

Abstract
Attaching and effacing (A/E) rabbit enteropathogenic Escherichia coli (REPEC) strains belonging to serogroup O103 are an important cause of diarrhea in weaned rabbits. Like human EPEC strains, they possess the locus of enterocyte effacement clustering the genes involved in the formation of the A/E lesions. In addition, pathogenic REPEC O103 strains produce an Esp-dependent but Eae (intimin)-independent alteration of the host cell cytoskeleton characterized by the formation of focal adhesion complexes and the reorganization of the actin cytoskeleton into bundles of stress fibers. To investigate the role of intimin and its translocated coreceptor (Tir) in the pathogenicity of REPEC, we have used a newly constructed isogenic tir null mutant together with a previously described eae null mutant. When human HeLa epithelial cells were infected, the tir mutant was still able to induce the formation of stress fibers as previously reported for the eae null mutant. When the rabbit epithelial cell line RK13 was used, REPEC O103 produced a classical fluorescent actin staining (FAS) effect, whereas both the eae and tir mutants were FAS negative. In a rabbit ligated ileal loop model, neither mutant was able to induce A/E lesions. In contrast to the parental strain, which intimately adhered to the enterocytes and destroyed the brush border microvilli, bacteria of both mutants were clustered in the mucus without reaching and damaging the microvilli. The role of intimin and Tir was then analyzed in vivo by oral inoculation of weaned rabbits. Although both mutants were still present in the intestinal flora of the rabbits 3 weeks after oral inoculation, neither mutant strain induced any clinical signs or significant weight loss in the inoculated rabbits whereas the parental strain caused the death of 90% of the inoculated rabbits. Nevertheless, an inflammatory infiltrate was present in the lamina propria of the rabbits infected with both mutants, with an inflammatory response greater for the eae null mutant. In conclusion, we have confirmed the role of intimin in virulence, and we have shown, for the first time, that Tir is also a key factor in vivo for pathogenicity.
Although Escherichia coli belongs to the normal microflora present in the gastrointestinal tracts of most mammals and birds, certain E. coli strains have been associated with intestinal or extraintestinal infections. Among these pathogenic E. coli strains, enteropathogenic E. coli (EPEC) is a major cause of infant diarrhea in developing countries (for a recent review, see reference 51) and is a significant category of diarrheagenic E. coli in different animal species. In addition, EPEC is an important cause of morbidity and mortality in weaned rabbits (5, 54, 56). EPEC is also pathogenic in neonatal calves (20, 53) and seems to be isolated more frequently in farms with recurrent diarrhea (7). In swine, EPEC is involved in cases of postweaning diarrhea (67). There is also increasing evidence for a diarrheagenic role of EPEC in dogs (16, 64). Finally, EPEC has been isolated from wild and domestic birds (21, 24, 66), although the role of these strains in avian diseases has yet to be defined.
EPEC and certain enterohemorrhagic E. coli (EHEC) strains produce a characteristic histopathological feature known as the attaching and effacing (A/E) lesion by subverting intestinal epithelial cell function (recently reviewed in reference 23). This striking phenotype is characterized by effacement of microvilli and intimate adherence between the bacteria and the epithelial cell membrane (49). Marked cytoskeletal changes, including accumulation of polymerized actin, are seen directly beneath the adherent bacteria and are detected through the use of the fluorescent actin staining (FAS) test (39). Other cytoskeleton components such as α-actinin and myosin light chain, but not vinculin, are also observed beneath the FAS-positive bacteria (19, 43). The FAS and A/E lesions are governed by a pathogenicity island called the locus of enterocyte effacement (LEE). The LEE was first described for human EPEC strain E2348/69 (44). The LEE encodes proteins with a range of functions, including a type III secretion system, various secreted effectors proteins, and their chaperones (14, 17). The central region of the LEE contains the eae (for E. coli attachment effacement) gene encoding the 94- to 97-kDa outer membrane protein known as intimin (33). This protein mediates close contact between the bacteria and the target cell upon interaction with its translocated receptor EspE, or Tir (for translocated intimin receptor), which is encoded by a gene upstream of eae (9, 37). Tir was identified initially as a 90-kDa tyrosine-phosphorylated protein in the target cell membrane and had been previously called Hp90 (58). The 78- to 80-kDa Tir/EspE proteins were shown to be secreted by the type III secretion system and translocated into the host cell, where they are localized in the cytoplasmic and plasma membrane fractions (9, 37).
The role of intimin in human disease was demonstrated by studies with human volunteers who ingested an isogenic eae null mutant of EPEC strain E2348/69 (12). To our knowledge, no other studies have demonstrated in a natural host the role of intimin in the pathogenesis of human or animal EPEC strains. Schauer and Falkow (62) also demonstrated that the intimin expressed by Citrobacter rodentium, the mouse homologue of EPEC, was essential for the formation of intestinal A/E lesions in infected mice. By contrast, further studies with EHEC O157:H7 strains have shown that intimin was required for these pathogens to intensively colonize the intestines and cause A/E lesions and diarrhea in calves and to cause colonic edema and A/E lesions in piglets (8, 13, 45). Although the role of intimin in pathogenicity is well documented, the role of Tir in vivo still has not been demonstrated by studies with animals or human volunteers.
The rabbit EPEC (REPEC) strains belonging to serotype O103:K−:H2 and to the rhamnose-negative biovar are the main cause of E. coli enteritis in weaned rabbits in western Europe (5, 6). These strains induced severe and lethal diarrhea upon oral inoculations with as little as 104 CFU (50). It is now established that these strains have pathogenic mechanisms that are analogous to those of human EPEC. Thus, REPEC can be considered one of the most relevant models for the study of the pathogenesis of A/E E. coli in a natural host (for a review, see reference 48). Although distinct in size and restriction polymorphism profile, the LEE of these strains is organized in similar clusters of genes homologous with those identified in other EPEC or EHEC strains (10, 41, 52). As with human EPEC E2348/69, the A/E lesions provoked by the REPEC O103 are characterized by an intimate adhesion to the cell in cup-like pedestals associated with a localized degeneration of host cell microvilli (1, 42, 52, 57). However, EPEC E2348/69 and REPEC O103 differ in their cytopathic effects (CPE) on mammalian cells in vitro. REPEC O103 strains induce only a weak FAS effect, whereas the human EPEC strain E2348/69 induces a strong FAS response. In addition, the REPEC O103 strains provoke a progressive and irreversible CPE which is not induced by the human EPEC strain E2348/69 (10). This CPE is characterized by a dramatic and progressive reorganization of the actin cytoskeleton into bundles of stress fibers and by the recruitment of focal adhesion plaques. This long-term cytoskeletal rearrangement is EspA, EspB, and EspD dependent but intimin independent (52). No single esp gene encodes the information needed to confer the CPE phenotype, since each espA, -B, and -D mutant could be fully complemented in trans by the corresponding cloned esp genes from both the parental REPEC strain and the CPE-negative human EPEC strain E2348/69 (52). The relevance of CPE in REPEC pathogenesis is not known, although the CPE leads in vitro to cell death 96 h after a 4-h interaction between the bacteria and the epithelial cells.
In this study, our purpose was to clarify the role of Tir and intimin in the virulence of REPEC serotype O103:H2, using both in vivo and in vitro models. We have observed that Tir and intimin were required for the capacity of REPEC to nucleate F-actin and induce A/E lesions but not for the reorganization of the actin cytoskeleton into bundles of stress fibers. The roles of intimin and Tir were then analyzed in vivo by oral infection of weaned rabbits, the natural host of REPEC. Neither mutant strain induced any symptoms or significant weight loss, although an inflammatory infiltrate was present in the lamina propria of the rabbits inoculated with isogenic eae and tir mutants.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
The wild-type and engineered enteropathogenic E. coli strains used in this study are listed in Table 1. The cloning vectors pBluescript II KS(+), pCR2-1, and pCR-XL-TOPO were obtained from Stratagene and Invitrogen, respectively. Plasmid pBRSK is a low-copy-number vector derived from pBR328 (63). Plasmid pKNG101 is a positive selection suicide vector containing strAB, sacBR, and a pir-dependent R6K replicon (34). Plasmids were maintained in laboratory strain XL1blue (Stratagene), except for suicide plasmids (pKNG101 and derivatives), which were maintained in CC118 λpir (30), and the cosmid pII5F, which was maintained in HB101 (18). Bacteria were isolated on Luria-Bertani (LB) agar and cultivated in LB broth with appropriate antibiotics at the following concentrations: carbenicillin, 50 μg ml−1; kanamycin, 50 μg ml−1; streptomycin, 50 μg ml−1; nalidixic acid, 25 μg ml−1; and chloramphenicol, 25 μg ml−1.
TABLE 1.
| Strain | Description | Source or reference |
|---|---|---|
| E22 | REPEC O103:K-H2, rhamnose-negative strain | 6 |
| E22ΔEae | E22 eae::aphT, Eae− | 52 |
| E22ΔTir | E22 tir::aphT, Tir− | This study |
| E22 (pBRSK) | E22 transformed with the low-copy-number vector pBRSK | This study |
| E22ΔEae (pBReaeREPEC) | E22ΔEae transformed with eae from E22 cloned into pBRSK vector | This study |
| E22ΔTir (pBRtirREPEC) | E22ΔTir transformed with tir from E22 cloned into pBRSK vector | This study |
| E2348/69 | Prototype O127:H6 human EPEC strain | 12 |
| BM21 | Laboratory K-12 strain | 55 |
Cell lines.
Rabbit kidney (RK13) cells were cultivated in Dulbecco's modified Eagle's medium with 10% fetal calf serum (FCS) (Gibco) and gentamicin (40 μg ml−1). HeLa cells (ATCC CCL2) were cultivated in Eagle's minimum essential medium (MEM) supplemented with 10% FCS, l-glutamine, and gentamicin (80 μg ml−1). Both cell lines were grown at 37°C in a 5% CO2–95% air atmosphere.
Fluorescence microscopy.
RK13 cells were seeded at 5 × 104 cells per well on Lab-Tek 8 chambers slides (Falcon) and grown overnight. Prior to infection, cells were washed twice with Earle balanced saline solution (Gibco), and the medium was replaced with (per well) 500 μl of MEM buffered with 25 mM HEPES (Gibco) complemented with 5% FCS and 1% mannose. Cells were inoculated with overnight static cultures of E. coli strains at a ratio of 500 bacteria per cell. After a 4-h interaction at 37°C, cell monolayers were washed five times with phosphate-buffered saline (PBS) (pH 7.4) and fixed with 3% paraformaldehyde in PBS for 1 h at 4°C. Cells were then permeabilized with 0.1% Triton X-100 in PBS for 5 min, and F-actin was labeled with rhodamine-phalloidin (Molecular Probes) according to the manufacturer's instructions. CPE was assessed on HeLa cells as previously described (10).
Recombinant DNA, genetic techniques, and nucleotide sequencing.
Routine recombinant DNA techniques were performed using standard procedures (59). Plasmids were introduced in REPEC strains by electroporation with a Gene Pulser II, set at 2.5 kV, 25 μF, and 200 Ω, in 0.2-cm cuvettes, according to the instructions of the manufacturer (Bio-Rad). The nucleotide sequence of double-stranded template DNA was determined using a Dye-Deoxy Terminator Cycle Sequencing Kit and an ABI 373A DNA sequencer (Applied Biosystems). PCR amplification of DNA fragments was carried out using a commercial kit (GenAmp; Perkin-Elmer Cetus) with high-fidelity Pfu DNA polymerase (Stratagene) according to the instructions of the manufacturers. The 1.7-kb PCR product obtained with primers Tir total-sens (5′ AGG ATA TAT GTA TGC CTA TTG GTA A-3′) and Tir-as (5′-CCC AAC CTC AAC TAA ATA CTC-3′) was used as a DNA probe for the detection of the tir gene.
Construction of nonpolar mutations in tir.
Plasmid pKTir2.1 was constructed by first cloning from cosmid pII5F a 3.1-kb BglII DNA fragment bearing tir into pKSII+ and then deleting a 1-kb HindIII DNA fragment containing sequence upstream from tir. The aphT gene without a transcription terminator was excised from pSB315 (25) by use of BamHI and inserted into the BamHI site of pKTir2.1 (at 588 bp downstream of the start codon of tir). The ApaI/SpeI fragment of pKTir2.1, bearing tir disrupted by aphT, was then cloned into pKNG101, giving plasmid pKNG tir::aphT. Suicide plasmid pKNG tir::aphT was introduced into E22 by electroporation. Mutants that had undergone allelic exchange leading to the replacement of the wild-type locus with the locus disrupted by aphT were selected on LB plates without NaCl containing 5% sucrose and kanamycin, as previously described (34). Mutations were confirmed by Southern blotting and PCR, as previously described (59). The resulting tir null mutant, called E22ΔTir, was trans complemented by the 2.1-kb HindIII/BglII insert from pKTir2.1 cloned into pBRSK. The resulting construction was called pBRtirREPEC.
Detection of expression of intimin and Tir proteins.
The detection of intimin in bacterial cultures by Western blotting was performed as previously described (40), using a polyclonal serum raised against the maltose-binding protein–Eae dog EPEC fusion protein (3). Cellular fractionation was performed as previously described (58). HeLa cells were cultivated in 5-cm-diameter tissue culture petri dishes. Bacteria were grown overnight in LB broth without shaking. HeLa cell monolayers were washed three times with Earle balanced saline solution, incubated in MEM-HEPES complemented with 5% FCS and 1% mannose, and infected with a bacterium-to-epithelial cell ratio of 1,000 to 1. After a 4-h interaction at 37°C in a 5% CO2–95% air atmosphere, monolayers were washed four times in PBS and scraped into 1 ml of PBS. After centrifugation, the cell pellets were lysed in 50 μl of lysis buffer (Triton X-100, 1%; NaF, 1 mM; Na3VO4, 0.4 mM;, and Complete protease inhibitor cocktail [Roche Molecular Biochemicals]) for 15 min at 4°C and centrifuged (3 min at 13,000 × g and 4°C). The supernatant (Triton X-100 soluble fraction) containing membrane and cytoplasmic proteins was mixed with 15 μl of 5× Laemmli sample buffer, and the pellet (insoluble fraction) containing both the cytoskeletal proteins and the adherent bacteria was mixed with 50 μl of 2.5× Laemmli sample buffer.
The soluble and insoluble fractions were resolved by sodium dodecyl sulfate–8% polyacrylamide gel electrophoresis (SDS–8% PAGE) and transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore). The membranes were blocked for 30 min at 37°C in Superblock (Pierce) and probed with a rabbit polyclonal serum raised against Tir (29) or with an antiphosphotyrosine monoclonal antibody (clone 4G10; Upstate Biotechnology, Inc.). Both antibodies were used at a dilution of 1/1,000 in Tris-buffered saline (TBS)–0.1% Tween 20–0.1% bovine serum albumin (Sigma) for 1 h at 37°C. After washing in TBS–0.1% Tween 20, the bound antibodies were reacted with alkaline phosphatase-conjugated secondary antibodies (1/5,000 in TBS–0.1% Tween 20–3% neonatal goat serum), and membranes were developed with the chemiluminescence substrate CDP-Star (Boehringer).
Complementation of REPEC O103 mutants.
A 3.7-kb PCR product of REPEC O103 E22 DNA, containing the 2,820-bp eae open reading frame, was obtained with primer OrfU-sens (5′-TAT GAT GAT CTA TGG CGT CTG T-3′) and EscD-asens (5′-TAT TTT CAA AAA GAA TGA TGT C-3′). These primers were designed by examination of the sequence of REPEC O103 strain 84/110/1 (accession number U59502). The PCR product was then cloned into pCR-XL-TOPO vector. A BamHI/NotI fragment bearing eae was then subcloned in pBRSK opened by BamHI and NotI. The resulting plasmid, called pBReaeREPEC, was introduced into E22ΔEae by electroporation. The ApaI/SpeI fragment of pKTir2.1 bearing tir was inserted into pBRSK opened by the same restriction enzymes. The resulting plasmid, called pBRtirREPEC, was introduced into E22ΔTir by electroporation. The transformants E22ΔEae(pBReaeREPEC) and E22ΔTir(pBRTtirREPEC) were selected in the presence of chloramphenicol and kanamycin.
Rabbit ligated intestinal loop assay and demonstration of A/E lesions.
The rabbit ligated intestinal loop assay was performed as previously described (27). Briefly, loops were created in 3-month-old New Zealand rabbits. One milliliter of an overnight LB bacterial culture (containing approximately 109 CFU) was injected into each ligated intestinal loop. On the following day, rabbits were euthanasied with sodium pentobarbital. Samples were taken for light and electron microscopic examination. Each strain was tested in at least three different animals.
Rabbit infection experiment.
To investigate the role of Eae and Tir in REPEC O103 virulence, we orally inoculated 32-day-old New Zealand weaned rabbits. The rabbits were divided into four groups and housed in cages of three animals. They were fed and watered daily with commercial feed supplemented with a coccidiostatic agent (Robenidine). Animals were inoculated orally with 2 × 107 CFU of strain E22 (17 animals), E22ΔEae (17 animals), E22ΔTir (16 animals), or the avirulent laboratory strain BM21 (9 animals). Each animal was weighed three times a week and checked daily for clinical symptoms, diarrhea, dehydration, and mortality. E. coli intestinal colonization was determined twice a week by dilution of fecal samples on MacConkey agar. For screening of inoculated strains, the following markers were used: E22, rhamnose negative and kanamycin sensitive; E22ΔEae and E22ΔTir, rhamnose negative and kanamycin resistant; and BM21, nalidixic acid resistant.
Tissue sampling for histopathological analysis.
Four groups of two 32-day-old New Zealand weaned rabbits were orally inoculated with 2 × 107 CFU of strain E22, E22ΔEae, E22ΔTir, or BM21 and housed as previously described. Animals were sacrificed at 5 days postinfection. Tissues from the ileum were removed immediately after euthanasia by intravenous overdosing with sodium pentobarbital. All tissue samples were scored blindly. They were analyzed by light microscopic observation.
Nucleotide sequence accession number.
The tir REPEC O103 nucleotide sequence is available in GenBank database. Its accession number is AF113597.
RESULTS
Construction of an isogenic tir null mutant of REPEC strain E22 and trans complementation of tir and eae REPEC O103 mutants.
Isogenic eae and tir mutants were used to analyze the role of Tir and intimin (Eae) in REPEC O103 pathogenesis (Fig. 1). After previously having constructed by allelic exchange an eae null mutant called E22ΔEae (52), we have constructed in this study an isogenic tir null mutant of REPEC strain E22. First, we undertook a genetic analysis to characterize the E22 tir gene. Southern blotting and hybridization analysis using a specific probe produced by PCR prompted us to clone a 2.1-kb HindIII/BglII fragment from cosmid pII5F isolated from a REPEC O103 genomic DNA library (18). Sequence analysis of the insert of the resulting plasmid pKTir2.1 (GenBank accession number AF043226), revealed the presence of a 1,614-bp open reading frame encoding a 538-amino-acid protein showing 100% amino acid identity with the Tir/EspE from bovine EHEC O26:H− strain 413/89-1 (AJ223063), human EHEC O26:H− strain 95ZG1 (AF070068), and rabbit EPEC O103:H2 strain 84/110/1 (U59502). The REPEC Tir contains the tyrosine residue which needs to be phosphorylated in human EPEC for actin nucleating activity (38).
FIG. 1.

Genetic and restriction maps of the REPEC O103 LEE locus, encoding Tir and intimin. Sites of insertional mutation in tir and eae and fragments cloned in plasmid pBRSK giving pBReaeREPEC and pBRtirREPEC and used to complement mutant strains E22ΔEae and E22ΔTir are shown.
A kanamycin resistance cassette without a transcription terminator was then inserted by allelic exchange into the E22 tir gene as described in Materials and Methods. The resulting mutant strain, E22ΔTir, was confirmed by PCR and Southern blot analysis. No significant difference in the growth rates of the parental strain E22 and the mutant was observed (data not shown). A similar strategy was used for the construction of E22ΔEae (52). E22ΔEae and E22ΔTir were then trans complemented by homologous parental genes, resulting in strains E22ΔEae(pBReaeREPEC) and E22ΔTir(pBRtirREPEC), respectively.
Immunoblots were used to analyze the production of intimin in REPEC O103 strains E22, E22ΔEae, and E22ΔEae (pBReaeREPEC). The absence of intimin production in E22ΔEae was confirmed, and production was fully restored by the introduction of the corresponding cloned wild-type allele in the mutant (Fig. 2). In order to confirm the deletion of Tir in E22ΔTir, we analyzed the Triton-soluble and -insoluble fractions of HeLa cells after infection with the REPEC O103 strains E22, E22ΔTir, and E22ΔTir(pBRtirREPEC). Triton-insoluble cellular extracts were separated by SDS-PAGE and probed with a Tir-specific antiserum or an antiphosphotyrosine monoclonal antibody. The Tir antiserum detected the presence of two major immunoreactive bands of approximately 75 and 85 kDa in the Triton-insoluble fraction of cells infected with E22. Both bands were totally absent after infection with E22ΔTir but were restored and even amplified in the trans-complemented E22ΔTir(pBRtirREPEC) strain (data not shown). We then probed the same fraction with a tyrosine phosphate-specific antibody. Only one major immunoreactive band of approximately 85 kDa was specifically detected in HeLa cell fractions infected with E22 or E22ΔTir(pBRtirREPEC) (Fig. 3). Similar results were obtained with the Triton-soluble fractions of infected HeLa cells (data not shown).
FIG. 2.

Western blot of total cell lysates of E22ΔEae(pBReaeREPEC) (lane 1), E22ΔEae (lane 2), and E22 (lane 3) grown in Dulbecco's modified Eagle's medium. Samples were resolved by SDS–8% PAGE and transferred to PVDF membranes. The membranes were probed with a serum raised against an Eae–maltose-binding protein fusion protein. Molecular mass markers are given in kilodaltons on the left.
FIG. 3.

Western blot of the Triton X-100-insoluble fraction of HeLa cells infected for 4 h with E22 (lane 1), E22ΔTir (lane 2), and E22ΔTir(pBRtirREPEC) (lane 3). Samples were resolved by SDS–8% PAGE and transferred to PVDF membranes. The membranes were probed with an antiphosphotyrosine monoclonal antibody. Molecular mass markers are given in kilodaltons on the left.
Taken together these results confirmed the phenotypes of both E22ΔEae and E22ΔTir mutants and suggested that Tir/EspE of REPEC O103:H2 was modified upon translocation into the eukaryotic cell and tyrosine phosphorylated, as was previously described for human EPEC strain E2348/69 and bovine EHEC strain 413/89-1 (9, 37).
REPEC O103 induces an eae- and tir-dependent FAS effect on RK13 epithelial cells.
Upon interaction with human HeLa cells, E22 showed a strong diffuse adhesion but did not induce the localized accumulation of F-actin typical of a FAS response, as previously described (52). However, the presence of a few HeLa cells with a weak F-actin condensation beneath some adhering bacteria suggested that the FAS-negative phenotype of the REPEC O103 strain E22 was not complete (data not shown). Considering the strong A/E lesions observed in vivo with the REPEC O103 strain, we hypothesized that its FAS-negative phenotype in human HeLa cells could be cell line dependent. Therefore, we tested the FAS capacity of the REPEC O103 strain E22 upon interaction with rabbit epithelial cells (RK13). After a 4-h interaction, the cells were stained with rhodamine-phalloidine and observed by phase-contrast and fluorescence microscopy to visualize the adhesion pattern and the reorganization of the underlying F-actin cytoskeleton. In contrast to the massive adhesion observed upon interaction with HeLa cells, the REPEC O103 strain E22 adhered in a weak, diffuse pattern to RK13 cells. However, they induced an accumulation of F-actin in the vicinity of intimate adhesion sites, typical of a FAS-positive response. When the RK13 cells were infected with the mutants, bacterial adhesion was greatly reduced. Only few bacteria adhered to the cells, and no accumulation of F-actin could be seen beneath their adhesion sites after a 4-h infection with either E22ΔEae or E22ΔTir, (Fig. 4 A, B, E, and F). The FAS phenotype and the adhesion pattern were fully restored upon trans complementation of E22ΔEae and E22ΔTir with a low-copy-number, pBRSK, expression vector bearing the homologous gene (Fig. 4C, D, G, and H). These data confirm that the FAS response requires Tir and intimin and suggest also that both Tir and intimin play an important role in promoting bacterial adherence to certain host cells.
FIG. 4.

REPEC O103 strains E22ΔEae (A and B) and E22ΔTir (E and F) lost their ability to focus F-actin beneath their adhesion sites. The FAS response was restored upon trans complementation of each mutant with its respective parental gene eae (C and D) or tir (G and H). RK13 cells were infected for 4 h with the challenged strains, and then the monolayers were washed, fixed, and permeabilized and F-actin was labeled with rhodamine-phalloidin. Fluorescence (A, C, E, and G) and corresponding phase-contrast (B, D, F, and H) micrographs were taken with a Leica X500 microscope.
REPEC O103 strain E22 induces an eae- and tir-independent long-term cytoskeletal rearrangement on HeLa epithelial cells.
REPEC strain E22 induces a progressive and irreversible CPE characterized by a massive multiplication of actin stress fibers and focal adhesion complexes, leading to cell swelling and cell death 5 days after infection (10, 52). In this study, we demonstrated that E22ΔTir was still CPE positive in HeLa cells 36 h after interaction. The alteration of the F-actin distribution (Fig. 5B) was similar to that in E22-infected cells (Fig. 5A). In addition, the minimum inoculum needed to transform at least 50% of the cells with either E22ΔTir or E22 was not significantly different (data not shown). These results are similar to those previously reported for the eae null mutant (52).
FIG. 5.

Alteration of actin cytoskeleton in HeLa cells 36 h after interaction with E22 (wild type) (A), E22ΔTir (B), and a K-12 laboratory strain (C). Both strains E22 (A) and E22ΔTir (B) induced a cell size increase with a multiplication of actin stress fibers characteristic of the CPE. Actin was labeled with rhodamine-phalloidine, and samples were observed with a Leica X300 microscope.
Tir and Eae are essential for the formation of A/E lesions by REPEC O103.
We then analyzed the triggering of A/E lesions by E22ΔEae and E22ΔTir in the rabbit ligated ileal loop model. Scanning electron microscopy analysis of ileal epithelium 24 h after inoculation showed that the E22 wild-type strain adhered massively and closely to the ileal enterocytes. This adhesion was characterized by microvillus effacement and the induction of cup-like structures underneath adhering bacteria. In contrast, we did not observe intimate adhesion and brush border microvillus alteration with the E22ΔEae or E22ΔTir strain, although these strains adhered in a diffuse pattern as did the parental strain (data not shown). Transmission electron microscopy analysis of rabbit ileal sections confirmed the induction of typical A/E lesions by the parental strain. Bacteria of strain E22 adhered intimately to the enterocytes and induced elongation, vesiculation, and effacement of the surrounding microvilli, leading to a complete degeneration of the epithelial brush border (Fig. 6A). The enterocytes presented a ragged surface with numerous adhering bacteria sitting on pedestal structures (Fig. 6A). With both E22ΔEae (Fig. 6B) and E22ΔTir (Fig. 6C), neither A/E and nor pathological damage was seen at the epithelial cell level. In ileal loops inoculated with these mutants strains, the brush border microvilli were intact and the bacteria appeared to be clustered in the mucoid material but were not attached intimately to the intestinal cells (Fig. 6B and C). These results confirm the essential role of Eae in the A/E process and show that Tir is also a key factor in the induction of A/E lesions in enterocytes in vivo.
FIG. 6.
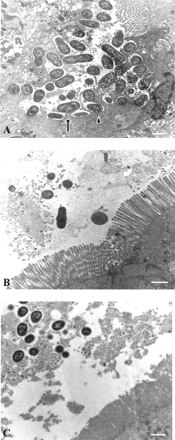
Transmission electron microscopy of rabbit ligated intestinal loops inoculated with E22 (A), E22ΔEae (B), and E22ΔTir (C). E22 (A) adhered in a diffuse pattern to the enterocytes and induced typical A/E lesions characterized by an intimate attachment of bacteria to the enterocyte surface on pedestal structures (arrow) or cup-like structures (arrowhead) and associated with a destruction of surrounding microvilli. In contrast, no A/E lesions were observed with E22ΔEae (B) and E22ΔTir (C). Brush border microvilli were intact, and adherent bacteria stayed clustered in the mucus without reaching the enterocytes (B and C). Samples were analyzed 24 h after infection. Bars, 1 μm.
Tir and Eae are essential virulence factors for the pathogenicity of REPEC O103.
The in vivo pathogenicities of E22ΔEae and E22ΔTir were then investigated in weaned rabbits, the natural host animals which develop severe diarrhea following infection with wild-type REPEC strains of serotype O103:H2. Nine rabbits of a negative control group were inoculated with BM21, a nonpathogenic laboratory strain. None of these rabbits developed any clinical symptoms, and all survived (Fig. 7). The rabbits grew normally, with an average weight gain of 45 g per day. BM21 was not recovered from feces at any time during the whole experiment (Fig. 8). Seventeen rabbits in a positive control group were inoculated with the wild-type strain E22. Among the 17 rabbits, 16 showed an impaired growth rate with weight loss. These 16 rabbits suffering from weight loss developed diarrhea, and 15 of them died between days 5 and 13 after inoculation (Fig. 7). The high mortality observed with rabbits inoculated with E22 was also associated with a massive excretion of bacteria of strain E22. The level of excretion was estimated to be 109 to 1010 CFU per g of feces during the peak of the infection and 108 and 105 CFU per g of feces at day 19 for the two survivors (Fig. 8). Two groups of 17 and 16 rabbits were inoculated with strains E22ΔEae and E22ΔTir, respectively. In contrast to the high mortality and morbidity observed with E22, the rabbits inoculated with E22ΔEae or E22ΔTir did not develop any clinical signs. The rabbits inoculated with mutant strains had an average weight gain (44 and 40 g per day for E22ΔEae and E22ΔTir, respectively) that was not statistically different from that observed with rabbits inoculated with the nonpathogenic strain BM21. Only transient and statistically nonsignificant weight loss was observed in two animals inoculated with E22ΔTir (data not shown). Rabbits inoculated with E22ΔEae or E22ΔTir excreted mutants at the same level (Fig. 8), reaching a peak of 108 CFU per g of feces at day 5 postinfection and persisting at 104 to 105 CFU on day 19 postinfection. Although E22ΔEae or E22ΔTir was able to persist in the gastrointestinal tracts of the rabbits, both mutants were unable to induce diarrhea, indicating that expression of both Eae and Tir is a prerequisite for REPEC pathogenesis.
FIG. 7.
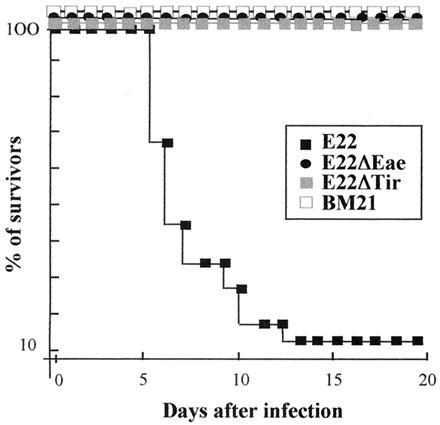
Percentages of rabbits that survived infection after oral inoculation of E22, E22ΔEae, E22ΔTir, and BM21.
FIG. 8.

Kinetics of fecal E. coli shedding in rabbits inoculated orally with wild-type rabbit REPEC strain E22, mutant strains E22ΔEae and E22ΔTir, and laboratory strain K-12 BM21. Solid bars represent the relative proportions of inoculated strains (E22, E22ΔEae, and E22ΔTir) compared to the normal E. coli population (white bars). The laboratory strain BM21 was not recovered from the feces of the control group throughout the experiment.
Nondiarrheagenic tir and eae mutants induce different inflammatory responses in the lamina propria without destroying the brush border.
To substantiate the absence of disease after inoculation of either E22ΔEae or E22ΔTir, histopathological analysis was performed on ileal tissue samples from inoculated rabbits sacrificed 5 days postinfection. Four groups of two weaned rabbits were orally inoculated with strains E22, E22ΔEae, E22ΔTir, and BM21, respectively. Rabbits inoculated with strain E22 displayed classical small intestine lesions of atrophic enteritis (Fig. 9B). The exterior of the villi was ragged or markedly scalloped, and villi were blunted, moderately atrophic, or fused. Epithelial cells on villi in the small intestine were short, rounded up, and in some cases exfoliating singly or in small clumps, causing focal microerosions. The microvillous border was indistinct and covered by a heavy layer of prominent gram-negative bacilli. The avirulent strain BM21 did not induce any histopathological modifications of the sections observed (Fig. 9A). The nondiarrheagenic Tir and Eae mutants induced intermediate lesions at the microscopic level. At 5 days after E22ΔTir infection, the epithelial lining was orthoplastic and goblet cells were present as observed normally. The lamina propria of the villi showed only a slight inflammatory infiltrate (Fig. 9C). In contrast, E22ΔEae-inoculated rabbits exhibited much more pronounced inflammatory lesions in the ileum. Small intestinal villi were moderately atrophic and focally scalloped, and their proprial core showed a moderate inflammatory infiltrate which extended slightly to the submucosa (Fig. 9D). High-power magnification of Gram-stained sections revealed that adhesion of either mutant strain to the brush border did not induce an A/E pattern.
FIG. 9.

Rabbit distal ileum tissue sections 5 days after inoculation. (A) Avirulent strain BM21; (B) E22; (C) E22ΔTir; (D) E22ΔEae. At low-power magnification (×100; hematoxylin-eosin staining), micrographs show the variation of the atrophy level of small intestinal villi, epithelial exfoliation, inflammatory infiltrate of the lamina propria, and edematous aspects of the submucosa, depending on the strain inoculated. (A) Normal histological structure of the small bowel villi. (B) Severely atrophic and fused villi, epithelial exfoliation, and marked inflammatory infiltrate of the lamina propria and submucosa. (C) Slight atrophy of the villi and inflammatory infiltrate of the proprial core of the villi. (D) Pronounced atrophy associated with polymorphous infiltrate extending to the oedematous submucosa. Higher magnification (insets; ×1,000; Gram staining) shows the normal appearance of the columnar enterocyte with a prominent brush border (A); for both mutants, the gram-negative bacilli observed are just at the extremity of the microvilli on the surface of the brush border, which seems to be relatively well preserved and not effaced (C and D). The enterocytes in the panel B inset are irregular and rounded up, and the microvillous border is indistinct and covered by a heavy layer of prominent gram-negative bacilli.
DISCUSSION
This study demonstrates that intimate adhesion mediated by the interaction of Eae and Tir is a prerequisite for the induction of diarrhea and eventually the death of rabbits infected with EPEC. Indeed, REPEC strains of the O103:H2 serotype are highly pathogenic in weaned rabbits inoculated by the oral route, but the single mutation of either the gene coding for the intimate adhesin (Eae) or that coding for its translocated receptor (Tir) was able to fully abolish the pathogenicity of REPEC at a clinical level. However, these nondiarrheagenic tir and eae mutants apparently still induced an inflammatory response in the lamina propria but without destroying the brush border and the general architecture. These results raise several questions on EPEC pathogenesis and provide new ideas for the development of live oral vaccines.
Inflammation of the lamina propria is a common feature of EPEC and EHEC infections (50). The inflammatory response observed during REPEC O103 infection required, at least partially, the production of EspA and EspB (1). Our results suggest that intimin is not required to induce an inflammatory response in the lamina propria, since the eae mutant still induced an inflammatory infiltrate (Fig. 9) with a reduction of villus size compared with control rabbits. The absence of clinical symptoms in rabbits infected with an eae mutant may be host specific (or strain specific). Indeed, the eae mutant of human EPEC strain E2348/69 retains its ability to induce mild diarrhea when tested in human volunteers (12). Of note, a large inoculum (up to 2 × 1010 CFU) was required in the human experimental model because lower doses resulted in lower attack rates. Our results showing that intimin is not required for inflammation are in contradiction with those obtained with the mouse homologue of EPEC, C. rodentium. Indeed, Higgins et al. showed that the inflammation and colonic hyperplasia observed in infected mice are mediated by intimin driving strong mucosal Th-1 responses (32). This discrepancy suggests that the two animal models might not be equivalent.
On the other hand, our results suggest a role for Tir in the induction of inflammation, since the inflammatory response was less pronounced in the ileal samples from rabbits inoculated with E22ΔTir. The intestinal villous architecture of E22ΔTir-infected rabbits was similar to that observed in control animals. Several in vitro studies substantiate a role of Tir rather than intimin in the inflammation. In human epithelial cell lines, the prototype human EPEC strain E2348/69 triggers activation of nuclear factor kappa B (NF-κB), which in turn initiates transcription of the gene encoding interleukin 8 (60). Activation of NF-κB is Eae independent but EspB dependent (61). No Tir mutant was available at that time, but it has now been shown that an EspB mutant is no longer able to translocate Tir in the host cell (37). In vivo, the activation of NF-κB could lead to the activation of transmigration of polymorphonuclear cells and contribute to diarrhea in rabbits. In addition, it was also speculated that an Eae-independent signal transduction event could result in fluid secretion without the loss of microvilli and was an alternative mechanism in the development of EPEC diarrhea (12, 61). Together, these results suggest that part of the inflammatory response in the lamina propria could be due to the insertion of Tir into the host epithelial cell membranes through the Esp-dependent translocation apparatus.
There is also an apparent discrepancy between the absence of clinical symptoms in rabbits infected with eae and tir mutants (this study) and several other reports that have documented a variety of signaling events in vitro within the host epithelial cell infected with eae (and tir) EPEC mutants. For instance, we have shown that REPEC O103 strains, as well as the REPEC O15 strains (RDEC-1) and two human EPEC isolates (10), were able to induce an irreversible CPE. This CPE is characterized by a dramatic progressive reorganization of the actin cytoskeleton into bundles of stress fibers and by the recruitment of vinculin, a protein specifically associated with focal adhesion complexes (10). This irreversible slow transformation results in cell death 5 days after the interaction and is EspA, -B, and -D dependent but Eae and Tir independent (reference 52 and this study). We do not know if such actin rearrangements occurred in vivo after infection with Tir and Eae mutants. Abe and colleagues (1) apparently did not observe bundles of stress fibers in cells infected with a similar wild-type REPEC O103 strain. However, the expression of this CPE is perhaps different in vivo and may not affect directly the epithelial cytoskeleton. Inoculation of rabbits with Esp, Tir, and Eae mutants using high doses of bacteria may provide some clues about the putative role in pathogenesis of the signaling events observed in vitro.
A common prerequisite for the development of any bacterial disease is the localization of the bacteria to a niche that is suitable for growth and pathogenesis. In the mammalian intestines, attachment is critical to avoid displacement from a preferred site by the continuous flow of the intestinal contents. Attachment is also hindered by competition with the multitude of indigenous microflora for binding sites on the intestinal epithelium. The initial step in bacterial attachment to the host epithelium is usually mediated by fimbriae. Therefore, full expression of virulence by EPEC strains of human or rabbit origin requires specific fimbriae, which mediate attachment of the bacteria to the intestinal tract. The human EPEC and the rabbit EPEC prototypes exhibit different patterns of adhesion upon interaction with HeLa or Hep-2 cell lines. EPEC shows a localized adherence pattern characterized by formation of microcolonies on epithelial cell monolayers, whereas REPEC O103 adheres massively in a diffuse pattern. Formation of EPEC localized colonies is mediated by a plasmid-encoded type IV pilus termed the bundle-forming-pilus (BFP) (26). REPEC O103 diffuse adherence depends on a chromosomally encoded fimbrial adhesin termed AF/R2 (18, 46). This adhesin shares homology with the K88 fimbrial adhesin, with the CS31A afimbrial adhesin, and with the recently described Ral fimbriae of a REPEC O15 strain (2). Both BFP and AF/R2 are virulence determinants, as strains with these adhesins deleted are partially devoid of in vivo pathogenicity (4, 55). BFP and AF/R2 nevertheless are not required for the induction of A/E lesions (reference 31 and unpublished data). Even though being impaired in virulence, strains mutated in BFP or AF/R2 retain the ability to colonize the digestive tract of their host, suggesting the involvement of other adhesins in this process. One of these adhesins may be intimin associated with Tir. By contrast, the ability of E22ΔTir and E22ΔEae to still colonize the gastrointestinal tract of the rabbit may be due (at least partially) to the expression of AF/R2.
The absence of intimate adhesion with E22ΔTir, even though the Esp proteins were still secreted, is in good agreement with the Eae receptor function of Tir. Nonetheless, a lectin-like binding module in the C-terminal domain of intimin has recently been identified, strongly suggesting that intimin may interact not only with Tir but also with a host cell receptor (29, 36). Indeed, the tissue tropism of EPEC and EHEC strains was shown to depend on the type of intimin they expressed in a gnotobiotic piglet model (65). Thus, it is difficult to imagine how intimin could cause EPEC to localize in a specific site in the bowel when the pathogen inserts its own receptor into host cells. One way to explain this result is the possibility that intimin binds to at least two different receptors: one of prokaryotic origin (Tir/EspE) and the other of eukaryotic origin (35). This hypothesis is substantiated by the finding that purified intimin binds to eukaryotic cells in the absence of signal transduction events mediated by EPEC (22). However, at least in rabbits, we did not observe a significant difference between the levels of shedding bacteria of strains E22ΔTir and E22ΔEae. We can speculate that in vivo Tir is required to stabilize the interaction of Eae with a low-affinity host intimin receptor. A careful analysis will be necessary to check whether E22ΔTir and E22ΔEae have different adhesion patterns on the mucosa (or on the mucus) according to the tissue or cell types.
EPEC is a major cause of infant diarrhea in developing countries (11, 51) and is pathogenic for several animal species. One of the striking clinical features of EPEC infections is the remarkable propensity of these strains to cause disease in young infants or young animals. Whether the low incidence of EPEC diarrhea in older humans or animals is due to acquired immunity or decreased inherent susceptibility is not known. In western Europe, EPEC strains belonging to the O103 serogroup and rhamnose-negative biotype cause mortality and considerable growth retardation in postweaning rabbits, leading to substantial economic losses (6). In this study, we have shown that both Tir and Eae mutants were harmless at a clinical level (no lethality, no diarrhea, and no weight loss) but were still present in the feces. We have monitored rabbits inoculated with these mutants up to 50 days postweaning without noticing any effects on the growth rate. At that age, the rabbits from the fattening units are usually slaughtered and sold. These results provide insights into possible protection of rabbits against REPEC infection with live attenuated bacterial strains. We have previously shown that oral inoculation of weaned rabbits with live nonpathogenic heterologous strains either harboring the same lipopolysaccharide or producing the same AF/R2 fimbrial adhesin provided nearly complete protection against challenge with highly pathogenic REPEC of serotype O103:H2 (55). We do not know if this protection was due to a protective local immune response (suggested by a specific immunoglobulin A response) and/or to an ecological effect of resistance to colonization against the challenge strain. Indeed, the immune response to EPEC infection remains poorly characterized. Similar experiments in adult human volunteers were less conclusive and less promising (15). Indeed, Donnenberg and collaborators (15) found no evidence of protective immunity against heterologous challenge but observed a significant effect of prior infection on the severity of illness upon reinfection with the homologous strain. It is important to note that a high inoculum (up to 2 × 1010 CFU) had to be used in this human experimental model, because lower doses resulted in lower attack rates. Those authors pointed out that natural infection in infants, which is spread from person to person, probably does not require such a high inoculum. Based on our results (this study and reference 47), we are currently developing a live vaccine based on mutations of both espB and tir. These mutants should heavily colonize the digestive tracts of animals and provide protection against the wild-type REPEC strains in rabbit-fattening units. In addition, the rabbit could also provide a good model to explore the possibility of inducing protective immunity against EPEC infection in humans.
In conclusion, intimate attachment and loss of microvilli seem to be prerequisites in induction of diarrhea by REPEC. However, the actual cause of diarrhea is still undetermined, and many of the observed host responses to EPEC infection could also lead to diarrhea (reviewed in references 28 and 35). It is therefore possible that an Esp-dependent but intimin-independent effect triggered by certain EPEC strains is important in vivo and could account, for instance, for the persistent diarrhea often associated with human EPEC infections. In addition, our results suggest that Tir may also play a crucial role in the inflammatory response induced by REPEC in rabbits. Experiments are currently being performed in our laboratory to analyze the basis of this response in rabbits.
ACKNOWLEDGMENTS
We thank Christian Tasca for helpful technical assistance and Frédéric Goffaux, Philippe Stordeur, and Hélène Bouchet for their assistance during the rabbit experiments. We are indebted to Josée Harel for the kind gift of the anti-intimin serum and to Gad Frankel for the kind gift of the anti-Tir antiserum. We thank John Fairbrother for critical reading of the manuscript. We also thank Istvan Toth and Veronika Huter for comments on the manuscript.
This work was supported in part by a grant from the Région Midi-Pyrénées (no. 9609691) and by a grant from the Institut National de la Recherche Agronomique (AIP Microbiologie). J.-P.N. was the recipient of a scholarship from the Institut National de la Recherche Agronomique and from Biové company. O.M. was the recipient of a scholarship from the Ecole Nationale Vétérinaire de Toulouse.
REFERENCES
- 1.Abe A, Heczko U, Hegele R G, Finlay B B. Two enteropathogenic Escherichia coli type III secreted proteins, EspA and EspB, are virulence factors. J Exp Med. 1998;188:1907–1916. doi: 10.1084/jem.188.10.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams L M, Simmons C P, Rezmann L, Strugnell R A, Robins-Browne R M. Identification and characterization of a K88- and CS31A-like operon of a rabbit enteropathogenic Escherichia coli strain which encodes fimbriae involved in the colonization of rabbit intestine. Infect Immun. 1997;65:5222–5230. doi: 10.1128/iai.65.12.5222-5230.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An H, Fairbrother J M, Dubreuil J D, Harel J. Cloning and characterization of the eae gene from a dog attaching and effacing Escherichia coli strain 4221. FEMS Microbiol Lett. 1997;148:239–245. doi: 10.1111/j.1574-6968.1997.tb10295.x. [DOI] [PubMed] [Google Scholar]
- 4.Bieber D, Ramer S W, Wu C Y, Murray W J, Tobe T, Fernandez R, Schoolnik G K. Type IV pili, transient bacterial aggregates, and virulence of enteropathogenic Escherichia coli. Science. 1998;280:2114–2118. doi: 10.1126/science.280.5372.2114. [DOI] [PubMed] [Google Scholar]
- 5.Blanco J E, Blanco M, Blanco J, Mora A, Balaguer L, Mourino M, Juarez A, Jansen W H. O serogroups, biotypes, and eae genes in Escherichia coli strains isolated from diarrheic and healthy rabbits. J Clin Microbiol. 1996;34:3101–3107. doi: 10.1128/jcm.34.12.3101-3107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camguilhem R, Milon A. Biotypes and O serogroups of Escherichia coli involved in intestinal infections of weaned rabbits: clues to diagnosis of pathogenic strains. J Clin Microbiol. 1989;27:743–747. doi: 10.1128/jcm.27.4.743-747.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.China B, Pirson V, Mainil J. Prevalence and molecular typing of attaching and effacing Escherichia coli among calf populations in Belgium. Vet Microbiol. 1998;63:249–259. doi: 10.1016/S0378-1135(98)00237-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dean-Nystrom E A, Bosworth B T, Moon H W, O'Brien A D. Escherichia coli O157:H7 requires intimin for enteropathogenicity in calves. Infect Immun. 1998;66:4560–4563. doi: 10.1128/iai.66.9.4560-4563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deibel C, Kramer S, Chakraborty T, Ebel F. EspE, a novel secreted protein of attaching and effacing bacteria, is directly translocated into infected host cells, where it appears as a tyrosine-phosphorylated 90 kDa protein. Mol Microbiol. 1998;28:463–474. doi: 10.1046/j.1365-2958.1998.00798.x. [DOI] [PubMed] [Google Scholar]
- 10.De Rycke J, Comtet E, Chalareng C, Boury M, Tasca C, Milon A. Enteropathogenic Escherichia coli O103 from rabbit elicits actin stress fibers and focal adhesions in HeLa epithelial cells, cytopathic effects that are linked to an analog of the locus of enterocyte effacement. Infect Immun. 1997;65:2555–2563. doi: 10.1128/iai.65.7.2555-2563.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donnenberg M S, Kaper J B. Enteropathogenic Escherichia coli. Infect Immun. 1992;60:3953–3961. doi: 10.1128/iai.60.10.3953-3961.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnenberg M S, Tacket C O, James S P, Losonsky G, Nataro J P, Wasserman S S, Kaper J B, Levine M M. Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J Clin Invest. 1993;92:1412–1417. doi: 10.1172/JCI116717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donnenberg M S, Tzipori S, McKee M L, O'Brien A D, Alroy J, Kaper J B. The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J Clin Invest. 1993;92:1418–1424. doi: 10.1172/JCI116718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donnenberg M S, Kaper J B, Finlay B B. Interactions between enteropathogenic Escherichia coli and host epithelial cells. Trends Microbiol. 1997;5:109–114. doi: 10.1016/S0966-842X(97)01000-7. [DOI] [PubMed] [Google Scholar]
- 15.Donnenberg M S, Tacket C O, Losonsky G, Frankel G, Nataro J P, Dougan G, Levine M M. Effect of prior experimental human enteropathogenic Escherichia coli infection on illness following homologous and heterologous rechallenge. Infect Immun. 1998;66:52–58. doi: 10.1128/iai.66.1.52-58.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drolet R, Fairbrother J M, Harel J, Helie P. Attaching and effacing and enterotoxigenic Escherichia coli associated with enteric colibacillosis in the dog. Can J Vet Res. 1994;58:87–92. [PMC free article] [PubMed] [Google Scholar]
- 17.Elliott S J, Wainwright L A, McDaniel T K, Jarvis K G, Deng Y K, Lai L C, McNamara B P, Donnenberg M S, Kaper J B. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol Microbiol. 1998;28:1–4. doi: 10.1046/j.1365-2958.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- 18.Fiederling F, Boury M, Petit C, Milon A. Adhesive factor/rabbit 2, a new fimbrial adhesin and a virulence factor from Escherichia coli O103, a serogroup enteropathogenic for rabbits. Infect Immun. 1997;65:847–851. doi: 10.1128/iai.65.2.847-851.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finlay B B, Rosenshine I, Donnenberg M S, Kaper J B. Cytoskeletal composition of attaching and effacing lesions associated with enteropathogenic Escherichia coli adherence to HeLa cells. Infect Immun. 1992;60:2541–2543. doi: 10.1128/iai.60.6.2541-2543.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer J, Maddox C, Moxley R, Kinden D, Miller M. Pathogenicity of a bovine attaching effacing Escherichia coli isolate lacking Shiga-like toxins. Am J Vet Res. 1994;55:991–9. [PubMed] [Google Scholar]
- 21.Foster G, Ross H M, Pennycott T W, Hopkins G F, McLaren I M. Isolation of Escherichia coli O86:K61 producing cyto-lethal distending toxin from wild birds of the finch family. Lett Appl Microbiol. 1998;26:395–398. doi: 10.1046/j.1472-765x.1998.00359.x. [DOI] [PubMed] [Google Scholar]
- 22.Frankel G, Candy D C, Fabiani E, Adu-Bobie J, Gil S, Novakova M, Phillips A D, Dougan G. Molecular characterization of a carboxy-terminal eukaryotic-cell-binding domain of intimin from enteropathogenic Escherichia coli. Infect Immun. 1995;63:4323–4328. doi: 10.1128/iai.63.11.4323-4328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frankel G, Phillips A D, Rosenshine L, Dougan G, Kaper J B, Knutton S. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol Microbiol. 1998;30:911–921. doi: 10.1046/j.1365-2958.1998.01144.x. [DOI] [PubMed] [Google Scholar]
- 24.Fukui H, Sueyoshi M, Haritani M, Nakazawa M, Naitoh S, Tani H, Uda Y. Natural infection with attaching and effacing Escherichia coli (O 103:H−) in chicks. Avian Dis. 1995;39:912–918. [PubMed] [Google Scholar]
- 25.Galán J E, Ginocchio C, Costeas P. Molecular and functional characterization of the Salmonella invasion gene invA: homology of InvA to members of a new protein family. J Bacteriol. 1992;174:4338–4349. doi: 10.1128/jb.174.13.4338-4349.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giron J A, Ho A S, Schoolnik G K. An inducible bundle-forming pilus of enteropathogenic Escherichia coli. Science. 1991;254:710–713. doi: 10.1126/science.1683004. [DOI] [PubMed] [Google Scholar]
- 27.Goffaux F, Mainil J, Pirson V, Charlier G, Pohl P, Jacquemin E, China B. Bovine attaching and effacing Escherichia coli possess a pathogenesis island related to the LEE of the human enteropathogenic Escherichia coli strain E2348/69. FEMS Microbiol Lett. 1997;154:415–421. doi: 10.1111/j.1574-6968.1997.tb12676.x. [DOI] [PubMed] [Google Scholar]
- 28.Goosney D L, de Grado M, Finlay B B. Putting E. coli on a pedestal: a unique system to study signal transduction and the actin cytoskeleton. Trends Cell Biol. 1999;9:11–14. doi: 10.1016/s0962-8924(98)01418-4. [DOI] [PubMed] [Google Scholar]
- 29.Hartland E L, Batchelor M, Delahay R M, Hale C, Matthews S, Dougan G, Knutton S, Connerton I, Frankel G. Binding of intimin from enteropathogenic Escherichia coli to Tir and to host cells. Mol Microbiol. 1999;32:151–158. doi: 10.1046/j.1365-2958.1999.01338.x. [DOI] [PubMed] [Google Scholar]
- 30.Herrero M, de Lorenzo V, Timmis K N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J Bacteriol. 1990;172:6557–6567. doi: 10.1128/jb.172.11.6557-6567.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hicks S, Frankel G, Kaper J B, Dougan G, Phillips A D. Role of intimin and bundle-forming pili in enteropathogenic Escherichia coli adhesion to pediatric intestinal tissue in vitro. Infect Immun. 1998;66:1570–1578. doi: 10.1128/iai.66.4.1570-1578.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Higgins L M, Frankel G, Connerton I, Goncalves N S, Dougan G, MacDonald T T. Role of bacterial intimin in colonic hyperplasia and inflammation. Science. 1999;285:588–591. doi: 10.1126/science.285.5427.588. [DOI] [PubMed] [Google Scholar]
- 33.Jerse A E, Yu J, Tall B D, Kaper J B. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc Natl Acad Sci USA. 1990;87:7839–7843. doi: 10.1073/pnas.87.20.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaniga K, Delor I, Cornelis G R. A wide-host-range suicide vector for improving reverse genetics in gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica. Gene. 1991;109:137–141. doi: 10.1016/0378-1119(91)90599-7. [DOI] [PubMed] [Google Scholar]
- 35.Kaper J B. EPEC delivers the goods. Trends Microbiol. 1998;6:169–173. doi: 10.1016/s0966-842x(98)01266-9. [DOI] [PubMed] [Google Scholar]
- 36.Kelly G, Prasannan S, Daniell S, Fleming K, Frankel G, Dougan G, Connerton I, Matthews S. Structure of the cell-adhesion fragment of intimin from enteropathogenic Escherichia coli. Nat Struct Biol. 1999;6:313–318. doi: 10.1038/7545. [DOI] [PubMed] [Google Scholar]
- 37.Kenny B, DeVinney R, Stein M, Reinscheid D J, Frey E A, Finlay B B. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell. 1997;91:511–520. doi: 10.1016/s0092-8674(00)80437-7. [DOI] [PubMed] [Google Scholar]
- 38.Kenny B. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modification. Mol Microbiol. 1999;31:1229–1241. doi: 10.1046/j.1365-2958.1999.01265.x. [DOI] [PubMed] [Google Scholar]
- 39.Knutton S, Baldwin T, Williams P H, McNeish A S. Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 1989;57:1290–1298. doi: 10.1128/iai.57.4.1290-1298.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knutton S, Adu-Bobie J, Bain C, Phillips A D, Dougan G, Frankel G. Down regulation of intimin expression during attaching and effacing enteropathogenic Escherichia coli adhesion. Infect Immun. 1997;65:1644–1652. doi: 10.1128/iai.65.5.1644-1652.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leroy S M, Lesage M C, Chaslus-Dancla E, Lafont J P. Presence of eaeA sequences in pathogenic and nonpathogenic Escherichia coli strains isolated from weaned rabbits. J Med Microbiol. 1994;40:90–94. doi: 10.1099/00222615-40-2-90. [DOI] [PubMed] [Google Scholar]
- 42.Licois D, Reynaud A, Federighi M, Gaillard-Martinie B, Guillot J F, Joly B. Scanning and transmission electron microscopic study of adherence of Escherichia coli O103 enteropathogenic and/or enterohemorrhagic strain GV in enteric infection in rabbits. Infect Immun. 1991;59:3796–3800. doi: 10.1128/iai.59.10.3796-3800.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manjarrez-Hernandez H A, Baldwin T J, Aitken A, Knutton S, Williams P H. Intestinal epithelial cell protein phosphorylation in enteropathogenic Escherichia coli diarrhoae. Lancet. 1992;339:521–523. doi: 10.1016/0140-6736(92)90340-9. [DOI] [PubMed] [Google Scholar]
- 44.McDaniel T K, Jarvis K G, Donnenberg M S, Kaper J B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci USA. 1995;92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKee M L, Melton-Celsa A R, Moxley R A, Francis D H, O'Brien A D. Enterohemorrhagic Escherichia coli O157:H7 requires intimin to colonize the gnotobiotic pig intestine and to adhere to HEp-2 cells. Infect Immun. 1995;63:3739–3744. doi: 10.1128/iai.63.9.3739-3744.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milon A, Esslinger J, Camguilhem R. Adhesion of Escherichia coli strains isolated from diarrheic weaned rabbits to intestinal villi and HeLa cells. Infect Immun. 1990;58:2690–2695. doi: 10.1128/iai.58.8.2690-2695.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milon A, Esslinger J, Camguilhem R. Oral vaccination of weaned rabbits against enteropathogenic Escherichia coli-like E. coli O103 infection: use of heterologous strains harboring lipopolysaccharide or adhesin of pathogenic strains. Infect Immun. 1992;60:2702–2709. doi: 10.1128/iai.60.7.2702-2709.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milon A, Oswald E, De Rycke J. Rabbit EPEC: a model for the study of enteropathogenic Escherichia coli. Vet Res. 1999;30:203–219. [PubMed] [Google Scholar]
- 49.Moon H W, Whipp S C, Argenzio R A, Levine M M, Giannella R A. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect Immun. 1983;41:1340–1351. doi: 10.1128/iai.41.3.1340-1351.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moon H W. Comparative histology of intestinal infections. Adv Exp Med Biol. 1997;412:1–21. doi: 10.1007/978-1-4899-1828-4_1. [DOI] [PubMed] [Google Scholar]
- 51.Nataro J P, Kaper J B. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nougayrede J P, Marches O, Boury M, Mainil J, Charlier G, Pohl P, De Rycke J, Milon A, Oswald E. The long-term cytoskeletal rearrangement induced by rabbit enteropathogenic Escherichia coli is Esp dependent but intimin independent. Mol Microbiol. 1999;31:19–30. doi: 10.1046/j.1365-2958.1999.01138.x. [DOI] [PubMed] [Google Scholar]
- 53.Pearson G R, Watson C A, Hall G A, Wray C. Natural infection with an attaching and effacing Escherichia coli in the small and large intestines of a calf with diarrhoea. Vet Rec. 1989;124:297–299. doi: 10.1136/vr.124.12.297. [DOI] [PubMed] [Google Scholar]
- 54.Peeters J E, Charlier G J, Halen P H. Pathogenicity of attaching effacing enteropathogenic Escherichia coli isolated from diarrheic suckling and weanling rabbits for newborn rabbits. Infect Immun. 1984;46:690–696. doi: 10.1128/iai.46.3.690-696.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pillien F, Chalareng C, Boury M, Tasca C, de Rycke J, Milon A. Role of adhesive factor/rabbit 2 in experimental enteropathogenic Escherichia coli O103 diarrhea of weaned rabbit. Vet Microbiol. 1996;50:105–115. doi: 10.1016/0378-1135(96)00012-0. [DOI] [PubMed] [Google Scholar]
- 56.Pohl P H, Peeters J E, Jacquemin E R, Lintermans P F, Mainil J G. Identification of eae sequences in enteropathogenic Escherichia coli strains from rabbits. Infect Immun. 1993;61:2203–2206. doi: 10.1128/iai.61.5.2203-2206.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robins-Browne R M, Tokhi A M, Adams L M, Bennett-Wood V, Moisidis A V, Krejany E O, O'Gorman L E. Adherence characteristics of attaching and effacing strains of Escherichia coli from rabbits. Infect Immun. 1994;62:1584–1592. doi: 10.1128/iai.62.5.1584-1592.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosenshine I, Donnenberg M S, Kaper J B, Finlay B B. Signal transduction between enteropathogenic Escherichia coli (EPEC) and epithelial cells: EPEC induces tyrosine phosphorylation of host cell proteins to initiate cytoskeletal rearrangement and bacterial uptake. EMBO J. 1992;11:3551–3560. doi: 10.1002/j.1460-2075.1992.tb05438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 60.Savkovic S D, Koutsouris A, Hecht G. Attachment of a noninvasive enteric pathogen, enteropathogenic Escherichia coli, to cultured human intestinal epithelial monolayers induces transmigration of neutrophils. Infect Immun. 1996;64:4480–4487. doi: 10.1128/iai.64.11.4480-4487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Savkovic S D, Koutsouris A, Hecht G. Activation of NF-kappaB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol. 1997;273:C1160–C1167. doi: 10.1152/ajpcell.1997.273.4.C1160. [DOI] [PubMed] [Google Scholar]
- 62.Schauer D B, Falkow S. The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect Immun. 1993;61:4654–4661. doi: 10.1128/iai.61.11.4654-4661.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmitt C K, Darnell S C, Tesh V L, Stocker B A, O'Brien A D. Mutation of flgM attenuates virulence of Salmonella typhimurium, and mutation of fliA represses the attenuated phenotype. J Bacteriol. 1994;176:368–377. doi: 10.1128/jb.176.2.368-377.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turk J, Maddox C, Fales W, Ostlund E, Miller M, Johnson G, Pace L, Turnquist S, Kreeger J. Examination for heat-labile, heat-stable, and Shiga-like toxins and for the eaeA gene in Escherichia coli isolates obtained from dogs dying with diarrhea: 122 cases (1992–1996) J Am Vet Med Assoc. 1998;212:1735–1736. [PubMed] [Google Scholar]
- 65.Tzipori S, Gunzer F, Donnenberg M S, de Montigny L, Kaper J B, Donohue-Rolfe A. The role of the eaeA gene in diarrhea and neurological complications in a gnotobiotic piglet model of enterohemorrhagic Escherichia coli infection. Infect Immun. 1995;63:3621–3627. doi: 10.1128/iai.63.9.3621-3627.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wada Y, Kondo H, Nakazawa M, Kubo M. Natural infection with attaching and effacing Escherichia coli and adenovirus in the intestine of a pigeon with diarrhea. J Vet Med Sci. 1995;57:531–533. doi: 10.1292/jvms.57.531. [DOI] [PubMed] [Google Scholar]
- 67.Zhu C, Harel J, Jacques M, Desautels C, Donnenberg M S, Beaudry M, Fairbrother J M. Virulence properties and attaching-effacing activity of Escherichia coli O45 from swine postweaning diarrhea. Infect Immun. 1994;62:4153–4159. doi: 10.1128/iai.62.10.4153-4159.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]