

Abstract
A method to identify molecular scaffolds potentially active against the Mycobacterium tuberculosis complex (MTBC) is developed. A set of structurally heterogeneous agents against MTBC was used to obtain a mathematical model based on topological descriptors. This model was statistically validated through a Leave-n-Out test. It successfully discriminated between active or inactive compounds over 86% in database sets. It was also useful to select new potential antituberculosis compounds in external databases. The selection of new substituted pyrimidines, pyrimidones and triazolo[1,5-a]pyrimidines was particularly interesting because these structures could provide new scaffolds in this field. The seven selected candidates were synthesized and six of them showed activity in vitro.
Keywords: MTBC, virtual screening, topological indices, linear discriminant analysis, pharmacological activity distribution diagrams, antimicrobial drugs, drug design
1. Introduction
Tuberculosis is one of the deadliest infections in the world, killing almost 1.45 million people annually [1,2]. Most of these deaths occur in poor countries, and a more rational distribution of wealth could prevent them. However, the increasing incidence of drug-resistant MTBC has caused its resurgence in developed countries and makes therapeutic alternatives necessary [3,4]. Although new compounds potentially active against these bacteria are under active research, very few new treatments have been developed in recent years [5,6,7,8,9]. Therefore, more extensive investigations are needed to find new molecular scaffolds capable of generating new inhibitors of mycobacteria.
The α-acetylenic ketones of type A (Scheme 1) have been shown to be highly versatile building blocks. For instance, these conjugated ynones have proven to be very suitable substrates for the synthesis of (E)-3-acylpropenoic acids [10], and of a wide range of heterocyclic systems [11,12,13,14,15,16,17,18,19,20,21], including the synthesis of the natural product L-lathyrine and related analogues [22,23]. Furthermore, when properly functionalized, compounds of type A have also proven to be valuable substrates for combinatorial and parallel synthesis on solid support of highly molecular diverse 2,4,6-trisubstituted pyrimidines [24,25,26,27]. These α-acetylenic ketones are produced readily, mainly by direct palladium-catalyzed coupling of acyl chlorides with 1-alkynes (Sonogashira reaction), [28,29] by reaction of alkynylzinc chlorides with acid halides, [30,31] or by reaction of lithium or magnesium acetylides with aldehydes followed by subsequent oxidation with oxalyl chloride in DMSO (Swern oxidation) [32], MnO2 [12,13,15,33], t-butyl hydroperoxide [34] or with IBX [33,34,35].
Scheme 1.

Cyclocondensation of α-acetylenic ketones A with 3-amino-5-benzylsulfanyl-1,2,4-triazole.
In this paper, we have further expanded the synthetic utility of these conjugated ynones A, and we wish to report the synthesis of novel 2,5,7-substituted triazolo[1,5-a]pyrimidines B by cyclocondensation of different α-acetylenic ketones A with 3-amino-5-benzylsulfanyl-1,2,4-triazole (Scheme 1).
The triazolo[1,5-a]pyrimidine nucleus is of considerable chemical and pharmacological interest. Antibiotic, anticholesteremic, antidiabetic, antiallergic, anti-inflammatory, antipyretic, antiphlogistic, analgesic and anticancer activities have been described for these types of compounds. They also serve in the treatment and prevention of circulatory diseases such as hypertension, heart diseases, stroke, hypercholesterol, arteriosclerosis and are effective coronary vasodilators and bronchodilators [36,37,38,39,40,41,42,43,44,45,46,47,48,49].
On the other hand, several triazolo[1,5-a]pyrimidine derivatives have found applications in the agrochemical industry. Additionally, 1,2,4-triazolo[1,5-a]pyrimidinesulfonamides are used as herbicides and plant growth inhibitors, and they show activity against acetolactate synthase [46,50,51].
Currently, there are many methods available to synthesize triazolo[1,5-a]pyrimidines, mainly they are cyclocondensations between 3-amino-1,2,4-triazoles and 1,3-bifunctional synthons such as 1,3-dicarbonyl compounds or their equivalents [52,53,54,55,56,57,58,59], vinylogous iminium salts [47,60,61], ketene dithioacetals [48,62,63], and 3-ketovinyl compounds [46]. We used α-acetylenic Ketones of types A as 1,3-bifunctional synthons.
Although a few pyrimidines [64], pyrimidones [65] and triazolo[1,5-a]pyrimidines [66] have already been synthetized and tested for anti-TB activity, their substitution patterns and synthesis methods are different from the compounds described here.
The identification of new targets requires knowledge of the specific biochemical pathways of mycobacteria, but many metabolic processes are still unknown and the structure-based design of new anti-TB agents is a complex task [67].
On the other hand, extra-mechanistic virtual screening methodologies have demonstrated their ability to model the presence of activity within structurally heterogeneous groups of compounds in different therapeutic areas [68,69,70,71,72,73,74,75,76,77] as well as in predicting toxicological properties [78,79] and drug-like characteristics [80]. In these models, structural similarity is the key. Molecules are characterized through structural invariants, that is, by descriptors that are independent of molecular conformation. Many of them are topological indices (TI) [81,82,83,84,85,86,87], which are capable of characterizing most of the molecular structure [88,89,90,91,92,93,94,95].
The aim of this study was to develop new discriminant models based on the molecular structures of antituberculosis agents, shown in Table S1 [96,97,98] and inactive substances, shown in Table S2, characterized by topological indices [99] (Table 1), in order to screen structural databases to identify new potentially useful scaffolds against MTBC.
Table 1.
Descriptors used.
| Symbol | Name | Definition | Reference |
|---|---|---|---|
| N | Molecular size | Number of non-hydrogen atoms. | [68] |
| Vk k = 3, 4 |
Vertices of degree k | Number of atoms having k bonds, σ or π, to non-hydrogen atoms. | [68] |
| R | Ramification | Number of single structural branches. | [68] |
| W | Wiener path number | Sum of the distances between any two atoms in terms of bonds. | [100] |
| L | Length | Maximal distance between atoms in terms of bonds. | [68] |
| PRk k = 0–3 |
Pairs of ramifications at distance k | Number of pairs of single branches at distance k in terms of bonds. | [68] |
|
kχt k = 0–4 t = p, c, pc |
Randić-like indices of order k and type path (p), cluster (c) and path-cluster (pc) |
δi, number of bonds, σ or π, of the atom i to non-hydrogen atoms. Sj, jth sub-structure of order k and type t. |
[71,81,82] |
|
kχtv k = 0–4 t = p, c, pc |
Kier-Hall indices of order k and type path (p), cluster (c) and path-cluster (pc) |
δiv, Kier-Hall valence of the atom i. Sj,jth sub-structure of order k and type t. |
[71,81,82] |
| Gk k = 1–5 |
Topological charge indices of order k |
, product of the adjacency and inverse squared distance matrices for the hydrogen-depleted molecular graph. D, distance matrix. δ, Kronecker delta. |
[68,101] |
| Gkv k = 1–5 |
Valence topological charge indices of order k |
product of the electronegativity-modified adjacency and inverse squared distance matrices for the hydrogen-depleted molecular graph. D, distance matrix. δ, Kronecker delta. |
[68,101] |
| Jk k = 1–5 |
Pondered topological charge indices of order k | [68,101] | |
| Jkv k = 1–5 |
Pondered valence topological charge indices of order k | [68,101] | |
|
kDt k = 0–4 t = p, c, pc |
Connectivity differences of order k and type path (p), cluster (c) and path-cluster (pc) | [68] | |
| Ek k = 1–5 |
Topological charge differences of order k | [102] | |
| Fk k = 1–5 |
Pondered topological charge differences of order k | [102] | |
|
kCt k = 0–4 t = p, c, pc |
Connectivity quotients of order k and type path (p), cluster (c) and path-cluster (pc) | [68] | |
|
kQt k = 0–4 t = p, c, pc |
Inverse connectivity quotients of order k and type path (p), cluster (c) and path-cluster (pc) | [102] | |
| CGk k = 1–5 |
Topological charge quotients of order k | [102] | |
| QGk k = 1–5 |
Inverse topological charge quotients of order k | [102] |
2. Results and Discussion
2.1. Antituberculosis Activity Modelling
Figure 1 illustrates the proposed virtual screening procedure. In this method, thresholds on descriptor values were combined with Linear Discriminant Analysis (LDA) to give a qualitative discriminant model. Table S3 in the Supplementary Materials collects all these values.
Figure 1.

Proposed virtual screening procedure.
Given a population, for example of molecules, that can be classified into several groups according to their experimental properties, for example a group of molecules with a pharmacological activity and another without this activity, LDA is a method to find linear combinations of independent variables (for example the aforementioned structural invariants) whose numerical values can be used to distinguish between these different categories. When two categories are defined, the classification is done by the so-called discriminant function (DF).
The following equation DF was obtained by stepwise LDA using the set shown in Table 2:
| DF = 0.88 − 11.99 J1 + 4.86 J1v − 11.11 J3v + 0.81 1D − 0.2 4Cc |
| N = 60 λ = 0.34 F = 21.12 |
where N = 60 represents 25 anti-MTBC drugs and 35 presumably inactive compounds.
Table 2.
Classification for each compound by DF.
| Active Group | Inactive Group | ||||||
|---|---|---|---|---|---|---|---|
| Compound | DF | Prob. a Active |
Predicted Activity b | Compound | DF | Prob. a Inactive |
Predicted Activity b |
| Streptonicozid | 3.14 | 1.000 | + | Butibufen | −3.14 | 1.000 | − |
| Tobramycin | 3.49 | 1.000 | + | Aldicarb | −2.74 | 1.000 | − |
| Kanamycin | 3.88 | 1.000 | + | Antrafenine | −2.55 | 1.000 | − |
| Amikacin | 4.14 | 1.000 | + | Carprofen | −1.89 | 0.997 | − |
| Dihydrostreptomycin | 2.80 | 0.999 | + | Beclobrate | −1.84 | 0.997 | − |
| Streptomycin | 2.32 | 0.997 | + | Benzoctamine | −1.77 | 0.996 | − |
| Ethambutol | 2.23 | 0.996 | + | Carmofur | −1.77 | 0.996 | − |
| Pyrazinamide | 1.97 | 0.992 | + | Aminothiazole | −1.73 | 0.996 | − |
| Enviomycin | 1.92 | 0.991 | + | Acifran | −1.72 | 0.996 | − |
| Ofloxacin | 1.82 | 0.988 | + | Brilliant Blue | −1.72 | 0.996 | − |
| Moxifloxacin | 1.66 | 0.982 | + | Amitraz | −1.68 | 0.995 | − |
| Gatifloxacin | 1.66 | 0.981 | + | Clofibrate | −1.62 | 0.994 | − |
| Rifampin | 1.60 | 0.978 | + | Paraoxon | −1.52 | 0.992 | − |
| Azithromycin | 1.49 | 0.971 | + | Piroxicam | −1.39 | 0.989 | − |
| Verazide | 1.44 | 0.967 | + | Carmustine | −1.11 | 0.977 | − |
| Isoniazid | 1.20 | 0.938 | + | Ornithine | −1.11 | 0.977 | − |
| Capreomycin | 1.16 | 0.930 | + | Alpidem | −1.10 | 0.976 | − |
| Sparfloxacin | 0.98 | 0.889 | + | Alprazolam | −1.04 | 0.972 | − |
| Clarithromycin | 0.93 | 0.874 | + | Bixin | −1.04 | 0.972 | − |
| Salinazid | 0.92 | 0.871 | + | Benzoic Acid | −1.03 | 0.971 | − |
| Tuberin | 0.91 | 0.869 | + | Amsacrine | −0.98 | 0.967 | − |
| Clofazimine | 0.13 | 0.432 | NC | Azaserine | −0.96 | 0.965 | − |
| Imipenem | 0.09 | 0.404 | NC | Theofibrate | −0.88 | 0.957 | − |
| Ph−Aminosalicylate | −0.200 | 0.230 | − | Azacosterol | −0.83 | 0.951 | − |
| PAS | −1.000 | 0.031 | − | Allicin | −0.77 | 0.943 | − |
| Prazepam | −0.77 | 0.943 | − | ||||
| Camazepam | −0.71 | 0.933 | − | ||||
| Aminopromazine | −0.57 | 0.904 | − | ||||
| Bromazepam | −0.45 | 0.870 | − | ||||
| Carnitine | −0.32 | 0.824 | − | ||||
| Acipimox | −0.28 | 0.809 | − | ||||
| Buspirone | −0.03 | 0.677 | − | ||||
| Azapicyl | 0.03 | 0.640 | − | ||||
| Acronine | 0.11 | 0.588 | NC | ||||
| Captodiamine | 0.28 | 0.470 | NC | ||||
a Prob., probability; b When probability active or probability inactive > 0.60, the compound is classified as active “+” or inactive “−”, respectively. In any other case, the compound is considered non-classified “NC”.
The topological descriptors selected in this equation were: the charge indices (J1, J1v, J3v); the difference index (1D = 1χ − 1χv); and the quotient index (4Cc = 4χc/4χcv), where mχt and mχtv are, respectively, single and valence Randić-Kier-Hall indices of order m and type t. Topological charge-transfer indices, J1, J1v and J3v, are measures of the contribution of molecular topological structure to the charge transfer at topological distance 1 and 3, respectively [68,101]. Difference and quotient indices are related to charge distributions within molecular fragments [68]. Thus, 1D is the net contribution of the heteroatoms to the electronic charge within fragments of order 1 (bonds). The 4Cc index can be related to electron densities of cluster-type fragments of order 4 (three atoms bonded to a central one) in which there is at least one heteroatom. The maximum correlation between pairs of these selected variables, for the group of actives, was as weak as 0.424, corresponding to the pair J1v and J3v. Thus, the variables can be considered as independent. Table S4 shows these intercorrelations.
Table 2 shows the results of the classification for each one of the compounds included in the LDA.
The linear equation gave good results since most compounds were classified with a probability over 86% (Table 2). When the probabilities are between 40% and 60%, the compounds were counted as non-classified (NC), and finally below 40%, they were considered as inactive. Under this framework, the error percentage in the active set was about 9%, whereas in the inactive was 0%. These probabilities are the so-called posterior probabilities, computed by the Bayes rule as the probability of classifying a case (molecule) conditioned to the model obtained. Let πk the prior probability: . The posterior probability P is given by: where is the class-conditional density of the case x in the class k. Assuming that this density for x, given every class k, follows a normal distribution, the density formula for a multivariate Gaussian distribution is applicable. Thus, where x and the mean are both column vectors, Cov is the covariance matrix and p is its dimension. The denominator involves the square root of the determinant of this matrix. The result of the matrix multiplications in the numerator is a scalar number.
The results of the internal validation are illustrated in Table 3, with the percentage of correct classification within each group. Five runs were performed. A number of compounds ranging from 9 to 16 were randomly extracted from the training to a test set. Wilks λ values are shown for each equation. The lower the Wilks λ value, the better the discrimination. Correct classification percentages are shown for training and test sets, for active and inactive compounds. The number of compounds classified as active (+) or inactive (−) appears in parentheses, where (a/b) = number of (+) compounds/number of (−) compounds. Average values are also shown, as well as the performance of DF function.
Table 3.
Results of the internal validation for DF.
| Run No. | λ | Training Group | Test Group | ||
|---|---|---|---|---|---|
| (+) | (−) | (+) | (−) | ||
| 1 | 0.28 | 90% (18/2) | 100% (0/26) | 60% (3/2) | 100% (0/9) |
| 2 | 0.31 | 87% (20/3) | 96% (1/27) | 50% (1/1) | 100% (0/7) |
| 3 | 0.35 | 86% (19/3) | 89% (3/24) | 100% (3/0) | 75% (2/6) |
| 4 | 0.33 | 82% (14/3) | 100% (0/27) | 88% (7/1) | 100% (0/8) |
| 5 | 0.33 | 91% (20/2) | 96% (1/27) | 100% (3/0) | 86% (1/6) |
| Average | - | 87% | 96% | 80% | 92% |
| DF | 0.34 | 84% (21/4) | 97% (1/34) | No | No |
(a/b) = number of (+) compounds/number of (−) compounds.
The results for the training and test groups are within the same range. The mean percentage of success obtained with the training group for DF (Table 3) was 87% for active (+) and 94% for inactive (−). For the test it was 80% and 92%, respectively. The results were similar to those obtained with the DF equation, which points out the validity of the LDA equation.
The results of the external validation test are shown in Table 4. As can be seen, for the active set there was a misclassified compound, namely ethionamide. The same is found in the opposite group, where glucosamine was misclassified.
Table 4.
Results of the external validation for DF.
| Active Group | Inactive Group | ||||||
|---|---|---|---|---|---|---|---|
| Compound | DF | Prob. a Active |
Predicted Activity b | Compound | DF | Prob. a Inactive |
Predicted Activity b |
| Morphazinamide | 4.32 | 1.000 | + | Canthaxanthin | −3.46 | 1.000 | − |
| Neomycin | 5.02 | 1.000 | + | Genite | −2.57 | 1.000 | − |
| Tubercidin | 2.85 | 0.999 | + | Altretamine | −1.91 | 0.997 | − |
| Ciprofloxacin | 2.15 | 0.995 | + | Etifoxin | −1.86 | 0.997 | − |
| Viomycin | 1.75 | 0.986 | + | Dichlone | −1.78 | 0.996 | − |
| Rifabutin | 0.73 | 0.799 | + | Feprazone | −1.47 | 0.991 | − |
| Ethionamide | −1.30 | 0.014 | − | Antipyrine | −0.44 | 0.868 | − |
| Benorylate | −0.25 | 0.792 | − | ||||
| Chloropal | 0.19 | 0.526 | NC | ||||
| Glucosamine | 1.77 | 0.014 | + | ||||
a Prob., probability; b When probability active or probability inactive > 0.60, the compound is classified as active “+” or inactive “−”, respectively. In any other case, the compound is considerate non-classified “NC”.
Figure 2 shows the PDD obtained from DF. As can be inferred from Figure 2, the optimal range of DF to find active compounds can be established between 0 and 4.5.
Figure 2.

PDD in the training set, obtained by DF equation. E, expectancy of activity or inactivity; black line, active group; grey line, inactive group.
2.2. Similarity-Based Virtual Screening
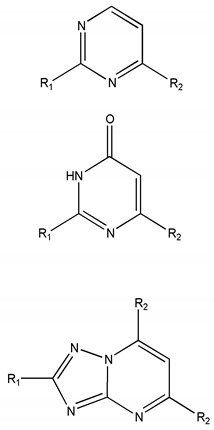
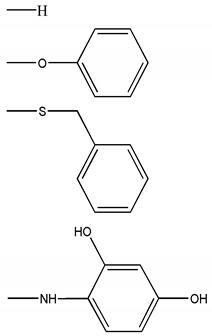
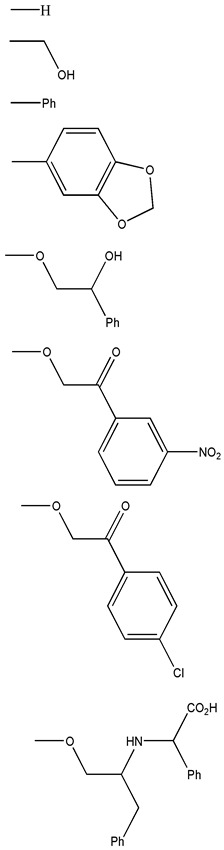
A virtual library containing new pyrimidine derivatives generated by combining the chemical scaffolds and substituents depicted in Table 5 was screened as described in Section 3.2. After building the database containing the substituted pyrimidines, the virtual screening was performed. Descriptors calculated for the entire database are available to the readers upon request to the corresponding author. Based on these parameters, a virtual screening was carried out so that those molecular structures with the values of the descriptors and DFs within the thresholds, shown in the Table S3, were selected as candidates. Table S5, under Supplementary Materials, shows the calculated descriptors for the selected compounds. Seven potentially active molecules were selected. These structures are presented in Figure 3. These compounds selected as possible candidates were synthesized and tested in vitro.
Table 5.
Scaffolds and fragments used for the generation of the virtual library.
| Chemical Scaffolds | R1 | R2 |
|---|---|---|
|
|
|
Figure 3.

Structures of the selected compounds.
2.3. Chemistry
Synthesis of 1,2,4-triazolo[1,5-a]pyrimidine derivatives. Reaction of the required magnesium acetylides, generated from alkynes 1a-b and i-PrMgCl in dry THF at 0 °C, with piperonal 2, afforded the expected propargylic alcohols 3a-b. Oxidation of 3a-b with MnO2 afforded α-acetylene ketones 4a and 4b in 79% and 37% overall yield, respectively (Scheme 2).
Scheme 2.

Synthesis of protected 1,2,4-triazolo[1,5-a]pyrimidine derivatives 6.
Cyclocondensation of 3-amino-5-benzylsulfanyl-1,2,4-triazole 5 with α-acetylene ketones 4a-b in dry DMF at 40 °C, gave the corresponding triazolo[1,5-a]pyrimidines 6a-b in moderate yields (73% and 38%, respectively) (Scheme 2).
Finally, deprotection of 6b in acidic conditions afforded 7 in 77% yield (Scheme 3).
Scheme 3.

Deprotection of 6b to give 7.
Synthesis of pyrimidine derivatives. Two of us reported on the synthesis of novel 4-alkoxypyrimidines starting from 2-alkylsulfanylpyrimidinones of type 8 [103,104]. The method is based on a selective O-alkylation reaction with bulky aliphatic alcohols using the Mitsunobu conditions (method A, Scheme 4) or in basic medium with sterically demanding agents like α–haloketones (method B, Scheme 4).
Scheme 4.

O-alkylation of 2-alkylsulfanylpyrimidinones 8.
Oxidation of the thioether moiety to the corresponding sulfone 10 using m-CPBA and nucleophilic displacement by different nucleophiles produced the corresponding highly molecular diverse pyrimidines of type 11. In addition, when 4-isopropoxipyrimidine 11a was treated with a 1:1 mixture of H2SO4/AcOH at 90 °C for 15 min, the selective cleavage of the 4-isopropoxy group took place yielding 2-aryloxypyrimidinone 12 (Scheme 5).
Scheme 5.

Oxidation of 9 to the corresponding sulfone 10, nucleophilic displacement to produce the corresponding pyrimidines 11, and displacement of the 4-isopropoxy group of 11a to yield 2-aryloxypyrimidinone 12.
On the other hand, the reaction of the deprotected amine 9e with phenylboronic acid and glyoxylic acid (Petasis reaction) [105,106] gave the desired α-phenyl glycine derivative 13 in moderated yield (Scheme 6).
Scheme 6.

Petasis reaction of the amine 9e to give α-phenyl glycine derivative 13.
Finally, we prepared the compound 14 by reducing the carbonyl group of compound 9d with NaBH4 in MeOH. This reduction afforded the hydroxy derivative 14 in 80% isolated yield (Scheme 7).
Scheme 7.

Reduction of 9d to obtain the hydroxy derivative 14.
Microbiological study. To check the predicted antituberculosis activity of the selected candidates, microbiological tests were performed. The result of in vitro susceptibility test of conventional drugs is shown in Table 6, and the experimental results of the selected compounds are illustrated in Table 7. Two compounds, 7 and 12, showed MIC50 = MIC90 = 32 mg/L, corresponding to 81.5 μM and 121.1 μM, respectively; four compounds, 6a, 9c, 11b and 13, showed MIC90 = 64 mg/L against M. tuberculosis, which corresponds to 146, 167.8, 160.1, 131.8 μM, respectively; and 14 was inactive at the assayed concentrations. The compound 11b showed the best MIC50 = 80 μM, but its MIC90 raised to 160,1 μM. Thus, the results pointed out that 7 was the most active agent, showing molar MIC90 four times lower than Ethambutol but within the same magnitude order.
Table 6.
Experimental MIC of conventional drugs against MTBC.
| Compound | MIC Range/mg/L | MIC50/mg/L | MIC90/mg/L | Molecular Mass/Da | MIC50/μM | MIC90/μM |
|---|---|---|---|---|---|---|
| Ethambutol | 1–8 | 4.0 | 4.0 | 204.3 | 19.6 | 19.6 |
| Isoniazid | 0.05–0.2 | 0.05 | 0.05 | 137.1 | 0.4 | 0.4 |
| Rifampin | 0.125–1 | 0.125 | 0.5 | 823.0 | 0.2 | 0.6 |
| Streptomycin | 0.125–0.5 | 0.25 | 0.5 | 581.6 | 0.4 | 0.9 |
MIC range: minimal and maximal inhibitory values found.
Table 7.
Experimental MIC of the selected compounds against MTBC.
| Compound | MIC Range/mg/L | MIC50/mg/L | MIC90/mg/L | Molecular Mass/Da | MIC50/μM | MIC90/μM |
|---|---|---|---|---|---|---|
| 6a | 64 | 64 | 64 | 438.5 | 146.0 | 146.0 |
| 7 | 32 | 32 | 32 | 392.4 | 81.5 | 81.5 |
| 9c | 64 | 64 | 64 | 381.4 | 167.8 | 167.8 |
| 11b | 16 - 64 | 32 | 64 | 399.8 | 80.0 | 160.1 |
| 12 | 32 | 32 | 32 | 264.3 | 121.1 | 121.1 |
| 13 | 32 - 64 | 64 | 64 | 485.6 | 131.8 | 131.8 |
| 14 | >128 | >128 | >128 | 414.5 | >308.8 | >308.8 |
MIC range: minimal and maximal inhibitory values found.
3. Materials and Methods
3.1. Antituberculosis Activity Modelling
A group of 32 compounds with known activity against MTBC were compiled from various sources [96,97,98]. Their structures are shown in Table S1 in the Supplementary Material. A set of 45 compounds with a different pharmacological activity was also used as inactive group. Their structures are shown in Table S2 in the Supplementary Material. The compounds were characterized by a set of 90 descriptors calculated using the DesMol program [99]. The descriptors used with their symbols, definitions and references are shown in Table 1. These were used to build a model capable of discriminating between active and inactive antituberculous compounds.
DFs were calculated with randomly selected subsets of 25 out of 32 active and 35 out of 45 inactive compounds by using BMDP New System [107]. Descriptor selection was based on the Fisher-Snedecor F parameter. The variables were introduced step by step in the DF: in each step, the variable that added the most to the separation of the groups was entered in the equation, or the variable that contributed the least to improving said separation was eliminated. The classification criterion was the shortest Mahalanobis distance. The discriminant ability of the DF was evaluated by two parameters, Wilks λ, and the percentage of correct classification in each group. The independent variables in this study were the calculated structural invariants, and the discrimination property was the presence of activity against MTBC.
The validation of the selected DF was performed by two methods. One internal leave-n-out test in which the program randomly chose and pulled out a ratio of compounds, and used them to evaluate the DF obtained with the rest; and another external test with a previously unused data set.
To choose the optimal ranges of values for this equation, the corresponding Pharmacological Distribution Diagram, PDD [108], was obtained. These diagrams are useful to determine the intervals of the equation in which the probability of finding new candidates is maximum. This is a graph similar to a histogram in which expectancies appear on the ordinate axis. For an arbitrary range of values of a given function, the activity expectancy is Ea = a/(i + 100), where a is the percentage of active compounds in the range and i is the corresponding percentage of inactive within the same range. The expectancy of inactivity is similarly defined as Ei = i/(a + 100). This plot provides a good visualization of the regions of minimal overlap between the active and inactive compounds and helps to select the optimal interval of the DF.
The 90 descriptors and the selected discriminant function were used as filters to select potential candidates in structural databases. For this, the maximum and minimum values of each descriptor were established as thresholds for the 90 variables, while for DF, the optimal interval was taken. Compounds exhibiting all 90 values within the thresholds and DF values within the optimal intervals were considered as candidates.
3.2. Similarity-Based Virtual Screening
A virtual library containing 320 structures of substituted pyrimidines was generated by combining the chemical scaffolds and substituents depicted in Table 5, using in-house software. The set of 90 descriptors and DFs were calculated for this library. A virtual screening was carried out based on these parameters to select as candidates those molecular structures whose values for the descriptors and DFs were within the thresholds.
3.3. Chemical Methods
DMF was dried over activated molecular sieves (4 Å). THF was dried over Na/benzophenone prior use. All the other commercially available chemicals were used as purchased without further purification. Reactions involving magnesium acetylides and synthesis of triazolo[1,5-a]pyrimidines were run under a dry Ar atmosphere. Melting points (capillary tube) were measured with an electrothermal digital melting point apparatus IA 91,000 and are uncorrected. IR spectra were recorded on a Mattson-Galaxy Satellite FT-IR. Additionally, 1H and 13C NMR spectra were recorded at 200 and 50 MHz, respectively, on a Brucker DPX200 Advance instrument with TMS as internal standard. MS spectra were recorded on a VG Quattro instrument in the positive ionization FAB mode, using 3-NBA or 1-thioglycerol as the matrix or in a Thermo Quest 2000 series apparatus for the EI (70 eV) mode. Analytical TLC was performed on precoated TLC plates, silica gel 60 F254 (Merck). Flash-chromatography (FC) purifications were performed on silica gel 60 (230–400 mesh, Merck).
Synthesis of propargylic alcohols 3a-b. General procedure.
To a cooled (0 °C) solution of the corresponding alkyne 1a-b in dry THF (2 mL/mmol), i-PrMgCl (2 M solution in THF) was added dropwise under Ar. The mixture was stirred at that temperature for 4 h. Then, a solution of the piperonal 2 (1.3 equiv) in dry THF (1 mL/mmol) was slowly added dropwise over a period of 15 min. The reaction mixture, under Ar, was stirred from 0 °C to r.t. until total consumption of 1a-b (5–12 h., monitored by TLC). The reaction was quenched with saturated solution of NH4Cl (3 mL/mmol) at r.t. and the organic solvent was eliminated under reduced pressure. The aqueous layer was extracted with AcOEt (3 × 3 mL/mmol) and the combined organic layers were dried over MgSO4, the solvent was evaporated and the resulting residue purified by flash-chromatography (n-hexane:AcOEt).
1-Benzo[1,3]dioxol-5-yl-3-phenyl-prop-2-yn-1-ol (3a). According to the general procedure described above, the reaction between 1a (4.01 g, 39.26 mmol) and piperonal 2 (7.65 g, 50.9 mmol) afforded 8.02 g (81%) of 3a as a white solid. m.p.: 60–61 °C. IR (KBr): ν 3439 (br., OH). 1H NMR (CDCl3): δ 7.5–7.4 (m, 2Harom), 7.3 (m, 3Harom), 7.2–7.1 (m, 2Harom), 6.85 (d, 1Harom, J = 8.0 Hz), 6.00 (s, 2H, OCH2O), 5.63 (s, 1H, CHC≡C), 2.59 (br., 1H, OH). 13C NMR (CDCl3): δ 147.8, 147.6, 134.6 (3s, 3Carom), 131.7, 128.6, 128.3 (3d, 5CHarom), 122.2 (s, Carom), 120.4, 108.1, 107.4 (3d, 3CHarom), 101.2 (t, CH2), 88.6 (s, C≡CPh), 86.5 (s, C≡CPh), 64.8 (d, CH). MS (FAB+) m/z: 253 ([M+1]+, 9). Anal. Calcd. for C16H12O3: C, 76.18; H, 4.79. Found: C, 76.39; H, 4.61%.
1-Benzo[1,3]dioxol-5-yl-3-(tetrahydro-pyran-2-yloxy)-prop-2-yn-1-ol (3b). According to the general procedure described above, the reaction between 1b (0.21 g, 1.48 mmol) and piperonal 2 (0.29 g, 1.92 mmol) afforded the crude of the compound 3b. The reaction mixture was used in the next step of synthesis without further purification.
Synthesis of α-acetylenic ketones 4a-b. General procedure.
Over a cooled (0 °C), mechanically stirred suspension of MnO2 (5 equiv) in CH2Cl2 (3 mL/mmol), a solution of the propargyl alcohol 3a or of the reaction mixture 3b in CH2Cl2 (3 mL/mmol) was added dropwise. The reaction mixture was stirred from 0 °C to r.t. until total consumption of 3a-b (3–12 h., monitored by TLC) and then filtered through a Celite® pad. The solvents were removed under reduced pressure and the resulting residue was purified by flash-chromatography (n-hexane:AcOEt).
1-Benzo[1,3]dioxol-5-yl-3-phenyl-propynone (4a). According to the general procedure described above, the reaction between 3a (1.98 g, 7.87 mmol) and MnO2 (3.84 g, 39.35 mmol) afforded after crystallization of the residue with MeOH:H2O instead of the chromatographic purification 1.91 g (97%) of 4a as a yellow solid. m.p.: 100–101 °C. IR (KBr): ν 1625 (s, C=O). 1H NMR (CDCl3): δ 7.92 (dd, 1Harom, J = 8.2 Hz, J′ = 1.8 Hz), 7.7–7.6 (m, 3Harom), 7.5–7.4 (m, 3Harom), 6.93 (d, 1Harom, J = 8.2 Hz), 6.10 (s, 2H, OCH2O). 13C NMR (CDCl3): δ 176.0 (s, C=O), 152.8, 148.1 (2s, 2Carom), 132.9 (d, 2CHarom), 131.9 (s, Carom), 130.6, 128.6, 127.2 (3d, 4CHarom), 120 (s, Carom), 108.2, 107.9 (2d, 2CHarom), 102.1 (t, CH2), 92.3 (s, C≡CPh), 86.7 (s, C≡CPh). MS (FAB+) m/z: 251 ([M+1]+, 100). Anal. Calcd. for C16H10O3: C, 76.79; H, 4.03. Found: C, 76.53; H, 4.24%. IR and NMR spectra can be seen in Figure S1.
1-Benzo[1,3]dioxol-5-yl-4-(tetrahydro-pyran-2-yloxy)-but-2-yn-1-one (4b). According to the general procedure described above, the reaction between the reaction mixture of 3b and MnO2 (0.72 g, 7.4 mmol) afforded 157 mg (overall yield = 37%) of 4b as a yellow oil. IR (NaCl): ν 1640 (m, C=O). 1H NMR (CDCl3): δ 7.82 (dd, 1Harom, J = 7.4 Hz, J′ = 1.8 Hz), 7.55 (d, 1Harom, J = 1.6 Hz), 6.89 (d, 1Harom, J = 8.2 Hz), 6.08 (s, 2H, OCH2O), 4.9–4.8 (m, 1H, CH), 4.56 (s, 2H, CH2OThp), 3.9–3.8 (m, 1H, CH2), 3.6–3.5 (m, 1H, CH2), 1.9–1.6 (m, 6H, 3CH2). 13C NMR (CDCl3): δ 175.6 (s, C=O), 152.9, 148.1 (2s, 2Carom), 131.5 (s, Carom), 127.4 (d, CHarom), 108.2, 107.9 (2d, 2CHarom), 102.1 (t, CH2), 97.3 (d, CH), 89.6 (s, C≡CCH2), 83.3 (s, C≡CCH2), 62.0, 54.0, 30.1, 25.2, 18.8 (5t, 5CH2). MS (FAB+) m/z: 289 ([M+1]+, 100). Anal. Calcd. for C16H16O5: C, 66.66; H, 5.59. Found: 66.94; H, 5.76%. IR and NMR spectra can be seen in Figure S2.
Synthesis of triazolo[1,5-a]pyrimidines 6a-b. General procedure.
A mixture of 3-amino-5-benzylsulfanyl-1,2,4-triazole 5, DBU and anhydrous MgSO4 (5 g/g) in dry DMF (3 mL/mmol) was heated at 40 °C under Ar for 30 min. Then, a solution of the corresponding α-acetylenic ketone 4a-b in dry DMF (3 mL/mmol) was slowly added using a syringe pump over a period of 5h. The reaction mixture, under Ar, was stirred at 40 °C until total consumption of 5 (1–2 h., monitored by TLC). The reaction was filtered and the organic solvent was eliminated under reduced pressure. The residue was dissolved with CH2Cl2 (15 mL/mmol) and was washed with saturated solution of NH4Cl (3 × 3 mL/mmol). The organic layer was dried over MgSO4, the solvent was evaporated and the resulting residue was purified by flash-chromatography (n-hexane:AcOEt).
5-Benzo[1,3]dioxol-5-yl-2-benzylsulfanyl-7-phenyl-1,2,4-triazolo[1,5-a]pyrimidine (6a). According to the general procedure described above, reaction between 5 (106 mg, 0.52 mmol), 4a (303 mg, 1.21 mmol, 2.5 equiv), DBU (81 μL, 0.53 mmol, 1.1 equiv) and anhydrous MgSO4 (0.5g) afforded 166 mg (73%) of 6a as a white solid. m.p.: 143–144 °C. 1H NMR (DMSO-d6): δ 8.3–7.1 (m, 14Harom), 6.21 (s, 2H, OCH2O), 4.59 (s, 2H, PhCH2S). 13C NMR (DMSO-d6): δ 166.4, 159.4, 158.1, 150.1, 148.1, 146.1, 137.7 (7s, 8Carom), 131.5 (d, CHarom), 130.1 (s, Carom), 129.7, 128.9, 128.4, 128.3, 127.2, 123.0, 108.5, 107.3, 105.6 (9d, 13CHarom), 101.8, 34.4 (2t, 2CH2). MS (FAB+) m/z: 439 ([M+1]+, 100). Anal. Calcd. for C25H18N4O2S: C, 68.48; H, 4.14; N, 12.78; S, 7.31. Found: C, 68.29; H, 4.27; N, 13.01; S, 7.02%. IR and NMR spectra can be seen in Figure S3.
5-Benzo[1,3]dioxol-5-yl-2-benzylsulfanyl-7-(tetrahydro-pyran-2-yloxymethyl)-1,2,4-triazolo[1,5-a]pyrimidine (6b). According to the general procedure described above, the reaction between 5 (255 mg, 1.24 mmol), 4b (698 mg, 2.42 mmol, 2 equiv), DBU (12 μL, 0.082 mmol, 5% mol.) and anhydrous MgSO4 (1.25 g,) afforded 225 mg (38%) of 6b as a yellow solid. m.p.: 62–64 °C. 1H NMR (CDCl3): δ 7.8–6.9 (m, 9Harom), 6.09 (s, 2H, OCH2O), 5.24 (d, 1H, J = 17 Hz, CH2OThp), 5.01 (d, 1H, J = 17 Hz, CH2OThp), 4.9 (m, 1H, CH), 4.60 (s, 2H, PhCH2S), 4.0–3.9 (m, 1H, CH2), 3.7–3.6 (m, 1H, CH2), 1.9–1.7 (m, 6H, 3CH2). 13C NMR (CDCl3): δ 168.3, 160.1, 155.8, 150.4, 148.5, 146.5, 137.2, 130.7 (8s, 8Carom), 129.1, 128.5, 127.4, 122.7, 108.4, 107.8, 102.7 (7d, 9CHarom), 101.7 (t, CH2), 99.14 (d, CH), 62.6, 62.5, 35.6, 30.2, 25.1, 19.3 (6t, 6CH2). MS (FAB+) m/z: 477 ([M+1]+, 100). Anal. Calcd. for C25H24N4O4S: C, 63.01; H, 5.08; N, 11.76; S, 6.73. Found: C, 63.22; H, 4.84; N, 11.92; S, 6.50%.
Synthesis of (5-benzo[1,3]dioxol-5-yl-2-benzylsulfanyl-1,2,4-triazolo[1,5-a]pyrimidin-7-yl)-methanol (7). A solution of 5-benzo[1,3]dioxol-5-yl-2-benzylsulfanyl-7-(tetrahydro-pyran-2-yloxymethyl)-1,2,4-triazolo[1,5-a]pyrimidine 6b (99 mg, 0.21 mmol) in (AcOH:THF:H2O) (4:2:1) (4 mL/mmol, 1 mL) was heated at 45 °C for 19 h. The organic solvent was eliminated under reduced pressure, was added saturated solution of NaHCO3 (3 mL/mmol) and the aqueous layer was extracted with AcOEt (3×3 mL/mmol). The combined organic layers were dried over MgSO4, the solvent was evaporated and the resulting residue purified by flash-chromatography (n-hexane:AcOEt) afforded 64 mg (77%) of 7 as a yellow solid. m.p.: 175–177 °C. IR (KBr): ν 3199 (br., OH). 1H NMR (DMSO-d6): δ 7.9–7.2 (m, 9Harom), 6.25 (s, 2H, OCH2O), 6.17 (t, 1H, J = 5.5 Hz, CH2OH), 5.05 (d, 2H, J = 5.5 Hz, CH2OH), 4.63 (s, 2H, PhCH2S). 13C NMR (DMSO-d6): δ 166.6, 159.3, 155.2, 150.5, 150.1, 148.3, 137.7, 130.2 (8s, 8Carom), 128.9, 128,4, 127.2, 122.7, 108.7, 107.0, 102.5 (7d, 9CHarom), 101.9, 57.7, 34.4 (3t, 3CH2). MS (FAB+) m/z: 393 ([M+1]+, 100). Anal. Calcd. for C20H16N4O3S: C, 61.21; H, 4.11; N, 14.28; S, 8.17. Found: C, 60.94; H, 4.36; N, 14.09; S, 8.39%.
Method A for the preparation of the alkoxypyrimidines 9a and 9e. Mitsunobu reaction.
A solution of DIAD (1.2 equiv) in dry THF (1 mL/mmol) is added dropwise to a solution of Ph3P (1.2 equiv), the appropriate 2-benzylsulfanylpyrimidinone 8a-b (1 equiv) and different alcohols in tetrahydrofuran (2 mL/mmol) at room temperature. The reaction mixture was stirred at room temperature until total disappearance of 8a-b (TLC monitoring). The solvent was evaporated until dryness and the crude product adsorbed over silica purified by flash chromatography (n-hexane:EtOAc).
2-Benzylsulfanyl-4-isopropoxy-6-phenyl-pyrimidine (9a). According to the general procedure described above, the reaction between 8b (500 mg, 1.70 mmol), TPP (675 mg, 2.55 mmol), and DIAD (0.50 mL, 2.55 mmol) in dry THF (6 mL), afforded 526 mg (92%) of 9a isolated as colorless solid after 2 h. m.p.: 81–82 °C. 1H NMR (CDCl3): δ 8.1–8.0 (s, 2Harom), 7.7–7.2 (m, 8Harom), 6.77 (s, 1H, Hpyrim), 5.46 (hept, 1H, J = 6.2 Hz, CH(CH3)2), 4.54 (s, 2H, PhCH2S), 1.38 (d, 6H, J = 6.2 Hz, CH(CH3)2). 13C NMR (CDCl3): δ 170.8, 169.4, 164.6 (3s, 3Cpyrim), 138.0, 136.8 (2s, 2Carom), 130.5, 128.8, 128.7, 128.5, 128.4, 127.0 (6d, 10CHarom), 99.7 (d, CHpyrim), 69.5 (d, CH), 35.4 (t, CH2), 21.9 (q, 2CH3). MS (EI) m/z: 336 ([M]·+, 90). Anal. Calcd. for C20H20N2OS: C, 71.40; H, 5.99; N, 8.33; S, 9.53. Found: C, 71.51; H, 6.17; N, 8.14; S, 9.25%.
[1-Benzyl-2-(2-benzylsulfanyl-pyrimidin-4-yloxy)-ethyl]-carbamic acid tert-butyl ester (9e). According to the general procedure described above, the reaction between 8a (1.00 g, 4.59 mmol), TPP (1.58 g, 5.96 mmol), N-Boc-phenylalaninol (1.50 g, 5.96 mmol) and DIAD (1.15 mL, 5.96 mmol) in dry THF (15 mL), afforded 1.47 g (71%) of 9e isolated as colorless solid after 5 h. m.p.: 108–110 °C. IR (KBr): ν 3388 (br., NH), 1685 (s, C=O). 1H NMR (CDCl3): δ 8.30 (d, 1H, J = 4.8 Hz, Hpyrim), 7.5–7.4 (m, 10Harom), 6.48 (d, 1H, J = 4.8 Hz, Hpyrim), 4.81 (br., 1H, NH), 4.37 (s, 2H, CH2), 4.30 (s, 2H, CH2), 4.25 (br., 1H, CH), 2.90 (m, 2H, PhCH2C), 1.39 (s, 9H, (CH3)3). 13C NMR (CDCl3): δ 171.3, 168.4 (2s, 2Cpyrim), 157.5 (d, CHpyrim), 152.2 (s, C=O), 137.8, 137.3 (2s, 2Carom), 129.3, 128.8, 128.5, 127.1, 126.6, 126.4 (6d, 10CHarom), 103.7 (d, CHpyrim), 79.6 (s, C), 64.1 (t, CH2), 50.8 (d, CH), 37.8, 35.2 (2t, 2CH2), 28.3 (q, 3CH3). MS (FAB+) m/z: 452 ([M+1]+, 17). Anal. Calcd. for C25H29N3O3S: C, 66.49; H, 6.47; N, 9.31; S, 7.10. Found: C, 66.30; H, 6.68; N, 9.07; S, 6.08%.
Method B for the preparation of the alkoxypyrimidines 9b-d.
To a solution of the corresponding 2-benzylsulfanylpyrimidinone 8a-b (1 equiv) in DMF (3 mL per mmol), 1.1 equiv of TMG were added. Then, the corresponding phenacyl bromide (1.1. equiv) was added dropwise. The reaction mixture was until total disappearance of 8a-b (TLC monitoring). The solvent was evaporated until dryness and the crude product adsorbed over silica purified by flash chromatography (n-hexane:EtOAc).
2-(2-Benzylsulfanyl-pyrimidin-4-yloxy)-1-(4-chloro-phenyl)-ethanone (9b). According to the general procedure described above, the reaction between 8a (600 mg, 2.8 mmol), (4-chloro-phenyl)-acetyl bromide (850 mg, 3.6 mmol) and TMG (0.45 mL, 3.6 mmol) in dry DMF (8 mL), afforded 856 mg (83%) of 9b isolated as colorless solid after 6 h. m.p.: 111–112 °C. IR (KBr): ν 1698 (s, C=O). 1H NMR (CDCl3): δ 8.34 (d, 1H, J = 5.6 Hz, Hpyrim), 7.9–7.3 (m, 9Harom), 6.64 (d, 1H, J = 5.6 Hz, Hpyrim), 5.54 (s, 2H, CH2O), 4.25 (s, 2H, PhCH2S). 13C NMR (CDCl3): δ 191.7 (s, C=O), 171.0, 167.6 (2s, 2Cpyrim), 157.8 (d, CHpyrim), 140.3, 137.0, 132.5 (3s, 3Carom), 129.2, 128.5, 128.4, 127.1, (4d, 9CHarom), 103.8 (d, CHpyrim), 67.4, 35.1 (2t, 2CH2). MS (EI) m/z: 372 ([M+2]·+, 19), 370 ([M]·+, 52). Anal. Calcd. for C19H15ClN2O2S: C, 61.53; H, 4.08; N, 7.55; S, 8.65. Found: C, 61.32; H, 3.98; N, 7.76; S, 8.46%.
2-(2-Benzylsulfanyl-pyrimidin-4-yloxy)-1-(3-nitro-phenyl)-ethanone (9c). According to the general procedure described above, the reaction between 8a (1.00 g, 4.6 mmol), (3-nitro-phenyl)-acetyl bromide (1.64 g, 5.7 mmol) and TMG (0.72 mL, 5.7 mmol) in dry DMF (15 mL), afforded 1.31 g (75%) of 9c isolated as colorless solid after 6 h. m.p.: 97–98 °C. IR (KBr): ν 1705 (s, C=O). 1H NMR (CDCl3): δ 8.74 (s, 1Harom), 8.46 (d, 1Harom, J = 8.2 Hz), 8.35 (d, 1H, J = 5.6 Hz, Hpyrim), 8.24 (d, 1Harom, J = 7.8 Hz), 7.71 (t, 1Harom), 7.3–7.2 (m, 5Harom), 6.65 (d, 1H, J = 5.6 Hz, Hpyrim), 5.60 (s, 2H, CH2O), 4.26 (s, 2H, PhCH2S). 13C NMR (CDCl3): δ 191.1 (s, C=O), 171.1, 167.4 (2s, 2Cpyrim), 158.0 (d, CHpyrim), 148.4, 137.0, 135.4 (3s, 3Carom), 133.3, 130.2, 128.5, 128.3, 128.0, 127.1, 122.7 (7d, 9CHarom), 103.8 (d, CHpyrim), 67.5, 35.0 (2t, 2CH2). MS (EI) m/z: 381 ([M]·+, 18). Anal. Calcd. for C19H15N3O4S: C, 59.83; H, 3.96; N, 11.02; S, 8.41. Found: C, 59.57; H, 4.11; N, 11.16; S, 8.14%.
2-(2-Benzylsulfanyl-6-phenyl-pyrimidin-4-yloxy)-1-phenyl-ethanone (9d). According to the general procedure described above, the reaction between 8b (500 mg, 1.7 mmol), phenyl-acetyl bromide (415 mg, 2.0 mmol) and TMG (0.26 mL, 2.0 mmol) in dry DMF (5 mL), afforded 512 mg (73%) of 9d isolated as colorless solid after 4 h. m.p.: 134–135 °C. IR (KBr): 1706 (m, C=O). 1H NMR (CDCl3): δ 8.1–8.0 (m, 4Harom), 7.6–7.5 (m, 6Harom), 7.4–7.1 (m, 5Harom), 7.05 (s, 1H, Hpyrim), 5.67 (s, 2H, OCH2), 4.39 (s, 2H, PhCH2S). 13C NMR (CDCl3): δ 192.9 (s, C=O), 170.7, 168.9, 165.2 (3s, 3Cpirim), 137.5, 136.4, 134.3 (3s, 3Carom), 133.8, 130.7, 128.8, 128.7, 128.6, 128.4, 127.7, 127.1, 127.0 (9d, 15CHarom), 99.1 (d, CHpyrim), 67.7, 35.2 (2t, 2CH2). MS (EI) m/z: 412 ([M]+, 25). Anal. Calcd. for C25H20N2O2S: C, 72.79; H, 4.89; N, 6.79; S, 7.77. Found: C, 72.57; H, 5.00; N, 6.56; S, 8.09%.
Oxidation of 9a,b to Sulfones 10a,b. General Procedure.
To a cooled (0 °C) solution of pyrimidine derivatives 9a,b (1 equiv) in CH2Cl2 (5 mL per mmol), 2.5 equiv of m-CPBA (60% purity) were added in small portions. The mixture was then stirred at 0 °C during 2 h, then diluted with CH2Cl2 (20 mL per mmol) and washed with aq. satd. NaHCO3 solution (2×5 mL per mmol) and brine (5 mL per mmol). The separated organic layer was dried (MgSO4), filtered and evaporated to give a residue which was purified by flash chromatography using n-hexane/EtOAc.
4-Isopropoxy-6-phenyl-2-phenylmethanesulfonyl-pyrimidine (10a). According to the general procedure described above, the reaction of 9a (450 mg, 1.34 mmol) and m-CPBA (1.15 g, 3.34 mmol) in CH2Cl2 (7 mL), afforded to 10a (432 mg, 88%) isolated as a colorless solid. m.p.: 124–125 °C. 1H NMR (CDCl3): δ 8.1–8.0 (m, 2Harom), 7.6–7.2 (m, 8Harom), 6.99 (s, 1H, Hpyrim), 5.65 (hept, 1H, J = 6.0 Hz, CH(CH3)2), 4.88 (s, 2H, PhCH2S), 1.43 (d, 6H, J = 6.2 Hz, CH(CH3)2). 13C NMR (CDCl3): δ 170.9, 165.5, 164.7 (3s, 3Cpyrim), 135.0 (s, Carom), 131.6, 131.3, 128.8, 128.7, 128.2, 127.2 (6d, 10CHarom), 127.1 (s, Carom), 106.0 (d, CHpyrim), 71.6 (d, CH), 57.3 (t, CH2), 21.7 (q, 2CH3). MS (EI) m/z: 368 ([M]·+, 67). Anal. Calcd. for C20H20N2O3S: C, 65.20; H, 5.47; N, 7.60; S, 8.70. Found: C, 65.29; H, 5.60; N, 7.48; S, 8.88%.
1-(4-Chloro-phenyl)-2-(2-phenylmethanesulfonyl-pyrimidin-4-yloxy)-ethanone (10b). According to the general procedure described above, the reaction of 9b (675 mg, 1.82 mmol) and m-CPBA (1.31 g, 4.55 mmol) in CH2Cl2 (9 mL), afforded to 10b (613 mg, 84%) isolated as a colorless solid. m.p.: 141–142 °C. IR (KBr): ν 1692 (s, C=O). 1H NMR (CDCl3): δ 8.65 (d, 1H, J = 5.8 Hz, Hpyrim), 7.92 (d, 2Harom, J = 6.8 Hz), 7.52 (d, 2Harom, J = 6.8 Hz), 7.3–7.2 (m, 5Harom), 7.12 (d, 1H, J = 5.6 Hz, Hpyrim), 5.72 (s, 2H, CH2O), 4.54 (s, 2H, PhCH2S). 13C NMR (CDCl3): δ 190.9 (s, C=O), 169.3, 163.9 (2s, 2Cpyrim), 158.3 (d, CHpyrim), 140.8, 132.2 (2s, 2Carom), 131.0, 129.4, 129.2, 128.8, 128.7 (5d, 9CHarom), 126.4 (s, Carom), 111.4 (d, CHpyrim), 68.3, 57.6 (2t, 2CH2). MS (FAB+) m/z: 405 ([M+3]+, 24), 403 ([M+1]+, 62). Anal. Calcd. for C19H15ClN2O4S: C, 56.65; H, 3.75; N, 6.95; S, 7.96. Found: C, 56.81; H, 3.54; N, 7.18; S, 8.15%.
Ipso-substitution reaction of pyrimidinyl sulfone derivatives 10a,b.
Synthesis of 4-isopropoxy-2-phenoxy-6-phenyl-pyrimidine (11a). To a solution of phenol (55 mg, 0.57 mmol) in dioxane (2 mL), the Cs2CO3 (210 mg, 0.59 mmol) was added. The reaction mixture was stirred at r.t. for 15–20 min. Then, the sulfone 10a (200 mg, 0.54 mmol) was added. After stirring at 60 °C for 3 hours, the solvent was removed in vacuo and the mixture was acidified with 2 N hydrochloric acid and extracted with ethyl acetate. The residue was purified using flash chromatography to give 11a (131 mg, 80%) as a colorless solid. m.p.: 68–69 °C. 1H NMR (CDCl3): δ 8.0–7.9 (m, 2Harom), 7.5–7.3 (m, 8Harom), 6.84 (s, 1H, Hpyrim), 5.28 (hept, 1H, J = 6.2 Hz, CH(CH3)2), 1.34 (d, 6H, J = 6.2 Hz, CH(CH3)2). 13C NMR (CDCl3): δ 171.9, 166.4, 165.0 (3s, 3Cpyrim), 153.2, 136.5 (2s, 2Carom), 130.6, 129.1, 128.7, 127.0, 124.8, 121.9 (6d, 10CHarom), 98.6 (d, CHpyrim), 69.9 (d, CH), 21.8 (q, 2CH3). MS (EI) m/z: 306 ([M]·+, 24). Anal. Calcd. for C19H18N2O2: C, 74.49; H, 5.92; N, 9.14. Found: C, 74.36; H, 5.75; N, 9.40%.
Synthesis of 1-(4-chloro-phenyl)-2-[2-(2,4-dimethoxy-phenylamino)-pyrimidin-4-yloxy]-ethanone (11b). To a solution of the sulfone 10b (125 mg, 0.31 mmol) in dioxane (2 mL), the 2,4-dimethoxyaniline (100 mg, 0.62 mmol) was added. The reaction mixture with good stirring was heated at 100 °C until total consumption of the starting material (30 h., TLC monitoring). The solvent was removed under reduced pressure and the residue purified by flash-chromatography (n-hexane:EtOAc) to give 11b (25 mg, 20%) as an orange crystalline solid. m.p.: 122–123 °C. IR (KBr): ν 3244 (br., NH), 1705 (m, C=O). 1H NMR (CDCl3): δ 8.20 (d, 1H, J = 5.4 Hz, Hpyrim), 7.96 (d, 2Harom, J = 8.2 Hz), 7.77 (d, 1Harom, J = 8.8 Hz), 7.54 (d, 2Harom, J = 8.2 Hz), 7.30 (s, 1Harom), 6.44 (d, 1Harom, J = 2.4 Hz), 6.36 (d, 1H, J = 5.2 Hz, Hpyrim), 5.90 (d, 1H, J = 8.2 Hz, NH), 5.58 (s, 2H, CH2O), 3.83 (s, 3H, CH3O), 3.72 (s, 3H, CH3O). 13C NMR (CDCl3): δ 192.2 (s, C=O), 168.5, 159.6 (2s, 2Cpyrim), 158.6 (d, CHpyrim), 155.4, 149.7, 140.2, 132.9 (4s, 4Carom), 129.3, 129.2 (2d, 4CHarom), 121.9 (s, Carom.), 120.3, 102.7 (2d, 2CHarom), 98.6 (d, CHpyrim), 98.5 (d, CHarom), 67.3 (t, CH2), 55.6, 55.4 (2q, 2CH3). MS (EI) m/z: 401 ([M+2]·+, 17), 399 ([M]·+, 52). Anal. Calcd. for C20H18ClN3O4: C, 60.08; H, 4.54; N, 10.51. Found: C, 60.29; H, 4.48; N, 10.32%.
Removal of the 4-isopropoxy group. Synthesis of 2-phenoxy-6-phenyl-3H-pyrimidin-4-one (12). The 4-isopropoxypyrimidine 11a (87 mg, 0.28 mmol) was added to a mixture of AcOH (0.6 mL) and con. H2SO4 (0.6 mL). The reaction mixture was stirred at 90 °C for 15 min. After cooling, the mixture was neutralized with aq. 5 N NaOH and extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine and the separated organic layer was dried (MgSO4), filtered and eliminated under reduced pressure to afford the pure pyrimidinone 12 (59 mg, 80%) as a colorless solid. m.p.: 263–264 °C. IR (KBr): ν 3060–2750 (br., NH), 1669 (s, C=O). 1H NMR (DMSO-d6): δ 12.75 (br., NH), 7.9–7.8 (m, 2Harom), 7.6–7.5 (m, 8Harom), 6.74 (s, 1H, Hpyrim). 13C NMR (DMSO-d6): δ 166.0, 161.4, 158.7 (3s, 3Cpyrim), 151.8, 135.8 (2s, 2Carom), 130.6, 129.5, 128.8, 126.6, 125.7, 121.7 (6d, 10CHarom), 102.4 (d, CHpyrim). MS (EI) m/z: 264 ([M]·+, 54). Anal. Calcd. for C16H12N2O2: C, 72.72; H, 4.58; N, 10.70. Found: C, 72.47; H, 4.81; N, 10.86%.
Synthesis of [1-benzyl-2-(2-benzylsulfanyl-pyrimidin-4-yloxy)-ethylamino]-phenyl-acetic acid (13): Petasis reaction.
To a stirred solution of glyoxylic acid monohydrate (74 mg, 0.78 mmol) in dichloromethane (5 mL) was added the primary amine 9e, previous deprotection of Boc using standard conditions, (275 mg, 0.78 mmol), followed by phenylboronic acid (99 mg, 0.78 mmol). After the flask was purged with nitrogen and sealed, the reaction mixture was stirred vigorously at room temperature for 3 days. The resulting precipitate was isolated by filtration and washed with to dichloromethane to give the pure pyrimidinone 13 (197 mg, 52%) as a colorless solid. m.p.: 134–135 °C. IR (KBr): ν 3217 (br, NH), 1713 (s, C=O). 1H NMR (DMSO-d6): δ 8.42 (d, 1H, J = 5.8 Hz, Hpyrim), 7.9–7.8 (m, 2H, NH + COOH), 7.5–7.2 (m, 15Harom), 6.69 (d, 1H, J = 5.8 Hz, Hpyrim), 4.63 (s, 1H, CHCOOH), 4.34 (s, 2H, PhCH2S), 4.31 (dd, 1H, J = 4.0 Hz, J′ = 11.0 Hz, OCH2), 4.17 (dd, 1H, J = 6.0 Hz, J′ = 11.0 Hz, OCH2), 3.15 (br., 1H, PhCH2CH), 3.02 (dd, 1H, J = 4.8 Hz, J′ = 13.6 Hz, PhCH2CH), 2.85 (dd, 1H, J = 7.5 Hz, J′ = 13.6 Hz, PhCH2CH). 13C NMR (DMSO-d6): δ 172.6, 170.0, 168.0 (3s, 3C), 158.0 (d, CHpyrim), 137.8, 137.7 (2s, 3Carom), 134.1, 130.0, 129.2, 128.8, 128.4, 127.8, 127.3, 127.0, 126.4 (9d, 15CHarom), 104.1 (d, CHpyrim), 66.9 (t, CH2), 62.4, 55.5 (2d, 2CH), 36.4, 34.2 (2t, 2CH2). MS (FAB+) m/z: 486 ([M+1]·+, 28). Anal. Calcd. for C28H27N3O3S: C, 69.25; H, 5.60; N, 8.65; S, 6.60. Found: C, 69.52; H, 5.84; N, 8.48; S, 6.31%.
Synthesis of 2-(2-benzylsulfanyl-6-phenyl-pyrimidin-4-yloxy)-1-phenyl-ethanol (14). To a stirred and cooled (0 °C) solution of pyrimidinone 9d (500 mg, 1.21 mmols) in MeOH (6 mL) was added NaBH4 (165 mg, 4.24 mmols) in small portions while stirring (vigorous evolution of gas observed). Stirring was continued for 2 h at 0 °C. The solution was evaporated to dryness, and the crude residue was partitioned between EtOAc (10 mL) and aq satd NH4Cl solution (15 mL). The organic layer was separated, washed with H2O (5 mL), dried (MgSO4), and evaporated to give a residue which was purified by flash chromatography using hexanes/EtOAc to afford pure (14) as a colorless solid (403 mg, 80%). m.p.: 140–141 °C. IR (KBr): 3400 (br., OH). 1H NMR (CDCl3): δ 8.1–8.0 (m, 2Harom), 7.5–7.3 (m, 13Harom), 6.90 (s, 1H, Hpyrim), 5.17 (dd, 1H, J = 8.4 Hz, J′ = 3.2 Hz, PhCH), 4.53 (s, 2H, PhCH2S), 4.47 (dd, 1H, J = 11.6 Hz, J′ = 8.4 Hz, CH2O), 4.03 (dd, 1H, J = 11.4 Hz, J′ = 3.2 Hz, CH2O), 2.90 (br., 1H, OH). 13C NMR (CDCl3): δ 170.9, 169.6, 165.1 (3s, 3Cpyrim), 139.8, 137.7, 136.4 (3s, 3Carom), 130.8, 128.8, 128.7, 128.6, 128.5, 128.4, 128.2, 127.1, 126.2 (9d, 15CHarom), 99.1 (d, CHpyrim), 72.5 (d, CH), 71.7, 35.4 (2t, 2CH2). MS (EI) m/z: 414 ([M]+, 33). Anal. Calcd. for C25H22N2O2S: C, 72.44; H, 5.35; N, 6.76; S, 7.74. Found: C, 72.62; H, 5.14; N, 6.52; S, 7.46%.
3.4. Microbiological Methods
Thirty-two isolates of M. tuberculosis from respiratory (n = 21) and non-respiratory (n = 11) clinical samples, identified by conventional methods [109] and by DNA hybridization probe (Accuprobe® Gen Probe Inc., San Diego, CA, USA) were selected from the laboratory collection of Dept. Microbiología, Hospital Clínico Universitario. Valencia, Spain. Susceptibility to first-line antituberculosis drugs (ethambutol, isoniazid, rifampin and streptomycin) was tested by a fluorometric method (Bactec® MGIT 960, Becton-Dickinson, Franklin Lakes, NJ, USA) and by a microdilution method [109].
The organisms were grown in modified Middlebrook 7H9 broth supplemented with 10% OADC enrichment (Difco Laboratories, Franklin Lakes, NJ, USA) for seven days at 37 °C. The inoculum size was obtained by dilution of M. tuberculosis isolates suspensions in 7H9 broth to yield an absorbance equivalent to that of a MacFarland nº 0.5 standard.
Antimicrobial susceptibility test was performed in 96-well microplates using serial twofold microdilution in 7H9 broth. Initial drug dilutions were prepared in deionized water or, if not soluble, dimethyl sulfoxide. Subsequent twofold dilutions were performed in 150 μL of modified 7H9 broth in the microplates to provide a final test range of 128 to 0.125 mg/L. Ten μL of a suspension of mycobacteria were added to the wells. Plates were covered with Parafilm “M”® (Laboratory Film, American National Can™, Chicago, IL, USA), and incubated for 12 days at 37 °C. Starting at day 13 of incubation, 20 μL of Resazurina® (Sigma 2127, St. Louis, MO, USA) with a concentration of 250 mg/L were added to the wells, and the microplates were reincubated at 37 °C for an additional period of 48 h. MIC50 and MIC90 were determined as the lowest concentrations of the compounds yielding no visible changes from blue to pink [110].
4. Conclusions
New chemical scaffolds have been identified that could render new lead drugs in this field, by using easy to calculate descriptors, such as structural invariants. The only substructure common to all the selected molecules is the pyrimidine ring, and the most frequent substituent is the benzylsulfanyl in position 2. The 1,2,4-triazolo[1,5-a]pyrimidine system is present in structures 7 and 6a, which have the same substituted groups in 2 and 5. Only one pyrimidone was selected (12). All these structures are proposed as new base structures in order to design new combinatorial synthesis projects.
Although several action mechanisms are represented in the training group, the usefulness of the method is appreciable. One possible explanation of this fact could be that the equation retains the structural features involved in all the mechanisms considered. In the opinion of the authors, this approach is not able to find new action mechanisms, but it is possible to obtain new unexpected molecular structures acting through the known mechanisms which combine the properties of the known compounds. This feature could be useful in order to avoid problems of resistance.
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms232315057/s1.
Author Contributions
Conceptualization and methodology, Á.G.-G., J.V.d.J.-O., J.G. and J.M.V.; molecular design, Á.G.-G., J.V.d.J.-O. and J.G.; organic synthesis, D.F., C.A. and J.M.V.; microbiological assays, Á.G.-G., M.d.R.G.S., C.M.-C. and R.B.; original draft preparation, Á.G.-G., J.V.d.J.-O., D.F., C.A., J.M.V. and M.d.R.G.S.; inal version, all authors. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available upon request to the authors.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research received no external funding.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Tuberculosis. [(accessed on 25 June 2022)]. Available online: https://www.who.int/westernpacific/health-topics/tuberculosis.
- 2.Dhote A., Kawishwar V. Utility of Concentration Bleach Method and Fluorescent Auramine Rhodamine Staining in Diagnosis of Mycobacterium tuberculosis on Formalin Fixed Tissue. New Horiz. Med. Med. Res. 2022;4:67–70. [Google Scholar]
- 3.Besalú E., Ponec R., de Julián-Ortiz J.V. Virtual generation of agents against Mycobacterium tuberculosis. A QSAR study. Mol. Divers. 2003;6:107–120. doi: 10.1023/B:MODI.0000006839.52374.d7. [DOI] [PubMed] [Google Scholar]
- 4.Acharya B., Acharya A., Gautam S., Ghimire S.P., Mishra G., Parajuli N., Sapkota B. Advances in Diagnosis of Tuberculosis: An Update into Molecular Diagnosis of Mycobacterium Tuberculosis. Molec. Biol. Rep. 2020;47:4065–4075. doi: 10.1007/s11033-020-05413-7. [DOI] [PubMed] [Google Scholar]
- 5.Hoffner S.E., Gezelius L., Olsson-Liljequist B. In vitro activity of fluorinated quinolones and macrolides against drug-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 1997;40:885–888. doi: 10.1093/jac/40.6.885. [DOI] [PubMed] [Google Scholar]
- 6.Tomioka H. Prospects for development of new antituberculous drug. Kekkaku. 2002;77:573–584. [PubMed] [Google Scholar]
- 7.Koketsu M., Tanaka K., Takenaka Y., Kwong C.D., Ishihara H. Synthesis of 1,3-thiazine derivates and their evaluation as potential antimicobacterial agents. Eur. J. Pharm. Sci. 2002;15:307–310. doi: 10.1016/S0928-0987(02)00014-3. [DOI] [PubMed] [Google Scholar]
- 8.Fernandes G.F.D.S., Jornada D.H., de Souza P.C., Chin C.M., Pavan F.R., Santos J.L.D. Current Advances in Antitubercular Drug Discovery: Potent Prototypes and New Targets. Curr. Med. Chem. 2015;22:3133–3161. doi: 10.2174/0929867322666150818103836. [DOI] [PubMed] [Google Scholar]
- 9.Fernandes G.F., Thompson A.M., Castagnolo D., Denny W.A., Dos Santos J.L. Tuberculosis Drug Discovery: Challenges and New Horizons. J. Med. Chem. 2022;65:7489–7531. doi: 10.1021/acs.jmedchem.2c00227. [DOI] [PubMed] [Google Scholar]
- 10.Obrecht D., Weiss B. A new method for the preparation of (E)-3-acylprop-2-enoics acids. Helv. Chim. Acta. 1989;72:117–122. doi: 10.1002/hlca.19890720115. [DOI] [Google Scholar]
- 11.Utimoto K., Miwa H., Nozaki H. Palladium-catalyzed synthesis of pyrroles. Tetrahedron Lett. 1981;22:4277–4278. doi: 10.1016/S0040-4039(01)82932-4. [DOI] [Google Scholar]
- 12.Obrecht D. Acid-catalyzed cyclization reactions of substituted acetylenic ketones: A new approach for the synthesis of 3-halofurans, flavones and styrylchromones. Helv. Chim. Acta. 1989;72:447–456. doi: 10.1002/hlca.19890720305. [DOI] [Google Scholar]
- 13.Masquelin T., Obrecht D. A facile preparation of 2- and 5-substituted 3-bromothiophenes. Tetrahedron Lett. 1994;35:9387–9390. doi: 10.1016/S0040-4039(00)78549-2. [DOI] [Google Scholar]
- 14.Garvey D.S., Wasicak J.T., Elliot R.L., Lebold S.A., Hettinger A.-M., Carrera G.M., Lin N.-H., He Y., Holladay M.W., Anderson D.J., et al. Ligands for brain cholinergic channel receptors: Synthesis and in vitro characterization of novel isoxazoles and isothiazoles as bioisosteric replacements for the pyridine ring in nicotine. J. Med. Chem. 1994;37:4455–4463. doi: 10.1021/jm00052a005. [DOI] [PubMed] [Google Scholar]
- 15.Masquelin T., Obrecht D. A new approach to the synthesis of N-protected 2- and 5-substituted 3-halopyrroles. Synthesis. 1995;1995:276–384. doi: 10.1055/s-1995-3893. [DOI] [Google Scholar]
- 16.Degl’Inocenti A., Scafato P., Capperucci A., Bartoletti L., Mordini A., Reginato G. Azide cyclocondensation with acetylenic silyl ketone: A general access to funtionalized-1,2,3-triazolylacylsilanes and –aldehydes. Tetrahedron Lett. 1995;36:9031–9034. [Google Scholar]
- 17.Masquelin T., Obrecht D. A novel access to 2,4-substituted quinolines from acetylenic ketones. Tetrahedron. 1997;53:641–646. doi: 10.1016/S0040-4020(96)01025-3. [DOI] [Google Scholar]
- 18.Falorni M., Giacomelli G., Spanedda A.M. Synthesis of chiral pyrazoles and isoxazoles as constrained amino acids. Tetrahedron Asymmetry. 1998;9:3039–3046. doi: 10.1016/S0957-4166(98)00305-X. [DOI] [Google Scholar]
- 19.Cabarrocas G., Rafel S., Ventura M., Villalgordo J.M. A new approach toward the stereoselective synthesis of novel quinolyl glycines: Synthesis of the enantiomerically pure quinolyl-β-amino alcohol precursors. Synlett. 2000;5:595–598. doi: 10.1002/chin.200034182. [DOI] [Google Scholar]
- 20.Cabarrocas G., Ventura M., Maestro M., Mahía J., Villalgordo J.M. Synthesis of Novel Optically Pure Quinolyl-β-Amino Alcohol Derivatives from 2-Amino Thiophenol and Chiral α-Acetylenic Ketones and Their IBX-Mediated Oxidative Cleavage to N-BOC Quinolyl Carboxamides. Tetrahedron Asymmetry. 2001;12:1851–1863. doi: 10.1016/S0957-4166(01)00308-1. [DOI] [Google Scholar]
- 21.Satheesha Rai N., Kalluraya B., Lingappa B., Shenoy S., Puranic V.G. Convenient Access to 1,3,4-Trisubstituted Pyrazoles Carrying 5-Nitrothiophene Moiety via 1,3-Dipolar Cycloaddition of Sydnones with Acetylenic Ketones and Their Antimicrobial Evaluation. Eur. J. Med. Chem. 2008;43:1715–1720. doi: 10.1016/j.ejmech.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Harigae R., Moriyama K., Togo H. Preparation of 3,5-Disubstituted Pyrazoles and Isoxazoles from Terminal Alkynes, Aldehydes, Hydrazines, and Hydroxylamine. J. Org. Chem. 2014;79:2049–2058. doi: 10.1021/jo4027116. [DOI] [PubMed] [Google Scholar]
- 23.Adlington R.M., Baldwin J.E., Catterick D., Pritchard G.J. The Synthesis of Pyrimidin-4-Yl Substituted α-Amino Acids. A Versatile Approach from Alkynyl Ketones. J. Chem. Soc. Perkin Trans. 1. 1999;8:855–866. doi: 10.1039/a806741d. [DOI] [Google Scholar]
- 24.Nájera C., Sydnes L.K., Yus M. Conjugated Ynones in Organic Synthesis. Chem. Rev. 2019;119:11110–11244. doi: 10.1021/acs.chemrev.9b00277. [DOI] [PubMed] [Google Scholar]
- 25.Chucholowski A., Masquelin T., Obrecht D., Stadlwieser J., Villalgordo J.M. Cheminform Abstract: Novel Solution- and Solid-Phase Strategies for the Parallel and Combinatorial Synthesis of Low-Molecular-Weight Compound Libraries. CHIMIA Int. J. Chem. 2010;28:525–530. doi: 10.1002/chin.199713273. [DOI] [Google Scholar]
- 26.Obrecht D., Abrecht C., Grieder A., Villalgordo J.M. A novel and efficient approach for the combinatorial synthesis of structurally diverse pyrimidines on solid support. Helv. Chim. Acta. 1997;80:65–72. doi: 10.1002/hlca.19970800106. [DOI] [Google Scholar]
- 27.Aparna E.P., Devaky K.S. Advances in the Solid-Phase Synthesis of Pyrimidine Derivatives. ACS Comb. Sci. 2019;21:35–68. doi: 10.1021/acscombsci.8b00172. [DOI] [PubMed] [Google Scholar]
- 28.Sonogashira K., Tohda Y., Hagihara N. Convenient synthesis of acetylenes. Catalytic substitutions of acetylenic hydrogen with bromo alkenes, iodo arenes and bromopyridines. Tetrahedron Lett. 1975;50:4467–4470. doi: 10.1016/S0040-4039(00)91094-3. [DOI] [Google Scholar]
- 29.Tohda Y., Sonogashira K., Hagihara N. A convenient synthesis of 1-alkynyl ketones and 2-alkynamides. Synthesis. 1977:777–778. doi: 10.1055/s-1977-24574. [DOI] [Google Scholar]
- 30.Corriu R.J.P., Bolin G., Iqbal J., Moreau J.J.E., Vernhet C. Aminosilanes in organic synthesis. Addition of organocopper reagents on γ-bis(trimethylsilyl)amino-α-acetylenic amides, esters and ketones. Stereochemistry and some synthetic uses. Tetrahedron. 1993;49:4603–4618. doi: 10.1016/S0040-4020(01)81289-8. [DOI] [Google Scholar]
- 31.Verkruijsse H.D., Heus-Kloos Y.A., Brandsma L. Efficient methods for the preparation of acetylenic ketones. J. Organomet. Chem. 1988;338:289–294. doi: 10.1016/0022-328X(88)80002-0. [DOI] [Google Scholar]
- 32.Larson D.P., Heathcock C.H. Total synthesis of tricolorin A. J. Org. Chem. 1997;62:8406–8418. doi: 10.1021/jo971413u. [DOI] [PubMed] [Google Scholar]
- 33.Palombi L., Arista L., Lattanzi A., Bonadies F., Scettri Q. Zeolite-catalized oxidation of benzylic and acetylenic alcohols with t-butyl hidroperoxide. Tetrahedron Lett. 1996;37:7849–7850. doi: 10.1016/0040-4039(96)01747-9. [DOI] [Google Scholar]
- 34.Serrat X., Cabarrocas G., Rafel S., Ventura M., Linden A., Villalgordo J.M. A highly efficient and straightforward stereoselective synthesis of novel chiral α-acetylenic ketones. Tetrahedron Asymmetry. 1999;10:3417–3430. doi: 10.1016/S0957-4166(99)00357-2. [DOI] [Google Scholar]
- 35.Frigerio M., Santagostino M., Sputore S., Palmisano G. Oxidation of alcohols with o-iodoxybenzoic acid in DMSO: A new insight into an old hypervalent iodine reagent. J. Org. Chem. 1995;60:7272–7276. doi: 10.1021/jo00127a036. [DOI] [Google Scholar]
- 36.El Ashry E.S.H., Rashed N. 1,2,4-triazolo- and tetrazolo[x,y-z]pyrimidines. Adv. Heterocycl. Chem. 1999;72:127–171. [Google Scholar]
- 37.El Ashry E.S.H., El Kilany Y., Rashed N. Assafir, H.R. Dimroth rearrangement: Translocation of heteroatoms in heterocyclic rings and its role in ring transformations of heterocycles. Adv. Heterocycl. Chem. 1999;75:79–167. [Google Scholar]
- 38.Pugmire R.J., Smith J.C., Grant D.M., Stanovnik B., Tisler M., Vercek B. Correlation of ring nitrogen substituents with carbon-13 nuclear magnetic resonance data in azoloazines. J. Heterocycl. Chem. 1987;24:805–809. doi: 10.1002/jhet.5570240351. [DOI] [Google Scholar]
- 39.Salas J.M., Romero M.A., Sánchez M.P., Quirós M. Metal complexes of 1,2,4-triazolo[1,5-a]pyrimidine derivatives. Coord. Chem. Rev. 1999;193:1119–1142. doi: 10.1016/S0010-8545(99)00004-1. [DOI] [Google Scholar]
- 40.Sato Y., Shimoji Y., Fujita H., Nishino H., Mizuno H., Kobayashi S., Kamakura S. Studies on cardiovascular agents. 6. Synthesis and coronary vasodilating and antihypertensive activities of 1,2,4-triazolo[1,5-a]pyrimidines fused to heterocyclic systems. J. Med. Chem. 1980;23:927–937. doi: 10.1021/jm00182a021. [DOI] [PubMed] [Google Scholar]
- 41.Tenor E., Ludwig R. Pharmaceutical-chemical research on s-triazolo[1,5-a]pyrimidines. Pharmazie. 1971;26:534–539. [PubMed] [Google Scholar]
- 42.Navarro J.A.R., Salas J.M., Romero M.A., Faure R. Influence of anions and crystallization conditions on the solid-state structure of some binuclear silver (I) complexes supported by triazolopyrimidine bridges. J. Chem. Soc. Dalton Trans. 1998;6:901–904. doi: 10.1039/a707789k. [DOI] [Google Scholar]
- 43.Velders A.H., Pazderski L., Ugozzoli F., Biagini-Cingi M., Manotti-Lanfredi A.M., Haasnot J.G., Reedijk J. Synthesis, characterization and crystal structure of trans-aquatrichlorobis(5,7-dimethyl-1,2,4-triazolo[1,5-a]pyrimidine-N3)ruthenium (III) monohydrate. Inorg. Chim. Acta. 1998;273:259–265. doi: 10.1016/S0020-1693(97)05969-0. [DOI] [Google Scholar]
- 44.Navarro J.A.R., Romero M.A., Salas J.M., Molina J., Tiekink E.R.T. Ternary copper (II) complexos with the versatile 4,7-dihydro-5-methyl-7-oxo-1,2,4-triazolo[1,5-a]pyrimidine ligand. Inorg. Chim. Acta. 1998;274:53–63. doi: 10.1016/S0020-1693(97)05988-4. [DOI] [Google Scholar]
- 45.Davies G.E. Anthibronchoconstrictor activity of two new phosphodiesterase inhibitors, a triazolopyrazine (ICI 58301) and a triazolopyrimidine (ICI 63197) J. Pharm. Pharmacol. 1973;25:681–689. doi: 10.1111/j.2042-7158.1973.tb10048.x. [DOI] [PubMed] [Google Scholar]
- 46.Fischer G. 1,2,4-Triazolo[1,5-a]pyrimidines. Adv. Heterocycl. Chem. 1993;57:81–138. [Google Scholar]
- 47.Gupton J.T., Petrich S.A., Hicks F.A., Wilkinson D.R., Vargas M., Hosein K.N., Sikorski J.A. The preparation of heterocyclic appended vinylogous iminium salts and their application to the regioselective preparation of biheterocyclic systems. Heterocycles. 1998;47:689–702. doi: 10.3987/COM-97-S(N)21. [DOI] [Google Scholar]
- 48.Tominaga Y., Sakai S., Kohra S., Tsuka J., Matsuda Y., Kobayashi G. Pyrimidine and fused pyrimidine derivatives. III. Synthesis of s-triazolo[1,5-a]pyrimidines derivatives by using ketene dithioacetals. Chem. Pharm. Bull. 1985;33:962–970. doi: 10.1248/cpb.33.962. [DOI] [Google Scholar]
- 49.Oukoloff K., Lucero B., Francisco K.R., Brunden K.R., Ballatore C. 1,2,4-Triazolo[1,5-a]Pyrimidines in Drug Design. Eur. J. Med. Chem. 2019;165:332–346. doi: 10.1016/j.ejmech.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Namgoong S.K., Lee H.J., Kim Y.S., Shin J.-H., Che J.-K., Jang D.Y., Kim G.S., Yoo J.K., Kang M.-K., Kil M.-W., et al. Synthesis of the quinoline-linked triazolopyrimidine analogues and their interections with the recombinant tobacco acetolactate synthase. Biochem. Biophys. Res. Commun. 1999;258:797–801. doi: 10.1006/bbrc.1999.0708. [DOI] [PubMed] [Google Scholar]
- 51.Johnson T.C., Mann R.K., Schmitzer P.R., Gast R.E. Bioactive Heterocyclic Compound Classes. Agrochemicals. 1st ed. Wiley-VCH Verlag & Co.; Weinheim, Germany: 2012. Acetohydroxyacid Synthase Inhibiting Triazolopyrimidine Herbicides; pp. 51–60. [Google Scholar]
- 52.Monte W.T., Kleschick W.A., Meikle R.W., Snider S.W., Bordner J. Methods for controlling the regioselection in the reaction of 3-amino-5-(benzylthiol-1,2,4-triazole with acetylacetaldehyde dimethyl acetal. J. Heterocycl. Chem. 1989;26:1393–1396. doi: 10.1002/jhet.5570260530. [DOI] [Google Scholar]
- 53.Kleschick W.A., Bordner J. Regioselection in the reaction of 3-amino-5-benzylthio-1,2,4-triazole with unsymmetrical 1,3-diketones. J. Heterocycl. Chem. 1989;26:1489–1493. doi: 10.1002/jhet.5570260545. [DOI] [Google Scholar]
- 54.Monte W.T., Kleschick W.A., Bordner J. Controlling substitution patterns on the 1,2,4-triazolo[1,5-a]pyrimidine ring. Selective removal of chlorine at the 7-position. J. Heterocycl. Chem. 1999;36:183–188. doi: 10.1002/jhet.5570360127. [DOI] [Google Scholar]
- 55.Shankar R.B., Pews R.G. Synthesis of 1,2,4-triazolo[1,5-a]pyrimidines-2-sulfonamides. J. Heterocycl. Chem. 1993;30:169–172. doi: 10.1002/jhet.5570300130. [DOI] [Google Scholar]
- 56.Okabe T., Bhooshan B., Novinson T., Hillyard I.W., Garner G.E., Robins R.K. Dialkyl bicyclic heterocycles with a bridgehead nitrogen as purine analogs possessing significant cardiac inotropic activity. J. Heterocycl. Chem. 1983;20:735–751. doi: 10.1002/jhet.5570200345. [DOI] [Google Scholar]
- 57.Williams L.A. Structure of certain polyazaindenes. VI. Structure of some products obtained from 3-amino-1,2,4-triazoles with acetylacetone and ethyl acetoacetate. J. Chem. Soc. 1960;1960:1829–1832. doi: 10.1039/jr9600001829. [DOI] [Google Scholar]
- 58.Novinson T., Springer R.H., O’Brien D.E., Scholten M.B., Miller J.P., Robins R.K. 2-(alkylthiol-1,2,4-triazolo[1,5-a]pyrimidines as adenosine 3′,5′-monophosphate phosphodiesterase inhibitors with potential as new cardiovascular agents. J. Med. Chem. 1982;25:420–426. doi: 10.1021/jm00346a017. [DOI] [PubMed] [Google Scholar]
- 59.Kidwai M., Chauhan R. Nafion-H® Catalyzed Efficient One-Pot Synthesis of Triazolo[5,1-b]Quinazolinones and Triazolo[1,5-a]Pyrimidines: A Green Strategy. J. Mol. Catal. A Chem. 2013;377:1–6. doi: 10.1016/j.molcata.2013.04.014. [DOI] [Google Scholar]
- 60.Petrick S.A., Qian Z., Santiago L.M., Grupton J.T. The application of unsymmetrical vinylogous iminium salts and related synthons to the preparation of monosubstituted triazolo[1,5-a]pyrimidines. Tetrahedron. 1994;50:12113–12124. doi: 10.1016/S0040-4020(01)89564-8. [DOI] [Google Scholar]
- 61.Petrick S.A., Qian Z., Santiago L.M., Gupton J.T. The application of symmetrical vinamidinium salts to the preparation of monosubstituted triazolo[1,5-a]pyrimidines. Heterocycles. 1995;40:729–742. [Google Scholar]
- 62.Tominaga Y., Kohra S., Honkawa H., Hosomi A. Synthesis of pyrimidine derivatives and their related compounds using ketene dithioacetals. Heterocycles. 1989;29:1409–1429. doi: 10.3987/REV-89-408. [DOI] [Google Scholar]
- 63.Gami S.P., Vilapara K.V., Khunt H.R., Babariya J.S., Naliapara Y.T. Synthesis and Antimicrobal Activities of Some Novel Triazolo[1,5-a]Pyrimidine Derivatives. Int. Lett. Chem., Phys. Astron. 2014;30:127–134. doi: 10.18052/www.scipress.com/ILCPA.30.127. [DOI] [Google Scholar]
- 64.Gasse C., Douguet D., Huteau V., Marchal G., Munier-Lehmann H., Pochet S. Substituted Benzyl-Pyrimidines Targeting Thymidine Monophosphate Kinase of Mycobacterium Tuberculosis: Synthesis and in Vitro Anti-Mycobacterial Activity. Bioorg. Med. Chem. 2008;16:6075–6085. doi: 10.1016/j.bmc.2008.04.045. [DOI] [PubMed] [Google Scholar]
- 65.Jadhav S.B., Fatema S., Bhagat S.S., Farooqui M. Thiazolo[3,2- a] Pyrimidones as a Novel Anti-TB Agents. J. Heterocycl. Chem. 2018;55:2893–2900. doi: 10.1002/jhet.3362. [DOI] [Google Scholar]
- 66.Bhatt J.D., Chudasama C.J., Patel K.D. Pyrazole Clubbed Triazolo[1,5-a]Pyrimidine Hybrids as an Anti-Tubercular Agents: Synthesis, in Vitro Screening and Molecular Docking Study. Bioorg. Med. Chem. 2015;23:7711–7716. doi: 10.1016/j.bmc.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 67.Anand P., Akhter Y. A Review on Enzyme Complexes of Electron Transport Chain from Mycobacterium tuberculosis as Promising Drug Targets. Int. J. Biol. Macromol. 2022;212:474–494. doi: 10.1016/j.ijbiomac.2022.05.124. [DOI] [PubMed] [Google Scholar]
- 68.Gálvez J., García-Domenech R., de Julián-Ortiz J.V., Soler R. Topological approach to drug design. J. Chem. Inf. Comp. Sci. 1995;35:272–284. doi: 10.1021/ci00024a017. Erratum in J. Chem. Inf. Comp. Sci. 1995, 35, 938. [DOI] [PubMed] [Google Scholar]
- 69.García-March F.J., García-Domenech R., Gálvez J., Antón-Fos G.M., de Julián-Ortiz J.V., Giner-Pons R., Recio-Iglesias M.C. Pharmacological Studies of 1-(p-Chlorophenyl)Propanol and 2-(1-Hydroxy-3-Butenyl)Phenol: Two New Non-Narcotic Analgesics Designed by Molecular Connectivity. J. Pharm. Pharmacol. 1997;49:10–15. doi: 10.1111/j.2042-7158.1997.tb06743.x. [DOI] [PubMed] [Google Scholar]
- 70.García-Domenech R., de Julián-Ortiz J.V. Antimicrobial activity characterization in a heterogeneous group of compounds. J. Chem. Inf. Comput. Sci. 1998;38:445–449. doi: 10.1021/ci9702454. [DOI] [PubMed] [Google Scholar]
- 71.De Julián-Ortiz J.V., Gálvez J., Muñoz-Collado C., García-Domenech R., Gimeno-Cardona C. Virtual Combinatorial Syntheses and Computational Screening of New Potential Anti-Herpes Compounds. J. Med. Chem. 1999;42:3308–3314. doi: 10.1021/jm981132u. [DOI] [PubMed] [Google Scholar]
- 72.Bruno-Blanch L., Galvez J., Garcia-Domenech R. Topological virtual screening: A way to find new anticonvulsant drugs from chemical diversity. Bioorg. Med. Chem. Lett. 2003;13:2749–2754. doi: 10.1016/S0960-894X(03)00535-3. [DOI] [PubMed] [Google Scholar]
- 73.De Julián-Ortiz J.V., Besalú E. Internal Test Sets Studies in a Group of Antimalarials. Int. J. Molec. Sci. 2006;7:456–468. doi: 10.3390/i8200456. [DOI] [Google Scholar]
- 74.García-Domenech R., Gálvez J., de Julián-Ortiz J.V., Pogliani L. Some New Trends in Chemical Graph Theory. Chem. Rev. 2008;108:1127–1169. doi: 10.1021/cr0780006. [DOI] [PubMed] [Google Scholar]
- 75.Garcia-Domenech R., Zanni R., Galvez-Llompart M., de Julian-Ortiz J.V. Modeling Anti-Allergic Natural Compounds by Molecular Topology. Comb. Chem. High Throughput Screening. 2013;16:628–635. doi: 10.2174/1386207311316080005. [DOI] [PubMed] [Google Scholar]
- 76.Zanni R., Galvez-Llompart M., Morell C., Rodríguez-Henche N., Díaz-Laviada I., Recio-Iglesias M.C., Garcia-Domenech R., Galvez J. Novel Cancer Chemotherapy Hits by Molecular Topology: Dual Akt and Beta-Catenin Inhibitors. PLoS ONE. 2015;10:e0124244. doi: 10.1371/journal.pone.0124244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castillo-Garit J.A., Flores-Balmaseda N., Álvarez O., Pham-The H., Pérez-Doñate V., Torrens F., Pérez-Giménez F. Computational Identification of Chemical Compounds with Potential Activity against Leishmania Amazonensis Using Nonlinear Machine Learning Techniques. Curr. Top. Med. Chem. 2019;18:2347–2354. doi: 10.2174/1568026619666181130121558. [DOI] [PubMed] [Google Scholar]
- 78.García-Domenech R., de Julián-Ortiz J.V., Duart M.J., García-Torrecillas J.M., Antón-Fos G.M., Ríos-Santamarina I., de Gregorio Alapont C., Gálvez J. Search of a topological pattern to evaluate toxicity of heterogeneous compounds. SAR QSAR Environ. Res. 2001;12:237–254. doi: 10.1080/10629360108035380. [DOI] [PubMed] [Google Scholar]
- 79.Lavado G.J., Baderna D., Carnesecchi E., Toropova A.P., Toropov A.A., Dorne J.L., Benfenati E. QSAR Models for Soil Ecotoxicity: Development and Validation of Models to Predict Reproductive Toxicity of Organic Chemicals in the Collembola Folsomia Candida. J. Hazard. Mater. 2022;423:127236. doi: 10.1016/j.jhazmat.2021.127236. [DOI] [PubMed] [Google Scholar]
- 80.Gálvez J., de Julián-Ortiz J.V., García-Domenech R. General topological patterns of known drugs. J. Mol. Graphics Modell. 2001;20:84–94. doi: 10.1016/S1093-3263(01)00103-6. [DOI] [PubMed] [Google Scholar]
- 81.Kier L.B., Murray W.J., Randić M., Hall L.H. Molecular Connectivity V: Connectivity series concept applied to density. J. Pharm. Sci. 1976;65:1226–1230. doi: 10.1002/jps.2600650824. [DOI] [PubMed] [Google Scholar]
- 82.Kier L.B., Hall L.H. Molecular Connectivity VII: Specific treatment of heteroatoms. J. Pharm. Sci. 1976;65:1806–1809. doi: 10.1002/jps.2600651228. [DOI] [PubMed] [Google Scholar]
- 83.Randić M. The connectivity index 25 years after. J. Mol. Graphics Modell. 2001;20:19–35. doi: 10.1016/S1093-3263(01)00098-5. [DOI] [PubMed] [Google Scholar]
- 84.Estrada E., Uriarte E. Recent advances on the role of topological indices in drug discovery research. Curr. Med. Chem. 2001;8:1573–1588. doi: 10.2174/0929867013371923. [DOI] [PubMed] [Google Scholar]
- 85.Selassie C.D., Mekapati S.B., Verma R.P. QSAR: Then and now. Curr. Top. Med. Chem. 2002;2:1357–1379. doi: 10.2174/1568026023392823. [DOI] [PubMed] [Google Scholar]
- 86.Ivanciuc O. Cheminform Abstract: Chemical Graphs, Molecular Matrices and Topological Indices in Chemoinformatics and Quantitative Structure-Activity Relationships. ChemInform. 2013;44:153–163. doi: 10.1002/chin.201349274. [DOI] [PubMed] [Google Scholar]
- 87.Havare Ö.Ç. Quantitative Structure Analysis of Some Molecules in Drugs Used in the Treatment of COVID-19 with Topological Indices. Polycyclic Aromat. Compd. 2021;42:1–12. doi: 10.1080/10406638.2021.1934045. [DOI] [Google Scholar]
- 88.Balaban A.T. Highly discriminating distance based topological index. Chem. Phys. Lett. 1982;89:399–404. doi: 10.1016/0009-2614(82)80009-2. [DOI] [Google Scholar]
- 89.Bonchev D. Overall connectivity: A next generation molecular connectivity. J. Mol. Graphics Modell. 2001;20:65–75. doi: 10.1016/S1093-3263(01)00101-2. [DOI] [PubMed] [Google Scholar]
- 90.Estrada E., Molina E. Novel local (fragment-based) topological molecular descriptors for QSpr/QSAR and molecular design. J. Mol. Graphics Modell. 2001;20:54–64. doi: 10.1016/S1093-3263(01)00100-0. [DOI] [PubMed] [Google Scholar]
- 91.Tropsha A., Zheng W. Identification of the descriptor pharmacophores using variable selection QSAR: Applications to database mining. Curr. Pharm. Des. 2001;7:599–612. doi: 10.2174/1381612013397834. [DOI] [PubMed] [Google Scholar]
- 92.Gozalbes R., Doucet J.P., Derouin F. Application of topological descriptors in QSAR and drug design: History and new trends. Curr. Drug. Targets Infect. Disord. 2002;2:93–102. doi: 10.2174/1568005024605909. [DOI] [PubMed] [Google Scholar]
- 93.Agatonovic-Kustrin S., Ling L.H., Tham S.Y., Alany R.G. Molecular descriptors that influence the amount of drugs transfer into human breast milk. J. Pharm. Biomed. Anal. 2002;29:103–119. doi: 10.1016/S0731-7085(02)00037-7. [DOI] [PubMed] [Google Scholar]
- 94.Basak S.C., Mills D.R., Balaban A.T., Gute B.D. Cheminform Abstract: Prediction of Mutagenicity of Aromatic and Heteroaromatic Amines from Structure: A Hierarchical QSAR Approach. ChemInform. 2010;32 doi: 10.1002/chin.200138234. [DOI] [PubMed] [Google Scholar]
- 95.Orosz Á., Héberger K., Rácz A. Comparison of Descriptor- and Fingerprint Sets in Machine Learning Models for ADME-Tox Targets. Front. Chem. 2022;10:852893. doi: 10.3389/fchem.2022.852893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jacobs M.R. Activity of Quinolones against Mycobacteria. Drugs. 1995;49((Suppl. 2)):67–75. doi: 10.2165/00003495-199500492-00011. [DOI] [PubMed] [Google Scholar]
- 97.García-Sánchez J.E., López R., Prieto J. Antimicrobianos en Medicina. Prous Science; Barcelona, Spain: 2006. [Google Scholar]
- 98.O’Neil M.J. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals. Royal Society of Chemistry; Cambridge, UK: 2013. [Google Scholar]
- 99.García-Domenech R. Unidad de Investigación de Diseño de Fármacos y Conectividad Molecular, Dep. Química-Física, Facultad de Farmacia, Universitat de València; Burjassot, Valencia, Spain: 2013. DesMol. [Google Scholar]
- 100.Wiener H. Structural Determination of Paraffin Boiling Points. J. Am. Chem. Soc. 1947;69:17–20. doi: 10.1021/ja01193a005. [DOI] [PubMed] [Google Scholar]
- 101.Gálvez J., García-Domenech R., Salabert M.T., Soler R. Charge indices. New topological descriptors. J. Chem. Inf. Comp. Sci. 1994;34:520–525. doi: 10.1021/ci00019a008. [DOI] [Google Scholar]
- 102.De Julián-Ortiz J.V., Besalú E., García-Domenech R. True prediction by consensus for small sets of cyclooxigenase-2 inhibitors. Indian J. Chem. 2003;42:1392–1404. [Google Scholar]
- 103.Font D., Heras M., Villalgordo J.M. Development of an efficient and straightforward methodology toward the synthesis of molecularly diverse 2,6-disubstituted 3,4-dihydropyrimidin-4(3h)-ones. Synthesis. 2002;2002:1833–1842. doi: 10.1002/chin.200301151. [DOI] [Google Scholar]
- 104.Font D., Heras M., Villalgordo J.M. Solution- and solid-phase parallel synthesis of 4-alkoxy-substituted pyrimidines with high molecular diversity. J. Comb. Chem. 2003;5:311–321. doi: 10.1021/cc020019t. [DOI] [PubMed] [Google Scholar]
- 105.Petasis N.A., Goodman A., Zavialov I.A. A new synthesis of α-arylglycines from aryl boronic acids. Tetrahedron. 1997;53:16463–16470. doi: 10.1016/S0040-4020(97)01028-4. [DOI] [Google Scholar]
- 106.Petasis N.A., Zavialov I.A. A new and practical synthesis of α-amino acids from alkenyl boronic acids. J. Am. Chem. Soc. 1997;119:445–446. doi: 10.1021/ja963178n. [DOI] [Google Scholar]
- 107.BMDP New System 2.0. Statistical Solutions Ltd.; Cork, Ireland: 2004. [Google Scholar]
- 108.Gálvez J., García-Domenech R., de Gregorio Alapont C., de Julián-Ortiz J.V., Popa L. Pharmacological distribution diagrams: A tool for the new drug design. J. Mol. Graphics. 1996;14:272–276. doi: 10.1016/S0263-7855(96)00081-1. [DOI] [PubMed] [Google Scholar]
- 109.Shinnick T.M., Good R.C. Mycobacterial taxonomy. Eur. J. Clin. Microbiol. Infect. Dis. 1994;13:884–901. doi: 10.1007/BF02111489. [DOI] [PubMed] [Google Scholar]
- 110.Franzblau S.G., Witzig R.S., McLaughinhlin J.C., Torres P., Madico G., Hernandez A., Degnan M.T., Cook M.B., Quenzer V.K., Ferguson R.M., et al. Rapid low technology MIC determination with clinical Mycobacterium isolates by using the microplate alamar blue assay. J. Clin. Microbiol. 1998;36:362–366. doi: 10.1128/JCM.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available upon request to the authors.