

Abstract
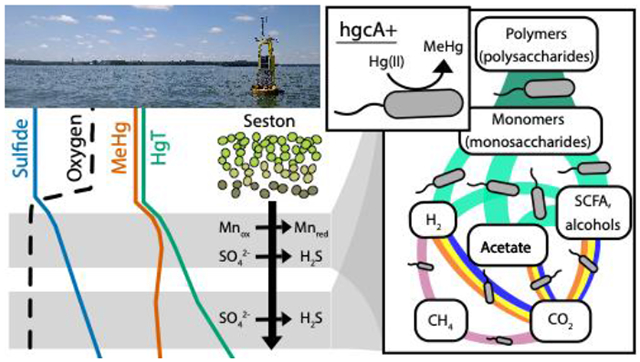
Mercury (Hg) methylation is a microbially mediated process that converts inorganic Hg into bioaccumulative, neurotoxic methylmercury (MeHg). The metabolic activity of methylating organisms is highly dependent on biogeochemical conditions, which subsequently influences MeHg production. However, our understanding of the ecophysiology of methylators in natural ecosystems is still limited. Here we identified potential locations of MeHg production in the anoxic, sulfidic hypolimnion of a freshwater lake. At these sites, we used shotgun metagenomics to characterize microorganisms with the Hg-methylation gene hgcA. Putative methylators were dominated by hgcA sequences divergent from those in well-studied, confirmed methylators. Using genome-resolved metagenomics, we identified organisms with hgcA (hgcA+) within the Bacteroidetes and the recently described Kiritimatiellaeota phyla. We identified hgcA+ genomes derived from sulfate-reducing bacteria, but these accounted for only 22% of hgcA+ genome coverage. The most abundant hgcA+ genomes were from fermenters, accounting for over half of the hgcA gene coverage. Many of these organisms also mediate hydrolysis of polysaccharides, likely from cyanobacterial blooms. This work highlights the distribution of the Hg-methylation genes across microbial metabolic guilds and suggests that primary degradation of polysaccharides and fermentation may play an important but unrecognized role in MeHg production in the anoxic hypolimnion of freshwater lakes.
Keywords: hgcAB, mercury methylation, methylmercury, freshwater, metagenomics
Graphical Abstract
Introduction
Environmental Hg levels have increased 3-4 times compared to pre-development times, largely due to human activity.1 Much of this anthropogenically released Hg is gaseous-elemental Hg, which can later be oxidized to Hg(II) and deposit to terrestrial and aquatic ecosystems.2 Microorganisms can then convert Hg(II) to methylmercury (MeHg) in various low redox environments, including sediments, periphyton, rice paddy soils, and sub- or anoxic regions of freshwater and marine water columns.3–9 MeHg bioaccumulates and biomagnifies up the food web, making Hg-methylation an important process in food web Hg accumulation.10 In freshwater lakes, MeHg accumulation has historically been attributed to production in sediments followed by diffusion across the sediment-water interface.5,11,12 However, Hg-methylation also occurs in the water column, under both anoxic and oxic conditions, and can account for the bulk of water column MeHg accumulation in some lakes.4,6,9,13–15
Biogeochemical conditions, like redox status and carbon bioavailability, can indirectly drive MeHg production by fueling metabolic activity of Hg-methylating microorganisms.12,16 Inhibition of sulfate reduction in cultured isolates and in situ assays have linked sulfate-reducing bacteria (SRBs) activity to MeHg production.11,12 Many subsequent studies have linked sulfate reduction to Hg-methylation across many ecosystems, suggesting that SRBs are the primary drivers of MeHg production in situ.17–20 Later studies identified iron-reducing bacteria (FeRB) and methanogenic archaea that can also produce MeHg, expanding the terminal electron-accepting processes (TEAPs) associated with MeHg production.21,22 We know little about how primary degradation of organic molecules, syntrophy, or fermentation influence MeHg production, but some fermentative and syntrophic microbes, such as Clostridia, are known to methylate.23,24 On the community level, Hg-methylation rates increase with increasing overall heterotrophic activity, suggesting that simply increasing carbon and energy flux through microbial communities can promote MeHg production.9,25 To date, most of our understanding of microbial Hg-methylation relies on reductionist monoculture experiments or on amendment/inhibition studies with environmental samples that lack information about the microbial community supporting MeHg production.
The identification of the hgcAB gene cluster has provided a molecular marker for MeHg production, allowing for the in-depth examination of microbial communities and conditions that promote Hg-methylation in the environment.23,26 Surveys of publicly available genomes, metagenomes and metagenome-assembled genomes have expanded the known phylogenetic and metabolic diversity of organisms with hgcA (hgcA+).23,27–32 Using polymerase chain reaction (PCR)-based amplicon sequencing, several studies have demonstrated that hgcA+ communities are phylogenetically distinct and linked to geochemical conditions across different environments.33–37 While this approach generally captures the deep diversity of hgcA sequences in complex communities, PCR primers are subject to amplification bias and do not provide additional phylogenetic or metabolic information.14,29,30 Shotgun metagenomics, which involves sequencing random small strands of DNA from a sample, reduces amplification biases and enables assembly of longer DNA segments that provide additional genetic context for identified genes. This method also offers a more robust identification of novel hgcA sequences from environmental samples, since computational tools such as Basic Local Alignment Search Tool (BLAST) and Hidden Markov Models (HMMs) are better equipped to identify divergent sequences.31,38,39 To provide further genetic context for hgcA, genome-resolved metagenomics can be used to generate population genomes (bins) from the assembled DNA, enabling phylogenetic identification using conserved genes and metabolic characterization.14,40,41 This approach has been used to identify prominent novel Hg-methylators from the Aminicenantes and Kiritimatiellaeota phyla in the water column of a sulfate-enriched lake.14 The ability to not only identify Hg-methylators but also describe their metabolic potential in situ makes genome-resolved metagenomics an important tool in closing the gap between culture work and in situ assays or observations and in understanding how nutrient and biogeochemical conditions influence Hg-methylation.
In this study we applied genome-resolved metagenomics to identify the metabolic pathways linking biogeochemical cycling to MeHg production in the hypolimnion of Lake Mendota, Wisconsin, USA, a large, well-studied, freshwater eutrophic lake. During stratification, >50% of the total Hg (HgT) in the hypolimnion is MeHg, yet it is unclear which microbial communities are contributing to these high MeHg levels. Mendota has elevated sulfate concentrations due to watershed geology, which supports sulfate reduction in the anoxic hypolimnion; thus, we hypothesized that the hgcA+ community is dominated by sulfate-reducing organisms.42,43 We used Hg speciation and redox profiles to identify sites with suspected in situ MeHg production and selected a subset of these for shotgun metagenomic sequencing. This approach allowed us to identify novel methylators and examine their metabolic pathways, place the methylators in the context of the broader microbial community, and ultimately provide insight into how biogeochemical cycles may influence MeHg production. Historically, TEAPs such as sulfate reduction and methanogenesis have been identified as drivers of MeHg production, but this work suggests that primary degradation and fermentation may drive in situ MeHg production in this system.
Materials and Methods
Field sampling.
Lake Mendota is a large dimictic lake located in Madison, Wisconsin, USA. Sampling was conducted at the deepest part of the lake, near the North Temperate Lakes Long-Term Ecological Research (NTL-LTER) buoy. The lake is approximately 24 meters deep at this site. Samples were collected approximately monthly in 2017 from the onset of hypolimnetic anoxia in June until stratification broke down and the water column mixed. Profiles of temperature, dissolved oxygen, and turbidity were collected with a multi-parameter sonde (YSI Incorporated, Yellow Springs, OH). Samples were collected through acid-washed Teflon sampling line using a peristaltic pump. Samples for total sulfide analysis were preserved in 1% zinc acetate. Water samples for dissolved metal (non-Hg) analysis were filtered through a 0.45 μm PES Acrodisc filter (Pall Corp., Port Washington, NY) and acidified to 1% hydrochloric acid (HCl). Hg samples were collected using trace-metal-clean methods in 2.5L bottles.44 These bottles were allowed to overflow before capping to minimize oxygen exposure within the sample, then were double-bagged and stored in a cooler. Water was filtered through ashed quartz fiber filter (QFF) within 24 hours and preserved to 1% HCl for filter-passing Hg and MeHg analysis.44 The QFF filters were frozen for particulate Hg analysis.44 DNA samples were collected by filtering 300-400 ml of sample water onto 0.22 μm pore-size PES filters (Pall Corp., Port Washington, NY) and were flash-frozen on liquid nitrogen within 90 seconds of collection.
Geochemical analyses.
Sulfide was quantified spectrophotometrically using the Cline method.45 Iron and manganese were quantified by inductively coupled plasma optical emission spectrometry. Processing and analysis of Hg samples was done at the U.S. Geological Survey (USGS) Mercury Research Laboratory (MRL), Middleton, WI. Filter-passing and particulate HgT samples were oxidized using bromine monochloride (BrCl) and quantified using tin reduction coupled to cold vapor atomic fluorescence spectrometry.46,47 Filter-passing and particulate MeHg samples were distilled to remove matrix interferences and then quantified by inductively coupled plasma mass spectrometry following US EPA method 1630 modified by quantification via isotope dilution.48–50 All HgT and MeHg analyses passed required quality assurance and control standards. Geochemical data can be found in Table S1a.
DNA extraction, sequencing, and assembly.
We selected five samples for shotgun metagenomic DNA sequencing and analysis. Three of these samples were selected to coincide with the chemocline and are referred to as CHE1, CHE2, and CHE3 (Table S2). The other two samples are from the deep euxinic hypolimnion and are referred to as EUX1 and EUX2 (Table S2). DNA was extracted by enzymatic and physical lysis followed by phenol-chloroform extraction and isopropanol precipitation.51 DNA library preparation was done at the Functional Genomics Lab and sequencing was done in the Vincent J. Coates Genomics Sequencing Lab (QB3-Berkeley, Berkeley, CA). Library preparation was done with a Kapa Biosystem Library Prep kit, targeting inserts ~600bp in length (Roche Sequencing and Life Science, Kapa Biosystems, Wilmington, MA). The five libraries were pooled and run on a single lane on an Illumina HiSeq4000 with 150bp paired-end sequencing (Illumina, Inc., San Diego, CA). Raw reads were trimmed using Sickle (v1.33) and assembled using metaSPADes (v3.12) (Table S3).52,53 Assembly-based analyses were run on all scaffolds >500bp long. Open reading frames (ORFs) were predicted using Prodigal (v2.6.2).54 Reads were mapped to the scaffolds of each assembly using BBMap (v35) with default settings.55 Scaffold abundance is defined as the mean value of the read coverage at each nucleic acid residue in a scaffold. Gene abundances are defined as the abundance of the corresponding scaffold. Scaffold abundances in each metagenome were normalized by calculating the ratio of reads in the smallest metagenome to the number of reads in that metagenome, then multiplying the abundance of each scaffold by this ratio.
Metagenomic binning and annotation.
Automatic binning was done for each assembly on scaffolds >1000bp in length. Bins were generated using Metabat2 (v2.12.1), MaxBin (v2.1.1), and CONCOCT (v0.4.1), then aggregated using Das Tool.56–59 Bins across assemblies were clustered into “high matching sets” (HMSs) if they shared at least 98% ANI over at least 50% of the genome. CheckM (v1.1.2) was used to estimate the completeness and redundancy of each bin.60 One bin from each HMS was selected for further analysis. We retrieved 228 medium quality bins that were more than 75% complete and less than 10% redundant (Table S6).61 These bins accounted for only 33% of the total number of reads. Bins were then decontaminated using Anvi’o (v5.2), reassembled with SPADes, and re-binned in Anvi’o.53,62 Taxonomy of each bin was estimated using GTDB-TK (v0.3.2).63 Preliminary metabolic annotations were done using MetaPathways.64 Annotations of metabolic genes of interest were confirmed using Hidden Markov Models (HMMs) from TIGRFAM and PFAM, gene neighborhoods, and phylogenies.38
hgcA identification.
A custom HMM for HgcA amino acid sequences was built with hmmbuild from hmmer (v3.1b2) using experimentally verified HgcA amino acid sequences (Table S4).23,65 Putative HgcA sequences were identified using hmmsearch (v3.1b2) (Table S5).65 Each hit was manually screened for the cap helix domain and at least 4 transmembrane domains.26 HgcA sequences were dereplicated across assemblies using CD-HIT (v4.8.1) with a 97% identity cut-off.66
Phylogenetic analyses.
Bin phylogenies were based on 16 ribosomal protein (rp16) sequences.67 For rp16 and HgcA phylogenies, amino acid sequences were aligned using MUSCLE (v3.8.31).68 Each rp16 gene was aligned individually, then all alignments were concatenated. Sequences with less than half of the aligned residues were manually removed. Alignments were inspected in Geneious and trimmed using BMGE1.1 with the BLOSUM30 substitution matrix.69 The final HgcA alignment included 181 residues and the final rp16 alignment included 2217 residues. RAxML (v8.2.11) was used to generate a maximum likelihood (ML) tree under the GAMMA distribution with the LG model.70 Branch support was generated by rapid bootstrapping. For HgcA phylogenies, RogueNaRok (v1.0) was used to remove “rogue taxa” interfering with proper tree generation.71 Rogue taxa were classified using pplacer and included in the analysis.72 The best-scoring ML tree for HgcA was mid-point rooted using the Phangorn R package and visualized using ggtree.73,74 For unbinned HgcA sequences, taxonomy was assigned to each HgcA sequence based on phylogenetic clustering with HgcA reference sequences from NCBI and bin phylogenies of binned HgcA sequences. HgcA sequences that did not fall into a monophyletic cluster are marked as “unknown”. The rp16 ML tree was rooted using an archaeal outgroup.
Data availability.
Trimmed metagenomes and metagenomic assemblies can be found under BioProject PRJNA646991. The scaffolds and the ORFs for the bins, can be found at the project page on the Open Science Framework (OSF), here: https://osf.io/9vwgt/. The nucleic acid and amino acid sequence files for the confirmed hgcA/HgcA sequences and the HgcA HMM used in this study can be found on the same OSF page.
Results and Discussion
Redox and Hg biogeochemistry in Lake Mendota.
Microbial anaerobic respiration is regulated by terminal electron acceptor availability, which continually evolves vertically in Mendota’s hypolimnion throughout the summer-fall season as negative redox conditions strengthen due to high biochemical oxygen demand (Figures 1, S1). We monitored limnological and biogeochemical conditions in the hypolimnion to identify likely TEAPs at play (Table S1). Anoxia developed in the hypolimnion as early as June, likely due to senescence and decomposition of biomass from spring phytoplankton blooms (Figure S1). Nitrate/nitrite levels reached 6 μM at the oxic/anoxic interface in August, but by September were nearly undetectable (Figure S1). Dissolved iron (Fe) transiently accumulated (5 μM) in the hypolimnion immediately following anoxia, but was quickly precipitated out by sulfide and was unlikely to serve as an electron acceptor in the water column (Figure S1).75
Figure 1:
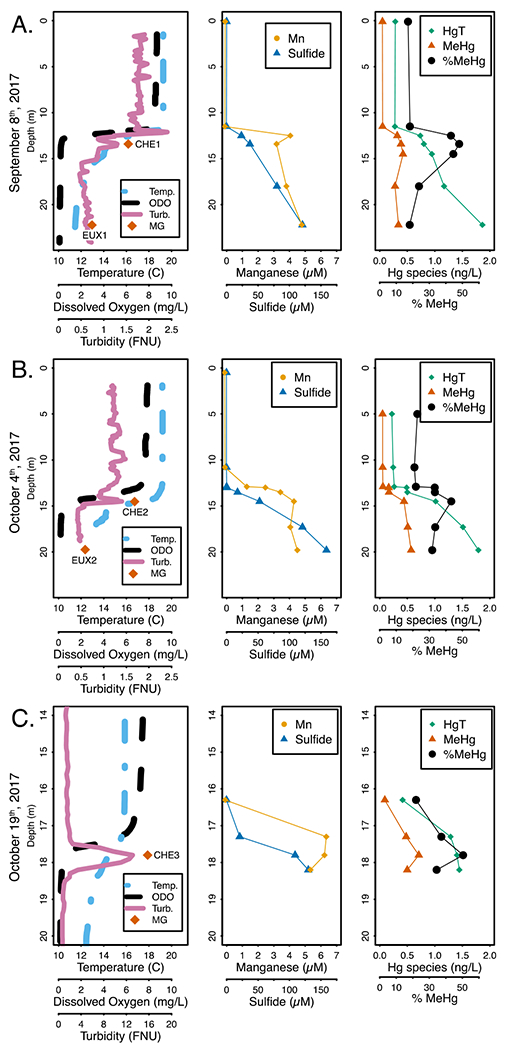
Physical and geochemical profiles of Lake Mendota from 2017 on September 8th (A), October 4th (B) and October 19th (C). Column 1: Parameters measured continuously with a sonde and includes orange diamonds where samples for metagenomic sequencing were collected, names denoted next to symbol. Column 2: Total sulfide and filter-passing manganese values at discrete depths. Column 3: HgT and MeHg values, as sum total of the dissolved and particulate fractions. Dissolved and particulate fractions are plotted individually in Figure S1. Note the changed scale for depth on the y-axis and for turbidity on the x-axis in the October 19th profiles (C). The metagenomic samples collected near the metalimnion for October 4th and October 19th were both collected coincident with the observed spike in turbidity. Abbreviations: Temp. - Temperature (°C), ODO - Optical dissolved oxygen in mg/L, Turb. - Turbidity in Formazin Nephelometric Units (FNU), MG - metagenome sample
Manganese (Mn) also accumulated shortly after anoxia developed and remained in the hypolimnion throughout the anoxic period, ranging from 4-6 μM. In June and August, the near-bottom hypolimnetic accumulation of Mn and linear profile suggests that Mn was being reduced in the surficial sediments and diffusing into the hypolimnion.76 During September and October, there was a peak in dissolved Mn near the oxic/anoxic interface (Figures S1, S2). Particulate Mn was detected (1.4 μM) just above the peak in dissolved Mn in late September (Figure S2). While this peak was not detected in October, this could be due to insufficient sampling resolution, since the profile from September suggests that particulate Mn is localized to a thin band in the water column (Figure S2). Together, these data suggest localized reduction, just below the oxic-anoxic interface, of settling Mn oxides that were produced by the downward migration of the thermocline, as previously shown to occur in Lake Mendota and other lakes.75,76 This indicates that Mn reduction could be an important TEAP near the oxic/anoxic interface during late anoxia.77
Sulfate reduction, commonly implicated in MeHg production, has been documented in the water column of Lake Mendota.43 During early stratification, under relatively high redox conditions, sulfate levels were approximately 180 μM throughout the water column (Figures S1, S2). Sulfide was first detected in August and accumulated to over 150 μM by October (Figures 1, S1, S2). Sulfate depletion mirrored sulfide accumulation, with sulfate levels falling to 21 μM in the deep hypolimnion by October. Sulfate levels have previously been shown to be limiting below 100 μM in Lake Mendota sediments, and other work has shown that SRB require >60 μM sulfate to outcompete methanogens.43,78 Taken together, these data suggest that sulfate reduction is likely an important TEAP in driving anaerobic metabolism throughout the hypolimnion during late fall, but may slow in the deep hypolimnetic waters during late stratification.
Once oxygen was depleted, both HgT and MeHg began accumulating in the hypolimnion (Figures 1, S1). We discuss here total MeHg and HgT levels, calculated by summing the dissolved and particulate fractions, but the dissolved and particulate fractions are shown in Figure S1. The lower hypolimnetic buildup of MeHg and HgT during June and August suggests that diffusion of Hg from sediments is important. This persists in September and October for HgT, which reached 1.86 ng/L in the deep hypolimnion. However, during this time, MeHg increased in the metalimnion up to 0.63 ng/L, while hypolimnetic MeHg remained between 0.4 and 0.5 ng/L. Correspondingly, the percent MeHg (the MeHg:HgT ratio) peaked at the oxic/anoxic interface (52%). The late fall peak in MeHg and percent MeHg near the oxic-anoxic interface is likely due to elevated MeHg production in the metalimnion relative to the hypolimnion. Other potential explanations of this MeHg enrichment include increased demethylation of MeHg in the deep hypolimnion, which to our knowledge has not been shown to occur in lakes; or MeHg diffusion from above, which is unlikely because MeHg levels are low in the epilimnion. Elevated MeHg production just beneath the oxycline has been shown in other freshwater lakes9,13 and in other redox transition zones, such as hyporheic zones and Sphagnum moss mats.19 There are likely two concurrent reasons for this elevated MeHg production in this region. First, high sulfide levels can strongly inhibit MeHg production by reducing the bioavailability of Hg to methylators.79 While sulfide concentrations are high enough in the deep hypolimnion to inhibit Hg-methylation, sulfide levels near the oxycline are relatively low. However, some of the highest MeHg levels recorded were at 17.8 m in October, when dissolved MeHg was 0.63 ng/L and sulfide was over 100 μM (Figure 1). Second, overall microbial metabolism is often elevated near strong redox gradients.9 During late stratification, the percent MeHg maxima also coincided with peaks in turbidity, which has been previously shown to co-localize with elevated microbial activity and MeHg production.15 It is most likely that a combination of these two factors (abiotic speciation and Hg-methylator activity) led to elevated MeHg production near the oxic-anoxic interface.
HgcA identification.
Metagenomic approaches were used to identify hgcA genes that corresponded with metalimnetic peaks of MeHg and hypolimnetic euxinic regions. We identified 108 unique hgcA genes on assembled scaffolds recovered from the five samples (Figure S3). While we used hgcA as our marker for Hg-methylation, the hgcB gene is also required for methylation activity.26 Ninety of the identified hgcA genes also had a downstream hgcB gene, confirming that these are likely functional Hg-methylation genes. Seven of the 18 hgcA+ scaffolds lacking hgcB ended just downstream of hgcA, and it is possible that hgcB did not assemble into the scaffold. The remaining 11 hgcA genes with no hgcB partner had a similar phylogenetic and coverage distribution to those with a downstream hgcB (Figure S4, Table S5). Notably, Hg-methylation has been experimentally verified in Desulfovibrio africanus sp. Walvis Bay and Desulfovibrio inopinatus, in which hgcA is separated from hgcB by a single gene and 29kbp, respectively.26,80,81 Since we cannot rule out that the corresponding hgcB gene is elsewhere in the genome for these 11 sequences, we included all 108 hgcA genes in our analysis.
While our biogeochemical data shows greater MeHg accumulation in the metalimnion than in the hypolimnion, hgcA abundance did not vary substantially between the metalimnion and hypolimnion (Figure 2b). This is consistent with a lack of correlation between hgcA abundance and Hg-methylation activity or MeHg levels in the literature.79 Relating overall abundance of hgcA genes from metagenomes to MeHg levels is problematic due to the fact that metagenomic data is compositional rather than absolute.82 In addition, culture experiments show there is a wide range of Hg-methylation activity by different hgcA+ organisms.23 Finally, as discussed above, abiotic factors such as sulfide complexion also likely play a large role in determining MeHg production in the water column.79
Figure 2.
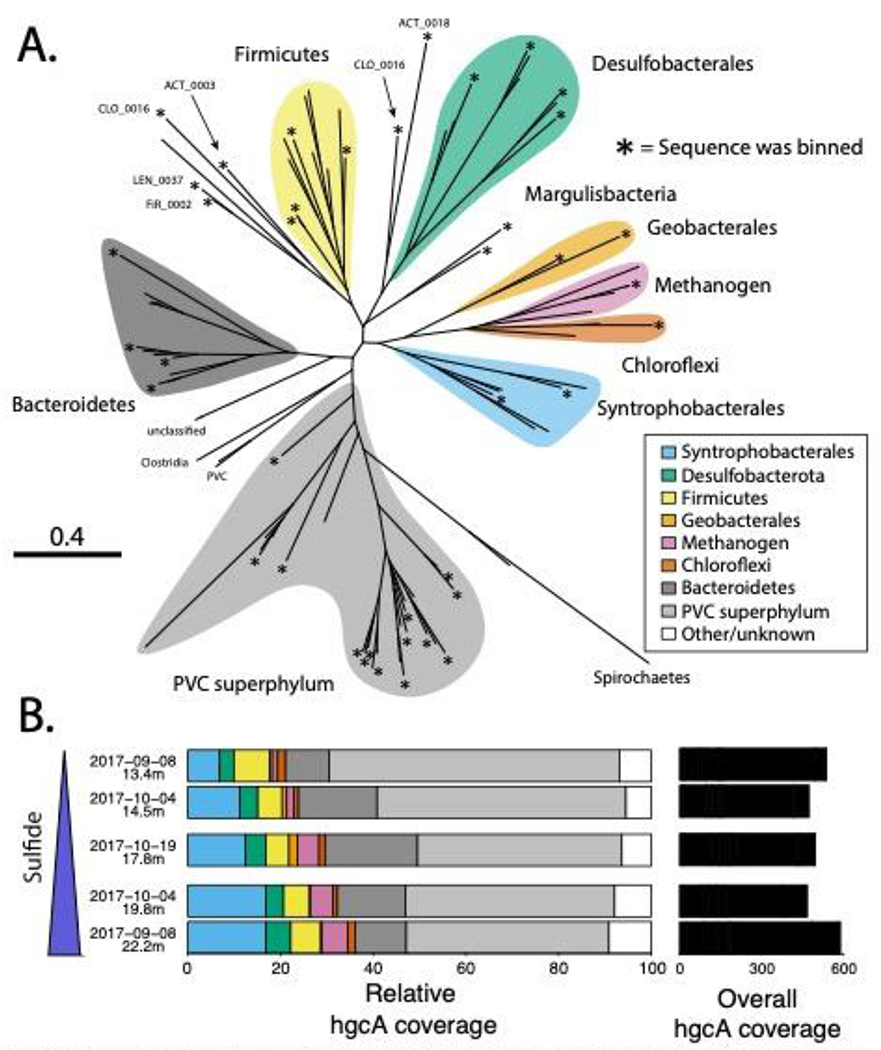
A) Phylogenetic tree of HgcA sequences front this study and B) fractional coverage of hgcA genes within predirted taxonomic groups. Unconfirmed methylators dominate hgcA sequence diversity in Lake Mendota, both numerically (A) and by coverage (B). For panel A, asterisks at the end of branches indicate sequence was binned, white all other branches are uncinned HgcA sequences from this study. Sequences were assigned a predicted taxonomic group based on phylogenetic clustery with HgcA reference sequences from NCBI and bin phytogenies of binned HgcA sequences (for delated tree with reference sequences, see Figure S3). Binned sequences outside of a monophyletic cluster are labeled with their bin name. In panel B, the back bars on the refer to the overall coverage of all hgcA sequences in each metagenome. Samples are arranged in order of increasing sulfide levels.
We then searched for the hgcA gene in the 228 reconstructed metagenomic bins. We identified 41 hgcA+ bins that were representative of the overall hgcA genetic diversity. All but three of these genomes had an hgcB sequence paired with hgcA, and no bins were found with hgcB but no hgcA. One of these bins (LEN_0031) included two copies of the hgcA gene. However, bins represent composite population genomes rather than individual genomes.83 Thus, we cannot confirm that the two hgcA sequences were present together in a single organism, which, to our knowledge, has not been demonstrated. These 41 bins accounted for 51% of the total hgcA coverage in our assemblies. This limited coverage highlights an inability to recover genomes for 13 out of the 30 most abundant hgcA sequences, including the most abundant hgcA gene (Figure S5). Efforts to recover these abundant hgcA+ bins through read subsampling, contig curation using assembly graphs, reassembly, and manual binning and curation were unsuccessful. Many of these scaffolds had highly abundant sequence nucleotide variants, suggesting there were multiple closely related strains, which can interfere with the binning process. While this means our view of the metabolic diversity of hgcA+ bins in these metagenomes is incomplete, we did successfully bin hgcA+ scaffolds corresponding to most of the HgcA phylogenetic clusters, suggesting that most of the methylator diversity is represented in our bins (Figure 2). The hgcA+ bins accounted for 17% of the total read coverage from all bins and included some of the most abundant bins from our metagenomes (Figure S6a). The hgcA+ bins were slightly less abundant than bins without hgcA (hgcA-) bins, but this could be due to the greater degree of manual curation of the hgcA+ bins (Figure S6b). Overall, the hgcA+ bins recruited 6% of the total number of reads from our metagenomic datasets. Because the hgcA+ bins accounted for only 51% of the total coverage of all recovered hgcA sequences, we estimate that hgcA+ genomes account for ~12% of the total metagenomic reads across our five samples, which is consistent with previous work applying this technique in similar systems.14
Phylogenetic diversity of hgcA+ community.
Most of the identified hgcA genes from this study are not representative of well-characterized and experimentally verified methylating organisms. Of the 108 HgcA sequences, only 43 clustered with HgcA sequences from experimentally verified methylators (Figures 2, S4). The majority of these sequences are associated with the Deltaproteobacteria class. The most abundant of these, accounting for 12% of the hgcA+ coverage, belong to the Syntrophobacterales order, which includes both syntrophic and sulfate-reducing organisms (Figures S4, S7). The two other Deltaproteobacteria orders are Geobacterales and Desulfobacterales, both of which are metabolically diverse but commonly associated with iron reduction and sulfate reduction, respectively. Notably, no sequences associated with Desulfovibrionales or Pseudodesulfovibrionales, two well-studied orders that include the model sulfate-reducing methylator Desulfovibrio desulfuricans ND132, were detected.84 We also detected hgcA genes from the other two common groups of confirmed methylators, the phylum Firmicutes and methanogenic Archaea. However, both of these also constituted a small percentage of the total hgcA coverage (<7% each). Overall, hgcA sequences associated with confirmed methylators only accounted for about 27% of the total hgcA coverage. While abundance does not necessarily correlate to activity, this suggests that novel unconfirmed methylators may play a larger than expected role in MeHg production in Lake Mendota.
The majority of hgcA read coverage was accounted for by two large hgcA clusters, neither of which are associated with experimentally verified methylators. Fourteen of these sequences, accounting for 13% of the total hgcA coverage, formed a monophyletic cluster with substantial bootstrap support (Figure S4). Taxonomic and phylogenetic analysis of the four bins with hgcA genes from this cluster placed them in the Bacteroidales order (phylum Bacteroidetes) (Table S6, Figures S7, S8). The other large cluster of HgcA sequences included 33 sequences and accounted for 50% of the total hgcA coverage. We could only recover a few genes from the NCBI NR database that clustered with these sequences, and none from reference isolate genomes (Figure S4). Phylogenetic analysis of the 15 bins with these hgcA genes identified them as members of the Planctomycetes-Verrucomicrobia-Chlamydia (PVC) superphylum, 11 of which are members of the recently proposed Kiritimatiellaeota phylum (Figures S7, S9).85 The PVC superphylum dominates the overall read coverage of our bins as well, with 79 PVC bins accounting for 42% of total bin coverage, with 30% coming from Kiritimatiellaeota alone (Figure S9, Table S6). There are very few publicly available Kiritimatiellaeota genomes and only three cultured representatives.85,86 Notably, a recent paper also identified several hgcA+ bins associated with the Kiritimatiellaeota phylum in a sulfate-enriched lake, but neither the HgcA sequences nor the rp16 sequences from those bins clustered closely with those from the current study (Figure S4).14 This suggests that the Kiritimatiellaeota phylum is far more diverse than our current reference databases indicate and that the hgcA gene could be widely distributed throughout it. For both the Kiritimatiellaeota and the Bacteroidales, the presence of hgcA within bins was not phylogenetically conserved (Figures S8, S9). We also identified several other novel putative methylators that were lower in number and abundance, including Margulisbacteria, Firestonebacteria, and Actinobacteria. The dominance of highly diverse and novel hgcA+ organisms in these samples highlights the value of using genome-resolved shotgun metagenomics (as compared to amplicon sequencing) to identify methylators in a new study system, as it allows for the identification of divergent hgcA lineages and taxonomic classification of the associated bins.
Metabolic potential of methylating bins.
Many of the hgcA+ lineages we identified can employ a wide variety of metabolic strategies, while others have few closely related reference genomes; thus, it was vital to examine their metabolic pathways to determine which TEAPs and other biogeochemical cycles could be potentially linked to Hg-methylation. While most of the literature has focused on both SRBs and methanogens as the dominant methylators, due to the sulfate/sulfide abundance in Lake Mendota and documentation of sulfate reduction in the water column,43 we hypothesized that most hgcA+ genomes in Lake Mendota harbor genes enabling sulfate reduction. The ability to respire sulfate to sulfide is encoded by the dsrABD, aprAB, sat, and qmoABC genes; bins with this complete set of genes were termed SR+.87,88 Three of the four Desulfobacterales hgcA+ bins and both of the Syntrophobacterales hgcA+ bins, including the most abundant hgcA+ bin (SYN_0007), were SR+ (Figures S10, S11). SR+ methylators are slightly more abundant in the euxinic samples (25-26% of hgcA+ bin coverage) than the chemocline samples (14-21%) (Figure 3). Overall, SR+ bins (hgcA+ and hgcA-) account for only 7% of the total bin coverage. Sulfate reduction is not the only respiratory pathway reliant on sulfur redox reactions, though. Three hgcA+ bins that are not SR+ contained polysulfide reductase (psr) homologues, which provide the ability to respire partially reduced sulfur compounds such as tetrathionate or thiosulfate (Figures S10, S12).89 However, these three bins were relatively low in abundance and may also rely on other TEAPs for respiration (Figure S10). Methanogenic methylators were even less abundant, with only a single bin (MET_0028) accounting for 2% of the hgcA+ bin coverage, mostly in the deep euxinic sites where MeHg production is suspected to be lower (Figure 3). No hgcA- methanogens were identified. MET_0028 is a member of the hydrogenotrophic Methanomicrobiales order.90 Overall, the fraction of methylators relying on metabolic pathways historically associated with Hg-methylation (sulfate reduction and methanogenesis) was far lower than we expected.
Figure 3.
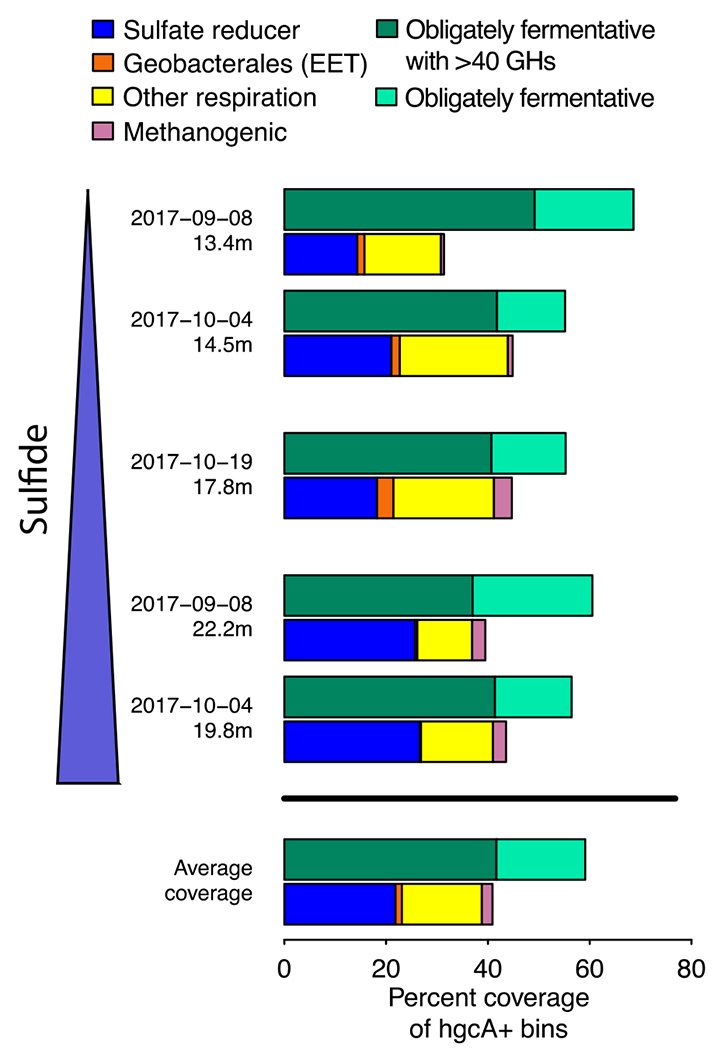
Fractional coverage of metabolic functional groups within hgcA+ community. Fermentative organisms are the most abundant hgcA+ organisms in Lake Mendota. Coverage of each functional group has been normalized to the coverage of all hgcA+ bins in each metagenome. Plots of coverage in the different metagenomes are arranged by decreasing redox potential, which corresponds to increasing sulfide concentrations. Abbreviations: GHs - glucoside hydrolases, EET - external electron transfer.
We also identified several hgcA+ bins corresponding to potential Mn-reducing organisms. While reduced Mn has been correlated to MeHg levels,91 Mn reduction has not, to our knowledge, been linked directly to MeHg production in situ and has even been proposed as a method for limiting MeHg production in sediments.92 Organisms that respire insoluble metal oxides often use porin-cytochrome C complexes (PCCs) to mediate extracellular electron transfer (EET).93 We recovered genomes for several Verrucomicrobia, Bacteroidetes, and Kiritimatiellaeota with PCC-like gene clusters, but they were not closely related to PCCs experimentally verified to conduct EET (Figures S10, S13). However, both hgcA+ Geobacterales bins had a PCC operon homologous to the extEFG operon from Geobacter sulfurreducens, which has been shown to mediate both Fe and Mn oxide reduction (Figures S10, S13).94 These bins both had low read coverage, but were most abundant in CHE3, where we saw evidence for enhanced Mn cycling and peaks in fraction MeHg (Figure 3). Notably, there is little evidence for Fe redox cycling, suggesting that these organisms were unlikely to rely on Fe reduction (Figure S2). While Mn levels are low in the water column relative to sulfate (<5 μM), previous work has shown that low Fe levels can drive substantial carbon oxidation in regions with a steep redox gradient.95 Combined with observations that Geobacterales methylators often produce MeHg at a high rates in culture, this suggests that Mn cycling at the oxic-anoxic interface may play a role in MeHg production in Lake Mendota.22,23,96
We also detected genes for nitrogen species reduction in hgcA+ bins (Figure S10). While nitrite-oxidizing Nitrospinae have been identified as potential methylators,39 reduction of nitrogen species has not, to our knowledge, been linked to MeHg production. In fact, nitrate amendment has been shown to reduce MeHg levels in lakes.97 While many bins, both hgcA+ and hgcA-, encoded genes required for dissimilatory nitrate/nitrite reduction to ammonia (DNRA), these proteins can detoxify nitrite or disperse reducing equivalents during fermentation in addition to respiration..98,99 This, in combination with low nitrate/nitrite levels in the water column during this time of year and the presence of other respiratory pathways in these bins suggest that nitrogen-based respiration does not play a major role in overall community metabolism or MeHg production in this system. However, we cannot rule out the potential role of cryptic N cycling, especially near the chemocline.
The remaining 27 hgcA+ bins are likely to be derived from fermentative or syntrophic organisms, based on their lack of canonical genes for TEAPs. Bins linked to obligate fermentation were also common in the total microbial community, as they represent 106 of the 228 bins, accounting for almost 50% of the bin coverage. This is likely an underestimate of the organisms relying on fermentation, as it does not include the many bins containing genes for dissimilatory nitrate/nitrite reduction or oxidases that were likely maintaining fermentative metabolism at these anoxic depths. These bins possess an array of genes for pyruvate fermentation and aldehyde and alcohol dehydrogenases for fermentative production of short chain fatty acids (Figure S14). They also had genes that could facilitate syntrophy through hydrogen or formate evolution.100 Hydrogenases used for H2 uptake and formate dehydrogenases were present in many respiratory bins (hgcA+ and hgcA-), further suggesting that this community may rely on syntrophic metabolism. Many of these fermentative/syntrophic bins correspond to organisms specialized in polysaccharide degradation, with 13 hgcA+ bins having at least 40 glycoside hydrolases (GHs). The highly abundant Kiritimatiellaeota appear particularly suited to polysaccharide degradation, with bins carrying up to 468 GHs. In fact, 100 total bins carried over 40 GH genes each, suggesting that primary polysaccharide degradation is a common metabolic strategy in the anoxic water column in Lake Mendota. Of these, 49 represent obligate fermenters, while 50 are thought to represent facultative aerobes. Together, these data indicate that fermentative and syntrophic processes may play a much larger role in MeHg production than we had hypothesized.
Our data show that hgcA is widely distributed throughout members of the anaerobic microbial food web. Overall, typical metabolic pathways associated with Hg-methylation such as sulfate reduction, methanogenesis, and iron reduction are outnumbered by fermentative, polysaccharide-degrading hgcA+ organisms, which predominated at both meta- and hypolimnetic sites. The dissolved organic carbon pool in Lake Mendota is dominated by autochthonous inputs and primary production at this time of year is controlled by cyanobacteria, which have a high proportion of exopolysaccharides in their biomass.42,101,102 Together, this suggests that the supply of large organic molecules, particularly polysaccharides from cyanobacterial blooms in the epilimnion, may contribute directly to MeHg production. A previous study in a eutrophic lake reported abundant polysaccharide-degrading, fermentative hgcA+ Kiritimatiellaeota, suggesting these organisms may link polysaccharide degradation to Hg-methylation in many eutrophic systems.14 Similar hgcA sequences were also identified in the Baltic sea on marine snow in oxygen-depleted waters, although at lower relative abundances.27 It is unclear whether these novel hgcA sequences will be amplified by existing primers so we cannot comment on their presence/absence in other systems where PCR-based amplicon sequencing methods are used. On the other hand, respiratory hgcA+ organisms are much less abundant in Lake Mendota and are dominated by SRB in the meta- and hypolimnion during late stratification, likely due to the elevated levels of sulfate in the lake. Sulfate reduction in general appears to be the dominant form of anaerobic respiration in the hypolimnion. At the onset of stratification, sulfate levels are approximately 160 μM, well above what they need to outcompete methanogens [Loveley and Klug, 1983]. Interestingly, Jones et al reported similar levels of hgcA+ SRB in the water column of two lakes heavily enriched in sulfate (~3mM and ~0.5mM), suggesting that in water columns both heavily and moderately impacted by sulfate loading, SRBs still account for a relatively small portion of the hgcA+ community.14 Other TEAPs, such as Mn reduction, may be linked to Hg-methylation under certain conditions in Lake Mendota as well, since the hgcA+ Geobacterales appeared in the metalimnion during late stratification where we saw evidence for enhanced Mn cycling. However, we still do not know which of these hgcA+ organisms are active methylators or how rapidly they produce MeHg. Additional work using more functional measurements such as metatranscriptomics or metaproteomics will help identify which of these hgcA+ organisms are metabolically active and expressing hgcA under in situ conditions.
It is also important to consider that each of these hgcA-carrying organisms is a member of the anaerobic microbial food web and is thus influenced by the overall levels of community metabolism. For example, while there is little information on mass flux constraints on carbon degradation in freshwater anoxic water columns, in other anoxic environments such as marine sediments, hydrolysis and primary fermentation are the rate-limiting steps in community metabolism.103–105 Additionally, syntrophic organisms require that respiratory partners consume their metabolic end-products, such as hydrogen.100 Thus, the supply of terminal electron acceptors and/or carbon substrates and the corresponding activity of flanking microbial community members controls the flux of carbon and energy through the anaerobic microbial food web, influencing the metabolism of individual hgcA+ organisms and presumably their methylation rates. This is supported by data that show that overall levels of heterotrophic activity correlate to MeHg production.9,25 This highlights the need for further research on complex natural communities to probe not only which organisms have and express the hgcAB genes or what metabolic pathways they have, but also how biogeochemical conditions and the overall flux of carbon and energy through different levels of the microbial anaerobic food web can directly and indirectly influence MeHg production in situ.
Supplementary Material
Table S1. A) Geochemical data table. B) Sonde profile data.
Table S2. Summary of metadata and geochemical data associated with metagenomic samples.
Table S3. Assembly statistics for each of the assemblies, after removing all scaffolds <500bp in length.
Table S4. Metadata for 30 HgcA amino acid sequences from confirmed methylators.
Table S5. Aggregated information for each hgcA gene from the dereplicated set. Classification of hgcA is based on the bin phylogenies, for the binned genes, and on the HgcA phylogenies, for the unbinned genes. “Rogue taxa” indicates that the HgcA sequence was interfering with phylogenetic reconstruction. These sequences were classified using pplacer with the hgcA phylogeny. The hgcB column indicates whether or not there was an hgcB gene immediately downstream of the hgcA gene on the scaffold. The abundance of each sequence is presented as the percentage of hgcA coverage within a metagenome that each gene accounts for.
Table S6. Bin information and statistics. Completeness and redundancy estimates are based on universal conserved proteins set in CheckM. Inferred taxonomy is based on a rp16-based ML-tree tree with a large reference data set. Coverage of each bin in each metagenome has been normalized to the number of reads in the smallest metagenome.
Synopsis:
Fermentative microorganisms dominate the mercury-methylating community in a sulfate-enriched lake.
Acknowledgements
The authors acknowledge extensive analytical support from the USGS Toxic Substances Hydrology Program. We also acknowledge the North Temperate Lakes Long Term Ecological Research (NTL-LTER) site, Lake Mendota Microbial Observatory field crews, and UW Center for Limnology for field and logistical support. In particular we thank graduate students Tylor Rosera, Stephanie Berg, and Marissa Kneer for sampling assistance, and Joseph Skarlupka for computational assistance. We also thank undergraduate researchers Mykala Sobieck for sampling design and sampling assistance, and Diana Mendez and Ariel Sorg for sampling assistance. Geochemical analyses were performed at the Water Science and Engineering Laboratory at the University of Wisconsin – Madison. Mercury analyses were performed in the Mercury Research Laboratory in the Upper Midwest Water Science Center at the U.S. Geological Survey in Middleton, WI. Computational work was performed in part using the Wisconsin Energy Institute computing cluster, which is supported by the Great Lakes Bioenergy Research Center as part of the U.S. Department of Energy Office of Science. Any use of trade, product, or firm names in this publication is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Funding Sources
Katherine D. McMahon received funding from the United States National Science Foundation Microbial Observatories program (MCB-0702395), the Long-Term Ecological Research Program (NTL–LTER DEB-1440297), and an INSPIRE award (DEB-1344254), the Wisconsin Alumni Research Foundation. Funding for sequencing was provided by the National Oceanic and Atmospheric Administration (NA10OAR4170070 via the Wisconsin Sea Grant College Program Project #HCE-22). Benjamin Peterson was supported by the National Science Foundation Graduate Research Fellowship Program during this research.
Footnotes
Supplementary Information (PDF):
Supplementary Methods includes additional details on sampling efforts, Hg analyses, DNA extractions, and bioinformatics workflows. Supplementary Figures include additional geochemical profiles, hgcAB alignments, detailed phylogenetic trees, and figures detailing metabolic pathways of methylators.
References
- 1.Amos HM, Jacob DJ, Streets DG, Sunderland EM. Legacy impacts of all-time anthropogenic emissions on the global mercury cycle. Global Biogeochem Cycles. 2013;27(2):410–421. doi: 10.1002/gbc.20040 [DOI] [Google Scholar]
- 2.UN. Global Mercury Assessment.; 2018.
- 3.Cleckner LB, Gilmour CC, Hurley JP, Krabbenhoft DP. Mercury methylation in periphyton of the Florida Everglades. Limnol Oceanogr. 1999;44(7):1815–1825. doi: 10.4319/lo.1999.44.7.1815 [DOI] [Google Scholar]
- 4.Gascón Díez E, Loizeau J-L, Cosio C, Bouchet S, Adatte T, Amouroux D, Bravo AG. Role of settling particles on mercury methylation in the oxic water column of freshwater systems. Environ Sci Technol. 2016;50(21):11672–11679. doi: 10.1021/acs.est.6b03260 [DOI] [PubMed] [Google Scholar]
- 5.Jensen S, Jernelöv A. Biological methylation of mercury in aquatic organisms. Nature. 1969;223:753–754. doi: 10.1038/223754b0 [DOI] [PubMed] [Google Scholar]
- 6.Lepak RF, Janssen SE, Yin R, Krabbenhoft DP, Ogorek JM, DeWild JF, Tate MT, Holsen TM, Hurley JP. Factors affecting mercury stable isotopic distribution in piscivorous fish of the Laurentian Great Lakes. Environ Sci Technol. 2018;52(5):2768–2776. doi: 10.1021/acs.est.7b06120 [DOI] [PubMed] [Google Scholar]
- 7.Rothenberg SE, Feng X. Mercury cycling in a flooded rice paddy. J Geophys Res. 2012;117(G3003). doi: 10.1029/2011JG001800 [DOI] [Google Scholar]
- 8.Sunderland EM, Krabbenhoft DP, Moreau JW, Strode SA, Landing WM. Mercury sources, distribution, and bioavailability in the North Pacific Ocean: Insights from data and models. Global Biogeochem Cycles. 2009;23(2):1–14. doi: 10.1029/2008GB003425 [DOI] [Google Scholar]
- 9.Watras CJ, Bloom NS, Claas SA, Morrison KA, Gilmour CC, Craig SR. Methylmercury production in the anoxic hypolimnion of a dimictic seepage lake. Water, Air, and Soil Pollution. 1995;80:735–745. [Google Scholar]
- 10.Bloom NS. On the chemical form of mercury in edible fish and marine invertebrate tissue. Can J Fish Aquat Sci. 1992;49(5):1010–1017. doi: 10.1139/f92-113 [DOI] [Google Scholar]
- 11.Compeau GC, Bartha R. Sulfate-reducing bacteria: Principal methylators of mercury in anoxic estuarine sediment. Appl Environ Microbiol. 1985;50(2):498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilmour CC, Henry EA, Mitchell R. Sulfate stimulation of mercury methylation in freshwater sediments. Environ Sci Technol. 1992;26(11):2281–2287. doi: 10.1021/es00035a029 [DOI] [Google Scholar]
- 13.Eckley CS, Watras CJ, Hintelmann H, Morrison K, Kent AD, Regnell O. Mercury methylation in the hypolimnetic waters of lakes with and without connection to wetlands in northern Wisconsin. Can J Fish Aquat Sci. 2005;62(2):400–411. doi: 10.1139/f04-205 [DOI] [Google Scholar]
- 14.Jones DS, Walker GM, Johnson NW, Mitchell CPJ, Coleman Wasik JK, Bailey JV. Molecular evidence for novel mercury methylating microorganisms in sulfate-impacted lakes. ISME J. February 2019. doi: 10.1038/s41396-019-0376-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watras CJ, Bloom NS. The vertical distribution of mercury species in Wisconsin lakes: Accumulation in plankton layers. In: Mercury Pollution: Integration and Synthesis. Lewis Publications; 1994:137–151. [Google Scholar]
- 16.Warner KA, Roden EE, Bonzongo J-C. Microbial mercury transformation in anoxic freshwater sediments under iron-reducing and other electron-accepting conditions. Environ Sci Technol. 2003;37(10):2159–2165. doi: 10.1021/es0262939 [DOI] [PubMed] [Google Scholar]
- 17.Jeremiason JD, Engstrom DR, Swain EB, Nater EA, Johnson BM, Almendinger JE, Monson BA, Kolka RK. Sulfate addition increases methylmercury production in an experimental wetland. Environ Sci Technol. 2006;40(12):3800–3806. doi: 10.1021/es0524144 [DOI] [PubMed] [Google Scholar]
- 18.King JK, Saunders FM, Lee RF, Jahnke RA. Coupling mercury methylation rates to sulfate reduction rates in marine sediments. Environmental Toxicology and Chemistry. 1999;18(7):1362–1369. doi: 10.1002/etc.5620180704 [DOI] [Google Scholar]
- 19.Regnell O, Watras CJ. Microbial mercury methylation in aquatic environments: A critical review of published field and laboratory studies. Environ Sci Technol. 2019;53(1):4–19. doi: 10.1021/acs.est.8b02709 [DOI] [PubMed] [Google Scholar]
- 20.Willacker JJ, Eagles-Smith CA, Ackerman JT. Mercury bioaccumulation in estuarine fishes: Novel insights from sulfur stable isotopes. Environ Sci Technol. 2017;51(4):2131–2139. doi: 10.1021/acs.est.6b05325 [DOI] [PubMed] [Google Scholar]
- 21.Hamelin S, Amyot M, Barkay T, Wang Y, Planas D. Methanogens: Principal methylators of mercury in lake periphyton. Environ Sci Technol. 2011;45(18):7693–7700. doi: 10.1021/es2010072 [DOI] [PubMed] [Google Scholar]
- 22.Kerin EJ, Gilmour CC, Roden E, Suzuki MT, Coates JD, Mason RP. Mercury methylation by dissimilatory iron-reducing bacteria. Appl Environ Microbiol. 2006;72(12):7919–7921. doi: 10.1128/AEM.01602-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilmour CC, Podar M, Bullock AL, Graham AM, Brown SD, Somenahally AC, Johs A, Hurt RA, Bailey KL, Elias DA. Mercury methylation by novel microorganisms from new environments. Environ Sci Technol. 2013;47(20):11810–11820. doi: 10.1021/es403075t [DOI] [PubMed] [Google Scholar]
- 24.Yu R-Q, Reinfelder JR, Hines ME, Barkay T. Syntrophic pathways for microbial mercury methylation. ISME J. 2018;12(7):1826–1835. doi: 10.1038/s41396-018-0106-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guimarães JRD, Mauro JBN, Meili M, Sundbom M, Haglund AL, Coelho-Souza SA, Hylander LD. Simultaneous radioassays of bacterial production and mercury methylation in the periphyton of a tropical and a temperate wetland. Journal of Environmental Management. 2006;81(2):95–100. doi: 10.1016/j.jenvman.2005.09.023 [DOI] [PubMed] [Google Scholar]
- 26.Parks JM, Johs A, Podar M, Bridou R, Hurt RA, Smith SD, Tomanicek SJ, Qian Y, Brown SD, Brandt CC, Palumbo AV, Smith JC, Wall JD, Elias DA, Liang L. The genetic basis for bacterial mercury methylation. Science. 2013;339(6125):1332–1335. doi: 10.1126/science.1230667 [DOI] [PubMed] [Google Scholar]
- 27.Capo E, Bravo AG, Soerensen AL, Bertilsson S, Pinhassi J, Feng C, Andersson AF, Buck M, Björn E. Deltaproteobacteria and Spirochaetes-like bacteria are abundant putative mercury methylators in oxygen-deficient water and marine particles in the Baltic Sea. Front Microbiol. 2020;11:1–11. doi: 10.3389/fmicb.2020.574080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilmour CC, Bullock AL, McBurney A, Podar M, Elias DA. Robust mercury methylation across diverse methanogenic Archaea. mBio. 2018;9(2):1–13. doi: 10.1128/mBio.02403-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gionfriddo CM, Wymore AM, Jones DS, Wilpiszeski RL, Lynes MM, Christensen GA, Soren A, Gilmour CC, Podar M, Elias DA. An improved hgcAB primer set and direct high-throughput sequencing expand Hg-methylator diversity in nature. Front Microbiol. 2020;11:1–23. doi: 10.3389/fmicb.2020.541554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDaniel EA, Peterson BD, Stevens SLR, Tran PQ, Anantharaman K, McMahon KD. Expanded phylogenetic diversity and metabolic flexibility of mercury-methylating microorganisms. Kent AD, ed. mSystems. 2020;5(4):1–21. doi: 10.1128/mSystems.00299-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Podar M, Gilmour CC, Brandt CC, Soren A, Brown SD, Crable BR, Palumbo AV, Somenahally AC, Elias DA. Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci Adv. 2015;1:1–12. doi: 10.1126/sciadv.1500675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villar E, Cabrol L, Heimbürger-Boavida L. Widespread microbial mercury methylation genes in the global ocean. Environ Microbiol Reports. February 2020:1–11. doi: 10.1111/1758-2229.12829 [DOI] [PubMed] [Google Scholar]
- 33.Bae H-S, Dierberg FE, Ogram A. Syntrophs dominate sequences associated with the mercury methylation-related gene hgcA in the Water Conservation Areas of the Florida Everglades. Appl Environ Microbiol. 2014;80(20):6517–6526. doi: 10.1128/AEM.01666-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bravo AG, Zopfi J, Buck M, Xu J, Bertilsson S, Schaefer JK, Poté J, Cosio C. Geobacteraceae are important members of mercury-methylating microbial communities of sediments impacted by waste water releases. ISME J. 2018;12(3):802–812. doi: 10.1038/s41396-017-0007-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christensen GA, Wymore AM, King AJ, Podar M, Hurt RA, Santillan EU, Soren A, Brandt CC, Brown SD, Palumbo AV, Wall JD, Gilmour CC, Elias DA. Development and validation of broad-range qualitative and clade-specific quantitative molecular probes for assessing mercury methylation in the environment. Appl Environ Microbiol. 2016;82(19):6068–6078. doi: 10.1128/AEM.01271-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y-R, Yu R-Q, Zheng Y-M, He J-Z. Analysis of the microbial community structure by monitoring an Hg methylation gene (hgcA) in paddy soils along an Hg gradient. Appl Environ Microbiol. 2014;80(9):2874–2879. doi: 10.1128/AEM.04225-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaefer JK, Kronberg R-M, Morel FMM, Skyllberg U. Detection of a key Hg methylation gene, hgcA , in wetland soils. Environ Microbiol Reports. 2014;6(5):441–447. doi: 10.1111/1758-2229.12136 [DOI] [PubMed] [Google Scholar]
- 38.Eddy SR. Hidden Markov Models. Current Opinion in Structural Biology. 1996;6:361–365. [DOI] [PubMed] [Google Scholar]
- 39.Gionfriddo CM, Tate MT, Wick RR, Schultz MB, Zemla A, Thelen MP, Schofield R, Krabbenhoft DP, Holt KE, Moreau JW. Microbial mercury methylation in Antarctic sea ice. Nature Microbiology. 2016;1(10):1–12. doi: 10.1038/nmicrobiol.2016.127 [DOI] [PubMed] [Google Scholar]
- 40.Lin H, Ascher DB, Myung Y, Lamborg CH, Hallam SJ, Gionfriddo CM, Holt KE, Moreau JW. Mercury methylation by metabolically versatile and cosmopolitan marine bacteria. bioRxiv. 2020;[preprint]. doi: 10.1101/2020.06.03.132969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428(6978):37–43. doi: 10.1038/nature02340 [DOI] [PubMed] [Google Scholar]
- 42.Brock TD. A Eutrophic Lake: Lake Mendota, Wisconsin. 1st ed. Springer; 1985. [Google Scholar]
- 43.Ingvorsen K, Zeikus JG, Brock TD. Dynamics of bacterial sulfate reduction in a eutrophic lake. Appl Environ Microbiol. 1981;42(6):1029–1036. doi: 10.1128/AEM.42.6.1029-1036.1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olson ML, DeWild JF. Techniques for the collection and species-specific analysis of low levels of mercury in water, sediment, and biota. In: U.S. Geological Survey Water-Resources Investigations Report. Vols 99-4018B. Washington, D.C.; 1999. [Google Scholar]
- 45.Cline JD. Spectrophotometric determine of hydrogen sulfide in natural waters. Limnol Oceanogr. 1969;14(3):454–458. doi: 10.4319/lo.1969.14.3.0454 [DOI] [Google Scholar]
- 46.U.S EPA Method 1631, Revision E: Mercury in Water by Oxidation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry. Washington, D.C.: U.S. Environmental Protection Agency; 2002. [Google Scholar]
- 47.Olund SD, DeWild JF, Olson ML, Tate MT. Methods for the preparation and analysis of solids and suspended solids for total mercury. In: U.S. Geological Survey Techniques of Water-Resources Investigations, Book 5, Chapter A8. Techniques and Methods.; 2004. [Google Scholar]
- 48.DeWild JF, Olson ML, Olund SD. Determination of Methyl Mercury by Aqueous Phase Ethylation, Followed by Gas Chromatographic Separation with Cold Vapor Atomic Fluorescence Detection. Open-File Report. Reston, VA: U. S. Geological Survey; 2002. [Google Scholar]
- 49.Horvat M, Bloom NS, Liang L. Comparison of distillation with other current isolation methods for the determination of methyl mercury compounds in low level environmental samples. Analytica Chimica Acta. 1993;281:135–152. doi: 10.1016/0003-2670(93)85348-N [DOI] [Google Scholar]
- 50.Lepak RF, Krabbenhoft DP, Ogorek JM, Tate MT, Bootsma HA, Hurley JP. Influence of Cladophora–quagga mussel assemblages on nearshore methylmercury production in Lake Michigan. Environ Sci Technol. 2015;49(13):7606–7613. doi: 10.1021/es506253v [DOI] [PubMed] [Google Scholar]
- 51.Lever MA, Torti A, Eickenbusch P, Michaud AB, Šantl-Temkiv T, Jørgensen BB. A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front Microbiol. 2015;6. doi: 10.3389/fmicb.2015.00476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joshi N, Fass J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files.; 2011. https://github.com/najoshi/sickle.
- 53.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: A new versatile metagenomic assembler. Genome Research. 2017;27(5):824–834. doi: 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11(1):119. doi: 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bushnell B. BBMap Short Read Aligner.; 2015. https://sourceforge.net/projects/bbmap/.
- 56.Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11(11):1144–1146. doi: 10.1038/nmeth.3103 [DOI] [PubMed] [Google Scholar]
- 57.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:1–13. doi: 10.7717/peerj.7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 2018;3(7):836–843. doi: 10.1038/s41564-018-0171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32(4):605–607. doi: 10.1093/bioinformatics/btv638 [DOI] [PubMed] [Google Scholar]
- 60.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–1055. doi: 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowers RM, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu W-T, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Consortium TGS, Lapidus A, Meyer F, Yilmaz P, Parks DH, Murat Eren A, Schriml L, Banfield JF, Hugenholtz P, Woyke T. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35(8):725–731. doi: 10.1038/nbt.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:1–29. doi: 10.7717/peerj.1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36(6):1925–1927. doi: 10.1093/bioinformatics/btz848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konwar KM, Hanson NW, Pagé AP, Hallam SJ. MetaPathways: A modular pipeline for constructing pathway/genome databases from environmental sequence information. BMC Bioinformatics. 2013;14(202):1–10. doi: 10.1186/1471-2105-14-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eddy SR. Hmmer.; 2015. http://hmmer.org/.
- 66.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152. doi: 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, Thomas BC, Singh A, Wilkins MJ, Karaoz U, Brodie EL, Williams KH, Hubbard SS, Banfield JF. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7(13219):1–11. doi: 10.1038/ncomms13219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edgar RC. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5(113):1–19. doi: 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10(210):1–21. doi: 10.1186/1471-2148-10-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aberer AJ, Krompass D, Stamatakis A. Pruning rogue taxa improves phylogenetic accuracy: An efficient algorithm and webservice. Systematic Biology. 2013;62(1):162–166. doi: 10.1093/sysbio/sys078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsen FA, Kodner RB, Armbrust EV. Pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11(538):1–16. doi: 10.1186/1471-2105-11-538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schliep KP. Phangorn: Phylogenetic analysis in R. Bioinformatics. 2011;27(4):592–593. doi: 10.1093/bioinformatics/btq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu G, Smith DK, Zhu H, Guan Y, Lam TT. Ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36. doi: 10.1111/2041-210X.12628 [DOI] [Google Scholar]
- 75.Stauffer RE. Cycling of manganese and iron in Lake Mendota, Wisconsin. Environ Sci Technol. 1986;20(5):449–457. doi: 10.1021/es00147a002 [DOI] [PubMed] [Google Scholar]
- 76.Chadwick SP, Babiarz CL, Hurley JP, Armstrong DE. Influences of iron, manganese, and dissolved organic carbon on the hypolimnetic cycling of amended mercury. Science of The Total Environment. 2006;368(1):177–188. doi: 10.1016/j.scitotenv.2005.09.039 [DOI] [PubMed] [Google Scholar]
- 77.Tebo BM, Rosson RA, Nealson KH. Potential for Manganese(II) Oxidation and Manganese(IV) Reduction to Co-Occur in the Suboxic Zone of the Black Sea. In: İzdar E, Murray JW, eds. Black Sea Oceanography. Dordrecht: Springer Netherlands; 1991:173–185. doi: 10.1007/978-94-011-2608-3_10 [DOI] [Google Scholar]
- 78.Lovley DR, Klug MJ. Sulfate reducers can outcompete methanogens at freshwater sulfate concentrations. Appl Environ Microbiol. 1983;45(1):187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsu-Kim H, Kucharzyk KH, Zhang T, Deshusses MA. Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: A critical review. Environ Sci Technol. 2013;47(6):2441–2456. doi: 10.1021/es304370g [DOI] [PubMed] [Google Scholar]
- 80.Goñi-Urriza M, Klopp C, Ranchou-Peyruse M, Ranchou-Peyruse A, Monperrus M, Khalfaoui-Hassani B, Guyoneaud R. Genome insights of mercury methylation among Desulfovibrio and Pseudodesulfovibrio strains. Research in Microbiology. 2020;171(1):3–12. doi: 10.1016/j.resmic.2019.10.003 [DOI] [PubMed] [Google Scholar]
- 81.Ranchou-Peyruse M, Monperrus M, Bridou R, Duran R, Amouroux D, Salvado JC, Guyoneaud R. Overview of mercury methylation capacities among anaerobic bacteria including representatives of the sulphate-reducers: Implications for environmental studies. Geomicrobiology Journal. 2009;26(1):1–8. doi: 10.1080/01490450802599227 [DOI] [Google Scholar]
- 82.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: And this is not optional. Front Microbiol. 2017;8:1–6. doi: 10.3389/fmicb.2017.02224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, Gregor I, Majda S, Fiedler J, Dahms E, Bremges A, Fritz A, Garrido-Oter R, Jørgensen TS, Shapiro N, Blood PD, Gurevich A, Bai Y, Turaev D, DeMaere MZ, Chikhi R, Nagarajan N, Quince C, Meyer F, Balvočiūtė M, Hansen LH, Sørensen SJ, Chia BKH, Denis B, Froula JL, Wang Z, Egan R, Don Kang D, Cook JJ, Deltel C, Beckstette M, Lemaitre C, Peterlongo P, Rizk G, Lavenier D, Wu Y-W, Singer SW, Jain C, Strous M, Klingenberg H, Meinicke P, Barton MD, Lingner T, Lin H-H, Liao Y-C, Silva GGZ, Cuevas DA, Edwards RA, Saha S, Piro VC, Renard BY, Pop M, Klenk H-P, Göker M, Kyrpides NC, Woyke T, Vorholt JA, Schulze-Lefert P, Rubin EM, Darling AE, Rattei T, McHardy AC. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat Methods. 2017;14(11):1063–1071. doi: 10.1038/nmeth.4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gilmour CC, Elias DA, Kucken AM, Brown SD, Palumbo AV, Schadt CW, Wall JD. Sulfate-reducing bacterium Desulfovibrio Desulfuricans ND132 as a model for understanding bacterial mercury methylation. Appl Environ Microbiol. 2011;77(12):3938–3951. doi: 10.1128/AEM.02993-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spring S, Bunk B, Spröer C, Schumann P, Rohde M, Tindall BJ, Klenk H-P. Characterization of the first cultured representative of Verrucomicrobia subdivision 5 indicates the proposal of a novel phylum. ISME J. 2016;10(12):2801–2816. doi: 10.1038/ismej.2016.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Vliet DM, Palakawong Na Ayudthaya S, Diop S, Villanueva L, Stams AJM, Sánchez-Andrea I. Anaerobic degradation of sulfated polysaccharides by two novel Kiritimatiellales strains isolated from black sea sediment. Front Microbiol. 2019;10:1–16. doi: 10.3389/fmicb.2019.00253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anantharaman K, Hausmann B, Jungbluth SP, Kantor RS, Lavy A, Warren LA, Rappé MS, Pester M, Loy A, Thomas BC, Banfield JF. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018;12(7):1715–1728. doi: 10.1038/s41396-018-0078-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbio. 2011;2:1–22. doi: 10.3389/fmicb.2011.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rothery RA, Workun GJ, Weiner JH. The prokaryotic complex iron–sulfur molybdoenzyme family. Biochimica et Biophysica Act. 2008;1778:1897–1929. doi: 10.1016/j.bbamem.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 90.Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6(8):579–591. doi: 10.1038/nrmicro1931 [DOI] [PubMed] [Google Scholar]
- 91.Alpers CN, Fleck JA, Marvin-DiPasquale M, Stricker CA, Stephenson M, Taylor HE. Mercury cycling in agricultural and managed wetlands, Yolo Bypass, California: Spatial and seasonal variations in water quality. Science of The Total Environment. 2014;484:276–287. doi: 10.1016/j.scitotenv.2013.10.096 [DOI] [PubMed] [Google Scholar]
- 92.Vlassopoulos D, Kanematsu M, Henry EA, Goin J, Leven A, Glaser D, Brown SS, O’Day PA. Manganese(IV) oxide amendments reduce methylmercury concentrations in sediment porewater. Environ Sci: Processes Impacts. 2018;20(12):1746–1760. doi: 10.1039/C7EM00583K [DOI] [PubMed] [Google Scholar]
- 93.Richardson DJ, Butt JN, Fredrickson JK, Zachara JM, Shi L, Edwards MJ, White G, Baiden N, Gates AJ, Marritt SJ, Clarke TA. The “porin-cytochrome” model for microbe-to-mineral electron transfer: Microbe-to-mineral electron transfer. Molecular Microbiology. 2012;85(2):201–212. doi: 10.1111/j.1365-2958.2012.08088.x [DOI] [PubMed] [Google Scholar]
- 94.Jiménez Otero F, Chan CH, Bond DR. Identification of different putative outer membrane electron conduits necessary for Fe(III) citrate, Fe(III) oxide, Mn(IV) oxide, or electrode reduction by Geobacter Sulfurreducens. J Bacteriol. 2018;200(19):1–20. doi: 10.1128/JB.00347-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Berg JS, Michellod D, Pjevac P, Martinez-Perez C, Buckner CRT, Hach PF, Schubert CJ, Milucka J, Kuypers MMM. Intensive cryptic microbial iron cycling in the low iron water column of the meromictic Lake Cadagno: A cryptic microbial iron cycle. Environmental Microbiology. 2016;18(12):5288–5302. doi: 10.1111/1462-2920.13587 [DOI] [PubMed] [Google Scholar]
- 96.Schaefer JK, Morel FMM. High methylation rates of mercury bound to cysteine by Geobacter sulfurreducens. Nature Geosci. 2009;2(2):123–126. doi: 10.1038/ngeo412 [DOI] [Google Scholar]
- 97.Todorova SG, Driscoll CT, Matthews DA, Effler SW, Hines ME, Henry EA. Evidence for regulation of monomethyl mercury by nitrate in a seasonally stratified, eutrophic lake. Environ Sci Technol. 2009;43(17):6572–6578. doi: 10.1021/es900887b [DOI] [PubMed] [Google Scholar]
- 98.Simon J. Enzymology and bioenergetics of respiratory nitrite ammonification. FEMS Microbiol Rev. 2002;26(3):285–309. doi: 10.1111/j.1574-6976.2002.tb00616.x [DOI] [PubMed] [Google Scholar]
- 99.Simon J, Kern M, Hermann B, Einsle O, Butt JN. Physiological function and catalytic versatility of bacterial multihaem cytochromes c involved in nitrogen and sulfur cycling. Biochemical Society Transactions. 2011;39(6):1864–1870. doi: 10.1042/BST20110713 [DOI] [PubMed] [Google Scholar]
- 100.Sieber JR, McInerney MJ, Gunsalus RP. Genomic insights into syntrophy: The paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol. 2012;66(1):429–452. doi: 10.1146/annurev-micro-090110-102844 [DOI] [PubMed] [Google Scholar]
- 101.Bertocchi C, Navarini L, Cesaro A, Anastasio M. Polysaccharides from cyanobacteria. Carbohydrate Polymers. 1990;12:127–153. doi: 10.1016/0144-8617(90)90015-K [DOI] [Google Scholar]
- 102.Beversdorf LJ, Miller TR, McMahon KD. The role of nitrogen fixation in cyanobacterial bloom toxicity in a temperate, eutrophic lake. PLoS ONE. 2013;8(2):1–11. doi: 10.1371/journal.pone.0056103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Billen G. Modelling the processes of organic matter degradation and nutrients recycling in sedimentary systems. In: Sediment Microbiology (Special Publications of the Society for General Microbiology, Book 7). 1st ed. Academic Press Inc; 1982:15–52. [Google Scholar]
- 104.Boschker HTS. Decomposition of organic matter in the littoral sediments of a lake. 1997.
- 105.Meyer-Reil L-A. Seasonal and spatial distribution of extracellular enzymatic activities and microbial incorporation of dissolved organic substrates in marine sediments. Appl Environ Microbiol. 1987;53(8):1748–1755. doi: 10.1128/AEM.53.8.1748-1755.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A) Geochemical data table. B) Sonde profile data.
Table S2. Summary of metadata and geochemical data associated with metagenomic samples.
Table S3. Assembly statistics for each of the assemblies, after removing all scaffolds <500bp in length.
Table S4. Metadata for 30 HgcA amino acid sequences from confirmed methylators.
Table S5. Aggregated information for each hgcA gene from the dereplicated set. Classification of hgcA is based on the bin phylogenies, for the binned genes, and on the HgcA phylogenies, for the unbinned genes. “Rogue taxa” indicates that the HgcA sequence was interfering with phylogenetic reconstruction. These sequences were classified using pplacer with the hgcA phylogeny. The hgcB column indicates whether or not there was an hgcB gene immediately downstream of the hgcA gene on the scaffold. The abundance of each sequence is presented as the percentage of hgcA coverage within a metagenome that each gene accounts for.
Table S6. Bin information and statistics. Completeness and redundancy estimates are based on universal conserved proteins set in CheckM. Inferred taxonomy is based on a rp16-based ML-tree tree with a large reference data set. Coverage of each bin in each metagenome has been normalized to the number of reads in the smallest metagenome.
Data Availability Statement
Trimmed metagenomes and metagenomic assemblies can be found under BioProject PRJNA646991. The scaffolds and the ORFs for the bins, can be found at the project page on the Open Science Framework (OSF), here: https://osf.io/9vwgt/. The nucleic acid and amino acid sequence files for the confirmed hgcA/HgcA sequences and the HgcA HMM used in this study can be found on the same OSF page.