature 476 (7359), 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q, 2013. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14 (6), 383–400. [DOI] [PubMed] [Google Scholar]
- Pardinas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, Legge SE, Bishop S, Cameron D, Hamshere ML, Han J, Hubbard L, Lynham A, Mantripragada K, Rees E, MacCabe JH, McCarroll SA, Baune BT, Breen G, Byrne EM, Dannlowski U, Eley TC, Hayward C, Martin NG, McIntosh AM, Plomin R, Porteous DJ, Wray NR, Caballero A, Geschwind DH, Huckins LM, Ruderfer DM, Santiago E, Sklar P, Stahl EA, Won H, Agerbo E, Als TD, Andreassen OA, Baekvad-Hansen M, Mortensen PB, Pedersen CB, Borglum AD, Bybjerg-Grauholm J, Djurovic S, Durmishi N, Pedersen MG, Golimbet V, Grove J, Hougaard DM, Mattheisen M, Molden E, Mors O, Nordentoft M, Pejovic-Milovancevic M, Sigurdsson E, Silagadze T, Hansen CS, Stefansson K, Stefansson H, Steinberg S, Tosato S, Werge T, Collier DA, Rujescu D, Kirov G, Owen MJ, O’Donovan MC, Walters JTR, 2018. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 50 (3), 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park GH, Noh H, Shao Z, Ni P, Qin Y, Liu D, Beaudreault CP, Park JS, Abani CP, Park JM, Le DT, Gonzalez SZ, Guan Y, Cohen BM, McPhie DL, Coyle JT, Lanz TA, Xi HS, Yin C, Huang W, Kim HY, Chung S, 2020. Activated microglia cause metabolic disruptions in developmental cortical interneurons that persist in interneurons from individuals with schizophrenia. Nat. Neurosci 23 (11), 1352–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, Smith SJ, Huguenard JR, Geschwind DH, Barres BA, Pasca SP, 2015. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12 (7), 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paşca SP, 2019. Assembling human brain organoids. Science 363 (6423), 126–127. [DOI] [PubMed] [Google Scholar]
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, Bernstein JA, Hallmayer J, Geschwind DH, Dolmetsch RE, 2011. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med 17 (12), 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul DS, Soranzo N, Beck S, 2014. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays 36 (2), 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G, 2011. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472 (7344), 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Wallace DC, 2018. Mitochondrial etiology of neuropsychiatric disorders. Biol. Psychiatry 83 (9), 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Bader JS, Avramopoulos D, 2021. Schizophrenia risk alleles often affect the expression of many genes and each gene may have a different effect on the risk: a mediation analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet 186 (4), 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM, 2011. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci 14 (3), 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PGC2, 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511 (7510), 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PGC3, Ripke S, Walters JTR, O’Donovan MC, 2020. Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia. MedRxiv. 10.1101/2020.09.12.20192922. [DOI] [Google Scholar]
- Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, Di Lullo E, Alvarado B, Bedolli M, Dougherty ML, Fiddes IT, Kronenberg ZN, Shuga J, Leyrat AA, West JA, Bershteyn M, Lowe CB, Pavlovic BJ, Salama SR, Haussler D, Eichler EE, Kriegstein AR, 2019. Establishing cerebral organoids as models of human-specific brain evolution. Cell 176 (4), 743–756 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouget JG, 2018. The emerging immunogenetic architecture of schizophrenia. Schizophr. Bull 44 (5), 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psych EC, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, Jaffe AE, Pinto D, Dracheva S, Geschwind DH, Mill J, Nairn AC, Abyzov A, Pochareddy S, Prabhakar S, Weissman S, Sullivan PF, State MW, Weng Z, Peters MA, White KP, Gerstein MB, Amiri A, Armoskus C, Ashley-Koch AE, Bae T, Beckel-Mitchener A, Berman BP, Coetzee GA, Coppola G, Francoeur N, Fromer M, Gao R, Grennan K, Herstein J, Kavanagh DH, Ivanov NA, Jiang Y, Kitchen RR, Kozlenkov A, Kundakovic M, Li M, Li Z, Liu S, Mangravite LM, Mattei E, Markenscoff-Papadimitriou E, Navarro FC, North N, Omberg L, Panchision D, Parikshak N, Poschmann J, Price AJ, Purcaro M, Reddy TE, Roussos P, Schreiner S, Scuderi S, Sebra R, Shibata M, Shieh AW, Skarica M, Sun W, Swarup V, Thomas A, Tsuji J, van Bakel H, Wang D, Wang Y, Wang K, Werling DM, Willsey AJ, Witt H, Won H, Wong CC, Wray GA, Wu EY, Xu X, Yao L, Senthil G, Lehner T, Sklar P, Sestan N, 2015. The PsychENCODE project. Nat. Neurosci 18 (12), 1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychiatric Genomics Consortium-Schizophrenia, 2011. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet 43 (10), 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychiatric Genomics Consortium-Schizophrenia, 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511 (7510), 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P, 2009. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460 (7256), 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Jacob F, Song MM, Nguyen HN, Song H, Ming GL, 2018. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc 13 (3), 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, Yoon KJ, Jeang W, Lin L, Li Y, Thakor J, Berg DA, Zhang C, Kang E, Chickering M, Nauen D, Ho CY, Wen Z, Christian KM, Shi PY, Maher BJ, Wu H, Jin P, Tang H, Song H, Ming GL, 2016. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165 (5), 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Su Y, Adam CD, Deutschmann AU, Pather SR, Goldberg EM, Su K, Li S, Lu L, Jacob F, Nguyen PTT, Huh S, Hoke A, Swinford-Jackson SE, Wen Z, Gu X, Pierce RC, Wu H, Briand LA, Chen HI, Wolf JA, Song H, Ming GL, 2020. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell 26 (5), 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrato G, Brown J, Arlotta P, 2016. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat. Med 22 (11), 1220–1228. [DOI] [PubMed] [Google Scholar]
- Rajarajan P, Borrman T, Liao W, Schrode N, Flaherty E, Casiño C, Powell S, Yashaswini C, LaMarca EA, Kassim B, Javidfar B, Espeso-Gil S, Li A, Won H, Geschwind DH, Ho S-M, MacDonald M, Hoffman GE, Roussos P, Zhang B, Hahn C-G, Weng Z, Brennand KJ, Akbarian S, 2018. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362 (6420), eaat4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajarajan P, Flaherty E, Akbarian S, Brennand KJ, 2020. CRISPR-based functional evaluation of schizophrenia risk variants. Schizophr. Res 217, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY, 2008. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455 (7215), 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Rodriguez M, Raurell-Vila H, Colli ML, Alvelos MI, Subirana-Granes M, Juan-Mateu J, Norris R, Turatsinze JV, Nakayasu ES, Webb-Robertson BM, Inshaw JRJ, Marchetti P, Piemonti L, Esteller M, Todd JA, Metz TO, Eizirik DL, Pasquali L, 2019. The impact of proinflammatory cytokines on the beta-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat. Genet 51 (11), 1588–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F, 2013. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8 (11), 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, Hanscom C, Pillalamarri V, Seabra CM, Abbott MA, Abdul-Rahman OA, Aberg E, Adley R, Alcaraz-Estrada SL, Alkuraya FS, An Y, Anderson MA, Antolik C, Anyane-Yeboa K, Atkin JF, Bartell T, Bernstein JA, Beyer E, Blumenthal I, Bongers EM, Brilstra EH, Brown CW, Bruggenwirth HT, Callewaert B, Chiang C, Corning K, Cox H, Cuppen E, Currall BB, Cushing T, David D, Deardorff MA, Dheedene A, D’Hooghe M, de Vries BB, Earl DL, Ferguson HL, Fisher H, FitzPatrick DR, Gerrol P, Giachino D, Glessner JT, Gliem T, Grady M, Graham BH, Griffis C, Gripp KW, Gropman AL, Hanson-Kahn A, Harris DJ, Hayden MA, Hill R, Hochstenbach R, Hoffman JD, Hopkin RJ, Hubshman MW, Innes AM, Irons M, Irving M, Jacobsen JC, Janssens S, Jewett T, Johnson JP, Jongmans MC, Kahler SG, Koolen DA, Korzelius J, Kroisel PM, Lacassie Y, Lawless W, Lemyre E, Leppig K, Levin AV, Li H, Li H, Liao EC, Lim C, Lose EJ, Lucente D, Macera MJ, Manavalan P, Mandrile G, Marcelis CL, Margolin L, Mason T, Masser-Frye D, McClellan MW, Mendoza CJ, Menten B, Middelkamp S, Mikami LR, Moe E, Mohammed S, Mononen T, Mortenson ME, Moya G, Nieuwint AW, Ordulu Z, Parkash S, Pauker SP, Pereira S, Perrin D, Phelan K, Aguilar RE, Poddighe PJ, Pregno G, Raskin S, Reis L, Rhead W, Rita D, Renkens I, Roelens F, Ruliera J, Rump P, Schilit SL, Shaheen R, Sparkes R, Spiegel E, Stevens B, Stone MR, Tagoe J, Thakuria JV, van Bon BW, van de Kamp J, van Der Burgt I, van Essen T, van Ravenswaaij-Arts CM, van Roosmalen MJ, Vergult S, Volker-Touw CM, Warburton DP, Waterman MJ, Wiley S, Wilson A, Yerena-de Vega MC, Zori RT, Levy B, Brunner HG, de Leeuw N, Kloosterman WP, Thorland EC, Morton CC, Gusella JF, Talkowski ME, 2017. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet 49 (1), 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, Andina M, King HO, Waisman A, Halassa MM, Huh JR, Choi GB, 2020. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature 577 (7789), 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie SK, Schreiner S, Witt H, Armoskus C, Lay FD, Camarena A, Spitsyna VN, Guo Y, Berman BP, Evgrafov OV, Knowles JA, Farnham PJ, 2018. Using 3D epigenomic maps of primary olfactory neuronal cells from living individuals to understand gene regulation. Sci. Adv 4 (12), eaav8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro M, Brigas HC, Temido-Ferreira M, Pousinha PA, Regen T, Santa C, Coelho JE, Marques-Morgado I, Valente CA, Omenetti S, Stockinger B, Waisman A, Manadas B, Lopes LV, Silva-Santos B, Ribot JC, 2019. Meningeal gammadelta T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci Immunol 4 (40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, Sessa A, Magagnotti C, Bachi A, Giarda E, Verpelli C, Kilstrup-Nielsen C, Sala C, Kalscheuer VM, Broccoli V, 2012. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol 14 (9), 911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, Wilson C, Koblan LW, Zeng J, Bauer DE, Doudna JA, Liu DR, 2020. Phage-assisted evolution of an adenine base editor with improved cas domain compatibility and activity. Nat. Biotechnol 38 (7), 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti A, Repetti GG, Sun C, Price FD, Reny DC, Rapino F, Weisinger K, Benkler C, Peterson QP, Davidow LS, Hansson EM, Rubin LL, 2016. Large-scale production of mature neurons from human pluripotent stem cells in a three-dimensional suspension culture system. Stem Cell Rep. 6 (6), 993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Concortium, S.G.o.P.G., 2013. Psychiatric genomics consortium quadruples schizophrenia GWAS sample size to 35,000 cases and 47,000 controls. In: Abstract. American Society of Human Genetics Annual Meeting 63. October 22–26. [Google Scholar]
- Robicsek O, Karry R, Petit I, Salman-Kesner N, Muller FJ, Klein E, Aberdam D, Ben-Shachar D, 2013. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol. Psychiatry 18 (10), 1067–1076. [DOI] [PubMed] [Google Scholar]
- Roth JG, Muench KL, Asokan A, Mallett VM, Gai H, Verma Y, Weber S, Charlton C, Fowler JL, Loh KM, Dolmetsch RE, Palmer TD, 2020. 16p11.2 microdeletion imparts transcriptional alterations in human iPSC-derived models of early neural development. elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Guennewig B, Kaczorowski DC, Barry G, Brennand KJ, 2016. Activity-dependent changes in gene expression in schizophrenia human-induced pluripotent stem cell neurons. JAMA psychiatry 73 (11), 1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer D, Ripke S, McQuillin A, Group B.a.S., 2018. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 173 (7), 1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacai H, Sakoori K, Konno K, Nagahama K, Suzuki H, Watanabe T, Watanabe M, Uesaka N, Kano M, 2020. Autism spectrum disorder-like behavior caused by reduced excitatory synaptic transmission in pyramidal neurons of mouse prefrontal cortex. Nat. Commun 11 (1), 5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara S, Yanagawa Y, O’Leary DD, Stevens CF, 2012. The fraction of cortical GABAergic neurons is constant from near the start of cortical neurogenesis to adulthood. J. Neurosci 32 (14), 4755–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, 2020. Core concept: how synaptic pruning shapes neural wiring during development and possibly, in disease. Proc. Natl. Acad. Sci 117 (28), 16096–16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim S, Asghar M, Chugh G, Taneja M, Xia Z, Saha K, 2010. Oxidative stress: a potential recipe for anxiety, hypertension and insulin resistance. Brain Res. 1359, 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK, 2014. CRISPR-cas systems for editing, regulating and targeting genomes. Nat. Biotechnol 32 (4), 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AR, Duan J, Levinson DF, Shi J, He D, Hou C, Burrell GJ, Rice JP, Nertney DA, Olincy A, Rozic P, Vinogradov S, Buccola NG, Mowry BJ, Freedman R, Amin F, Black DW, Silverman JM, Byerley WF, Crowe RR, Cloninger CR, Martinez M, Gejman PV, 2008. No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am. J. Psychiatry 165 (4), 497–506. [DOI] [PubMed] [Google Scholar]
- Scala F, Kobak D, Bernabucci M, Bernaerts Y, Cadwell CR, Castro JR, Hartmanis L, Jiang X, Laturnus S, Miranda E, Mulherkar S, Tan ZH, Yao Z, Zeng H, Sandberg R, Berens R, Tolias AS, 2020. Phenotypic Variation Within and Across Transcriptomic Cell Types in Mouse Motor Cortex, biorxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C, 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511 (7510), 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C, Balschun D, Skryabin BV, Dieck S, Smalla KH, Montag D, Leblond CS, Faure P, Torquet N, Le Sourd AM, Toro R, Grabrucker AM, Shoichet SA, Schmitz D, Kreutz MR, Bourgeron T, Gundelfinger ED, Boeckers TM, 2012. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486 (7402), 256–260. [DOI] [PubMed] [Google Scholar]
- Schrode N, Ho SM, Yamamuro K, Dobbyn A, Huckins L, Matos MR, Cheng E, Deans PJM, Flaherty E, Barretto N, Topol A, Alganem K, Abadali S, Gregory J, Hoelzli E, Phatnani H, Singh V, Girish D, Aronow B, McCullumsmith R, Hoffman GE, Stahl EA, Morishita H, Sklar P, Brennand KJ, 2019. Synergistic effects of common schizophrenia risk variants. Nat. Genet 51 (10), 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Lee T, 1975. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 188 (4194), 1217–1219. [DOI] [PubMed] [Google Scholar]
- Sefik E, Purcell RH, Merritt-Garza M, Karne S, Randall J, The Emory 3q, P.collab, Walker EF, Bassell GJ, Mulle JG, 2020. Convergent and Distributed Effects of the Schizophrenia-associated 3q29 Deletion on the Human Neural Transcriptome. bioRxiv, 2020.2005.2025.111351. [Google Scholar]
- Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, Fu T, Worringer K, Brown HE, Wang J, Kaykas A, Karmacharya R, Goold CP, Sheridan SD, Perlis RH, 2019. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci 22 (3), 374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellgren CM, Sheridan SD, Gracias J, Xuan D, Fu T, Perlis RH, 2017. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry 22 (2), 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sercel AJ, Carlson NM, Patananan AN, Teitell MA, 2021. Mitochondrial DNA dynamics in reprogramming to pluripotency. Trends Cell Biol 31 (4), 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343 (6166), 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Noh H, Bin Kim W, Ni P, Nguyen C, Cote SE, Noyes E, Zhao J, Parsons T, Park JM, Zheng K, Park JJ, Coyle JT, Weinberger DR, Straub RE, Berman KF., Apud J, Ongur D, Cohen BM, McPhie DL, Rapoport JL, Perlis RH, Lanz TA, Xi HS, Yin C, Huang W, Hirayama T, Fukuda E, Yagi T, Ghosh S, Eggan KC, Kim HY, Eisenberg LM, Moghadam AA, Stanton PK, Cho JH, Chung S, 2019. Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat. Neurosci 22 (2), 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE, 2013. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503 (7475), 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B, 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488 (7409), 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV, 2009. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460 (7256), 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Poterba T, Curtis D, Akil H, Al Eissa M, Barchas JD, Bass N, Bigdeli TB, Breen G, Bromet EJ, Buckley PF, Bunney WE, Bybjerg-Grauholm J, Byerley WF, Chapman SB, Chen WJ, Churchhouse C, Craddock N, Curtis C, Cusick CM, DeLisi L, Dodge S, Escamilla MA, Eskelinen S, Fanous AH, Faraone SV, Fiorentino A, Francioli L, Gabriel SB, Gage D, Gagliano Taliun SA, Ganna A, Genovese G, Glahn DC, Grove J, Hall M-H, Hamalainen E, Heyne HO, Holi M, Hougaard DM, Howrigan DP, Huang H, Hwu H-G, Kahn RS, Kang HM, Karczewski K, Kirov G, Knowles JA, Lee FS, Lehrer DS, Lescai F, Malaspina D, Marder SR, McCarroll SA, Medeiros H, Milani L, Morley CP, Morris DW, Mortensen PB, Myers RM, Nordentoft M, Brien NL, Olivares AM, Ongur D, Ouwehand WH, Palmer DS, Paunio T, Quested D, Rapaport MH, Rees E, Rollins B, Satterstrom FK, Schatzberg A, Scolnick E, Scott L, Sharp SI, Sklar P, Smoller JW, Sobell JL, Solomonson M, Stevens CR, Suvisaari J, Tiao G, Watson SJ, Watts NA, Blackwood DH, Borglum A, Cohen BM, Corvin AP, Esko T, Freimer NB, Glatt SJ, Hultman CM, McQuillin A, Palotie A, Pato CN, Pato MT, Pulver AE, St. Clair D, Tsuang MT, Vawter MP, Walters JT, Werge T, Ophoff RA, Sullivan PF, Owen MJ, Boehnke M, Donovan M, Neale BM, Daly MJ, 2020. Exome Sequencing Identifies Rare Coding Variants in 10 Genes Which Confer Substantial Risk for Schizophrenia. medRxiv, 2020.2009.2018.20192815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Walters JTR, Johnstone M, Curtis D, Suvisaari J, Torniainen M, Rees E, Iyegbe C, Blackwood D, McIntosh AM, Kirov G, Geschwind D, Murray RM, Di Forti M, Bramon E, Gandal M, Hultman CM, Sklar P, Study I, Consortium UK, Palotie A, Sullivan PF, O’Donovan MC, Owen MJ, Barrett JC, 2017. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet 49 (8), 1167–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, Giusti-Rodriguez P, Hodge RD, Miller JA, Munoz-Manchado AB, O’Donovan MC, Owen MJ, Pardinas AF, Ryge J, Walters JTR, Linnarsson S, Lein ES, , Major Depressive Disorder Working Group of the Psychiatric Genomics C, Sullivan PF, Hjerling-Leffler J, 2018. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet 50 (6), 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM, Protect Study G, 2010. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord 25 (11), 1670–1674. [DOI] [PubMed] [Google Scholar]
- Snyder SH, 1976. The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am. J. Psychiatry 133 (2), 197–202. [DOI] [PubMed] [Google Scholar]
- Soliman MA, Aboharb F, Zeltner N, Studer L, 2017. Pluripotent stem cells in neuropsychiatric disorders. Mol. Psychiatry 22 (9), 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Yang X, Ren X, Maliskova L, Li B, Jones IR, Wang C, Jacob F, Wu K, Traglia M, Tam TW, Jamieson K, Lu SY, Ming GL, Li Y, Yao J, Weiss LA, Dixon JR, Judge LM, Conklin BR, Song H, Gan L, Shen Y, 2019. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet 51 (8), 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieler D, Kaffe M, Knauf F, Bessa J, Tena JJ, Giesert F, Schormair B, Tilch E, Lee H, Horsch M, Czamara D, Karbalai N, von Toerne C, Waldenberger M, Gieger C, Lichtner P, Claussnitzer M, Naumann R, Muller-Myhsok B, Torres M, Garrett L, Rozman J, Klingenspor M, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Beckers J, Holter SM, Meitinger T, Hauck SM, Laumen H, Wurst W, Casares F, Gomez-Skarmeta JL, Winkelmann J, 2014. Restless legs syndrome-associated intronic common variant in Meis1 alters enhancer function in the developing telencephalon. Genome Res. 24 (4), 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Genetic R, Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de Frutos R, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Yoon JL, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St Clair D, Goldstein DB, Stefansson K, Collier DA, Outcome in P, 2009. Common variants conferring risk of schizophrenia. Nature 460 (7256), 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, Green Snyder L, D’Angelo D, Chen Q, Chung WK, Sherr EH, 2016. 16p11.2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. A 170 (11), 2943–2955. [DOI] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, Song H, 2017. Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci 20 (3), 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CR, Mielnik CA, Funk A, O’Donovan SM, Bentea E, Pletnikov M, Ramsey AJ, Wen Z, Rowland LM, McCullumsmith RE, 2019. Measurement of lactate levels in postmortem brain, iPSCs, and animal models of schizophrenia. Sci. Rep 9 (1), 5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Borglum AD, Breen G, Cichon S, Edenberg HJ, Faraone SV, Gelernter J, Mathews CA, Nievergelt CM, Smoller JW, O’Donovan MC, Psychiatric Genomics C, 2018. Psychiatric genomics: an update and an agenda. Am. J. Psychiatry 175 (1), 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg M, Pinson H, Smith RS, Winden KD, Venugopal P, Tai DJC, Gusella JF, Talkowski ME, Walsh CA, Tegmark M, Sahin M, 2021. 16p11.2 deletion is associated with hyperactivation of human iPSC-derived dopaminergic neuron networks and is rescued by RHOA inhibition in vitro. Nat. Commun 12 (1), 2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkiewicz JP, O’Dushlaine C, Chen G, Chambert K, Moran JL, Neale BM, Fromer M, Ruderfer D, Akterin S, Bergen SE, Kahler A, Magnusson PK, Kim Y, Crowley JJ, Rees E, Kirov G, O’Donovan MC, Owen MJ, Walters J, Scolnick E, Sklar P, Purcell S, Hultman CM, McCarroll SA, Sullivan PF, 2014. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry 19 (7), 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC, 2007. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318 (5847), 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S, 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131 (5), 861–872. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S, 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4), 663–676. [DOI] [PubMed] [Google Scholar]
- Tansey KE, Rees E, Linden DE, Ripke S, Chambert KD, Moran JL, McCarroll SA, Holmans P, Kirov G, Walters J, Owen MJ, O’Donovan MC, 2016. Common alleles contribute to schizophrenia in CNV carriers. Mol. Psychiatry 21 (8), 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, Bertagnolli D, Goldy J, Shapovalova N, Parry S, Lee C, Smith K, Bernard A, Madisen L, Sunkin SM, Hawrylycz M, Koch C, Zeng H, 2016. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 19 (2), 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, Penn O, Bakken T, Menon V, Miller J, Fong O, Hirokawa KE, Lathia K, Rimorin C, Tieu M, Larsen R, Casper T, Barkan E, Kroll M, Parry S, Shapovalova NV, Hirschstein D, Pendergraft J, Sullivan HA, Kim TK, Szafer A, Dee N, Groblewski P, Wickersham I, Cetin A, Harris JA, Levi BP, Sunkin SM, Madisen L, Daigle TL, Looger L, Bernard A, Phillips J, Lein E, Hawrylycz M, Svoboda K, Jones AR, Koch C, Zeng H, 2018. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563 (7729), 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme SJ, Maher BJ, Christian KM, 2016. Using induced pluripotent stem cells to investigate complex genetic psychiatric disorders. Curr. Behav. Neurosci. Rep 3 (4), 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke S, Walters JT, O’Donovan MC, 2020. Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia. medRxiv. 10.1101/2020.09.12.20192922. [DOI] [Google Scholar]
- Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Drager N, Leng K, Nalls MA, Singleton AB, Xu K, Faghri F, Kampmann M, 2021. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci 24 (7), 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley KG, Brennand KJ, Huckins LM, 2020. Massively parallel techniques for cataloguing the regulome of the human brain. Nat. Neurosci 23 (12), 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima M, Akamatsu W, Okada Y, Ohnishi T, Balan S, Hisano Y, Iwayama Y, Toyota T, Matsumoto T, Itasaka N, Sugiyama S, Tanaka M, Yano M, Dean B, Okano H, Yoshikawa T, 2016. Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Transl. Psychiatry 6 (11), e934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA, 2001. Control of synapse number by glia. Science 291 (5504), 657–661. [DOI] [PubMed] [Google Scholar]
- Urresti J, Zhang P, Moran-Losada P, Yu NK, Negraes PD, Trujillo CA, Antaki D, Amar M, Chau K, Pramod AB, Diedrich J, Tejwani L, Romero S, Sebat J, Yates Iii JR, Muotri AR, Iakoucheva LM, 2021. Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol Psychiatry 26 (12), 7560–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bon BWM, Coe BP, Bernier R, Green C, Gerdts J, Witherspoon K, Kleefstra T, Willemsen MH, Kumar R, Bosco P, Fichera M, Li D, Amaral D, Cristofoli F, Peeters H, Haan E, Romano C, Mefford HC, Scheffer I, Gecz J, de Vries BBA, Eichler EE, 2016. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21 (1), 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mierlo HC, Schot A, Boks MPM, de Witte LD, 2020. The association between schizophrenia and the immune system: review of the evidence from unbiased ‘omic-studies’. Schizophr. Res 217, 114–123. [DOI] [PubMed] [Google Scholar]
- Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LT, Fernandez N, Ballester B, Andrau JC, Spicuglia S, 2015. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat. Commun 6, 6905. [DOI] [PubMed] [Google Scholar]
- Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, Paulsen B, Nguyen L, Adiconis X, Regev A, Levin JZ, Arlotta P, 2019. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570 (7762), 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M, 2010. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463 (7284), 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelaar CF, Mandal S, Lerch S, Birkner K, Birkenstock J, Buhler U, Schnatz A, Raine CS, Bittner S, Vogt J, Kipnis J, Nitsch R, Zipp F, 2018. Fast direct neuronal signaling via the IL-4 receptor as therapeutic target in neuroinflammation. Sci. Transl. Med 10 (430). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, Xu M, Gandal MJ, Lou S, Zhang J, Park JJ, Yan C, Rhie SK, Manakongtreecheep K, Zhou H, Nathan A, Peters M, Mattei E, Fitzgerald D, Brunetti T, Moore J, Jiang Y, Girdhar K, Hoffman GE, Kalayci S, Gumus ZH, Crawford GE, Psych EC, Roussos P, Akbarian S, Jaffe AE, White KP, Weng Z, Sestan N, Geschwind DH, Knowles JA, Gerstein MB, 2018. Comprehensive functional genomic resource and integrative model for the human brain. Science 362 (6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R, 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153 (4), 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mirabella VR, Dai R, Su X, Xu R, Jadali A, Bernabucci M, Singh I, Chen Y, Tian J, Jiang P, Kwan KY, Pak C, Liu C, Comoletti D, Hart RP, Chen C, Südhof TC, Pang ZP, 2021. Analyses of the Autism-associated Neuroligin-3 R451C Mutation in Human Neurons Reveals a Gain-of-Function Synaptic Mechanism. bioRxiv, 2021.2012.2007.471501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ye F, Wen Z, Guo Z, Yu C, Huang WK, Rojas Ringeling F, Su Y, Zheng W, Zhou G, Christian KM, Song H, Zhang M, Ming GL, 2019. Structural interaction between DISC1 and ATF4 underlying transcriptional and synaptic dysregulation in an iPSC model of mental disorders. Mol. Psychiatry 26 (4), 1346–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Christian KM, Song H, Ming GL, 2016. Modeling psychiatric disorders with patient-derived iPSCs. Curr. Opin. Neurobiol 36, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, Kim NS, Yoon KJ, Shin J, Zhang C, Makri G, Nauen D, Yu H, Guzman E, Chiang CH, Yoritomo N, Kaibuchi K, Zou J, Christian KM, Cheng L, Ross CA, Margolis RL, Chen G, Kosik KS, Song H, Ming GL, 2014. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515 (7527), 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S, Zhang H, Zhang S, Kozlova A, Sanders A, Pang Z, Gejman P, Duan J, 2019. 46 modelling the schizophrenia-associated loss-of-function mutation of SETD1A in human stem cell-derived BRAIN organoids. Eur. Neuropsychopharmacol 29, S84–S85. [Google Scholar]
- Whitehurst T, Howes O, 2022. The role of mitochondria in the pathophysiology of schizophrenia: a critical review of the evidence focusing on mitochondrial complex one. Neurosci. Biobehav. Rev 132, 449–464. [DOI] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E, 2012. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486 (7402), 261–265. [DOI] [PubMed] [Google Scholar]
- Wright R, Rethelyi JM, Gage FH, 2014. Enhancing induced pluripotent stem cell models of schizophrenia. JAMA psychiatry 71 (3), 334–335. [DOI] [PubMed] [Google Scholar]
- Wroblewska A, Dhainaut M, Ben-Zvi B, Rose SA, Park ES, Amir ED, Bektesevic A, Baccarini A, Merad M, Rahman AH, Brown BD, 2018. Protein barcodes enable high-dimensional single-cell CRISPR screens. Cell 175 (4), 1141–1155 e1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Kosten TR, Zhang XY, 2013. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 46, 200–206. [DOI] [PubMed] [Google Scholar]
- Xu P, Liu Z, Liu Y, Ma H, Xu Y, Bao Y, Zhu S, Cao Z, Wu Z, Zhou Z, Wei W, 2021. Genome-wide interrogation of gene functions through base editor screens empowered by barcoded sgRNAs. Nat. Biotechnol 39 (11), 1403–1413. [DOI] [PubMed] [Google Scholar]
- Yang N, Chanda S, Marro S, Ng YH, Janas JA, Haag D, Ang CE, Tang Y, Flores Q, Mall M, Wapinski O, Li M, Ahlenius H, Rubenstein JL, Chang HY, Buylla AA, Sudhof TC, Wernig M, 2017. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 14 (6), 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Guo W, Zhang S, Yu H, Yan H, Zhang H, Sanders AR, Yue W, Duan J, 2021. Cell type-specific and cross-population polygenic risk score analyses of MIR137 gene pathway in schizophrenia. iScience 24 (7), 102785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EL, Greenberg ME, 2018. Activity-regulated transcription: bridging the gap between neural activity and behavior. Neuron 100 (2), 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Kang E, Yu C, Qian X, Jacob F, Yu C, Mao M, Poon RYC, Kim J, Song H, Ming GL, Zhang M, 2017. DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis. Neuron 96 (5), 1041–1054 e1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, Sudhof TC, 2016. Autism-associated SHANK3 haploinsufficiency causes ih channelopathy in human neurons. Science 352 (6286), aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Chen W, Chao ES, Soriano S, Wang L, Wang W, Cummock SE, Tao H, Pang K, Liu Z, Pereira FA, Samaco RC, Zoghbi HY, Xue M, Schaaf CP, 2018. Otud7a knockout mice recapitulate many neurological features of 15q13.3 microdeletion syndrome. Am. J. Hum. Genet 102 (2), 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Pearse TL, Morrow EM, 2016. Modeling developmental neuropsychiatric disorders with iPSC technology: challenges and opportunities. Curr. Opin. Neurobiol 36, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella M, Vitriolo A, Andirko A, Martins PT, Sturm S, O’Rourke T, Laugsch M, Malerba N, Skaros A, Trattaro S, Germain PL, Mihailovic M, Merla G, Rada-Iglesias A, Boeckx C, Testa G, 2019. Dosage analysis of the 7q11.23 Williams region identifies BAZ1B as a major human gene patterning the modern human face and underlying self-domestication. Sci. Adv 5 (12), eaaw7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslavsky K, Zhang WB, McCready FP, Rodrigues DC, Deneault E, Loo C, Zhao M, Ross PJ, El Hajjar J, Romm A, Thompson T, Piekna A, Wei W, Wang Z, Khattak S, Mufteev M, Pasceri P, Scherer SW, Salter MW, Ellis J, 2019. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci 22 (4), 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffler J, Arenas E, Ernfors P, Marklund U, Linnarsson S, 2018. Molecular architecture of the mouse nervous system. Cell 174 (4), 999–1014 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, Robinson D, Yu J, Gallego J, Fleischhacker WW, Kahn RS, Crespo-Facorro B, Vazquez-Bourgon J, Kane JM, Malhotra AK, Lencz T, 2019. Schizophrenia polygenic risk score as a predictor of antipsychotic efficacy in first-episode psychosis. Am. J. Psychiatry 176 (1), 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang H, Forrest MP, Zhou Y, Bagchi VA, Kozlova A, Santos MD, Piguel NH, Dionisio LE, Sanders AR, Pang ZP, He X, Penzes P, Duan J, 2021. Multiple genes in cis mediate the effects of a single chromatin accessibility variant on aberrant synaptic development and function in human neurons. bioRxiv, 2021.2012.2011.472229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang H, Zhou Y, Qiao M, Zhao S, Kozlova A, Shi J, Sanders AR, Wang G, Luo K, Sengupta S, West S, Qian S, Streit M, Avramopoulos D, Cowan CA, Chen M, Pang ZP, Gejman PV, He X, Duan J, 2020. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science 369 (6503), 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang X, Purmann C, Ma S, Shrestha A, Davis KN, Ho M, Huang Y, Pattni R, Wong WH, Bernstein JA, Hallmayer J, Urban AE, 2021b. Network effects of the 15q13.3 microdeletion on the transcriptome and epigenome in human-induced neurons. Biol. Psychiatry 89 (5), 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, Xu W, Yang N, Danko T, Chen L, Wernig M, Sudhof TC, 2013. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78 (5), 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilocchi M, Broderick K, Phanse S, Aly KA, Babu M, 2020. Mitochondria under the spotlight: on the implications of mitochondrial dysfunction and its connectivity to neuropsychiatric disorders. Comput. Struct. Biotechnol. J 18, 2535–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Zizhen, N. TN, van Velthoven Cindy T.J., Goldy Jeff, Sedeno-Cortes Adriana E., F. B, Bertagnolli Darren, Casper Tamara, Crichton Kirsten, Ding Song-Lin, O. F, Garren Emma, Glandon Alexandra, Gray James, Graybuck Lucas T., D. H, Kroll Matthew, Lathia Kanan, Levi Boaz, McMillen Delissa, Mok Stephanie, T. P, Ren Qingzhong, Rimorin Christine, Shapovalova Nadiya, Sulc Josef, S. SM, Tieu Michael, Torkelson Amy, Tung Herman, Ward Katelyn, Dee Nick, S. KA, Tasic Bosiljka, Zeng Hongkui, 2020. A Taxonomy of Transcriptomic Cell Types Across the Isocortex and Hippocampal Formation. bioRxiv e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Zizhen, L. H, Xie Fangming, Fischer Stephan, Booeshaghi A.Sina, Adkins Ricky, S., A. AI, Ament Seth A., Pinto-Duarte Antonio, Bartlett Anna, Behrens M. Margarita, Van den Berge Koen, B. D, Biancalani Tommaso, Bravo Héctor Corrada, Casper Tamara, Colantuoni Carlo, C. H, Crichton Kirsten, Crow Megan, Dee Nick, Dougherty Elizabeth L., Wayne I, Doyle SD, Fang Rongxin, Felix Victor, Fong Olivia, Giglio Michelle, Goldy Jeff, Hawrylycz Mike H.R.d.B., Herb Brian R., Ronna Hertzano, Hou Xiaomeng, Hu Qiwen, Crabtree Jonathan, K. J, Kroll Matthew, Lathia Kanan, Li Yang Eric, Lucero Jacinta D., Luo Chongyuan, M. A, McMillen Delissa, Nadaf Naeem, Nery Joseph R., Niu Sheng-Yong, Orvis Joshua, O. JK, Pham Thanh, Poirion Olivier, Preissl Sebastian, Purdom Elizabeth, Rimorin Christine, R. D, Rivkin Angeline C., Smith Kimberly, Street Kelly, Sulc Josef, Thuc Nghi Nguyen, T. M, Torkelson Amy, Tung Herman, Vaishnav Eeshit Dhaval, Valentine Svensson, V. CR, Ntranos Vasilis, van Velthoven Cindy, Wang Xinxin, White Owen R., Huang Z.Josh, K PV, Pachter Lior, Ngai John, Regev Aviv, Tasic Bosiljka, Welch Joshua, D., G. J, Macosko Evan Z., Ren Bing, Ecker Joseph R., Zeng Hongkui, Mukamel Eran A., 2020. An Integrated Transcriptomic and Epigenomic Atlas of Mouse Primary Motor Cortex Cell Types. bioRxiv. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.