

Abstract
There is a possible accelerated biological aging in patients with substance use disorders (SUD). The evaluation of epigenetic clocks, which are accurate estimators of biological aging based on DNA methylation changes, has been limited to blood tissue in patients with SUD. Consequently, the impact of biological aging in the brain of individuals with SUD remains unknown. In this study, we evaluated multiple epigenetic clocks (DNAmAge, DNAmAgeHannum, DNAmAgeSkinBlood, DNAmPhenoAge, DNAmGrimAge, and DNAmTL) in individuals with SUD (n=42), including alcohol (n=10), opioid (n=19), and stimulant use disorder (n=13), and controls (n=10) in postmortem brain (prefrontal cortex) and blood tissue obtained from the same individuals. We found a higher DNAmPhenoAge (beta=0.191, p-value=0.0104) and a nominally lower DNAmTL (beta=−0.149, p-value=0.0603) in blood from individuals with SUD compared to controls. SUD subgroup analysis showed a nominally lower brain DNAmTL in subjects with alcohol use disorder, compared to stimulant use disorder and controls (beta=0.0150, p-value=0.087). Cross-tissue analyses indicated a lower blood DNAmTL and a higher blood DNAmAge compared to their respective brain values in the SUD group. This study highlights the relevance of tissue specificity in biological aging studies and suggests that peripheral measures of epigenetic clocks in SUD may depend on the specific type of drug used.
Keywords: epigenetic clocks, DNA methylation, cross-tissue, stimulants use disorder, opioid use disorder
1. Introduction
Substance use disorders (SUD) have been associated with an increased risk of premature all-cause mortality and an early onset of age-related diseases, such as cardiac, cerebrovascular, kidney, and liver diseases (Chesney et al., 2014, Hjemsæter et al., 2019). A possible increase in aging acceleration in patients with SUD, i.e., an outpace of biological age over chronological age, is considered one of the main contributors to the adverse outcomes in SUD (Bachi, Sierra, Volkow, Goldstein, & Alia-Klein, 2017). While studies of accelerated biological aging have been most extensively done using telomere length as the biological measure (Monroy-Jaramillo, Dyukova, & Walss-Bass, 2018), DNA methylation-based epigenetic clocks, better known as epigenetic clocks, have more recently been demonstrated as highly accurate molecular correlates of biological age (Bell et al., 2019).
Several epigenetic clocks have been developed in the last years, including DNAmAge, DNAmAgeHannum and DNAmAgeSkinBlood. The available epigenetic clocks were built based on the DNA methylation of different CpG sites, varying from 3 to 353 CpG sites (Field et al., 2018). Overall, epigenetic clocks have exhibited high accuracy in the estimation of biological age in different tissues including whole blood, skeletal muscle, and bone (Voisin et al., 2020; Gopalan, Gaige, & Henn, 2019; Fransquet, Wrigglesworth, Woods, Ernst, & Ryan, 2019). Furthermore, the most recent epigenetic clocks, i.e., DNAmPhenoAge (Levine et al., 2018) and DNAmGrimAge, besides estimating biological age, have been shown to predict clinical health measures, including blood pressure (Quach et al., 2017), cognitive and physical functioning (Marioni et al., 2015), frailty (Breitling et al., 2016), and the incidence of age-related diseases such as cancer (Ambatipudi et al., 2017; Durso et al., 2017), ischemic stroke (Soriano-Tárraga et al., 2016), and mortality (Fransquet, Wrigglesworth, Woods, Ernst, & Ryan, 2019; Shireby et al., 2020). Furthermore, epigenetic clocks have allowed the identification of individuals with substantial deviations from their actual chronological age, better known as ‘accelerated biological aging’ (Fransquet, Wrigglesworth, Woods, Ernst, & Ryan, 2019).
The hypothesis of an increased age acceleration in SUD is supported by the association between SUD, particularly alcohol and smoking use disorder, with leukocyte telomere shortening (Yang et al., 2013) and accelerated epigenetic aging, as demonstrated by measures of epigenetic clocks in blood (Luo et al., 2020; Rosen et al., 2018; Gao, Zhang, Breitling, & Brenner, 2016). Given that DNA methylation is highly tissue-specific, it is crucial to evaluate epigenetic clocks in different tissues in individuals with SUD (Gao, Zhang, Breitling, & Brenner, 2016). The cross-tissue study of epigenetic clocks could provide insights on tissue-specific aging alterations associated with SUD.
Moreover, the simultaneous assessment of epigenetic clocks in both brain and peripheral tissues may ultimately help to determine the potential use of peripheral estimates, such as those evaluated in whole blood, as surrogates of brain measures. In this study, we assessed and compared several epigenetic clocks that capture different aging aspects in individuals with and without SUD in both postmortem brain and blood tissues. First, we evaluated single tissue, i.e., brain and blood, separately, to identify epigenetic clocks and biological aging differences between individuals with SUD and non-psychiatric controls. Then, we evaluated the cross-tissue differences in these epigenetic aging estimates in brain and blood samples from the same individuals. Finally, we evaluated brain-blood correlations on epigenetic aging estimates to evaluate the use of blood measures as proxies for brain measures.
2. Materials and methods
2.1. Brain and blood tissue samples
Postmortem brain and peripheral blood tissue from 42 individuals with SUD and 11 controls were obtained from The University of Texas Health Science Center at Houston (UTHealth) Brain Collection in collaboration with the Harris County Institute of Forensic Science (HCIFS), with Institutional Review Board approval. Informed consent was obtained from the next of kin to include the samples in the present study. Of note, both brain and blood samples were obtained from most individuals. Upon receipt of the brain, the right hemisphere was coronally sectioned, immediately frozen, and stored at −80° C. Dissections of Brodmann area 9 (BA9), defined within the dorsolateral prefrontal cortex between the superior frontal gyrus and the cingulate sulcus, were obtained using a 4mm cortical punch, yielding approximately 100mg of tissue. We considered dorsolateral prefrontal cortex of particular interest for SUD as it is crucially involved in cognitive processes implicated in this group of disorders, i.e., decision-making, inhibition, craving, and memory (Zhai et al., 2021). Furthermore, patients with SUD have exhibited functional and structural alterations in this area in neuroimaging studies (Goldstein & Volkow, 2011; Beylergil et al., 2017; Lin et al., 2018), and postmortem studies have identified molecular alterations in this region in individuals with SUD (Zhang et al., 2014; Ribeiro et al., 2017). Postmortem interval (PMI) was calculated from the estimated time of death until tissue preservation, and cerebellar pH was calculated as previously described (Monoranu et al., 2009). Peripheral blood samples were collected into EDTA-containing tubes, and then stored as whole blood at −80 °C until further use.
For each subject, demographic information, autopsy and toxicology reports, and medical and psychiatric notes were obtained. A structured interview (psychological autopsy) was performed with each subject’s next-of-kin to obtain detailed information regarding psychiatric clinical phenotypes (evidence of depression, mania, psychosis, etc.), age at onset of drug use, types of drugs used, drinking history, and co-morbidities. After review of all the available case information by an independent panel of three trained clinicians, a consensus diagnosis of a specific SUD, or non-psychiatric control, was reached for each subject. For brain tissue analysis, the specific SUD diagnoses were alcohol use disorder (AUD, n=10), opioid use disorder (OUD, n=19), and stimulant use disorder (amphetamines and cocaine, StUD, n=13). For peripheral blood samples, the specific SUD diagnoses were AUD (n=7), OUD (n=20), and StUD (n=13). Classification of the individuals in the mentioned groups was made based on the primary diagnosis as determined in the consensus diagnosis. The primary diagnosis was associated with the underlying primary disease of the deceased. Three individuals in the OUD group and two from the stimulant use disorder group had an additional SUD or secondary diagnosis. A summary of the demographic characteristics of the subgroups analyzed in brain and blood tissue is shown in Supplementary Table S6 and Supplementary Table S7, respectively.
2.2. Statistical analysis
Differences on continuous demographic variables between the main groups, i.e., SUD and controls, were assessed by t-tests or Wilcoxon rank-sum tests, according to their distribution determined by Shapiro–Wilk test. While differences among the subgroups were assessed by analysis of variance, differences on categorical variables between the main groups and the subgroups of different SUDs were assessed by Fisher’s exact and chi-squared tests, respectively.
2.3. DNA isolation and microarray hybridization
Total DNA was isolated from brain and peripheral blood samples with the DNeasy Blood & Tissue kit (cat. 69504) (Qiagen, USA) according to the manufacturer’s instructions. Isolated DNA samples were quantified on NanoDrop (Thermo, Waltham, MA, USA) and 500 ng of DNA from each sample were bisulfite-converted using the EZ DNA Methylation™ Kit (Zymo Research, Irvine, CA, USA). Then, samples DNA methylation of the samples was evaluated using the Infinium Human Methylation EPIC BeadChip (Illumina, San Diego, CA, USA). For genotyping, DNA samples (200 ng) were hybridized into the Illumina Global Screening Array-24 (Illumina, USA) according to standard protocols.
Briefly, the processing of samples for both microarrays consisted in the amplification of DNA, its fragmentation and hybridization into the BeadChip followed by fluorescence staining. As part of the standard microarray processing, an assessment of control metrics monitoring the various experimental steps such as bisulfite conversion and fluorescence staining was performed. Also, sample-independent controls were included in the assay to monitor and verify every step in the hybridization and visualization processes. Sample-dependent behavior such as call rate and intensity, were also evaluated (Infinium Methylation Beadchip - www.illumina.com; Infinium Global Screening - www.illumina.com). Microarrays were scanned with an iScan Microarray Scanner (Illumina, San Diego, CA, USA) for microarray signal detection, according to their respective protocols.
2.4. DNA methylation age estimates
We used the New DNA Methylation Age Calculator (https://horvath.genetics.ucla.edu; Horvath, 2013) to calculate DNA methylation age estimates and other measures of age acceleration. DNA methylation beta values were normalized by the Beta MIxture Quantile dilation (BMIQ) method (Teschendorff et al., 2013) using the ChAMP package (Tian et al., 2017) as suggested by the developer. The DNA methylation age estimates evaluated in this study were DNAmAge, DNAmAgeHannum, DNAmAgeSkinBlood, DNAmPhenoAge, DNAmGrimAge, and a DNAm-based estimate of telomere length (DNAmTL) (Horvath et al., 2018; Levine et al., 2018; Lu, Seeboth, et al., 2019; Horvath, 2013; Hannum et al., 2013; Kwiatkowska et al., 2020).
The evaluated DNA methylation age estimates are based on DNA methylation levels of different CpG sites sets and had been correlated with different biological traits and clinical measures (Roshandel et al., 2020; Bergsma & Rogaeva, 2020). Thus, each one of them is expected to capture different aging aspects. Hence, it has been suggested that the evaluation of multiple DNA methylation age estimates is likely to better describe the aging complexity (Bergsma & Rogaeva, 2020). Both, the multi-tissue clock DNAmAge (based on 353 CpG sites) and the blood-based DNAmAgeHannum (71 CpG sites) stand out by their correlation with chronological age (r = 0.96 for Horvath and r = 0.91 for Hannum) and all-cause mortality (Horvath & Raj, 2018). Despite their similar correlates, DNAmAge and DNAmAgeHannum have only six CpG sites in common (Jylhävä, Pedersen & Hägg, 2017). Although DNAmAgeSkinBlood (391 CpG sites) was developed to estimate aging in human fibroblasts, keratinocytes, buccal cells, endothelial cells, lymphoblastoid cells, skin, blood, and saliva samples, it has also exhibited high age correlations in brain, liver, and bone tissues (Horvath et al., 2018).
The called ‘second-generation’ DNA methylation clocks, DNAmPhenoAge and DNAmGrimAge, include DNAm correlates of lab tests for their calculation, which has resulted in a higher accuracy in the prediction of morbidity and mortality compared to previous epigenetic clocks (Levine, 2020; McCrory et al., 2020). For example, DNAmGrimAge (1030 CpG sites) calculation is based on age, and DNAm surrogates of smoking pack-years and seven blood proteins -adrenomedullin, beta-2 microglobulin, cystatin C, growth differentiation factor 15, leptin, plasminogen activation inhibitor 1, tissue inhibitor metalloproteinase 1- (Lu et al., 2019). While, DNAmPhenoAge (513 CpG sites) is based on DNAm surrogates of albumin, alkaline phosphatase, creatinine, C-reactive protein, lymphocyte percent, mean cell volume, red cell distribution width, white cells count, and serum glucose (Levine et al., 2018).
In addition, we evaluated a DNAm-based estimate of telomere length (DNAmTL) (140 CpG sites), which is one of the most widely used aging estimators. Of note, DNAmTL exhibited a higher accuracy than southern blot-measured telomere length in the prediction of coronary heart disease (Lu et al., 2019). Aging Acceleration (AA) is the difference between DNA methylation age and chronological age and was estimated by calculating the residuals from linear regressions of each epigenetic clock on the chronological age. A positive value of epigenetic age acceleration indicates that the DNA methylation-predicted age is older than chronological age i.e., the analyzed tissue has aged faster than expected (Mendelson, 2018).
Furthermore, we calculated extrinsic epigenetic age acceleration (EEAA), which is an estimate of immune system aging, and intrinsic epigenetic age acceleration (IEAA), an estimate of cellular age acceleration independent of cell proportions (Chen et al., 2016; Horvath et al., 2016). Also, we estimated DNA methylation-based cell proportion for the different blood cell types (monocytes, CD4 + T-lymphocytes, B-lymphocytes, granulocytes, natural killer cells, and CD8 + T-lymphocytes) and neuronal cells in blood and brain samples, respectively (Horvath & Levine, 2015; Guintivano, Aryee, & Kaminsky, 2013; Houseman et al., 2012). We compared the estimates of cell proportions between the SUD and control groups using a two-side Wilcoxon rank sum test for each cellular type.
In addition, we estimated the smoking status (i.e., current, former, and never smokers) and smoking score from the analyzed individuals using the EpiSmokEr R package (Bollepalli et al., 2019). Briefly, such estimations were performed based on blood DNA methylation profiles of targeted CpG sites known to be strongly associated with smoking. Smoking status was estimated based on the DNA methylation levels of 121 CpGs, while the smoking score was calculated based on 187 smoking-associated CpGs, as described in Elliott et al. 2014.
Since the evaluated age estimates are based on DNA methylation changes, we estimated the statistical power of the analyzed sample to detect differences between the SUD and control groups with the pwr.t2n.test function of the R package pwr (Champely et al., 2020). For calculating the statistical power of detecting differences among the SUD subgroups we used the pwr.anova.test function. The power analyses for the detection of cross-tissue correlations (r=0.75) and differences were performed with the pwr.r.test and pwr.t.test (for paired samples) functions, respectively. Power analyses were calculated for a large effect size of 1.0 (chosen to facilitate interpretation) and a significance p-value of 0.05. The effect size was provided as Cohen’s d, which is the expected difference between the two group means divided by their pooled standard deviation (Cohen, 1988). Also, we performed a sensitivity analysis to test whether our results were different after excluding individuals with an additional SUD (or secondary diagnosis) by t-tests or Wilcoxon rank-sum tests, according to their distribution determined by Shapiro–Wilk tests.
2.5. Correlations between epigenetic variables and age
Distributions and homoscedasticity of the epigenetic variables were evaluated by Shapiro–Wilk and Levene’s tests, respectively. Those variables with a distribution other than normal, were log-transformed. In case of epigenetic variables with negative numbers, we log-transformed the absolute value of the variable plus one and multiplied those values that were originally negative by minus one (John & Draper, 1980). First, correlations between brain and blood epigenetic variables and chronological age were tested for the SUD and control groups separately, as well as for each subgroup per tissue, using either Pearson or Spearman tests, depending on their distribution, with the Hmisc R package. Then, we plotted the correlation results using the corrplot package (Taiyun, 2021). Finally, we compared the correlation coefficients between both groups of those significant correlations using the Fisher r-to-z transformation with the compcorr function from the DiffCorr R package (Fukushima, 2013).
2.6. Correlations among epigenetic variables
The correlations between the epigenetic aging variables in both brain and blood tissues were tested for the SUD and control groups separately. For this, we used either Pearson or Spearman tests, depending on their distribution, using the cor.test function of the R stats package (R Core Team, 2013). Again, we compared the correlation coefficients of those significant correlations between both groups using the Fisher r-to-z transformation with the compcorr function from the DiffCorr R package (Fukushima, 2013).
2.7. Single-tissue measures of epigenetic aging variables among groups
Differences on epigenetic aging variables in blood and brain tissue were compared, separately, between the main groups, i.e., SUD and controls, by linear regression models using chronological age, sex, blood cell count estimates (for blood), neuronal proportion, PMI and tissue pH (for brain), and the top three ancestry principal components as covariates, as they accounted for the highest proportion of the ancestry variability, with 34.84%, 26.69%, and 13.43%, respectively. Furthermore, we performed an additional analysis including the estimated smoking score as covariate. We performed a quality control of the genetic data prior to the estimation of ancestry principal components. This quality control was performed using PLINK 2.0 (Purcell et al., 2007) and consisted in the exclusion of SNPs of with a variant calling >95% and a minor allele frequency (MAF) greater than 5%. Those individuals with >10% of missing genotypes and related individuals were excluded (PI_HAT ≥0.5). All individuals passed the latter quality control filter. Finally, in order to include only linkage disequilibrium (LD)-independent SNPs in the ancestry principal components analysis, we performed a LD pruning considering a window size of 50 kb, a variant count to shift the window at the end of each step of 5, and a pair pairwise correlation threshold of 0.2. Principal ancestry component analysis was performed with smartPCA from the EIGENSOFT package (Price et al., 2006; Patterson, Price, & Reich, 2006), using the genome-wide genotype information from each subject. Ancestry-informative markers were obtained from the Human Genome Diversity Project (HGDP) (Cavalli-Sforza, 2005).
We then evaluated differences on epigenetic aging variables among the SUD subgroups using linear regression models with the same covariates as in the main groups’ comparisons. Beta coefficients, which correspond to the degree of change in the epigenetic age variable for every 1-unit change in the predictor variables, were calculated with the lm.beta package (Behrendt, 2014). P-values were corrected for multiple comparisons using the Benjamini-Hochberg method for controlling the false discovery rate (FDR) – which is the expected proportion of positive tests that are false, i.e., incorrect rejections of the null hypothesis (Benjamini & Hochberg, 1995). Briefly, this method ranks the individual p-values corresponding to each tested hypothesis in ascending order. Then, each FDR-corrected p-value is recalculated considering its ranking place, the number of total tests performed (or hypothesis testes) and the selected FDR (Haynes, 2013). For this study, the FDR value was 0.05 and we considered as nominally significant a FDR value of 0.1 or lower.
2.8. Comparisons of epigenetic variables across tissues
For the cross-tissue analysis we included data from 39 individuals with SUD and 10 controls, selected based on the availability of matching samples from both brain and blood tissues. No significant differences were found regarding age, sex, PMI, pH, and ethnicity proportions between individuals with SUD and controls included in the cross-tissue analyses using a linear model recoding the original groups (SUD and control) to a new variable including the examined tissue, i.e., SUD brain, SUD blood, Control brain, and Control blood. Sex, chronological age, and the top three ancestry principal components were used as covariates. Furthermore, we performed an additional analysis including the estimated smoking score as covariate. Interaction terms among between the variables included in the model were tested and discarded if their associated p-value was lower than 0.05. A summary of demographic characteristics from the individuals included in the cross-tissue analyses in shown in Table 2.
Table 2.
Sample characteristics of individuals included in the cross-tissue (brain and blood) analysis.
| Variable | SUD | Control | p-value |
|---|---|---|---|
| N | 39 | 10 | -- |
| Age, years | 45 ± 12.49 | 48.1 ± 12.87 | 0.4716 |
| Sex (M: F) | 27:12 | 9:1 | 0.1844 |
| PMI, hours | 26.28 ± 8.54 | 26.99 ± 10.44 | 0.9708 |
| pH | 6.62 ± 0.27 | 6.48 ± 0.27 | 0.1884 |
| Ethnicity† | 26/2/11 | 6/2/2 | 0.2993 |
| Psychiatric diagnosis‡ | 0/7/13/19 | 10/0/0/0 | |
| Predicted smoking status§ | 28/4/7 | 5/3/2 | 0.243 |
| Smoking score | 3.25 ± 8.95 | 5.74 ±6.28 | 0.3203 |
Continuous data are presented as mean ± standard deviation. Abbreviations: Substance use disorder (SUD); Male (M), female (F); Post-mortem interval (PMI).
Caucasian/Hispanic/African American
No comorbidities/Alcohol use disorder/ Stimulants use disorder (amphetamines and cocaine)/ Opioid use disorder
Current/Former/Never smoker
We evaluated cross-tissue correlations between blood and brain epigenetic variables within each group, i.e., SUD and control groups, with either Pearson or Spearman tests, depending on their distribution, using the cor.test function of the R stats package (R Core Team, 2013). Then, we compared the correlation coefficients between both groups of those significant correlations using the Fisher r-to-z transformation with the compcorr function from the DiffCorr R package (Fukushima, 2013).
3. Results
Brain BA9 samples from 53 individuals and blood samples from 51 individuals were analyzed. BA9 was selected based on the reported disruption of cognitive functions and executive processes regulated by the prefrontal cortex such as attention, inhibitory control, working memory, and cognitive flexibility, in individuals with SUD (Sullivan & Pfefferbaum, 2019; Zahr, Pfefferbaum, & Sullivan, 2017). No significant differences were found regarding chronological age, sex, PMI, tissue pH, and ethnicity proportions between SUD and control groups in either brain or blood. A summary of the demographic characteristics from the sample analyzed in each tissue is shown in Table 1. Sample characteristics of the subgroups is shown in Supplementary Table S2 and S3. Detailed individual-level information regarding cause of death and toxicology at time of death is shown in Supplementary Table 1 (Supplementary File 1).
Table 1.
Sample characteristics of individuals included in single tissue analyses
| Tissue | Brain | Blood | ||||
|---|---|---|---|---|---|---|
| Variable | SUD | Control | p-value | SUD | Control | p-value |
| N | 42 | 11 | -- | 40 | 11 | -- |
| Age, years | 45.21 ± 13.05 | 50 ± 13.74 | 0.2359 | 45.12 ±13.34 | 48.90 ± 12.5 | 0.3358 |
| Sex (M: F) | 29:13 | 9:2 | 0.4821 | 28:12 | 10:1 | 0.3084 |
| PMI, hours | 26.86 ± 9.11 | 26.79 ± 9.93 | 0.9225 | -- | -- | |
| pH | 6.62 ± 0.27 | 6.47 ± 0.26 | 0.1357 | -- | -- | |
| Ethnicity† | 28/2/12 | 7/2/2 | 0.294 | 0/2/38 | 0/2/9 | 0.4197 |
| Psychiatric diagnosis‡ | 0/10/13/19 | 11 /0 /0 /0 | 0/7/13/20 | 11/0/0/0 | ||
| Predicted smoking status§ | 28/4/7 | 5/3/2 | 0.243 | 29/4/7 | 5/4/2 | 0.09202 |
| Smoking score | 3.25 ± 8.95 | 5.74 ±6.28 | 0.3203 | 2.97 ± 8.54 | 5.62 ± 6.25 | 0.242 |
Continuous data are presented as mean ± standard deviation. Abbreviations: Substance use disorder (SUD); Male (M), female (F); Post-mortem interval (PMI).
Caucasian/Hispanic/African American
No comorbidities/Alcohol use disorder/ Stimulants use disorder (amphetamines and cocaine)/ Opioid use disorder
Current/Former/Never smoker. Note: In the brain group, we report the estimates only from those individuals with blood DNA methylation data from which smoking status and smoking score were available.
Statistical power of the analyzed sample to detect differences between the SUD and control groups was 0.825 and 0.821 for brain and blood tissues, respectively. For subgroup analyses the calculated power was 0.99 for both brain and blood tissues. For cross-tissue differences, power was estimated as 0.99, and for cross-tissue correlations we estimated a power of 0.99 for detecting a correlation of 0.75. Power estimations were calculated for an effect size of 1.0 provided as Cohen’s d, and a significance p-value of 0.05.
No significant differences were found between the SUD and control groups regarding the proportion of CD4 + T-lymphocytes (p-value = 0.2568), B-lymphocytes (p-value = 0.5733), granulocytes (p-value = 0.7923), natural killer cells (p-value = 0.8011), CD8 + T-lymphocytes (p-value = 0.936), and monocytes (p-value = 0.1722) in blood tissue samples. Similarly, we did not find significant differences in the estimation of neuronal cells proportion (p-value = 0.1336) between the SUD and control groups. Descriptive statistics (median and interquartile range) of the estimated cell proportions are shown in the Supplementary Table S1. Finally, we did not identify significant differences in the proportion of individuals with the predicted smoking status (p-value for blood tissue = 0.09; p-value for brain tissue = 0.243) nor the smoking score of the included individuals (p-value for blood tissue = 0.242; p-value for brain tissue = 0.320).
3.1. Correlations of epigenetic variables with chronological age
Results of the correlations between epigenetic clocks and chronological age correlations in brain and blood are shown in Figure 1. All evaluated epigenetic variables were normally distributed according to the Shapiro-Wilk’s test, except for DNAmAge-based epigenetic age acceleration (AAR) (Shapiro-Wilk’s P = 0.0005) and PhenoAge-based epigenetic age acceleration (AAPheno) (Shapiro-Wilk’s P = 0.002) in blood. After log-transformation, both variables fitted to a normal distribution (AAR- Shapiro-Wilk’s P = 0.5881; AAPheno - Shapiro-Wilk’s P = 0.4231).
Figure 1.
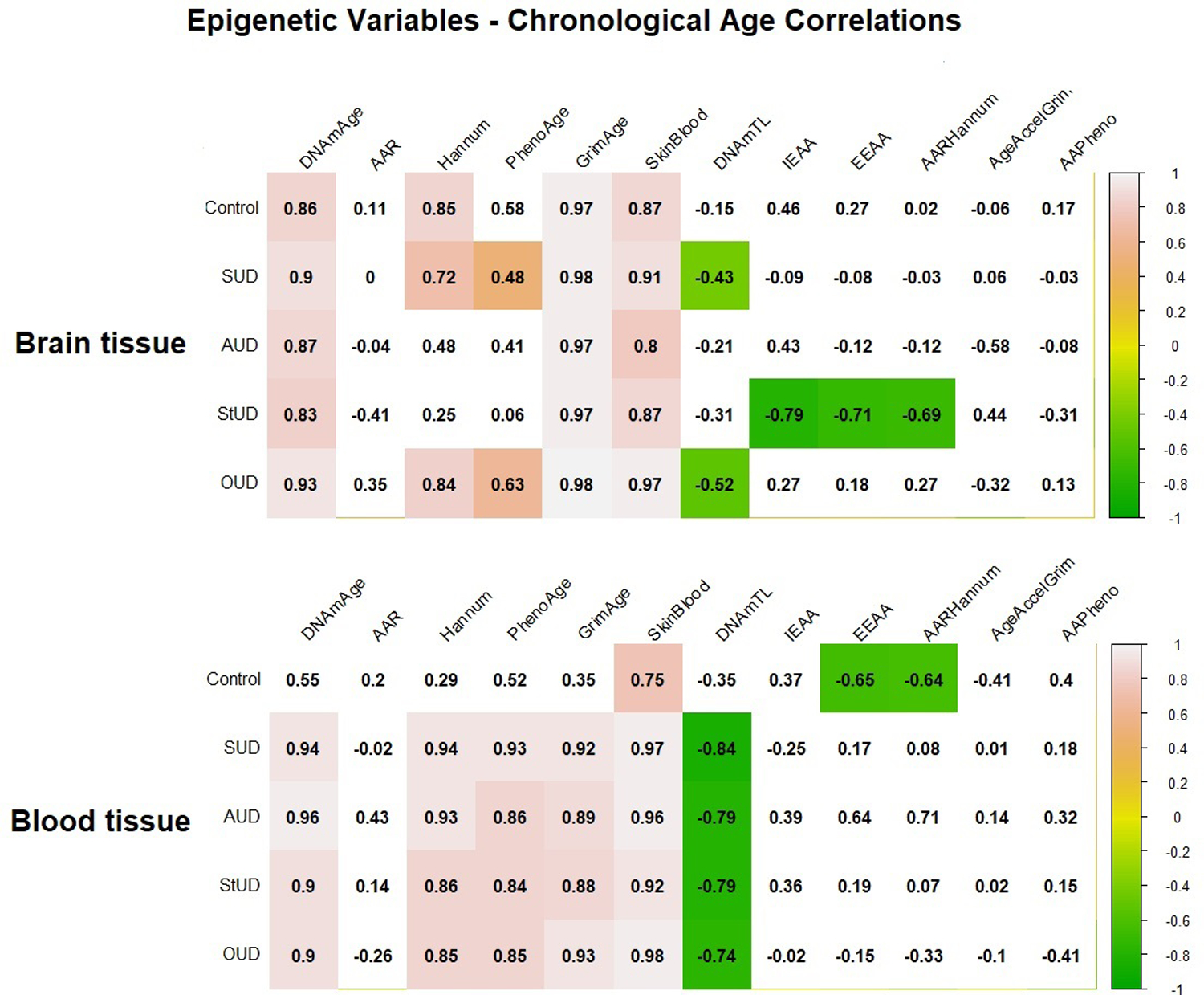
Matrix plot of correlations between epigenetic variables and chronological age in brain and blood tissues. Cells with a significant correlation (p-value <0.05) are colored according to their correlation coefficient for epigenetic variable-chronological age correlation, with green and pink indicating a negative and a positive correlation, respectively. The correlation coefficient is indicated in the number within each cell. Abbreviations: Substance use disorder (SUD); Alcohol use disorder (AUD); Stimulant use disorder (StUD); Opioid use disorder (OUD); DNAmAge-based epigenetic age acceleration (AAR); Extrinsic epigenetic age acceleration (EEAA); Intrinsic epigenetic age acceleration (IEAA); Hannum-based epigenetic age acceleration (AAHannum); GrimAge-based epigenetic age acceleration (AAGrim); PhenoAge-based epigenetic age acceleration (AAPheno).
In brain tissue, DNAmGrimAge had the highest correlation with chronological age, with a correlation coefficient ranging from 0.97 in the control, AUD and StUD groups, to 0.98 in the SUD and OUD groups. We also observed substance-specific correlations in our sample; specifically, a negative correlation between IEAA, EEAA, and AAHannum and chronological age in the StUD subgroup, as well as between chronological age and DNAmTL in the OUD group.
In blood tissue, DNAmAgeSkinBlood exhibited the highest correlation with chronological age with a correlation coefficient ranging from 0.75 in the control group to 0.98 in the OUD subgroup. We found a significant negative correlation between blood DNAmTL and chronological age in the SUD group and in all SUD subgroups, and there was a significant difference in the correlation coefficients between the control and the SUD groups (z=2.0716, p-value = 0.03). However, the differences were not significant when comparing the control with the individual SUD subgroups, i.e., AUD (z=1.12, p-value = 0.26), StUD (z=1.55, p-value = 0.11) and OUD (z=1.18, p-value = 0.23).
3.2. Correlations among epigenetic variables
We found positive correlations between DNAmAge, DNAmAgeHannum, DNAmPhenoAge, DNAmGrimAge, and DNAmSkinBlood in the blood samples from both control and SUD individuals. We found positive correlations between these same age measures in brain samples of control and SUD subjects, except for DNAmPhenoAge, which was only found in brains of the SUD group.
Except for DNAmAgeHannum, we found negative correlations between DNAmTL and all evaluated epigenetic clocks in both brain and blood of the SUD group, as well as in blood from the control group. These negative correlations between DNAmTL and the epigenetic clocks were not significant in the brain samples of the control group. The correlations among the epigenetic variables in the SUD and control groups are plotted in Figure 2.
Figure 2.
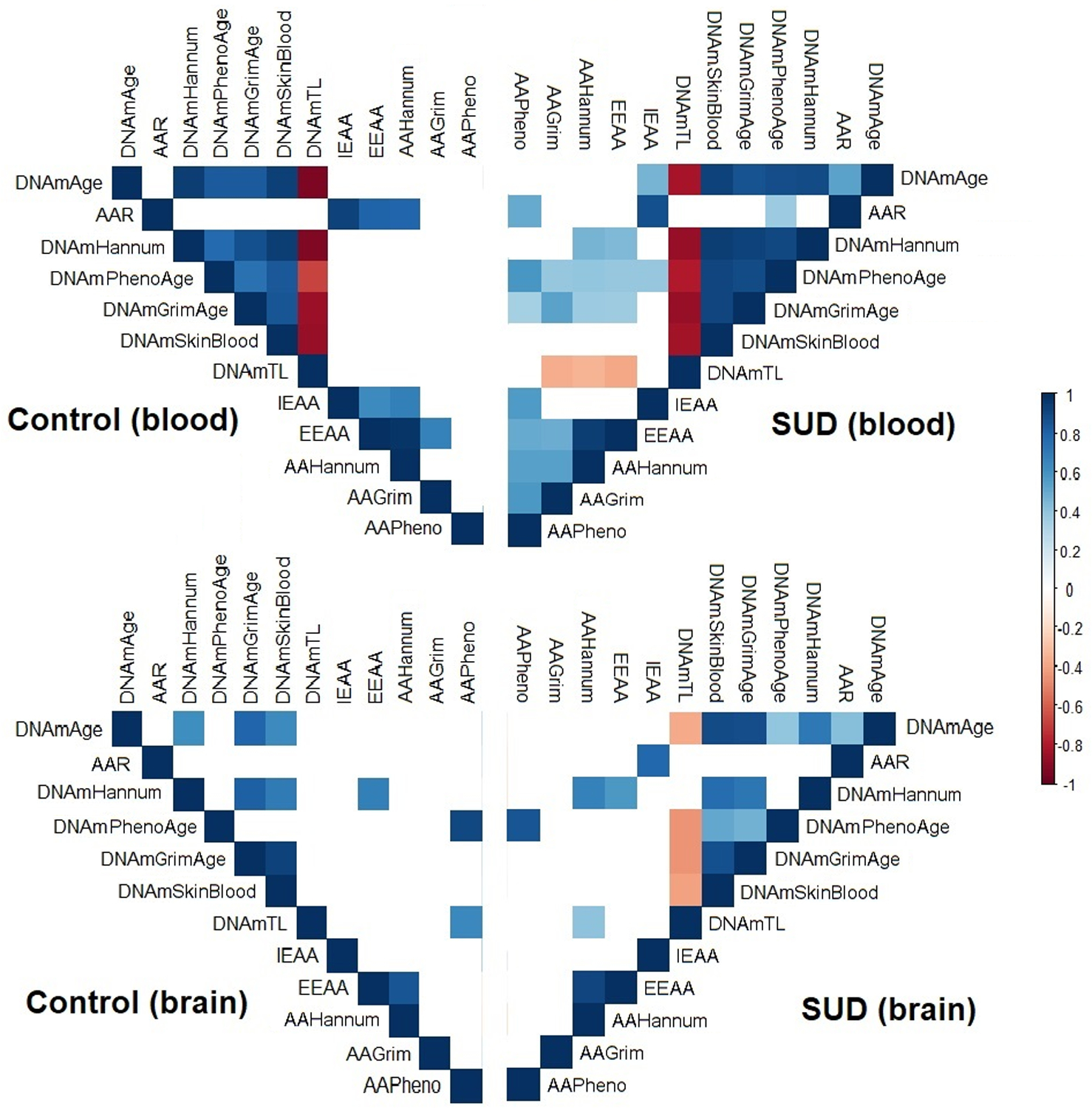
Matrix plot of the correlations between epigenetic variables. The colors represent the degree of pairwise correlation regarding Pearson’s rank correlation coefficient (rho). Blank squares indicate the absence of correlation (p-value > 0.05). Abbreviations: Substance use disorder (SUD); DNAmAge-based epigenetic age acceleration (AAR); Extrinsic epigenetic age acceleration (EEAA); Intrinsic epigenetic age acceleration (IEAA); Hannum-based epigenetic age acceleration (AAHannum); GrimAge-based epigenetic age acceleration (AAGrim); PhenoAge-based epigenetic age acceleration (AAPheno).
In the SUD subgroup analysis, we observed a similar pattern of correlations in the blood samples as in the whole SUD group, with positive correlations between DNAmAge, DNAmAgeHannum, DNAmPhenoAge, DNAmGrimAge, and DNAmSkinBlood and negative correlations between DNAmTL and all evaluated epigenetic clocks. In contrast, in the brain samples there was a very different pattern of correlation among the epigenetic variables. Most noteworthy, there was an absence of significant correlations between the epigenetic variables compared to those found in the blood tissue samples. The correlations between the epigenetic variables in the subgroups are plotted in Figure 3.
Figure 3.
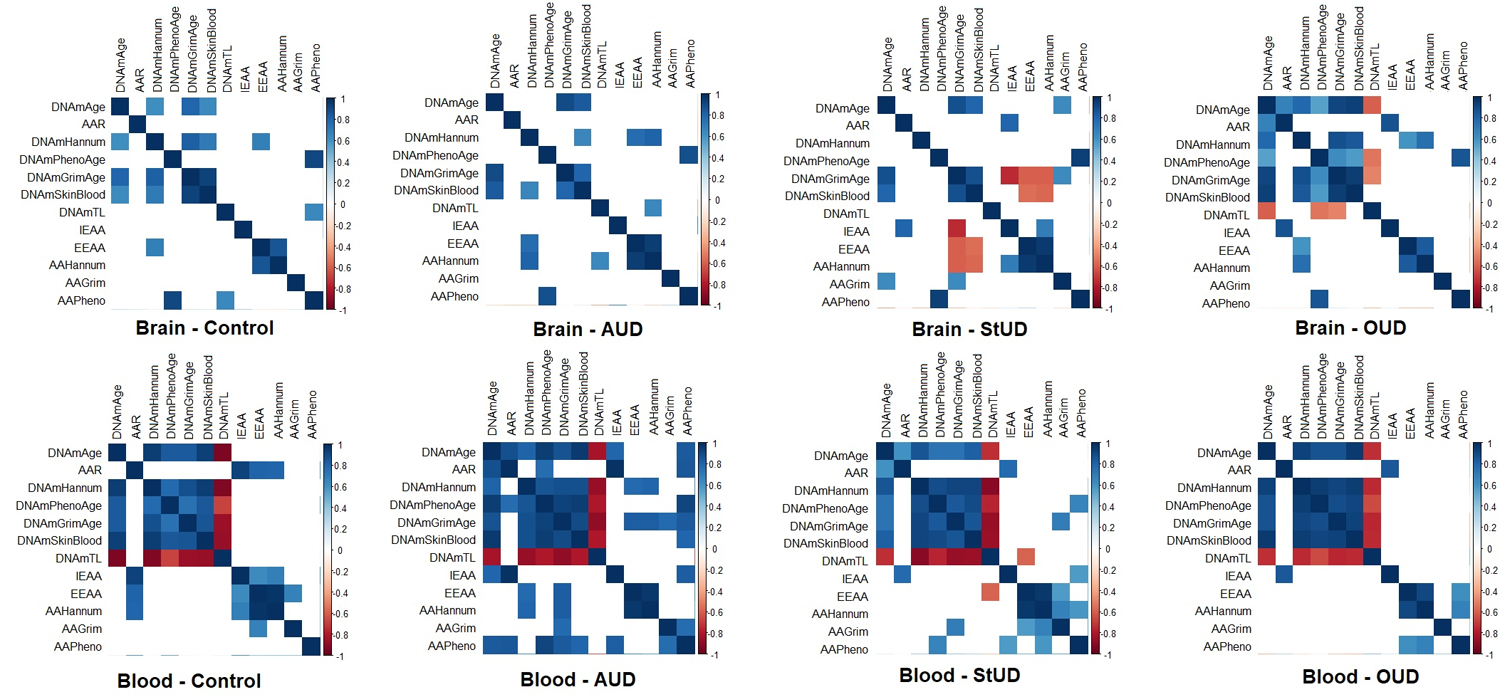
Matrix plot of the correlations between epigenetic variables per subgroups. The colors represent the degree of pairwise correlation regarding Pearson’s rank correlation coefficient (rho). Blank squares indicate the absence of correlation (p-value > 0.05). Abbreviations: Alcohol use disorder (AUD); Stimulants use disorder (StUD); Opioid use disorder (OUD); DNAmAge-based epigenetic age acceleration (AAR); Extrinsic epigenetic age acceleration (EEAA); Intrinsic epigenetic age acceleration (IEAA); Hannum-based epigenetic age acceleration (AAHannum); GrimAge-based epigenetic age acceleration (AAAGrim); PhenoAge-based epigenetic age acceleration (AAPheno).
3.3. Single-tissue measures of epigenetic aging variables
In brain samples, we found no significant differences in epigenetic aging variables between SUD subjects and controls after controlling for chronological age, PMI, sex, age, smoking score, top three ancestry principal components, pH, and neuronal proportion. Regarding blood tissue, we found a higher DNAmPhenoAge (FDR corrected p-value= 0.0104) and a nominally significant lower DNAmTL (FDR corrected p-value= 0.0603) in individuals with SUD compared to controls after controlling for chronological age, sex, smoking score, the top three ancestry principal components and blood cell count estimates (Figure 4). The mean and standard deviation of the evaluated epigenetic variables per group, as well as the beta and FDR-corrected p-values associated to each comparison as well as results excluding smoking score as covariate are shown in Supplementary Tables S4 and S5. We did not detect significant differences in the mean of the evaluated epigenetic aging estimates when excluding individuals with an additional SUD (or secondary diagnosis), as shown in Supplementary Table S6.
Figure 4.
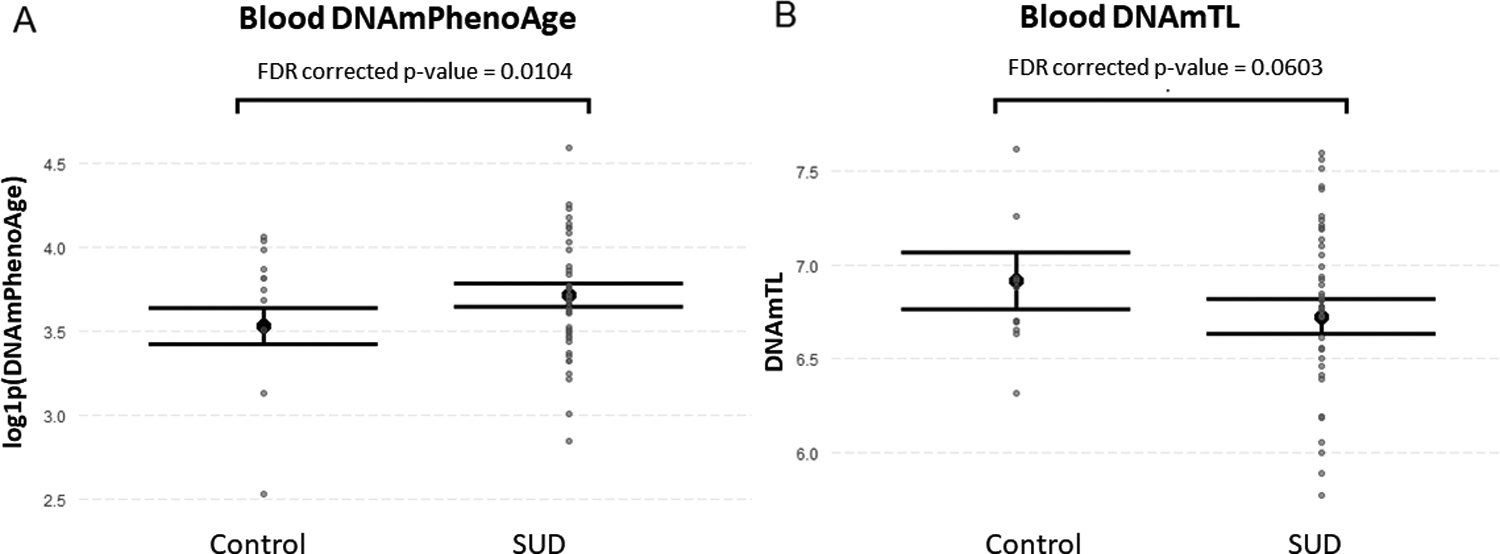
Epigenetic variables with significant differences between SUD and control groups in blood tissue. A) Blood DNAmPhenoAge was higher in individuals with SUD compared to the controls (beta=0.191, FDR corrected p-value= 0.0104) after controlling for chronological age, sex, smoking score, three top ancestry principal components and blood cell count estimates. B) Blood DNAmTL was nominally lower in individuals with SUD compared to the controls (beta=−0.149, FDR corrected p-value= 0.0603). Data are presented as means with their standard error. Abbreviations: Alcohol use disorder (AUD); Stimulant use disorder (StUD); Opioid use disorder (OUD).
When comparing epigenetic variables in BA9 among SUD subgroups, we found a lower brain DNAmTL in AUD compared to Controls and StUD (FDR corrected p-value = 0.087) (Figure 5). Nominal differences on other epigenetic variables (DNAmAgeHannum, DNAmPhenoAge, DNAmTL, EEAA, and AAHannum) in blood tissue were identified among the subgroups before multiple-comparison correction; however, these were not significant after FDR correction. These results are available in Supplementary Table S7.
Figure 5.
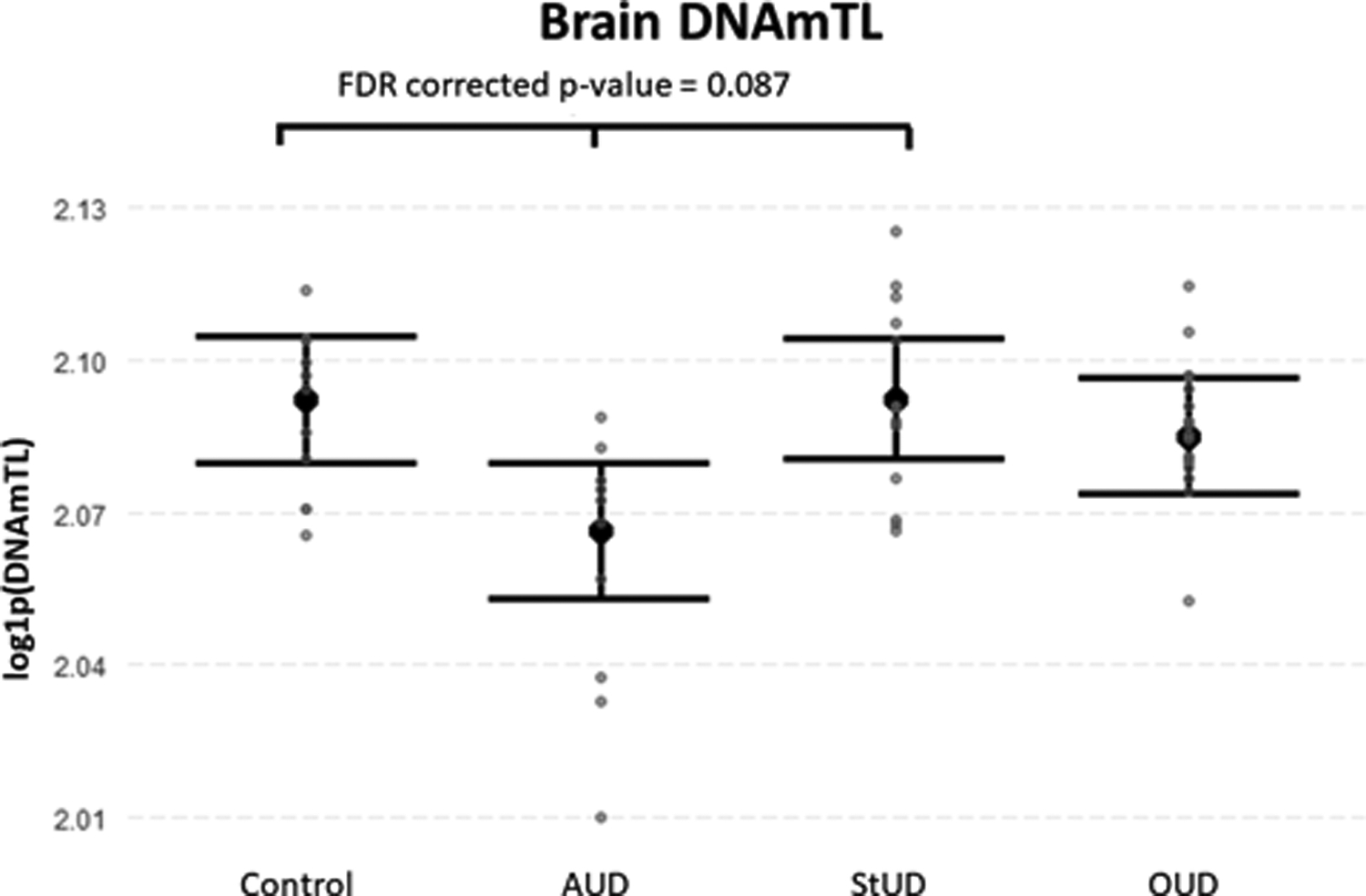
DNAm-based estimate of telomere length (DNAmTL) among SUD subgroups in brain tissue. DNAmTL was higher in the Controls and StUD subgroups compared to the AUD subgroup (FDR corrected p-value = 0.087) after controlling for chronological age, sex, smoking score, three top ancestry principal components and blood cell count estimates. Data are presented as means with their standard error. Abbreviations: Alcohol use disorder (AUD); Stimulants use disorder (StUD); Opioid use disorder (OUD).
3.4. Comparisons of epigenetic variables across tissues
We found significant differences between brain and blood measures for DNAmAgeHannum, DNAmPhenoAge, and DNAmGrimAge and AAGrim within both control and SUD groups after controlling by sex, chronological age, smoking score, and the top three ancestry principal components (Figure 6). Also, we found a significantly lower blood DNAmTL and a higher blood DNAmAge, compared to their respective brain values, in the SUD group (Figure 6A, J). The complete results from these comparisons as well as results excluding smoking score as covariate are shown in Supplementary Tables S8 and S9.
Figure 6.
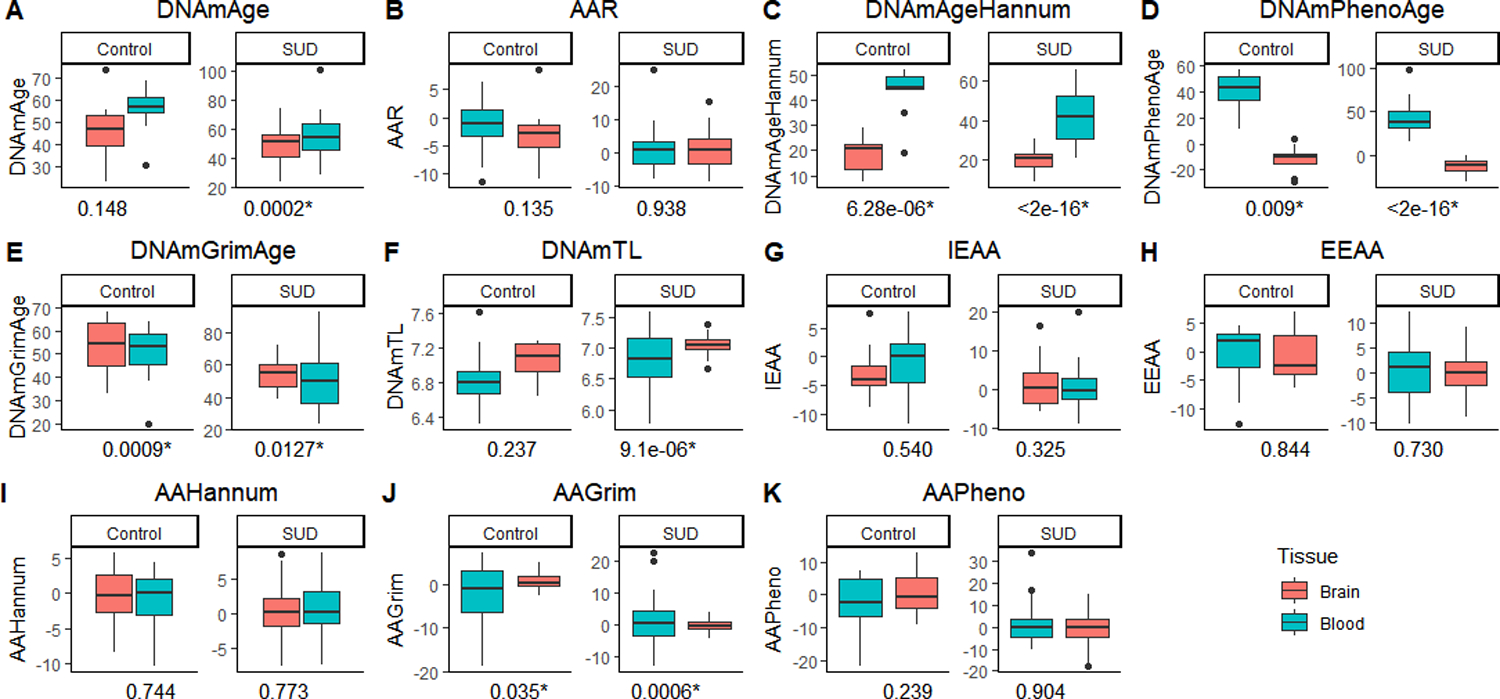
Boxplots of epigenetic variables in individuals with SUD and controls. The horizontal line in each box indicates the median, boxes indicate 25–75% interquartile ranges, whiskers indicate 1.5 (IQR) boundaries. The p-value associated to each comparison is indicated under each boxplot. Asterisk indicates a significant difference between the groups (FDR corrected < .01). The y-axis indicates the scale of each epigenetic variable. Abbreviations: DNAmAge-based epigenetic age acceleration (AAR); Extrinsic epigenetic age acceleration (EEAA); Intrinsic epigenetic age acceleration (IEAA); Hannum-based epigenetic age acceleration (AAHannum); GrimAge-based epigenetic age acceleration (AAGrim); PhenoAge-based epigenetic age acceleration (AAPheno).
3.5. Cross-tissue correlations (main groups)
We found a positive correlation between brain and blood DNAmAge, DNAmAgeHannum, DNAmPhenoAge, DNAmGrimAge and DNAmAgeSkinBlood in the control group. In the SUD group, we did not find any significant correlation between brain and blood epigenetic variables. We found a significant difference between both groups regarding correlation coefficients for DNAmAge (z = 3.8578, p-value = 0.0001). Furthermore, correlations between brain-blood epigenetic variables varied among the SUD subgroups in their correlation coefficients and statistical significance, with OUD showing the highest number of positive correlations, and StUD the least. Complete results from cross-tissue correlations are shown in Figure 7.
Figure 7.
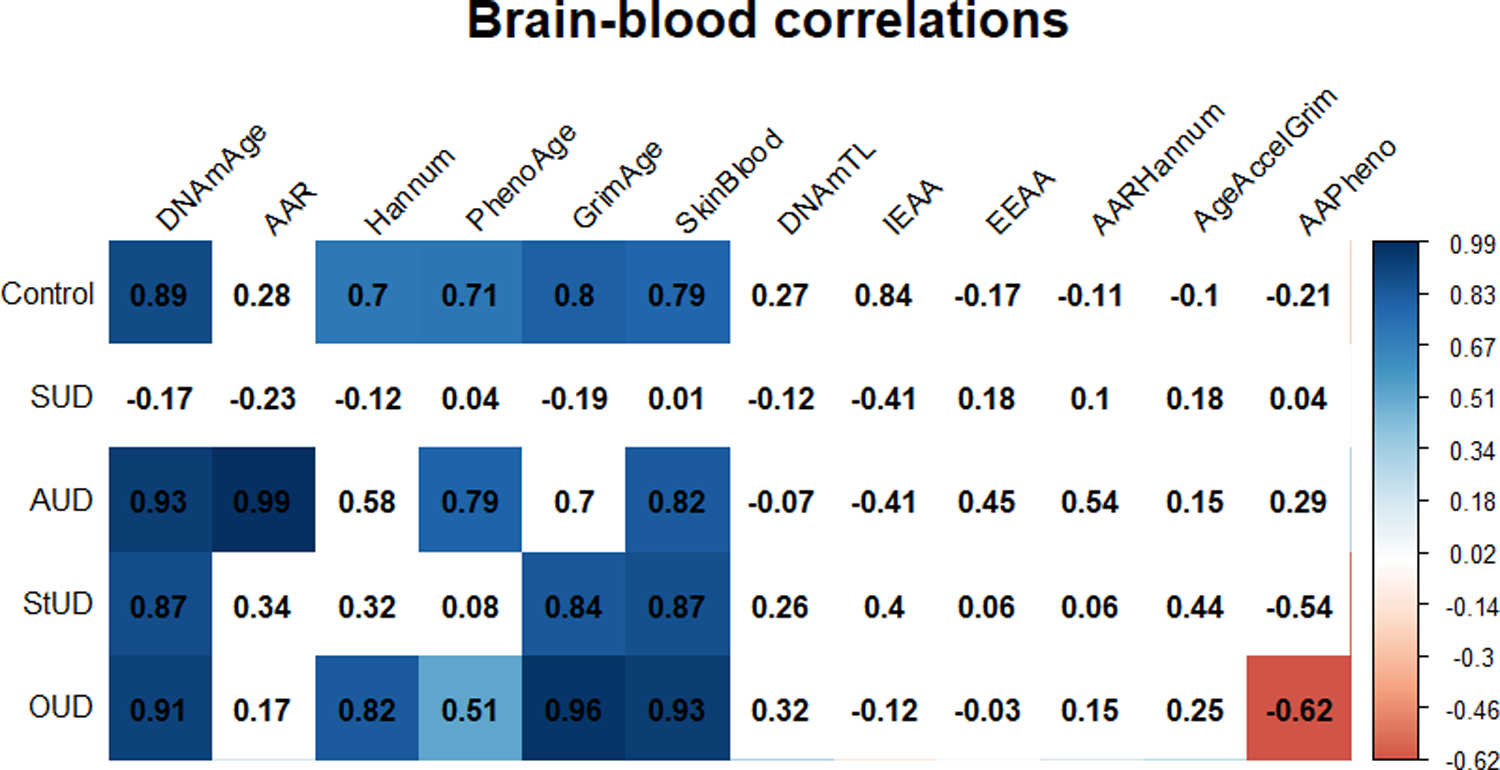
Matrix plot of correlations between brain and blood epigenetic variables. Cells with a significant correlation (p-value <0.05) are colored according to their correlation coefficient for epigenetic variable-chronological age correlation, with red and blue indicating a negative and a positive correlation, respectively. The correlation coefficient is indicated in the number within each cell. Abbreviations: Substance use disorder (SUD); Alcohol use disorder (AUD); Stimulants use disorder (StUD); Opioid use disorder (OUD); DNAmAge-based epigenetic age acceleration (AAR); Extrinsic epigenetic age acceleration (EEAA); Intrinsic epigenetic age acceleration (IEAA); Hannum-based epigenetic age acceleration (AAHannum); GrimAge-based epigenetic age acceleration (AAGrim); PhenoAge-based epigenetic age acceleration (AAPheno).
4. Discussion
Comparisons of epigenetic clocks across tissues obtained from the same subjects may provide insight on the neural substrates of accelerated aging and the feasibility of using peripheral measures to estimate aging effects in the brain. Previous studies have evaluated associations between brain and blood epigenetic clocks in patients with schizophrenia (McKinney, Lin, Ding, Lewis, & Sweet, 2018; Stevenson et al., 2020). To the best of our knowledge, this is the first cross-tissue study investigating the relationship between brain and blood epigenetic age measures in postmortem tissues from individuals with SUD.
We evaluated potential differences and correlations on epigenetic aging using several different epigenetic clocks. The available epigenetic clocks have been developed using different methods and are composed of different measures (Yang et al., 2020). For example, DNAmGrimAge consists of the DNA methylation surrogates of seven plasma proteins, while DNAmHannum is defined by the DNA methylation values of 71 CpG sites. Furthermore, they have been associated with different diseases and health outcomes, as mentioned previously in the Methods section. Therefore, they are likely to capture different aspects of aging (Bergsma & Rogaeva, 2020; Horvath & Raj, 2018). This study permitted us to evaluate potential variations in these different aging mechanisms across tissues, and their association with SUD.
Even though DNAmGrimAge was trained with blood data (Lu, Quach, et al., 2019), this epigenetic clock exhibited the highest correlation with chronological age in brain tissue. This is consistent with the fact that the estimation of DNAmGrimAge uses age, along with the DNA methylation estimates of smoking pack-years and seven serum proteins, as parameters in its calculation (Gutman et al., 2020). DNAmGrimAge has been shown to outperform other epigenetic clocks in their association with all-cause mortality and incidence of age-related diseases, i.e., chronic obstructive pulmonary disease, type 2 diabetes, and ischemic heart disease (Hillary et al., 2020; Protsenko et al., 2020). Also, AgeAccelGrim was found to be positively correlated with PTSD symptom severity scores, outperforming other epigenetic clocks (Yang et al., 2020). It has been hypothesized that DNAmGrimAge could be more sensitive in studies of accelerated cellular aging in psychiatric conditions compared to other epigenetic measures (Protsenko et al., 2020). In blood, DNAmAgeSkinBlood exhibited the highest correlation with chronological age. This is in line with this measure being considered a highly sensitive age estimator and previous reports of high correlations between this epigenetic clock in peripheral blood and chronological age (Horvath et al., 2018).
The observed negative correlation between IEAA, EEAA, and AAHannum and chronological age in the StUD subgroup was remarkable. Although these epigenetic measures may not necessarily correlate with chronological age (Chen et al., 2016), their negative correlation in our study suggests that young individuals with StUD may have a higher vulnerability to the effects of stimulants on epigenetic aging. In regards to group comparisons, we identified a significantly higher blood DNAmPhenoAge in subjects with SUD compared to controls. Luo et al., (Luo et al., 2020) reported a higher epigenetic age acceleration derived from this epigenetic clock in the blood of patients with AUD compared to controls. Our SUD subgroup analyses indicated a trend for higher blood DNAmPhenoAge in patients with AUD, StUD and OUD compared to controls. Although not significant for specific SUDs, likely due to the small sample size of our study, our findings provide additional evidence for the presence of possible alterations in blood DNAmPhenoAge in individuals with SUD.
Similar to our results, Montalvo-Ortiz et al., did not detect significant differences in blood AAR between European-American women with and without OUD (Montalvo-Ortiz et al., 2019). However, we did not detect differences on brain AAR and AAPheno between OUD and other subgroups as those reported in previous studies in brain tissue (Kozlenkov et al., 2017; Shu et al., 2021). Such discrepancies could be due to the small number of individuals with OUD included in our sample (20 for brain tissue) compared to that included in the Kozlenkov et al. and Shu et al. studies with 37 and 72 individuals with OUD, respectively. Of note, the mentioned OUD studies did not evaluate differences on the epigenetic clocks where we detected differences among the subgroups and evaluated only epigenetic acceleration measures. Thus, we are not able to compare our results regarding other epigenetic aging measures to them. Future studies examining both epigenetic clocks and its associated age acceleration in larger samples will provide insight into the epigenetic aging alterations related with OUD.
The DNAmPhenoAge clock is based on chronological age and nine clinical measures including albumin, alkaline phosphatase, creatinine, C-reactive protein, lymphocyte percent, mean cell volume, red cell distribution width, white cells count, and serum glucose (Levine et al., 2018). Thus, the higher blood DNAmPhenoAge in individuals with SUD could reflect alterations of clinical measures secondary to liver damage (albumin and alkaline phosphatase), metabolic (glucose serum), immune (lymphocyte percent, mean cell volume, red cell distribution width and white cells count), and inflammatory disturbances (C-reactive protein). In this regard, there is evidence of alterations in blood cells counts (Soder et al., 2020; Haghpanah, Afarinesh, & Divsalar, 2010), C-reactive protein (Costello, Copeland, Shanahan, Worthman, & Angold, 2013), mean corpuscular volume (Ng et al., 2019), red cell distribution width (Tajuddin, Nalls, Zonderman, & Evans, 2017), and fasting blood glucose in patients with SUD (Ojo, Ojo, Adebowale, & Wang, 2018). Liver damage has been extensively demonstrated in patients with AUD, and its presence has been recently associated with an accelerated epigenetic aging (Luo et al., 2020; Osna, Donohue, & Kharbanda, 2017). However, hepatic alterations have also been associated with other SUDs (Pateria, de Boer, & MacQuillan, 2013), which could be a consequence of the drug abuse, but also of the high incidence of hepatitis C virus infection in patients with SUD, particularly in those individuals using parenteral drugs, i.e., opioids (Zeremski & Martinez, 2017, Verna, Schluger, & Brown, 2019). Our findings of increased DNAmPhenoAge in blood but not in brain could reflect the importance of this measure in reflecting consequences of drug abuse related to peripheral tissues. In this regard, DNAmPhenoAge may capture aspects of immunosenescence in blood (Levine et al., 2018). The DNAmPhenoAge clock has also been associated with age-related diseases and all-cause mortality, which are known to occur at a higher incidence in individuals with SUD compared with individuals without this disorder (Hjemsæter et al., 2019). Future studies with larger sample sizes are needed to further investigate the factors implicated in the DNAmPhenoAge variations in patients with SUD.
We found a nominally lower blood DNAmTL in individuals with SUD, as well as a negative correlation between blood DNAmTL and chronological age in the SUD group, and within all the SUD subgroups. These findings are in line with previous reports of telomere shortening in patients with SUD (for review see (Monroy-Jaramillo, Dyukova, & Walss-Bass, 2018, Navarro-Mateu et al., 2019), which is hypothesized to be secondary to a drug-induced increase in oxidative stress (Yang et al., 2013). Importantly, cross-tissue comparisons indicated a significant difference between brain and blood DNAmTL measures in the SUD group, whereas in the control group this difference was not significant. The subgroup analysis indicated a nominally lower brain DNAmTL in individuals with AUD compared to StUD, OUD, and controls. Further, we found a negative correlation between DNAmTL and epigenetic clocks (DNAmAge, DNAmAgeHannum, DNAmPhenoAge, DNAmGrimAge, and DNAmSkinBlood) in both brain and blood of the SUD group, including the SUD subgroups, while in the control group these correlations were found only in the blood tissue. These results suggest possible substance- and tissue-specific effects on accelerated aging, which could be secondary to vulnerability to DNA methylation changes captured by the DNAmTL estimator. DNAmTL has been found to be fairly correlated with quantitative polymerase chain reaction and Southern blotting TL measures, with a correlation coefficient of 0.39 and 0.40, respectively (Lu, Seeboth, et al., 2019). Therefore, our findings are consistent with previous reports of negative correlations between leucocyte telomere length and epigenetic age measures (Bergsma & Rogaeva, 2020; Vetter et al., 2019; Vyas et al., 2019) and are in line with increased accelerated aging causing a decrease in telomere length in subjects with SUDs.
The presence of positive and significant brain-blood correlations in controls indicate that peripheral measures of the epigenetic clocks might provide a good estimate of brain aging in subjects with no history of substance use disorders. However, for subjects with SUD, we found that blood-brain correlations depend on the specific type of drug used, which might suggest a tissue-specific effect of the particular substance being used. Thus, correlates of epigenetic variables in peripheral tissue and their use as surrogates for brain measures in specific SUD should be further studied with larger cohorts.
Reduced DNAmTL in individuals with SUD was one of the most relevant findings of our study, as it was found in both brain and blood tissue. A mechanism potentially involved in DNAmTL reduction is telomerase activity. Telomerase activity is crucial for telomere restoration and was found to be lower in individuals with heroin use disorder compared with healthy controls (Cheng et al., 2013). Furthermore, telomerase activity was correlated with atrophy and functional changes (i.e., connectivity with other brain regions) in the dorsolateral prefrontal cortex, which is the brain area analyzed in this study (Cheng et al., 2013; Levandowski et al., 2016). Other possible contributors to reduced DNAmTL are oxidative stress and subsequent activation of an inflammatory response (Bachi, Sierra, Volkow, Goldstein, & Alia-Klein, 2017; Monroy-Jaramillo, Dyukova, & Walss-Bass, 2018).
Drug-induced oxidative stress has been hypothesized to be involved in the SUD aging acceleration. Oxidative stress can enhance an inflammatory response in brain and peripheral organs (heart, kidney and liver) (Bachi, Sierra, Volkow, Goldstein, & Alia-Klein, 2017)and also leads to modifications in macromolecules such as RNA, DNA, proteins and lipids, ultimately leading to cell death and tissue injury (Bachi, Sierra, Volkow, Goldstein, & Alia-Klein, 2017; Ames & Shigenaga 1992; Pizzino et al., 2017). These cell and tissue changes have been associated with age-related diseases including as cardiac, cerebrovascular, kidney, and liver diseases (Liguori et al., 2018). Further research is needed to confirm our findings on epigenetic clocks in brain and blood of individuals with SUD and to elucidate the specific mechanisms involved in these changes.
The limitations of the present study should be acknowledged. First, our sample size is relatively small and might have hampered the identification of correlations in specific groups due to a possible type II error. Future studies with larger sample sizes are required to confirm our findings and to further elucidate the mechanisms of accelerated aging in SUD. In this regard, longitudinal studies on epigenetic clocks in SUD are particularly relevant to better understand SUD-associated biological aging alterations, its course, and related factors. For their nature, longitudinal studies are only feasible using blood samples. Thus, correlations of brain measures with epigenetic clock measures in blood at different time points on epigenetic clocks require further research. Second, both brain and blood samples were obtained postmortem. Thus, the onset and duration of the SUD, which might influence our results, could not be considered due to the limitations of the psychological autopsy approach. Furthermore, the classification of the individuals in the comparison groups was based only on the primary diagnosis determined by consensus diagnosis. Although we performed sensitivity analyses to test whether our results were different after excluding individuals with an additional SUD (or secondary diagnosis) and we did not detect a significant difference in these results, we cannot rule out the potential effect of comorbid SUD in our results. Also, the information we included regarding smoking status (smoking score) was estimated based on DNA methylation information. Even though this estimation has shown an adequate performance, the use of more precise data, as those obtained from medical records, could benefit future studies. Future research considering the comorbid use of other drugs, including self-reported tobacco consumption, will be necessary to estimate their effect in the epigenetic aging of individuals with SUD. Finally, our analyses were limited to a single brain region (BA9). Further studies analyzing other brain regions relevant for SUD, such as other areas of the prefrontal cortex (orbitofrontal and ventromedial prefrontal cortex), basal ganglia, and amygdala (Substance Abuse and Mental Health Services Administration, 2016) will be helpful to delineate the extension of aging brain changes associated with SUD.
Conclusions
This study suggests that epigenetic clocks measured in peripheral blood may be good estimators of brain aging in controls subjects. Moreover, our results highlight the relevance of tissue specificity in studies of epigenetic aging measures and suggest that peripheral measures of epigenetic clocks in SUD may depend on the specific type of drug used.
Supplementary Material
Acknowledgements
We are grateful for the invaluable donations and participation from families, as well as for the generous collaboration of the medical examiners at the Harris County Institute of Forensic Sciences.
Funding:
This study was funded by the National Institute on Drug Abuse (NIDA) and the Fogarty Foundation (R01DA044859 to CWB). GRF is funded by the National Institute of Mental Health (NIMH, K01 MH121580).
References
- Ambatipudi S, Horvath S, Perrier F, Cuenin C, Hernandez-Vargas H, Le Calvez-Kelm F, … Herceg Z (2017). DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. European Journal of Cancer, 75, 299–307. doi: 10.1016/j.ejca.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames BN, & Shigenaga MK (1992). Oxidants are a major contributor to aging. Annals of the New York Academy of Sciences, 663, 85–96. doi: 10.1111/j.1749-6632.1992.tb38652.x [DOI] [PubMed] [Google Scholar]
- Bachi K, Sierra S, Volkow ND, Goldstein RZ, & Alia-Klein N (2017). Is biological aging accelerated in drug addiction? Current Opinion in Behavioral Sciences, 13, 34–39. doi: 10.1016/j.cobeha.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrendt S (2014). Add Standardized Regression Coefficients to lm-Objects [R package lm.beta version 1.5–1]. Retrieved May 31, 2021, from https://CRAN.R-project.org/package=lm.beta
- Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, … Rakyan VK (2019). DNA methylation aging clocks: Challenges and recommendations. Genome Biology, 20(1), 249. doi: 10.1186/s13059-019-1824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, & Hochberg Y (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological), 57(1), 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Bergsma T, & Rogaeva E (2020). DNA methylation clocks and their predictive capacity for aging phenotypes and healthspan. Neuroscience Insights, 15, 263310552094222. doi: 10.1177/2633105520942221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beylergil SB, Beck A, Deserno L, Lorenz RC, Rapp MA, Schlagenhauf F, … Obermayer K (2017). Dorsolateral prefrontal cortex contributes to the impaired behavioral adaptation in alcohol dependence. NeuroImage: Clinical, 15, 80–94. doi: 10.1016/j.nicl.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollepalli S, Korhonen T, Kaprio J, Anders S, & Ollikainen M (2019). EpiSmokEr: A robust classifier to determine smoking status from DNA methylation data. Epigenomics, 11(13), 1469–1486. doi: 10.2217/epi-2019-0206 [DOI] [PubMed] [Google Scholar]
- Breitling LP, Saum K-U, Perna L, Schöttker B, Holleczek B, & Brenner H (2016). Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clinical Epigenetics, 8(1), 21. doi: 10.1186/s13148-016-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli-Sforza LL (2005). The human genome diversity project: Past, present and future. Nature Reviews Genetics, 6(4), 333–340. doi: 10.1038/nrg1596. [DOI] [PubMed] [Google Scholar]
- Champely S, Ekstrom C, Dalgaard P, Gill J, Weibelzahl S, Anandkumar A, … Rosario HD (2020). Pwr: Basic functions for power analysis (Version 1.3–0). Retrieved from https://CRAN.R-project.org/package=pwr
- Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai P-C, … Horvath S (2016). DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging, 8(9), 1844–1865. doi: 10.18632/aging.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng GLF, Zeng H, Leung M-K, Zhang H-J, Lau BWM, Liu Y-P, Liu G-X, Sham PC, Chan CCH, So K-F, & Lee TMC (2013). Heroin abuse accelerates biological aging: A novel insight from telomerase and brain imaging interaction. Translational Psychiatry, 3(5), e260–e260. doi: 10.1038/tp.2013.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesney E, Goodwin GM, & Fazel S (2014). Risks of all-cause and suicide mortality in mental disorders: A meta-review. World Psychiatry, 13(2), 153–160. doi: 10.1002/wps.20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J (1988). Statistical power analysis for the behavioral sciences (Second. ed). New York: Academic Press. [Google Scholar]
- Costello EJ, Copeland WE, Shanahan L, Worthman CM, & Angold A (2013). C-reactive protein and substance use disorders in adolescence and early adulthood: A prospective analysis. Drug and Alcohol Dependence, 133(2), 712–717. doi: 10.1016/j.drugalcdep.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durso DF, Bacalini MG, Sala C, Pirazzini C, Marasco E, Bonafé M, … Nardini C (2017). Acceleration of leukocytes’ epigenetic age as an early tumor and sex-specific marker of breast and colorectal cancer. Oncotarget, 8(14), 23237–23245. doi: 10.18632/oncotarget.15573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, Davey Smith G, Hughes AD, Chaturvedi N, & Relton CL (2014). Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clinical Epigenetics, 6(1), 4. doi: 10.1186/1868-7083-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransquet PD, Wrigglesworth J, Woods RL, Ernst ME, & Ryan J (2019). The epigenetic clock as a predictor of disease and mortality risk: A systematic review and meta-analysis. Clinical Epigenetics, 11(1), 62. doi: 10.1186/s13148-019-0656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field AE, Robertson NA, Wang T, Havas A, Ideker T, & Adams PD (2018). DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Molecular Cell, 71(6), 882–895. doi: 10.1016/j.molcel.2018.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima A (2013). DiffCorr: An R package to analyze and visualize differential correlations in biological networks. Gene, 518(1), 209–214. doi: 10.1016/j.gene.2012.11.028. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Breitling LP, & Brenner H (2016). Relationship of tobacco smoking and smoking-related DNA methylation with epigenetic age acceleration. Oncotarget, 7(30), 46878–46889. doi: 10.18632/oncotarget.9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RZ, & Volkow ND (2011). Dysfunction of the prefrontal cortex in addiction: Neuroimaging findings and clinical implications. Nature Reviews Neuroscience, 12(11), 652–669. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan S, Gaige J, & Henn BM (2019). DNA methylation-based forensic age estimation in human bone [Preprint]. Genetics. doi: 10.1101/801647. [DOI] [Google Scholar]
- Guintivano J, Aryee MJ, & Kaminsky ZA (2013). A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics, 8(3), 290–302. doi: 10.4161/epi.23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman D, Rivkin E, Fadida A, Sharvit L, Hermush V, Rubin E, … Atzmon G (2020). Exceptionally long-lived individuals (Elli) demonstrate slower aging rate calculated by dna methylation clocks as possible modulators for healthy longevity. International Journal of Molecular Sciences, 21(2), 615. doi: 10.3390/ijms21020615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghpanah T, Afarinesh M, & Divsalar K (2010). A review on hematological factors in opioid-dependent people (Opium and heroin) after the withdrawal period. Addiction & Health, 2(1–2), 9–16. [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, … Zhang K (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular Cell, 49(2), 359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes W (2013). Benjamini–hochberg method. In Dubitzky W, Wolkenhauer O, Cho K-H, & Yokota H (Eds.), Encyclopedia of Systems Biology (pp. 78–78). New York, NY: Springer New York. doi: 10.1007/978-1-4419-9863-7_1215. [DOI] [Google Scholar]
- Hillary RF, Stevenson AJ, McCartney DL, Campbell A, Walker RM, Howard DM, … Marioni RE (2020). Epigenetic measures of ageing predict the prevalence and incidence of leading causes of death and disease burden. Clinical Epigenetics, 12(1), 115. doi: 10.1186/s13148-020-00905-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjemsæter AJ, Bramness JG, Drake R, Skeie I, Monsbakken B, Benth JŠ, & Landheim AS (2019). Mortality, cause of death and risk factors in patients with alcohol use disorder alone or poly-substance use disorders: A 19-year prospective cohort study. BMC Psychiatry, 19(1), 101. doi: 10.1186/s12888-019-2077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S (2013). DNA methylation age of human tissues and cell types. Genome Biology, 14(10), R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, & Levine AJ (2015). Hiv-1 infection accelerates age according to the epigenetic clock. Journal of Infectious Diseases, 212(10), 1563–1573. doi: 10.1093/infdis/jiv277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Gurven M, Levine ME, Trumble BC, Kaplan H, Allayee H, … Assimes TL (2016). An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biology, 17(1), 171. doi: 10.1186/s13059-016-1030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, & Raj K (2018). DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nature Reviews Genetics, 19(6), 371–384. doi: 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, … Raj K (2018). Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging, 10(7), 1758–1775. doi: 10.18632/aging.101508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, … Kelsey KT (2012). DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics, 13(1), 86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illumina. Infinium HD Methylation Assay. (September, 2020) https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/infinium_assays/infinium_hd_methylation/infinium-methylation-assay-reference-guide-15019519-07.pdf
- Illumina. Infinium Global Screening Array-24 v3.0 BeadChip. (August 19, 2019) https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/infinium-global-screening-array-data-sheet-370-2016-016.pdf
- John JA, & Draper NR (1980). An alternative family of transformations. Applied Statistics, 29(2), 190. doi: 10.2307/2986305. [DOI] [Google Scholar]
- Jylhävä J, Pedersen NL, & Hägg S (2017). Biological age predictors. EBioMedicine, 21, 29–36. doi: 10.1016/j.ebiom.2017.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlenkov A, Jaffe A, Timashpolsky A, Apontes P, Rudchenko S, Barbu M, … Dracheva S (2017). DNA methylation profiling of human prefrontal cortex neurons in heroin users shows significant difference between genomic contexts of hyper- and hypomethylation and a younger epigenetic age. Genes, 8(6), 152. doi: 10.3390/genes8060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska KM, Bacalini MG, Sala C, Kaziyama H, de Andrade DC, Terlizzi R, … Pirazzini C (2020). Analysis of epigenetic age predictors in pain-related conditions. Frontiers in Public Health, 8, 172. doi: 10.3389/fpubh.2020.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Moon S, Park M-J, Koh I-U, Choi N-H, Yu H-Y, … Kim B-J (2020). Integrated analysis of tissue-specific promoter methylation and gene expression profile in complex diseases. International Journal of Molecular Sciences, 21(14), 5056. doi: 10.3390/ijms21145056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levandowski ML, Tractenberg SG, de Azeredo LA, De Nardi T, Rovaris DL, Bau CHD, Rizzo LB, Maurya PK, Brietzke E, Tyrka AR, & Grassi-Oliveira R (2016). Crack cocaine addiction, early life stress and accelerated cellular aging among women. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 71, 83–89. doi: 10.1016/j.pnpbp.2016.06.009. [DOI] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, … Horvath S (2018). An epigenetic biomarker of aging for lifespan and healthspan. Aging, 10(4), 573–591. doi: 10.18632/aging.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME (2020). Assessment of epigenetic clocks as biomarkers of aging in basic and population research. The Journals of Gerontology: Series A, 75(3), 463–465. 10.1093/gerona/glaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, & Abete P (2018). Oxidative stress, aging, and diseases. Clinical Interventions in Aging, 13, 757–772. doi: 10.2147/CIA.S158513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Wang P, Wu H, Ko C, Yang Y, & Yen C (2018). Altered gray matter volume and disrupted functional connectivity of dorsolateral prefrontal cortex in men with heroin dependence. Psychiatry and Clinical Neurosciences, 72(6), 435–444. doi: 10.1111/pcn.12655. [DOI] [PubMed] [Google Scholar]
- Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, … Horvath S (2019). DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging, 11(2), 303–327. doi: 10.18632/aging.101684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AT, Seeboth A, Tsai P-C, Sun D, Quach A, Reiner AP, … Horvath S (2019). DNA methylation-based estimator of telomere length. Aging, 11(16), 5895–5923. doi: 10.18632/aging.102173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo A, Jung J, Longley M, Rosoff DB, Charlet K, Muench C, … Lohoff FW (2020). Epigenetic aging is accelerated in alcohol use disorder and regulated by genetic variation in APOL2. Neuropsychopharmacology, 45(2), 327–336. doi: 10.1038/s41386-019-0500-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, … Deary IJ (2015). The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. International Journal of Epidemiology, 44(4), 1388–1396. doi: 10.1093/ije/dyu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrory C, Fiorito G, Hernandez B, Polidoro S, O’Halloran AM, Hever A, … Kenny RA (2020). Association of 4 epigenetic clocks with measures of functional health, cognition, and all-cause mortality in The Irish Longitudinal Study on Ageing (Tilda) [Preprint]. Genomics. doi: 10.1101/2020.04.27.063164. [DOI] [Google Scholar]
- McKinney BC, Lin H, Ding Y, Lewis DA, & Sweet RA (2018). DNA methylation age is not accelerated in brain or blood of subjects with schizophrenia. Schizophrenia Research, 196, 39–44. doi: 10.1016/j.schres.2017.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson MM (2018). Epigenetic Age Acceleration: A Biological Doomsday Clock for Cardiovascular Disease?. Circulation. Genomic and precision medicine, 11(3), e002089. doi: 10.1161/CIRCGEN.118.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monoranu CM, Apfelbacher M, Grünblatt E, Puppe B, Alafuzoff I, Ferrer I, … Roggendorf W (2009). Ph measurement as quality control on human post mortem brain tissue: A study of the brainnet europe consortium. Neuropathology and Applied Neurobiology, 35(3), 329–337. doi: 10.1111/j.1365-2990.2008.01003a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroy-Jaramillo N, Dyukova E, & Walss-Bass C (2018). Telomere length in psychiatric disorders: Is it more than an ageing marker? The World Journal of Biological Psychiatry, 19(sup2), S2–S20. doi: 10.1080/15622975.2016.1273550. [DOI] [PubMed] [Google Scholar]
- Montalvo-Ortiz JL, Cheng Z, Kranzler HR, Zhang H, & Gelernter J (2019). Genomewide study of epigenetic biomarkers of opioid dependence in european- american women. Scientific Reports, 9(1), 4660. doi: 10.1038/s41598-019-41110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Mateu F, Rubio-Aparicio M, Cayuela P, Álvarez F-J, Roca-Vega A, Chirlaque MD, … Sánchez-Meca J (2019). The association of telomere length with substance use disorders: Systematic review and meta-analysis protocol. Systematic Reviews, 8(1), 298. doi: 10.1186/s13643-019-1199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M-H, Chen VC-H, Ting H, Lin T-Y, Chang S-H, & Gossop M (2019). Macrocytosis among patients with heroin use disorder. Neuropsychiatric Disease and Treatment, 15, 2293–2298. doi: 10.2147/NDT.S211649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo O, Ojo O, Adebowale F, & Wang X-H (2018). The effect of dietary glycaemic index on glycaemia in patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Nutrients, 10(3), 373. doi: 10.3390/nu10030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osna NA, Donohue TM, & Kharbanda KK (2017). Alcoholic liver disease: Pathogenesis and current management. Alcohol Research: Current Reviews, 38(2), 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pateria P, de Boer B, & MacQuillan G (2013). Liver abnormalities in drug and substance abusers. Best Practice & Research Clinical Gastroenterology, 27(4), 577–596. doi: 10.1016/j.bpg.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price AL, & Reich D (2006). Population structure and eigenanalysis. PLoS Genetics, 2(12), e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, & Bitto A (2017). Oxidative stress: Harms and benefits for human health. Oxidative Medicine and Cellular Longevity, 2017, 1–13. doi: 10.1155/2017/8416763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, & Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics, 38(8), 904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Protsenko E, Yang R, Nier B, Reus V, Hammamieh R, Rampersaud R, … Wolkowitz OM (2020). “GrimAge,” an epigenetic predictor of mortality, is accelerated in Major Depressive Disorder [Preprint]. Psychiatry and Clinical Psychology. doi: 10.1101/2020.12.25.20248290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PTSD Systems Biology Consortium, Yang R, Wu GWY, Verhoeven JE, Gautam A, Reus VI, … Wolkowitz OM (2020). A DNA methylation clock associated with age-related illnesses and mortality is accelerated in men with combat PTSD. Molecular Psychiatry. doi: 10.1038/s41380-020-0755-z. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, … Sham PC (2007). Plink: A tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics, 81(3), 559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, … Horvath S (2017). Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging, 9(2), 419–446. doi: 10.18632/aging.101168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL. http://www.R-project.org/ [Google Scholar]
- Ribeiro EA, Scarpa JR, Garamszegi SP, Kasarskis A, Mash DC, & Nestler EJ (2017). Gene network dysregulation in dorsolateral prefrontal cortex neurons of humans with cocaine use disorder. Scientific Reports, 7(1), 5412. 10.1038/s41598-017-05720-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen AD, Robertson KD, Hlady RA, Muench C, Lee J, Philibert R, … Lohoff FW (2018). DNA methylation age is accelerated in alcohol dependence. Translational Psychiatry, 8(1), 182. doi: 10.1038/s41398-018-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshandel D, Chen Z, Canty AJ, Bull SB, Natarajan R, & Paterson AD, DCCT/EDIC Research Group. (2020). DNA methylation age calculators reveal association with diabetic neuropathy in type 1 diabetes. Clinical Epigenetics, 12(1), 52. doi: 10.1186/s13148-020-00840-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shireby GL, Davies JP, Francis PT, Burrage J, Walker EM, Neilson GWA, … Mill J (2020). Recalibrating the epigenetic clock: Implications for assessing biological age in the human cortex. Brain, 143(12), 3763–3775. doi: 10.1093/brain/awaa334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu C, Sosnowski DW, Tao R, Deep-Soboslay A, Kleinman JE, Hyde TM, … Maher BS (2021). Epigenome-wide study of brain DNA methylation following acute opioid intoxication. Drug and Alcohol Dependence, 221, 108658. doi: 10.1016/j.drugalcdep.2021.108658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soder HE, Berumen AM, Gomez KE, Green CE, Suchting R, Wardle MC, … Lane SD (2020). Elevated neutrophil to lymphocyte ratio in older adults with cocaine use disorder as a marker of chronic inflammation. Clinical Psychopharmacology and Neuroscience, 18(1), 32–40. doi: 10.9758/cpn.2020.18.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano-Tárraga C, Giralt-Steinhauer E, Mola-Caminal M, Vivanco-Hidalgo RM, Ois A, Rodríguez-Campello A, … Jiménez-Conde J (2016). Ischemic stroke patients are biologically older than their chronological age. Aging, 8(11), 2655–2666. doi: 10.18632/aging.101028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson AJ, McCartney DL, Shireby GL, Hillary RF, King D, Tzioras M, … Spires-Jones TL (2020). A comparison of blood and brain-derived ageing and inflammation-related DNA methylation signatures and their association with microglial burdens [Preprint]. Neuroscience. doi: 10.1101/2020.11.30.404228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration (US); Office of the Surgeon General. (2016). Facing Addiction in America: The Surgeon General’s Report on Alcohol, Drugs, and Health [Internet]. Washington (DC): US Department of Health and Human Services. Chapter 2, The neurobiology of substance use, misuse, and addiction. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK424849/ [PubMed] [Google Scholar]
- Sullivan EV, & Pfefferbaum A (2019). Brain-behavior relations and effects of aging and common comorbidities in alcohol use disorder: A review. Neuropsychology, 33(6), 760–780. doi: 10.1037/neu0000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiyun. (2021). Taiyun/corrplot [R]. Retrieved from https://github.com/taiyun/corrplot (Original work published 2011); Wei T, & Simko V (2017). R package “corrplot”: Visualization of a Correlation Matrix. https://github.com/taiyun/corrplot.
- Tajuddin SM, Nalls MA, Zonderman AB, & Evans MK (2017). Association of red cell distribution width with all-cause and cardiovascular-specific mortality in African American and white adults: A prospective cohort study. Journal of Translational Medicine, 15(1), 208. doi: 10.1186/s12967-017-1313-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, & Beck S (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics, 29(2), 189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Valencia A, editor. Bioinformatics. 2017;33:3982–4. doi: 10.1093/bioinformatics/btx513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verna EC, Schluger A, & Brown RS (2019). Opioid epidemic and liver disease. JHEP Reports, 1(3), 240–255. doi: 10.1016/j.jhepr.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter VM, Meyer A, Karbasiyan M, Steinhagen-Thiessen E, Hopfenmüller W, & Demuth I (2019). Epigenetic clock and relative telomere length represent largely different aspects of aging in the berlin aging study ii(Base-ii). The Journals of Gerontology: Series A, 74(1), 27–32. doi: 10.1093/gerona/gly184. [DOI] [PubMed] [Google Scholar]
- Voisin S, Harvey NR, Haupt LM, Griffiths LR, Ashton KJ, Coffey VG, … Eynon N (2020). An epigenetic clock for human skeletal muscle. Journal of Cachexia, Sarcopenia and Muscle, 11(4), 887–898. doi: 10.1002/jcsm.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas CM, Hazra A, Chang S-C, Qiu W, Reynolds CF, Mischoulon D, … Okereke OI (2019). Pilot study of DNA methylation, molecular aging markers and measures of health and well-being in aging. Translational Psychiatry, 9(1), 118. doi: 10.1038/s41398-019-0446-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Wu G, Verhoeven JE, Gautam A, Reus VI, Kang JI, Flory JD, Abu-Amara D, PTSD Systems Biology Consortium, Hood L, Doyle FJ 3rd, Yehuda R, Marmar CR, Jett M, Hammamieh R, Mellon SH, & Wolkowitz OM (2020). A DNA methylation clock associated with age-related illnesses and mortality is accelerated in men with combat PTSD. Molecular psychiatry. doi: 10.1038/s41380-020-0837-y. [DOI] [PubMed] [Google Scholar]
- Yang Z, Ye J, Li C, Zhou D, Shen Q, Wu J, … Liu Y (2013). Drug addiction is associated with leukocyte telomere length. Scientific Reports, 3(1), 1542. doi: 10.1038/srep01542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Pfefferbaum A, & Sullivan EV (2017). Perspectives on fronto-fugal circuitry from human imaging of alcohol use disorders. Neuropharmacology, 122, 189–200. doi: 10.1016/j.neuropharm.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai T, Salmeron BJ, Gu H, Adinoff B, Stein EA, & Yang Y (2021). Functional connectivity of dorsolateral prefrontal cortex predicts cocaine relapse: Implications for neuromodulation treatment. Brain Communications, 3(2), fcab120. doi: 10.1093/braincomms/fcab120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wang F, Xu H, Liu Y, Liu J, Zhao H, & Gelernter J (2014). Differentially co-expressed genes in postmortem prefrontal cortex of individuals with alcohol use disorders: Influence on alcohol metabolism-related pathways. Human Genetics, 133(11), 1383–1394. doi: 10.1007/s00439-014-1473-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeremski M, & Martinez A (2017). Liver disease and fibrosis assessment in substance use-related disorders. Clinical Pharmacology in Drug Development, 6(2), 164–168. doi: 10.1002/cpdd.312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.