

Abstract
Expression of the transmembrane protein Tim-3 is increased on dysregulated T cells undergoing chronic activation, including during chronic infection and in solid tumors. Thus, Tim-3 is generally thought of as an inhibitory protein. We and others previously reported that under some circumstances, Tim-3 exerts paradoxical costimulatory activity in T cells (and other cells), including enhancement of the phosphorylation of ribosomal S6 protein. Here, we examined the upstream signaling pathways that control Tim-3–mediated increases in phosphorylated S6 in T cells. We also defined the localization of Tim-3 relative to the T cell immune synapse and its effects on downstream signaling. Recruitment of Tim-3 to the immune synapse was mediated exclusively by the transmembrane domain, replacement of which impaired the ability of Tim-3 to costimulate T cell receptor (TCR)–dependent S6 phosphorylation. Furthermore, enforced localization of the Tim-3 cytoplasmic domain to the immune synapse in a chimeric antigen receptor still enabled T cell activation. Together, our findings are consistent with a model whereby Tim-3 enhances TCR-proximal signaling under acute conditions.
INTRODUCTION
T cell activation and effector function are orchestrated by a complex series of cell-cell interactions between T cells and antigen-presenting cells (APCs). The most important interaction is between the T cell receptor (TCR) for antigen and the major histocompatibility complex (MHC) proteins, which present peptide antigens to the T cell. Recognition of a peptide-MHC by a TCR is translated into biochemical signaling events by the TCR-associated CD3 γ, δ, ε, and ζ signaling chains (1, 2). Whereas these events are critical for T cell activation, there are numerous other receptor-ligand interactions that also regulate, either positively or negatively, the ultimate effects on T cell activation and function. The negative regulators in particular have garnered much attention, and blocking antibodies to some of these [including, cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) and programmed death 1 (PD-1)] are now clinically approved for use in selected solid tumors (3, 4). In a parallel series of studies, the use of engineered T cells expressing chimeric antigen receptors (CARs) directed at specific tumor antigens has gained substantial traction in the treatment of specific hematological malignancies (5). The efficacy of the latter approach is also affected by both the expression of endogenous positive and negative regulators in the CAR T cells, as well as the inclusion of different costimulatory signaling domains in the CAR itself.
The protein Tim-3 {T cell [or transmembrane (TM)] immunoglobulin and mucin domain 3} was originally described as a marker for T helper 1 cells (6). Subsequent studies revealed that Tim-3 is also expressed on acutely activated CD8+ T cells and a subset of regulatory T cells, in addition to various non–T cells, including some macrophages, dendritic cells, natural killer cells, and mast cells (7–9). However, most of the attention paid to Tim-3 has focused on the high expression of Tim-3 on “exhausted” CD8+ T cells, which is observed under conditions of chronic infection or within the tumor microenvironment. These cells comprise a subset of the cells that express the immune checkpoint molecule PD-1, which was previously described as the most robust marker for exhausted T cells; monoclonal antibodies (mAbs) that interfere with the interactions between PD-1 and its ligands (PD-L1 and PD-L2) have demonstrated efficacy in a subset of patients with cancer. Furthermore, T cells expressing high amounts of both PD-1 and Tim-3 appear to be even more dysfunctional than those expressing PD-1 alone, as was originally shown for chronic viral infections (10–13). Consistent with this notion, dual blockade of PD-1 and Tim-3 has a modestly enhanced ability to “rescue” the function of a population of exhausted T cells (10, 11, 14). Several antibodies directed against Tim-3 are now in clinical trials, and combination therapies are being actively explored (15). Thus, a prominent model that has emerged is that Tim-3 primarily functions as a negative regulator of T cell activation, in a manner similar to PD-1. Correlative evidence for this model was provided by the discovery of patients bearing germline loss-of-function mutations in HAVCR2, the gene encoding Tim-3 (16).
Despite these data supporting the notion that Tim-3 is a major negative regulator, a number of observations complicate such a straightforward model. First, there has yet to emerge a clear-cut mechanism by which Tim-3 might transmit an inhibitory signal, for example, through the recruitment of phosphatases as has been described for PD-1 or by competition with costimulatory receptors, as does CTLA-4. Second, at least four ligands have been described to interact with Tim-3, including galectin-9, phosphatidylserine (PS), high-mobility group box 1 (HMGB1), and carcinoembryonic antigen–related cell-adhesion molecule 1 (CEACAM1) (7–9), and these also interact with other and distinct receptors (15, 17–20), making the definition of important receptor-ligand interactions difficult at best. Third, studies from multiple laboratories on both T cells and non–T cells have directly or indirectly demonstrated an ability of Tim-3 to transmit positive, activation-promoting, signals (21–27). Our own work has suggested a model by which the expression of Tim-3 might either promote the development of T cell exhaustion by enhancing or sustaining TCR signaling or else represent a “last-ditch” effort by exhausted T cells to maintain some function (22, 28). Mechanistic studies of PD-1 blockade demonstrated that the main target of this therapy is a pool of Tcf1-expressing, “stem-like” CD8+ T cells that express intermediate amounts of PD-1 but no Tim-3 (29–32), whereas the few cells that express Tim-3 alone are capable of robust proliferation (33). In addition, tumor-infiltrating CD8+ T cells expressing Tim-3 and high amounts of PD-1 adopt a distinct epigenetic program that serves as a barrier to rejuvenation by checkpoint blockade (34).
A previous study provided evidence, through standard confocal microscopy, that Tim-3 is recruited to the immune synapse (35), a structure that forms between T cells and APCs during productive antigen recognition. In addition, this study suggested that the recruitment of Tim-3 to the immune synapse contributed to its negative regulatory function, although the precise mechanism was not defined. Here, we have examined the relationship between Tim-3 and the immune synapse using both imaging cytometry with T cell–APC conjugates and total interference reflection fluorescence (TIRF) microscopy with lipid bilayers. We report that Tim-3 was recruited to the immune synapse, although this lagged behind the recruitment of the TCR/CD3 complex. Furthermore, we demonstrated that recruitment of Tim-3 to the immune synapse was mainly mediated by its TM domain. By substituting the TM, we showed that recruitment of Tim-3 to the immune synapse correlated with its previously reported costimulatory activity. Last, we showed that even under conditions of enforced recruitment to the immune synapse with a CAR, the cytoplasmic domain of Tim-3 provided a costimulatory, but not inhibitory, signal for T cell activation. Thus, our data are consistent with a model whereby the primary proximal effect of Tim-3 signaling is to support T cell activation.
RESULTS
Enhancement of S6 phosphorylation by Tim-3 is mediated by both MEK-ERK and PI3K-Akt signaling
We previously showed that ectopic expression of Tim-3 in T cells can enhance the phosphorylation of ribosomal protein S6 (to generate pS6) in conjunction with TCR signaling (22, 24). S6 phosphorylation can be promoted by activation of MAPK/ERK kinase-extracellular signal-regulated kinase (MEK-ERK) or phosphoinositide 3-kinase (PI3K)-Akt signaling or both (36). To assess whether Tim-3–mediated activation of ERK, Akt, or both was associated with the enhancement of S6 phosphorylation, we used a highly selective inhibitor of MEK1 and MEK2 (U0126) or an allosteric inhibitor of Akt (Akti1/2). We confirmed that U0126 and Akti1/2 substantially blocked the generation of pERK and pAkt, respectively, in a dose-dependent manner (fig. S1A). We next cotransfected Jurkat cells with a green fluorescent protein (GFP)–encoding plasmid (as a control for transfection) and either empty vector or plasmid encoding Flag-tagged Tim-3. We then stimulated these cells with a mAb against the TCR, with or without inhibitors, and detected Tim-3 with an anti-Flag antibody (Fig. 1A). As we reported previously (24), Tim-3 expression enhanced TCR-induced S6 phosphorylation in the transfected (GFP+) population. We also observed that either U0126 or Akti1/2 treatment inhibited S6 phosphorylation, although neither was sufficient to completely block the enhancement of S6 phosphorylation by Tim-3 expression. However, combined treatment with both inhibitors reduced S6 phosphorylation more than either inhibitor alone and repressed the costimulatory activity of Tim-3, indicating that both the MEK-ERK and PI3K-Akt pathways play a role in the regulation of S6 phosphorylation by Tim-3 (Fig. 1, B and C).
Fig. 1. Tim-3 enhancement of S6 phosphorylation requires both MEK-ERK and PI3K-Akt signaling.
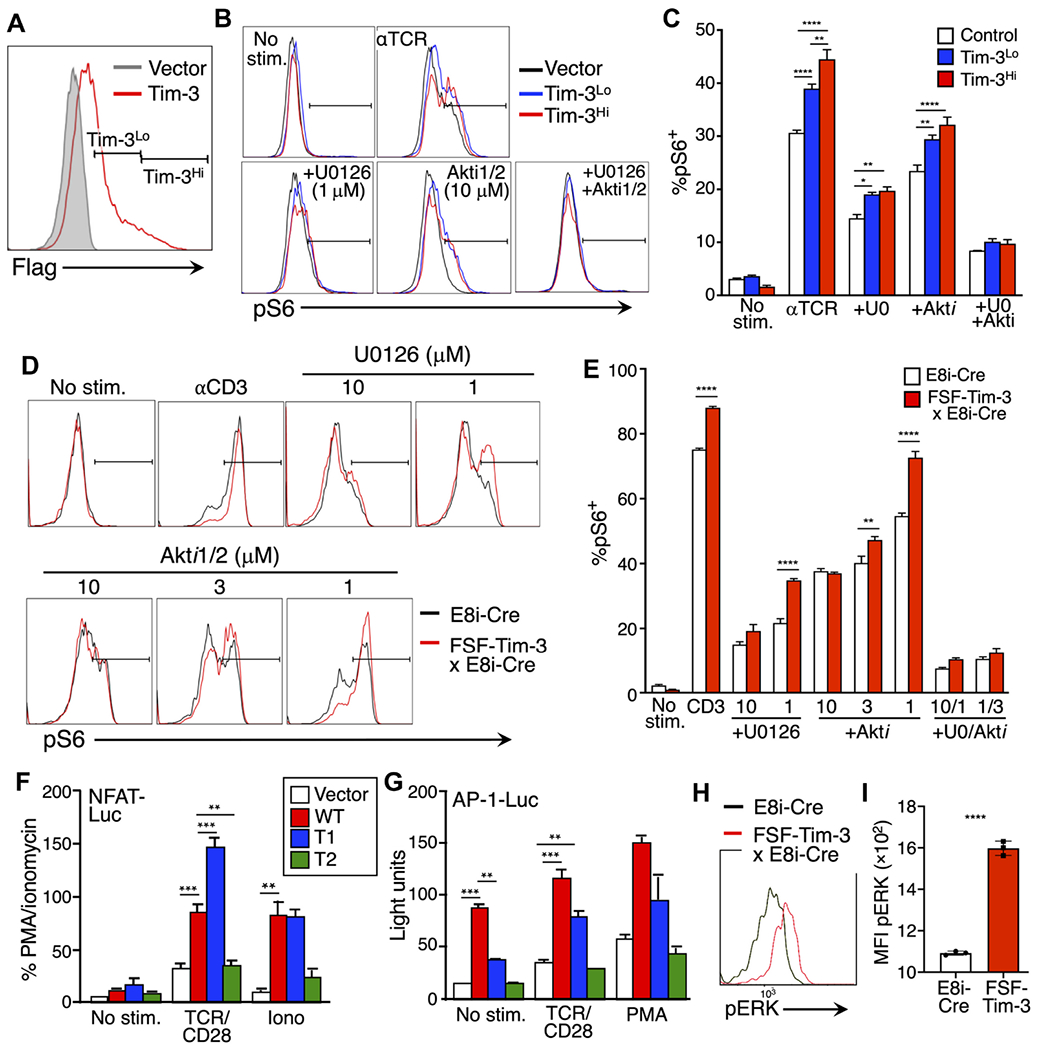
(A) Expression of Flag-tagged Tim-3 on transfected Jurkat cells was assessed by flow cytometry. (B) Jurkat cells were pretreated with vehicle, U0126, or Akti1/2 as indicated before being stimulated with anti-TCR for 30 min. The cells were then analyzed by flow cytometry to detect pS6 (Ser236/236) within control, Tim-3Lo, or Tim-3Hi cells. (C) Quantification of the percentage of pS6+ cells based on the flow cytometry analysis shown in (B). (D) Primary T cells were pretreated with vehicle, U0126, or Akti1/2 as indicated before being stimulated with anti-CD3 for 4 hours. The cells were then analyzed by flow cytometry to detect pS6 within CD8+ T cells from E8i-Cre or E8i-Cre/FSF-Tim-3 mice. (E) Quantification of the percentage of pS6+ cells based on the flow cytometry analysis shown in (D). (F and G) Luciferase reporter assays were performed on Jurkat cells transfected with vector control or the indicated Tim-3 constructs together with either the NFAT-luciferase (F) or AP-1-luciferase (G) reporter constructs. Cells were stimulated as indicated and luciferase activity was quantified in a luminometer. PMA, phorbol 12-myristate 13-acetate. (H and I) Analysis of pERK expression in WT (E8i-Cre only) or Tim-3 Tg T cells directly ex vivo. Representative flow cytometry histograms are shown in (H), and aggregated quantified data of three independent samples are shown in (I). Data in each panel are representative of at least three experiments. Data in the bar graphs are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Tukey’s multiple comparisons test of triplicate samples. Data in (A) to (C) are representative of seven experiments each, whereas data in (D) to (I) are representative of three experiments.
Jurkat cells lack expression of phosphatase and tensin homolog (PTEN) (37), rendering these cells hyper-responsive to TCR stimulation. To confirm our earlier findings in primary T cells, we used CD8-specific inducible Tim-3 mice (fig. S1B). These mice were obtained by crossing transgenic Rosa26 knock-in “flox-stop-flox” Tim-3 (FSF-Tim-3) mice with CD8+ T cell–specific E8i-Cre mice (22). Naïve T cells from E8i-Cre and E8i-Cre x FSF-Tim-3 mice were pretreated with the MEK and Akt inhibitors before being stimulated through the TCR. Consistent with our previous findings (22), S6 phosphorylation was enhanced in CD8+ T cells from E8i-Cre x FSF-Tim-3 mice (Fig. 1, D and E). Treatment with either inhibitor at high concentrations was sufficient to block the enhancement of S6 phosphorylation by Tim-3, whereas combined treatment with moderate concentrations of both inhibitors resulted in a greater decrease in S6 phosphorylation than was achieved with either inhibitor alone. We also confirmed the effects of these inhibitors on S6 phosphorylation in D10 cells, a murine T cell line (38). Consistent with the earlier results, combined treatment with both inhibitors blocked the TCR-induced phosphorylation of S6 (fig. S1, C to E). These results suggest that costimulation of TCR-induced S6 protein phosphorylation by Tim-3 requires both the MEK-ERK and PI3K-Akt pathways to enhance TCR signaling. Given the connection of both mechanistic target of rapamycin (mTOR) and ERK signaling to T cell proliferation (39–41), we wanted to determine whether there was an effect of transgenic Tim-3 expression on T cell proliferation. This was assessed by in vitro stimulation of dye-labeled cells. Thus, we observed only minor differences in the proliferation of CD8+ T cells at either 2 or 4 days after stimulation, comparing cells from control E8i-Cre only mice with those carrying both the Cre and FSF-Tim-3 transgenes (fig. S1, F and G). There was a small, but statistically significant, increase in the degree of proliferation of Tim-3 Tg T cells at 2 days after stimulation, with the converse (that is, relatively more proliferation of the E8i-Cre only cells) observed at day 4 (fig. S1G), suggesting an early enhancement of T cell proliferation by Tim-3.
To further analyze the contribution of Tim-3 to the activation of MEK-ERK signaling, we transfected Jurkat cells with luciferase reporter constructs driven by nuclear factor of activated T cells (NFAT) or activator protein 1 (AP-1) elements, which are activated by Ca2+ or Ras-MAPK signaling, respectively (42). We also transfected these cells with plasmids encoding wild-type (WT) or truncated forms of Tim-3 (fig. S2A). Thus, as we previously showed with a composite NFAT/AP-1 reporter (24), WT Tim-3 and a construct with a partially truncated cytoplasmic tail [truncation 1 (T1)] efficiently costimulated the activation of an NFAT-driven reporter in the presence of TCR/CD28 or ionomycin stimulation (Fig. 1F). By contrast, a Tim-3 construct with a larger cytoplasmic tail truncation (T2) did not costimulate NFAT-dependent transcription. We also assessed the effects of the same Tim-3 constructs on an AP-1 reporter. Unlike the results obtained with the NFAT/AP-1 or NFAT reporters, a pure AP-1 reporter was activated by cotransfection with WT Tim-3 alone, without a need for additional stimulation (Fig. 1G). This suggested an ability of Tim-3 to activate upstream MEK-ERK signaling in the basal state. We addressed this further by examining ERK phosphorylation directly in the FSF-Tim-3 mice described earlier. Thus, CD8+ T cells expressing transgenic Tim-3 displayed higher basal phosphorylation of ERK than did control E8i-Cre T cells (Fig. 1, H and I). These results suggest that Tim-3 has an intrinsic ability to activate MEK-ERK signaling, whereas its costimulatory activity may function more through the Ca2+-NFAT pathway.
Imaging cytometry analysis reveals the recruitment of Tim-3 to the immune synapse
Costimulatory and coinhibitory receptors often colocalize with the TCR at the immune synapse (43). A previous study suggested that Tim-3 is recruited to the immune synapse in activated T cells (35). To better understand the mechanisms underlying costimulation by Tim-3 in T cells, we examined Tim-3 localization during immune synapse formation in Jurkat cells transfected with plasmid encoding Flag-tagged Tim-3, using the Amnis ImageStream flow cytometry system. Raji cells (a human B cell lymphoma line) pulsed with staphylococcal enterotoxin E (SEE) were used as APCs, and immune synapse formation was confirmed on the basis of CD3 recruitment to the Jurkat cell–Raji cell interface (Fig. 2, A and B). We observed an increase in the proportion of Tim-3 recruited to the immune synapse over time, although Tim-3 displayed a lower overall degree of recruitment, with somewhat slower kinetics, compared with the behavior of CD3 (Fig. 2, A and C). To determine whether the same pattern of recruitment of Tim-3 was also seen in primary murine T cells, we used P14 TCR transgenic mice, with or without expression of Tim-3 from a Rosa26 knock-in cassette (22). CD8+ T cells from these mice were stimulated with autologous T cell–depleted splenocytes, with or without the cognate peptide antigen, the gp33 peptide from lymphocytic choriomeningitis virus (LCMV). Consistent with the results obtained with Jurkat cells, we observed enhanced recruitment of Tim-3 to the immune synapse of CD8+ P14 TCR Tg x Tim-3 Tg T cells in the presence of cognate peptide (Fig. 2, D and F). As expected, CD3 was also recruited to the contact site between the T cell and the APC, in an antigen-dependent fashion (Fig. 2, D and E).
Fig. 2. Recruitment of Tim-3 to the immune synapse of Jurkat and P14 Tg T cells.
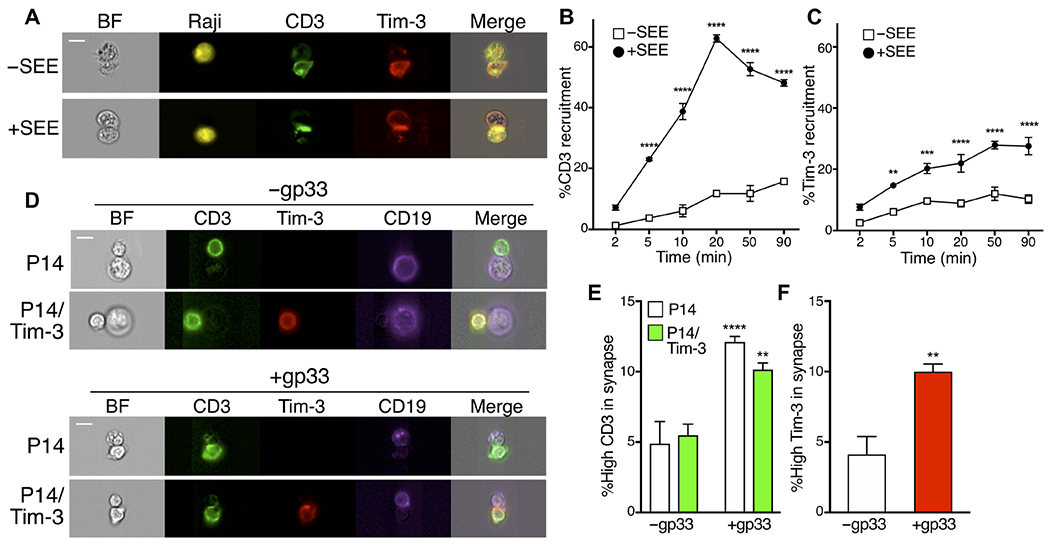
(A) Jurkat cells transfected with plasmid encoding Flag-tagged Tim-3 (red) were mixed with Raji cells (yellow) that were either unpulsed or pulsed with SEE at a ratio of 1:1 for 20 min and then were analyzed by ImageStream. BF, brightfield. (B and C) Cell-cell conjugates were incubated for the indicated times. The percentage of either CD3 or Tim-3 recruitment was quantified from the ratio of the intensity of either CD3 (green) or Tim-3 (red) within the interface mask of cell conjugates containing one Jurkat cell and a Raji cell to that of the whole cell mask, using IDEAS software. Each point represents an average of 291 conjugates. (D to F) CD8+ T cells from control P14 TCR Tg mice or those expressing Cre-inducible Tim-3 were mixed with purified B cells from the same mice, with or without gp33 peptide, and cell conjugates were analyzed as described for (B) and (C). Each bar represents an average of 418 cell conjugates in the absence of antigen and 5500 cells in the presence of antigen. Scale bars, 10 μm. Data in (A) to (C) are representative of two experiments, whereas data in (D) to (F) are representative of four experiments. **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Tukey’s multiple comparisons test (B, C, and E) or Student’s t test (F).
The extracellular and intracellular domains of Tim-3 are dispensable for its recruitment to the immune synapse
We next sought to determine the roles of specific Tim-3 domains in its recruitment to the immune synapse. Using various mutants of Tim-3, we previously showed that the cytoplasmic tail of Tim-3 is required for its costimulatory activity, at least at the level of anti-TCR–induced activation of NFAT/AP-1 and nuclear factor κB transcriptional reporters (24). To address the relationship between Tim-3 signaling and its localization, we used Flag-tagged Tim-3 variants lacking either the sequence encompassing three tyrosines at positions 271, 272, and 274 (T1) or a more truncated variant also lacking tyrosines at positions 256 and 263 (T2) (fig. S2A). We previously showed that the T1 construct retains the ability to costimulate NFAT/AP-1 activity, whereas the T2 construct loses this activity (24). Consistent with those data, we found that the T1 mutant still retained the ability to enhance S6 phosphorylation comparably to WT Tim-3, whereas the T2 truncation lost the ability to enhance S6 phosphorylation (fig. S2, E and F). However, we noted no significant differences in the localization of these constructs to the immune synapse upon stimulation with SEE-pulsed Raji cells, compared to that of full-length Tim-3 (Fig. 3, A to C). The T2 construct still retains 38 amino acids downstream of the TM domain. To more rigorously address the role of the intracellular domain in the recruitment of Tim-3 to the immune synapse, we used a Flag-tagged Tim-3 variant lacking the entire cytoplasmic region (ΔCyto; fig. S2A). However, there was no significant difference in the localization of the ΔCyto construct to the immune synapse compared to that of full-length Tim-3 (Fig. 3, D to F). As expected, the ΔCyto form of Tim-3 was efficiently expressed at the surface of transfected cells (fig. S2, B and C). Thus, the recruitment of Tim-3 to the immune synapse does not require its cytoplasmic domain.
Fig. 3. The IgV domain and cytoplasmic tail are dispensable for the recruitment of Tim-3 to the immune synapse.
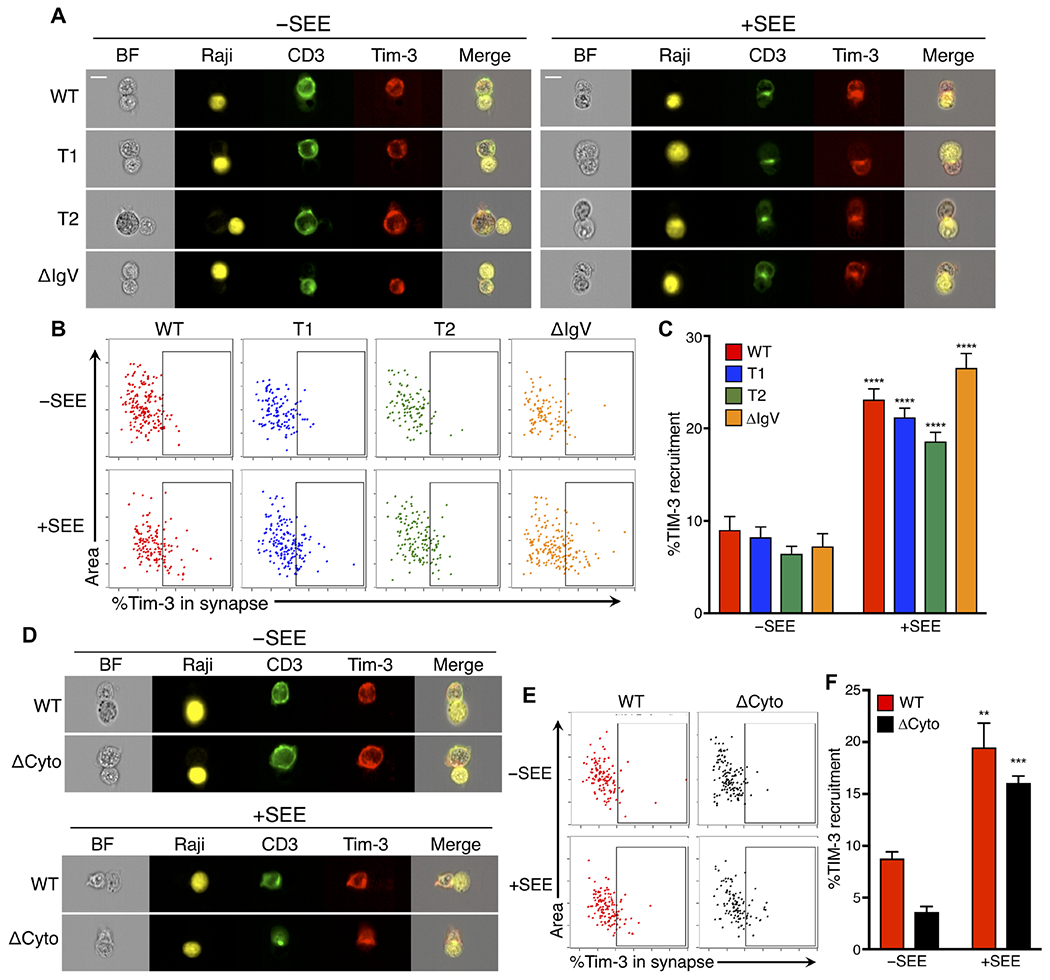
(A) Representative images of Jurkat cells transfected with plasmids encoding Flag-tagged WT Tim-3, Tim-3 partial cytoplasmic truncations (T1 and T2), or ΔIgV constructs (red) and mixed with Raji cells (yellow), which were either unpulsed or pulsed with SEE, at a ratio of 1:1 for 20 min. (B) Representative dot plots analyzing the ratio of the intensity of WT or mutant Tim-3 constructs in the interface mask between a transfected Jurkat cell and a Raji cell to that of the whole cell mask. (C) Quantification of the percentage of Tim-3 recruitment in cells expressing the indicated Tim-3 constructs. Each bar represents an average of 147 cell conjugates. (D) Representative images of Jurkat cells transfected with plasmids expressing either Flag-tagged WT or ΔCyto Tim-3 (red) and mixed with Raji cells (yellow), which were either unpulsed or pulsed with SEE, at a ratio of 1:1 for 20 min. (E) Representative dot plots analyzing the ratio of the fluorescence intensity of WT or ΔCyto Tim-3 in the interface mask between a transfected Jurkat cell and a Raji cell to that of the whole cell mask. (F) Quantification of the percentage of Tim-3 recruitment. Each bar represents an average of 71 cells. Data are means ± SEM. **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus −SEE, Tukey’s multiple comparisons test. Differences between the different Tim-3 constructs were not significant (P > 0.05). Scale bars, 10 μm. Experiments comparing WT, T1, and T2 constructs were performed at least three times, whereas those with the ΔCyto and ΔIgV constructs were performed twice.
Previous studies showed that receptor-ligand binding, for example, the binding of B7 to CD28, regulates the recruitment of immune costimulatory and coinhibitory molecules to the immune synapse (44, 45). The Tim-3 immunoglobulin V (IgV) domain binds to various and diverse ligands, such as galectin-9, PS, HMGB1, and Ceacam-1 (46–49), which constitute all known Tim-3 ligands. We therefore used a Flag-tagged Tim-3 variant lacking the IgV domain (ΔIgV) (fig. S2A) to address the role of ligand binding in Tim-3 recruitment to the immune synapse. As expected, this construct was expressed at the surface of transfected cells (fig. S2, B and C). As with the cytoplasmic tail deletions, we observed that deletion of the IgV domain did not impair Tim-3 recruitment to the immune synapse (Fig. 3, A to C), suggesting that Tim-3 recruitment to the immune synapse may not require ligand binding, because deletion of the IgV domain eliminates the ability of Tim-3 to bind to all of its known ligands.
The TM domain of Tim-3 regulates its costimulatory activity and recruitment to the immune synapse
Previous studies suggested that the TM domain of some cell surface molecules can regulate recruitment to lipid rafts, cholesterol-rich membrane subregions that accumulate at the immune synapse. We examined the role of the TM domain of Tim-3 in its recruitment to the immune synapse in experiments with a Tim-3-CD71 TM chimeric protein (CD71tm), in which we replaced the TM domain of Tim-3 with the corresponding domain of CD71 (fig. S2A). This construct was expressed at the surface of transfected cells comparably to WT Tim-3 (fig. S2, B and C) and appeared to maintain its glycosylation, as revealed by Western blotting analysis of samples with or without prior treatment with glycosidase (fig. S2D). The CD71 TM domain mediates its exclusion from lipid rafts in the context of a chimeric receptor (50). We observed a decrease in Tim-3 localization at the immune synapse upon stimulation with SEE-pulsed Raji cells in transfected Jurkat cells expressing CD71tm Tim-3 compared with that in cells expressing WT Tim-3 (Fig. 4, A to C). Thus, the TM domain of Tim-3 positively regulates the efficiency of its localization to the immune synapse.
Fig. 4. Role of the TM domain in Tim-3 localization and function during immune synapse formation.
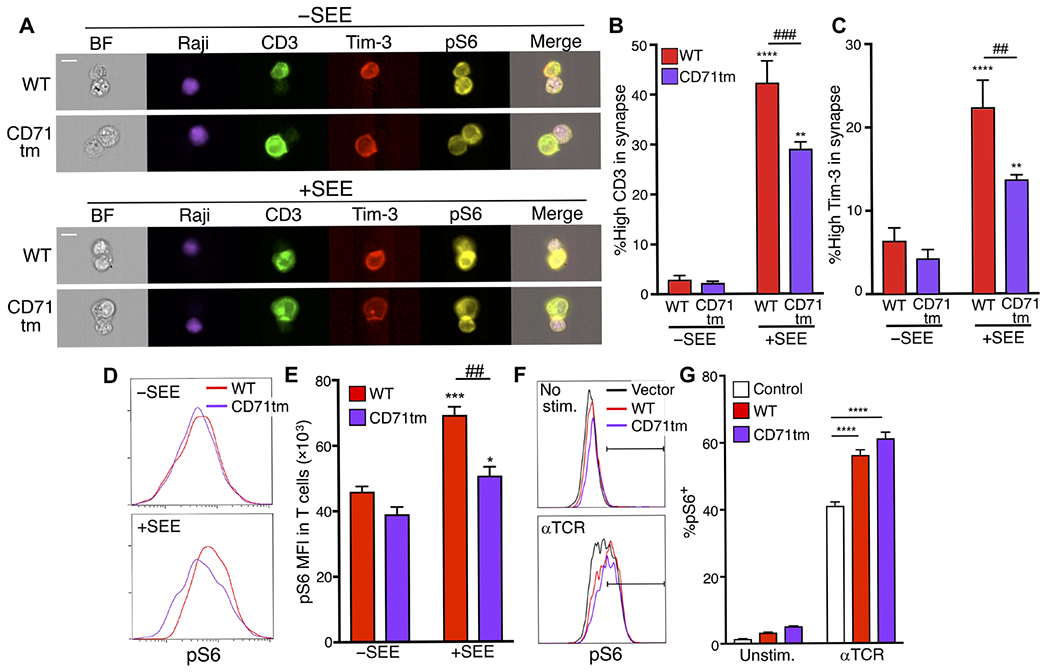
(A) Representative images of pS6 (yellow) in Jurkat cells transfected with plasmids encoding either Flag-tagged WT or CD71tm Tim-3 (red) and mixed with Raji cells (purple), which were either unpulsed of pulsed with SEE, at a ratio of 1:1, for 20 min. (B and C) Quantification of the percentage of CD3 (B) and Tim-3 (C) enriched in the immune synapses of multiple Jurkat cell–Raji cell conjugates in the presence or absence of SEE. Each bar represents 85 cell conjugates without SEE and 203 cell conjugates with SEE. (D) Representative histogram analyzing pS6 intensity in transfected Jurkat cells in cell conjugates with Raji cells in the presence or absence of SEE. (E) Quantification of the mean fluorescence intensity (MFI) of pS6 across multiple cell conjugates. (F) Flow cytometry analysis of pS6 within control, WT Tim-3-, or Tim-3 CD71tm–expressing transfected Jurkat cells stimulated with anti-TCR for 30 min. (G) Quantification of the percentage of pS6+ cells from the experiments shown in (F). Each panel is representative of at least two independent experiments. Data are means ± SEM. *P < 0.05, ***P < 0.001, and ****P < 0.0001 versus −SEE; ##P < 0.01 and ###P < 0.001 by Tukey’s multiple comparisons test. Scale bars, 10 μm. Data in (A) to (C) are representative of three experiments, whereas data in (D) to (G) are representative of two experiments.
To assess the role of the Tim-3 TM domain in T cell activation during immune synapse formation, we stimulated transfected Jurkat cells with Raji cells pulsed with SEE. Thus, we observed that transfected Jurkat cells expressing WT Tim-3 showed increased pS6 abundance compared to that in cells expressing CD71tm Tim-3, after conjugation with SEE-pulsed Raji cells (Fig. 4, D and E). To assess whether replacement of the TM domain of Tim-3 affected T cell activation more generally, we stimulated Jurkat cells expressing either Flag-tagged WT Tim-3 or CD71tm Tim-3 with anti-TCR mAb. However, in this context, we found similar pS6 amounts between cells expressing the WT and CD71tm forms of Tim-3 (Fig. 4, F and G), indicating that CD71tm Tim-3 retained the ability to enhance S6 phosphorylation after antibody stimulation, which does not result in immune synapse formation. These data suggest that localization of Tim-3 at the immune synapse is required for its costimulatory effect on T cell activation, specifically under conditions inducing immune synapse formation.
In the earlier experiments, we used imaging cytometry to assess the localization of Tim-3 and the formation of immune synapse between T cells and APCs. This approach has the advantages of visualizing immune synapse formation in a native T cell–APC setting, as well as being amenable to high-throughput data collection. Nonetheless, we wanted to also assess Tim-3 localization in a system that enables imaging of these structures at higher resolution and in real-time. We therefore chose the lipid bilayer system described by Dustin et al. (51). We paired this with TIRF microscopy and a fluorogen-activating protein (FAP) chimeric approach, which together enabled high signal:noise imaging of protein localization at the plasma membrane (52). Primary murine T cells were activated in vitro and transduced with lentivirus encoding WT or CD71tm Tim3-FAP fusion proteins (fig. S3A). The expression of the constructs in the packaging cells and transduced T cells was examined (fig. S3, B and C). Transduced cells were sorted and imaged on lipid bilayers containing intercellular adhesion molecule–1 (ICAM-1) and anti-TCRβ Fab fragment. Consistent with earlier results, we observed recruitment of WT Tim-3 to the immune synapse, as defined by the presence of TCR/CD3 (Fig. 5A). We next imaged the CD71tm variant of Tim-3 with the same system. Again, consistent with results obtained from experiments with T cell–APC conjugates, the CD71tm Tim-3 construct was recruited less efficiently to the immune synapse formed on lipid bilayers, with small Tim-3 foci that appeared less organized than those containing WT Tim-3 (Fig. 5B). We quantified immune synapse formation by measuring the areas of recruited CD3 and Tim-3 to the site of contact with the bilayer. This central region of the immune synapse is sometimes referred to as the central supramolecular activating cluster (c-SMAC). Cells expressing the CD71tm form of Tim-3 displayed significantly smaller c-SMAC structures, both at the level of Tim-3 and CD3, suggesting a dominant inhibitory effect of this construct on immune synapse formation.
Fig. 5. Lipid bilayer imaging of immune synapses with WT versus CD71tm Tim-3.
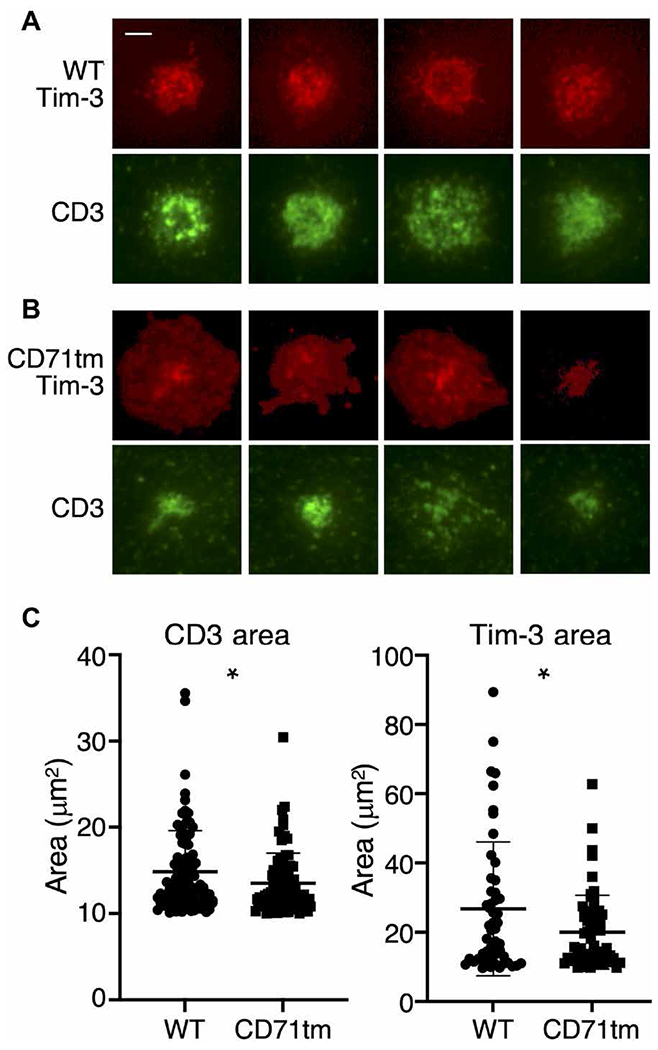
(A and B) Representative images of murine CD8+ T cells expressing FAP-tagged WT Tim-3 (A) or CD71tm Tim-3 (B). Tim-3 localization was determined on the basis of FAP staining after the addition of dye. TCR/CD3 localization was tracked by a labeled anti-TCR (H57) Fab fragment (green). Images were acquired at 15 min after the addition of the T cells to the bilayer. Scale bar, 2 μm. (C) Quantitation of the average diameters of immune synapse c-SMACs containing Tim-3 and CD3 from 50 to 60 cells (Tim-3) or 80 to 90 cells (CD3). Statistical significance was derived from Student’s t tests of WT versus CD71tm. *P < 0.05. Data are representative of three experiments.
Effects of enforced localization of the Tim-3 cytoplasmic domain to the immune synapse in a CAR
As discussed earlier, multiple ligands have been reported to bind to the IgV domain of Tim-3, although none of these is exclusive to Tim-3. The earlier experiments here were conducted in the absence of added ligands to the ecto domain of Tim-3, although we cannot rule out the presence of such ligands in our cell cultures. We therefore considered the possibility that ligand engagement of Tim-3 could affect its ability to localize to the immune synapse, provide a costimulatory signal, or both. Whereas several ligands for Tim-3 have been described (8, 53), including galectin-9, HMGB1, PS, and CEACAM1, none of these is exclusive to Tim-3. We therefore tested the effects of enforced localization of Tim-3 to the immune synapse using a CAR approach. To this end, we compared the activity of an anti-CD20 CAR containing the cytoplasmic domains of TCRζ and CD28 (54, 55) with one containing the cytoplasmic domains of TCRζ and Tim-3. Initially, we expressed these receptors in Jurkat cells, which were cultured with CD20-expressing Raji cells to generate cell-cell conjugates, as described earlier. As expected, cells expressing either of these CARs formed discrete immune synapses when bound to cells expressing cognate antigens (Fig. 6A).
Fig. 6. Enforced immune synapse localization of the human Tim-3 cytoplasmic tail supports T cell activation through a CAR.
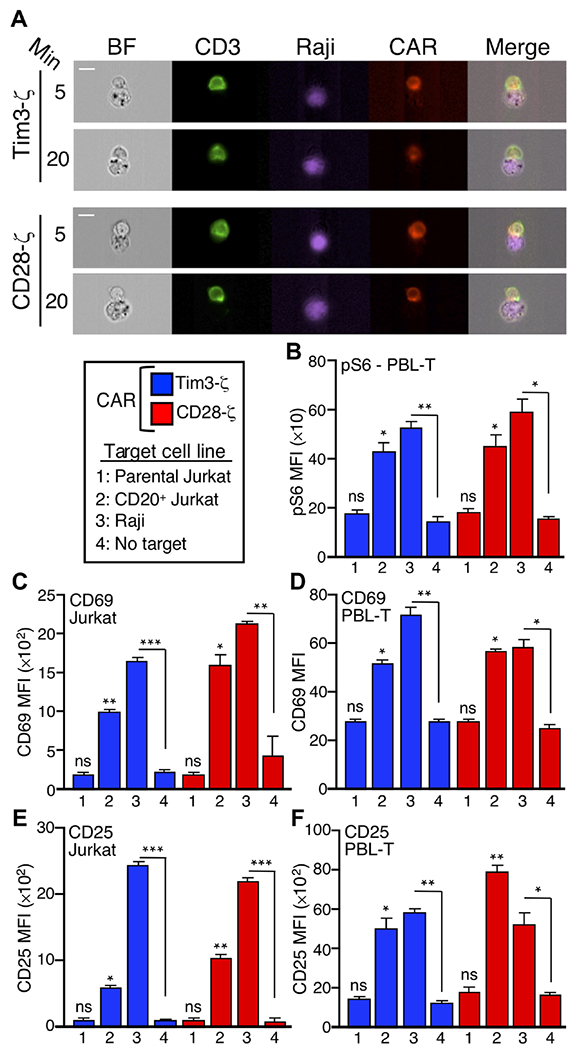
(A) Imaging cytometry analysis of immune synapse formation by the CD28-ζ or Tim3-ζ CARs expressed in Jurkat cells that were mixed with Raji cells (purple) for the indicated times. (B) Peripheral blood T cells expressing either the Tim3-ζ or CD28-ζ CAR were assayed by intracellular flow cytometry to detect pS6 after stimulation for 30 min with the indicated target cells. (C to F) Jurkat cells (C and E) or human peripheral blood T cells (D and F) expressing either the CD28 or Tim-3 CAR were stimulated with the indicated target cells (B) and analyzed 18 hours later by flow cytometry for the cell surface expression of CD69 (C and D) or CD25 (E and F). Each bar represents an average of 450 conjugates. Data are means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 by Tukey’s multiple comparisons test, based on three independent experiments. ns, not significant. All statistics indicate comparison with no-target conditions. Data are representative of two experiments. Scale bars, 10 μm.
We next assessed the functional consequences of this enforced localization of the Tim-3 cytoplasmic tail to the immune synapse. We mixed Jurkat cells expressing either the CD28- or Tim3-containing CAR with Raji cells (CD20+) as targets. Given our earlier findings and previous studies (22, 24) that showed that Tim-3 is capable of costimulating the TCR-mediated phosphorylation of the ribosomal S6 protein, we investigated whether this activity was retained by the Tim-3 CAR. Consistent with our functional data, the Tim-3 CAR mediated an increase in S6 phosphorylation, similarly to the CD28 CAR (Fig. 6B). This was observed with two different target cell lines expressing CD20: either Raji cells (which express endogenous CD20) or Jurkat cells that were transfected to express CD20. Using this approach, we also found that the Tim3-containing CAR was about as efficient as a CD28-containing CAR at mediating an increase in the cell surface expression of the early activation marker CD69 when the CARs were expressed in either Jurkat cells or primary human peripheral blood lymphocyte T cells (PBL-T) (Fig. 6, C and D). This activity was dependent on the presence of the CD20 antigen on the target cells, because it was not observed in the absence of target cells or in the presence of parental Jurkat cells that do not express CD20. We also assessed the effects of the two CARs on the induced expression of CD25, which is increased with somewhat delayed kinetics relative to that of CD69. Consistent with the effects shown earlier, the CD28 and Tim-3 CARs mediated an antigen-specific increase in CD25 abundance to similar extents in both Jurkat cells and primary human T cells (Fig. 6, E and F). Thus, in the context of a synthetic antigen receptor, which eliminates any potential confounding effects of Tim-3 ligands, the cytoplasmic tail of Tim-3 is still permissive for T cell activation, similar to a standard CD28-containing CAR.
DISCUSSION
Despite studies linking the expression of Tim-3 to T cell dysfunction, the molecular mechanisms underlying the biological effects of Tim-3 remain unresolved. In particular, mechanisms by which Tim-3 might directly inhibit TCR or costimulatory signaling are not well understood. Work presented here is consistent with previous reports from us and others that Tim-3 can paradoxically mediate positive, costimulatory effects on T cell activation, at least under some circumstances. These effects are tightly correlated with enhanced phosphorylation of the ribosomal S6 protein. Because S6 phosphorylation can be increased by either MEK-ERK or Akt-mTOR signaling, we used specific inhibitors to probe the roles of these pathways in the costimulatory effect of Tim-3 on S6 phosphorylation. Our data suggest that both of these pathways are involved in Tim-3–mediated costimulation. This is consistent with previous findings from our group showing that Tim-3 can also enhance TCR-proximal signaling at the level of phospholipase C–γ1 phosphorylation (24). Furthermore, we also found that expression of Tim-3 itself appears to be sufficient for at least a partial increase in ERK signaling and AP-1 activation, something that we have not observed with other downstream pathways involving Akt, pS6, and NFAT, which all require additional stimulation through TCR and CD28.
Proper localization of numerous proteins during T cell recognition of antigens and APCs is important for early T cell activation. For example, a protein kinase C θ (PKCθ) mutant that fails to accumulate at the immune synapse impairs T cell activation, even upon stimulation with anti-CD3 and anti-CD28 mAbs (56). In agreement with a previous report from Clayton et al. (35) with human PBL T cells, we observed stimulation-dependent recruitment of Tim-3 to the immune synapse in both Jurkat cells and primary murine T cells engaged by antigen-pulsed APCs. Here, we extended these findings by examining the role of specific domains within Tim-3 for this recruitment, in parallel with experiments quantifying the effects on downstream signaling. Recruitment of Tim-3 to the immune synapse was not dependent on the extracellular or intracellular domains of Tim-3 but rather appeared to be primarily determined by the TM domain. Thus, previous data demonstrated the TM domain–dependent recruitment of CD148 to lipid rafts, which could be disrupted by replacing the TM domain of CD148 with that of CD71 (57). Because the recruitment of Tim-3 to the immune synapse is associated with lipid raft recruitment (35), we replaced the TM domain of Tim-3 with that of CD71. Although this did not affect the cell surface expression of Tim-3, it significantly impaired both the recruitment of Tim-3 to the immune synapse and the ability of Tim-3 to costimulate TCR-dependent increases in S6 phosphorylation.
One of the unresolved aspects of Tim-3 biology concerns the roles of the multiple ligands that have been reported to interact with it. Thus, galectin-9, HMGB1, PS, and CEACAM-1 have all been reported as Tim-3 ligands (8, 9, 53). However, none of these is exclusive to Tim-3, because all of these putative ligands can also interact with other cell surface receptors (9, 17). We cannot assume that none of the Tim-3 ligands is present in our experiments, because the APCs used for immune synapse formation may express one or more of them. Relevant for this point, a Tim-3 construct lacking the IgV domain was still recruited to the immune synapse to a similar degree as WT Tim-3. We noted a trend toward increased recruitment of the ΔIgV construct. Although this did not reach statistical significance in our studies, it may be that the IgV limits the recruitment of Tim-3 to the immune synapse under some circumstances, through relief of either steric hindrance or an inhibitory effect of ligand binding. In any case, these results do strongly suggest that ligand binding is not required for the localization of Tim-3 in the immune synapse.
Lipid raft–associated proteins are recruited into the immune synapse (58). Note that several TM proteins with immune function partition into lipid rafts in a manner dependent on their specific TM domains (59–61). Thus, when we replaced the TM domain of Tim-3 with that of CD71, which is excluded from lipid rafts (50), we observed significantly decreased recruitment of Tim-3 to the immune synapse. It is still not clear how Tim-3 would partition into lipid rafts, as it is not known to be acylated. However, the TM domain does contain a GxxxG motif, which forms a helical structure that cannot only mediate helix-helix associations but also interact with membrane cholesterol, in the case of amyloid precursor protein (62). A similar domain is found in the TM domain of CD71. However, the context (that is, the surrounding sequence) is distinct between the TM domains of Tim-3 and CD71, and on the basis of previous work, this could influence the differential effects of these domains on immune synapse recruitment (63). An unexpected aspect of the CD71 TM Tim-3 construct was that it was associated with the less robust recruitment of CD3 to the immune synapse. We do not yet know whether this was a direct effect or rather just a reflection of relatively lower immune synapse recruitment in comparison to cells ectopically expressing Tim-3. We previously reported that Tim-3 recruits a substantial fraction of Fyn to its cytoplasmic tail (24), and another study suggested a role for Fyn recruitment in stabilizing the TCR in immune synapses (64). Thus, it is possible that nonsynapse-localized Tim-3 with a CD71 TM domain is sequestering Fyn away from active synapses. Nonetheless, this was not sufficient to completely shut down CD3 signaling, because the anti-CD3–induced activation of S6 was not affected by the CD71 TM mutant.
Returning to the question of stimulatory versus inhibitory signaling by Tim-3, the results presented here are consistent with previous studies from our group (22, 24) and others (23, 27, 65), suggesting a costimulatory role for Tim-3 in T cells. As suggested by others, interactions of Tim-3 in trans with other TM proteins, such as CEACAM1 (46) or phosphatases (35), could modulate the signaling function of Tim-3 toward an inhibitory pathway. Nonetheless, some of our cytoplasmic tail deletion mutants of Tim-3 (the T2 and ΔCyto constructs) lost their costimulatory activity but still localized to the immune synapse. Furthermore, even enforced localization of Tim-3 to the immune synapse through the use of a CAR still enabled efficient T cell activation upon stimulation with cells expressing the ligand for the CAR. This finding supports our conclusion that the cytoplasmic tail of Tim-3 contains intrinsic costimulatory activity.
Most studies of Tim-3 in T cells have focused on exhausted T cells, and endogenous Tim-3 is not expressed to any detectable degree by naïve T cells in either mice or humans. Thus, how do our findings relate to the in vivo function of Tim-3? We previously speculated that enhanced TCR signaling by Tim-3 may be important for the acquisition of T cell exhaustion, a process known to be tightly linked to chronic stimulation (28). On the basis of our recent work (22) and that of Gorman and Colgan (65), Tim-3 is also transiently increased in abundance under conditions of acute T cell activation, so the protein may be playing a role during the effector phase. Similarly, we also found that Tim-3 is maintained, albeit at low abundance, on memory T cells, where it may play a role in the reactivation of these cells (22). Tim-3 is constitutively expressed by mast cells and some cells of the monocytic lineage (for example, CD103+ dendritic cells). Thus, the signaling pathways downstream of Tim-3 that we have described here and elsewhere are potentially also of relevance for those cells, including under steady-state conditions.
Although much of the evidence is indirect, many studies have concluded that Tim-3 is an inhibitory molecule. Going forward, it will be important to reconcile findings of the positive effects of Tim-3 signaling with previous reports of its inhibitory function within T cells. This is an important issue, because antibody therapies directed at Tim-3 are being developed, with some already in clinical trials, for immune checkpoint blockade. In addition, our finding that the cytoplasmic tail of Tim-3 still enables the positive function of a CAR opens the way for more detailed study of the possible therapeutic benefits of CAR-T cells expressing such constructs.
MATERIALS AND METHODS
Cell lines
Jurkat and D10 T cell and Raji B cell lines were maintained in RPMI medium supplemented with 10% bovine growth serum (BGS; Hyclone), penicillin, streptomycin, L-glutamine, nonessential amino acid, sodium pyruvate, Hepes, and 2-mercaptoethanol (2-ME). Recombinant human interleukin-2 (IL-2) (25 U/ml) was also supplemented for D10 cells. Human embryonic kidney (HEK) 293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% BGS, penicillin, streptomycin, and l-glutamine.
Mice
FSF-Tim3 knock-in mice were generated as previously described (22). P14 TCR transgenic mice were obtained from Jackson Laboratories. E8i-Cre mice were obtained from Dario Vignali (University of Pittsburgh) and were originally generated by Yasutomo and colleague (University of Tokushima), as described previouslys (66). WT C57BL/6 mice were either obtained from Jackson Laboratories or bred in-house. Mice were age-matched within experiments. Animals were maintained in the facilities of the University of Pittsburgh Division of Laboratory Animal Resources. All animal studies were performed in accordance with University of Pittsburgh Institutional Animal Care and Use Committee guidelines.
Cell transfections and activation
Jurkat and D10 T cells were resuspended in RPMI medium without supplements and electroporated with control plasmid, the Flag-tagged WT Tim-3 or Flag-tagged mutant Tim-3 constructs, together with the pMaxGFP plasmid in a Bio-Rad GenePulser at 260 V and 960 μF for Jurkat T cells or at 250 V and 950 μF for D10 cells. Transfected cells were starved of serum in phosphate-buffered saline (PBS) with 0.1% bovine serum albumin (BSA) for 1 hour at 37°C. Cells were also pretreated with U0126 (Millipore Sigma #662005) and/or Akti1/2 (Abcam #ab142088) before being stimulated with the anti-Jurkat TCR Vβ8 mAb C305. After stimulation, cells were washed in staining buffer (1% BGS-supplemented PBS) and incubated with anti–Flag-PE (clone L5; BioLegend #637310) and Ghost Dye Violet 510 (Tonbo Biosciences #13-0870-T100) at 4°C for extracellular staining. Cells were then fixed and permeabilized with the BD Cytofix/Cytoperm kit (BD Biosciences #554714). Fixed and permeabilized cells were then incubated at 4°C with APC-conjugated anti-pS6 antibody (Ser235/236) (clone D57.2.2E; Cell Signaling Technology #14733). Samples were acquired on a BD LSRII flow cytometer, and the data were analyzed with FlowJo software.
Luciferase assays
Jurkat cells were transfected by electroporation, as described earlier, with empty vector (pCDEF3) or various Tim-3 constructs, together with luciferase reporter driven by multimerized NFAT or AP-1 elements. On the next day, cells (1 × 105 per well) were cultured in 96-well U-bottom plates for 6 hours with the stimuli indicated in the figure legends, after which the plates were placed at −80°. Luciferase analysis was performed as previously described (67). Briefly, plates were thawed at room temperature, which was followed by the addition of lysis buffer containing NP-40 and assay buffer containing Mg2+/adenosine triphosphate. Luciferase activity was determined in a 96-well luminometer (Promega) with injection of luciferin.
Western blotting
Cells were lysed in ice-cold NP-40 lysis buffer [1% NP-40, 1 mM EDTA, 20 mM tris-HCl (pH 7.4), and 150 mM NaCl] with protease inhibitors. Proteins were resolved by 10% SDS–polyacrylamide gel electrophoresis and were transferred onto polyvinylidene difluoride (PVDF) membranes, which were then blocked in 4% BSA. The membranes were then incubated with the primary antibodies overnight. This was followed by incubating the membrane with horseradish peroxidase (HRP)–conjugated secondary antibodies for 2 hours before signals were detected with the SuperSignal West Pico ECL substrate (Thermo Fisher Scientific #34577) and imaged on a Protein Simple FluorChem M. The following antibodies were used for Western blotting: direct-blot HRP anti-pERK antibody (Thr202/Tyr204) (clone 4B11B69, BioLegend #675505); anti-Erk1/2 (Cell Signaling Technology #9102); anti-pAkt antibody (Thr308; clone 244F9, Cell Signaling Technology #4056); and anti-Akt (clone 2, PKBα/Akt, BD Biosciences #610877).
Immunoprecipitations (IPs) and glycosidase treatment
HEK 293T cells (1 × 106) were transfected in six-well plates with the appropriate plasmids with TransIT-LT1 transfection reagent (Mirus) according to the manufacturer’s protocol. Transfection efficiency was determined by flow cytometry. Transfected cells (5 × 105 per well) were lysed using for 1 hour with ice-cold 1% NP-40 lysis buffer [20 mM tris-HCl (pH 7.5), 150 mM NaCl, and 1 mM EDTA] supplemented with complete mini (Roche) and HALT (Invitrogen) protease inhibitors. Protein G agarose beads were washed and blocked with 1% BSA for 30 min. Whole-cell lysates and beads were incubated with 1 μg of anti-Flag antibody M2 with rotation at 4°C overnight. Beads were pelletted by centrifugation and washed three times with lysis buffer or with deglycosylation buffer. Deglycosylation was performed on beads with Protein Deglycosylation Mix II (New England Biolabs; NEB) in denaturing conditions according to the manufacturer’s protocol. Beads were boiled with 2× Sample buffer (Bio-Rad) with 2-ME for 5 min at 95°C. Samples were spun down, and the protein supernatant was removed. Supernatant samples were resolved on 10% NuPAGE gels and transferred to PVDF membranes. The membranes were blocked with 5% BSA in tris-buffered saline with Tween 20 buffer and incubated with anti-mouse Tim-3 goat polyclonal antibody (R&D) and detected with anti–goat-HRP antibody and SuperSignal West Pico Chemiluminescent reagent (Thermo Fisher Scientific).
Jurkat cell–Raji cell conjugate formation
Raji cells were labeled with either CellTracker Orange CMRA Dye (Thermo Fisher Scientific #C34551) or Cell Proliferation Dye eFluor 450 (Thermo Fisher Scientific #65-0842-85) and then pulsed with SEE (2 μg/ml; Toxin Technology #ET404) for 1 hour at 37°C. Transfected Jurkat cells were mixed with Raji cells, either unpulsed or pulsed with SEE, at a ratio of 1:1 and incubated at 37°C for the times indicated in the figure legends.
Primary T cell–B cell conjugate formation
B cells from the spleens and lymph node of naïve mice were purified by magnetic separation with a pan-B cell isolation kit (Miltenyi Biotec #130-095-813) and then incubated overnight with lipopolysaccharide (30 μg/ml) (Sigma-Aldrich #L2630) and 10 μM LCMV gp33-41 peptide (AnaSpec #AS-61296). CD8+ T cells from the spleens and lymph nodes of either P14 or FSF-Tim-3/E8i-Cre/P14 mice were purified by magnetic separation with a naïve CD8+ T cell isolation kit (Miltenyi Biotec #130-104-075) and then mixed with gp33-pulsed B cells at a ratio of 1:1 and incubated at 37°C for the times indicated in the figure legends.
Imaging flow cytometry
Cell-cell conjugates were fixed with 2% paraformaldehyde and then incubated with antibodies at 4°C for extracellular staining. For intracellular staining, cells were permeabilized with 0.2% saponin and then incubated with anti-pS6 antibody (Ser235/236) antibody at 4°C. Imaging cytometry data on fixed and stained cells were acquired on an Amnis ImageStreamX Mark II running INSPIRE software (Millipore Sigma). Initially, 1 × 104 events were acquired, before downstream gating and analysis, which was performed with IDEAS software (Millipore Sigma). First, focused cells, based on the gradient root mean square feature, were identified. Doublets were identified by the aspect ratio and area of the bright field and then were gated by the aspect ratio and staining intensity of the B cells. Gated cells were further refined by the aspect ratio and staining intensity of T cells to identify T cell–B cell doublets. For Jurkat cell–Raji cell conjugates, an interface mask covering the immune synapse was made from bright-field and Raji staining. The percentages of CD3 and Tim-3 recruitment to the immune synapse were quantified from the ratio of the intensity of either CD3 or Tim-3 in the interface mask to that of the whole cell mask. The intensity of pS6 in Jurkat cells was quantified in the mask made by subtracting the B cell mask from the bright-field mask. For primary T cell–B cell conjugates, an interface mask was made from bright-field and CD8 staining. The intensity of pS6 in CD8+ T cells was quantified in the mask made from CD3 staining. Approximately 100 to 300 cells per point are represented in the Jurkat cell–Raji cell conjugate data, and 100 to 2000 cells are represented in the primary T cell–B cell conjugate data. The following antibodies were used for the conjugation assay: anti-human CD3–fluorescein isothiocyanate (FITC) (clone OKT3; Tonbo Biosciences #35-0037-T100), anti–Flag-APC (clone L5; BioLegend #637308), anti-mouse CD3-FITC (clone 145-2C11; Tonbo Biosciences #35-0031-U500), anti-mouse CD19-violetFluor 450 (clone 1D3; Tonbo Biosciences #75-0193-U100), and anti-pS6 (Ser235/236)–phycoerythrin (PE) (clone D57.2.2E; Cell Signaling Technology #5316S).
TIRF imaging of T cell immune synapses on lipid bilayers
WT and CD71tm mutant Tim-3 constructs were subcloned into murine stem cell virus (MSCV)-based retroviral vectors containing the FAP sequence in-frame, which is followed by an internal ribosomal entry site and the coding sequence of mouse Thy1.1. These constructs were then used to transfect 293T cells, together with the pCL-Eco packaging plasmid, with the TransIT-LT1 transfection reagent (Mirus). CD8+ T cells were purified from C57BL/6 mice with a mouse CD8+ T cell isolation kit (Miltenyi). Cells were stimulated for 27 hours on plates coated with anti-CD3/CD28 mAbs and recombinant human IL-2 (100 U/ml). Activated T cells were harvested and transduced with FAP-encoding virus from the 293T cell transfectants. Transduced Thy1.1+ cells were purified by sorting and kept on ice until imaging. Lipid bilayers were constructed as described previously (68), with some modifications (69). Briefly, liposomes composed of 90% dioleoylphosphocholine, 10% DOGS (1,2-dioleoyl-sn-glycero-3-{[N(5-amino-1carboxypentyl) iminodiacetic acid]succinyl}), and 0.2% biotin-CAP-PE (Avanti Polar Lipids) were deposited on glass coverslips cleaned with piranha solution (50:50 mixture of 30% H2O2 and 96% H2SO4). Alexa 488 streptavidin, Alexa 647 streptavidin, or unconjugated streptavidin and biotinylated TCRβ mAb were sequentially loaded onto the bilayer, whereas unlabeled poly–His-tagged ICAM-1 produced in a baculovirus system was loaded to facilitate T cell adhesion. Transduced T cells were sorted, rested for 24 to 48 hours, incubated with MGnBu (200 nM) in RPMI without serum or phenyl red for 15 min at 37°C, washed, resuspended in RPMI without serum or phenyl red, and stimulated on a planar lipid bilayer. Imaging was performed with a Nikon Ti inverted microscope equipped with a motorized TIRF arm and a 100× 1.40 numerical aperture objective. Images were acquired at 100-ms intervals continuously for up to 15 to 20 min with a Zyla sCMOS camera (Andor) equipped with bandpass emission filters for 4′,6-diamidino-2-phenylindole, FITC, tetramethyl rhodamine isothiocyanate, and Cy5 spectral profiles. Images were analyzed with NIS elements software version 5.21.00. Images were separated by layer, and a threshold for each layer was set: The AF488 threshold was set between 200 and 1000 fluorescence intensity units, and the AF647 threshold was set between 150 and 40,000 fluorescence intensity units. Areas for individual channels and intersecting areas were measured with automated measurements.
Construction and characterization of CARs
To generate the TIM-3 CAR constructs (pHR-EF1-aCD19-TIM3ζ-T2A-TagBFP and pHR-EF1-aCD20-TIM3ζ-T2A-TagBFP), DNA encoding the human TIM-3 cytoplasmic domain was codon-optimized, synthesized (by Integrated DNA Technologies), and subcloned into the pHR-EF1-aCD19-28ζ or pHR-EF1-αCD20-28ζ CAR lentiviral expression vectors, respectively, to replace the CD28 cosignaling domain by isothermal assembly. To generate virus, HEK 293T cells (American Type Culture Collection) were transfected with pVSV-G (VSV glycoprotein expression plasmid), pMD2.G, and the CAR expression plasmid by calcium phosphate transfection (Clontech). Sixteen hours later, the cells were washed with PBS and incubated in complete DMEM media containing 6 mM sodium butyrate (Sigma-Aldrich). Supernatants were collected at 24 and 48 hours and were subsequently combined and filtered through a 45-μm vacuum filter. Viral particles were concentrated by ultracentrifugation for 1.5 hours at 120,000g, and viral pellets were resuspended in 0.05 ml of supplemented RPMI medium and frozen at −80°C. For CAR-T pS6 analysis, 100,000 Jurkat cells or primary T cells expressing the TIM-3 or CD28 CAR were incubated with an equal number of the appropriate antigen-positive or antigen-negative target cell line in a V-bottom plate for 30 min at 37°C and 5% CO2. Cells then underwent intracellular staining and flow cytometry as described previously (22). For CAR-T surface marker analysis, 100,000 Jurkat cells or primary T cells expressing the TIM-3 or CD28 CAR were incubated with an equal number of the appropriate antigen-positive or antigen-negative target cell line in a V-bottom plate for 18 hours at 37°C and 5% CO2. Cells then underwent cell surface antibody staining and flow cytometry analysis (22).
Supplementary Material
Acknowledgments:
We thank the University of Pittsburgh Department of Immunology flow cytometry core and Center for Biologic Imaging for valuable technical assistance. We also thank D. Vignali for providing mice. J. Lohmueller also acknowledges the support of O. J. Finn.
Funding:
This work was supported by PHS awards AI138504 and CA206517 to L.P.K. S.K. was supported in part by an unrestricted award from Asahi Kasei Pharma. J. Lohmueller was supported in part by PHS award CA210039, the Michael G. Wells Prize, and start-up funds from the University of Pittsburgh. Imaging flow cytometry was performed on an ImageStreamX MarkII, which was acquired with a shared instrument grant from the NIH (1S10OD019942).
Competing interests:
The work of S.K. was supported in part by Asahi Kasei, who had no input into the manuscript. The Tim-3 CAR construct is the subject of a provisional patent application (U.S. provisional application no. 62/923,201) by J. Lohmueller and L.P.K. The other authors declare that they have no competing interests.
Footnotes
SUPPLEMENTARY MATERIALS
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Full-length, truncated, and CD71tm Tim-3 constructs have been deposited with Addgene and are available through them. Chimeric antigen receptor constructs are available upon request from the authors.
REFERENCES AND NOTES
- 1.Courtney AH, Lo WL, Weiss A, TCR signaling: Mechanisms of initiation and propagation. Trends Biochem. Sci 43, 108–123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaud G, Lesourne R, Love PE, Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol 18, 485–497 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Hargadon KM, Johnson CE, Williams CJ, Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol 62, 29–39 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.June CH, Sadelain M, Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, Freeman GJ, Kuchroo VK, Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Gorman JV, Colgan JD, Regulation of T cell responses by the receptor molecule Tim-3. Immunol. Res 59, 56–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson AC, Joller N, Kuchroo VK, Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee H, Kane LP, Immune regulation by Tim-3. F1000Res 7, 316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM, Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med 207, 2175–2186 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R, Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U.S.A 107, 14733–14738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kassu A, Marcus RA, D’Souza MB, Kelly-McKnight EA, Golden-Mason L, Akkina R, Fontenot AP, Wilson CC, Palmer BE, Regulation of virus-specific CD4+ T cell function by multiple costimulatory receptors during chronic HIV infection. J. Immunol 185, 3007–3018 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vali B, Jones RB, Sakhdari A, Sheth PM, Clayton K, Yue FY, Gyenes G, Wong D, Klein MB, Saeed S, Benko E, Kovacs C, Kaul R, Ostrowski MA, HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression. Eur. J. Immunol 40, 2493–2505 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC, Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med 207, 2187–2194 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR, TIM-3, a promising target for cancer immunotherapy. Onco. Targets. Ther 11, 7005–7009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gayden T, Sepulveda FE, Khuong-Quang DA, Pratt J, Valera ET, Garrigue A, Kelso S, Sicheri F, Mikael LG, Hamel N, Bajic A, Dali R, Deshmukh S, Dervovic D, Schramek D, Guerin F, Taipale M, Nikbakht H, Majewski J, Moshous D, Charlebois J, Abish S, Bole-Feysot C, Nitschke P, Bader-Meunier B, Mitchell D, Thieblemont C, Battistella M, Gravel S, Nguyen VH, Conyers R, Diana JS, McCormack C, Prince HM, Besnard M, Blanche S, Ekert PG, Fraitag S, Foulkes WD, Fischer A, Neven B, Michonneau D, de Saint Basile G, Jabado N, Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat. Genet 50, 1650–1657 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Su EW, Bi S, Kane LP, Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology 21, 1258–1265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Segawa K, Nagata S, An apoptotic ‘eat me’ signal: Phosphatidylserine exposure. Trends Cell Biol 25, 639–650 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME, HMGB1 as biomarker and drug target. Pharmacol. Res 111, 534–544 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Dankner M, Gray-Owen SD, Huang YH, Blumberg RS, Beauchemin N, CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology 6, e1328336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, Bruce JN, Kane LP, Kuchroo VK, Hafler DA, Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318, 1141–1143 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Avery L, Filderman J, Szymczak-Workman AL, Kane LP, Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. U.S.A 115, 2455–2460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorman JV, Starbeck-Miller G, Pham NL, Traver GL, Rothman PB, Harty JT, Colgan JD, Tim-3 directly enhances CD8 T cell responses to acute Listeria monocytogenes infection. J. Immunol 192, 3133–3142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, Smithgall TE, Kuchroo VK, Kane LP, Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol 31, 3963–3974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, Kane LP, Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. J. Exp. Med 212, 2289–2304 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakae S, Iikura M, Suto H, Akiba H, Umetsu DT, Dekruyff RH, Saito H, Galli SJ, TIM-1 and TIM-3 enhancement of Th2 cytokine production by mast cells. Blood 110, 2565–2568 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabins NC, Chornoguz O, Leander K, Kaplan F, Carter R, Kinder M, Bachman K, Verona R, Shen S, Bhargava V, Santulli-Marotto S, TIM-3 engagement promotes effector memory T cell differentiation of human antigen-specific CD8 T cells by activating mTORC1. J. Immunol 199, 4091–4102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferris RL, Lu B, Kane LP, Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol 193, 1525–1530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue H-H, Ahmed R, Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, Pradervand S, Thimme R, Zehn D, Held W, T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC, Checkpoint blockade immunotherapy induces dynamic changes in PD-1−CD8+ tumor-infiltrating T cells. Immunity 50, 181–194.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W, Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Li J, Shayan G, Avery L, Jie HB, Gildener-Leapman N, Schmitt N, Lu BF, Kane LP, Ferris RL, Tumor-infiltrating Tim-3+ T cells proliferate avidly except when PD-1 is co-expressed: Evidence for intracellular cross talk. Onco. Targets. Ther 5, e1200778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, Wherry EJ, Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clayton KL, Haaland MS, Douglas-Vail MB, Mujib S, Chew GM, Ndhlovu LC, Ostrowski MA, T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J. Immunol 192, 782–791 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anjum R, Blenis J, The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol 9, 747–758 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y, Schwartzberg PL, Wange RL, Deficiency of PTEN in jurkat T cells causes constitutive localization of itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol. Cell. Biol 20, 6945–6957 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kane LP, Mollenauer MN, Weiss A, A proline-rich motif in the C terminus of Akt contributes to its localization in the immunological synapse. J. Immunol 172, 5441–5449 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Cantrell D, Signaling in lymphocyte activation. Cold Spring Harb. Perspect. Biol 7, a018788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hwang JR, Byeon Y, Kim D, Park SG, Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med 52, 750–761 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith-Garvin JE, Koretzky GA, Jordan MS, T cell activation. Annu. Rev. Immunol 27, 591–619 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rincon M, Flavell RA, Davis RJ, Signal transduction by MAP kinases in T lymphocytes. Oncogene 20, 2490–2497 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Flies DB, Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol 13, 227–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pentcheva-Hoang T, Chen L, Pardoll DM, Allison JP, Programmed death-1 concentration at the immunological synapse is determined by ligand affinity and availability. Proc. Natl. Acad. Sci. U.S.A 104, 17765–17770 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP, B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21, 401–413 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen BS, Melum E, Pertel T, Clayton KL, Raab M, Chen Q, Beauchemin N, Yazaki PJ, Pyzik M, Ostrowski MA, Glickman JN, Rudd CE, Ploegh HL, Franke A, Petsko GA, Kuchroo VK, Blumberg RS, CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517, 386–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK, The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol 6, 1245–1252 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M, Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol 13, 832–842 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim Y-L, Lee H-H, Karisola P, Pichavant M, Kaplan GG, Umetsu DT, Freeman GJ, Casasnovas JM, T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol 184, 1918–1930 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho NH, Kingston D, Chang H, Kwon EK, Kim JM, Lee JH, Chu H, Choi MS, Kim S, Jung JU, Association of herpesvirus saimiri tip with lipid raft is essential for downregulation of T-cell receptor and CD4 coreceptor. J. Virol 80, 108–118 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dustin ML, Starr T, Varma R, Thomas VK, Supported planar bilayers for study of the immunological synapse. Curr. Protoc. Immunol Chapter 18, Unit 18.13 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JA, Woolford CA, Yan Q, Vasilev KV, Berget PB, Bruchez MP, Jarvik JW, Waggoner A, Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol 26, 235–240 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, Kane LP, Lu B, TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int. J. Mol. Sci 18, 645 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jensen M, Tan G, Forman S, Wu AM, Raubitschek A, CD20 is a molecular target for scFvFc:zeta receptor redirected T cells: Implications for cellular immunotherapy of CD20+ malignancy. Biol. Blood Marrow Transplant 4, 75–83 (1998). [DOI] [PubMed] [Google Scholar]
- 55.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M, Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat. Biotechnol 20, 70–75 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Kong K-F, Yokosuka T, Canonigo-Balancio AJ, Isakov N, Saito T, Altman A, A motif in the V3 domain of the kinase PKC-Θ determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol 12, 1105–1112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin J, Weiss A, The tyrosine phosphatase CD148 is excluded from the immunologic synapse and down-regulates prolonged T cell signaling. J. Cell Biol 162, 673–682 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harder T, Engelhardt KR, Membrane domains in lymphocytes - from lipid rafts to protein scaffolds. Traffic 5, 265–275 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Benslimane N, Hassan GS, Yacoub D, Mourad W, Requirement of transmembrane domain for CD154 association to lipid rafts and subsequent biological events. PLOS ONE 7, e43070 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nadiri A, Polyak MJ, Jundi M, Alturaihi H, Reyes-Moreno C, Hassan GS, Mourad W, CD40 translocation to lipid rafts: Signaling requirements and downstream biological events. Eur. J. Immunol 41, 2358–2367 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Perschl A, Lesley J, English N, Hyman R, Trowbridge IS, Transmembrane domain of CD44 is required for its detergent insolubility in fibroblasts. J. Cell Sci 108 (Pt. 3), 1033–1041 (1995). [DOI] [PubMed] [Google Scholar]
- 62.Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR, The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teese MG, Langosch D, Role of GxxxG motifs in transmembrane domain interactions. Biochemistry 54, 5125–5135 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Tang Q, Subudhi SK, Henriksen KJ, Long CG, Vives F, Bluestone JA, The Src family kinase Fyn mediates signals induced by TCR antagonists. J. Immunol 168, 4480–4487 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Gorman JV, Colgan JD, Acute stimulation generates Tim-3-expressing T helper type 1 CD4 T cells that persist in vivo and show enhanced effector function. Immunology 154, 418–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maekawa Y, Minato Y, Ishifune C, Kurihara T, Kitamura A, Kojima H, Yagita H, Sakata-Yanagimoto M, Saito T, Taniuchi I, Chiba S, Sone S, Yasutomo K, Notch2 integrates signaling by the transcription factors RBP-J and CREB1 to promote T cell cytotoxicity. Nat. Immunol 9, 1140–1147 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Kane LP, Shapiro VSS, Stokoe D, Weiss A, Induction of NF-κB by the Akt/PKB kinase. Curr. Biol 9, 601–604 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM, TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature 463, 963–967 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD, Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc. Natl. Acad. Sci. U.S.A 104, 20296–20301 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Full-length, truncated, and CD71tm Tim-3 constructs have been deposited with Addgene and are available through them. Chimeric antigen receptor constructs are available upon request from the authors.