

Abstract
HIV-1 capsid (CA) performs multiple roles in the viral life cycle and is a promising target for antiviral development. In this work, we describe the design, synthesis, assessment of antiviral activity, and mechanistic investigation of 20 piperazinone phenylalanine derivatives with a terminal indole or benzene ring. Among them, F2-7f exhibited moderate anti-HIV-1 activity with an EC50 value of 5.89 μM, which was slightly weaker than the lead compound PF74 (EC50 = 0.75 μM). Interestingly, several compounds showed a preference for HIV-2 inhibitory activity, represented by 7f with an HIV-2 EC50 value of 4.52 μM and nearly 5-fold increased potency over anti-HIV-1 (EC50 = 21.81 μM), equivalent to PF74 (EC50 = 4.16 μM). Furthermore, F2-7f preferred to bind to the CA hexamer rather than to the monomer, similar to PF74, according to surface plasmon resonance results. Molecular dynamics simulation indicated that F2-7f and PF74 bound at the same site. Additionally, we computationally analyzed the ADMET properties for 7f and F2-7f. Based on this analysis, 7f and F2-7f were predicted to have improved drug-like properties and metabolic stability over PF74, and no toxicities were predicted based on the chemotype of 7f and F2-7f. Finally, the experimental metabolic stability results of F2-7f in human liver microsomes and human plasma moderately correlated with our computational prediction. Our findings show that F2-7f is a promising small molecule targeting the HIV-1 CA protein with considerable development potential.
Keywords: HIV-1, PF74, capsid modulators, target identification, mechanistic investigation
1. Introduction
Acquired immunodeficiency syndrome (AIDS) is a series of syndromes characterized by T cell immune deficiency caused by human immunodeficiency virus (HIV) infection [1]. Since the first case of AIDS was reported in 1981, approximately 40 million people have died of AIDS worldwide [2]. The rapid spread of AIDS and its high mortality rate threaten human health and social development. HIV contains two species: HIV-1 is the predominant pathogenic pathogen, but the danger of HIV-2 infection is increasing and has been discovered globally [3,4]. Current combination antiretroviral therapy (cART) and, in light of frequent mutations within the retrovirus, available antivirals can delay disease progression and suppress viral load in patients; however, a cure is currently not possible due to latent viral pools induced by cART [5,6,7]. Therefore, focusing on potential new targets and developing novel HIV inhibitors with new mechanisms of action are of great significance for developing antiviral drugs.
HIV-1 capsid (CA) is a structural protein encoded within the HIV-1 Gag gene [8]. The capsid protein forms a higher order structure that encapsulates and protects the core proteins, viral genome, and various enzymes essential for viral replication [9,10]. Because of its important role in the early and late stages of viral replication, CA has become a promising new target for anti-HIV-1 drug design [11,12]. The mature HIV-1 CA is a conical ‘fullerene-like’ (cone) lattice shell comprising approximately 1500 CA monomers, which are arranged in approximately 250 hexamers and 12 pentamers [13,14]. The mature CA monomer consists of an N-terminal domain (NTD), a C-terminal domain (CTD), and a flexible linker in the middle [15]. CA cone stability highly depends on intermolecular contacts such as NTD-NTD, NTD-CTD, and CTD-CTD between monomers of the hexamer or pentamer [16]. Notably, the NTD-CTD interprotomer pocket is the binding site of host factor cleavage and polyadenylation specificity factor 6 (CPSF6), nucleoporin 153 (NUP153), and Sec24C [17,18,19]. Interference with interactions between CA and host factors can affect HIV-1 replication. Furthermore, the NTD-CTD interface is also critical for CA assembly, which can affect the intrinsic flexibility of CA and subsequently destroy the integrity of virus particles [20,21]. Therefore, the NTD-CTD interface is an effective site for developing novel and potent CA modulators.
PF74, a small molecule modulator targeting the NTD-CTD interface of the adjacent subunit of CA hexamers, can inhibit viral replication through multiple effects and has received extensive attention [22,23]. PF74 comprises a phenylalanine core, an indole substituent, and a linker between them. Structural studies have shown that the phenylalanine core of PF74 interacts with Asn53, Asn57, Lys70, Ile73, and Tyr130. Within this interaction, the amide forms multiple, key hydrogen bonds with Asn57. The methyl indole moiety extends to the NTD-CTD interface and interacts with Gln63, Gln67, and Lys70 of the NTD moiety and Arg173 of the CTD moiety (Figure 1) [24,25]. However, the low antiviral activity and inferior metabolic stability of PF74 make it unsuitable for clinical applications [26]. Extensive decoration of the PF74 skeleton by Gilead Sciences generated lenacapavir (LEN; GS-6207), which has picomolar anti-HIV-1 activity (EC50 = 105 pM), favorable metabolic stability, and is approved by the European Community (EC) [27]. Nevertheless, limited by its complicated structure and labor-intensive synthesis, LEN displays extremely poor water solubility, resulting in mainly injectable use, and drug-resistant strains have also already appeared in clinical trials [28]. Further structural optimization of PF74 to identify novel CA modulators is needed to address these inadequacies. Crystallographic studies of PF74 bound within the interprotomer pocket of HIV-1 CA hexamers provide valuable information on binding site flexibility and size, which can guide the design of further modifications of PF74, especially at the tolerable indole, as well as the design of novel chemotypes. In this study, we chose to retain the privileged (intolerant) phenylalanine skeleton of PF74 and introduce a methoxy group at the para position of the aniline ring, which proved to have more favorable antiviral activity [29]. Fewer interactions between the indole ring of PF74 and CTD might be the reason for its low antiviral activity. Therefore, we extended the linker butane-2-one fragment to a 4-acetyl-1- (2-propionyl) piperazine-2-one fragment rich in hydrogen bond acceptors to maintain interactions with Lys70 and Arg173. Then, different substituted indole groups (series I) and aniline groups (series II) were introduced into the left wing, which were expected to form more interactions with the critical amino acids Gln67, Glu71, Tyr169, and Lys182 at the NTD-CTD interface (Figure 1), thus improving antiviral activity as well as drug-like properties.
Figure 1.

Design principle of piperazinone phenylalanine derivatives with terminal indole or benzene ring as HIV-1 CA modulators. The figure was generated in PyMOL (www.pymol.org, accessed on 20 October 2022).
Herein, our screening identified 20 piperazinone phenylalanine derivatives with a terminal indole or benzene ring that functioned as HIV-1 CA modulators. The antiviral activities of all newly synthesized compounds were tested using MTT assays, and structure-activity relationships (SARs) were established. We then performed surface plasmon resonance (SPR) and molecular dynamics simulation to investigate the mechanism of action of the representative compounds. Furthermore, the ADMET properties of the representative compounds and PF74 were computationally predicted. Finally, we experimentally assessed the metabolic stability of F2-7f in human liver microsomes (HLMs) and human plasma.
2. Results and Discussion
2.1. Chemistry
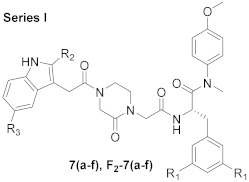
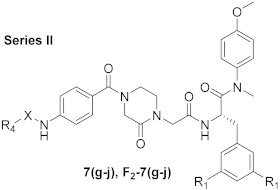
The synthetic route is shown in Scheme 1. N-(tert-butoxycarbonyl)-L-phenylalanine (1) and N-(tert-butoxycarbonyl)-3,5-difluoro-L-phenylalanine (F2-1) were treated with 4-methoxy-N-methylaniline and 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluoro (HATU) in the presence of N, N-diisopropylethyl-amine (DIEA) and dichloromethane (DCM) to obtain 2 and F2-2, followed by the removal of tert-butyloxycarbonyl (Boc) protection using trifluoroacetic acid (TFA), yielding the free amines 3 and F2-3. The acylation of 3 and F2-3 with bromoacetic acid in DCM resulted in the crucial intermediates 4 and F2-4. Further treatment of 4 and F2-4 with 1-Boc-3-oxopiperazine via a nucleophilic substitution (SN2) reaction afforded the intermediates 5 and F2-5. The removal of Boc protection of 5 and F2-5 yielded the free amines 6 and F2-6, which were treated with the corresponding substituted indoleacetic acids and the corresponding substituted benzoic acids in DCM under the action of HATU and DIEA to obtain the target compounds 7 (a–j) and F2-7 (a–j).
Scheme 1.

Reagents and conditions: (i) 4-Methoxy-N-methylaniline, HATU, DIEA, 0 °C to r.t.; (ii) TFA, DCM, r.t.; (iii) Bromoacetic Acid, HATU, DIEA, 0 °C to r.t.; (iv) 1-Boc-3-oxopiperazine, Cs2CO3, DMF, 45 °C; (v) TFA, DCM, r.t.; (vi) HATU, DIEA, 0 °C to r.t.
2.2. Antiviral Activity in MT-4 Cells against HIV-1 (IIIB) and HIV-2 (ROD) Replication
All newly synthesized compounds in this study were evaluated for their antiviral activity in an MT-4 cell-based MTT assay containing wild-type (WT) HIV-1 (IIIB) and HIV-2 (ROD), with PF74 serving as an in-line control. The test compounds’ selectivity index (SI value, CC50/EC50) was determined by assessing compound toxicity in MT-4 cells using the MTT assay. Table 1 and Table 2 display the results of the analysis.
Table 1.
Anti-HIV activity and cytotoxicity of series I in MT-4 cells.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound ID | R1 | R2 | R3 | EC50 a (μM) | CC50 b (μM) | SI c | ||
| IIIB | ROD | IIIB | ROD | |||||
| 7a | H | CH3 | H | 14.28 ± 0.76 | 7.74 ± 0.79 | 107.56 ± 3.26 | 7.73 | 13.90 |
| 7b | H | H | OCH3 | 14.58 ± 0.39 | 16.95 ± 3.58 | 104.21 ± 3.37 | 7.15 | 6.15 |
| 7c | H | H | Br | >16.37 | >16.37 | 16.37 ± 3.44 | <1 | <1 |
| 7d | H | H | H | 14.97 ± 0.24 | 4.89 ± 1.39 | 112.41 ± 3.22 | 7.51 | 22.99 |
| 7e | H | H | OH | 198.10 ± 9.74 | 186.48 ± 4.42 | >209.29 | >1.06 | >1.12 |
| 7f | H | H | F | 21.81 ± 4.81 | 4.52 ± 0.87 | 90.06 ± 15.72 | 4.13 | 19.92 |
| F2-7a | F | CH3 | H | 20.83 ± 5.09 | 19.66 ± 6.59 | 76.85 ± 19.98 | 3.69 | 3.91 |
| F2-7b | F | H | OCH3 | >15.94 | >15.94 | 15.94 ± 3.83 | <1 | <1 |
| F2-7c | F | H | Br | >3.15 | >3.15 | 3.15 ± 0.40 | <1 | <1 |
| F2-7d | F | H | H | 29.37 ± 7.03 | 17.61 ± 9.14 | 57.84 ± 21.81 | 1.97 | 3.28 |
| F2-7e | F | H | OH | >131.51 | ≥49.55 | 131.51 ± 32.96 | <1 | ≤2.65 |
| F2-7f | F | H | F | 5.89 ± 2.03 | 5.07 ± 0.63 | 16.36 ± 3.38 | 2.78 | 3.23 |
| PF74 | 0.75 ± 0.33 | 4.16 ± 2.02 | 32.27 ± 2.94 | 43.03 | 7.76 | |||
a EC50: concentration of the test compound to attain 50% protection against HIV-induced cytotoxicity, determined by the MTT assay. b CC50: concentration of the test compound to reduce the viability of mock-infected cell cultures by 50%, determined by the MTT assay; values were averaged from at least three independent experiments. c SI: selectivity index, the ratio of CC50/EC50.
Table 2.
Anti-HIV activity and cytotoxicity of series II in MT-4 cells.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound ID | R1 | X | R4 | EC50 a (µM) | CC50 b (µM) | SI c | ||
| IIIB | ROD | IIIB | ROD | |||||
| 7g | H | CO | CH3 | 203.14 ± 10.98 | 184.07 ± 19.33 | >213.43 | >1.05 | >1.16 |
| 7h | H | SO2 | CH3 | 192.39 ± 3.17 | >201.06 | >201.06 | >1.05 | ND |
| 7i | H | CO | cyclopropyl | >167.50 | 15.02 ± 2.27 | 167.50 ± 19.81 | <1 | 11.15 |
| 7j | H | CO | CF3 | 157.50 ± 6.77 | 14.95 ± 4.83 | >195.42 | >1.24 | >13.08 |
| F2-7g | F | CO | CH3 | 177.40 ± 16.55 | 192.14 ± 4.57 | 196.03 ± 6.80 | 1.11 | 1.02 |
| F2-7h | F | SO2 | CH3 | 160.53 ± 15.43 | 183.40 ± 2.77 | >190.06 | >1.18 | >1.04 |
| F2-7i | F | CO | cyclopropyl | 14.36 ± 1.31 | 15.10 ± 0.68 | 88.56 ± 11.27 | 6.17 | 5.87 |
| F2-7j | F | CO | CF3 | 13.74 ± 0.70 | 16.55 ± 6.13 | 150.38 ± 25.46 | 10.95 | 9.09 |
| PF74 | 0.75 ± 0.33 | 4.16 ± 2.02 | 32.27 ± 2.94 | 43.03 | 7.76 | |||
a EC50: concentration of the test compound to attain 50% protection against HIV-induced cytotoxicity, determined by the MTT assay. b CC50: concentration of the test compound to reduce the viability of mock-infected cell cultures by 50%, determined by the MTT assay; values were averaged from at least three independent experiments. c SI: selectivity index, the ratio of CC50/EC50.
Most of the compounds in series I showed moderate anti-HIV-1 activity. Unsubstituted compounds and those with an electron-donating substituent of the indole ring led to mid-range efficacy and low toxicity, as in compounds 7d (EC50 = 14.97 ± 0.24 μM), 7a (EC50 = 14.28 ± 0.76 μM) and 7b (EC50 = 14.58 ± 0.39 μM). The efficacy of F2-7f (EC50 = 5.89 ± 2.03 μM) was further improved by introducing two F atoms on the indole ring. Interestingly, this series displayed potent anti-HIV-2 activity, with 7d (EC50 = 4.89 ± 1.39 μM), 7f (EC50 = 4.52 ± 0.87 μM) and F2-7f (EC50 = 5.07 ± 0.63 μM) comparable to PF74 (EC50 = 4.16 ± 2.02 μM). When R1 was unsubstituted, the anti-HIV-2 activity was greater than that when R1 was substituted with F, such as 7a (EC50 = 7.74 ± 0.79 μM) > F2-7a (EC50 = 19.66 ± 6.59 μM), 7b (EC50 = 16.95 ± 3.58 μM) > F2-7b (EC50 > 15.94 μM), 7d > F2-7d (EC50 = 17.61 ± 9.14 μM), 7e (EC50 = 186.48 ± 4.42 μM) > F2-7e (EC50 ≥ 49.55 μM), and 7f > F2-7f. Remarkably, compounds 7d and 7f showed good selectivity for HIV-2.
However, the substitution of R3 with Br or OH resulted in a marked decrease in efficacy, such as compounds 7c, F2-7c, 7e, and F2-7e, indicating that a slight change in the substitutes targeting CTD of CA had a considerable effect on the anti-HIV activity of the compounds.
When R1 was substituted with F, the compound toxicities of series I were increased, such as F2-7a (CC50 = 76.85 ± 19.98 μM) > 7a (CC50 = 107.56 ± 3.26 μM), F2-7b (CC50 = 15.94 ± 3.83 μM) > 7b (CC50 = 104.21 ± 3.37 μM), F2-7c (CC50 = 3.15 ± 0.40 μM) > 7c (CC50 = 16.37 ± 3.44 μM), F2-7d (CC50 = 57.84 ± 21.81 μM) > 7d (CC50 = 112.41 ± 3.22 μM), F2-7e (CC50 = 131.51 ± 32.96 μM) > 7e (CC50 > 209.29 μM), and F2-7f (CC50 = 16.36 ± 3.38 μM) > 7f (CC50 = 90.06 ± 15.72 μM). It was worth noting that when R3 was Br, the compounds lost their efficacy against HIV-1 and HIV-2, which may have been due to high toxicity.
In series II, the rule of anti-HIV-1 activity for these compounds was that F-substituted R1 was superior to unsubstituted R1, such as F2-7g (EC50 = 177.40 ± 16.55 μM) > 7g (EC50 = 203.14 ± 10.98 μM), F2-7h (EC50 = 160.53 ± 15.43 μM) > 7h (EC50 = 192.39 ± 3.17 μM), F2-7i > 7i (EC50 > 167.50 μM), and F2-7j > 7j (EC50 = 157.50 ± 6.77 μM). When X was SO2, the anti-HIV-1 activity was greater than that when X was CO, such as 7h > 7g and F2-7h > F2-7g. Compounds F2-7i (EC50 = 14.36 ± 1.31 μM) and F2-7j (EC50 = 13.74 ± 0.70 μM) with cyclopropyl and CF3 substitution at R4 had the best efficacy with low toxicity. For anti-HIV-2 activity, the R1 substituent showed the opposite rule with anti-HIV-1 activity; that is, unsubstituted R1 was superior to F-substituted R1. Specifically, 7g (EC50 = 184.07 ± 19.33 μM) > F2-7g (EC50 = 192.14 ± 4.57 μM), 7i (EC50 = 15.02 ± 2.27 μM) > F2-7i (EC50 = 15.10 ± 0.68 μM), and 7j (EC50 = 14.95 ± 4.83 μM) > F2-7j (EC50 = 16.55 ± 6.13 μM). When R4 was cyclopropyl (7i) and CF3 (7j), the compounds showed obvious selectivity for HIV-2.
Notably, the compounds with more fluorine atoms at the terminal of the two series had the most potent anti-HIV-1 activity, such as F2-7f and F2-7j. It is speculated that the F atom forms a hydrogen bond with the surrounding key amino acid residues. In addition, the anti-HIV activity of series I was generally better than that of series II, indicating that the indole derivatives for the NTD-CTD interface were more conducive to antiviral activity than the aniline derivatives. Preliminary SARs analysis of newly synthesized compounds revealed that the linker of PF74 derivatives and the substituent components was particularly important for anti-HIV activity, which could serve as guidance for further research exploring the NTD-CTD interface.
Overall, the study of these compounds showed that the alteration of substituents had a significant impact on antiviral activity and selectivity, and F2-7f represented a promising lead compound for further optimization.
2.3. Surface Plasmon Resonance (SPR) Assay on CA Protein
Next, we selected the representative compound F2-7f for direct binding evaluation with monomeric and hexameric HIV-1 CA protein using SPR. We utilized the previously reported SPR method to test the binding affinity and off-rates of F2-7f to CA monomers and CA hexamers, using the lead compound PF74 as an internal control [30].
The SPR results are shown in Figure 2 and Figure 3 and Table 3, providing evidence for direct binding with both monomeric and hexameric CA proteins. Based on the equilibrium dissociation constant (KD), these compounds preferred to bind to the CA hexamer rather than to the CA monomer. The affinity of PF74 (hexamer: KD = 0.159 ± 0.041 μM; monomer: KD = 3.410 ± 1.310 μM) to CA was greater than that of F2-7f (hexamer: KD = 7.203 ± 1.101 μM; monomer: KD = 16.063 ± 1.316 μM), which was consistent with their antiviral activity (PF74, EC50 = 0.75 ± 0.33 μM > F2-7f, EC50 = 5.89 ± 2.03 μM) in vitro. Simultaneously, for CA hexamers, the koff values of F2-7f were approximately 8.5 times higher than that of PF74, indicating faster dissociation. As found in a previous study, the increase in koff value was positively correlated with the decrease in antiviral activity [31], which suggested that the lower affinity and faster off-rate of F2-7f for HIV-1 CA might be the reason for its reduced anti-HIV-1 activity. Therefore, SPR experiments proved that these newly synthesized compounds could be defined as HIV-1 CA modulators.
Figure 2.

SPR sensorgram depicting the interaction of F2-7f and PF74 with two HIV-1 CA variants (monomer and disulfide-stabilized hexamer).
Figure 3.

SPR isotherms of F2-7f (A) binding to two variants (monomer and disulfide-stabilized hexamer) of the CA protein using PF74 (B) as the reference. Isotherms are an average of 3 replicates with error bars indicating the standard deviation (SD).
Table 3.
SPR results of F2-7f and PF74 binding to monomeric and hexameric CA constructs.
| Compound ID | KD a (μM) | Ratio b | koff c (s−1) | |
|---|---|---|---|---|
| Monomer | Hexamer | Hexamer | ||
| F2-7f | 16.063 ± 1.316 | 7.203 ± 1.101 | 2.230 | 0.151 ± 0.028 |
| PF74 | 3.410 ± 1.310 | 0.159 ± 0.041 | 21.446 | 0.0177 ± 0.0004 |
a All values represent the average response from at least three replicates. Error bars indicate standard deviation (SD); b Ratio = KDMonomer/KDHexamer. c koff = off-rate measured for HIV-1 CA hexamer.
2.4. Molecular Dynamics (MD) Simulation with F2-7f Bound HIV-1 CA Hexamer
The most active molecule, F2-7f, was selected to investigate its binding to the ligand site of HIV-1 CA. Figure 4A shows the root mean square deviation (RMSD) values of HIV-1 CA residues from the first frame of the MD simulation. The figure highlights that the protein had largely deviated from the X-ray structure and formed diverse protein conformations. The root mean square fluctuation (RMSF) of amino acids was investigated to identify the deviated amino acids (Figure 4B). Most amino acids had largely deviated from the X-ray structure, indicating that HIV-1 CA had multiple conformations during MD simulation. This deviation of amino acids indicated that HIV-1 CA had different binding modes with F2-7f. To investigate the potential binding modes of F2-7f to the binding site, the RMSD values of F2-7f were calculated, as shown in Figure 4C. The figure showed that F2-7f had deviated from the docked conformation and clustered in different conformations.
Figure 4.

MD simulation of F2-7f bound HIV-1 CA. (A) RMSD of HIV-1 CA monomer amino acids refers to the first framework of the MD simulation. (B) RMSF of the backbone Cα atoms for amino acids of HIV-1 CA monomer. (C) RMSD (heavy atoms) of the bound F2-7f.
The MD trajectory was clustered based on F2-7f to explore its interactions with the binding site. The clustering procedure returned 10 clusters with one dominant. Figure 5 shows the representative structure of the most populated cluster. The conformation of the HIV-1 CA representative structure had a folded structure, which was different from the X-ray structure. The binding site in the representative structure was at the same location as the X-ray structure.
Figure 5.

Binding of F2-7f to HIV-1 CA in the most populated cluster.
The representative structure of the dominant cluster was investigated to determine the bonding forces between HIV-1 CA and F2-7f. Methoxybenzene was embedded between Leu56 and Lys70, where it could be involved in aromatic-aliphatic hydrophobic interactions. Also, the benzene ring of methoxybenzene could be involved in ion-induced dipole with the charged nitrogen of Lys70. 3,5-difluorobenzene was involved in hydrophobic interactions with Ala105. Thr107 formed a hydrogen bond with the oxygen atom of the amide (24.5%), as shown in Figure 5. Glu71 formed a hydrogen bond with the indole ring of F2-7f in 20% of the MD simulations, which might explain why F2-7f was the most potent compound in this study. It could be seen that F2-7f and PF74 bound in the same site, but the conformation of F2-7f was partially changed relative to that of PF74, resulting in the absence of some key interactions, which might explain its reduced antiviral activity.
2.5. In Silico Predicted ADMET Properties of Representative Compounds
2.5.1. Drug-Like Properties and Metabolic Stability
One of the main drawbacks of PF74 is its low metabolic stability and poor drug-like property profile, which limits its clinical use. Therefore, we evaluated selected compounds from this study for their drug-like properties and metabolic stability and compared them with PF74 (Figure 6). We utilized in silico prediction of drug-like metrics as implemented in the oral non–central nervous system (CNS) drug profile in StarDrop 7 software (Optibrium, Ltd., Cambridge, UK) [32]. This profile consists of several models, and a probabilistic scoring algorithm combines the model predictions in the oral non-CNS drug profile into an overall score. Scores range from 0 to 1, with 0 suggesting extremely non–drug-like and 1 suggesting the perfect drug.
Figure 6.

(A) Plot showing the StarDrop V7 (Optibrium, Ltd., Cambridge, UK)–derived logS versus a multimetric oral non-CNS profile score. Importance for the score: logS = 0.9, HIA = 0.85, logP = 0.6, logD = 0.6, hERG pIC50 = 0.4, 2D6 affinity category = 0.3, 2C9 pKi = 0.3, P-gp category = 0.3, PPB90 category = 0.2, BBB category = 0.11, BBB log(brain:blood) = 0.11 (B) P-gp category, (C) PPB90 = Plasma protein binding > 90%, (D) HIA = human intestinal absorption category, and probability of prediction of 7f, F2-7f and PF74. The size of the circle correlates with the corresponding score or probability.
The oral non-CNS drug profile in Optibrium’s StarDrop software combines logS (intrinsic aqueous solubility); classification for human intestinal absorption; logP (octanol/water); hERG (human ether-a-go-go-related gene) pIC50 (mammalian cells); cytochrome P450 CYP2D6 classification; cytochrome P450 CYP2C9 pKi values; classification of P-glycoprotein transport; classification of blood-brain barrier (BBB) penetration; and predicted BBB penetration value (Figure 6).
Based on this analysis, F2-7f and especially 7f displayed improved aqueous solubility compared to PF74, as judged by the logS values. This can contribute to improved overall bioavailability. F2-7f and 7f showed improved oral non-CNS drug profile scores, primarily due to improved solubility and low plasma protein binding compared to PF74.
The poor metabolic stability of PF74 limits its use in clinical applications [31]. For orally administered drugs, the intestinal wall and portal circulation to the liver represents first-pass metabolism and can limit compound concentrations in the bloodstream. Therefore, we next sought to investigate in silico whether or not our compounds had improved predicted metabolic stability over PF74. We employed a computational analysis first demonstrated to be an accurate indicator of metabolic stability by the Cocklin group [33,34,35]. We utilized the P450 module in StarDrop 7 software (Optibrium, Ltd., Cambridge, UK) to predict each compound’s major metabolizing Cytochrome P450 isoforms using the WhichP450™ model. Subsequently, we predicted the affinity to that isoform using the HYDE function in See SAR (BioSolveIT Gmbh, Sankt Augustin, Germany) [34,36,37]. The results of this analysis are shown in Figure 7.
Figure 7.

Computational prediction of metabolic stability. (A) Prediction of the major metabolizing CYP isoforms. All compounds are predicted to be metabolized primarily by the 3A4 isoform, including PF74. (B) Overall composite site lability (CSL) score and number of labile sites within the compound (for metabolism). A lower CSL score indicates a more stable molecule. The prediction was achieved using the StarDrop (version 7) P450 module. (C) Predicted affinity of docked 7f, F2-7f, and PF74 to Cytochrome P450 3A4 (PDB ID: 4D78). The lower boundary for predicted 3A4 affinity utilized the hydrogen bond and dehydration scoring function (HYDE) implemented in SeeSAR12.1.
The main metabolizing isoform for all compounds, including PF74, is the CYP3A4 isoform [12] (Figure 7A). In addition, F2-7f and 7f were also predicted to be metabolized to a greater extent than PF74 by the 2D6 isoform and were within the high-affinity category for 2D6, according to the analysis in StarDrop.
We next investigated the predicted metabolic lability of our compounds and PF74 with the CY3A4 isoform by comparing the overall composite site lability (CSL) score and number of labile sites. The CSL score can reflect the overall efficiency of metabolism of the molecule by combining the labilities of individual sites within the compound. The number of labile sites between our compounds and PF74 was not significantly different; however, the CSL score indicated increased metabolic stability for PF74 (Figure 7B).
In addition to the CSL score and number of labile sites, which assumed that all compounds bind with similar affinity to the CYP3A4 isoform, other factors such as compound reduction rate and actual binding affinity to the CYP3A4 isoform can influence metabolic stability. In addition, intrinsic compound properties, such as size and lipophilicity, can also infer affinity. Therefore, we performed predictive binding affinity calculations using the hydrogen bond and dehydration (HYDE) energy scoring function in SeeSAR 12.1 (BioSolveIT Gmbh, Sankt Augustin, Germany) [38] using the structure of the human CYPA4 bound to an inhibitor (PDB ID 4D78) [39]. The HYDE scoring function in SeeSAR provides a range of affinities, including an upper and lower limit. We used the lower limit as the affinity predictor to compare F2-7f, 7f, and PF74 (Figure 7C), which resulted in an affinity of 657 μM for F2-7f, 179 μM for 7f, and 2 nM for PF74. Although F2-7f and 7f had less favorable CSL scores and F2-7f had two labile sites (terminal methoxy and a carbon between the two F atoms within the difluorobenzene), the much lower predicted CYP3A4 affinities for F2-7f and 7f might have compensated for the higher CSL and number of labile sites.
Combining the results from these predictions (CSL scores, labile sites, and predicted CYP3A4 affinity), this analysis indicated that compounds F2-7f and 7f have similar metabolic stability as PF74.
2.5.2. Genotoxicity and Hepatotoxicity
To obtain a toxicity profile for the lead compounds, we included genotoxicity and hepatotoxicity endpoints in our multiparameter optimization for F2-7f, 7f, and PF74 using the Derek Nexus module within StarDrop V7 software. Derek Nexus utilizes a knowledge- and rule-based expert system for semi-quantitative estimations of DNA-reactive moieties within molecules. None of the lead compounds nor PF74 showed any concerning likelihood of genotoxicity or hepatotoxicity (Figure 8). To evaluate in silico the accuracy of the prediction, we used positive controls in our prediction. Ethyl methanesulfonate (EMS) [40] and lumiracoxib [41] are known to have in vivo genotoxic and hepatotoxic effects.
Figure 8.

Genotoxicity and hepatotoxicity endpoints for selected compounds. The highlighted table shows the StarDrop V7 (Optibrium, Ltd., Cambridge, UK)–derived toxicity endpoints using the Derek Nexus module with knowledge-based prediction. Structural alerts within the molecule with a level of likelihood for concern are based on precedence from experimental data. Likelihood level 0 = No report; 1 = Inactive; 2 = Equivocal; 3 = Plausible; 4 = Probable. Ethyl methanesulfonate (EMS) was used as a positive control for genotoxicity, and lumiracoxib was a positive control for hepatotoxicity endpoints.
2.6. Metabolic Stability in the Presence of Human Liver Microsomes and Human Plasma
Equipped with the computational predictions, we next performed metabolic stability assays in human liver microsomes (HLMs) and human plasma. Firstly, testosterone, diclofenac, and propafenone with moderate metabolic stability were selected as control drugs, and we tested the metabolic stability of F2-7f and PF74 in HLMs. As shown in Table 4, F2-7f and PF74 could be rapidly metabolized in HLMs with a half-life of 0.5 min. The intrinsic clearance (CLint) of F2-7f was slightly lower than that of PF74 (2759.1 and 2862.5 μL/min/mg, respectively). The results of the human plasma stability assay for F2-7f and PF74 are shown in Table 5. After 120 min of incubation, the residual amount of the original F2-7f decreased to 86.9%, and the residual amount of the original PF74 decreased to 85.2%, indicating that the metabolic stability of F2-7f in human plasma was slightly improved compared to that of PF74. This concurred with the lower plasma protein binding prediction probability for F2-7f (PPB90, Figure 6C). Overall, the experimental data moderately correlated with the computational prediction, and improving the metabolic stability of PF74-like small molecules remains an urgent issue for future optimization efforts.
Table 4.
Metabolic stability assay in human liver microsomes.
| Sample | HLM (Final Concentration of 0.5 mg Protein/mL) | |||||
|---|---|---|---|---|---|---|
| R2 a | T1/2 b (min) | CLint(mic)
c (μL/min/mg) |
CLint(liver)
d (mL/min/kg) |
Remaining (T = 60 min) |
Remaining (NCF e = 60 min) |
|
| F2-7f | 1.0000 | 0.5 | 2759.1 | 2483.2 | 0.0% | 103.5% |
| PF74 | 1.0000 | 0.5 | 2862.5 | 2576.2 | 0.0% | 112.6% |
| Testosterone | 0.9982 | 16.7 | 82.8 | 74.5 | 7.9% | 90.7% |
| Diclofenac | 0.9947 | 3.7 | 372.0 | 334.8 | 0.0% | 96.7% |
| Propafenone | 0.9350 | 5.0 | 279.5 | 251.5 | 0.0% | 93.6% |
a R2 is the correlation coefficient of the linear regression for determination of the kinetic constant. b T1/2 is half-life, and CLint(mic) is the intrinsic clearance. c CLint(mic) = (0.693/half-life)/mg microsome protein per mL. d CLint(liver) = CLint(mic) × mg microsomal protein/g liver weight × g liver weight/kg body weight. e NCF: no cofactor. No NADPH regenerating system was added to the NCF sample (replaced by buffer) during the 60 min incubation. Less than 60% remaining indicates that a non-NADPH-dependent reaction occurred.
Table 5.
Result of human plasma stability assay.
| Compound ID | Batch | Time Point (min) | % Remaining a | T1/2 (min) |
|---|---|---|---|---|
| Human | Human | |||
| F2-7f | / | 0 | 100.0 | >289.1 |
| 10 | 98.0 | |||
| 30 | 97.8 | |||
| 60 | 90.9 | |||
| 120 | 86.9 | |||
| PF74 | / | 0 | 100.0 | >289.1 |
| 10 | 84.7 | |||
| 30 | 81.7 | |||
| 60 | 80.7 | |||
| 120 | 85.2 | |||
| Propantheline bromide | ODR-4547 | 0 | 100.0 | 11.2 |
| 10 | 56.5 | |||
| 30 | 19.4 | |||
| 60 | 1.9 | |||
| 120 | 0.1 |
a % remaining = 100 × (PAR at appointed incubation time/PAR at time T0). PAR is the peak area ratio of test compound to internal standard. Accuracy should be within 80−120% of the indicated value.
3. Experimental Section
3.1. Chemistry
All melting points (mp) of the new compounds were determined on a micro melting point apparatus. 1H NMR and 13C NMR spectra were obtained in DMSO-d6 on a Bruker AV-400 spectrometer or Bruker AV-600 spectrometer using tetramethylsilane (TMS) as the internal reference. Chemical shifts were reported in δ values (ppm) and J values were expressed in hertz (Hz). Thin layer chromatography (TLC) for monitoring reactions or purifying products was performed on silica gel GF254 or Huanghai HSGF254, 0.15–0.2 mm, respectively. Spots were visualized with iodine vapor or by irradiation with UV light (λ = 254 nm or λ = 365 nm). Mass spectrometry (MS) was carried out using a Standard G1313A LC autosampler instrument. Flash column chromatography was performed on a column packed with silica gel 60 (200–300 mesh). Solvents were of reagent grade and, if needed, were purified and dried using standard methods. Rotary evaporators were involved in concentrating the reaction solutions under reduced pressure. The solvents dichloromethane, TEA, methanol, etc. were obtained from Sinopharm Chemical Reagent Co., Ltd. (SCRC, Shanghai, China) and were of AR grade. The key reactants, including 4-methoxy-N-methylaniline (CAS: 5961-59-1), N-(tert-butoxycarbonyl)-L-phenylalanine (CAS: 13734-34-4), and N-(tert-butoxycarbonyl)-3,5-difluoro-L-phenylalanine (CAS: 205445-52-9), were purchased from Shanghai Haohong Scientific Co., Ltd. (Shanghai, China). The purity of all target compounds was analyzed by high-performance liquid chromatography (HPLC) and was >95%.
3.1.1. Procedure for the Synthesis of Intermediates
The procedure for the synthesis of key intermediates is provided in the Supplementary Materials [29,30].
3.1.2. General Procedure for the Synthesis of 7 (a-f) and F2-7 (a-f)
Corresponding substituted indoleacetic acids (1.2 eq.) were first dissolved in 20 mL dichloromethane (DCM), and then HATU (1.5 eq.) was added. The reaction mixture was stirred at 0 °C for 0.5 h. Then, the key intermediates 6 and F2-6 (1 eq.) were added dropwise into the solution, and DIEA (2 eq.) was added. The reaction mixture was restored at room temperature for 3 h. After monitoring the reaction by TLC, the excess solvent was evaporated under reduced pressure, and the residue was redissolved in water and extracted with DCM (3 × 20 mL). Subsequently, the organic layers were combined and washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to obtain the corresponding crude products. These products were purified by flash column chromatography to provide target compounds 7 (a-f) and F2-7 (a-f).
(S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(4-(2-(2-methyl-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-3-phenylpropanamide (7a)
White solid. Yield: 72%. Purity: 99%. Mp: 109–111 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H, NH), 8.35 (d, J = 7.9 Hz, 1H, NH), 7.40 (d, J = 7.7 Hz, 1H, Ph-H), 7.23 (d, J = 7.9 Hz, 1H, Ph-H), 7.20–7.06 (m, 5H, Ph-H), 6.97 (d, J = 8.4 Hz, 3H, Ph-H), 6.91 (d, J = 7.0 Hz, 1H, Ph-H), 6.84 (d, J = 6.6 Hz, 2H, Ph-H), 4.47 (q, J = 8.5 Hz, 1H, CH), 4.14 (s, 1H, CH), 3.99 (d, J = 5.6 Hz, 1H, CH), 3.96–3.83 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.72 (d, J = 7.9 Hz, 2H, CH2), 3.68 (s, 1H, CH), 3.60 (s, 1H, CH), 3.10 (s, 3H, NCH3), 3.08 (s, 2H, CH2), 2.85 (dd, J = 13.5, 4.7 Hz, 1H, PhCH), 2.64 (dd, J = 13.1, 9.7 Hz, 1H, PhCH), 2.31 (s, 3H, CH3). 13C NMR (150 MHz, DMSO-d6) δ 171.40 (C=O), 169.91 (C=O), 167.58 (C=O), 159.04 (C=O), 137.93, 135.98, 135.60, 129.32, 129.12, 128.82, 128.57, 126.85, 120.47, 118.68, 118.15, 115.18, 110.78, 55.91, 51.81, 47.21, 37.79, 11.92. HRMS (ESI) calculated for C34H37N5O5 [M+H]+ 596.2867, found 596.2872.
(S)-2-(2-(4-(2-(5-methoxy-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (7b)
White solid. Yield: 69%. Purity: 99%. Mp: 142–144 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.77 (d, J = 8.9 Hz, 1H, NH), 8.40 (d, J = 7.9 Hz, 1H, NH), 7.24 (d, J = 8.7 Hz, 1H, Ph-H), 7.19 (s, 1H, Ph-H), 7.18–7.09 (m, 5H, Ph-H), 7.06 (s, 1H, Ph-H), 6.98 (d, J = 8.3 Hz, 2H, Ph-H), 6.85 (d, J = 6.6 Hz, 2H, Ph-H), 6.73 (d, J = 8.7 Hz, 1H, Ph-H), 4.54–4.41 (m, 1H, CH), 4.18 (s, 1H, CH), 4.03 (t, J = 6.7 Hz, 1H, CH), 3.99–3.84 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.77 (s, 2H, CH2), 3.74 (s, 3H, OCH3), 3.71(m, 1H, CH), 3.63 (s, 1H, CH), 3.11 (s, 5H, NCH3, CH2), 2.86 (dd, J = 13.4, 4.3 Hz, 1H, PhCH), 2.71–2.59 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.42 (C=O), 169.78 (C=O), 167.64 (C=O), 159.02 (C=O), 137.94, 135.94, 131.73, 129.32, 129.14, 128.60, 126.88, 124.69, 115.16, 112.46, 111.66, 101.08, 55.90, 55.83, 51.88, 51.84, 37.78, 37.73, 30.81. HRMS (ESI) calculated for C34H37N5O6 [M+H]+ 612.2817, found 612.2819.
(S)-2-(2-(4-(2-(5-bromo-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (7c)
White solid. Yield: 70%. Purity: 99%. Mp: 115–116 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.11 (d, J = 8.8 Hz, 1H, NH), 8.37 (d, J = 7.8 Hz, 1H, NH), 7.74 (s, 1H, Ph-H), 7.30 (t, J = 10.3 Hz, 2H, Ph-H), 7.14 (d, J = 13.2 Hz, 6H, Ph-H), 6.97 (d, J = 8.3 Hz, 2H, Ph-H), 6.84 (d, J = 6.6 Hz, 2H, Ph-H), 4.54–4.42 (m, 1H, CH), 4.20 (s, 1H, CH), 4.01 (q, J = 9.9 Hz, 1H, CH), 3.97–3.88 (m, 2H, CH2), 3.86 (s, 2H, CH2), 3.79 (s, 3H, OCH3), 3.73 (s, 1H, CH), 3.62 (s, 1H, CH), 3.15 (s, 2H, CH2), 3.11 (s, 3H, NCH3), 2.85 (dd, J = 13.3, 4.1 Hz, 1H, PhCH), 2.71–2.59 (m, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.40 (C=O), 169.51 (C=O), 167.61 (C=O), 159.04 (C=O), 137.93, 135.98, 135.33, 129.68, 129.33, 129.11, 128.57, 126.86, 125.85, 123.96, 121.68, 115.18, 113.81, 111.59, 108.20, 55.91, 51.80, 49.23, 46.23, 42.91, 37.79, 30.38. HRMS (ESI) calculated for C33H34BrN5O5 [M+H]+ 660.1816, found 660.1819.
(S)-2-(2-(4-(2-(1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (7d)
White solid. Yield: 75%. Purity: 99%. Mp: 170–171 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 7.6 Hz, 1H, NH), 8.38 (d, J = 7.8 Hz, 1H, NH), 7.55 (d, J = 7.4 Hz, 1H, Ph-H), 7.34 (d, J = 8.0 Hz, 1H, Ph-H), 7.21 (d, J = 11.6 Hz, 1H, Ph-H), 7.13 (dt, J = 15.4, 6.2 Hz, 5H, Ph-H), 7.06 (d, J = 7.5 Hz, 1H, Ph-H), 6.97 (d, J = 8.0 Hz, 3H, Ph-H), 6.84 (d, J = 6.7 Hz, 2H, Ph-H), 4.46 (q, J = 7.5 Hz, 1H, CH), 4.17 (s, 1H, CH), 4.00 (q, J = 13.0, 10.9 Hz, 1H, CH), 3.96–3.83 (m, 2H, CH2), 3.80 (s, 2H, CH2), 3.78 (s, 3H, OCH3), 3.72 (s, 1H, CH), 3.61 (s, 1H, CH), 3.10 (s, 5H, NCH3, CH2), 2.85 (dd, J = 13.3, 4.3 Hz, 1H, PhCH), 2.71–2.59 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.42 (C=O), 169.73 (C=O), 167.63 (C=O), 159.02 (C=O), 137.94, 136.60, 135.95, 129.32, 129.14, 128.60, 127.67, 126.88, 124.07, 121.53, 119.22, 118.86, 115.16, 111.81, 55.90, 51.83, 48.52, 47.36, 46.16, 42.93, 30.79. HRMS (ESI) calculated for C33H35N5O5 [M+H]+ 582.2711, found 582.2716.
(S)-2-(2-(4-(2-(5-hydroxy-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (7e)
White solid. Yield: 78%. Purity: 99%. Mp: 131–132 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (d, J = 5.8 Hz, 1H, NH), 8.60 (s, 1H, OH), 8.39 (d, J = 7.8 Hz, 1H, NH), 7.24–7.06 (m, 7H, Ph-H), 6.98 (d, J = 8.2 Hz, 2H, Ph-H), 6.92–6.81 (m, 3H, Ph-H), 6.60 (d, J = 8.6 Hz, 1H, Ph-H), 4.54–4.42 (m, 1H, CH), 4.16 (s, 1H, CH), 4.02 (p, J = 11.4, 9.9 Hz, 1H, CH), 3.96–3.84 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 3.73 (d, J = 15.2 Hz, 2H, CH2), 3.69 (s, 1H, CH), 3.62 (s, 1H, CH), 3.11 (s, 5H, NCH3, CH2), 2.86 (dd, J = 13.0, 3.9 Hz, 1H, PhCH), 2.71–2.60 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.42 (C=O), 169.76 (C=O), 167.64 (C=O), 159.02 (C=O), 150.78, 137.94, 135.94, 131.18, 129.32, 129.15, 128.60, 128.34, 126.89, 124.45, 115.17, 112.09, 111.95, 107.19, 103.24, 55.90, 51.82, 48.52, 47.36, 46.14, 37.79. HRMS (ESI) calculated for C33H35N5O6 [M+H]+ 598.2660, found 598.2658.
(S)-2-(2-(4-(2-(5-fluoro-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (7f)
White solid. Yield: 72%. Purity: 97%. Mp: 95–96 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.00 (d, J = 8.4 Hz, 1H, NH), 8.37 (d, J = 8.0 Hz, 1H, NH), 7.37–7.27 (m, 3H, Ph-H), 7.14 (dd, J = 19.0, 6.6 Hz, 5H, Ph-H), 6.97 (d, J = 8.5 Hz, 2H, Ph-H), 6.91 (t, J = 9.1 Hz, 1H, Ph-H), 6.84 (d, J = 6.9 Hz, 2H, Ph-H), 4.47 (q, J = 8.2 Hz, 1H, CH), 4.19 (s, 1H, CH), 4.07–3.97 (m, 1H, CH), 3.97–3.84 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.76 (s, 2H, CH2), 3.75–3.69 (m, 1H, CH), 3.65–3.58 (m, 1H, CH), 3.13 (s, 2H, CH2), 3.11 (s, 3H, NCH3), 2.85 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.65 (dd, J = 13.1, 9.6 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.40 (C=O), 169.59 (C=O), 167.60 (C=O), 159.04 (C=O), 157.19 (d, 1JC-F = 231.0 Hz), 137.93, 135.98, 133.29, 129.32, 129.11, 128.57, 126.85, 126.23, 115.18, 112.73 (d, 3JC-F = 10.0 Hz), 109.61 (d, 2JC-F = 26.2 Hz), 108.53, 103.95 (d, 2JC-F = 23.4 Hz), 55.91, 51.80, 48.59, 46.21, 42.93, 37.79. HRMS (ESI) calculated for C33H34FN5O5 [M+H]+ 600.2617, found 600.2619.
(S)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methyl-2-(2-(4-(2-(2-methyl-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)propenamide (F2-7a)
White solid. Yield: 68%. Purity: 99%. Mp: 106–108 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H, NH), 8.43 (d, J = 7.9 Hz, 1H, NH), 7.41 (d, J = 7.7 Hz, 1H, Ph-H), 7.24 (t, J = 6.3 Hz, 3H, Ph-H), 7.03 (d, J = 8.0 Hz, 3H, Ph-H), 7.01–6.95 (m, 1H, Ph-H), 6.91 (t, J = 7.3 Hz, 1H, Ph-H), 6.50 (d, J = 7.1 Hz, 2H, Ph-H), 4.46 (s, 1H, CH), 4.20–4.11 (m, 1H, CH), 4.06–3.99 (m, 1H, CH), 3.94 (d, J = 20.4 Hz, 2H, CH2), 3.80 (s, 3H, OCH3), 3.75 (s, 1H, CH), 3.72 (s, 2H, CH2), 3.68–3.56 (m, 1H, CH), 3.17 (s, 2H, CH2), 3.14 (s, 3H, NCH3), 2.92–2.83 (m, 1H, PhCH), 2.75–2.64 (m, 1H, PhCH), 2.32 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 170.88 (C=O), 169.88 (C=O), 167.76 (C=O), 165.80 (C=O), 162.50 (dd, 1JC-F = 245.6 Hz, 3JC-F = 13.4 Hz), 159.16, 142.54 (t, 3JC-F = 9.5 Hz), 135.82, 135.56, 133.31, 128.80, 120.47, 118.67, 118.18, 115.27, 112.35 (d, 2JC-F = 24.6 Hz), 110.78, 104.08, 102.50 (t, 2JC-F = 25.7 Hz), 55.92, 51.49, 48.57, 47.39, 46.23, 42.74, 37.75, 29.64, 11.92. HRMS (ESI) calculated for C34H35F2N5O5 [M+H]+ 632.2679, found 632.2679.
(S)-3-(3,5-difluorophenyl)-2-(2-(4-(2-(5-methoxy-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methylpropanamide (F2-7b)
White solid. Yield: 73%. Purity: 99%. Mp: 108–109 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (d, J = 7.0 Hz, 1H, NH), 8.48 (d, J = 7.9 Hz, 1H, NH), 7.28 (d, J = 8.1 Hz, 3H, Ph-H), 7.22 (d, J = 9.9 Hz, 1H, Ph-H), 7.16–7.03 (m, 4H, Ph-H), 6.77 (d, J = 8.6 Hz, 1H, Ph-H), 6.54 (d, J = 7.1 Hz, 2H, Ph-H), 4.50 (s, 1H, CH), 4.30–4.17 (m, 1H, CH), 4.08 (t, J = 5.2 Hz, 1H, CH), 4.03–3.90 (m, 2H, CH2), 3.84 (s, 3H, OCH3), 3.82 (s, 1H, CH), 3.79 (s, 2H, CH2), 3.79 (s, 3H, OCH3), 3.67 (ddt, J = 26.0, 13.8, 6.8 Hz, 1H, CH), 3.27–3.19 (m, 2H, CH2), 3.18 (s, 3H, NCH3), 2.97–2.87 (m, 1H, PhCH), 2.79–2.69 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 170.88 (C=O), 169.82 (C=O), 167.79 (C=O), 165.75 (C=O), 162.51 (dd, 1JC-F = 245.4 Hz, 3JC-F = 13.6 Hz), 159.16, 153.54, 142.53 (t, 3JC-F = 9.4 Hz), 135.82, 131.73, 129.17, 128.01, 124.69, 115.27, 112.35 (d, 2JC-F = 24.4 Hz), 111.67, 107.85, 102.49 (t, 2JC-F = 25.8 Hz), 101.09, 55.92, 55.82, 51.49, 48.62, 47.54, 46.15, 42.96, 37.76, 37.14, 30.80. HRMS (ESI) calculated for C34H35F2N5O6 [M+H]+ 648.2628, found 648.2625.
(S)-2-(2-(4-(2-(5-bromo-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (F2-7c)
White solid. Yield: 72%. Purity: 99%. Mp: 120–121 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (d, J = 7.1 Hz, 1H, NH), 8.43 (d, J = 7.9 Hz, 1H, NH), 7.74 (s, 1H, Ph-H), 7.30 (t, J = 9.8 Hz, 2H, Ph-H), 7.24 (d, J = 7.9 Hz, 2H, Ph-H), 7.18 (d, J = 8.6 Hz, 1H, Ph-H), 7.02 (d, J = 8.1 Hz, 3H, Ph-H), 6.49 (d, J = 7.1 Hz, 2H, Ph-H), 4.45 (s, 1H, CH), 4.26–4.14 (m, 1H, CH), 4.03 (d, J = 8.0 Hz, 1H, CH), 3.91 (t, J = 12.1 Hz, 2H, CH2), 3.82 (m, 1H, CH), 3.79 (s, 3H, OCH3), 3.76 (s, 2H, CH2), 3.71–3.55 (m, 1H, CH), 3.18 (d, J = 26.9 Hz, 2H, CH2), 3.13 (s, 3H, NCH3), 2.92–2.82 (m, 1H, PhCH), 2.69 (dd, J = 13.2, 9.8 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 170.89 (C=O), 169.47 (C=O), 165.73 (C=O), 165.73 (C=O), 162.50 (dd, 1JC-F = 245.8 Hz, 3JC-F = 13.6 Hz), 159.16, 142.54 (t, 3JC-F = 9.4 Hz), 135.82, 135.28, 129.67, 129.18, 125.87, 123.95, 121.71, 115.27, 113.82, 112.36 (dd, 2JC-F = 19.7 Hz), 111.53, 108.10, 102.52 (t, 2JC-F = 25.8 Hz), 55.92, 51.50, 47.51, 46.17, 42.85, 37.75, 37.12, 30.35. HRMS (ESI) calculated for C33H32BrF2N5O5 [M+H]+ 696.1628, found 696.1632.
(S)-2-(2-(4-(2-(1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (F2-7d)
White solid. Yield: 71%. Purity: 96%. Mp: 91–92 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.88 (d, J = 8.7 Hz, 1H, NH), 8.38 (dd, J = 7.7, 3.0 Hz, 1H, NH), 7.59–7.52 (m, 1H, Ph-H), 7.38–7.31 (m, 1H, Ph-H), 7.22 (t, J = 10.8 Hz, 3H, Ph-H), 7.07 (t, J = 7.5 Hz, 1H, Ph-H), 7.01 (d, J = 8.3 Hz, 3H, Ph-H), 6.99–6.94 (m, 1H, Ph-H), 6.50 (d, J = 7.7 Hz, 2H, Ph-H), 4.51–4.42 (m, 1H, CH), 4.24–4.12 (m, 1H, CH), 4.06–3.97 (m, 1H, CH), 3.92 (q, J = 16.8 Hz, 2H, CH2), 3.80 (s, 1H, CH), 3.79 (s, 3H, OCH3), 3.75 (s, 2H, CH2), 3.71–3.57 (m, 1H, CH), 3.17 (d, J = 21.0 Hz, 2H, CH2), 3.13 (s, 3H, NCH3), 2.90–2.83 (m, 1H, PhCH), 2.74–2.65 (m, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 170.86 (C=O), 169.78 (C=O), 169.75 (C=O), 165.78 (C=O), 162.53 (dd, 1JC-F = 246.0 Hz, 3JC-F = 13.1 Hz), 159.18, 142.53 (t, 3JC-F = 9.4 Hz), 136.64, 135.86, 129.13, 127.70, 124.05, 121.51, 119.19, 118.86, 115.30, 112.36 (dd, 2JC-F = 19.8 Hz), 111.80, 108.12, 102.45 (t, 2JC-F = 25.9 Hz), 55.93, 51.44, 48.64, 47.50, 46.20, 42.99, 37.76, 37.26, 30.76. HRMS (ESI) calculated for C33H33F2N5O5 [M+H]+ 618.2523, found 618.2526.
(S)-3-(3,5-difluorophenyl)-2-(2-(4-(2-(5-hydroxy-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methylpropanamide (F2-7e)
White solid. Yield: 71%. Purity: 96%. Mp: 131–132 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H, NH), 8.59 (s, 1H, OH), 8.41 (d, J = 7.8 Hz, 1H, NH), 7.23 (d, J = 7.6 Hz, 2H, Ph-H), 7.11 (t, J = 8.9 Hz, 2H, Ph-H), 7.02 (d, J = 7.8 Hz, 3H, Ph-H), 6.87 (s, 1H, Ph-H), 6.59 (d, J = 8.6 Hz, 1H, Ph-H), 6.49 (d, J = 7.3 Hz, 2H, Ph-H), 4.45 (s, 1H, CH), 4.23–4.09 (m, 1H, CH), 4.05–3.98 (m, 1H, CH), 3.98–3.85 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.73 (s, 1H, CH), 3.69 (d, J = 10.0 Hz, 2H, CH2), 3.66–3.53 (m, 1H, CH), 3.22–3.14 (m, 2H, CH2), 3.13 (s, 3H, NCH3), 2.91–2.82 (m, 1H, PhCH), 2.74–2.64 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 170.87 (C=O), 169.75 (C=O), 167.73 (C=O), 165.75 (C=O), 162.51 (dd, 1JC-F = 246.0 Hz, 3JC-F = 13.2 Hz), 159.16, 150.77, 142.53 (t, 3JC-F = 9.5 Hz), 135.82, 131.18, 129.17, 128.34, 124.45, 115.28, 112.36 (d, 2JC-F = 24.7 Hz), 112.09, 111.94, 107.07, 103.25, 102.51 (t, 2JC-F = 25.6 Hz), 55.92, 51.49, 48.60, 47.54, 46.13, 42.93, 37.76, 37.15, 31.13. HRMS (ESI) calculated for C33H33F2N5O6 [M+H]+ 634.2472, found 634.2477.
(S)-3-(3,5-difluorophenyl)-2-(2-(4-(2-(5-fluoro-1H-indol-3-yl)acetyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methylpropanamide (F2-7f)
White solid. Yield: 71%. Purity: 96%. Mp: 86–88 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.99 (d, J = 10.8 Hz, 1H, NH), 8.39 (d, J = 7.8 Hz, 1H, NH), 7.37–7.27 (m, 3H, Ph-H), 7.23 (d, J = 7.4 Hz, 2H, Ph-H), 7.01 (d, J = 8.5 Hz, 3H, Ph-H), 6.91 (t, J = 9.0 Hz, 1H, Ph-H), 6.50 (d, J = 7.2 Hz, 2H, Ph-H), 4.46 (s, 1H, CH), 4.24–4.14 (m, 1H, CH), 4.07–3.98 (m, 1H, CH), 3.98–3.86 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.76 (s, 2H, CH2), 3.70–3.57 (m, 1H, CH), 3.18 (d, J = 29.2 Hz, 2H, CH2), 3.13 (s, 3H, NCH3), 2.87 (dd, J = 13.4, 3.6 Hz, 1H, PhCH), 2.74–2.66 (m, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 170.86 (C=O), 169.58 (C=O), 167.75 (C=O), 165.77 (C=O), 162.53 (dd, 1JC-F = 246.0 Hz, 3JC-F = 13.2 Hz), 159.18, 157.19 (d, 1JC-F = 231.2 Hz), 142.53 (t, 3JC-F = 9.4 Hz), 135.86, 133.30, 129.13, 128.02 (d, 3JC-F = 10.2 Hz), 126.24, 115.30, 112.72 (d, 3JC-F = 9.8 Hz), 112.35 (dd, 2JC-F = 19.6 Hz), 109.61 (d, 2JC-F = 26.0 Hz), 108.52, 103.95 (d, 2JC-F = 23.3 Hz), 102.45 (t, 2JC-F = 25.7 Hz), 55.93, 51.44, 47.48, 46.21, 42.95, 37.76, 37.25. HRMS (ESI) calculated for C33H32F3N5O5 [M+H]+ 636.2428, found 636.2427.
3.1.3. General Procedure for the Synthesis of 7 (g-j) and F2-7 (g-j)
Corresponding substituted indoleacetic acids (1.2 eq.) were first dissolved in 20 mL DCM, and then HATU (1.5 eq.) was added. The reaction mixture was stirred at 0 °C for 0.5 h. Then, the key intermediates 6 and F2-6 (1 eq.) were added dropwise into the solution and DIEA (2 eq.) was added. The reaction mixture was restored to room temperature for 3 h. After monitoring the reaction by TLC, the excess solvent was evaporated under reduced pressure, and the residue was redissolved in water and extracted with DCM (3 × 20 mL). Subsequently, the organic layers were combined and washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to obtain the corresponding crude products. These products were purified by flash column chromatography to provide target compounds 7 (g-j) and F2-7 (g-j).
(S)-2-(2-(4-(4-acetamidobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (7g)
White solid. Yield: 75%. Purity: 95%. Mp: 116–118 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H, NH), 8.40 (d, J = 8.0 Hz, 1H, NH), 7.66 (d, J = 8.5 Hz, 2H, Ph-H), 7.39 (d, J = 8.4 Hz, 2H, Ph-H), 7.16 (dq, J = 12.8, 6.5 Hz, 5H, Ph-H), 6.98 (d, J = 8.9 Hz, 2H, Ph-H), 6.85 (d, J = 6.8 Hz, 2H, Ph-H), 4.47 (td, J = 8.7, 5.1 Hz, 1H, CH), 4.09 (s, 2H, CH2), 4.01–3.86 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.73–3.54 (m, 2H, CH2), 3.22–3.13 (m, 2H, CH2), 3.11 (s, 3H, NCH3), 2.90–2.82 (m, 1H, PhCH), 2.65 (dd, J = 13.3, 9.4 Hz, 1H, PhCH), 2.07 (s, 3H, COCH3). 13C NMR (100 MHz, DMSO-d6) δ 171.39 (C=O), 169.11 (C=O), 169.07 (C=O), 167.55 (C=O), 165.21 (C=O), 159.04, 141.43, 137.96, 135.97, 130.81, 129.55, 129.33, 129.13, 128.69, 128.58, 126.85, 118.84, 115.18, 55.91, 51.84, 48.68, 37.79, 24.53. HRMS (ESI) calculated for C32H35N5O6 [M+H]+ 586.2660, found 586.2663.
(S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(4-(4-(methylsulfonamido)benzoyl)-2-oxopiperazin-1-yl)acetamido)-3-phenylpropanamide (7h)
White solid. Yield: 80%. Purity: 99%. Mp: 106–108 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H, NH), 8.40 (d, J = 8.0 Hz, 1H, NH), 7.43 (d, J = 8.3 Hz, 2H, Ph-H), 7.26 (d, J = 8.5 Hz, 2H, Ph-H), 7.16 (dq, J = 13.6, 6.8 Hz, 5H, Ph-H), 6.98 (d, J = 8.9 Hz, 2H, Ph-H), 6.85 (d, J = 6.8 Hz, 2H, Ph-H), 4.52–4.41 (m, 1H, CH), 4.09 (s, 2H, CH2), 4.01–3.86 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.75–3.52 (m, 2H, CH2), 3.16 (d, J = 7.5 Hz, 2H, CH2), 3.11 (s, 3H, NCH3), 3.06 (s, 3H, SO2CH3), 2.90–2.81 (m, 1H, PhCH), 2.65 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.40 (C=O), 168.89 (C=O), 167.56 (C=O), 165.17 (C=O), 159.03, 140.62, 137.95, 135.95, 130.10, 129.32, 129.23, 129.13, 128.59, 126.86, 118.85, 115.17, 55.90, 51.86, 48.68, 37.79. HRMS (ESI) calculated for C31H35N5O7S [M+H]+ 622.2330, found 622.2335.
(S)-N-(4-(4-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-3-oxopiperazine-1-carbonyl)phenyl)cyclopropanecarboxamide (7i)
White solid. Yield: 69%. Purity: 99%. Mp: 108–110 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H, NH), 8.42 (d, J = 8.0 Hz, 1H, NH), 7.68 (d, J = 8.5 Hz, 2H, Ph-H), 7.40 (d, J = 8.3 Hz, 2H, Ph-H), 7.16 (dq, J = 10.1, 5.1, 3.8 Hz, 5H, Ph-H), 6.98 (d, J = 8.8 Hz, 2H, Ph-H), 6.84 (d, J = 6.9 Hz, 2H, Ph-H), 4.46 (td, J = 8.8, 5.1 Hz, 1H, CH), 4.09 (s, 2H, CH2), 4.00–3.87 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.72–3.53 (m, 2H, CH2), 3.16 (q, J = 7.8, 5.0 Hz, 2H, CH2), 3.11 (s, 3H, NCH3), 2.90–2.81 (m, 1H, PhCH), 2.65 (dd, J = 13.4, 9.5 Hz, 1H, PhCH), 1.79 (td, J = 11.2, 10.3, 5.1 Hz, 1H, CH), 0.82 (d, J = 5.2 Hz, 4H, CH2×2). 13C NMR (100 MHz, DMSO-d6) δ 172.47 (C=O), 171.40 (C=O), 169.08 (C=O), 167.56 (C=O), 165.21 (C=O), 159.03, 141.44, 137.96, 135.95, 129.44, 129.32, 129.13, 128.75, 128.59, 126.86, 118.83, 115.17, 55.90, 51.86, 48.67, 37.79, 15.11, 7.86. HRMS (ESI) calculated for C34H37N5O6 [M+H]+ 612.2817, found 636.2821.
(S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(2-oxo-4-(4-(2,2,2-trifluoroacetamido)benzoyl)piperazin-1-yl)acetamido)-3-phenylpropanamide (7j)
White solid. Yield: 76%. Purity: 98%. Mp: 123–125 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.45 (s, 1H, NH), 8.41 (d, J = 8.0 Hz, 1H, NH), 7.78 (d, J = 8.5 Hz, 2H, Ph-H), 7.51 (d, J = 8.2 Hz, 2H, Ph-H), 7.16 (dq, J = 12.3, 7.9, 7.2 Hz, 5H, Ph-H), 6.98 (d, J = 8.8 Hz, 2H, Ph-H), 6.85 (d, J = 6.8 Hz, 2H, Ph-H), 4.48 (dq, J = 8.3, 5.3 Hz, 1H, CH), 4.11 (s, 2H, CH2), 4.02–3.86 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 3.72–3.46 (m, 2H, CH2), 3.22–3.14 (m, 2H, CH2), 3.11 (s, 3H, NCH3), 2.91–2.82 (m, 1H, PhCH), 2.66 (dd, J = 13.3, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 172.22 (C=O), 169.44 (C=O), 168.38 (C=O), 159.84 (C=O), 156.13 (C=O), 155.77, 139.11, 138.76, 136.74, 133.15, 130.13, 129.95, 129.50, 129.41, 127.68, 121.93, 118.40, 115.97, 56.70, 52.69, 49.47, 38.59, 38.55. HRMS (ESI) calculated for C32H32F3N5O6 [M+H]+ 640.2377, found 640.2378.
(S)-2-(2-(4-(4-acetamidobenzoyl)-2-oxopiperazin-1-yl)acetamido)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (F2-7g)
White solid. Yield: 79%. Purity: 95%. Mp: 105–106 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H, NH), 8.43 (d, J = 8.0 Hz, 1H, NH), 7.67 (d, J = 8.6 Hz, 2H, Ph-H), 7.40 (d, J = 8.4 Hz, 2H, Ph-H), 7.25 (d, J = 8.7 Hz, 2H, Ph-H), 7.03 (d, J = 8.8 Hz, 3H, Ph-H), 6.50 (d, J = 6.5 Hz, 2H, Ph-H), 4.47 (td, J = 9.0, 4.5 Hz, 1H, CH), 4.10 (s, 2H, CH2), 4.02–3.89 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 3.75–3.57 (m, 2H, CH2), 3.28–3.20 (m, 2H, CH2), 3.13 (s, 3H, NCH3), 2.88 (dd, J = 13.2, 4.0 Hz, 1H, PhCH), 2.70 (dt, J = 13.5, 7.0 Hz, 1H, PhCH), 2.07 (s, 3H, COCH3). 13C NMR (100 MHz, DMSO-d6) δ 171.67 (C=O), 169.97 (C=O), 169.88 (C=O), 168.51 (C=O), 166.02 (C=O), 163.30 (dd, 1JC-F = 245.7 Hz, 3JC-F = 13.3 Hz), 159.97, 143.40 (d, 3JC-F = 9.4 Hz), 142.24, 136.62, 131.64, 130.28, 129.97, 129.51, 119.63, 116.09, 113.16 (d, 2JC-F = 24.6 Hz), 103.16 (d, 2JC-F = 25.7 Hz), 56.73, 52.30, 49.49, 38.57, 37.96, 25.34. HRMS (ESI) calculated for C32H33F2N5O6 [M+H]+ 622.2472, found 622.2476.
(S)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methyl-2-(2-(4-(4-(methylsulfonamido)benzoyl)-2-oxopiperazin-1-yl)acetamido)propenamide (F2-7h)
White solid. Yield: 81%. Purity: 95%. Mp: 114–116 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H, NH), 8.44 (d, J = 8.0 Hz, 1H, NH), 7.44 (d, J = 8.3 Hz, 2H, Ph-H), 7.26 (t, J = 8.3 Hz, 4H, Ph-H), 7.03 (d, J = 8.9 Hz, 3H, Ph-H), 6.50 (d, J = 6.6 Hz, 2H, Ph-H), 4.47 (td, J = 8.9, 4.5 Hz, 1H, CH), 4.10 (s, 2H, CH2), 3.93 (t, J = 9.7 Hz, 2H, CH2), 3.80 (s, 3H, OCH3), 3.74–3.56 (m, 2H, CH2), 3.28–3.20 (m, 2H, CH2), 3.13 (s, 3H, NCH3), 3.07 (s, 3H, SO2CH3), 2.88 (dd, J = 13.4, 4.0 Hz, 1H, PhCH), 2.70 (dd, J = 13.5, 9.6 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 170.86 (C=O), 168.89 (C=O), 167.70 (C=O), 165.19 (C=O), 162.50 (dd, 1JC-F = 245.8 Hz, 3JC-F = 13.6 Hz), 159.16, 142.54 (d, 3JC-F = 9.5 Hz), 140.63, 135.82, 131.35, 130.05, 129.17, 118.80, 118.09, 115.28, 112.33 (d, 2JC-F = 25.0 Hz), 102.49 (t, 2JC-F = 25.4 Hz), 55.92, 51.49, 48.70, 37.75, 37.15. HRMS (ESI) calculated for C31H33F2N5O7S [M+H]+ 658.2142, found 658.2146.
(S)-N-(4-(4-(2-((3-(3,5-difluorophenyl)-1-((4-methoxyphenyl)(methyl)amino)-1-oxopropan-2-yl)amino)-2-oxoethyl)-3-oxopiperazine-1-carbonyl)phenyl)cyclopropanecarboxamide (F2-7i)
White solid. Yield: 73%. Purity: 97%. Mp: 119–121 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H, NH), 8.42 (d, J = 8.0 Hz, 1H, NH), 7.68 (d, J = 8.5 Hz, 2H, Ph-H), 7.40 (d, J = 8.4 Hz, 2H, Ph-H), 7.24 (d, J = 8.6 Hz, 2H, Ph-H), 7.03 (d, J = 8.8 Hz, 3H, Ph-H), 6.50 (d, J = 6.6 Hz, 2H, Ph-H), 4.47 (td, J = 8.9, 4.5 Hz, 1H, CH), 4.10 (s, 2H, CH2), 4.01–3.88 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 3.75–3.59 (m, 2H, CH2), 3.23 (t, J = 4.9 Hz, 2H, CH2), 3.13 (s, 3H, NCH3), 2.88 (dd, J = 13.3, 4.0 Hz, 1H, PhCH), 2.75–2.65 (m, 1H, PhCH), 1.80 (p, J = 6.2 Hz, 1H, CH), 0.89–0.78 (m, 4H, CH2×2). 13C NMR (100 MHz, DMSO-d6) δ 172.49 (C=O), 170.86 (C=O), 169.08 (C=O), 167.70 (C=O), 165.22 (C=O), 162.49 (dd, 1JC-F = 245.9 Hz, 3JC-F = 13.4 Hz), 159.16, 142.54 (d, 3JC-F = 9.6 Hz), 141.44, 135.81, 129.38, 129.16, 128.74, 118.82, 115.27, 112.35 (d, 2JC-F = 24.7 Hz), 102.48 (t, 2JC-F = 25.6 Hz), 55.91, 51.50, 48.69, 37.75, 37.16, 15.10, 7.87. HRMS (ESI) calculated for C34H35F2N5O6 [M+H]+ 648.2628, found 648.2627.
(S)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methyl-2-(2-(2-oxo-4-(4-(2,2,2-trifluoroacetamido)benzoyl)piperazin-1-yl)acetamido)propenamide (F2-7j)
White solid. Yield: 78%. Purity: 96%. Mp: 104–106 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H, NH), 8.43 (d, J = 8.0 Hz, 1H, NH), 7.78 (d, J = 8.5 Hz, 2H, Ph-H), 7.51 (d, J = 8.4 Hz, 2H, Ph-H), 7.25 (d, J = 8.6 Hz, 2H, Ph-H), 7.03 (d, J = 8.8 Hz, 3H, Ph-H), 6.50 (d, J = 6.8 Hz, 2H, Ph-H), 4.47 (td, J = 8.9, 4.4 Hz, 1H, CH), 4.11 (s, 2H, CH2), 4.01–3.89 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 3.74–3.50 (m, 2H, CH2), 3.28–3.20 (m, 2H, CH2), 3.14 (s, 3H, NCH3), 2.88 (dd, J = 13.0, 3.5 Hz, 1H, PhCH), 2.76–2.65 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 170.87 (C=O), 168.64 (C=O), 167.71 (C=O), 162.49 (dd, 1JC-F = 245.9 Hz, 3JC-F = 13.4 Hz), 159.16 (C=O), 155.32, 154.95, 142.59 (d, 3JC-F = 9.6 Hz), 138.31, 135.81, 129.17, 128.70, 121.11, 120.94, 117.58, 115.27, 114.71, 112.33 (d, 2JC-F = 24.8 Hz), 102.49 (t, 2JC-F = 25.9 Hz), 55.91, 51.51, 48.69, 37.76, 37.13. HRMS (ESI) calculated for C32H30F5N5O6 [M+H]+ 676.2189, found 676.2194.
3.2. In Vitro Anti-HIV Assay with MT-4 Cells
The protocol for the in vitro anti-HIV assay with MT-4 cells is provided in the Supplementary Materials.
3.3. Analysis of Binding to HIV-1 CA Proteins via Surface Plasmon Resonance
The protocol for analysis of binding to CA proteins via surface plasmon resonance is provided in the Supplementary Materials.
3.4. Molecular Dynamics Simulation
The protocol for the molecular dynamics simulation is provided in the Supplementary Materials.
3.5. In Silico ADMET Analysis
The protocol for the in silico ADMET analysis is provided in the Supplementary Materials.
3.6. Metabolic Stability in Human Liver Microsomes
The protocol for the metabolic stability assay using human liver microsomes is provided in the Supplementary Materials.
3.7. Metabolic Stability in Human Plasma
The protocol for the metabolic stability assay using human plasma is provided in the Supplementary Materials.
4. Conclusions
The multiple key roles of HIV-1 CA in the viral life cycle make it a novel and attractive target for drug design. However, due to the structural plasticity of CAs, especially between interprotomer pockets, rational drug design and optimization remain challenging. To address this issue, we thoroughly explored the crystallographic information available for the PF74-CA complex, focusing on key residues Gln67, Glu71, Tyr169, and Lys182 of the NTD-CTD interface as new binding sites for structural optimization to obtain compounds with improved antiviral activity and metabolic stability. In this study, we designed and synthesized 20 piperazinone phenylalanine derivatives with a terminal indole or benzene ring, preliminarily verifying the HIV-1 CA protein as the target of these compounds via SPR and MD simulation. According to our SAR studies, compounds with higher numbers of fluorine atoms at the terminal benzene of the two series showed the most potent anti-HIV-1 activity, as shown by F2-7f (EC50 = 5.89 μM) and F2-7j (EC50 = 13.74 μM). Mechanistically, our MD simulation results confirmed this finding, as compounds with terminal F atoms, such as F2-7f, had a 20% probability of forming a hydrogen bond with key residue Glu71. However, we acknowledge that the terminal electron-withdrawing F atoms introduce metabolic lability at the benzene ring, as our in silico analysis predicted.
Nevertheless, the anti-HIV-1 activities of the newly synthesized compounds were lower than that of PF74 (EC50 = 0.75 μM), which might be due to a partial change in their binding modes to CA, resulting in the disappearance of two key hydrogen bonds between the compound and Asn57. Therefore, in our follow-up work, we will employ a conformational restriction strategy to maintain the conformation of the phenylalanine core to maintain its critical interactions. Notably, the newly synthesized compounds displayed significant anti-HIV-2 activity, with compound 7f (EC50 = 4.52 μM) comparable to PF74 (EC50 = 4.16 μM).
We used a previously reported workflow to predict the ADMET properties of representative compounds 7f and F2-7f. Compared with PF74, F2-7f exhibited an improved oral non-CNS drug profile score but no significant improvements in metabolic stability. Moreover, the knowledge-based in silico prediction indicated that the compounds should not induce any genotoxicity and hepatotoxicity issues. Finally, we experimentally verified the in silico metabolic stability results utilizing HLMs and human plasma stability. Although lacking significant metabolic improvements, F2-7f represents a new chemotype with the potential for further modifications to improve anti-HIV potency and metabolic stability.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules27238415/s1. References [29,30,32,34,35,37,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, S.X. and P.Z.; writing—original draft preparation, S.X. and L.S.; writing—review and editing, T.H., X.Z., D.D., X.J., F.Z., A.D., S.C. and P.Z.; synthesis, methodology, investigation, and data collation, S.X., L.S., T.H. and X.S.; biological studies: E.D.C. and C.P.; SPR and ADMET analysis: A.D.; molecular dynamic simulation, W.A.Z.; supervision, project administration, and funding acquisition, S.C., X.L. and P.Z. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
Funding Statement
We gratefully acknowledge financial support from the National Natural Science Foundation of China (NSFC No. 82173677, 82204196), Science Foundation for Outstanding Young Scholars of Shandong Province (ZR2020JQ31), Qilu Young Scholars Program of Shandong University, Taishan Scholar Program at Shandong Province, Shandong Provincial Natural Science Foundation (ZR2022QH015) and NIH/NIAID grant R01AI150491 (Cocklin, PI, Salvino, Co-I). We would like to thank the OpenEye Scientific Software, Inc. for providing a free academic license.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zhan P., Pannecouque C., De Clercq E., Liu X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016;59:2849–2878. doi: 10.1021/acs.jmedchem.5b00497. [DOI] [PubMed] [Google Scholar]
- 2. [(accessed on 12 October 2022)]. Available online: https://www.who.int/health-topics/hiv-aids#tab=tab_1.
- 3.Wang Z., Cherukupalli S., Xie M., Wang W., Jiang X., Jia R., Pannecouque C., De Clercq E., Kang D., Zhan P., et al. Contemporary Medicinal Chemistry Strategies for the Discovery and Development of Novel HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors. J. Med. Chem. 2022;65:3729–3757. doi: 10.1021/acs.jmedchem.1c01758. [DOI] [PubMed] [Google Scholar]
- 4.Rasmussen D.N., Vieira N., Hønge B.L., da Silva Té D., Jespersen S., Bjerregaard-Andersen M., Oliveira I., Furtado A., Gomes M.A., Sodemann M., et al. HIV-1 and HIV-2 Prevalence, Risk Factors and Birth Outcomes Among Pregnant Women in Bissau, Guinea-Bissau: A Retrospective Cross-sectional Hospital Study. Sci. Rep. 2020;10:12174. doi: 10.1038/s41598-020-68806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan M.A., Gupta K.K., Singh S.K. A Review on Pharmacokinetics Properties of Antiretroviral Drugs to Treat HIV-1 Infections. Curr. Comput. Aid. Drug Des. 2021;17:850–864. doi: 10.2174/1573409916666201006143007. [DOI] [PubMed] [Google Scholar]
- 6.Menéndez-Arias L., Delgado R. Update and Latest Advances in Antiretroviral Therapy. Trends Pharmacol. Sci. 2022;43:16–29. doi: 10.1016/j.tips.2021.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Du J., Guo J., Kang D., Li Z., Wang G., Wu J., Zhang Z., Fang H., Hou X., Huang Z., et al. New Techniques and Strategies in Drug Discovery. Chin. Chem. Lett. 2020;31:695–1708. doi: 10.1016/j.cclet.2020.03.028. [DOI] [Google Scholar]
- 8.Sun L., Zhang X., Xu S., Huang T., Song S., Cherukupalli S., Zhan P., Liu X. An Insight on Medicinal Aspects of Novel HIV-1 Capsid Protein Inhibitors. Eur. J. Med. Chem. 2021;217:113380. doi: 10.1016/j.ejmech.2021.113380. [DOI] [PubMed] [Google Scholar]
- 9.Rossi E., Meuser M.E., Cunanan C.J., Cocklin S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life. 2021;11:100. doi: 10.3390/life11020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aiken C., Rousso I. The HIV-1 Capsid and Reverse Transcription. Retrovirology. 2021;18:29. doi: 10.1186/s12977-021-00566-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novikova M., Zhang Y., Freed E.O., Peng K. Multiple Roles of HIV-1 Capsid during the Virus Replication Cycle. Virol. Sin. 2019;34:119–134. doi: 10.1007/s12250-019-00095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L., Casey M.C., Vernekar S.K.V., Sahani R.L., Kirby K.A., Du H., Zhang H., Tedbury P.R., Xie J., Sarafianos S.G., et al. Novel PF74-like Small Molecules Targeting the HIV-1 Capsid Protein: Balance of Potency and Metabolic Stability. Acta. Pharm. Sin. B. 2021;11:810–822. doi: 10.1016/j.apsb.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell E.M., Hope T.J. HIV-1 Capsid: The Multifaceted Key Player in HIV-1 Infection. Nat. Rev. Microbiol. 2015;13:471–483. doi: 10.1038/nrmicro3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mattei S., Glass B., Hagen W.J., Kräusslich H.G., Briggs J.A. The Structure and Flexibility of Conical HIV-1 Capsids Determined within Intact Virions. Science. 2016;354:1434–1437. doi: 10.1126/science.aah4972. [DOI] [PubMed] [Google Scholar]
- 15.Domenech R., Neira J.L. The HIV-1 Capsid Protein as a Drug Target: Recent Advances and Future Prospects. Curr. Prot. Pept. Sci. 2013;14:658–668. doi: 10.2174/13892037113146660084. [DOI] [PubMed] [Google Scholar]
- 16.Yufenyuy E.L., Aiken C. The NTD-CTD Intersubunit Interface Plays a Critical Role in Assembly and Stabilization of the HIV-1 Capsid. Retrovirology. 2013;10:29. doi: 10.1186/1742-4690-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Achuthan V., Perreira J.M., Ahn J.J., Brass A.L., Engelman A.N. Capsid-CPSF6 Interaction: Master Regulator of Nuclear HIV-1 Positioning and Integration. J. Life Sci. 2019;1:39–45. doi: 10.36069/jols/20190604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S., Patel J.S., Yang J., Crabtree A.M., Rubenstein B.M., Lund-Andersen P.K., Ytreberg F.M., Rowley P.A. Defining the HIV Capsid Binding Site of Nucleoporin 153. mSphere. 2022;7:e0031022. doi: 10.1128/msphere.00310-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rebensburg S.V., Wei G., Larue R.C., Lindenberger J., Francis A.C., Annamalai A.S., Morrison J., Shkriabai N., Huang S.W., KewalRamani V., et al. Sec24C is an HIV-1 Host Dependency Factor Crucial for Virus Replication. Nat. Microbiol. 2021;6:435–444. doi: 10.1038/s41564-021-00868-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen B. HIV Capsid Assembly, Mechanism, and Structure. Biochemistry. 2016;55:2539–2552. doi: 10.1021/acs.biochem.6b00159. [DOI] [PubMed] [Google Scholar]
- 21.Xu S., Sun L., Dick A., Zalloum W.A., Huang T., Meuser M.E., Zhang X., Tao Y., Cherukupalli S., Ding D., et al. Design, Synthesis, and Mechanistic Investigations of Phenylalanine Derivatives Containing a Benzothiazole Moiety as HIV-1 Capsid Inhibitors with Improved Metabolic Stability. Eur. J. Med. Chem. 2022;227:113903. doi: 10.1016/j.ejmech.2021.113903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blair W.S., Pickford C., Irving S.L., Brown D.G., Anderson M., Bazin R., Cao J., Ciaramella G., Isaacson J., Jackson L., et al. HIV Capsid is a Tractable Target for Small Molecule Therapeutic Intervention. PLoS Pathog. 2010;6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X., Sun L., Meuser M.E., Zalloum W.A., Xu S., Huang T., Cherukupalli S., Jiang X., Ding X., Tao Y., et al. Design, Synthesis, and Mechanism Study of Dimerized Phenylalanine Derivatives as Novel HIV-1 Capsid Inhibitors. Eur. J. Med. Chem. 2021;226:113848. doi: 10.1016/j.ejmech.2021.113848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gres A.T., Kirby K.A., KewalRamani V.N., Tanner J.J., Pornillos O., Sarafianos S.G. STRUCTURAL VIROLOGY. X-ray Crystal Structures of Native HIV-1 Capsid Protein Reveal Conformational Variability. Science. 2015;349:99–103. doi: 10.1126/science.aaa5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L., Huang T., Dick A., Meuser M.E., Zalloum W.A., Chen C.H., Ding X., Gao P., Cocklin S., Lee K.H., et al. Design, Synthesis and Structure-activity Relationships of 4-phenyl-1H-1,2,3-triazole Phenylalanine Derivatives as Novel HIV-1 Capsid Inhibitors with Promising Antiviral Activities. Eur. J. Med. Chem. 2020;190:112085. doi: 10.1016/j.ejmech.2020.112085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu S., Sun L., Ding D., Zhang X., Liu X., Zhan P. Metabolite Identification of HIV-1 Capsid Modulators PF74 and 11L in Human Liver Microsomes. Metabolites. 2022;12:752. doi: 10.3390/metabo12080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Link J.O., Rhee M.S., Tse W.C., Zheng J., Somoza J.R., Rowe W., Begley R., Chiu A., Mulato A., Hansen D., et al. Clinical Targeting of HIV Capsid Protein with a Long-acting Small Molecule. Nature. 2020;584:614–661. doi: 10.1038/s41586-020-2443-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Margot N.A., Naik V., Vander V.L., Anoshchenko O., Singh R., Dvory-Sobol H., Rhee M.S., Callebaut C. Resistance Analyses in Highly Treatment-Experienced People with HIV Treated with the Novel Capsid HIV Inhibitor Lenacapavir. J. Infect. Dis. 2022 doi: 10.1093/infdis/jiac364. ahead of print . [DOI] [PubMed] [Google Scholar]
- 29.Sun L., Dick A., Meuser M.E., Huang T., Zalloum W.A., Chen C.H., Cherukupalli S., Xu S., Ding X., Gao P., et al. Design, Synthesis, and Mechanism Study of Benzenesulfonamide-Containing Phenylalanine Derivatives as Novel HIV-1 Capsid Inhibitors with Improved Antiviral Activities. J. Med. Chem. 2020;63:4790–4810. doi: 10.1021/acs.jmedchem.0c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu S., Sun L., Zalloum W.A., Zhang X., Huang T., Ding D., Tao Y., Zhao F., Gao S., Kang D., et al. From Design to Biological Mechanism Evaluation of Phenylalanine-bearing HIV-1 Capsid Inhibitors Targeting a Vital Assembly Interface. Chin. Chem. Lett. 2022 doi: 10.1016/j.cclet.2022.06.034. in press . [DOI] [Google Scholar]
- 31.Xu J.P., Francis A.C., Meuser M.E., Mankowski M., Ptak R.G., Rashad A.A., Melikyan G.B., Cocklin S. Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J. Drug Des. Res. 2018;5:1070. [PMC free article] [PubMed] [Google Scholar]
- 32.Segall M., Champness E., Obrezanova O., Leeding C. Beyond Profiling: Using ADMET Models to Guide Decisions. Chem. Biodivers. 2009;6:2144–2151. doi: 10.1002/cbdv.200900148. [DOI] [PubMed] [Google Scholar]
- 33.Tuyishime M., Danish M., Princiotto A., Mankowski M.K., Lawrence R., Lombart H.G., Esikov K., Berniac J., Liang K., Ji J., et al. Discovery and Optimization of Novel Small-molecule HIV-1 Entry Inhibitors Using Field-based Virtual Screening and Bioisosteric Replacement. Bioorg. Med. Chem. Lett. 2014;24:5439–5445. doi: 10.1016/j.bmcl.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karadsheh R., Meuser M.E., Cocklin S. Composition and Orientation of the Core Region of Novel HIV-1 Entry Inhibitors Influences Metabolic Stability. Molecules. 2020;25:1430. doi: 10.3390/molecules25061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meuser M.E., Reddy P.A.N., Dick A., Maurancy J.M., Salvino J.M., Cocklin S. Rapid Optimization of the Metabolic Stability of a Human Immunodeficiency Virus Type-1 Capsid Inhibitor Using a Multistep Computational Workflow. J. Med. Chem. 2021;64:3747–3766. doi: 10.1021/acs.jmedchem.0c01810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyzack J.D., Hunt P.A., Segall M.D. Predicting Regioselectivity and Lability of Cytochrome P450 Metabolism Using Quantum Mechanical Simulations. J. Chem. Inf. Model. 2016;56:2180–2193. doi: 10.1021/acs.jcim.6b00233. [DOI] [PubMed] [Google Scholar]
- 37.Hunt P.A., Segall M.D., Tyzack J.D. WhichP450: A Multi-class Categorical Model to Predict the Major Metabolising CYP450 Isoform for a Compound. J. Comput. Aid. Mol. Des. 2018;32:537–546. doi: 10.1007/s10822-018-0107-0. [DOI] [PubMed] [Google Scholar]
- 38.Reulecke I., Lange G., Albrecht J., Klein R., Rarey M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem. 2008;3:885–897. doi: 10.1002/cmdc.200700319. [DOI] [PubMed] [Google Scholar]
- 39.Kaur P., Chamberlin A.R., Poulos T.L., Sevrioukova I.F. Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J. Med. Chem. 2016;59:4210–4220. doi: 10.1021/acs.jmedchem.5b01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sega G.A. A Review of The Genetic Effects of Ethyl Methanesulfonate. Mutat. Res. 1984;134:113–142. doi: 10.1016/0165-1110(84)90007-1. [DOI] [PubMed] [Google Scholar]
- 41.Pillans P.I., Ghiculescu R.A., Lampe G., Wilson R., Wong R., Macdonald G.A. Severe Acute Liver Injury Associated with Lumiracoxib. J. Gastroenterol. Hepatol. 2012;27:1102–1105. doi: 10.1111/j.1440-1746.2011.07036.x. [DOI] [PubMed] [Google Scholar]
- 42.Kortagere S., Madani N., Mankowski M.K., Schon A., Zentner I., Swaminathan G., Princiotto A., Anthony K., Oza A., Sierra L.J., et al. Inhibiting Early-stage Events in HIV-1 Replication by Small-molecule Targeting of the HIV-1 Capsid. J. Virol. 2012;86:8472–8481. doi: 10.1128/JVI.05006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Studier F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 44.Jacques D.A., McEwan W.A., Hilditch L., Price A.J., Towers G.J., James L.C. HIV-1 Uses Dynamic Capsid Pores to Import Nucleotides and Fuel Encapsidated DNA Synthesis. Nature. 2016;536:349. doi: 10.1038/nature19098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ester M., Kriegel H.P., Sander J., Xu X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In: Simoudis E., Han J., Fayyad U., editors. Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining. AAAI Press; Menlo Park, CA, USA: 1996. pp. 226–231. [Google Scholar]
- 46.Hawkins P.C.D., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. OMEGA, Version 3.0.0.1. OpenEye Scientific; Santa Fe, NM, USA: 2010. [Google Scholar]
- 47.Hawkins P.C., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and the Cambridge Structural Database. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.OEDOCKING 3.0.1: OpenEye Scientific Software, Santa Fe, NM. [(accessed on 12 October 2022)]. Available online: http://www.eyesopen.com.
- 49.Kelley B.P., Brown S.P., Warren G.L., Muchmore S.W. POSIT: Flexible Shape-Guided Docking for Pose Prediction. J. Chem. Inf. Model. 2015;55:1771–1780. doi: 10.1021/acs.jcim.5b00142. [DOI] [PubMed] [Google Scholar]
- 50.McGann M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011;51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 51.McGann M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided Mol. Des. 2012;26:897–906. doi: 10.1007/s10822-012-9584-8. [DOI] [PubMed] [Google Scholar]
- 52.Case D.A., Betz R.M., Botello-Smith W., Cerutti D.S., Cheatham T.E., Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., et al. Amber 2016. University of California; San Francisco, CA, USA: 2016. [Google Scholar]
- 53.Shao J., Tanner S.W., Thompson N., Cheatham T.E. Clustering molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 54.Huey R., Morris G., Olson A.J., Goodsell D.S. A Semiempirical Free Energy Force Field with Charge-based Desolvation. J. Comput. Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 55.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris G.M., Huey R., Olson A.J. Using AutoDock for Ligand-receptor Docking. Curr. Protoc. Bioinform. 2008;24:8–14. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 57.Bikadi Z., Hazai E. Application of the PM6 Semi-empirical Method to Modeling Proteins Enhances Docking Accuracy of AutoDock. J. Cheminform. 2009;1:15. doi: 10.1186/1758-2946-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.