

Abstract
Studying the correlation between cerebrospinal fluid (CSF) metabolites and the Alzheimer’s Disease (AD) biomarkers may offer a window to the alterations of the brain metabolome and unveil potential biological mechanisms underlying AD. In this analysis, 308 CSF metabolites from 338 individuals of Wisconsin Registry for Alzheimer’s Prevention and Wisconsin Alzheimer’s Disease Research Center were included in a principal component analysis (PCA). The resulted principal components (PCs) were tested for association with CSF total tau (t-tau), phosphorylated tau (p-tau), amyloid β 42 (Aβ42), and Aβ42/40 ratio using linear regression models. Significant PCs were further tested with other CSF NeuroToolKit (NTK) and imaging biomarkers. Using a Bonferroni corrected p <0.05, five PCs were significantly associated with CSF p-tau and t-tau and three PCs were significantly associated with CSF Aβ42. Pathway analysis suggested that these PCS were enriched in six pathways, including metabolism of caffeine and nicotinate and nicotinamide. This study provides evidence that CSF metabolites are associated with AD pathology through core AD biomarkers and other NTK markers and suggests potential pathways to follow up in future studies.
Keywords: Alzheimer’s disease, CSF metabolomics, principal component analysis, total tau, phosphorylated tau, amyloid β 42/40
1. Introduction
Alzheimer’s disease (AD), a neurodegenerative disorder, is characterized by the progressive decline in cognition and functional status. The pathology of AD may begin 20 years before the clinical symptoms appear and two established pathological changes in AD brains are the extracellular deposits of amyloid β (Aβ), which form amyloid plaques, and intraneuronal aggregates of hyperphosphorylated and misfolded tau, giving rise to neurofibrillary tangles (DeTure and Dickson, 2019). Besides these two changes and their corresponding cerebrospinal fluid (CSF) biomarkers of Aβ 42, phosphorylated-tau (p-tau), and total-tau (t-tau), recent studies have shown that CSF biomarkers of synaptic dysfunction, astrocyte activation, microglial activation, and inflammation are also associated with neurodegeneration and AD (Antonell et al., 2019; Milà‐Alomà et al., 2020). However, the potential mechanisms linking these CSF biomarkers to neurodegeneration and AD have not been established.
Advancements in omics technologies enable the use of novel approaches to study the biological changes involved in AD. One promising approach is metabolomics, which can identify and quantify a large number of small molecules (<1500 Da), such as amino acids, carbohydrates, and lipids, simultaneously in a biological sample (Hasin et al., 2017). Metabolites are relevant to the current physiological state of a cell (Enche Ady et al., 2017). Fluctuations in metabolite levels may be involved in the disease process and can give insight into the underlying mechanisms of the disorder (Toledo et al., 2017). As the downstream alterations resulting from transcripts or proteins, metabolites can build a bridge to link those upstream molecular changes to AD pathophysiology. On the other hand, metabolites can also be influenced by modifiable risk factors such as diet, physical activity, or pollutants. Thus, metabolites also open a window for us to learn how modifiable risk factors influence the biological molecules and lead to AD.
So far, several studies have examined targeted or untargeted human metabolomics in blood, post-mortem brain tissue, CSF, and saliva that associate with clinical AD status or AD biomarkers (Koal et al., 2015; Paglia et al., 2016; Guiraud et al., 2017; Toledo et al., 2017; Huan et al., 2018). Multiple metabolic pathways, such as those involving: alanine, aspartate, and glutamate; glycerophospholipids; sphingolipids; homocysteine-methionine; and polyamines, have been reported to be associated with clinical AD, mild cognitive impairment (MCI), or AD biomarkers (Wilkins and Trushina, 2018). However, among those metabolomic studies, few of them (Guiraud et al., 2017; Koal et al., 2015) have been conducted in CSF, with a particular dearth of untargeted CSF metabolomics studies, since untargeted metabolomics aims at discovery and relies on a large sample size, which has been hard to achieve in CSF thus far. An untargeted approach measures a larger number of metabolites and is useful for hypothesis generation by conducting comprehensive analyses to identify previously undiscovered metabolites and metabolic pathways that are associated with disease (Schrimpe-Rutledge et al., 2016; Wilkins and Trushina, 2018). Since the CSF surrounds the extracellular space of the brain, it is an ideal fluid to study the pathological changes occurring in the central nervous system in the early stages of AD. Correlating metabolites in the CSF with established and developing AD biomarkers may: elucidate additional changes that are associated with early AD disease pathology, enhancing our knowledge of the disease; develop new biomarkers for preclinical and clinical AD diagnosis; and provide candidates for AD treatment.
In this study, we aimed to determine which CSF metabolites and pathways are associated with a comprehensive panel of AD biomarkers in two cohorts: the Wisconsin Registry for Alzheimer’s Prevention (WRAP) and the Wisconsin Alzheimer’s Disease Research Center (ADRC). We first reduced the dimension of untargeted CSF metabolites by using principal components analysis (PCA) and then tested the associations between the resulting principal components (PCs) and AD biomarkers. The biological functions of significant PCs were then analyzed using pathway analyses.
2. Methods
2.1. Participants
WRAP began recruitment in 2001 as a prospective cohort study, with initial follow up four years after baseline, and subsequent ongoing follow up every two years (Johnson et al., 2018). WRAP is comprised of initially cognitively-unimpaired, asymptomatic, middle-aged (between 40 and 65) adults enriched for a parental history of clinical AD (Johnson et al., 2018). At each visit, the participants undergo comprehensive medical and cognitive evaluations. Follow up is discontinued if dementia develops. Additional details of the study design and methods of WRAP have been described previously (Johnson et al., 2018). From the WRAP cohort, we identified 133 self-reported non-Hispanic white individuals with CSF biomarker and metabolomic data. The sample size for other racial/ethnic groups was too small (n<10) to calculate accurate coefficients, so these participants were excluded from the analyses.
The Wisconsin ADRC’s clinical core cohorts started in 2009 and are comprised of well-characterized participants who undergo cognitive testing and physical exams every two years (Bettcher et al., 2018). The Wisconsin ADRC has a cohort of initially cognitively-unimpaired, asymptomatic middle-aged (between 45 and 65) adults with a similar study design to WRAP (the Investigating Memory in Preclinical AD-Causes and Treatments (IMPACT) cohort) (Darst et al., 2017; Racine et al., 2016; Vogt et al., 2018). From the IMPACT cohort, we identified 205 self-reported non-Hispanic white participants with CSF biomarker and metabolomic data. As with WRAP, the sample size for other racial/ethnic groups was too small (n<10) to calculate accurate coefficients, so these participants were excluded from the analyses.
Data from the 133 WRAP participants and 205 Wisconsin ADRC IMPACT participants were combined for the PCA analysis. This study was conducted with the approval of the University of Wisconsin Institutional Review Board, and all participants provided signed informed consent before participation.
2.2. CSF sample collection and CSF biomarkers quantification
Fasting CSF samples were collected via lumbar puncture using a Sprotte 25- or 24-gauge spinal needle at the L3/4 or L4/5 interspace with gentle extraction into polypropylene syringes. More details can be found in the previous study (Darst et al., 2017). Importantly, the CSF collection for WRAP and the Wisconsin ADRC follows the same protocol and the lumbar puncture for both studies is performed by the same group of well-trained individuals.
All CSF samples were batched together and assayed for biomarkers using the Roche NeuroToolKit (NTK), a panel of automated Elecsys® and prototype immunoassays (Roche Diagnostics International Ltd, Rotkreuz, Switzerland) at the Clinical Neurochemistry Laboratory, University of Gothenburg, using the same lot of reagents, under strict quality control procedures. The immunoassays of Elecsys β-amyloid(1–42), β-amyloid(1–40), Phospho-Tau (181P) and Total-Tau, as well as S100 calcium-binding protein B (S100b) and interleukin-6 (IL-6) were performed on a cobas e 601 analyzer (Roche Diagnostics International Ltd, Rotkreuz, Switzerland) (Van Hulle et al., 2020). The remaining NTK panel was assayed on a cobas e 411 analyzer (Roche Diagnostics International Ltd, Rotkreuz, Switzerland) including α-synuclein, glial fibrillary acidic protein (GFAP), chitinase-3-like protein 1 (YKL-40), soluble triggering receptor expressed on myeloid cells 2 (sTREM2), neurofilament light protein (NfL), and neurogranin (Van Hulle et al., 2020).
2.3. Neuroimaging biomarkers
A subset of WRAP and Wisconsin ADRC participants underwent Pittsburgh Compound B (PiB) positron emission tomography (PET) and T1-weighted magnetic resonance imaging (MRI). The details regarding PiB radiopharmaceutical synthesis, PET and MRI acquisition protocols, and image processing have been previously described (Bendlin et al., 2012; Johnson et al., 2014; Sprecher et al., 2015). Briefly, tissue segmentation, spatial normalization, and anatomical delineation for PET analysis was performed on T1-weighted MRI using SPM12’s unified segmentation algorithm (Ashburner and Friston, 2005) (www.fil.ion.ucl.ac.uk/spm) and software written in MATLAB. Gray matter, white matter, and CSF volumes were extracted from the native space tissue segments using the Tissue Volumes module in SPM. Total intracranial volume (ICV) was calculated by adding up the grey matter volume, white matter volume, and CSF volume. The total brain volume (TBV) was calculated as the sum of grey and white matter adjusted for ICV in analyses. Hippocampal volumes (HV) were obtained using FSL FIRST (Patenaude et al., 2011) and were adjusted for ICV in analyses.
PET regions of interest were obtained by applying the inverse deformation field from SPM12’s segmentation to the Automated Anatomical Labeling Atlas (Tzourio-Mazoyer et al., 2002), and restricting the resultant ROIs to voxels with P(GM)>0.30. [C-11]PiB were dynamically acquired 0–70 minutes post-injection on either a Siemens EXACT HR+ or Siemens Biograph Horizon PET/CT tomograph. The reconstructed PET time series were smoothed, interframe realigned, dynamically denoised, and registered to T1-weighted MRI. Amyloid plaque burden (i.e., global PiB distribution volume ratio [DVR]) was assessed by calculating the mean distribution volume ratio across eight bilateral regions of interest using Logan graphical analysis with the cerebellar gray matter as reference region (k2bar = 0.169 min−1) (Lopresti et al., 2005).
2.4. CSF metabolomic profiling and quality control
CSF metabolomic analyses and quantification were performed in one batch by Metabolon (Durham, NC) using an untargeted approach, based on Ultrahigh Performance Liquid Chromatography‐Tandem Mass Spectrometry platform (Bridgewater BR, 2014). Details of the metabolomic profiling were described in an earlier study (Darst et al., 2019).
The number of metabolites present in CSF is less than that found in other fluids, such as blood. A total of 412 CSF metabolites were identified and quality control procedures were performed (Supplemental Figure 1). First, 46 metabolites missing for at least 80% (Denburg et al., 2021) of the individuals were excluded. The original missing percentages are provided in Supplemental Table 1. Then the values for each of the remaining metabolites were scaled so that the median equaled 1. Since all the endogenous metabolites should be present in the human body, missing values indicate a value below the technical detection level. For the 325 endogenous metabolites, the missing values were then imputed to half of the lowest level of detection. However, for xenobiotic metabolites, e.g., drug metabolites, missing values likely indicate the absence of the metabolite in the sample; these metabolites were imputed to 0.0001 (a value near zero that could be accepted in the analytic software), as recommended by Metabolon. No metabolites with zero variability between individuals and 2 metabolites with an interquartile range of zero were excluded. Log10 transformation was applied to normalize the data. After quality control, 308 metabolites with known biochemical names (i.e., metabolites that could be chemically identified using existing metabolite libraries) remained for this investigation. Since both WRAP and the Wisconsin ADRC are longitudinal studies, we generally had 2–3 measures of each metabolite per individual, each approximately two years apart. The average of the multiple measures of each metabolite within each individual were calculated and used for the PCA.
2.5. Statistical analysis
2.5.1. PCA and association tests
In order to group correlated CSF metabolites and reduce the data dimension and multiple testing burden due to hundreds of CSF metabolites and more than ten AD endophenotypes in our analysis, PCA was applied by using the within-individual averaged values of each of the 308 CSF metabolites. The PCA was conducted using the R function “prcomp”. All the PCs that explained up to 90% of the variance (Jolliffe, 2005) in CSF metabolites were extracted for association tests. After PCA, the associations between these PCs and the last visit value of four core AD biomarkers: p-tau, t-tau, Aβ42, and Aβ42/40, were tested using the linear regression method while adjusting for age at last visit of biomarkers, sex, years of education, and cohort. A sensitivity analysis using a fixed-effects meta-analysis model weighted by the sample size of each of the two cohorts, instead of using combined samples, was also conducted to calculate the pooled estimates and p values. Another sensitivity analysis was conducted by excluding the 8 samples that were not collected after fasting for at least 12 hours. Then, the significant PCs were tested with nine other NTK biomarkers: Aβ40 as a marker of amyloid production; S100b, YKL-40, and GFAP as markers of astrocyte activation; sTREM2 and IL-6 as markers of microglial activation and inflammation; and NfL, neurogranin, and α-synuclein as markers of synaptic damage and neuronal degeneration, as well as three imaging markers: MRI TBV, MRI HV, and PiB DVR. The values of all biomarkers were log10 transformed and the pairwise correlations among all NTK biomarkers are shown in Supplemental Figure 2. All tests used the Bonferroni adjustment to correct p values for multiple testing. The Bonferroni-adjusted significance threshold was p<0.05.
2.5.2. Biological relevance of significant PCs
For each significant PC, the metabolites were ranked by the absolute values of their loadings. Potential functional pathways of each PC were identified from the Kyoto Encyclopedia of Genes and Genomes (KEGG) Homo sapiens pathway database (Kanehisa and Goto, 2000; Okuda et al., 2008) by conducting pathway analyses of the top 10% of the ranked metabolites in a web-based software, MetaboAnalyst5.0 (Chong et al., 2018). By inputting the metabolites’ human metabolome database (HMDB) IDs and using the default hypergeometric test and the relative-betweenness centrality, which is a measure of centrality in a graph based on the shortest paths that passes through the vertex, enriched pathways were identified. Pathways were considered as important if the impact score, which represents an objective estimate of the importance of a given pathway relative to a global metabolic network, was >0.1 (Wang et al., 2016). The position of these important metabolites in the KEGG pathway was examined.
2.5.3. Caffeine metabolism analysis
Pathway analysis suggested that four of the most significant PCs were enriched for metabolites in the caffeine metabolism pathway (impact score >0.1). Consequently, the associations between caffeine intake and those four significant PCs were tested in WRAP (caffeine intake was not measured in the Wisconsin ADRC). Caffeine intake was assessed via questionnaire. All participants were asked “During the past month, how often did you drink any caffeinated beverages? (e.g., coffee, tea, soft drinks)” with possible responses including “1 = less than once per day, 2 = 1–2 per day, 3 = 3–5 per day, 4 = 6 or more per day.” A mediation analysis from baseline caffeine intake through the PC enriched in the caffeine metabolism pathway to the last visit of p-tau and t-tau was conducted in the WRAP cohort using linear regression models adjusting for the mean age, sex, years of education, and body mass index (BMI).
In the combined cohort, the associations between the two tau outcomes and the mean values of the five caffeine metabolism metabolites in the four significant PCs, theobromine, caffeine, paraxanthine, 7-methylxanthine, and 1-methylxanthine, was tested. Additionally, the pairwise ratios of the four caffeine metabolism metabolites that formed a feedback loop (theobromine/caffeine, caffeine/paraxanthine, paraxanthine/7-methylxanthine, and 7-methylxanthine/theobromine) were also tested for association with tau. The linear regression model was used for each metabolite or metabolite ratio adjusted for age at last visit of biomarkers, sex, years of education, and cohort.
3. Results
3.1. Participant characteristics
We included 338 participants from both WRAP and the Wisconsin ADRC IMPACT cohort for the main analysis. Table 1 shows the sample characteristics of WRAP, Wisconsin ADRC IMPACT, and all participants. In general, 133 (39.3%) were from WRAP and 205 (60.7%) were from Wisconsin ADRC IMPACT. After combining the cohorts, two thirds of the participants were female and the mean age at the last CSF collection (used for the AD biomarkers) was 61.5 (SD=6.9) years. The mean years of education was 16.3 (SD=2.4). The mean Mini-Mental State Examination (MMSE) total scores at the last CSF collection for WRAP and Wisconsin ADRC IMPACT participants were 29.5 (SD=0.9, min=26, max=30) and 29.4 (SD=0.8, min=27, max=30), respectively.
Table 1.
Sample characteristics of 338 combined WRAP and Wisconsin ADRC IMPACT participants.
| Characteristics | WRAP (n=133) | IMPACT (n=205) | Total (N=338) | |||
|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |
| Gender | ||||||
| Male | 51 | 38.3 | 61 | 29.8 | 112 | 33.1 |
| Female | 82 | 61.7 | 144 | 70.2 | 226 | 66.9 |
| Caffeine intake* | NA |
NA |
||||
| Less than once per day | 33 | 25.0 | ||||
| 1–2 per day | 59 | 44.7 | ||||
| 3–5 per day | 35 | 26.5 | ||||
| 6 or more per day | 5 | 3.8 | ||||
| Mean | SD | Mean | SD | Mean | SD | |
| Age of CSF collection + | 65.4 | 6.7 | 58.9 | 5.7 | 61.5 | 6.9 |
| MMSE at CSF collection + | 29.5 | 0.9 | 29.4 | 0.8 | 29.4 | 0.8 |
| Age at MRI + | 66.9 | 6.6 | 61.7 | 6.1 | 63.5 | 6.8 |
| Age at PiB DVR + | 67.1 | 6.7 | 63.7 | 4.8 | 66.4 | 6.5 |
| Years of education | 16.5 | 2.6 | 16.2 | 2.3 | 16.3 | 2.4 |
| p-tau + | 18.9 | 7.5 | 15.8 | 5.8 | 17.0 | 6.7 |
| t-tau + | 212.9 | 75.1 | 182.9 | 67.3 | 194.7 | 71.9 |
| Aβ42 + | 868.6 | 390.0 | 917.6 | 363.5 | 898.3 | 374.3 |
| Aβ42/40 + | 0.06 | 0.02 | 0.07 | 0.01 | 0.06 | 0.02 |
Within WRAP, one participant did not report caffeine intake, n=132. The summary statistics of caffeine intake was based on the baseline visit.
NA: the caffeine intake data were not available in the IMPACT cohort.
For the AD biomarkers, the last available visit was used.
3.2. PCA and association test results
From the PCA, the first 30 PCs explained just over 90% of the variance of all 308 metabolites (Supplemental Table 2). Among these 30 PCs, PC13, PC16, PC20, PC22, and PC29 were significantly associated with CSF p-tau and t-tau after adjusting for multiple testing (estimates of the effect, p-values, and Bonferroni-adjusted p-values are shown in Table 2). PC8, PC16, and PC22 were significantly associated with CSF Aβ42. None of the PCs were associated with Aβ42/40. Figure 1 shows the metabolites ranked by the absolute value of their loading for each significant PC (metabolites in the top 10% of loadings are displayed). Some metabolites had high loadings for multiple PCs (e.g., kynurenate for PC8, PC20, and PC22). The estimates and significance of the effect from meta-analyses of the WRAP and Wisconsin ADRC IMPACT cohorts and the sensitivity analyses, which excluded 8 samples not collected after fasting for at least 12 hours, were similar and can be found in Supplemental Tables 4 and 5.
Table 2.
Results from linear regression models for four core AD CSF biomarkers.
| p-tau> | t-tau | Aß42 | Aß42/40 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| PC | Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p |
| 1 | −0.5238 | 1.53E-01 | 1.00E+00 | −0.4901 | 1.91E-01 | 1.00E+00 | 0.1963 | 4.71E-01 | 1.00E+00 | 0.5357 | 2.04E-01 | 1.00E+00 |
| 2 | −0.6802 | 9.61E-02 | 1.00E+00 | −0.4695 | 2.60E-01 | 1.00E+00 | 0.2779 | 3.59E-01 | 1.00E+00 | 0.6674 | 1.55E-01 | 1.00E+00 |
| 3 | 0.5286 | 1.13E-01 | 1.00E+00 | 0.5640 | 9.78E-02 | 1.00E+00 | 0.2209 | 3.72E-01 | 1.00E+00 | −0.1761 | 6.44E-01 | 1.00E+00 |
| 4 | −0.7265 | 1.79E-02 | 5.38E-01 | −0.6778 | 3.25E-02 | 9.75E-01 | −0.5178 | 2.43E-02 | 7.30E-01 | −0.1791 | 6.11E-01 | 1.00E+00 |
| 5 | 0.8594 | 4.02E-03 | 1.21E-01 | 0.8667 | 4.47E-03 | 1.34E-01 | 0.5028 | 2.34E-02 | 7.01E-01 | −0.0255 | 9.41E-01 | 1.00E+00 |
| 6 | −0.4597 | 1.15E-01 | 1.00E+00 | −0.4448 | 1.37E-01 | 1.00E+00 | −0.4066 | 6.08E-02 | 1.00E+00 | −0.1109 | 7.41E-01 | 1.00E+00 |
| 7 | 0.0622 | 8.21E-01 | 1.00E+00 | 0.1649 | 5.57E-01 | 1.00E+00 | 0.3540 | 8.20E-02 | 1.00E+00 | 0.4180 | 1.86E-01 | 1.00E+00 |
| 8 | −0.7601 | 2.92E-03 | 8.77E-02 | −0.7680 | 3.19E-03 | 9.57E-02 | −0.6323 | 8.03E-04 | 2.41E-02 | −0.2317 | 4.32E-01 | 1.00E+00 |
| 9 | −0.2758 | 2.45E-01 | 1.00E+00 | −0.2240 | 3.54E-01 | 1.00E+00 | 0.1087 | 5.36E-01 | 1.00E+00 | 0.1861 | 4.92E-01 | 1.00E+00 |
| 10 | 0.3075 | 1.76E-01 | 1.00E+00 | 0.4192 | 7.07E-02 | 1.00E+00 | 0.0008 | 9.96E-01 | 1.00E+00 | −0.1394 | 5.91E-01 | 1.00E+00 |
| 11 | −0.2276 | 2.95E-01 | 1.00E+00 | −0.2408 | 2.77E-01 | 1.00E+00 | 0.1113 | 4.89E-01 | 1.00E+00 | 0.5172 | 3.77E-02 | 1.00E+00 |
| 12 | 0.4771 | 1.44E-02 | 4.32E-01 | 0.5012 | 1.23E-02 | 3.70E-01 | 0.4438 | 2.21E-03 | 6.63E-02 | 0.2316 | 3.02E-01 | 1.00E+00 |
| 13 | −0.6286 | 7.42E-04 | 2.22E-02 | −0.6503 | 6.15E-04 | 1.85E-02 | −0.3306 | 1.69E-02 | 5.08E-01 | 0.0404 | 8.51E-01 | 1.00E+00 |
| 14 | 0.1981 | 2.46E-01 | 1.00E+00 | 0.1655 | 3.55E-01 | 1.00E+00 | 0.2351 | 7.00E-02 | 1.00E+00 | 0.1484 | 4.50E-01 | 1.00E+00 |
| 15 | −0.0790 | 6.45E-01 | 1.00E+00 | −0.1106 | 5.28E-01 | 1.00E+00 | −0.1188 | 3.49E-01 | 1.00E+00 | −0.2497 | 2.03E-01 | 1.00E+00 |
| 16 | −0.6785 | 2.48E-05 | 7.43E-04 | −0.7216 | 1.06E-05 | 3.18E-04 | −0.3970 | 9.07E-04 | 2.72E-02 | 0.0535 | 7.74E-01 | 1.00E+00 |
| 17 | −0.0901 | 5.64E-01 | 1.00E+00 | −0.0787 | 6.21E-01 | 1.00E+00 | −0.0113 | 9.22E-01 | 1.00E+00 | 0.1135 | 5.25E-01 | 1.00E+00 |
| 18 | −0.2563 | 9.06E-02 | 1.00E+00 | −0.2856 | 6.43E-02 | 1.00E+00 | −0.2501 | 2.54E-02 | 7.62E-01 | −0.1071 | 5.39E-01 | 1.00E+00 |
| 19 | −0.1066 | 4.46E-01 | 1.00E+00 | −0.1279 | 3.70E-01 | 1.00E+00 | −0.0807 | 4.36E-01 | 1.00E+00 | −0.0932 | 5.62E-01 | 1.00E+00 |
| 20 | −0.4409 | 5.04E-04 | 1.51E-02 | −0.4517 | 4.72E-04 | 1.42E-02 | −0.0971 | 3.05E-01 | 1.00E+00 | 0.2028 | 1.67E-01 | 1.00E+00 |
| 21 | −0.1416 | 2.70E-01 | 1.00E+00 | −0.1000 | 4.50E-01 | 1.00E+00 | 0.0278 | 7.72E-01 | 1.00E+00 | 0.0714 | 6.27E-01 | 1.00E+00 |
| 22 | −1.0058 | 1.61E-16 | 4.84E-15 | −1.0686 | 7.23E-18 | 2.17E-16 | −0.5130 | 3.47E-08 | 1.04E-06 | 0.0160 | 9.13E-01 | 1.00E+00 |
| 23 | 0.3025 | 1.38E-02 | 4.14E-01 | 0.2765 | 2.81E-02 | 8.43E-01 | 0.0230 | 8.02E-01 | 1.00E+00 | −0.2818 | 4.57E-02 | 1.00E+00 |
| 24 | −0.0896 | 4.54E-01 | 1.00E+00 | −0.1217 | 3.19E-01 | 1.00E+00 | −0.1339 | 1.31E-01 | 1.00E+00 | −0.1206 | 3.79E-01 | 1.00E+00 |
| 25 | 0.1733 | 1.30E-01 | 1.00E+00 | 0.1587 | 1.74E-01 | 1.00E+00 | 0.1556 | 6.62E-02 | 1.00E+00 | 0.0774 | 5.57E-01 | 1.00E+00 |
| 26 | −0.0200 | 8.54E-01 | 1.00E+00 | −0.0514 | 6.42E-01 | 1.00E+00 | −0.0424 | 5.98E-01 | 1.00E+00 | −0.0856 | 4.91E-01 | 1.00E+00 |
| 27 | 0.1214 | 2.63E-01 | 1.00E+00 | 0.0959 | 3.87E-01 | 1.00E+00 | −0.0214 | 7.90E-01 | 1.00E+00 | −0.1003 | 4.18E-01 | 1.00E+00 |
| 28 | 0.0674 | 5.24E-01 | 1.00E+00 | 0.0792 | 4.63E-01 | 1.00E+00 | 0.0166 | 8.32E-01 | 1.00E+00 | −0.0932 | 4.42E-01 | 1.00E+00 |
| 29 | −0.3401 | 7.13E-04 | 2.14E-02 | −0.3382 | 9.87E-04 | 2.96E-02 | −0.0536 | 4.75E-01 | 1.00E+00 | 0.1359 | 2.42E-01 | 1.00E+00 |
| 30 | 0.1393 | 1.69E-01 | 1.00E+00 | 0.1815 | 7.84E-02 | 1.00E+00 | 0.1705 | 2.24E-02 | 6.73E-01 | 0.1706 | 1.42E-01 | 1.00E+00 |
The adjusted p values were calculated based on the Bonferroni adjustment for 30 PCs.
Figure 1.
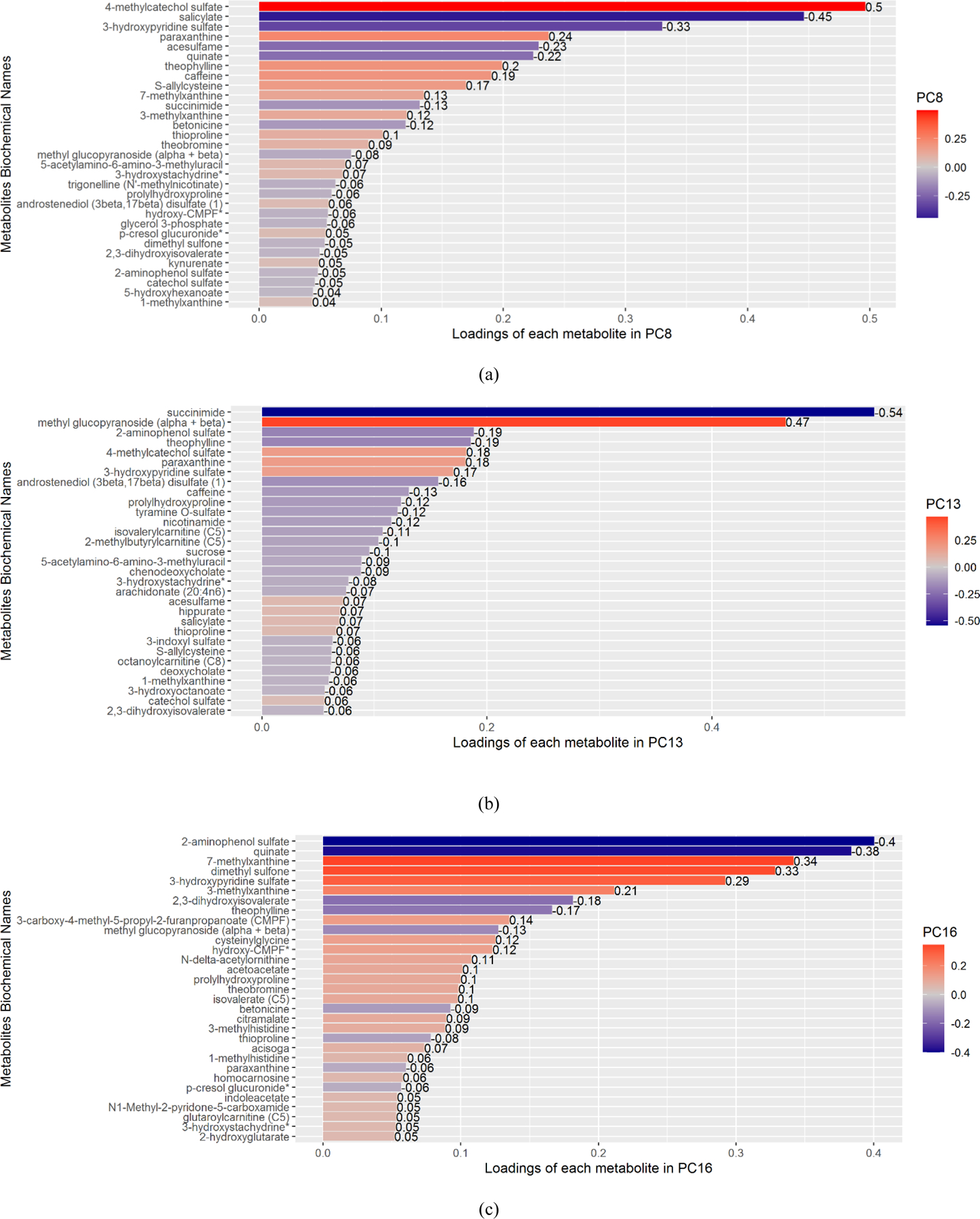
The bar plots of metabolites whose ranked absolute values of loadings were in the top 10% for PC8 (a), PC13 (b), PC16 (c), PC20 (d), PC22 (e), and PC29 (f).
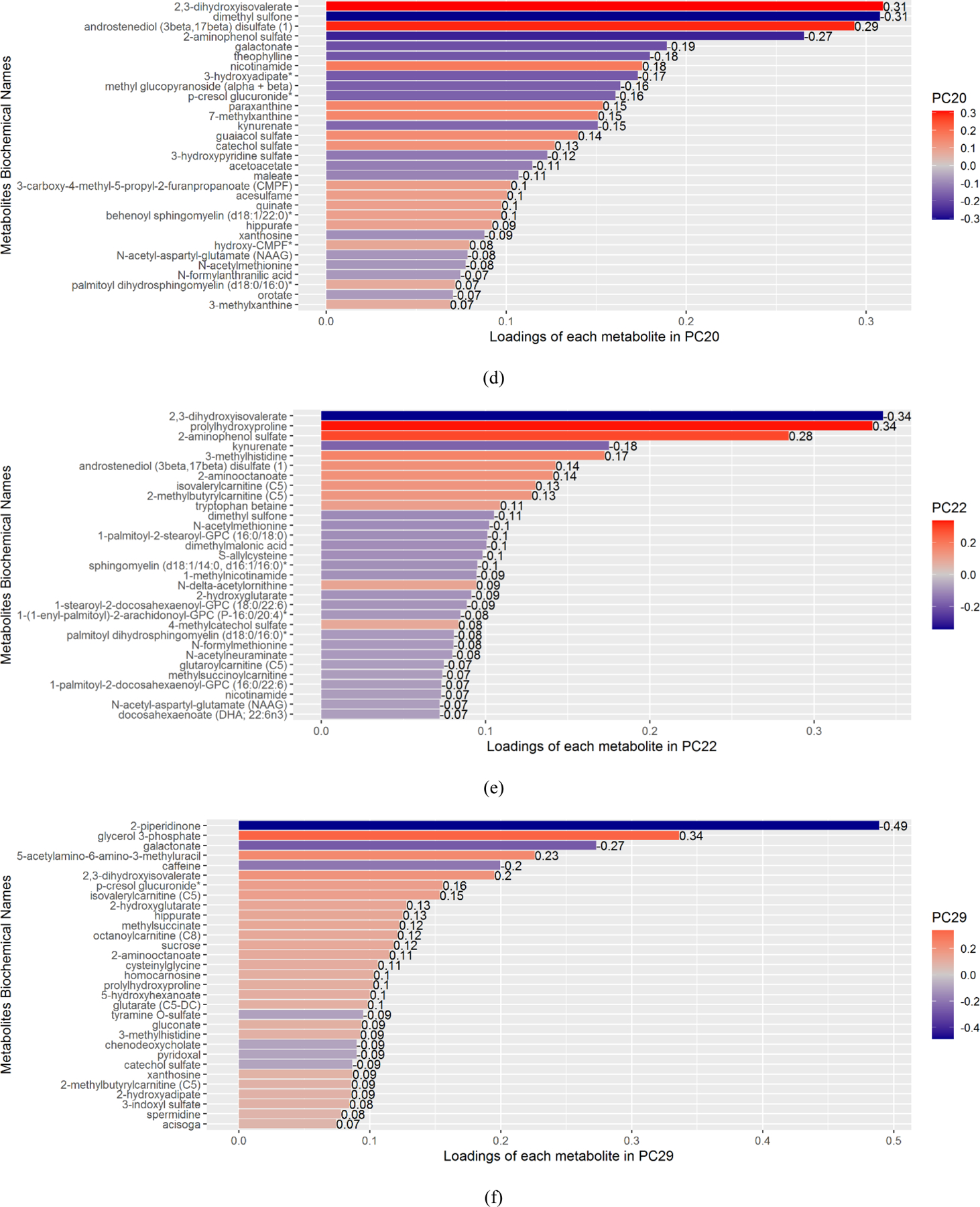
As shown in Table 3 and Figure 2, among the 6 significant PCs, PC13 was significantly associated with Aβ40, GFAP, sTREM2, neurogranin, and α-synuclein after adjusting for multiple testing. PC 16 was associated with Aβ40, GFAP, neurogranin, and α-synuclein. PC20 was associated with Aβ40 and neurogranin. PC22 was associated with Aβ40, S100b, YKL40, GFAP, sTREM2, neurogranin, and α-synuclein. PC29 was associated with YKL-40, GFAP, and MRI TBV.
Table 3.
Results of the association tests between 6 significant PCs and NTK CSF biomarkers and imaging markers.
| PC | Aß40 | S100b | YKL-40 | GFAP | sTREM2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p | |
| PC8 | −1.0386 | 8.19E-05 | 4.91E-04 | −0.6541 | 9.50E-02 | 5.70E-01 | −0.6743 | 2.22E-02 | 1.33E-01 | −0.5209 | 5.38E-02 | 3.23E-01 | −0.4676 | 1.10E-01 | 6.61E-01 |
| PC13 | −0.7386 | 1.20E-04 | 7.18E-04 | −0.556 | 5.20E-02 | 3.12E-01 | −0.6452 | 2.68E-03 | 1.61E-02 | −0.5767 | 3.36E-03 | 2.02E-02 | −0.7602 | 3.44E-04 | 2.06E-03 |
| PC16 | −0.7849 | 2.10E-06 | 1.26E-05 | −0.5102 | 3.98E-02 | 2.39E-01 | −0.4179 | 2.55E-02 | 1.53E-01 | −0.4597 | 7.12E-03 | 4.27E-02 | −0.4600 | 1.30E-02 | 7.80E-02 |
| PC20 | −0.3539 | 7.43E-03 | 4.46E-02 | −0.2341 | 2.30E-01 | 1.00E+00 | −0.3198 | 2.93E-02 | 1.76E-01 | −0.0725 | 5.91E-01 | 1.00E+00 | −0.1122 | 4.42E-01 | 1.00E+00 |
| PC22 | −1.0232 | 6.68E-16 | 4.01E-15 | −0.568 | 3.60E-03 | 2.16E-02 | −0.9452 | 4.24E-11 | 2.55E-10 | −0.4465 | 8.87E-04 | 5.32E-03 | −0.7373 | 2.98E-07 | 1.79E-06 |
| PC29 | −0.2421 | 2.08E-02 | 1.25E-01 | −0.2432 | 1.16E-01 | 6.95E-01 | −0.3454 | 2.93E-03 | 1.76E-02 | −0.3116 | 3.36E-03 | 2.02E-02 | −0.2827 | 1.42E-02 | 8.53E-02 |
| PC | IL-6 | NfL | Neurogranin | α-synuclein | |||||||||||
| Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p | ||||
| PC8 | −0.0005 | 9.98E-01 | 1.00E+00 | −0.2898 | 2.30E-01 | 1.00E+00 | −0.6745 | 2.91E-03 | 1.74E-02 | −0.5632 | 4.30E-03 | 2.58E-02 | |||
| PC13 | 0.2389 | 8.89E-02 | 5.34E-01 | −0.4175 | 1.77E-02 | 1.06E-01 | −0.5589 | 7.19E-04 | 4.31E-03 | −0.5954 | 3.19E-05 | 1.92E-04 | |||
| PC16 | 0.2260 | 6.35E-02 | 3.81E-01 | −0.2815 | 6.57E-02 | 3.94E-01 | −0.5824 | 4.55E-05 | 2.73E-04 | −0.4340 | 5.00E-04 | 3.00E-03 | |||
| PC20 | −0.0766 | 4.26E-01 | 1.00E+00 | −0.0897 | 4.56E-01 | 1.00E+00 | −0.3721 | 9.44E-04 | 5.66E-03 | −0.2101 | 3.27E-02 | 1.96E-01 | |||
| PC22 | −0.1110 | 2.50E-01 | 1.00E+00 | −0.6176 | 1.87E-07 | 1.12E-06 | −0.9353 | 4.32E-18 | 2.59E-17 | −0.6923 | 4.03E-13 | 2.42E-12 | |||
| PC29 | −0.0370 | 6.28E-01 | 1.00E+00 | −0.1841 | 5.31E-02 | 3.19E-01 | −0.2736 | 2.21E-03 | 1.33E-02 | −0.1716 | 2.79E-02 | 1.68E-01 | |||
| PC | MRI TBV | MRI HV | PiB DVR | ||||||||||||
| Estimate | p | adjusted p | Estimate | p | adjusted p | Estimate | p | adjusted p | |||||||
| PC8 | −4.2023 | 2.00E-02 | 1.20E-01 | 0.2984 | 7.15E-01 | 1.00E+00 | 0.8142 | 3.32E-01 | 1.00E+00 | ||||||
| PC13 | −1.1585 | 3.88E-01 | 1.00E+00 | 0.3675 | 5.44E-01 | 1.00E+00 | 0.9058 | 8.70E-02 | 5.22E-01 | ||||||
| PC16 | −1.1467 | 3.24E-01 | 1.00E+00 | −0.8184 | 1.19E-01 | 7.11E-01 | 0.0664 | 9.02E-01 | 1.00E+00 | ||||||
| PC20 | −1.3188 | 1.56E-01 | 9.35E-01 | −0.3608 | 3.90E-01 | 1.00E+00 | −0.0315 | 9.40E-01 | 1.00E+00 | ||||||
| PC22 | −0.0783 | 9.33E-01 | 1.00E+00 | −0.8996 | 3.28E-02 | 1.97E-01 | 0.6842 | 1.52E-01 | 9.10E-01 | ||||||
| PC29 | −1.9791 | 6.67E-03 | 4.00E-02 | −0.1509 | 6.48E-01 | 1.00E+00 | 0.3121 | 3.64E-01 | 1.00E+00 | ||||||
The adjusted p values were calculated based on the Bonferroni adjustment for 6 PCs.
Figure 2. Plot of rounded Bonferroni-corrected p-values of the association between the 6 significant PCs and CSF and imaging biomarkers.
The colors of the cells indicate the p-value magnitudes.
3.3. Biological relevance of significant PCs
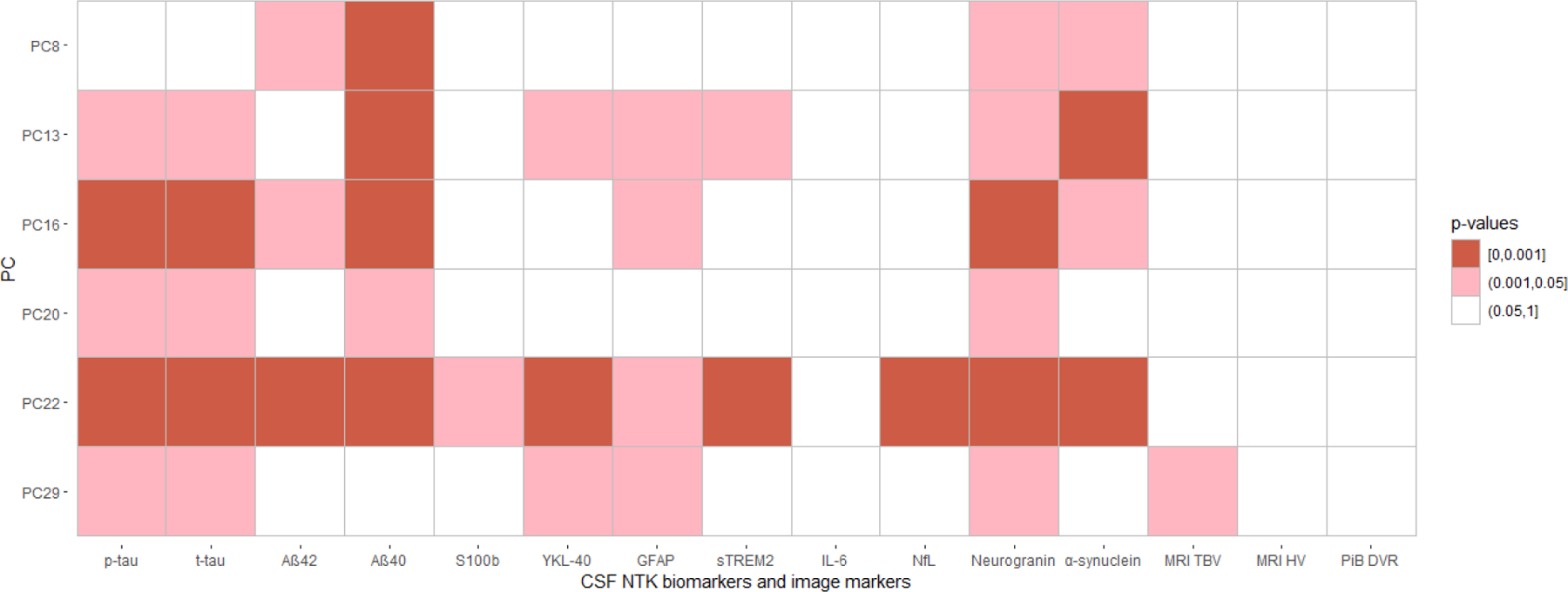
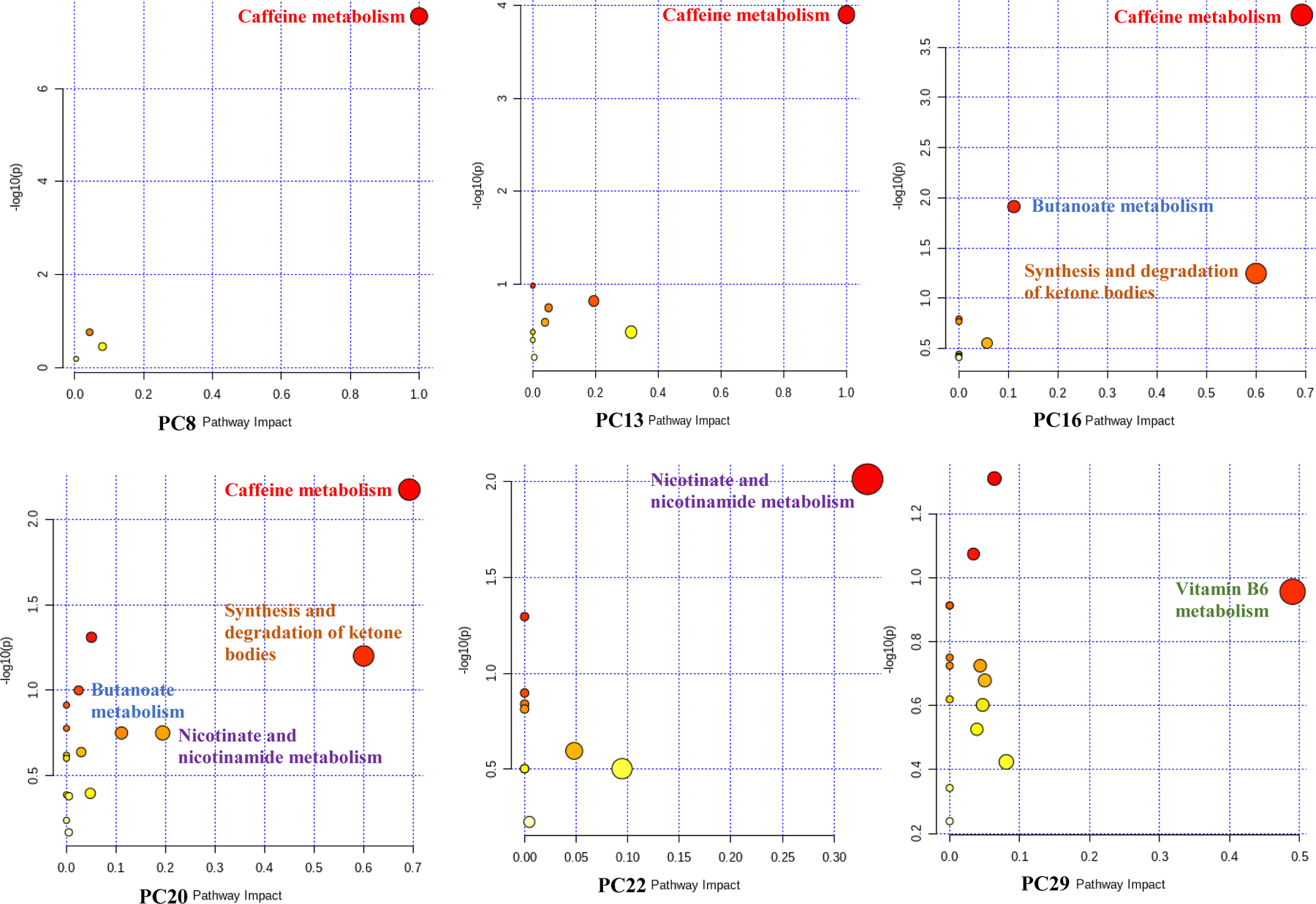
We conducted a pathway analysis using metabolites with an assigned human metabolite database (HMDB) ID and ranked absolute value of loadings in the top 10% for each significant PC (Figure 1). We identified six unique pathways showing enrichment in PCs associated with AD biomarkers (Figure 3). Four pathways were enriched in more than one significant PC: caffeine metabolism (including paraxanthine, caffeine, theobromine, 1-methylxanthine and 7-methylxanthine), which was enriched in PC8, PC13, PC16, and PC20; nicotinate and nicotinamide metabolism (including nicotinamide and 1-methylnicotinamide), which was enriched in PC13, PC20, and PC22; and butanoate metabolism (including acetoacetate and 2-hydroxyglutarate), and synthesis and degradation of ketone bodies (including acetoacetate), which were both enriched in PC16 and PC20 (Table 4). The positions of the five metabolites included in the caffeine metabolism pathway are shown in Figure 4 (Kanehisa and Goto, 2000; Okuda et al., 2008), in which caffeine, theobromine, paraxanthine, and 7-methylxanhine compose a metabolic feedback loop.
Figure 3. Pathway analysis results for all significant PCs.
The x-axis represents the pathway impact and the y-axis represents the pathway enrichment. Larger sizes and darker colors represent higher pathway impact and enrichment, respectively. The text of the pathway name is color coded such that each pathway has a unique color across the figures to easily see which pathways are enriched in multiple PCs.
Table 4.
The significant pathways from pathway analyses for the six significant PCs.
| PC | Pathway | Total | Raw p | FDR p | Impact | Hits | Hit metabolites |
|---|---|---|---|---|---|---|---|
| PC8 | Caffeine metabolism | 10 | 2.81E-08 | 2.36E-06 | 1.00 | 5 | Paraxanthine 1-Methylxanthine Theobromine 7-Methylxanthine Caffeine |
| PC13 | Caffeine metabolism | 10 | 1.26E-04 | 1.06E-02 | 1.00 | 3 | Paraxanthine 1-Methylxanthine Caffeine |
| Arachidonic acid metabolism | 36 | 3.31E-01 | 1.00 | 0.31 | 1 | Arachidonate | |
| Nicotinate and nicotinamide metabolism | 15 | 1.53E-01 | 1.00 | 0.19 | 1 | Nicotinamide | |
| PC16 | Caffeine metabolism | 10 | 1.78E-04 | 1.49E-02 | 0.31 | 3 | Paraxanthine Theobromine 7-Methylxanthine |
| Butanoate metabolism | 15 | 1.36E-02 | 5.71E-01 | 0.11 | 2 | Acetoacetate 2-Hydroxyglutarate |
|
| Synthesis and degradation of ketone bodies | 5 | 5.99E-02 | 1.00 | 0.60 | 1 | Acetoacetate | |
| PC20 | Caffeine metabolism | 10 | 6.69E-03 | 5.62E-01 | 0.69 | 2 | Paraxanthine 7-Methylxanthine |
| Synthesis and degradation of ketone bodies | 5 | 6.30E-02 | 1.00 | 0.60 | 1 | Acetoacetate | |
| Nicotinate and nicotinamide metabolism | 15 | 1.78E-01 | 1.00 | 0.19 | 1 | Nicotinamide | |
| Butanoate metabolism | 15 | 1.78E-01 | 1.00 | 0.11 | 1 | Acetoacetate | |
| PC22 | Nicotinate and nicotinamide metabolism | 15 | 9.70E-03 | 8.15E-01 | 0.33 | 2 | 1-Methylnicotinamide Nicotinamide |
| PC29 | Vitamin B6 metabolism | 9 | 1.11E-01 | 1.00 | 0.49 | 1 | Pyridoxal |
Total means the total number of metabolites in this pathway according to KEGG. Raw p is the original p value calculated from the enrichment analysis; FDR p is the p value adjusted using False Discovery Rate; Impact is the pathway impact value calculated from the pathway topology analysis. Hits represents the number of metabolites out of the Total that were in the top 10% of loadings for the PC in present study for the corresponding pathway.
Figure 4. The positions of 5 hit metabolites in the KEGG caffeine metabolism pathway.
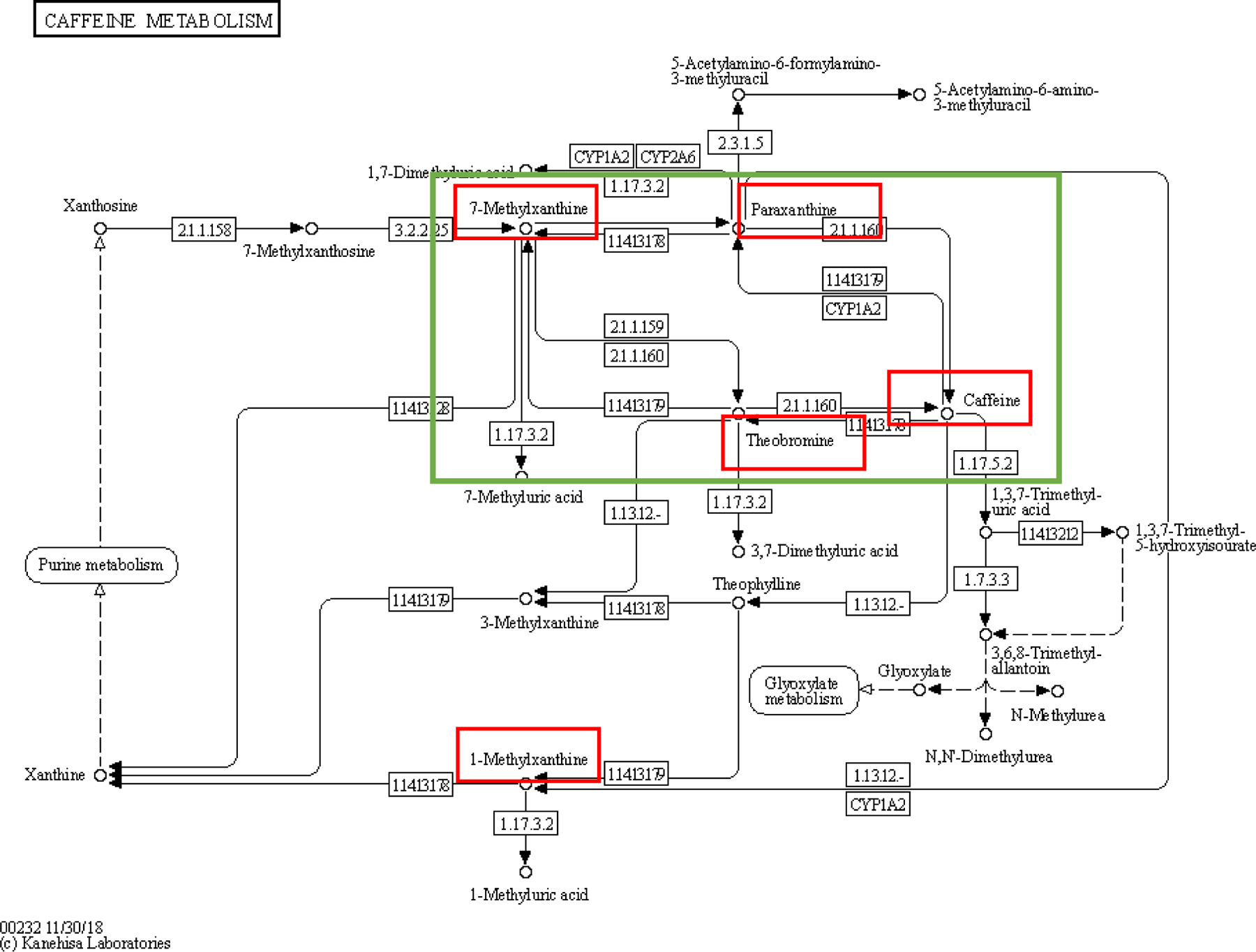
(Okuda et al., 2008). The green box shows a feedback loop composed of 7-methylxanthine, paraxanthine, theobromine, and caffeine. The metabolites in red boxes are the five hit metabolites in the caffeine metabolism pathway from the pathway analyses.
3.4. Caffeine metabolism analysis
In the WRAP cohort, among the four PCs that were enriched for the caffeine metabolism pathway, PC16 was significantly associated with caffeine intake at baseline (Table 5, p=1.49E-02). While there was suggestive evidence for mediation of the association between caffeine intake and both p-tau and t-tau by PC16, this was not statistically significant (p=0.096 and p=0.08, respectively; Supplemental Figure 3). In the combined cohort, the metabolite, 7-methylxanthine, was associated with both p-tau (p=0.01) and t-tau (p=0.03), but no ratios were associated with the two tau outcomes (Supplemental Table 6).
Table 5.
The association between PCs enriched in caffeine metabolism and caffeine intake in WRAP.
| PC | Caffeine intake | ||
|---|---|---|---|
| Estimate | p | adjusted p | |
| PC8 | −0.1368 | 3.95E-01 | 1.00E+00 |
| PC13 | 0.1204 | 3.06E-01 | 1.00E+00 |
| PC16 | −0.2873 | 3.72E-03 | 1.49E-02 |
| PC20 | 0.0026 | 9.75E-01 | 1.00E+00 |
The adjusted p values were calculated based on the Bonferroni adjustment for 4 PCs.
4. Discussion
In this analysis, we aimed to identify potential CSF metabolites and pathways associated with AD pathology and to understand their roles in AD pathogenesis. Using untargeted CSF metabolomics data from the combined WRAP and Wisconsin ADRC IMPACT cohorts and PCA, we were able to reduce the whole CSF untargeted metabolomics data to 30 PCs that explained the majority of the variance in CSF metabolites. Among the 30 PCs, six were associated with CSF p-tau and t-tau, Aβ42, other NTK biomarkers such as α-synuclein, NfL, and neurogranin, and total brain volume. Based on the significant correlation between PC13 and markers of astrocyte activation and synapse function, the metabolites and pathways in PC13 may be involved in the role astrocytes play in synapse function. PC22 was most strongly correlated with p-tau and t-tau, but also with markers of axonal injury and synaptic dysfunction. The significant PCs provide potential metabolites of interest such as paraxanthine, 1-methylxanthine, theobromine, 7-methylxanthine, caffeine, acetoacetate, and nicotinamide, and pathways such as metabolism of caffeine, nicotinate and nicotinamide, and butanoate, and synthesis and degradation of ketone bodies for AD pathology. These metabolites and pathways may be worthy of further research.
Caffeine metabolism is driven primarily by cytochrome P450 1A2 (CYP1A2) enzyme-mediated demethylation, resulting in the formation of three major metabolites. Of these, paraxanthine accounts for 80–85% of the metabolites, with the remainder consisting primarily of theobromine and theophylline (Jandova et al., 2019) After caffeine enters the human body, it is quickly absorbed in the gastrointestinal system and distributed into the blood and body tissues (Oñatibia‐Astibia et al., 2017). Importantly, caffeine can cross the blood-brain-barrier (BBB) and enter the brain by simple or facilitated diffusion (Liu et al., 2005). Accumulating evidence suggests a protective role of caffeine intake in AD. In a systematic review of 28 epidemiological reports and experimental studies based on animal and cell AD models (Panza et al., 2015), both cross-sectional (Valls-Pedret et al., 2012; Wu et al., 2011) and longitudinal studies (Lindsay, 2002; Vercambre et al., 2013) have suggested that caffeine intake is associated with reduced risk of cognitive impairment, dementia, and AD.
Several studies have tested the relationship between caffeine intake and AD pathology (i.e., brain amyloidosis and tau pathology). Previous mouse model studies have suggested several potential mechanisms of caffeine’s protective effect against AD. First, long-term caffeine intake may increase the production of CSF, thus improving CSF turnover and clearance of potentially toxic metabolites such as Aβ (Han et al., 2009). Second, in an APP transgenic mouse model, long-term caffeine intake could limit the brain production of Aβ due to reduced expression of both β-secretase and presenilin-1/γ secretase (Arendash et al., 2006). Third, caffeine has antioxidant and anti-inflammatory capacities and the ability to block the disruption of the BBB and A1 and A2A receptors, which are the adenosine receptors where caffeine binds in the brain (Arendash et al., 2006). Finally, chronic caffeine intake could prevent the development of spatial memory deficits and improved memory has been associated with reduced hippocampal tau phosphorylation and proteolytic fragments in the hTau mouse model (Laurent et al., 2014). In our mediation analysis, although the mediation effects were only marginally significant, they provide some evidence that the caffeine metabolism pathway may be mediating the association between caffeine intake and tau pathology and are worth of future replication in a larger sample.
Less research has been conducted to analyze other methylxanthines such as paraxanthine and theobromine’s effect on AD. In a longitudinal study of 299 participants of the Amsterdam Dementia Cohort, high levels of theobromine were detected in CSF and were associated with clinical progression to dementia (de Leeuw et al., 2020). A mouse model study of Parkinson’s disease has shown that paraxanthine exerts a neuroprotective role in dopaminergic neurons by stimulating the ryanodine receptor (Xu et al., 2010). In our analysis, we found that CSF 7-methylxanthine (PC8, PC16, and PC20) was significantly negatively associated with both p-tau and t-tau, providing new evidence for a putative role of methylxanthines in AD pathophysiology.
In a study of 88 patients with AD or MCI, a significant positive correlation was found between the concentrations of theobromine, both in the CSF and in the plasma, with CSF Aβ42 in the AD patients (Travassos et al., 2015). In a diet experiment of mice, a significant decrease of cognitive impairment was found when mice were fed with a diet rich in theobromine, polyphenols, and polyunsaturated acids, and the potential mechanism may involve the modulation of the catecholaminergic and cholinergic systems (Fernández-Fernández et al., 2015). To our knowledge, our study is the first to analyze the association between CSF caffeine metabolites and p-tau and t-tau in humans with a relatively large sample size. The significant associations are worth replicating and exploring in future in vivo studies.
Acetoacetate in the butanoate metabolism and synthesis and degradation of ketone bodies pathways may also be involved in AD pathology. Acetoacetate is produced in the liver and released into the bloodstream as an energy source during periods of fasting, exercise, or as a result of type 1 diabetes mellitus, when glucose levels are low. It can cross the BBB and be used by the brain for energy. From a six-week intervention study of a Mediterranean-ketogenic diet, which aimed at shifting the main fuel used by the body from glucose to ketone bodies, in participants with subjective memory complaints and MCI, CSF Aβ42 was increased, CSF tau was decreased (only in MCI) and the CSF Aβ42/tau ratio was increased (greater in MCI) (Neth et al., 2020). In the current study, acetoacetate was not associated with any of the AD biomarkers after Bonferroni correction, indicating that a set of metabolites in these pathways may be important, not a single metabolite.
Animal model experiments have studied the functions of nicotinamide, in the nicotinate and nicotinamide metabolism pathway. For example, nicotinamide treatment has been shown to reduce levels of oxidative stress, apoptosis, and PARP-1 activity in an Aβ42-induced rat model of AD (Turunc et al., 2013). Nicotinamide also has the ability to restore cognition in AD transgenic mice by reducing a specific phospho-species of tau (Thr231) (Green et al., 2008). Another study has shown that the nicotinamide loaded functionalized solid lipid nanoparticles can improve cognition in an AD rat model by reducing tau hyperphosphorylation (Vakilinezhad et al., 2018). These preclinical findings suggest that oral nicotinamide may be considered for evaluation in clinical trials on an effective treatment for AD patients. Our results suggested that 1-methylnicotinamide and nicotinamide were enriched in the nicotinamide metabolism in PC22, and PC22 was associated with CSF biomarkers such as p-tau, t-tau, Aβ42, α-synuclein, YKL-40, and GFAP in human. Another metabolite, kynurenate, which is synthesized from kynurenine, was in the top 10% of loadings for PC8, PC20, and PC22 (Figure 1) and the kynurenine pathway leads to the production of nicotinamide.
The sample size of the WRAP and Wisconsin ADRC IMPACT cohorts is relatively small separately, however, we combined the two cohorts and also conducted a meta-analysis to confirm the results. Another strength of the study is that we included a rich set of CSF AD biomarkers from the NTK as well as imaging biomarkers for AD. Both WRAP and the Wisconsin ADRC are longitudinal, with up to three metabolomics data points per participant (average of 2 time points). Although we took the average of the values for each metabolite in this analysis, further analyses that use trajectories of the longitudinal measures can be done in the future, particularly once data for additional time points are available. Since the mediation results were suggestive, inclusion of the IMPACT cohort would have increased our power to detect mediation if such a relationship exists. However, the IMPACT cohort did not collect information on caffeine intake, so we were unable to replicate the mediation analysis from caffeine intake through PC16 to tau outcomes. Replication of this potential mediation effect in an independent cohort should be pursued in a future study. The analysis conducted in both the WRAP and IMPACT cohorts only included non-Hispanic white individuals due to a small sample size in other racial/ethnic groups. Thus, the results may not be generalizable to other groups. Finally, two of the metabolites in the significant pathways had a relatively large number of missing values: nicotinamide (50%) and paraxanthine (55%), making replication in additional samples an important next step.
In summary, our study provides different groups of CSF metabolites that are associated with a variety of CSF AD biomarkers. The promising caffeine metabolism pathway and its metabolites identified in this study provide new insights into how caffeine consumption may help prevent AD through tau pathology, astrocyte activation, and synaptic function.
Supplementary Material
Acknowledgements
The authors especially thank the WRAP and Wisconsin ADRC participants and staff for their contributions to the studies. Without their efforts, this research would not be possible. This study was supported by the National Institutes of Health (NIH) grants [R01AG27161 (Wisconsin Registry for Alzheimer Prevention: Biomarkers of Preclinical AD), R01AG054047 (Genomic and Metabolomic Data Integration in a Longitudinal Cohort at Risk for Alzheimer’s Disease), R21AG067092 (Identifying Metabolomic Risk Factors in Plasma and Cerebrospinal Fluid for Alzheimer’s Disease), and P30AG062715 (Wisconsin Alzheimer’s Disease Research Center Grant)], the Helen Bader Foundation, Northwestern Mutual Foundation, Extendicare Foundation, State of Wisconsin, the Clinical and Translational Science Award (CTSA) program through the NIH National Center for Advancing Translational Sciences (NCATS) grant [UL1TR000427], and the University of Wisconsin-Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation. This research was supported in part by the Intramural Research Program of the National Institute on Aging. Computational resources were supported by core grants to the Center for Demography and Ecology [P2CHD047873] and the Center for Demography of Health and Aging [P30AG017266].
HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council [#2018–02532], the European Research Council [#681712], the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement [#ALFGBG-720931], the Alzheimer Drug Discovery Foundation (ADDF), USA [#201809–2016862], and the UK Dementia Research Institute at UCL.
KB is supported by the Swedish Research Council (#2017–00915), ADDF, USA [#RDAPB-201809–2016615], the Swedish Alzheimer Foundation [#AF-742881], Hjärnfonden, Sweden [#FO2017–0243], the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement [#ALFGBG-715986], and European Union Joint Program for Neurodegenerative Disorders [JPND2019–466-236].
Thanks Roche for providing the NTK kits for this study. COBAS, COBAS E and ELECSYS are trademarks of Roche.
Abbreviations
- AD
Alzheimer’s disease
- CSF
cerebrospinal fluid
- t-tau
total tau
- p-tau
phosphorylated tau
- Aβ
amyloid β
- WRAP
Wisconsin Registry for Alzheimer’s Prevention
- Wisconsin-ADRC
Wisconsin Alzheimer’s Disease Research Center
- PCA
principal components analysis
- PCs
principal components
- IMPACT
investigating memory in preclinical AD-causes and treatments
- NTK
NeuroToolKit
- S100b
S100 calcium-binding protein B
- IL-6
interleukin-6
- GFAP
glial fibrillary acidic protein
- YKL-40
chitinase-3-like protein 1
- sTREM2
soluble triggering receptor expressed on myeloid cells 2
- NfL
neurofilament light protein
- PiB
Compound B
- PET
positron emission tomography
- MRI
magnetic resonance
- ICV
intracranial volume
- TBV
total brain volume
- HV
hippocampal volumes
- DVR
distribution volume ratio
- KEGG
Kyoto encyclopedia of genes and genomes
- HMDB
human metabolome database
Footnotes
Conflicts of interest
HZ has served at scientific advisory boards for Alector, Eisai, Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies and CogRx, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. KB has served as a consultant or at advisory boards for Abcam, Axon, Biogen, Lilly, MagQu, Novartis and Roche Diagnostics, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. GK and NW are full-time employees of Roche DiagnosticsGmbH. IS is a full-time employee and shareholder of Roche Diagnostics International Ltd.
References
- Antonell A, Tort-Merino A, Ríos J, Balasa M, Borrego-Écija S, Auge JM, Muñoz-García C, Bosch B, Falgàs N, Rami L, Ramos-Campoy O, Blennow K, Zetterberg H, Molinuevo JL, Lladó A, Sánchez-Valle R, 2019. Synaptic, axonal damage and inflammatory cerebrospinal fluid biomarkers in neurodegenerative dementias. Alzheimer’s & Dementia 10.1016/j.jalz.2019.09.001 [DOI] [PubMed] [Google Scholar]
- Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J, 2006. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain β-amyloid production. Neuroscience 142, 941–952. 10.1016/j.neuroscience.2006.07.021 [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ, 2005. Unified segmentation. NeuroImage 26, 839–851. 10.1016/j.neuroimage.2005.02.018 [DOI] [PubMed] [Google Scholar]
- Bendlin BB, Carlsson CM, Johnson SC, Zetterberg H, Blennow K, Willette AA, Okonkwo OC, Sodhi A, Ries ML, Birdsill AC, Alexander AL, Rowley HA, Puglielli L, Asthana S, Sager MA, 2012. CSF T-Tau/Aβ42 Predicts White Matter Microstructure in Healthy Adults at Risk for Alzheimer’s Disease. PLoS ONE 7. 10.1371/journal.pone.0037720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettcher BM, Johnson SC, Fitch R, Casaletto KB, Heffernan KS, Asthana S, Zetterberg H, Blennow K, Carlsson CM, Neuhaus J, Bendlin BB, Kramer JH, 2018. Cerebrospinal Fluid and Plasma Levels of Inflammation Differentially Relate to CNS Markers of Alzheimer’s Disease Pathology and Neuronal Damage. JAD 62, 385–397. 10.3233/JAD-170602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgewater BR EA, 2014. High Resolution Mass Spectrometry Improves Data Quantity and Quality as Compared to Unit Mass Resolution Mass Spectrometry in High-Throughput Profiling Metabolomics. Metabolomics 04. 10.4172/2153-0769.1000132 [DOI] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J, 2018. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46, W486–W494. 10.1093/nar/gky310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darst BF, Koscik RL, Racine AM, Oh JM, Krause RA, Carlsson CM, Zetterberg H, Blennow K, Christian BT, Bendlin BB, Okonkwo OC, Hogan KJ, Hermann BP, Sager MA, Asthana S, Johnson SC, Engelman CD, 2017. Pathway-specific polygenic risk scores as predictors of β-amyloid deposition and cognitive function in a sample at increased risk for Alzheimer’s disease. J Alzheimers Dis 55, 473–484. 10.3233/JAD-160195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darst BF, Lu Q, Johnson SC, Engelman CD, 2019. Integrated analysis of genomics, longitudinal metabolomics, and Alzheimer’s risk factors among 1,111 cohort participants. Genetic Epidemiology 43, 657–674. 10.1002/gepi.22211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw FA, van der Flier WM, Tijms BM, Scheltens P, Mendes VM, Manadas B, Bierau J, van Wijk N, van den Heuvel EGHM, Mohajeri MH, Teunissen CE, Kester MI, 2020. Specific Nutritional Biomarker Profiles in Mild Cognitive Impairment and Subjective Cognitive Decline Are Associated With Clinical Progression: The NUDAD Project. Journal of the American Medical Directors Association 21, 1513.e1-1513.e17. 10.1016/j.jamda.2019.12.009 [DOI] [PubMed] [Google Scholar]
- Denburg MR, Xu Y, Abraham AG, Coresh J, Chen J, Grams ME, Feldman HI, Kimmel PL, Rebholz CM, Rhee EP, Vasan RS, Warady BA, Furth SL, 2021. Metabolite Biomarkers of CKD Progression in Children. CJASN 16, 1178–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTure MA, Dickson DW, 2019. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegeneration 14, 32. 10.1186/s13024-019-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enche Ady CNA, Lim SM, Teh LK, Salleh MZ, Chin A-V, Tan MP, Poi PJH, Kamaruzzaman SB, Abdul Majeed AB, Ramasamy K, 2017. Metabolomic-guided discovery of Alzheimer’s disease biomarkers from body fluid: Metabolomic-based Alzheimer’s Disease Biomarkers. J Neuro Res 95, 2005–2024. 10.1002/jnr.24048 [DOI] [PubMed] [Google Scholar]
- Fernández-Fernández L, Esteban G, Giralt M, Valente T, Bolea I, Solé M, Sun P, Benítez S, Ramón Morelló J, Reguant J, Ramírez B, Hidalgo J, Unzeta M, 2015. Catecholaminergic and cholinergic systems of mouse brain are modulated by LMN diet, rich in theobromine, polyphenols and polyunsaturated fatty acids. Food & Function 6, 1251–1260. 10.1039/C5FO00052A [DOI] [PubMed] [Google Scholar]
- Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM, 2008. Nicotinamide Restores Cognition in Alzheimer’s Disease Transgenic Mice via a Mechanism Involving Sirtuin Inhibition and Selective Reduction of Thr231-Phosphotau. J. Neurosci 28, 11500–11510. 10.1523/JNEUROSCI.3203-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiraud SP, Montoliu I, Da Silva L, Dayon L, Galindo AN, Corthésy J, Kussmann M, Martin F-P, 2017. High-throughput and simultaneous quantitative analysis of homocysteine–methionine cycle metabolites and co-factors in blood plasma and cerebrospinal fluid by isotope dilution LC–MS/MS. Anal Bioanal Chem 409, 295–305. 10.1007/s00216-016-0003-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M-E, Kim H-J, Lee Y-S, Kim D-H, Choi J-T, Pan C-S, Yoon S, Baek S-Y, Kim B-S, Kim J-B, Oh S-O, 2009. Regulation of cerebrospinal fluid production by caffeine consumption. BMC Neurosci 10, 110. 10.1186/1471-2202-10-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y, Seldin M, Lusis A, 2017. Multi-omics approaches to disease. Genome Biology 18, 83. 10.1186/s13059-017-1215-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T, Tran T, Zheng J, Sapkota S, MacDonald SW, Camicioli R, Dixon RA, Li L, 2018. Metabolomics Analyses of Saliva Detect Novel Biomarkers of Alzheimer’s Disease. Journal of Alzheimer’s Disease 65, 1401–1416. 10.3233/JAD-180711 [DOI] [PubMed] [Google Scholar]
- Jandova Z, Gill SC, Lim NM, Mobley DL, Oostenbrink C, 2019. Binding Modes and Metabolism of Caffeine. Chem. Res. Toxicol 32, 1374–1383. 10.1021/acs.chemrestox.9b00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Christian BT, Okonkwo OC, Oh JM, Harding S, Xu G, Hillmer AT, Wooten DW, Murali D, Barnhart TE, Hall LT, Racine AM, Klunk WE, Mathis CA, Bendlin BB, Gallagher CL, Carlsson CM, Rowley HA, Hermann BP, Dowling NM, Asthana S, Sager MA, 2014. Amyloid burden and neural function in people at risk for Alzheimer’s Disease. Neurobiology of Aging 35, 576–584. 10.1016/j.neurobiolaging.2013.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Koscik RL, Jonaitis EM, Clark LR, Mueller KD, Berman SE, Bendlin BB, Engelman CD, Okonkwo OC, Hogan KJ, Asthana S, Carlsson CM, Hermann BP, Sager MA, 2018. The Wisconsin Registry for Alzheimer’s Prevention: A review of findings and current directions. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 10, 130–142. 10.1016/j.dadm.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe I, 2005. Principal Component Analysis, in: Encyclopedia of Statistics in Behavioral Science John Wiley & Sons, Ltd. 10.1002/0470013192.bsa501 [DOI] [Google Scholar]
- Kanehisa M, Goto S, 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koal T, Klavins K, Seppi D, Kemmler G, Humpel C, 2015. Sphingomyelin SM(d18:1/18:0) is Significantly Enhanced in Cerebrospinal Fluid Samples Dichotomized by Pathological Amyloid-β 42, Tau, and Phospho-Tau-181 Levels. Journal of Alzheimer’s Disease 44, 1193–1201. 10.3233/JAD-142319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent C, Eddarkaoui S, Derisbourg M, Leboucher A, Demeyer D, Carrier S, Schneider M, Hamdane M, Müller CE, Buée L, Blum D, 2014. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiology of Aging 35, 2079–2090. 10.1016/j.neurobiolaging.2014.03.027 [DOI] [PubMed] [Google Scholar]
- Lindsay J, 2002. Risk Factors for Alzheimer’s Disease: A Prospective Analysis from the Canadian Study of Health and Aging. American Journal of Epidemiology 156, 445–453. 10.1093/aje/kwf074 [DOI] [PubMed] [Google Scholar]
- Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, Hosea N, Liu J, Nelson FR, Szewc MA, Deusen JV, 2005. Use of a Physiologically Based Pharmacokinetic Model to Study the Time to Reach Brain Equilibrium: An Experimental Analysis of the Role of Blood-Brain Barrier Permeability, Plasma Protein Binding, and Brain Tissue Binding. J Pharmacol Exp Ther 313, 1254–1262. 10.1124/jpet.104.079319 [DOI] [PubMed] [Google Scholar]
- Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, Meltzer CC, Schimmel K, Tsopelas ND, DeKosky ST, Price JC, 2005. Simplified Quantification of Pittsburgh Compound B Amyloid Imaging PET Studies: A Comparative Analysis. Journal of Nuclear Medicine 46, 1959–1972. [PubMed] [Google Scholar]
- Milà‐Alomà M, Salvadó G, Gispert JD, Vilor‐Tejedor N, Grau‐Rivera O, Sala‐Vila A, Sánchez‐Benavides G, Arenaza‐Urquijo EM, Crous‐Bou M, González‐de‐Echávarri JM, Minguillon C, Fauria K, Simon M, Kollmorgen G, Zetterberg H, Blennow K, Suárez‐Calvet M, Molinuevo JL, 2020. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer’s continuum. Alzheimer’s & Dementia 16, 1358–1371. 10.1002/alz.12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neth BJ, Mintz A, Whitlow C, Jung Y, Solingapuram Sai K, Register TC, Kellar D, Lockhart SN, Hoscheidt S, Maldjian J, Heslegrave AJ, Blennow K, Cunnane SC, Castellano C-A, Zetterberg H, Craft S, 2020. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiology of Aging 86, 54–63. 10.1016/j.neurobiolaging.2019.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Yamada T, Hamajima M, Itoh M, Katayama T, Bork P, Goto S, Kanehisa M, 2008. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res 36, W423–426. 10.1093/nar/gkn282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oñatibia‐Astibia A, Franco R, Martínez‐Pinilla E, 2017. Health benefits of methylxanthines in neurodegenerative diseases. Molecular Nutrition & Food Research 61, 1600670. 10.1002/mnfr.201600670 [DOI] [PubMed] [Google Scholar]
- Paglia G, Stocchero M, Cacciatore S, Lai S, Angel P, Alam MT, Keller M, Ralser M, Astarita G, 2016. Unbiased Metabolomic Investigation of Alzheimer’s Disease Brain Points to Dysregulation of Mitochondrial Aspartate Metabolism. J. Proteome Res 15, 608–618. 10.1021/acs.jproteome.5b01020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, Solfrizzi V, Barulli MR, Bonfiglio C, Guerra V, Osella A, Seripa D, Sabbà C, Pilotto A, Logroscino G, 2015. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: A systematic review. J Nutr Health Aging 19, 313–328. 10.1007/s12603-014-0563-8 [DOI] [PubMed] [Google Scholar]
- Patenaude B, Smith SM, Kennedy DN, Jenkinson M, 2011. A Bayesian model of shape and appearance for subcortical brain segmentation. NeuroImage 56, 907–922. 10.1016/j.neuroimage.2011.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine AM, Koscik RL, Berman SE, Nicholas CR, Clark LR, Okonkwo OC, Rowley HA, Asthana S, Bendlin BB, Blennow K, Zetterberg H, Gleason CE, Carlsson CM, Johnson SC, 2016. Biomarker clusters are differentially associated with longitudinal cognitive decline in late midlife. Brain 139, 2261–2274. 10.1093/brain/aww142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA, 2016. Untargeted metabolomics strategies – Challenges and Emerging Directions. J Am Soc Mass Spectrom 27, 1897–1905. 10.1007/s13361-016-1469-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher KE, Bendlin BB, Racine AM, Okonkwo OC, Christian BT, Koscik RL, Sager MA, Asthana S, Johnson SC, Benca RM, 2015. Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiology of Aging 36, 2568–2576. 10.1016/j.neurobiolaging.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Arnold M, Kastenmüuller G, Chang R, Baillie RA, Han X, Thambisetty M, Tenenbaum JD, Suhre K, Thompson JW, St. John-Williams L, MahmoudianDehkordi S, Rotroff DM, Jack JR, Motsinger-Reif A, Risacher SL, Blach C, Lucas JE, Massaro T, Louie G, Zhu H, Dallmann G, Klavins K, Koal T, Kim S, Nho K, Shen L, Casanova R, Varma S, Legido-Quigley C, Moseley MA, Zhu K, Henrion MYR, van der Lee SJ, Harms AC, Demirkan A, Hankemeier T, van Duijn CM, Trojanowski JQ, Shaw LM, Saykin AJ, Weiner MW, Doraiswamy PM, Kaddurah-Daouk R, 2017. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement 13, 965–984. 10.1016/j.jalz.2017.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- M. Travassos, Santana I, Baldeiras I, Tsolaki M, Gkatzima O, Sermin G, Yener GG, Simonsen A, Hasselbalch SG, Kapaki E, Mara B, Cunha RA, Agostinho P, Blennow K, Zetterberg H, Mendes VM, Manadas B, de Mendon A, 2015. Does Caffeine Consumption Modify Cerebrospinal Fluid Amyloid-β Levels in Patients with Alzheimer’s Disease? Journal of Alzheimer’s Disease 47, 1069–1078. 10.3233/JAD-150374 [DOI] [PubMed] [Google Scholar]
- Turunc E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A, 2013. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ(1–42)-induced rat model of Alzheimer’s disease. Free radical research 48. 10.3109/10715762.2013.857018 [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M, 2002. Automated Anatomical Labeling of Activations in SPM Using a Macroscopic Anatomical Parcellation of the MNI MRI Single-Subject Brain. NeuroImage 15, 273–289. 10.1006/nimg.2001.0978 [DOI] [PubMed] [Google Scholar]
- Vakilinezhad MA, Amini A, Akbari Javar H, Baha’addini Beigi Zarandi BF, Montaseri H, Dinarvand R, 2018. Nicotinamide loaded functionalized solid lipid nanoparticles improves cognition in Alzheimer’s disease animal model by reducing Tau hyperphosphorylation. DARU J Pharm Sci 26, 165–177. 10.1007/s40199-018-0221-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valls-Pedret C, Lamuela-Raventós RM, Medina-Remón A, Quintana M, Corella D, Pintó X, Martínez-González MÁ, Estruch R, Ros E, 2012. Polyphenol-Rich Foods in the Mediterranean Diet are Associated with Better Cognitive Function in Elderly Subjects at High Cardiovascular Risk. Journal of Alzheimer’s Disease 29, 773–782. 10.3233/JAD-2012-111799 [DOI] [PubMed] [Google Scholar]
- Van Hulle C, Jonaitis EM, Betthauser TJ, Batrla R, Wild N, Kollmorgen G, Andreasson U, Okonkwo O, Bendlin BB, Asthana S, Carlsson CM, Johnson SC, Zetterberg H, Blennow K, 2020. An examination of a novel multipanel of CSF biomarkers in the Alzheimer’s disease clinical and pathological continuum. Alzheimers Dement 10.1002/alz.12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercambre M-N, Berr C, Ritchie K, Kang JH, 2013. Caffeine and Cognitive Decline in Elderly Women at High Vascular Risk. Journal of Alzheimer’s Disease 35, 413–421. 10.3233/JAD-122371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt NM, Romano KA, Darst BF, Engelman CD, Johnson SC, Carlsson CM, Asthana S, Blennow K, Zetterberg H, Bendlin BB, Rey FE, 2018. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Research & Therapy 10, 124. 10.1186/s13195-018-0451-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li J, Zhang A-H, 2016. Urine metabolic phenotypes analysis of extrahepatic cholangiocarcinoma disease using ultra-high performance liquid chromatography-mass spectrometry. RSC Advances 6, 63049–63057. 10.1039/C6RA09430A [DOI] [Google Scholar]
- Wilkins JM, Trushina E, 2018. Application of Metabolomics in Alzheimer’s Disease. Frontiers in Neurology 8. 10.3389/fneur.2017.00719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M-S, Lan T-H, Chen C-M, Chiu H-C, Lan T-Y, 2011. Socio-demographic and health-related factors associated with cognitive impairment in the elderly in Taiwan. BMC Public Health 11, 22. 10.1186/1471-2458-11-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Xu Y-H, Chen J-F, Schwarzschild MA, 2010. Neuroprotection by caffeine: time course and role of its metabolites in the MPTP model of Parkinson’s disease. Neuroscience 167, 475–481. 10.1016/j.neuroscience.2010.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.