

Abstract
Background
Congenital heart defects (CHDs) are the most common congenital malformations, including structural malformations in the heart and great vessels. CHD complications such as low birth weight, prematurity, pregnancy termination, mortality, and morbidity depend on the type of defect.
Methods
In the present research, genetic analyses via whole-exome sequencing (WES) was performed on 3 unrelated pedigrees with CHDs. The candidate variants were confirmed, segregated by PCR-based Sanger sequencing, and evaluated by bioinformatics analysis.
Results
A novel stop-gain c.C244T:p.R82X variant in the FLT4 gene, as well as a nonsynonymous c.C1403T:p.T468M variant in the PTPN11 gene, was reported by WES. FLT4 encodes a receptor tyrosine kinase involved in lymphatic development and is known as vascular endothelial growth factor 3.
Conclusions
We are the first to report a novel c.C244T variant in the FLT4 gene associated with CHDs. Using WES, we also identified a nonsynonymous variant affecting protein-tyrosine phosphatase, the non-receptor type 11 (PTPN11) gene. The clinical implementation of WES can determine gene variants in diseases with high genetic and phenotypic heterogeneity like CHDs.
Keywords: Congenital heart defect, CHDs, Whole-exome sequencing, FLT4, PTPN11, VEGFR3
Introduction
Congenital heart defects (CHDs) are common defects that are present at birth, accounting for 4 to 8 cases per 1000 births (about one-third of neonatal mortalities) [1, 2]. Moreover, CHDs include structural malformations in the heart and great vessels that occur during the development of the fetus [3]. These diseases have various symptoms depending on the types of defects, which are thought to have resulted from a combination of genetic and environmental factors [4]. Notably, atrioventricular septal defects, ventricular septal defects, atrial septal defects, patent ductus arteriosus, and the tetralogy of Fallot are the most common types of CHDs [5]. The tetralogy of Fallot (OMIM disease 187,500) is the most common cyanotic heart defect, accounting for approximately 5% to 10% of CHD cases [6]. With an estimated incidence of 3/10000 births, the tetralogy of Fallot presents with a combination of abnormalities such as pulmonary valve stenosis, right ventricular hypertrophy, ventricular septal defects, and the overriding aorta [6, 7]. Pulmonary valve stenosis (OMIM disease 265,500) is mostly congenital and comprises 7% to 12% of CHD cases [8, 9]. It is a relatively common defect of the pulmonic valve that is most often associated with congenital cardiac syndromes like the tetralogy of Fallot, congenital rubella, and Noonan syndrome [9]. Ventricular septal defects constitute one of the most common CHDs, and they appear with or without several complex abnormalities that can be detected between the prenatal period and adulthood [10]. Pulmonary atresia with ventricular septal defects (178,370) is a complex cyanotic CHD with various clinical symptoms based on the anatomy of the central pulmonary arteries and pulmonary distribution [11]. Aortic valve stenosis is the most common valvular cardiac defect and affects nearly 2% of the population aged over 65 years [12]. Although this progressive disease usually occurs in persons over 65 years of age, it can also occur in younger individuals with rheumatic heart disease or congenital valve abnormalities [13]. Many genes have been reported to be involved in CHD etiology [14]. Vascular endothelial growth factor 3 (VEGFR3) is encoded by the fms-like tyrosine kinase-4 (FLT4; 136,352) gene, involved in the development of the lymphatic system. FLT4 variants cause Milroy disease (153,100), one of the main forms of hereditary primary lymphedema, and they also predispose to the tetralogy of Fallot, indicating the role of VEGFR3 in the primary development of the heart [15]. The protein-tyrosine phosphatase, non-receptor type 11 (PTPN11; 176,876) gene encodes the non-receptor type protein tyrosine phosphatase SHP-2 (src homology region 2 domain phosphatase–2), which accounts for nearly 50% of cases with Noonan syndrome. The syndrome (OMIM 163,950) is associated with a wide spectrum of CHDs such as pulmonary valve stenosis, hypertrophic cardiomyopathy, aortic coarctation, and atrioventricular septal defects [16, 17].
In this research, we utilized whole-exome sequencing [18] to determine the genetic causes of CHDs in 3 unrelated pedigrees and succeeded in identifying a novel heterozygous pathogenic stop-gain variant in exon 3 of the FLT4 gene in an Iranian family with CHDs. Moreover, with the aid of WES, we identified a pathogenic nonsynonymous variant (c.C1403T:p.T468M) in exon 12 of the PTPN11 gene in another Iranian family with CHDs.
Methods
Ethics approval and consent to participate
The present research was conducted in accordance with the Declaration of Helsinki. Ethical approval was obtained from the Ethics Committees of Rajaie Cardiovascular Medical and Research Center, Iran University of Medical Sciences, Tehran, Iran (IR.RHC.REC.1399.017). Informed consent was obtained from the probands’ parents and/or legal guardians for study participation.
Study subjects and samples
Three families with CHDs were selected for genetic analysis. The genetic causes of CHDs were detected in 2 pedigrees (Fig. 1(. For further information on the cardiovascular anatomy, computed tomography angiography was performed with a Siemens SOMATOM Definition Flash 128-Slice Dual Source CT Scanner (Fig. 2). After CHD diagnosis, written informed consent was obtained from the families. Next, peripheral blood collection was performed on each study subject. Genomic DNA was then obtained from the whole-blood sample with a Cinna Pure DNA Kit (CinnaGen, Tehran, Iran). Afterward, the extracted DNA was evaluated with a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA).
Fig. 1.

The image presents the pedigree of 2 families with congenital heart defects (CHDs), sequencing chromatograms, and the conservation analysis of the mutated amino acids in the FLT4 and PTPN11 genes. A The image depicts Family I. A1 The image demonstrates the pedigree of Family I with individuals suffering from CHDs. The black arrow indicates the proband, and the affected and unaffected individuals are presented with filled and clean symbols, respectively. A2 The Sanger-based sequencing results in Family I show a heterozygous variant in the FLT4 gene in the proband (III-3) and her affected brother (III-2). The c.C244T variant in the FLT4 gene was detected in her healthy mother (II-6), while her father (II-5) did not carry this variant. The candidate variant was validated by Sanger-based sequencing using the reverse primer. A3 In Family I, the conservation of the p.R82X variant is shown. The variant site is highly conserved in various species. B The image depicts Family II. B1 The image demonstrates the pedigree of Family II with individuals suffering from CHDs. The black arrow indicates the proband, and the affected and unaffected individuals are represented by filled and clean symbols, respectively. B2 The Sanger-based sequencing results in Family II show a heterozygous variant in the PTPN11 gene in the proband (III-2) and her affected sister (III-1). The c.C1403T variant in the PTPN11 gene was detected in her healthy father (II-4), while her mother (II-5) did not carry this variant. The candidate variant was validated by Sanger-based sequencing using the forward primer. B3 In Family II, the conservation of the p.T468M variant is shown. The variant site is highly conserved in various species
Fig. 2.
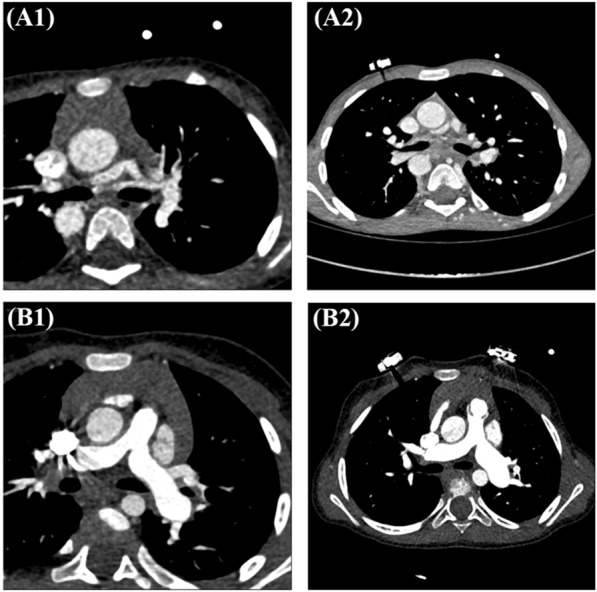
Computed tomography angiography results are presented herein. A1 The axial view shows a dilated aorta, an atretic main pulmonary artery, and hypoplastic confluent pulmonary arterial branches. A2 The axial view demonstrates a dilated Aorta, pulmonary atresia, and hypoplastic pulmonary arterial branches. B1 The axial view illustrates severe PS and ASD. B2 The axial view reveals severe pulmonary valve stenosis, a ventricular septal defect, and an atrial septal defect
WES, validation, and search strategy
WES was performed on DNA samples from each pedigree’s proband (Fig. 1A1, B1) at Macrogen (Seoul, South Korea) with a SureSelect XT Library Prep Kit (Agilent Technologies, CA, USA) on an Illumina HiSeq 4000 (Illumina, San Diego, USA). With the aid of the BWA software package, qualified reads were aligned to the hg19 version of the human reference genome. Subsequently, the Genome Analysis Toolkit (GATK) was applied to access variant calling. Functional annotation of the detected variants was performed using the ANNOVAR software tool. The effects of the variants on protein functions were predicted using MutationTaster, MutationAssessor, PolyPhen, SIFT, and CADD. In the next stage, variants with a minor allele frequency below 5% in the 1000 Genomes Project, ESP6500, and ExAC databases were filtered out. Next, all the candidate variants were validated by targeted polymerase chain reaction (PCR) amplification and Sanger sequencing on available family members (Applied Biosystems 3500G, Foster City, CA, USA). The specific primers were designed with the Geneious software (Geneious 10.2.2, Biomatters Ltd).
In the first family study, 2 specific primers were constructed: 1 forward primer (5′-GGTCATCTGCATCCACTCC-3′) and 1 reverse primer (5′-ATGCGTGCCTTGATGTACTTGT-3′). In the second family study, 2 specific primers were constructed: 1 forward primer (5ʹ-ACTCTGTATGGTATGTGCTGTTG-3′) and 1 reverse primer (5′-GAAGTGGCAGAAGTCAGGATT-3′). A literature search was conducted by searching Google Scholar, ClinVar, HGMD, and PubMed to gather the detected variants in the identified genes related to CHDs. The search was performed utilizing the keywords that included the clinical significance of FLT4 variants, the clinical significance of PTPN11 variants, FLT4 variants, and PTPN11 variants.
Homology modeling and docking
FLT4
FLT4 encodes the VEGFR3 protein, which plays a significant role in embryonic blood vascular development and postnatal angiogenesis. Defects in any members of VEGF signaling, as a novel and plausible pathomechanism of the tetralogy of Fallot, are related to cardiovascular defects. The stop-gain variant in VEGFA and loss of function in FLT4 is a candidate for the tetralogy of Fallot pathogenesis. VEGFR3 homodimers respond to signaling induced by vascular endothelial growth factor C (VEGFC) and regulate the development of the lymphatic vasculature. In return, VEGFR3/VEGFR2 heterodimers are induced by VEGFA and VEGFC and are involved in the regulation of sprouting angiogenesis and vascular network formation [19]. According to a previous study, in the absence of VEGFR3 in knockout murine samples, a higher abundance of VEGFC might cause signaling only through VEGFR2, leading to the disruption of blood cell formation and angiogenesis during embryogenesis [20]. The effects of normal/mutant VEGFR3 on VEGF signaling are illustrated in Figs. 3 and 4.
Fig. 3.

The image illustrates the binding pattern of the normal/mutant vascular endothelial growth factor 3 (VEGFR3) with vascular endothelial growth factor C (VEGFC). A The image presents the 3D interaction between the normal VEGFR3 (green) and VEGFC (violet). B The image illustrates the 3D interaction between the mutant VEGFR3 (green) and VEGFC (violet). These visualizations were obtained using the PyMOL software
Fig. 4.

The image presents a 2D image of ligand–protein interactions. A The image illustrates the interaction between the normal VEGFR3 and VEGFC. B The image depicts the interaction between the mutant VEGFR3 and VEGFC. These images were generated by LigPlus + . (The green dashed lines represent hydrogen bonds with bond distance.)
PTPN11
PTPN11 encodes the SHP-2 protein, which, as a phosphatase, has a critical role in the activation of the RAS mitogen-activated protein kinase (MAPK) pathway by phosphorylation mechanism. The RAS–MAPK signaling pathway is involved in embryonic development during weeks 2 to 8, the critical cardiac development period. Hence, any disturbance to this pathway is likely to cause heart defects [21]. The SHP-2 protein has an auto-inhibited state in the closed structure by N-SH2–PTP inter-domain interaction. The substitution of Met for Thr468 in the PTP domain weakens intramolecular N-SH2–PTP interactions, destabilizes the SHP-2 closed structure, and increases the SHP-2 open form, enhancing the binding of N-SH2 with phosphorylated tyrosine in partners. The major SHP-2 binding partner is Grb2-associated binder-1 (Gab-1), which is bound by SHP-2 to its phosphorylated tyrosine through the N-SH2 domain. T468M SHP-2 lengthens the binding of SHP-2 to Grb1, which probably leads to the further phosphorylation and activation of the RAS–ERK1/2 pathway [22, 23]. The effects of normal/mutant SHP-2 on the RAS–MAPK signaling pathway are depicted in Figs. 5 and 6.
Fig. 5.

The binding pattern of the normal/mutant src homology region 2 domain phosphatase-2 (SHP-2) with Grb2-associated binder-1 (Gab-1) is presented herein. A The image shows the 3D interaction between the normal SHP-2 (N-SH2 domain: violet, C-SH2 domain: limo green, PTP domain: teal) and Gab-1 (red). B The image demonstrates the 3D interaction between the mutant SHP-2 (N-SH2 domain: violet, C-SH2 domain: limo green, PTP domain: teal) and Gab-1 (red). These visualizations were obtained using the PyMOL software
Fig. 6.

The 2D image of ligand–protein interactions is illustrated herein. A The image demonstrates the interaction between the normal SHP-2 and Gab-1. B The image shows the interaction between the mutant SHP-2 and Gab-1. These images were generated by LigPlus + . (The green dashed lines represent hydrogen bonds with bond distance.)
Modeling and docking methods
In the present study, the Protein Data Bank (PDB) (https://www.rcsb.org/) was drawn upon to obtain the crystal structure of SHP-2 (normal and mutant), Gab-1, and VEGFC. Additionally, through the use of the homology-modeling server of SWISS-MODEL (https://swissmodel.expasy.org/), 3D structures of VEGFR3 (normal and mutant) were created. The protein structures were corrected by ViewerLite (v.1.5.1). In other words, all heteroatoms, consisting of water molecules, ions, and native ligands, were deleted, and polar hydrogens were added. Then, the energy of the models was minimized using the YASARA minimization server (http://www.yasara.org/). The SCE files obtained from the YASARA minimization server were imported into YASARA View (v.20.12.24) to be saved as PDB files. For the prediction of the binding site, the server of the Computed Atlas of Surface Topography of proteins (CASTp) (http://sts.bioe.uic.edu/) was employed. Docking was also performed by using the HADDOCK web server (https://wenmr.science.uu.nl/haddock2.4/). Thereafter, the interactions among the conformations were analyzed using PyMOL (v.2.5.2) and LigPlus + (v.2.2.4). After the completion of docking, the lowest HADDOCK score and the root mean square deviation conformation were considered the most suitable docking pose for the compounds.
Results
Clinical analysis
The computed tomography angiography findings indicated that both patients of Family I (Fig. 2A1, A2) had severe cyanosis in the neonatal period. Echocardiography revealed pulmonary atresia and a large ventricular septal defect in both cases. Multiple coronal, axial, and sagittal images revealed a large subaortic ventricular septal defect and pulmonary atresia with hypoplastic pulmonary artery branches and multiple aortopulmonary collaterals in both patients. The patients of Family II (Fig. 2 B1, B2) had a heart murmur in infancy. Echocardiography revealed pulmonary stenosis, a large ventricular septal defect, and an atrial septal defect in the first case and pulmonary valve stenosis and an atrial septal defect in the second case.
Molecular analysis
The current study enrolled 3 distinct pedigrees with various patterns of inheritance. The novel pathogenic c.C244T:p.R82X variant in the FLT4 gene was detected in the first pedigree, and it segregated with CHDs in this family. In the second pedigree, the heterozygous c.C1403T:p.T468M variant in the PTPN11 gene was identified. The 2 pedigrees are presented in Fig. 1. The proband of the third pedigree was a 3-year-old girl with heart murmurs and cyanosis. The initial analysis showed that the identified variants in this pedigree were variants of uncertain significance. These variants in 5 genes (AXIN2, EGF, PFKL, PKHD1L1, and PIEZO1) did not segregate with the examined defect in this pedigree.
The reported pathogenic variants in the FLT4 gene related to lymphedema and Milroy disease are summarized in Table 1 [19, 24–39], and the reported pathogenic variants in the PTPN11 gene related to Noonan syndrome and LEOPARD syndrome are summarized in Table 2 [40–55]. The functional significance of the identified genetic variants was assessed through bioinformatics analysis using CADD, SIFT, PROVEAN, MutationTaster, and PolyPhen-2. According to the collected data, the p.Q736X variant in the FLT4 gene had the highest CADD number in Table 1. Likewise, the p.D61N, p.E110K, p.F285L, p.F285C, p.F285S, p.A461S, p.A461T, p.S502L and P.F285I variants in the PTPN11 gene had the highest CADD number in Table 2.
Table 1.
Bioinformatics analysis of pathogenic reported variants in FLT4 (NM_182925.5) related to congenital heart defects
| No. | Exon | HGVS DNA | HGVS Protein | Chromosome 5 Position (hg19) | dbSNP | CADD | SIFT | Polyphen-2 | PROVEAN | Mutation taster | ClinVar | Condition | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 18 | c.2554G > A | p. G852S | 180,046,758 | NA | 26.8 | D | PD | D | DC | NA | MD | [24] |
| 2 | 18 | c.2554G > C | p. G852R | 180,046,758 | NA | 26.7 | D | PD | D | DC | NA | MD | [24] |
| 3 | 18 | c.2555G > T | p. G852V | 180,046,757 | NA | 26.1 | D | PD | D | DC | NA | MD | [24] |
| 4 | 18 | c.2560G > C | p. G854R | 180,046,752 | rs144045237 | 29.2 | D | PD | D | DC | NA | MD | [24,25] |
| 5 | 18 | c.2569G > A | p. G857R | 180,046,743 | rs267606818 | 27.9 | D | PD | D | DC | P | MD | [24,26] |
| 6 | 26 | c.3433C > T | p.R1145C | 180,039,610 | rs202140363 | 25.9 | D | PD | N | DC | NA | MD | [24] |
| 7 | 19 | c.2740G > C | p. G914R | 180,046,274 | NA | 32 | D | PD | D | DC | NA | MD | [24] |
| 8 | 19 | c.2744C > A | p. A915E | 180,046,270 | NA | 32 | D | PD | D | DC | NA | MD | [24] |
| 9 | 19 | c.2749A > C | p. T917P | 180,046,265 | NA | 29.4 | D | PD | D | DC | NA | MD | [24] |
| 10 | 20 | c.2797G > C | p. G933R | 180,046,074 | NA | 32 | D | PD | D | DC | NA | MD | [24,25] |
| 11 | 20 | c.2800A > T | p. N934Y | 180,046,071 | NA | 32 | D | PD | D | DC | NA | MD | [24] |
| 12 | 20 | c.2828G > C | p. R943P | 180,046,043 | NA | 31 | D | PD | D | DC | NA | MD | [24] |
| 13 | 22 | c.3054C > A | p. S1018R | 180,043,942 | NA | 25.4 | D | PD | D | DC | NA | MD | [24] |
| 14 | 22 | c.3056 T > C | p. F1019S | 180,043,940 | NA | 30 | D | PD | D | DC | UVS | MD | [24] |
| 15 | 22 | c.3075G > A | p. M1025I | 180,043,921 | NA | 29.8 | D | PD | D | DC | NA | MD | [24] |
| 16 | 23 | c.3097 T > G | p. C1033G | 180,043,489 | NA | 27.9 | D | PD | D | DC | NA | MD | [24] |
| 17 | 23 | c.3104A > G | p. H1035R | 180,043,482 | rs121909653 | 25.4 | D | PD | D | DC | P | MD | [24] |
| 18 | 23 | c.3109G > C | p. D1037H | 180,043,477 | NA | 28.9 | D | PD | D | DC | NA | MD | [24] |
| 19 | 23 | c.3111C > A | p. D1037E | 180,043,475 | NA | 24.5 | D | PD | D | DC | NA | MD | [24] |
| 20 | 23 | c.3119C > A | p. A1040D | 180,043,467 | NA | 27.6 | D | PD | D | DC | NA | MD | [24] |
| 21 | 23 | c.3122G > A | p. R1041Q | 180,043,464 | NA | 29.8 | D | PD | D | DC | NA | MD | [24] |
| 22 | 23 | c.3122G > C | p. R1041P | 180,043,464 | rs121909650 | 28.9 | D | PD | D | DC | P | MD | [24,26,27] |
| 23 | 23 | c.3157A > T | p. I1053F | 180,043,429 | NA | 27.1 | D | PD | D | DC | NA | MD | [24] |
| 24 | 23 | c.3170G > A | p. G1057D | 180,043,416 | NA | 26.7 | D | PD | D | DC | NA | MD | [24] |
| 25 | 24 | c.3250G > A | p. E1084K | 180,041,149 | NA | 31 | D | PD | D | DC | NA | MD | [24] |
| 26 | 24 | c.3308 T > C | p. L1103P | 180,041,091 | NA | 26.1 | D | PD | D | DC | NA | MD | [24] |
| 27 | 24 | c.3310C > T | p. L1104F | 180,041,089 | NA | 25.3 | D | PD | D | DC | NA | MD | [24] |
| 28 | 24 | c.3318G > T | p. E1106D | 180,041,081 | rs201431522 | 25.3 | D | PD | D | DC | NA | MD | [24] |
| 29 | 24 | c.3323 T > C | p. F1108S | 180,041,076 | NA | 29.1 | D | PD | D | DC | NA | MD | [24] |
| 30 | 25 | c.3391G > C | p. G1131R | 180,040,051 | NA | 27.5 | D | PD | D | DC | P | MD | [24] |
| 31 | 17 | c.2531G > C | p. R844P | 180,047,184 | NA | 29.5 | D | PD | D | DC | NA | MD | [24] |
| 32 | 18 | c.2560G > A | p. G854S | 180,046,752 | NA | 28.5 | D | PD | D | DC | NA | MD | [24] |
| 33 | 18 | c.2563G > A | p. A855T | 180,046,749 | rs121909657 | 24.6 | D | PD | D | DC | P | MD | [24,27,74] |
| 34 | 18 | c.2629G > A | p. A877T | 180,046,683 | NA | 25.9 | D | PD | D | DC | NA | MD | [24] |
| 35 | 18 | c.2632G > A | p. V878M | 180,046,680 | rs121909654 | 25.6 | D | PD | D | DC | P | MD | [24,27,29] |
| 36 | 19 | c.2677C > G | p. L893V | 180,046,337 | NA | 25.0 | D | PD | D | DC | NA | MD | [24,30,31] |
| 37 | 19 | c.2737_2739del | p.Leu913del | 180,046,275 | NA | 22.5 | NA | NA | NA | DC | NA | MD | [24] |
| 38 | 19 | c.2743G > C | p.A915P | 180,046,271 | NA | 32 | D | PD | D | DC | NA | MD | [24,25] |
| 39 | 19 | c.2748C > G | p.C916W | 180,046,266 | NA | 27.3 | D | PD | D | DC | NA | MD | [24,25] |
| 40 | 22 | c.3059A > T | p.Q1020L | 180,043,937 | NA | 27.4 | D | PD | D | DC | NA | MD | [24,32] |
| 41 | 22 | c.3070G > A | p.G1024R | 180,043,926 | NA | 29.6 | D | PD | D | DC | NA | MD | [24] |
| 42 | 22 | c.3071G > A | p.G1024E | 180,043,925 | NA | 28.2 | D | PD | D | DC | NA | MD | [24] |
| 43 | 23 | c.3105C > G | p.H1035Q | 180,043,481 | NA | 25.1 | D | PD | D | DC | NA | MD | [24,29,30] |
| 44 | 23 | c.3109G > T | p.D1037Y | 180,043,477 | NA | 31 | D | PD | D | DC | NA | MD | [24] |
| 45 | 23 | c.3121C > T | p.R1041W | 180,043,465 | rs1451816005 | 27.9 | D | PD | D | DC | LP | MD | [24,25] |
| 46 | 23 | c.3125A > G | p.N1042S | 180,043,461 | NA | 25.0 | D | PD | D | DC | NA | MD | [24] |
| 47 | 23 | c.3131 T > C | p.L1044P | 180,043,455 | rs121909651 | 27.2 | D | PD | D | DC | P | MD | [24,26] |
| 48 | 23 | c.3151G > A | p.V1051M | 180,043,435 | NA | 25.1 | D | PD | D | DC | NA | MD | [24] |
| 49 | 23 | c.3164A > C | p.D1055A | 180,043,422 | NA | 26.9 | D | PD | D | DC | NA | MD | [24] |
| 50 | 23 | c.3164A > T | p.D1055V | 180,043,422 | NA | 27.7 | D | PD | D | DC | NA | MD | [24] |
| 51 | 24 | c.3257 T > C | p.I1086T | 180,041,142 | rs121909655 | 26.4 | D | PD | D | DC | P | MD | [24,29] |
| 52 | 24 | c.3316G > A | p.E1106K | 180,041,083 | rs121909656 | 27.2 | D | PD | D | DC | P | MD | [24,33] |
| 53 | 25 | c.3341C > T | p.P1114L | 180,040,101 | rs121909652 | 29.8 | D | PD | D | DC | P | MD | [24,26,27,30] |
| 54 | 25 | c.3344A > G | p.Y1115C | 180,040,098 | NA | 28.3 | D | PD | D | DC | NA | MD | [24] |
| 55 | 25 | c.3410C > T | p.P1137L | 180,040,032 | rs1762335528 | 27.6 | D | PD | D | DC | LP | MD | [24,25,30,31] |
| 56 | 3 | c.244C > T | p.R82X | 180,057,711 | NA | 34 | NA | NA | NA | DC | P | TOF | [24] |
| 57 | 8 | c.1083C > A | p.Y361X | 180,055,902 | NA | 36 | NA | NA | NA | DC | NA | TOF | [24] |
| 58 | 15 | c.2206C > T | p.Q736X | 180,047,969 | NA | 42 | NA | NA | NA | DC | NA | TOF | [24] |
| 59 | 21 | c.2995C > T | p.Q999X | 180,045,776 | NA | 35 | NA | NA | NA | DC | NA | TOF | [24] |
| 60 | 21 | c.2860C > T | p.P954S | 180,045,911 | rs34255532 | 21.3 | D | B | N | DC | NA | MD | [24] |
| 61 | 18 | c.2587 T > C | p.S863P | 180,046,725 | NA | 25.6 | T | PD | D | DC | NA | Lymphedema | [24] |
| 62 | 20 | c.2819G > C | p.R940P | 180,046,052 | NA | 32 | D | PD | D | DC | NA | Lymphedema | [30] |
| 63 | 25 | c.3391G > A | p.G1131S | 180,040,051 | rs1554109707 | 27.7 | D | PD | D | DC | LP | Lymphedema | [30] |
| 64 | 13 | c.1921C > T | p.P641S | 180,048,641 | rs55667289 | 22.1 | T | B | N | DC | NA | PCL | [26] |
| 65 | 20 | c.2777 T > C | p.I926T | 180,046,094 | NA | 32 | D | PD | D | DC | NA | PCL | [34] |
| 66 | 19 | c.2740G > T | p.G914W | 180,046,274 | NA | 32 | D | PD | D | DC | NA | PCL | [34] |
| 67 | 18 | c.2575G > A | p.V859M | 180,046,737 | NA | 26.2 | D | PD | D | DC | NA | PCL | [34] |
| 68 | 19 | c.2650G > A | p.G884S | 180,046,364 | NA | 29.9 | D | PD | D | DC | NA | MD | [35] |
| 69 | 28 | c.3704C > G | p.S1235C | 180,037,008 | NA | 29.9 | T | PD | N | DC | NA | MD | [36] |
| 70 | 23 | c.3163G > C | p.D1055H | 180,043,423 | NA | 27.7 | D | PD | D | DC | NA | MD | [37] |
| 71 | 24 | c.3315G > C | p.W1105C | 180,041,084 | NA | 28.7 | D | PD | D | DC | NA | MD | [37] |
| 72 | 24 | c.3295 T > C | p.S1099P | 180,041,104 | NA | 27.2 | D | PD | D | DC | NA | MD | [37] |
| 73 | 18 | c.2515G > C | p.E839Q | 180,047,200 | NA | 27.5 | D | PD | D | DC | NA | MD | [37] |
| 74 | 10 | c.1289C > T | p.S430F | 180,053,001 | NA | 23.6 | T | PD | D | DC | NA | MD | [37] |
| 75 | 20 | c.2771 T > C | p.M924T | 180,046,100 | NA | 29.5 | D | PD | D | DC | NA | MD | [37] |
| 76 | 24 | c.3296C > T | p.S1099F | 180,041,103 | NA | 29.2 | D | PD | D | DC | NA | MD | [37] |
| 77 | 24 | c.3230C > T | p.P1077L | 180,041,169 | NA | 28.6 | D | PD | D | DC | NA | MD | [37] |
| 78 | 18 | c.2615G > A | p.S872N | 180,046,697 | NA | 22.6 | D | PD | N | DC | NA | MD | [37] |
| 79 | 24 | c.3233 T > A | p.L1078Q | 180,041,166 | NA | 28.0 | D | PD | D | DC | NA | MD | [37] |
| 80 | 23 | c.3175G > C | p.A1059P | 180,043,411 | NA | 26.8 | D | PD | D | DC | P | MD | [37] |
| 81 | 24 | c.3316G > C | p.E1106Q | 180,041,083 | NA | 26.2 | D | PD | D | DC | NA | MD | [37] |
| 82 | 18 | c.2546G > A | p.R849K | 180,046,766 | NA | 14.84 | T | B | N | DC | NA | MD | [37] |
| 83 | 18 | c.2554G > T | p.G852C | 180,046,758 | NA | 27.8 | D | PD | D | DC | NA | MD | [37] |
| 84 | 2 | c.137G > A | p.S46N | 180,058,700 | NA | 22.3 | D | B | N | DC | NA | MD | [37] |
| 85 | 22 | c.3073A > T | p.M1025L | 180,043,923 | NA | 26.5 | D | B | D | DC | NA | MD | [37] |
| 86 | 23 | c.3111 C > G | p.D1037E | 180,043,475 | NA | 24.5 | D | PD | D | DC | NA | MD | [38] |
| 87 | 24 | c.3323_3325delTCT | p.F1108del | 180,041,074 | rs587776833 | 21.1 | NA | NA | NA | DC | P | MD | [24,25,29] |
| 88 | 17 | c.2542 + 2delT | NA | 180,047,171 | NA | 24.3 | NA | NA | NA | NA | MD | [24,35] | |
| 89 | 24 | c.3243G > T | M1081I | 180,041,156 | NA | 26.9 | D | PD | D | DC | NA | MD | [39] |
| 90 | 9 | c.1258 + 6_1258 + 10 delTCAGG | NA | 180,053,101 | rs1247895470 | 3.209 | NA | NA | NA | DC | NA | MD | [37] |
| 91 | 12 | c.1622dupG | p. Q542PTer3 | 180,049,766 | rs1581655293 | 33 | NA | NA | NA | DC | NA | TOF | [37] |
| 92 | 2 | c.89delC | p. P30R Ter3 | 180,058,748 | rs755445139 | 29.1 | NA | NA | NA | DC | NA | TOF | [37] |
CHD congenital heart disease, MD Milroy disease, TOF tetralogy of Fallot, PCL primary congenital lymphedema, DC disease causing, D deleterious, T tolerated, N neutral, B benign, PD probably damaging, P pathogenic, NA not available/not applicable
CADD, Phred ≤ 20: damaging; Phred > 20: natural
SIFT, score ≤ 0.05: deleterious; score > 0.05: tolerable
Polyphen-2, score = 0–0.15: benign; score = 0.15–0.85: possibly damaging; score = 0.85–1: probably damaging
PROVEAN, score ≤ − 2.5: deleterious; score > − 2.5: natural
Table 2.
Bioinformatics analysis of pathogenic reported variants in PTPN11 (NM_002834) related to congenital heart defects
| No. | Exon | HGVS DNA | HGVS protein | Chromosome 12 position (hg19) | dbSNP | CADD | SIFT | Polyphen-2 | PROVEAN | MutationTaster | ClinVar | Condition (CHD or cardiomyopathy) | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | c.5C > T | p.T2I | 112,856,920 | rs267606990 | 23.8 | T | B | N | DC | P | NS (PS) | [40] |
| 2 | 2 | c.124A > G | p.T42A | 112,884,189 | rs397507501 | 22.7 | T | PD | D | DC | P | NS (PS, ASD) | [40,41] |
| 3 | 3 | c.184 T > G | p.Y62D | 112,888,168 | rs121918460 | 24.9 | D | PD | D | DC | P | NS (AVCD, ASD, PS), PST, PDA | [40–44] |
| 4 | 3 | c.188A > G | p.Y63C | 112,888,172 | rs121918459 | 28.1 | D | PD | D | DC | P | NS (AVCD, HCM, PS) | [40–42] |
| 5 | 3 | c.215C > G | p.A72G | 112,888,199 | rs121918454 | 23.7 | D | PD | D | DC | P | NS (HCM, PS) | [40,41] |
| 6 | 3 | c.A236C | p.Q79P | 112,888,220 | NA | 24.0 | D | PD | D | DC | NA | NS (PS) | [40,43] |
| 7 | 3 | c.317A > C | p.D106A | 112,888,301 | rs397507517 | 25.3 | D | PD | D | DC | P | NS (ASD, PS) | [40,41,43,45] |
| 8 | 7 | c.836A > G | p.Y279C | 112,910,827 | rs121918456 | 28.9 | D | PD | D | DC | P | LS (PVS, HCM), NS (PS,HCM) | [40–42,46–48] |
| 9 | 8 | c.922A > G | p.N308D | 112,915,523 | rs28933386 | 23.6 | D | PD | D | DC | P | NS (PS, HCM,VSD) | [40–42,49,50] |
| 10 | 8 | c.923A > G | p.N308S | 112,915,524 | rs121918455 | 22.7 | T | PD | D | DC | P | PS, NS (HCM) | [40,41,51] |
| 11 | 12 | c.1403C > T | p.T468M | 112,926,270 | rs121918457 | 28.7 | D | PD | D | DC | P | LS (HCM, AVCD), NS (PS, HCM) | [40,41,46–48] |
| 12 | 13 | c.1508G > C | p.G503A | 112,926,888 | rs397507546 | 27.4 | T | PD | D | DC | P | NS (PS) | [40] |
| 13 | 13 | c.1510A > G | p.M504V | 112,926,890 | rs397507547 | 26.8 | T | PD | D | DC | P | NS (PS,HCM) | [[40–43] |
| 14 | 14 | c.1678C > T | p.L560F | 112,940,026 | rs397516797 | 22.7 | D | B | N | DC | LB | NS (HCM) | [40] |
| 15 | 5 | c.540C > T | p.D180 = | 112,892,382 | rs753269427 | 9.256 | T | NA | N | DC | LB | CHD | [52] |
| 16 | 2 | c.127C > T | p.L43F | 112,884,192 | NA | 27.8 | D | PD | D | DC | NA | CHD | [52] |
| 17 | 3 | c.155C > T | p.T52I | 112,888,139 | rs397507503 | 28.0 | D | PD | D | DC | P | NS (PS) | [41] |
| 18 | 3 | c.172A > G | p.N58D | 112,888,156 | rs397507505 | 26.6 | T | PD | D | DC | P | NS (PS) | [41,51] |
| 19 | 3 | c.172A > C | p.N58H | 112,888,156 | rs397507505 | 25.7 | T | PD | D | DC | P | NS (PS) | [41] |
| 20 | 3 | c.174C > A | P.N58K | 112,888,158 | rs397507506 | 25.0 | T | PD | D | DC | P | NS (PS) | [41] |
| 21 | 3 | c.179G > C | p.G60A | 112,888,163 | rs397507509 | 26.5 | D | PD | D | DC | P | NS (PS, ASD) | [41,43] |
| 22 | 3 | c.178G > A | p.G60S | 112,888,162 | rs397507507 | 29.7 | D | PD | D | DC | P | NS (PS) | [41] |
| 23 | 3 | c.182A > G | p.D61G | 112,888,166 | rs121918461 | 28.2 | D | PD | D | DC | P | NS (PS,ASD) | [41,43] |
| 24 | 3 | c.181G > A | p.D61N | 112,888,165 | rs397507510 | 32 | D | PD | D | DC | P | ASD, NS (PS) | [41–43,51] |
| 25 | 3 | c.214G > T | p.A72S | 112,888,198 | rs121918453 | 25.0 | D | PD | D | DC | P | NS (PS,HCM) | [41,51] |
| 26 | 3 | c.218C > T | p.T73I | 112,888,202 | rs121918462 | 27.4 | D | PD | D | DC | DC | NS (PS,ASD) | [41,43] |
| 27 | 3 | c.217_218delinsCT | p.T73L | 112,888,201 | rs397516802 | 28.0 | D | PD | D | DC | P | NS (PS) | [41] |
| 28 | 3 | c.228G > C | p.E76D | 112,888,212 | rs397507514 | 22.3 | D | PD | D | DC | P | NS | [41,51] |
| 29 | 3 | c.236A > G | p.Q79R | 112,888,220 | rs121918466 | 26.0 | D | PD | D | DC | P | NS (PS) | [41–43] |
| 30 | 4 | c.328G > A | p.E110K | 112,888,312 | rs397507518 | 32 | D | PD | D | DC | P | NS | [41] |
| 31 | 4 | c.417G > C | p.E139D | 112,891,083 | rs397507520 | 25.0 | D | PD | D | DC | P | NS (PS), HIS | [41,53] |
| 32 | 7 | c.767A > G | p.Q256R | 112,910,758 | rs397507523 | 24.3 | T | B | D | DC | P | NS (PS) | [41] |
| 33 | 7 | c.781C > T | p.L261F | 112,910,772 | rs397507525 | 22.6 | T | B | N | DC | P | NS | [41] |
| 34 | 7 | c.802G > T | p.G268C | 112,910,793 | rs397507527 | 31 | D | PD | D | DC | P | NS | [41] |
| 35 | 7 | c.844A > G | p.I282V | 112,910,835 | rs397507529 | 23.4 | T | B | N | DC | P | NS (PS) | [41] |
| 36 | 7 | c.853 T > C | p.F285L | 112,910,844 | rs397507531 | 32 | D | PD | D | DC | P | NS (PS) | [41] |
| 37 | 8 | c.854 T > G | p.F285C | 112,915,455 | rs121918463 | 32 | D | PD | D | DC | P | NS (PS) | [41] |
| 38 | 8 | c.854 T > C | p.F285S | 112,915,455 | rs121918463 | 32 | D | PD | D | DC | P | NS (PS),VSD, ASD | [41,51,53] |
| 39 | 12 | c.1381G > T | p.A461S | 112,926,248 | rs121918468 | 32 | D | PD | D | DC | P | NS (HCM) | [41] |
| 40 | 12 | c.1381G > A | p.A461T | 112,926,248 | rs121918468 | 32 | D | PD | D | DC | P | NS (HCM) | [41] |
| 41 | 13 | c.1471C > T | p.P491S | 112,926,851 | rs397507539 | 23.1 | T | B | N | DC | P | NS (PS) | [41] |
| 42 | 13 | c.1471C > A | p.P491T | 112,926,851 | rs397507539 | 23.1 | T | PD | N | DC | P | NS (PS) | [41] |
| 43 | 13 | c.1504 T > G | p.S502A | 112,926,884 | rs121918458 | 26.9 | T | PD | D | DC | P | NS (PS) | [41] |
| 44 | 13 | c.1505C > T | p.S502L | 112,926,885 | rs397507544 | 32 | T | PD | D | DC | P | NS (PS) | [41] |
| 45 | 13 | c.1528C > G | p.Q510E | 112,926,908 | rs397507549 | 26.5 | D | PD | D | DC | P | NS (PS, HCM),VSD | [41,48,54] |
| 46 | 13 | c.1529A > C | p.Q510P | 112,926,909 | rs121918470 | 27.9 | D | PD | D | DC | P | NS (PS) | [41] |
| 47 | 13 | c.1529A > G | p.Q510R | 112,926,909 | rs121918470 | 27.5 | D | PD | D | DC | P | NS (PS, HCM) | [41,42] |
| 48 | 3 | c.166A > G | p.I56V* | 112,888,150 | rs397507504 | 23.6 | D | PD | N | DC | P | HCM | [42] |
| 49 | 3 | c.205G > C | p.E69Q | 112,888,189 | rs397507511 | 25.5 | D | PD | D | DC | P | NS | [42,45] |
| 50 | 7 | c.846C > G | p.I282M* | 112,910,837 | rs397507530 | 23.2 | D | PD | D | DC | P | NS | [42] |
| 51 | 3 | c.181G > C | p.D61H | 112,888,165 | rs397507510 | 29.2 | D | PD | D | DC | P | NS (AVSD) | [53] |
| 52 | 13 | c.1472C > T | p.P491L | 112,926,852 | rs397507540 | 23.7 | T | PD | D | DC | P | NS | [45] |
| 53 | 3 | c.317A > G | p.D106G | 112,888,301 | NA | 29.3 | D | PD | D | DC | P | NS | [45] |
| 54 | 13 | c.1492C > T | P.R498W | 112,926,872 | rs397507541 | 31 | D | PD | D | DC | P | LS (HCM) | [46] |
| 55 | 13 | c.1493G > T | P.R498L | 112,926,873 | rs397507542 | 31 | D | PD | D | DC | P | LS (HCM) | [46] |
| 56 | 13 | c.1517A > C | P.Q506P | 112,926,897 | rs397507548 | 27.2 | D | PD | D | DC | P | LS (PS) | [46] |
| 57 | 7 | c.836A > C | P.Y279S | 112,910,827 | rs121918456 | 27.7 | D | PD | D | DC | P | LS (HCM) | [46,47] |
| 58 | 7 | c.853 T > A | p.F285I | 112,910,844 | rs397507531 | 32 | D | PD | D | DC | P | NS | [51] |
| 59 | 12 | c.1391G > C | p.G464A | 112,926,258 | rs121918469 | 28.4 | D | PD | D | DC | P | NS | [51] |
| 60 | 13 | c.1507G > A | p.G503R | 112,926,887 | rs397507545 | 31 | T | PD | D | DC | P | NS (PS) | [41,51] |
| 61 | 13 | c.1508G > A | p. G503E | 112,926,888 | rs397507546 | 29.3 | T | PD | D | DC | P | NS (PS) | [51] |
| 62 | 13 | c.1510A > T | p.M504L | 112,926,890 | NA | 23.3 | T | B | N | DC | NA | NS | [51] |
| 63 | 13 | c.1471C > G | p.P491A | 112,926,851 | rs397507539 | 22.6 | T | B | N | DC | P | NS (PS, ASD) | [49] |
| 64 | 13 | c.1530G > C | p.Q510H | 112,926,910 | rs397507550 | 24.5 | D | PD | D | DC | P | LS (PS, ASD, HCM) | [54,55] |
| 65 | 13 | c.1510A > C | p.M504L | 112,926,890 | NA | 23.1 | T | B | N | DC | NA | NS | [43] |
CHD congenital heart disease, NS Noonan syndrome, LS LEOPARD syndrome, PS pulmonary valve stenosis, ASD atrial septal defects, VSD ventricular septal defect, AVSD atrioventricular septal defect, AVCD atrioventricular canal defect, PST paroxysmal supraventricular tachycardia, PDA patent ductus arteriosus, HCM hypertrophic cardiomyopathy, PVS pulmonary vein stenosis, HIS hypertrophy interventricular septum, DC disease causing, D deleterious, T tolerated, N neutral, B, benign, PD probably damaging, P pathogenic, B benign, NA not available/not applicable
CADD, Phred ≤ 20: damaging; Phred > 20: natural
SIFT, score ≤ 0.05: deleterious; score > 0.05: tolerable
Polyphen-2, score = 0–0.15: benign; score = 0.15–0.85: possibly damaging; score = 0.85–1: probably damaging
PROVEAN, score ≤ − 2.5: deleterious; score > − 2.5: natural
Family I
The proband (III-3) was a 2-year-old girl diagnosed with CHD (Fig. 1A1). She was referred to Rajaie Cardiovascular Medical and Research Center to undergo WES. Computed tomography angiography was performed (Fig. 2A1). The proband of the first pedigree had severe cyanosis. A diagnostic workup showed that her 8-year-old brother (III-2) also had the same CHD. In both patients of this family, dysmorphology was evaluated by a medical geneticist, and no syndromic features were diagnosed. The inheritance pattern of the studied family was autosomal dominant (Fig. 1A1). Filtering the WES data yielded 2 variants as final candidate variants, probably responsible for CHDs in the family. In addition, PCR-based Sanger sequencing was performed on all available family members to confirm the identified variants observed in the family and to find a heterozygous stop-gain variant (NM_182925.5: c.C244T: p.R82X) in exon 3 of the FLT4 gene in the studied proband and her affected brother. The heterozygous c.C244T variant in the FLT4 gene was detected in her healthy mother (II-6), while her healthy father (II-5) lacked this variant (Fig.1 A2). The other nonsynonymous variant in the TRRAP gene did not segregate with the defects in the family. WES on the proband’s genomic DNA identified the variant in the FLT4 gene as the genetic cause of CHDs in the family. According to the American College of Medical Genetics and Genomics 2015 (ACMG) [56], the c.C244T variant is a pathogenic variant (criteria: PVS1, PM2, PP1, PP4, and PP5). The stop-gain variant was considered the cause of the disease by MutationTaster and CADD. The sequence alignments of proteins showed that the stop-gain variant occurred within a highly conserved amino acid across various species, which supports its essential performance (Fig. 1A3).
Family II
The proband (III-2) was a 3-year-old girl diagnosed with CHD (Fig. 1B1). She was referred to Rajaie Cardiovascular Medical and Research Center to undergo WES. The computed tomography angiography results are displayed in Fig. 2B1. The proband of the second pedigree was cyanotic. The diagnostic workup showed that her 6-year-old sister (III-1) had the same CHD. In both patients of this family, dysmorphology was evaluated by a medical geneticist, and no syndromic features were diagnosed. The inheritance pattern of the second family was autosomal dominant (Fig. 1B1). Six variants were identified as final candidate variants, probably responsible for CHDs in the family. PCR-based Sanger sequencing confirmed the presence of the nonsynonymous variant (NM_002834: c.C1403T:p.T468M) in exon 12 of the PTPN11 gene in the studied proband and her affected sister (Fig. 1B2). The affected proband (III-2) and her affected sister (III-1) were heterozygous for this locus, whereas her unaffected father (II-4) was heterozygous for this locus, and his unaffected mother (II-5) lacked the variant (Fig. 1B2). The other variants in 5 genes (AKAP12, DNAH11, HNRNPC, NPHP4, and FBN1) did not segregate with the defects in this family. WES on the proband’s genomic DNA detected a heterozygous nonsynonymous variant (c.C1403T:p.T468M) in the PTPN11 gene, which co-segregated with the defect within the family. According to the ACMG 2015 [56], the c.C1403T variant is a pathogenic variant (criteria: PS3, PM1, PM2, PM5, PP1, PP2, PP3, PP4, and PP5). The nonsynonymous variant was considered the cause of the disease by SIFT, PolyPhen, PROVEAN, MutationTaster, and CADD. The amino acid residue 468 of the human protein-tyrosine phosphatase, non-receptor type 11 is highly conserved among vertebrates (Fig.1B3).
Protein structure modeling
Docking was performed between the normal/mutant VEGFR3 and VEGFC by obtaining the 3D structure of the normal/mutant VEGFR3 from the SWISS-MODEL server and downloading the 3D structure of VEGFC from PDB (ID: 2X1W, resolution: 2.7 Å). Further, the HADDOCK scores of the normal and mutant VEGFR3 with VEGFC were − 55.0 ± 2.5 and − 61.6 ± 18.5, respectively. The docking results indicated that the binding affinity of the mutant VEGFR3 to VEGFC was reduced. The binding site of the normal VEGFR3 was surrounded by 16 amino acids, which formed 21 hydrogen bonds with VEGFC. Nonetheless, in the Arg82Ter mutant VEGFR3, only 3 amino acids (Glu37, Ser38, and Ile41) formed 5 hydrogen bonds with VEGFC. This stop-gain variant reduced the hydrogen bond and the affinity with VEGFC (Figs. 3 and 4). Docking was performed between the normal/mutant SHP-2 and Gab-1 by downloading the 3D structures of the human SHP-2 (normal ID: 7jvn, resolution: 1.92 Å and mutant ID: 4ohl, resolution: 2.4 Å) and Gab-1 (ID: 4qsy, resolution: 2.1 Å) from PDB. The HADDOCK scores of the normal/mutant SHP-2 with Gab-1 were − 28.0 ± 7.5 and − 25.1 ± 4.1, respectively. The normal SHP-2 had 4 hydrogen bonds with Gab-1, and the T468M mutant SHP-2 had 10 hydrogen bonds. This variant led to more hydrogen bonds and hydrophobic interactions at the surface of SHP-2 with Gab-1 and a higher binding affinity. This increased affinity weakened the N-SH2–PTP inter-domain interaction in the T468M mutant SHP-2 and enhanced the binding of N-SH2 with Gab-1 (Figs. 5 and 6).
Discussion
In the present study, WES unraveled a novel stop-gain variant, which affected the FLT4 gene, as well as a known nonsynonymous c.C1403T:p.T468M variant in the PTPN11 gene. VEGFs regulate angiogenesis and vasculogenesis by binding to the receptor tyrosine kinases VEGFR1, VEGFR2, and VEGFR3 [57]. The FLT4 gene encodes VEGFR3, which plays a unique role in the survival, proliferation, and migration of lymphatic endothelial cells. In adults, VEGFR3 is predominantly expressed in lymphatic endothelial cells and is crucial to lymphatic vessel growth [58]. Furthermore, this receptor is expressed in vascular endothelial cells during embryonic development, which is crucial to the development of blood vessels [59]. This human gene has 31 exons, which encode a protein with an extracellular region consisting of 7 immunoglobulin-like domains, 1 transmembrane region, 2 intracellular tyrosine kinase domains, and 1 c-terminal tail [24]. The NOTCH1 and FLT4 genes are the genes most frequently implicated in the etiology of the tetralogy of Fallot, with their variants accounting for almost 7% of all cases [60]. In a prior study, loss-of-function variants in the FLT4 gene were found in 2.3% of the recruited patients with the tetralogy of Fallot [27].
A cohort study by Page et al. showed that the variants of the FLT4 gene contributed to the incidence of the tetralogy of Fallot, accounting for 2.4% of the studied patients. In that study, 22 FLT4 variants were reported in 21 patients with the tetralogy of Fallot. In addition, 16 of the variants identified were loss of function: 4 splice variants (c.3002-1C > T, c.3002-2 T > C, c.2300C > G, and c.2849del21), 6 indels resulting in frameshifts and premature truncation (p.P363fsX25, p.Q423fsX3, p.L636fsX3, p.Y853fsX20, p.N905fsX20, and p.Y1337fsX19), and 6 premature termination codons (p.Y361X, p.Y369X, p.E896X, p.Q920X, p.R1031X, and p.Q1126X) [59].
A genome-sequencing study conducted by Reuter et al. reported 9 novel variants in the FLT4 gene related to the tetralogy of Fallot [19]. Elsewhere, a frameshift deletion in the FLT4 gene in a patient with the tetralogy of Fallot was detected by WES [44]. A link has been reported between FLT4 loss-of-function mutations and the tetralogy of Fallot, and VEGF signaling seems to be a new mechanism contributing to the pathogenesis of the disease [19, 59, 61]. The FLT4 mutations identified in patients with the tetralogy of Fallot are mainly missense or truncating variants in extracellular domains. Nevertheless, all the FLT4 mutations (missense or small in-frame deletions) known to cause Milroy disease have been located in 2 intracellular kinase domains (exons 17–26) and are assumed to interfere with the tyrosine kinase activation of the VEGFR3 receptor [15, 58]. The mutations within the tyrosine kinase domains of the VEGFR3 receptor lead to decreased tyrosine kinase activity [28]. The mutant receptor is maintained longer on the cell surface than is the wild type; thus, the amount of the mutant receptor on the surface of the endothelial cell is higher, which probably leads to lymphedema by reducing the relative rate of ligand binding to the active wild type [58]. Page et al. maintained that CHDs went unreported in patients with VEGFR3 variants causing Milroy disease [59]. In the current investigation, we conducted WES on a patient with CHD and succeeded in identifying a heterozygous stop-gain variant in the FLT4 gene. Further, the pathogenic stop-gain variant (c.C244T: p.R82X) in the FLT4 gene altered an arginine amino acid to a premature termination codon at codon 82, leading to the truncation of the VEGFR3 protein. The identified variant was not reported in the Genome Aggregation Database (gnomAD), the 1000 Genomes Project, ExAC, and ESP6500. This variant has been categorized in the Catalogue of Somatic Mutations in Cancer (COSMIC) as COSM126706. The C/T transition at position 244 in exon 3 predicted Arg82Ter within the VEGFR3 protein. The mutated residue was located in the VEGFR3 protein and disturbed its function by abnormally shortening it, with the amino acids lost affecting its main activity. Moreover, Arg82 is highly conserved among species, suggesting that the mutated amino acid is essential for protein function (Fig. 1A3). The detected variant in the FLT4 gene can change the arginine amino acid to a premature termination codon at position 82 in the VEGFR3 protein, denoting a feasible mechanism for the pathologies associated with pulmonary atresia and ventricular septal defects. Most FLT4 variants are associated with the tetralogy of Fallot; consequently, this CHD presents with a combination of defects such as ventricular septal defects, pulmonary valve stenosis, right ventricular hypertrophy, and the overriding aorta. In the present study, both patients of Family I had the identified FLT4 variant, which is associated with large ventricular septal defects and pulmonary atresia with hypoplastic pulmonary artery branches and multiple aortopulmonary collaterals. This finding may indicate the diversity of the FLT4 phenotype.
The PTPN11 gene encodes SHP-2, which participates in signaling cascades downstream for cytokines and growth factors [62]. The mutations of the PTPN11 gene have been identified in Noonan syndrome and LEOPARD syndrome [63]. In addition, the PTPN11 gene is a candidate gene for contributions to the risk of the nonsyndromic tetralogy of Fallot [64]. Pulmonary valve stenosis is the most common CHD in Noonan syndrome patients with PTPN11 mutations [40]. The PTPN11 gene consists of 16 exons, which translate into a 593-amino acid protein with 4 distinct domains: 2 tandem SH2 domains (N-SH2 and C-SH2) on the N-terminal side, 1 protein-tyrosine phosphatase (PTP) domain, and 1 C-terminal hydrophilic tail on the C-terminal side [65]. The PTP domain and the 2 SH2 domains are the functional domains of the SHP-2 protein, and PTP activity is regulated by the SH2 domains (particularly N-SH2) [65, 66]. The N-SH2 domain works as a molecular switch in the SHP-2 protein, which interacts with the PTP domain in the inactive conformation, blocking the catalytic site [67]. PTPN11 missense mutations were demonstrated to be gain-of-function changes that disrupted the intramolecular interaction between the PTP and N-SH2 domains, leading to increased SHP-2 activity [66]. The PTPN11 mutations related to the N-SH2 and PTP domains can stabilize the active conformation of the SHP-2 protein [62]. The reported mutations in the PTPN11 gene are clustered in 7 exons (2, 3, 4, 7, 8, 12, and 13) [68]. According to a study by Athota et al., the highest numbers of pathogenic variants were detected in exons 3, 8, and 13 [65]. The PTPN11 mutations related to LEOPARD syndrome cluster in exons 7 and 12, and those related to Noonan syndrome cluster in exons 3 and 8 [69].
In the current study, using the WES facility, we presented an Iranian family with a known variant in the PTPN11 gene. A medical geneticist confirmed nonsyndromic symptoms in the investigated family, who exhibited no features of Noonan syndrome. Furthermore, the c.C1403T variant in the PTPN11 gene led to the substitution of methionine for threonine at position 468. The identified variant in the PTPN11 gene was not reported in the 1000 Genomes Project and ESP6500. The variant has been cataloged in dbSNP as rs121918457 with a minor allele frequency of 0.000008236 in ExAC, with a minor allele frequency of 0.000004066 in gnomAD, and as COSM170715 in COSMIC. The C/T transition at position 1403 in exon 12 predicted the substitution of Met for Thr468 within the PTP domain of the SHP-2 protein. This residue is located in the protein tyrosine signature motif (positions 457–469), involved in phosphate binding. The mutated residue is located in the PTP domain, which plays a significant role in the main activity of the SHP-2 protein. The normal function of the SHP-2 protein can be consequently disturbed by the variant. Furthermore, the fact that Thr468 is highly conserved among species suggests that the mutated amino acid could be essential for protein function (Fig. 1B3). The detected variant in the PTPN11 gene can change the amino acid at position 468 in the PTP domain, hinting at a feasible mechanism for the pathologies associated with pulmonary valve stenosis, ventricular septal defects, and atrial septal defects. In this study, the identified PTPN11 variant was associated with nonsyndromic CHDs.
Concerning Family I, we examined the proband (III-3), her healthy parents, and her affected brother (III-2) with pulmonary atresia and a ventricular septal defect (Fig. 1A1). Regarding Family II, we examined the proband (III-2), her healthy parents, and her affected sister (III-1) with pulmonary stenosis, an atrial septal defect, and a ventricular septal defect (Fig. 1B1). In Family I, the 2 affected siblings had the same variant, while their mother was healthy despite having the same variant. In Family II, the 2 affected siblings had the same variant, while their father was healthy despite having the same variant. Phenotypic variability is observed in the members of such families with the same variants. Different phenotypes in individuals with the same variants can be caused by the incomplete penetrance of the phenotype. In both families, the asymptomatic parents of the affected children were unaffected carriers of the variants, indicating incomplete penetrance. Previous research suggests that the identified variants in the FLT4 and PTPN11 genes could cause the incomplete penetrance of the phenotypes [25, 59, 70]. Patients with the tetralogy of Fallot have inherited a variant of the FLT4 gene from an asymptomatic parent, indicating that the mutant allele has reduced penetrance. Although the tetralogy of Fallot is seldom inherited in a Mendelian fashion, the penetrance of susceptibility variants is influenced by environmental and genetic factors [15, 59]. In the current study, we detected a novel pathogenic variant in the FLT4 gene, which was associated with pulmonary atresia and ventricular septal defects in the first family. Identification of this novel variant via WES can improve the genetic diagnosis of cardiovascular diseases such as pulmonary atresia and ventricular septal defects. Notably, in the current investigation, we utilized molecular modeling to examine the molecular mechanism of the detected variant in the PTPN11 gene for pathologies associated with pulmonary valve stenosis, ventricular septal defects, and atrial septal defects. Given the genetic heterogeneity of CHDs, finding the exact genetic cause is challenging. Despite a pattern of autosomal dominant inheritance in most families, genetic counseling in familial CHDs is complicated by reduced penetrance and variable expressivity [71]. Due to the hitherto unknown mechanisms involved in CHDs and the diversity in the nonsyndromic CHD spectrum, the molecular diagnosis of patients with such cardiac defects via WES could be deemed an appropriate, cost-effective approach [72, 73]. Additionally, our evaluation of these 2 families showed that WES was efficient in the accurate diagnosis of the genetic causes of CHDs with incomplete penetrance.
Conclusions
The current study presents the first report of a novel pathogenic c.C244T variant in the FLT4 gene, resulting in CHDs in an Iranian family. In addition, a heterozygous nonsynonymous c.C1403T variant in the PTPN11 gene was identified through WES. Notably, WES is a preferable diagnostic implement for evaluating the complex genetics of CHDs.
Acknowledgements
The authors wish to acknowledge the kind contribution of the family described herein. This research was funded by the Cardiogenetics Research Center, Rajaie Cardiovascular Medical and Research Center, Tehran, Iran.
Author contributions
SK and TT drafted the work. SK, AT, and MM designed the project. SK performed WES. TT, MP, NN, and SGH collected the data. MM and AT surveyed the patients clinically. All the authors read and approved the final manuscript.
Funding
The authors received no specific funding for this research.
Availability of data and materials
All data generated or analyzed during this study are included in this manuscript. The accession number of the identified variant in ClinVar is as follows: NM_182925.5 (FLT4):c.244C > T (p.Arg82Ter): VCV001177461.1.
Declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of Rajaie Cardiovascular Medical and Research Center, Iran University of Medical Sciences, Tehran, Iran (IR.RHC.REC.1399.017). Informed consent was obtained from the probands’ parents and/or legal guardians for study participation. The age of the participant’s family’s patients was less than 10 years.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Xie D, et al. Perinatal outcomes and congenital heart defect prognosis in 53313 non-selected perinatal infants. PLoS ONE. 2017 doi: 10.1371/journal.pone.0177229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie D, et al. Epidemiology and major subtypes of congenital heart defects in Hunan Province, China. Medicine. 2018 doi: 10.1097/MD.0000000000011770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48:455–463. doi: 10.1093/ije/dyz009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiao F, Hu P, Xu Z. Application of next-generation sequencing for the diagnosis of fetuses with congenital heart defects. Curr Opin Obstet Gynecol. 2019;31:132–138. doi: 10.1097/GCO.0000000000000520. [DOI] [PubMed] [Google Scholar]
- 5.Pierpont M E, et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American heart association. Circulation. 2018;138:e653–e711. doi: 10.1161/CIR.0000000000000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llamosas-Falcón L, et al. Tetralogy of Fallot in Spain: a nationwide registry-based mortality study across 36 years. Orphanet J Rare Dis. 2019;14:1–8. doi: 10.1186/s13023-019-1056-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manshaei R, et al. Genes and pathways implicated in tetralogy of Fallot revealed by ultra-rare variant burden analysis in 231 genome sequences. Fronti Genet. 2020 doi: 10.3389/fgene.2020.00957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pignatelli R H, Noel C, Reddy S C B. Imaging of the pulmonary valve in the adults. Curr Opin Cardiol. 2017;32:529–540. doi: 10.1097/HCO.0000000000000436. [DOI] [PubMed] [Google Scholar]
- 9.Tsunoda S, et al. Isolated pulmonary valvular stenosis with equilateral triangle orifice. J Cardiol Cases. 2020;21:145–148. doi: 10.1016/j.jccase.2019.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang, J., Ko, J. M., Guileyardo, J. M. & Roberts, W. C. A review of spontaneous closure of ventricular septal defect. Baylor University Medical Center Proceedings (2015) 28, 516-520 [DOI] [PMC free article] [PubMed]
- 11.Lertsakulpiriya K, et al. Current era outcomes of pulmonary atresia with ventricular septal defect: a single center cohort in Thailand. Sci Rep. 2020;10:1–8. doi: 10.1038/s41598-020-61879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czarny M J, Resar J R. Diagnosis and management of valvular aortic stenosis. Clin Med Insights: Cardiol. 2014 doi: 10.4137/CMC.S15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel A, Kirtane A J. Aortic valve stenosis. JAMA Cardiol. 2016;1:623–623. doi: 10.1001/jamacardio.2016.2060. [DOI] [PubMed] [Google Scholar]
- 14.Edwards J J, Gelb B D. Genetics of congenital heart disease. Curr Opin Cardiol. 2016;31:235. doi: 10.1097/HCO.0000000000000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monaghan R M, Page D J, Ostergaard P, Keavney B D. The physiological and pathological functions of VEGFR3 in cardiac and lymphatic development and related diseases. Cardiovasc Res. 2021;117:1877–1890. doi: 10.1093/cvr/cvaa291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tartaglia M, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. AmJ Hum Genet. 2002;70:1555–1563. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tartaglia M, Gelb B D, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab. 2011;25:161–179. doi: 10.1016/j.beem.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowczenio D M, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35:E2403–E2412. doi: 10.1002/humu.22619. [DOI] [PubMed] [Google Scholar]
- 19.Reuter MS, Jobling R, Chaturvedi RR, Manshaei R, Costain G, Heung T, Curtis M, Hosseini SM, Liston E, Lowther C. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genetics in Medicine (2019) 21, 1001-1007. [DOI] [PMC free article] [PubMed]
- 20.Hamada K, et al. VEGF-C signaling pathways through VEGFR-2 and VEGFR-3 in vasculoangiogenesis and hematopoiesis. Blood. 2000;96:3793–3800. doi: 10.1182/blood.V96.12.3793. [DOI] [PubMed] [Google Scholar]
- 21.Xu Z Q, et al. PTPN11 gene mutations and its association with the risk of congenital heart disease. Dis Markers. 2022 doi: 10.1155/2022/8290779. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Yu Z-H, et al. Molecular basis of gain-of-function LEOPARD syndrome-associated SHP2 mutations. Biochemistry. 2014;53:4136–4151. doi: 10.1021/bi5002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang F, et al. Gain-of-function E76K-mutant SHP2 promotes cell proliferation, metastasis, and tumor growth in glioblastoma through activation of the ERK/CREB pathway. Onco Targets Ther. 2019;12:9435. doi: 10.2147/OTT.S222881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon K, et al. FLT 4/VEGFR 3 and Milroy disease: novel mutations, a review of published variants and database update. Hum Mutat. 2013;34:23–31. doi: 10.1002/humu.22223. [DOI] [PubMed] [Google Scholar]
- 25.Evans A, et al. Identification of eight novel VEGFR-3 mutations in families with primary congenital lymphoedema. J Med Genet. 2003;40:697–703. doi: 10.1136/jmg.40.9.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michelini S, et al. Clinical and genetic study of 46 Italian patients with primary lymphedema. Lymphology. 2012;45:3–12. [PubMed] [Google Scholar]
- 27.Jin S C, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. doi: 10.1038/ng.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karkkainen M J, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- 29.Ghalamkarpour A, et al. Sporadic in utero generalized edema caused by mutations in the lymphangiogenic genes VEGFR3 and FOXC2. J Pediatr. 2009;155:90–93. doi: 10.1016/j.jpeds.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 30.Mendola A, et al. Mutations in the VEGFR3 signaling pathway explain 36% of familial lymphedema. Molecular syndromology. 2013;4:257–266. doi: 10.1159/000354097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butler M G, Dagenais S L, Rockson S G, Glover T W. A novel VEGFR3 mutation causes Milroy disease. Am J Med Genet Part A. 2007 doi: 10.1002/ajmg.a.31703. [DOI] [PubMed] [Google Scholar]
- 32.Liu N, Yu Z, Luo Y, Sun D, Yan Z. A novel FLT4 gene mutation and MR lymphangiography in a Chinese family with Milroy disease. Lymphology. 2015;48:93–96. [PubMed] [Google Scholar]
- 33.Melikhan-Revzin S, Kurolap A, Dagan E, Mory A, Gershoni-Baruch R. A novel missense mutation in FLT4 causes autosomal recessive hereditary lymphedema. Lymphat Res Biol. 2015;13:107–111. doi: 10.1089/lrb.2014.0044. [DOI] [PubMed] [Google Scholar]
- 34.Futatani T, et al. Molecular characterization of two novel VEGFR3 mutations in Japanese families with Milroy’s disease. Pediatr Int. 2008;50:116–118. doi: 10.1111/j.1442-200X.2007.02505.x. [DOI] [PubMed] [Google Scholar]
- 35.Ghalamkarpour A, et al. Hereditary lymphedema type I associated with VEGFR3 mutation: the first de novo case and atypical presentations. Clin Genet. 2006;70:330–335. doi: 10.1111/j.1399-0004.2006.00687.x. [DOI] [PubMed] [Google Scholar]
- 36.Carver C, et al. Three children with Milroy disease and de novo mutations in VEGFR3. Clin Genet. 2007;71:187–189. doi: 10.1111/j.1399-0004.2007.00741.x. [DOI] [PubMed] [Google Scholar]
- 37.Liu N, Gao M. FLT4 mutations are associated with segmental lymphatic dysfunction and initial lymphatic aplasia in patients with Milroy disease. Genes. 2021;12:1611. doi: 10.3390/genes12101611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DiGiovanni R, Erickson R, Ohlson E, Bernas M, Witte M H. A novel FLT4 gene mutation identified in a patient with Milroy disease. Lymphology. 2014;47:44–47. [PubMed] [Google Scholar]
- 39.Daniel-Spiegel E, et al. Hydrops fetalis: an unusual prenatal presentation of hereditary congenital lymphedema. Prenat Diagn. 2005;25:1015–1018. doi: 10.1002/pd.1237. [DOI] [PubMed] [Google Scholar]
- 40.Sarkozy A, et al. Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J Med Genet. 2003;40:704–708. doi: 10.1136/jmg.40.9.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ezquieta B, et al. Alterations in RAS-MAPK genes in 200 Spanish patients with Noonan and other neuro-cardio-facio-cutaneous syndromes Genotype and cardiopathy. Rev Esp Cardiol. 2012;65:447–455. doi: 10.1016/j.recesp.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 42.Atik T, et al. Mutation spectrum and phenotypic features in Noonan syndrome with PTPN11 mutations: definition of two novel mutations. Ind J Pediatr. 2016;83:517–521. doi: 10.1007/s12098-015-1998-6. [DOI] [PubMed] [Google Scholar]
- 43.Narayanan D L, et al. Hotspots in PTPN11 gene among Indian children With Noonan syndrome. Indian Pediatr. 2017;54:638–640. doi: 10.1007/s13312-017-1125-z. [DOI] [PubMed] [Google Scholar]
- 44.Szot J O, et al. A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ Genom Precis Med. 2018;11:e001978. doi: 10.1161/CIRCGEN.117.001978. [DOI] [PubMed] [Google Scholar]
- 45.Bertelloni S, et al. IGF-I generation test in prepubertal children with Noonan syndrome due to mutations in the PTPN11 gene. Hormones. 2013;12:86–92. doi: 10.1007/BF03401289. [DOI] [PubMed] [Google Scholar]
- 46.Sarkozy A, et al. Clinical and molecular analysis of 30 patients with multiple lentigines LEOPARD syndrome. J Med Genet. 2004;41:e68–e68. doi: 10.1136/jmg.2003.013466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keren B, et al. PTPN11 mutations in patients with LEOPARD syndrome: a French multicentric experience. J Med Genet. 2004;41:e117–e117. doi: 10.1136/jmg.2004.021451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi K, et al. A novel mutation in the PTPN11 gene in a patient with Noonan syndrome and rapidly progressive hypertrophic cardiomyopathy. Eur J Pediatr. 2005;164:497–500. doi: 10.1007/s00431-005-1679-y. [DOI] [PubMed] [Google Scholar]
- 49.Ueda K, Yaoita M, Niihori T, Aoki Y, Okamoto N. Craniosynostosis in patients with RASopathies: accumulating clinical evidence for expanding the phenotype. Am J Med Genet A. 2017;173:2346–2352. doi: 10.1002/ajmg.a.38337. [DOI] [PubMed] [Google Scholar]
- 50.Arvaniti A, Samakouri M, Keskeridou F, Veletza S. Concurrence of anorexia nervosa and Noonan syndrome. Eur Eat Disord Rev. 2014;22:83–85. doi: 10.1002/erv.2261. [DOI] [PubMed] [Google Scholar]
- 51.Ferrero G B, et al. Clinical and molecular characterization of 40 patients with Noonan syndrome. Eur J Med Genet. 2008;51:566–572. doi: 10.1016/j.ejmg.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 52.Weismann C G, et al. PTPN11 mutations play a minor role in isolated congenital heart disease. Am J Med Genet A. 2005;136:146–151. doi: 10.1002/ajmg.a.30789. [DOI] [PubMed] [Google Scholar]
- 53.Croonen E A, et al. Prenatal diagnostic testing of the Noonan syndrome genes in fetuses with abnormal ultrasound findings. Eur J Hum Genet. 2013;21:936–942. doi: 10.1038/ejhg.2012.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wakabayashi Y, et al. Implantable cardioverter defibrillator for progressive hypertrophic cardiomyopathy in a patient with LEOPARD syndrome and a novel PTPN11 mutation Gln510His. Am J Med Genet A. 2011;155:2529–2533. doi: 10.1002/ajmg.a.34194. [DOI] [PubMed] [Google Scholar]
- 55.Gezdirici A, et al. How necessary is to analyze PTPN11 gene in fetuses with first trimester cystic hygroma and normal karyotype? J Matern Fetal Neonatal Med. 2017;30:938–941. doi: 10.1080/14767058.2016.1191463. [DOI] [PubMed] [Google Scholar]
- 56.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korpelainen E I, Alitalo K. Signaling angiogenesis and lymphangiogenesis. Curr Opin Cell Biol. 1998;10:159–164. doi: 10.1016/S0955-0674(98)80137-3. [DOI] [PubMed] [Google Scholar]
- 58.Sui Y, et al. A family with Milroy disease caused by the FLT4/VEGFR3 gene variant c. 2774 T> A. BMC Med Genomics. 2021;14:1–8. doi: 10.1186/s12920-021-00997-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Page D J, et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circ Res. 2019;124:553–563. doi: 10.1161/CIRCRESAHA.118.313250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matos-Nieves A, Yasuhara J, Garg V. Another notch in the genetic puzzle of tetralogy of fallot. Circ Res. 2019 doi: 10.1161/CIRCRESAHA.118.314520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diab N S, et al. Molecular genetics and complex inheritance of congenital heart disease. Genes. 2021;12:1020. doi: 10.3390/genes12071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Essawi M L, et al. Mutational analysis of the PTPN11 gene in Egyptian patients with Noonan syndrome. J Formos Med Assoc. 2013;112:707–712. doi: 10.1016/j.jfma.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 63.Sarkozy A, et al. A novel PTPN11 gene mutation bridges Noonan syndrome, multiple lentigines/LEOPARD syndrome and Noonan-like/multiple giant cell lesion syndrome. Eur J Hum Genet. 2004;12:1069–1072. doi: 10.1038/sj.ejhg.5201290. [DOI] [PubMed] [Google Scholar]
- 64.Goodship J A, et al. A common variant in the PTPN11 gene contributes to the risk of tetralogy of Fallot. Circ Cardiovasc Genet. 2012;5:287–292. doi: 10.1161/CIRCGENETICS.111.962035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Athota J P, et al. Molecular and clinical studies in 107 Noonan syndrome affected individuals with PTPN11 mutations. BMC Med Genet. 2020;21:1–10. doi: 10.1186/s12881-020-0986-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hung C-S, et al. Mutational analysis of PTPN11 gene in Taiwanese children with Noonan syndrome. J Formos Med Assoc. 2007;106:169–172. doi: 10.1016/S0929-6646(09)60235-7. [DOI] [PubMed] [Google Scholar]
- 67.Hof P, Pluskey S, Dhe-Paganon S, Eck M J, Shoelson S E. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–450. doi: 10.1016/S0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 68.Sznajer Y, et al. The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics. 2007;119:e1325–e1331. doi: 10.1542/peds.2006-0211. [DOI] [PubMed] [Google Scholar]
- 69.Formigari R, et al. Genetic syndromes and congenital heart defects: how is surgical management affected? Eur J Cardiothorac Surg. 2009;35:606–614. doi: 10.1016/j.ejcts.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 70.Sobreira N L, et al. Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet. 2010;6:e1000991. doi: 10.1371/journal.pgen.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jia Y, et al. The diagnostic value of next generation sequencing in familial nonsyndromic congenital heart defects. Am J Med Genet A. 2015;167:1822–1829. doi: 10.1002/ajmg.a.37108. [DOI] [PubMed] [Google Scholar]
- 72.Kalayinia S, Maleki M, Mahdavi M, Mahdieh N. A novel de novo dominant mutation of NOTCH1 gene in an Iranian family with non-syndromic congenital heart disease. J Clin Lab Anal. 2020;34:e23147. doi: 10.1002/jcla.23147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalayinia S, Goodarzynejad H, Maleki M, Mahdieh N. Next generation sequencing applications for cardiovascular disease. Ann Med. 2018;50:91–109. doi: 10.1080/07853890.2017.1392595. [DOI] [PubMed] [Google Scholar]
- 74.Karkainnen M, et al. Missense mutations interfere with VEGFR-3 signaling in primary lymphedema. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this manuscript. The accession number of the identified variant in ClinVar is as follows: NM_182925.5 (FLT4):c.244C > T (p.Arg82Ter): VCV001177461.1.