

SUMMARY
The underlying cellular events driving kidney fibrogenesis and metabolic dysfunction are incompletely understood. Here, we employed single-cell combinatorial indexing RNA-sequencing to analyze 24 mouse kidneys from two fibrosis models. We profiled 309,666 cells in one experiment, representing 50 cell types/states encompassing epithelial, endothelial, immune and stromal populations. Single-cell analysis identified diverse injury states of the proximal tubule, including two distinct early-phase populations with dysregulated lipid and amino acid metabolism, respectively. Lipid metabolism was defective in the chronic phase but was transiently activated in the very early stages of ischemia-induced injury, where we discovered increased lipid deposition and increased fatty acid β-oxidation. Perilipin 2 was identified as a surface marker of intracellular lipid droplets and its knockdown in vitro disrupted cell energy state maintenance during lipid accumulation. Surveying epithelial cells across nephron segments identified shared and unique injury responses. Stromal cells exhibited high heterogeneity and contributed to fibrogenesis by epithelial-stromal crosstalk.
Keywords: single-cell combinatorial indexing, kidney, fibrosis, acute kidney injury, chronic kidney disease, lipid metabolism, lipid droplet, PLIN2, cell differentiation, tissue regeneration
Graphical Abstract
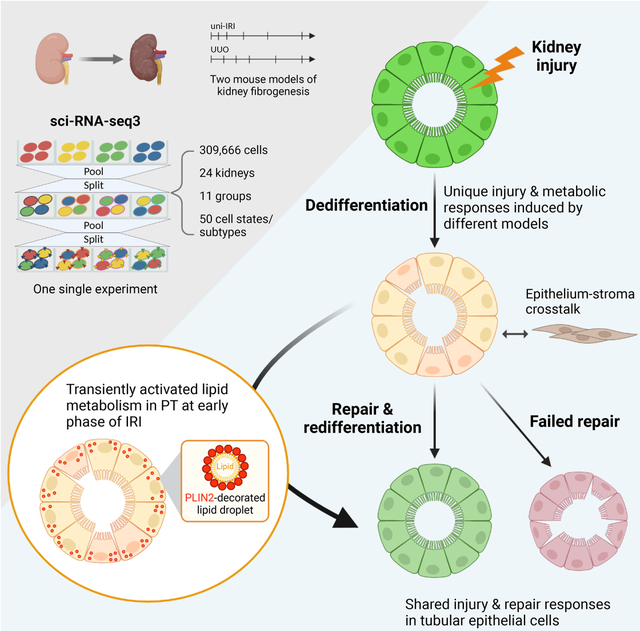
In brief
Li et al. profile the full-time courses of mouse kidney fibrogenesis using single-cell combinatorial indexing RNA-sequencing. They describe diverse injury states of proximal tubular cells, including one cell state with enhanced lipid metabolism at an early phase of ischemia-induced injury. This single-cell atlas defines kidney epithelial injury responses in fibrosis.
INTRODUCTION
Chronic kidney disease (CKD) affects ~10% of the population worldwide and ultimately can lead to kidney failure (Hill et al., 2016; Kalantar-Zadeh et al., 2021). With no cure and relatively few therapies that slow progression, people with CKD suffer considerable morbidity and mortality. Across all CKDs, regardless of the underlying cause, dysregulated epithelial metabolism is increasingly recognized as an important pathological feature that drives interstitial fibrosis (Kang et al., 2014; Slee, 2012; Tran et al., 2016; Zhu et al., 2021). Understanding the earliest cellular events driving kidney fibrogenesis will improve our knowledge of CKD pathophysiology and may identify new, effective therapeutic targets.
Single-cell RNA sequencing (scRNA-seq) allows for the unbiased characterization of cell transcriptomics and has been widely applied to decipher cell fate dynamics and metabolic heterogeneity (Evers et al., 2019; Kuppe et al., 2021; Park et al., 2018; Stuart and Satija, 2019). The most commonly used platform for scRNA-seq is based upon droplet microfluidics, but due to several technical limitations including low throughput, difficulty in analyzing multiple samples or timepoints, batch effects and incompatibility with fixed samples, many studies analyze a limited number of samples, providing only a “snapshot” of a specific biological condition (Li and Humphreys, 2021; Stoeckius et al., 2018; Weinreb et al., 2018). Previous work has characterized CKD and kidney fibrosis at single cell resolution (Dhillon et al., 2021; Lu et al., 2021; Wu et al., 2019; Zhang et al., 2021), but these studies typically lack multiple timepoints especially in early stages. Even though sample multiplexing approaches exist (e.g., CITE-seq (Stoeckius et al., 2017)), this method does not easily scale up, which is detrimental for analysis of the kidney due to diversity of cell types and states that arise during injury and progression of fibrosis (Balzer et al., 2021; Gerhardt et al., 2021; Kirita et al., 2020; Wu et al., 2019). As an example, proximal tubule (PT) cells constitute ~50% of the total kidney cell number, so rare cell types and states may be underrepresented in a lower complexity scRNA-seq dataset. While multiple scRNA-seq experiments and large-scale data integration could resolve these limitations, batch effect correction would be required (Tran et al., 2020).
Here, we optimized single-cell combinatorial indexing RNA-seq (sci-RNA-seq) (Cao et al., 2020, 2019) in order to decipher the molecular events driving kidney fibrogenesis. We leveraged the high throughput, high sample multiplexing capacity, and low costs of sci-RNA-seq to characterize two mouse models of kidney injury and fibrosis, unilateral ischemia-reperfusion injury (uni-IRI) and unilateral ureteral obstruction (UUO), at multiple time points. sci-RNA-seq is compatible with tissue fixation, which stabilizes RNA immediately after tissue collection preventing degradation, allowing multi-site sample acquisition and facilitating storage of samples from multiple timepoints prior to processing. We have generated an atlas of kidney fibrogenesis (data visualizer available at http://humphreyslab.com/SingleCell/) from a single experiment with 11 biological conditions and 24 samples. This approach enabled the elimination of batch effects and profiling of 309,666 cells. We report that uni-IRI and UUO induced two distinct PT cell states after injury with unique transcriptomic signatures and fate outcomes. Further investigation of these two cell states highlighted their distinct mechanisms of metabolic regulation, including activated lipid metabolism in the earliest stages of uni-IRI where we identified PLIN2+ lipid droplets. Additionally, we describe both shared and unique epithelial responses to injury and repair across nephron segments, as well as kidney stromal heterogeneity and intercellular communication dynamics during kidney fibrosis. This atlas of kidney fibrogenesis serves as a unique resource, and reveals previously unappreciated epithelial cell states.
RESULTS
Generation of two mouse models of kidney fibrogenesis
We performed uni-IRI and UUO surgeries on 8–9 week old adult male C57BL6/J mice and collected samples at multiple timepoints during disease progression (0, 6 hours and 2, 7, 14, 28 days post uni-IRI or 0, 2, 4, 6, 10, 14 days post UUO; n = 2 for each timepoint) (Figure 1A). To validate each sample prior to scRNA-seq, we first stained kidney injury and fibrosis markers by immunofluorescence (Figure 1B). In mice with uni-IRI, the kidney injury marker HAVCR1 was strongly upregulated after 2 days post injury (uni-IRI D2) and its expression gradually decreased during the repair phase after uni-IRI D7. The fibrosis marker Collagen Type I (COL1) started to accumulate at uni-IRI D2 and was highly abundant at uni-IRI D14. By uni-IRI D28, the expression of HAVCR1 was close to baseline while COL1 expression was only partially resolved, suggesting the acute kidney injury (AKI) to CKD transition (Figure 1B). By contrast, UUO kidneys had sustained HAVCR1 expression and increased upregulation of COL1 over the full time course (Figure 1B), reflecting the more aggressive fibrotic burden in this model. Successful induction of injury and fibrogenesis on mouse kidneys was also confirmed by quantitative PCR (qPCR) where we measured Havcr1 and myofibroblast marker genes Acta2 and Col1a1 and observed similar expression patterns (Figure S1A).
Figure 1. A single-cell transcriptomics landscape of mouse kidney fibrogenesis profiled with sci-RNA-seq3.
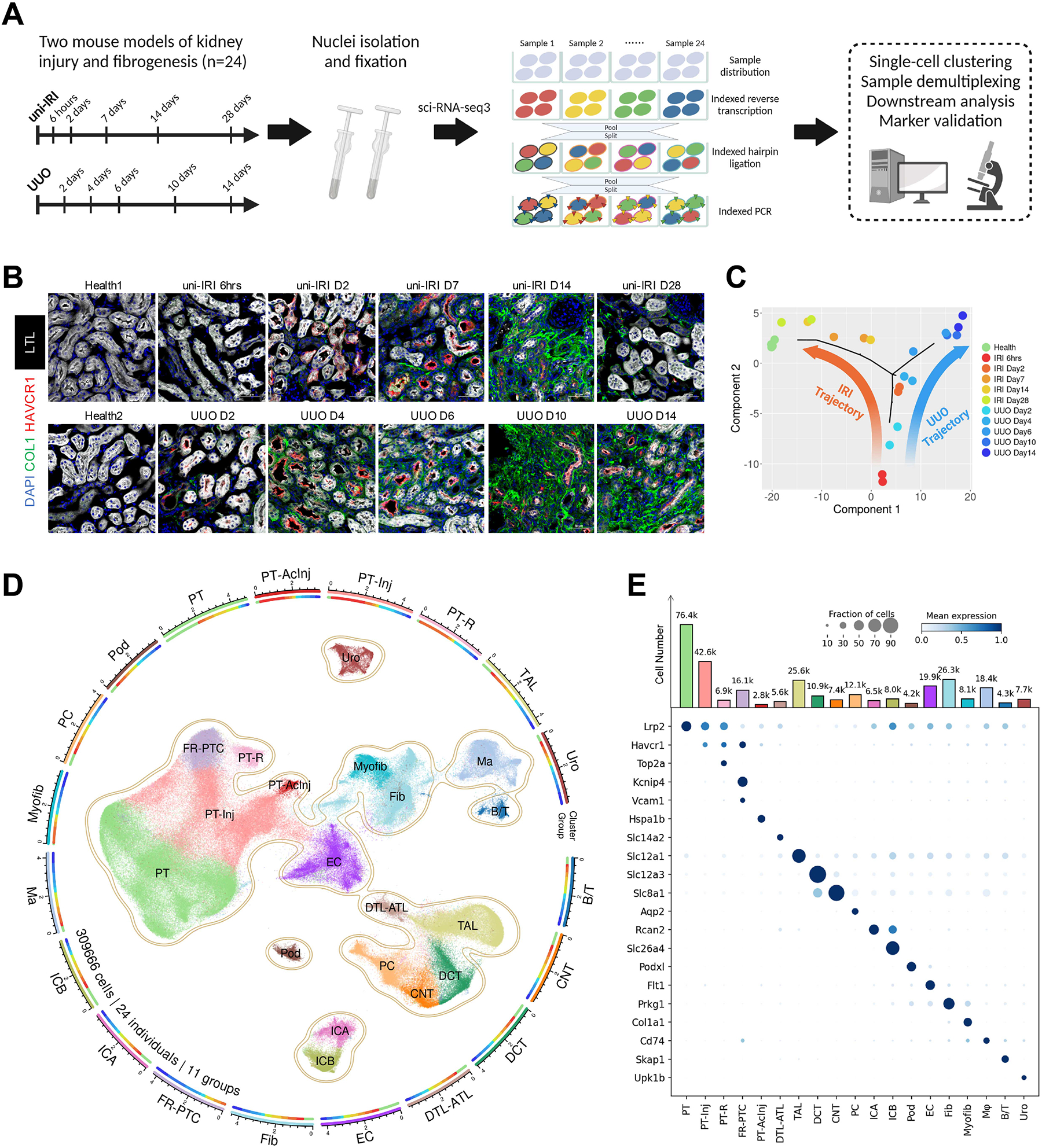
(A) Summary of experimental methodology. n = 2 per timepoint. Nuclei were extracted from all kidney samples and profiled with a three-level combinatorial indexing sequencing strategy. Cells were demultiplexed based on 1st indexing barcodes to identify sample origins in data analysis. Figure created with BioRender.com.
(B) Immunofluorescence staining of HAVCR1 (red), Collagen Type I (green), Lotus Tetragonolobus Lectin (LTL; white) and DAPI (blue) on tissue sections collected from all healthy and diseased conditions of our study cohort. Scale bar: 50 μm.
(C) Pseudobulk trajectory projection (using Monocle2) of all sample conditions in the study cohort revealing distinct transcriptomic signature of uni-IRI and UUO. Each dot represents a sample (n = 24 in total).
(D) An atlas of mouse kidney fibrogenesis. A UMAP presentation (center) shows 309,666 cells profiled from 24 individual mouse kidneys of 11 healthy or diseased conditions. The surrounding circular layouts indicate the cell number of each population (log10-transformed scale bar), 19 major cell types (outer layout) and distributions of 11 group conditions in each cell type (inner layout; color legend same as Figure 1C). PT, proximal tubule; PT-Inj, injured PT; PT-R, repairing PT; FR-PTC, failed repair PT cells; PT-AcInj, acute injury PT; DTL, descending limb of loop of Henle (LoH); ATL, thin ascending limb of LoH; TAL, thick ascending limb of LoH; DCT, distal convoluted tubule; CNT, connecting tubule; PC, principal cell of collecting duct; ICA, type A intercalated cell of collecting duct; ICB, type B intercalated cell of collecting duct; Pod, podocyte; EC, endothelial cell; Fib, fibroblast; Myofib, myofibroblast; Ma, macrophage (Mφ); B/T, immune cell; Uro, urothelium.
(E) Dot plot showing expression pattern of cluster-specific marker genes and bar plot showing the number of cells of each cluster. In the dot plot, the diameter of the dot corresponds to the proportion of cells expressing the indicated gene and the density of the dot corresponds to average expression relative to all cell types.
Characterization of kidney fibrogenesis with sci-RNA-seq3
Nuclear suspensions were prepared from each sample, fixed, and snap-frozen. This enabled us to process all 24 samples simultaneously in a single experiment to achieve sample multiplexing using the sci-RNA-seq3 protocol (Cao et al., 2020, 2019), which employed a combinatorial indexing strategy based on reverse transcription (RT), hairpin ligation, and indexed PCR. In sci-RNA-seq3, the nuclei from each sample were divided into several wells of four 96-well plates, and thus the first barcode introduced by RT allowed sample identification (Figure 1A). In addition to the multiplexing capacity, high throughput, and relatively low cost of sci-RNA-seq3, common laboratory supplies could be used and the protocol was modifiable. Early results revealed several challenges in applying the original sci-RNA-seq3 protocol to kidney, including low nuclei extraction yield, nuclei aggregation in the suspension, reduced library quality due to non-uniform transposase activity and incomplete purification. We therefore separately optimized each of these steps in our modified protocol, including performing a Tn5 transposase activity test which significantly improved library yield and quality. The changes to the original protocol are summarized in Table S1 and in more detail in Methods. We included a species-mixing control with nuclei harvested from human HEK-293T and mouse C3H/10T1/2 cultured cells in order to evaluate doublet frequency.
We sequenced the entire sci-RNA-seq3 library on one NovaSeq 6000 flow cell. Over half of the reads (60.4%) mapped to intronic regions, as expected for single-nucleus sequencing (Wu et al., 2019). After demultiplexing, we first assessed doublets by analyzing the species mixing samples. This revealed a very low cell collision rate of 1.3% (Figure S1B). For the remaining mouse kidney samples, we generated a total of 413,681 raw cell transcriptomes at a minimum threshold of 200 uniform molecular identifiers (UMI) per cell. We detected an average of 1165 UMIs/cell (Table S1). After quality control procedures including removal of predicted doublets and artifacts (see Methods), we proceeded to analyze 309,666 high-quality cells. We first projected the pseudobulk transcriptomes of all 24 samples into two dimensions in an unsupervised fashion. This revealed clearly distinct trajectories between the uni-IRI and UUO samples and low variation between biological replicates. The uni-IRI 6hrs and UUO D2 samples were similar, but later samples diverged substantially (Figure 1C). Even though uni-IRI and UUO are both models of kidney fibrosis, the distinct trajectories suggested quite different cellular mechanisms.
The large size of this scRNA-seq dataset allowed for a detailed characterization of cellular heterogeneity in healthy and fibrotic kidneys. Cell clustering of the 309,666 cells revealed 19 major cell clusters, including cells of the proximal tubule (PT), loop of Henle, and podocytes (Figure 1D). We performed subclustering analysis on all major cell clusters, which identified a total of 50 cell types or states (summarized in Table S2) including low abundance cell types such as juxtaglomerular apparatus (JGA), dendritic cell subtypes (Figure S1C), and vascular cells (Figure S1D). The 19 major clusters were annotated based on expression of known marker genes (Figure 1E) and data integration with previous cell atlas resources such as our bilateral IRI (bi-IRI) scRNA-seq dataset (Kirita et al., 2020) (Figure S1E). Correlation analysis across cell types indicated high transcriptomic similarity between fibroblasts (Prkg1/Gpc6 high) and myofibroblasts (Col1a1/Col1a2 high) and across distal nephron epithelia (TAL, DCT, CNT, PC, ICA, ICB) (Figure S1F). We next identified cells according to sample condition (health, uni-IRI or UUO) which revealed that nearly all cells from disease timepoints distributed distinctly from the healthy cells – reflecting that fibrosis affects the entire organ (Figure S1G–H). Myofibroblasts and immune cells were quite sparse in health but underwent proliferative expansion during disease (Figure S1I–J).
Diverse Proximal Tubule Injury States
Our initial clustering suggested considerable heterogeneity in PT cell states during fibrosis (Figure 1D). We therefore performed unsupervised subclustering on PT cells alone (130,503 cells after quality control; Figure 2A). This revealed the expected healthy S1/S2/S3 PT clusters, as well as multiple injury states (high expression of Havcr1 and Nrg1), including acute injury (PT-AcInj), repairing (PT-R), a state we have previously characterized as failed repair proximal tubule cells (FR-PTC) (Kirita et al., 2020; Muto et al., 2021) and two apparent intermediate PT injury states, located between healthy cells and FR-PTC in the UMAP space, which we annotated as Type1 and Type2 injured PT cells. All injured PT cell states were also characterized by downregulation of healthy PT marker genes such as solute-linked carriers (e.g., Slc34a1, Slc5a12, Slc7a13), suggesting cell dedifferentiation. The significances of PT-AcInj, PT-R and FR-PTC have been described in previous scRNA-seq studies (Gerhardt et al., 2021; Kirita et al., 2020; Lu et al., 2021; Rudman-Melnick et al., 2020) and were benchmarked in our large-scale dataset. Specifically, PT-AcInj cells expressed genes encoding heat-shock proteins (e.g., Hspa1b, Hsp90aa1) and showed high activity of Hsf1 in the transcription factor activity analysis (Figure 2B–C and S2A). The PT-R cluster strongly expressed genes associated with cell proliferation (e.g., Top2a, Mki67, Lmnb1) and scored highly by cell-cycle scoring analysis (Figure S2B). FR-PTC were characterized by expression of known marker genes Vcam1 and Kcnip4. Smad1, an essential component of TGF-β signaling (Zhang et al., 2015), exhibited high transcription factor activity in FR-PTC (Figure 2C–D) and single-cell pathway activity analysis revealed that NF-κB and TNF-α pathways were also highly active (Figure 2D), indicating that these cells were proinflammatory and profibrotic, confirming prior results (Markó et al., 2016; Ramseyer and Garvin, 2013; Shimizu et al., 2011; Zager et al., 2005).
Figure 2. Diverse cell states of injured proximal tubule.
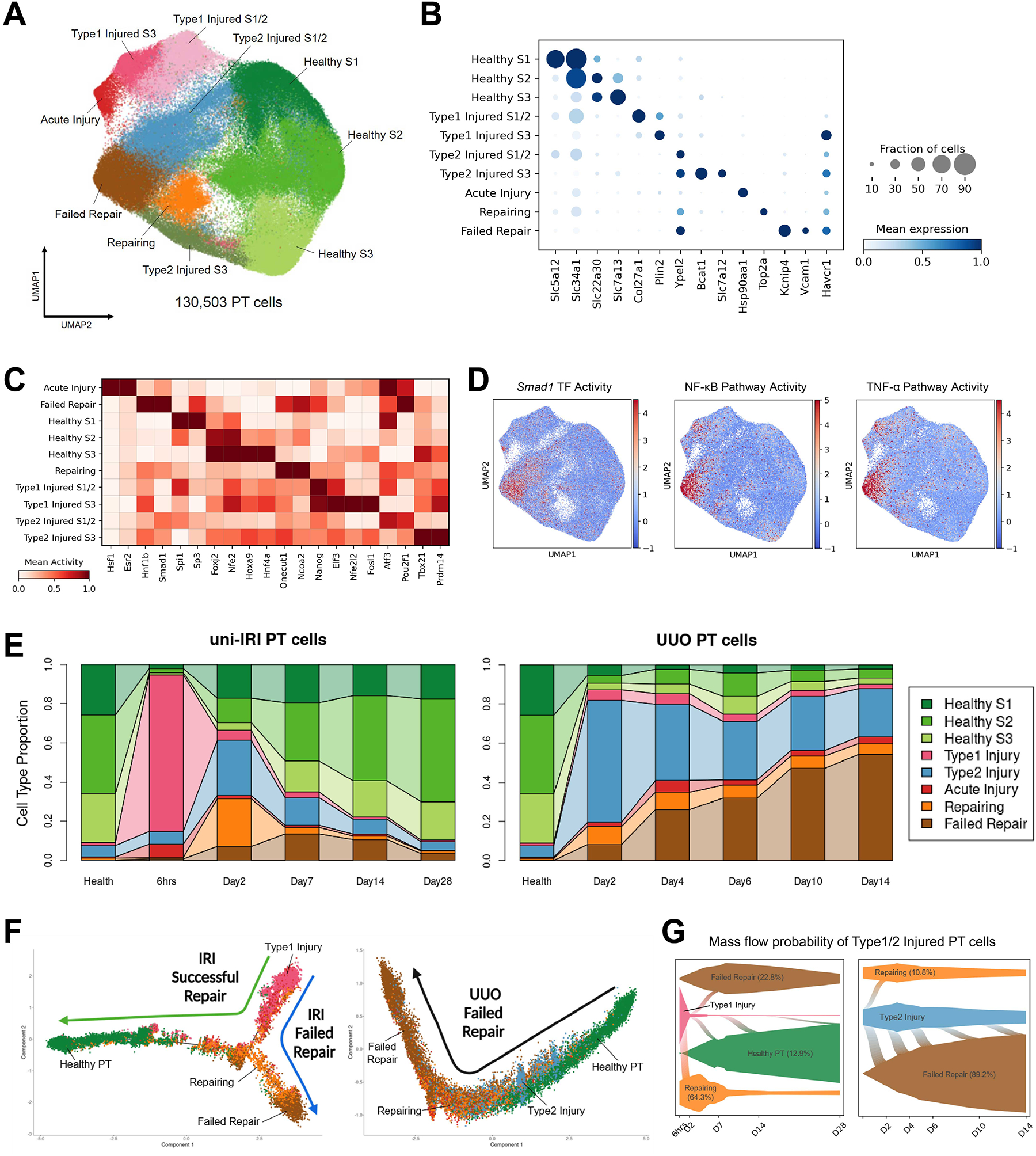
(A) UMAP plot of all PT cells after quality control in subclustering analysis. S1, S2 and S3 indicate the three anatomical segments of PT.
(B) Dot plot showing expression of marker genes of each PT cell clusters, including 3 clusters in healthy states and 7 injured cell states expressing Havcr1.
(C) Heat map showing cluster-specific transcription factor activity predicted by gene regulatory network analysis. Color density corresponds to average activity of the indicated gene relative to all PT cells.
(D) Transcription factor activity and single-cell pathway analysis showing activities of Smad1 and NF-κB/TNF-α pathways are enriched in the FR-PTC cluster.
(E) Connected bar plots displaying the proportional abundance of each cell cluster in each disease condition. Injured S1/2 and S3 cells are combined for the convenience of data visualization.
(F) Pseudotemporal ordering of cells sampled from uni-IRI and UUO subsets colored by cluster identity (color legend same as Figure 2E), using Monocle2.
(G) Single-cell fate mapping of Type1 and Type2 injured PT cells (color legend same as Figure 2E), using CellRank. Flows connecting two cell types describe lineage transition and the flow width indicate predicted probability. See also Figure S2K.
By contrast, we struggled to annotate the Type1 and Type2 PT cell clusters to previously published work, possibly because these cells were enriched in early and middle stages of kidney fibrogenesis (i.e., IRI 6hrs, UUO D2-D4) that have not been previously analyzed in scRNA-seq studies. Surveying the proportion of each PT cell type across conditions (Figure 2E and S2C) revealed that Type1 injured PT was primarily found in uni-IRI (occurrence frequency in uni-IRI: UUO ~ 11:1) and Type2 injured PT was more specific to UUO samples (abundance of Type1: Type2 injured PT ~ 10:1). More specifically, in uni-IRI, Type1 injured cells comprised 80% of all PT cells at 6 hrs after injury, with this proportion falling rapidly to 5% of the total by D2. In UUO, Type2 injured cells also appeared in the early timepoint (D2) and dominated the PT population (62% of the total cells), but their frequency did not fall as quickly as Type1 injured cells in uni-IRI (Figure 2E). This analysis also highlighted distinct outcomes of PT successful repair vs. failed repair in the two mouse models: the frequency of healthy PT cells was reduced remarkably by both uni-IRI or UUO surgeries, but only in uni-IRI did these cells return to their prior uninjured state (Figure 2E). While a small percentage of FR-PTC (~4%) remained at -IRI D28, FR-PTC constituted a large and increasing proportion of all cells as the time course proceeded in UUO (~55% at UUO D14) (Figure 2E).
Two Types of Cell States of Injured Proximal Tubule
We next asked whether the ability of PT to successfully repair in uni-IRI but not in UUO might be related to Type1 vs. Type2 injury. Gene ontology (GO) enrichment analysis on differentially expressed genes (DEGs) for the two populations highlighted wound healing, cell junction organization and cell-cell adhesion in Type1 injured PT and epithelial morphogenesis and MAPK signaling in the Type2 group (Figure S2D). Regulation of cell motility was a shared term in both groups (Figure S2D). Type1 injured PT cells were primarily observed early – at 6hrs post uni-IRI with defining DEGs such as Plin2 and Col27a1 (Figure 2B and S2E). We found that Elf3, a transcription factor that has been reported to be upregulated in both mouse and human AKI samples (Famulski et al., 2012; Rudman-Melnick et al., 2020), showed high gene activity in Type1 injured PT cells (Figure 2C and S2F). This PT subpopulation also exhibited activated EGFR signaling (Figure S2F), a pathway known to promote PT recovery after AKI (Tang et al., 2013). For Type2 injured PT, we observed some cluster-specific DEGs including Slc6a6, Bcat1 and Slc7a12, but others that were in common with the FR-PTC cluster (e.g., Sema5a, Dcdc2a, Ypel2) (Figure S2E), hinting at a lineage relationship between Type2 PT and FR-PTC. Correlation analysis confirmed high similarity between Type2 PT and FR-PTC compared with the other cell states (Figure S2G). Mapping the Type1 and Type2 subclusters back onto the entire dataset revealed that they constituted the major cluster annotated as “PT-Inj” (Figure S2H). Collectively, these results led us to hypothesize that the Type1 PT injury state is protective while the Type2 state leads to FR-PTC driving fibrogenesis.
To better characterize potential lineage relationships between Type1/Type2 injured PT and other PT subpopulations, we leveraged single-cell trajectory inference analysis (Qiu et al., 2017) and identified two major trajectories starting from Type1 injured PT in uni-IRI (Figure 2F, left panel): these cells first became repairing cells and then either differentiated into healthy PT cells (successful repair trajectory) or FR-PTC (failed repair trajectory). Consistent with this result, the successful repair trajectory upregulated healthy PT marker genes but the FR-PTC lineage failed to do so (Figure S2I). By contrast only one major trajectory was observed in UUO: it started from healthy cells and ended at FR-PTC with Type2 injured PT cells located in between (Figure 2F, right panel). In this UUO trajectory, expression of FR-PTC markers gradually increased over pseudotime (Figure S2I). This analysis suggested that the Type1 injury state was a bipotential one with the ability to become either healthy cells or FR-PTC, whereas the Type2 category was unipotential with the ability only to differentiate into FR-PTC. We also performed trajectory analysis on dataset combining uni-IRI and UUO cells, which presented consistent results (Figure S2J). To further support this hypothesis, we conducted a computational cell fate mapping analysis, which simulated a birth-death process based on the Markov chains model (Lange et al., 2022) (see Methods). We observed that in uni-IRI, Type1 injured PT cells showed the highest probability of contributing to the repairing cell lineage (Figure 2G, left panel), which could further develop into either healthy cells or FR-PTC (Figure S2K). By contrast, the majority of Type2 injured PT cells in UUO differentiated into FR-PTC (Figure 2G, right panel). Even though a small proportion of Type2 injured PT cells were predicted to acquire a repairing cell state, most of these ultimately adapted the FR-PTC phenotype (Figure S2K).
Dysregulated Proximal Tubule Lipid Metabolisms during Fibrogenesis
Next, we aimed to explore metabolic mechanisms underlying the distinct fate outcomes of Type1 and Type2 injured PT cells. Protein-protein interaction enrichment analysis on the top DEGs for Type1 injured PT cells highlighted terms associated with lipid metabolism, including mitochondrial long chain fatty-acid β-oxidation (FAO), regulation of lipid transport and lipid localization (Figure 3A). Previous studies have demonstrated defective FAO metabolism of PT cells in CKD, which could be reversed by restoring the capacity of FAO (Kang et al., 2014; Pei et al., 2020; Stadler et al., 2015; Wu et al., 2020). Consistently, by scoring genes involved in FAO across all PT cells, we observed a reduced FAO activity in the middle stages of uni-IRI (i.e., D2/7/14) and all UUO samples (Figure 3B and S3A). FR-PTC exhibited the lowest FAO score when compared with healthy PT (Figure 3B) and the proportion of FR-PTC correlated negatively with FAO activity (Figure S3B), highlighting the central role of this population in CKD.
Figure 3. Dysregulated lipid metabolism in proximal tubule cells during fibrogenesis and activated fatty acid oxidation after short-term lipid deposition.
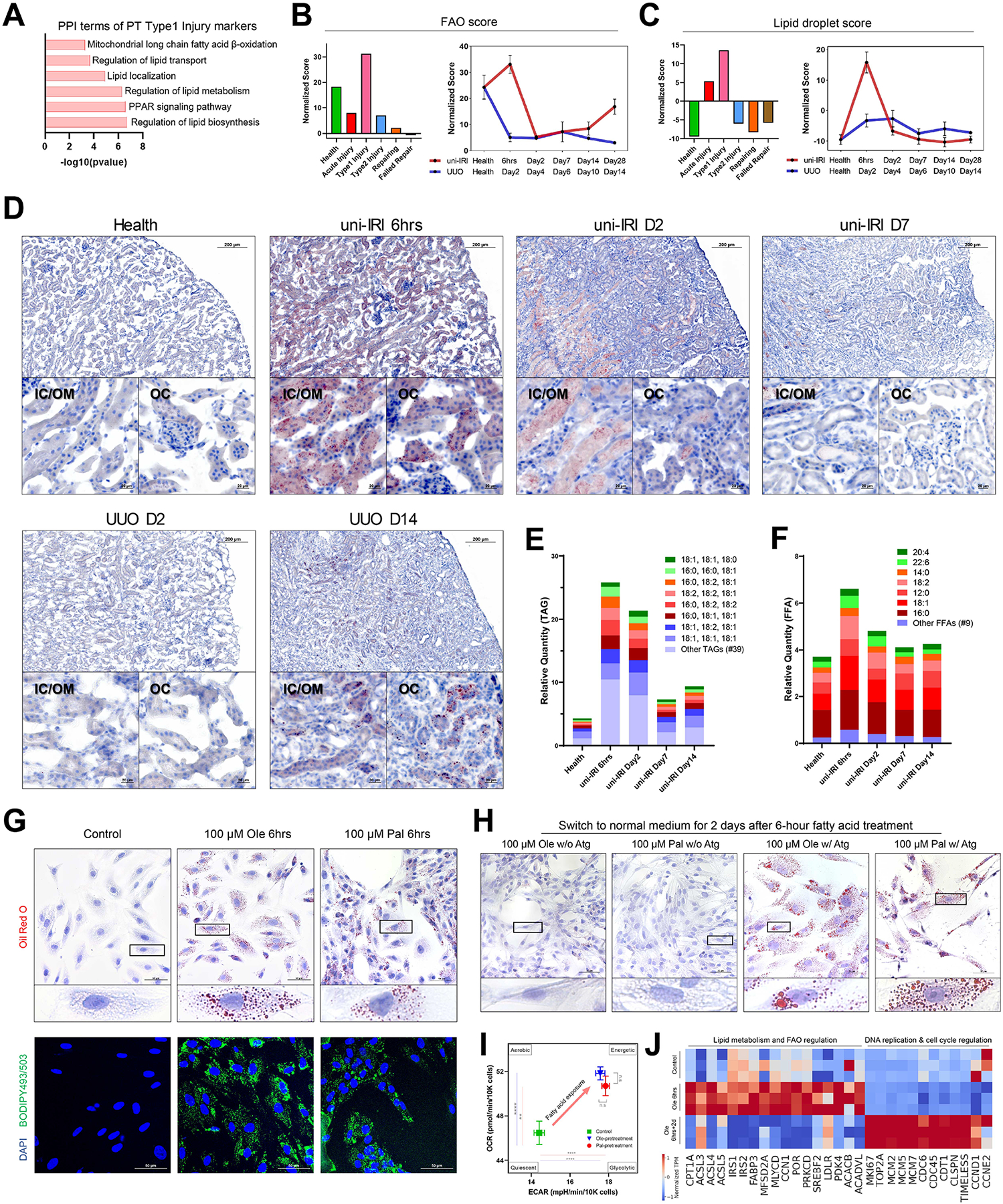
(A) Protein-protein interaction (PPI) enrichment analysis on upregulated differentially expressed genes of Type1 injured PT cells showing terms associated with lipid metabolism.
(B-C) Gene module activity scores of FAO (B) and lipid droplets (C) in different PT clusters (all time points) (left panels) and across the time courses of uni-IRI and UUO (right panels), where each dot indicates mean score of two samples of a group condition and data are shown as the mean ± SEM.
(D) Oil Red O staining on multiple group conditions identifying transient accumulation of lipids at uni-IRI 6hrs, clearance of lipids after uni-IRI D2 and lipid accumulation at late stages of UUO. Regions of inner cortex or outer medulla (IC/OM) and outer cortex (OC) are shown. Red color indicates lipids and blue indicates cell nucleus. See also Figure S3C.
(E) Relative quantity of triglyceride (TAG) species in mouse kidney tissues of different group conditions. The most abundant 8 TAG species are presented and the other species are combined and annotated as ‘Other TAGs’. See also Figure S3D.
(F) Relative quantity of free fatty acid (FFA) species in mouse kidney tissues of different group conditions. The most abundant 7 FFA species are presented and the other species are combined and annotated as ‘Other FFAs’. See also Figure S3D.
(G) Oil Red O staining (upper panels) and BODIPY493/503 staining (lower panels) on RPTECs after 6-hour treatment of 100 μM BSA-conjugated oleate (Ole) or palmitate (Pal) fatty acids. Scale bars: 50 μm. Zoom-in figures of a single cell are also presented for Oil Red O staining, which demonstrates accumulation of lipid droplets after treatment.
(H) Oil Red O staining on RPTECs which were exposed to culture medium without fatty acid supplements after 6-hour of 100 μM oleate or palmitate fatty acid treatment, with (w/) or without (w/o) Atglistatin (Atg) treatment. Scale bars: 50 μm. Zoom-in figures of a single cell are also presented.
(I) Energy map presenting an increased OCR and ECAR at the basal condition after 6-hour 100 μM oleate or palmitate fatty acid pretreatment on RPTECs. OCR and ECAR readouts are normalized by cell numbers. Data are shown as the mean ± SEM. The four energy states were annotated as previously described. **p < 0.01, ****p < 0.0001 and n.s (not significant) by Student’s t test.
(J) Heat map showing expression of genes involved in lipid metabolism & FAO regulation and DNA replication & cell cycle regulation in RPTECs with control and 6-hour 100 μM oleic acid (Ole 6hrs) treatments and harvested at 2 days after the 6-hour treatment (Ole 6hrs+2d). Each group has three biological replicates. Transcript per million (TPM) is normalized for visualization.
By contrast, we noticed an unexpected increase in the FAO score at uni-IRI 6hrs (Figure 3B), which implied activated lipid metabolism in Type1 injured PT cells at this early phase. In addition, we observed activated peroxisome proliferator-activated receptor signaling, reflected by significantly increased expression of FAO rate limiting genes such as Cpt1a, Acox1, Hadha and Hadhb (Figure S3A) (Mann-Whitney-U test with the Benjamini-Hochberg correction).
We hypothesized that the upregulated FAO gene expression at uni-IRI 6hrs would be accompanied by increased lipid deposition. A variety of proteins are required for maintenance of cytoplasmic lipid droplets, so we examined the expression of genes involved in formation and maintenance of lipid droplets. Consistent with this hypothesis, the lipid droplet score was significantly increased at uni-IRI 6hrs but returned to baseline for all subsequent timepoints (Figure 3C). The expression of lipid droplet genes Plin2, Fabp4, Acsl4 and Ehd1 were upregulated in the Type1 injured PT cells compared to the healthy (Figure S3A).
Next, we analyzed lipid content at multiple timepoints identifying a striking increase in Oil Red O-positive lipid droplets throughout both cortical and medullary tubules at uni-IRI 6hrs compared to healthy kidney (Figure 3D). Interestingly, at uni-IRI D2, most lipids were cleared from cortical tubular cells though we also observed mild persistence of intraluminal, extracellular lipids in outer medullary casts (Figure 3D). Oil Red O-positive lipid droplets were undetectable at uni-IRI D7 and later timepoints (Figure 3D and S3C). On the other hand, UUO lacked the early lipid droplet accumulation but gradually accumulated tubular lipids over time (Figure 3D and S3C).
To quantitatively determine this transient lipid accumulation, we measured the abundance of triglycerides (TAGs), free fatty acids (FFAs) and cholesterol in uni-IRI mouse kidney tissues from multiple timepoints with mass spectrometry. With a total of 47 TAG species analyzed, this lipidomics analysis revealed a ~6-fold increased abundance of total TAGs at uni-IRI 6hrs compared to healthy tissues (Figure 3E). ~70% of the accumulated TAGs were a combination of palmitate (16:0), oleate (18:1) and linoleate (18:2). Consistent with our Oil Red O staining, TAG was still abundant at uni-IRI D2, but it decreased nearly to baseline by uni-IRI D7 and D14 (Figure 3E). We also observed a 1.8-fold increased abundance of total FFAs at uni-IRI 6hrs compared to healthy (Figure 3F). Among the 16 FFA species analyzed, palmitic acid (16:0) and oleic acid (18:1) were the two major FFAs in both healthy and diseased mouse kidneys, constituting ~50% of total FFAs (Figure 3F). Lipidomic analysis was also performed on UUO D10 and UUO D14 samples, where we identified ~2-fold increased TAG abundance but almost no differences in FFA abundance (Figure S3D). In addition, we did not identify obvious changes in cholesterol abundance across samples (Figure S3E).
Therefore, our results suggested a transient upregulation of genes involved in FAO and lipid metabolism, accompanied by cytoplasmic lipid accumulation, at the earliest timepoints after uni-IRI.
Fatty acid exposure in vitro leads to lipid accumulation and FAO burst
To study how PT cells respond to short-term lipid accumulation, we established an in vitro model by treating primary human renal proximal tubule epithelial cells (RPTECs) with oleic or palmitic acids for 6 hours. Oil Red O and BODIPY 493/503 staining confirmed a striking increase in intracellular lipid deposition after 6-hour exposure of oleic or palmitic acids (Figure 3G). A longer exposure (2–6 days) to fatty acids resulted in an increased size of Oil Red O+ lipid aggregates (Figure S3H). We also treated RPTECs with fluorescently labeled palmitic acid (BODIPY C16) for 6 hours and validated that RPTECs actively transported fatty acids leading to intracellular lipid accumulation (Figure S3F). Both 6-hour oleic and palmitic acid exposure led to significant upregulation of CD36, which encodes a plasma membrane receptor for long-chain fatty acid transport and CPT1A, which encodes a mitochondrial membrane enzyme for long-chain fatty acyl-CoA transport (Figure S3G).
Previous studies reported mitochondrial dysregulation in PT cells in kidney diseases (Chung et al., 2019; Mori et al., 2021; Zhan et al., 2013). We stained for mitochondria and observed that most mitochondrion had a thread-like appearance in the steady state, but an increased fraction of mitochondria became fragmented into a sphere-like appearance (i.e., mitochondrial fission) after 6-hour oleic or palmitic acid treatment (Figure S3I). On the other hand, we did not observe significant changes in reactive oxygen species, a feature of mitochondrial damage, after the 6-hour fatty acid treatment (Figure S3J).
Next, to answer whether the accumulated lipid droplets could be oxidized later, we exposed RPTECs to lipids for 6 hours then washed them away and replaced with normal culture medium (without fatty acid supplements) for 2 days. We observed very little Oil Red O staining at 2 days post culture medium renewal (Figure 3H), indicating that the deposited lipids induced by 6-hour fatty acid treatment were largely cleared from cells by this timepoint. We asked whether this lipid clearance was the consequence of oxidation or due to other mechanisms such as lipid secretion. The presence of the lipolysis inhibitor Atglistatin during the two day chase prevented clearance of lipid droplets for both palmitic and oleic acids (Figure 3H), while 2-day treatment of Atglistatin alone did not induce significant lipid accumulation (Figure S3K). These results strongly suggest that RPTEC clearance of lipid accumulation occurs through FAO.
To directly measure FAO and determine whether a dysregulation of glucose metabolism might also be involved, we measured the real-time oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) on RPTECs after 6-hour fatty acid treatment, in a similar approach as described before (Kang et al., 2014). We identified a significantly higher OCR in cells pretreated with oleic or palmitic acids than control cells (Figure 3I and S3L), suggesting enhanced FAO activity. Injection of etomoxir, a CPT1 inhibitor, and oligomycin, an ATP synthase inhibitor, both reduced OCR, confirming that the increased OCR observed was the consequence of increased FAO (Figure S3L). In addition, an increased ECAR was also identified in cells pretreated with fatty acids (Figure 3I and S3L). These results suggest that 6-hour fatty acid exposure increases both FAO and glycolysis activity, characteristics of an energetically active cell state (Hocaoglu et al., 2021).
Next, to study cellular responses to lipid accumulation at the gene expression level, we performed bulk RNA-seq on RPTECs treated with oleic acids for 6 hours (Ole_6hrs). We also sequenced cells that were exposed to medium without fatty acid supplements for 2 days after the 6-hour treatment (Ole_6hrs+2d) to study the long-term effect of lipid accumulation. GO analysis indicated that the upregulated DEGs of the Ole_6hrs group compared to control cells were enriched in intracellular lipid droplets (FDR = 3.84×10−3). Consistently, genes associated with FAO and lipid metabolism were significantly upregulated in the Ole_6hrs group (GO term FDR = 7.94×10−5), including CPT1A and genes encoding Long-chain acyl-CoA synthetases (ACSLs) (Figure 3J). Glucose metabolic process was also an upregulated GO term in the enrichment analysis (FDR = 5.88×10−3), supporting our ECAR measurement mentioned above. Interestingly, we found that genes involved in DNA replication and cell cycle regulation were significantly upregulated in the Ole_6hrs+2d group (GO term FDR = 6.36×10−14), including MKI67, TOP2A and genes encoding minichromosome maintenance (MCM) proteins (Figure 3J). Thus, our results suggested that fatty acid exposure and lipid accumulation in RPTECs promotes cell proliferation, consistent with our single-cell fate mapping analysis identifying that Type1 injured PT cells (enriched at uni-IRI 6hrs) were precursors of Mki67-expressing Repairing PT cells (enriched at uni-IRI D2) (Figure 2F–G).
PLIN2 marks lipid droplets in Type1 injured proximal tubular cells and maintains cell energy state
PLIN2, also known as perilipin 2 or adipose differentiation-related protein, is a lipid droplet surface protein and an essential component of the PPAR signaling pathway (Kimmel and Sztalryd, 2016). Our scRNA-seq data identified Plin2 as a marker gene of Type1 injured PT cells (Figure 2B and 4A). Reanalyzing a recently published spatial transcriptomic analysis of mouse kidney bi-IRI (Dixon et al., 2021) revealed the transiently increased expression of Plin2 throughout the kidney cortex at 12hrs post-surgery (Figure 4B). To further validate Plin2 as a marker gene for Type1 injured PT cell state, we performed immunofluorescence and identified that PLIN2 localized to intracellular basolateral droplets at uni-IRI 6hrs but no other timepoints (Figure 4C–D). PLIN2 expression was absent in the time course of UUO, including at UUO 6hrs (Figure 4C). To further validate that PLIN2 expression co-localized with lipid compounds, we stained PLIN2 with lipid probe BODIPY 493/503 on the uni-IRI 6hrs tissue and found that PLIN2 coated the surface of BODIPY+ lipid particles (Figure 4E). PLIN2 also co-localized with oxidized low-density lipoprotein (oxLDL) (Figure 4F). Together, our results identified PLIN2 as a marker Type1 injured PT cells and a surface protein of intracellular lipid droplets. Surveying a previous RNA-seq study on folic acid-induced mouse nephropathy (Craciun et al., 2016) identified upregulation of Plin2 at one day post injury and then gradual decreased expression (Figure S4A), suggesting that increased Plin2 expression could be induced by other types of kidney injury.
Figure 4. PLIN2 marks lipid droplets in Type1 injured proximal tubule and maintains cell energy state.
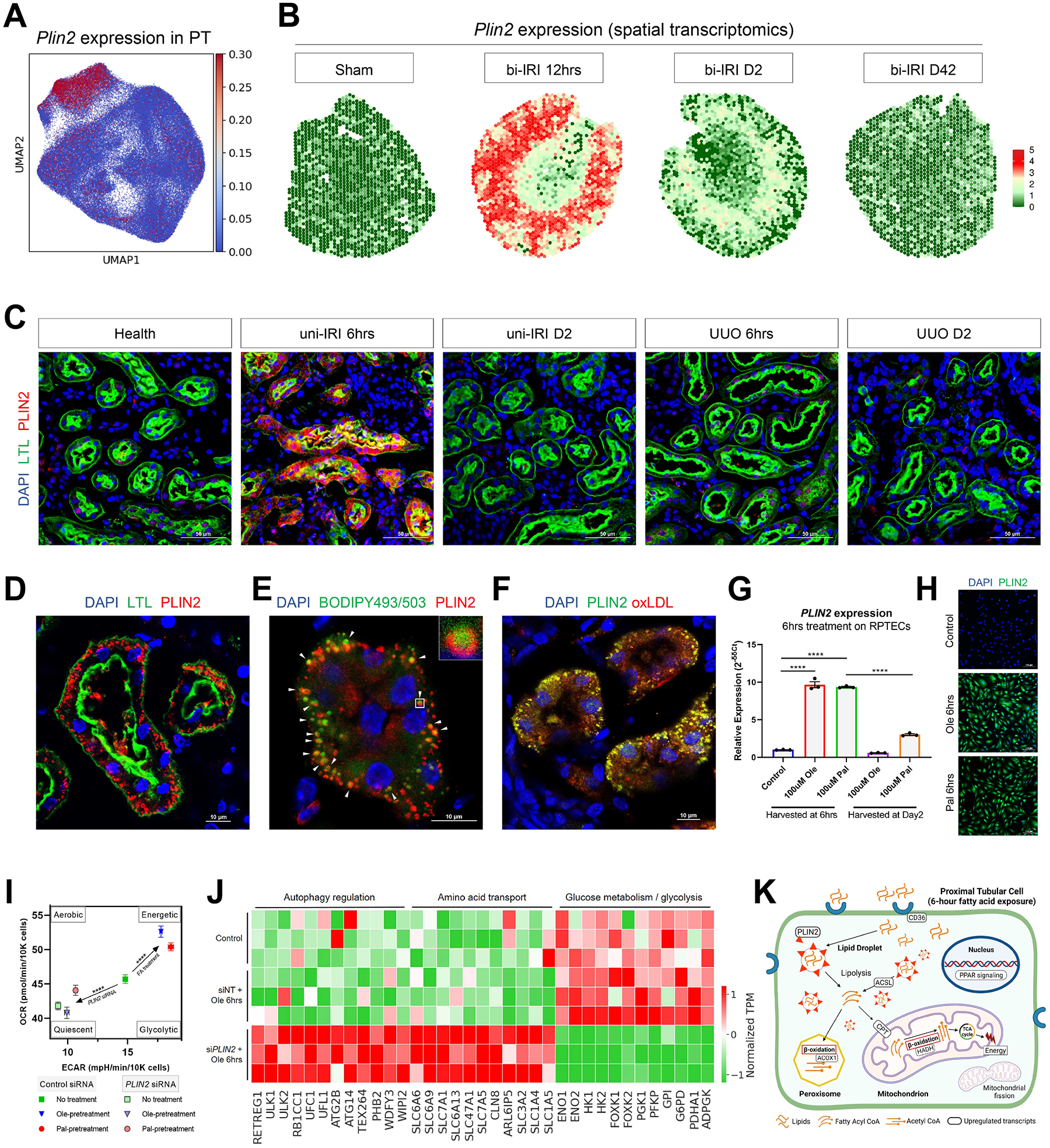
(A) Specific expression of Plin2 in Type1 injured PT cells.
(B) Revisiting a spatial transcriptomics dataset on female bi-IRI kidneys identifying transient upregulation of Plin2 at kidney cortex in early stages. Each spot of a tissue section is colored by gene expression.
(C) Specific upregulation of PLIN2 in PT at uni-IRI 6hrs validated by immunofluorescence staining of PLIN2 (red), LTL (green) and DAPI (blue) on multiple group conditions. Scale bars: 50 μm.
(D) Immunofluorescence staining of PLIN2 (red), LTL (green) and DAPI (blue) on a tissue section collected from uni-IRI 6hrs showing presence of intracellular PLIN2+ droplets. Scale bar: 10 μm.
(E) Immunofluorescence staining of PLIN2 (red), BODIPY493/503 (green) and DAPI (blue) on a uni-IRI 6hrs tissue section showing localization of PLIN2 at the surface of lipid droplets. Scale bar: 10 μm. A single droplet was encircled and presented in the top-right panel.
(F) Immunofluorescence staining of PLIN2 (green), oxidized low-density lipoprotein (oxLDL; red) and DAPI (blue) on a uni-IRI 6hrs tissue section showing PLIN2 colocalizes with oxLDL. Scale bar: 10 μm.
(G) Relative expression of PLIN2 in RPTECs after 6-hour oleate (Ole) or palmitate (Pal) fatty acid exposure or at 2 days after removal of the fatty acids from culture medium, measured by qPCR. Data are shown as the mean ± SEM. ****p < 0.0001 by Student’s t test.
(H) Immunostaining of PLIN2 (green) and DAPI (blue) on RPTECs after 6-hour oleate or palmitate fatty acid treatment. Scale bar: 100 μm.
(I) Energy map presenting an increased OCR and ECAR after 6-hour fatty acid pretreatment, as well as a decreased OCR and ECAR after PLIN2 knockdown on RPTECs. OCR and ECAR readouts are normalized by cell numbers. Data are shown as the mean ± SEM. Comparisons were made between No treatment and combined oleate & palmitate fatty acid-pretreatment (Control siRNA), and between No treatment with Control siRNA and No treatment with PLIN2 siRNA. The four energy states were annotated as previously described. ****p < 0.0001 by Student’s t test.
(J) Heat map showing expression of genes involved in autophagy, amino acid transport and glucose metabolism in RPTECs with non-targeting siRNA (control) and 6-hour 100 μM oleic acid (siNT+Ole 6hrs) treatments and in cells treated with PLIN2 siRNA and 6-hour 100 μM oleic acid (siPLIN2+Ole 6hrs). Each group has three biological replicates. TPM expression is normalized for visualization.
(K) Proposed model of activated lipid metabolisms in Type1 injured PT cells. CD36 is a transporter of long-chain fatty acids. Intracellular fatty acids aggregate and form lipid droplets with PLIN2 as a surface protein. Fatty acyl-CoA (Coenzyme A) is converted from lipids through ACSL-mediated lipolysis or lipophagy and used in ACOX1-mediated peroxisomal β-oxidation or CPT-mediated mitochondrial β-oxidation, which can generate acetyl CoA, the substrate of tricarboxylic acid (TCA) cycle to produce energy. PPAR signaling is the main regulator of the lipid metabolism pathway and mitochondrial fission is involved in lipid accumulation. Figure created with BioRender.com.
Next, we sought to investigate the mechanism of Plin2 upregulation in Type1 injured PT cells. Previous studies have reported that PLIN2 expression can be increased either by cellular uptake of fatty acids or ER (endoplasmic reticulum) stress (Chen et al., 2017; Dalen et al., 2006; Gao and Serrero, 1999). Therefore, we exposed free fatty acids or chemical ER stress inducers on RPTECs. We observed ~ 10-fold increased expression of PLIN2 after a 6-hour treatment of oleic or palmitic fatty acids by qPCR analysis (Figure 4G). PLIN2 expression was significantly reduced when cells were exposed to culture medium without fatty acid supplements for 2 days (Figure 4G), suggesting a positive correlation between PLIN2 expression level and abundance of lipids. 6-hour treatment of ER stress inducers including Tunicamycin and Thapsigargin could also increase PLIN2 expression, but the fold change (~ 1.5-fold) was much lower than observed with fatty acid treatments (Figure S4B). We also performed immunofluorescence on fatty-acid-treated RPTECs and confirmed strongly upregulated PLIN2 protein expression in almost all cells (Figure 4H). With the bulk RNA-seq data on RPTECs mentioned above, we surveyed the gene expression of all PLIN family members (Figure S4C) and found that PLIN4 was another significantly upregulated gene (1.9-fold) after 6-hour oleic acid treatment but it was much more lowly expressed compared to PLIN2 (average transcript per million (TPM) of PLIN2: 1104.90; average TPM of PLIN4: 0.93). Therefore, in vitro modelling of PLIN2 activation was consistent with our in vivo observation in uni-IRI mouse surgery, indicating that fatty acid exposure is sufficient to induce PLIN2 upregulation in PT cells.
To further investigate the functional significance of PLIN2 in response to lipid accumulation, we performed PLIN2 gene knockdown with small interfering RNA (siRNA) on RPTECs. Successful gene knockdown was validated with qPCR analysis, revealing ~15–20 folds decreased expression of PLIN2 in cells treated with PLIN2 siRNA (siPLIN2) both with and without fatty acid exposure compared with corresponding controls (Figure S4D). Importantly, siPLIN2 treatment did not significantly alter fatty acid uptake and lipid accumulation as demonstrated by Oil Red O staining (Figure S4E). Next, to ask whether PLIN2 was important for cellular metabolic activities, we imposed 6-hour fatty acid treatment on RPTECs with or without siPLIN2 and measured real-time OCR and ECAR. In the absence of fatty acid pretreatment, we observed a significantly reduced OCR and ECAR in cells treated with siPLIN2 compared with cells treated with non-targeting control siRNA (siNT) (Figure 4I and S4F). With 6-hour oleic or palmitic acid pretreatment, both OCR and ECAR were significantly increased in siNT-treated cells (Figure 4I and S4F), consistent with our results presented in Figure 3I. By contrast, for cells treated with siPLIN2, the decreased OCR was only partially reversed by 6-hour palmitic acid pretreatment and could not be increased by oleic acid exposure (Figure 4I and S4F), implying defective lipid metabolism after PLIN2 knockdown. Both decreased OCR and ECAR suggested that siPLIN2 knockdown drove a metabolically quiescent cell state.
Next, we exposed siPLIN2-treated RPTECs to oleic acids for 6 hours (siPLIN2+Ole6hrs) and performed RNA-seq to determine the transcriptomic variations caused by gene knockdown. Compared with the siNT+Ole6hrs group, the siPLIN2-treated cells upregulated genes associated with autophagy and reticulophagy (GO term FDR = 4.28×10−3) such as genes encoding autophagy activating kinases ULK1/2 (Figure 4J). Previous studies have reported autophagy can induced by cell stress and nutrient deprivation in kidney and persistent activation of autophagy after kidney injury leads to maladaptive repair (Tang et al., 2020), implying that normal PLIN2 function could be essential for successful repair of Type1 injured PT cells. We also identified a decreased glucose metabolism gene profile and increased expression of genes involved in amino acid transport in siPLIN2+Ole6hrs cells compared to siNT+Ole6hrs (Figure 4J). The reduced expression of genes responsible for glycolysis (GO term FDR = 4.22×10−3) such as ENO1/2 and HK1/2 was consistent with our observation of decreased ECAR after PLIN2 knockdown (Figure 4I).
In the above analysis, we found that genes associated with DNA replication and cell cycle regulation were upregulated in Ole_6hrs+2d cells compared to control (Figure 3J). We wondered whether PLIN2 knockdown undermined this cellular event as it disrupted the metabolic cellular response after 6-hour fatty acid exposure. Thus, cells exposed to normal culture medium for 2 days after the 6-hour oleic acid treatment (siPLIN2+Ole6hrs+2d) were analyzed by RNA-seq. Compared with siNT+Ole6hrs+2d, we found that DNA replication was a downregulated GO term in siPLIN2+Ole6hrs+2d cells (FDR = 7.72×10−5), reflected by decreased expression of genes associated with DNA primase activity (e.g., GINS1/2 and PRIM2), DNA polymerase regulation (e.g., RFC2/5 and PCNA) and genes encoding MCM proteins (Figure S4G). Therefore, this RNA-seq analysis indicated that PLIN2 knockdown reduced activities of DNA replication and cell proliferation after 6-hour fatty acid uptake.
Taken together, our results highlight PLIN2 as a marker of intracellular lipid droplets in Type1 injured PT cells and knockdown studies show that PLIN2 regulates energy homeostasis in PT cells.
Metabolic Variations in Type2 Injured Proximal Tubule Cells
To better characterize the metabolic consequences of Type2 injured PT, we performed protein-protein interaction enrichment analysis and identified amino acid metabolism as a downregulated pathway compared to healthy PT (Figure S5A), consistent with previous studies reporting defective amino acid metabolisms in CKD (Garibotto Giacomo et al., 2010; Kang et al., 2014). We also identified several genes associated with amino acid transport and catalysis that were among top upregulated markers of S3 segment cells of Type2 injured PT, including Bcat1, Slc6a6 and Slc7a12 (Figure 5A). A survey of the Human Protein Atlas (Uhlén et al., 2015) confirmed that the proteins encoded by these genes were expressed in the renal tubule. Revisiting a published RNA-seq work on PT-enriched transcripts of UUO mice (Wu et al., 2020) further validated increased expression of Bcat1, Slc6a6 and Slc7a12, as well as other DEGs of Type2 injured PT, in UUO D5/10 mouse kidneys than contralateral control kidneys (Figure S5B).
Figure 5. Dysregulation of genes involved in amino acid metabolisms in Type2 injured PT cells.
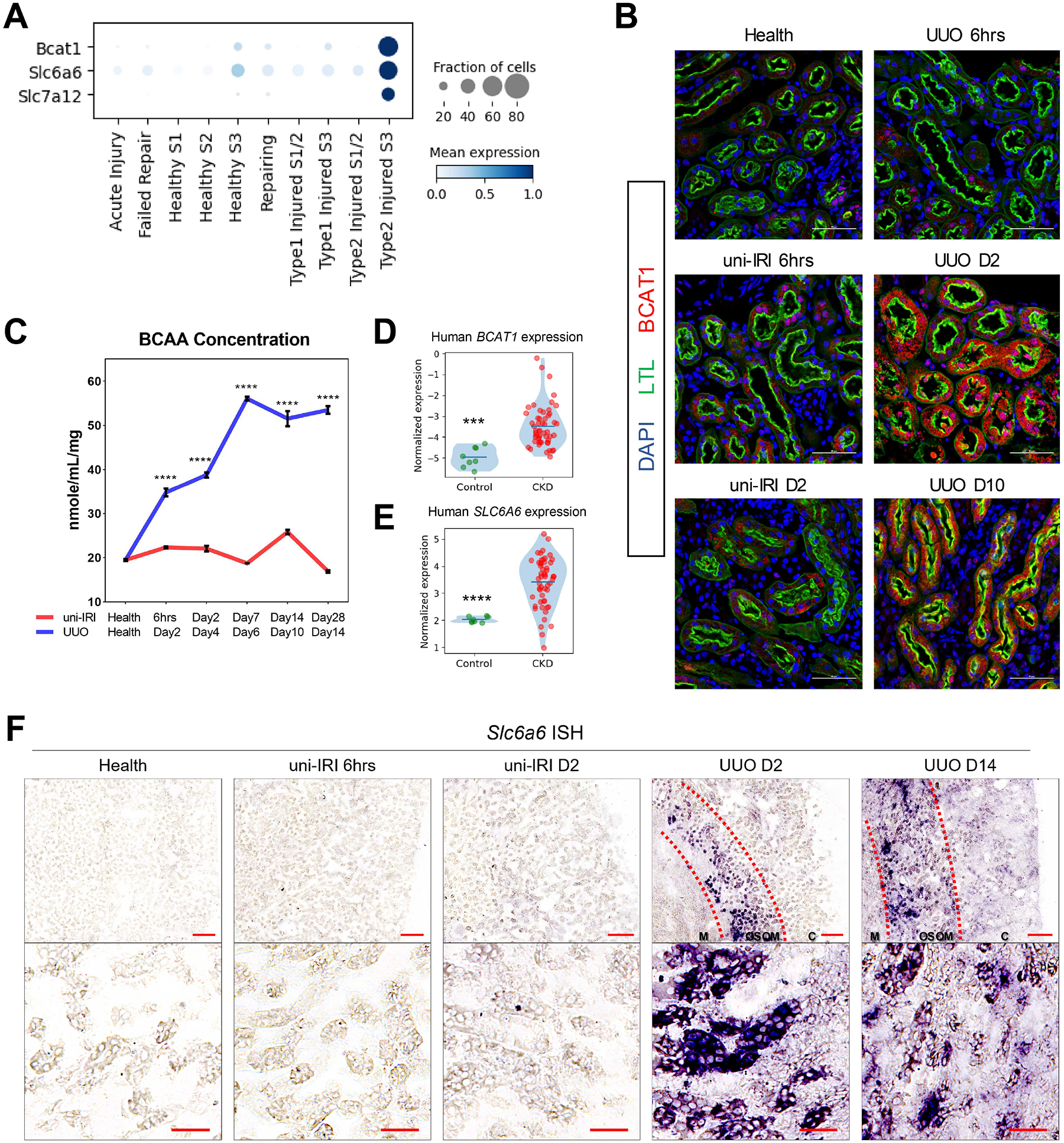
(A) Dot plot showing that three amino acid metabolism-associated genes, Bcat1, Slc6a6 and Slc7a12, were specifically upregulated in Type2 injured PT.
(B) Specific upregulation of BCAT1 in PT after UUO validated by immunofluorescence staining of BCAT1 (red), LTL (green) and DAPI (blue) on multiple group conditions. Outer medulla regions are presented. Scale bars: 50 μm.
(C) Concentrations of branched-chain amino acids (BCAA) measured in mouse cortical tissues across all group conditions of this study cohort showing significantly increased BCAA concentration in UUO kidneys. Data are shown as the mean ± SEM. ****p < 0.0001 by Student’s t test.
(D-E) Violin plots showing increased expression of BCAT1 (D) and SLC6A6 (E) in biopsy samples of patients with CKD than controls.
(F) Representative images of RNA in situ hybridization staining of Slc6a6 on multiple group conditions revealing gene upregulation in UUO. Scale bars: 200 μm for the upper panel and 50 μm for the lower panel. Cortical (C), outer stripe of the outer medulla (OSOM) and medullary (M) regions were highlighted. See also Figure S5E.
Bcat1 is branched chain amino acid transaminase 1 and it is responsible for transamination of branched chain amino acids (BCAAs, including leucine, isoleucine and valine) resulting in production of branched chain keto acids (BCKAs) and glutamate (Adeva et al., 2011). Immunostaining results verified that the expression of BCAT1 was specific to PT cells and mostly limited to UUO (Figure 5B), where Type2 injured cells were highly abundant. The upregulation of BCAT1, compared to healthy controls, was also observed in UUO D10/14 (Figure 5B and S5C), in which the Type2 state still existed. In addition, we found that genes that are responsible for BCKA catalysis, including Bckdha, Bckdhb and Ppm1k, were downregulated in injured PT compared to healthy PT (Figure S5D), confirming a recent report (Piret et al., 2021). Next, we measured the concentration of BCAAs in mouse kidney cortical tissues across multiple timepoints. We identified an increased BCAA accumulation during the UUO time course and the concentration in the uni-IRI time course was not changed too much (Figure 5C). Re-analyzing a previous human dataset (Nakagawa et al., 2015) confirmed a significantly increased BCAT1 expression in patients with CKD than controls (Figure 5D).
SLC6A6, also known as TauT, is a transporter of the sulfur-containing amino acid taurine, and the accumulation of taurine has been described in patients with kidney failure (Mozaffari, 2003; Suliman et al., 2002). We found the expression of SLC6A6 was also significantly elevated in patients with CKD than healthy controls (Figure 5E). Next, we validated the increased expression of Slc6a6 in Type2 injured PT by RNA in situ hybridization (RNA ISH), in which the expression of Slc6a6 was not observed in healthy and uni-IRI samples, but was upregulated as early as at UUO D2 in outer stripe of the outer medulla of kidney, where S3 segment of PT cells are supposed to locate, (Figure 5F). We also co-stained Slc6a6 with Havcr1 and validated the expression of Slc6a6 in Havcr1-expressing injured PT (Figure S5E). Slc7a12 encodes a transporter for cationic amino acids and recent work demonstrated that Slc7a12 was present in kidney PT in disease and the upregulation was accompanied by emergence of Vcam1-expressing FR-PTC (Gerhardt et al., 2021), concordant with our analysis identifying the high probability of Type2 injured PT differentiating into FR-PTC (Figure 2F–G). Taken together, these results suggest that Type2 PT cells are characterized by dysregulated amino acid metabolisms.
Shared and Unique Cellular Response of Tubular Epithelia in Fibrogenesis
Our analysis revealed that most injured PT cells in uni-IRI repair (Figure 2E, left panel), whereas they do not in UUO (Figure 2E, right panel). We next sought to compare shared and unique responses to injury across tubular epithelial cell types. We identified multiple subtypes of loop of Henle (LoH) (Figure 6A) and cells of the distal nephron, including distal convoluted tubule (DCT), connecting tubule (CNT) and principal cells (PC) of collecting duct (Figure 6B) and Type A and Type B intercalated cells of collecting duct (Figure S6A). Subclustering of LoH showed thick ascending limb (TAL) to be the abundant population, and it expressed marker genes such as Slc12a1 and Umod (Figure 6C). TAL cells could also be stratified by their cortical (Kng2/Thsd4 high) or medullary (Mrps6/Tmem207 high), as could principal cells of the collecting duct (medullary PC2: Pcdh7 high; cortical PC3: Mgat4c high) (Figure S6B). Interestingly, we found another group of TAL expressing healthy marker genes at a lower level and showing enhanced expression of a well-known injury marker Lcn2 (also known as Ngal) that we annotated as injured TAL (TAL-inj). TAL-inj also showed upregulation of Kctd1, a gene that regulates reabsorption of paracellular urinary Ca2+/Mg2+ and performs a protective role in kidney fibrosis (Marneros, 2021, 2020). Compared to healthy TAL, cells of TAL-inj showed increased expression of genes associated with profibrotic and proinflammatory signaling, such as Tgfbr1, Map3k1, Stat3 and Myh9 (Figure S6C). GO enrichment analysis presented terms that also appeared in injured PT, such as cell junction organization, actin cytoskeleton regulation, cell migration and epithelial cell differentiation (Figure S6D). Using a similar approach, we identified injured DCT (DCT-inj) and CNT (CNT-inj) both of which showed downregulated healthy marker genes (e.g., Slc12a3 for DCT and Slc8a1 for CNT) and increased expression of fibrotic genes (Figure 6D and S6E). We identified Trpv5, a gene encoding a calcium channel essential for Ca2+ reabsorption in kidney (De Groot et al., 2008), as upregulated in both DCT-inj and CNT-inj.
Figure 6. Shared and unique injury responses of renal tubular epithelial cells.
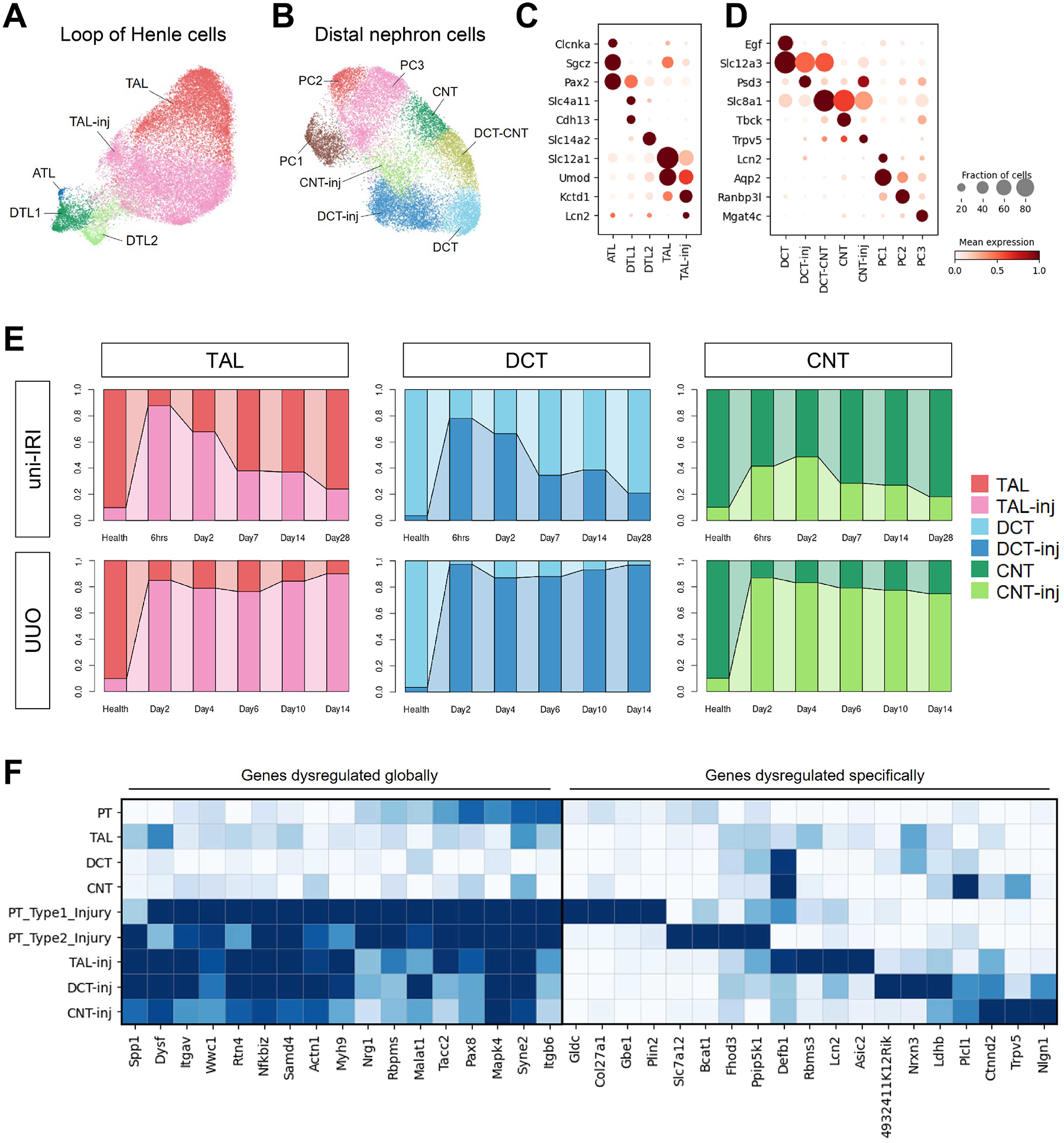
(A-B) UMAP plots of cells of LoH (A) and DCT, CNT and PC (B) in subclustering analysis. Abbreviations of cell types have been described in Figure 1D.
(C-D) Dot plots showing expression of genes specific to cell clusters identified in Figure 6A–B. Visualization was performed on dataset combining both uni-IRI and UUO subsets.
(E) Connected bar plots displaying the proportional abundance of healthy and injured TECs (including TAL, DCT and CNT) in each group condition, which identifies a shared injury response of TECs in an insult-dependent manner.
(F) Heat maps presenting expression of genes that are either co-varied across all injured TECs or dysregulated in a cell-type-specific manner compared to the healthy state of each TEC.
We next surveyed the transition between these epithelial cells in health and disease across the full time course of either uni-IRI and UUO. We found that injury cell states (i.e., TAL-inj, DCT-inj and CNT-inj) were largely absent in healthy kidneys (Figure 6E), but as expected, their numbers increased after either insult. Similar to PT, injured TAL, DCT and CNT took on a transient injury state but then repaired at later timepoints, whereas these same cell types remained injured through the UUO time course (Figure 6E).
We examined the DEGs for each injured subtype compared to its healthy state and identified those that were common to all injury states (Figure 6F, left panel). For example, Spp1 encodes osteopontin, a pleiotropic glycoprotein, which is induced in both AKI and CKD and is important for tubulogenesis (Kaleta, 2019; Khamissi et al., 2022; Wu et al., 2022). Here, its upregulation was observed not only in TAL/DCT/CNT, but also in PT, though the expression is more increased in Type2 injured PT than the Type1 state. We also identified Nrg1, which modifies EGFR signaling, as a gene that co-varied across injury states (Harskamp et al., 2016). Some identified genes have poorly understood functions in epithelia, such as Syne2, which contributes to maintenance of the nuclear envelope structure, though its role in cell proliferation in skin wound healing has also been noted (Rashmi et al., 2012). We also identified Wwc1 and its protein product (also known as KIBRA, kidney and brain expressed protein) as an upstream regulator of Hippo pathway and KIBRA overexpression can disrupt cytoskeleton of podocytes via inhibiting YAP signaling (Meliambro et al., 2017). Each injured nephron segment also expressed transcripts unique to that segment (Figure 6F, right panel), such as upregulation of Rbms3 (encoding a c-Myc single-strand binding protein (Penkov et al., 2000)) in TAL-inj and decreased expression of Plcl1 (encoding a regulator of GABA(A) receptors (Kanematsu et al., 2007)) in CNT-inj.
Heterogeneity of Kidney Stroma
In response to injury, kidney resident pericytes and fibroblasts proliferate and differentiate into myofibroblasts with increased cell motility and extracellular matrix (ECM) deposition, contributing to kidney fibrosis (Kuppe et al., 2021; Sato and Yanagita, 2017). But it remains unclear whether the fibroblasts or myofibroblasts are homogenous populations or heterogeneous groups with subtypes performing distinct functions (Humphreys, 2018). We therefore next aimed to characterize kidney stromal cell heterogeneity.
Subclustering of fibroblasts and myofibroblasts led to identification of multiple subtypes of kidney stroma including Ren1-expressing JGA cells (Figure 7A and S7A). Three clusters showed elevated expression of Acta2 and Col1a1, classic myofibroblast marker genes (Myo-2/3/4) (Figure 7A and S7A). One subpopulation (Myo-1) exhibited high transcriptomic similarity with Myo-2/3/4 and showed increased expression of multiple myosin genes (Figure S7B), suggesting an enhanced capacity for cell migration and contraction, so we considered this a myofibroblast subtype as well. We annotated the remaining clusters as fibroblasts (Fib-1/2/3) due to presence of fibroblast marker genes and their high abundance in healthy kidneys (Figure 7B). Time course analysis revealed that in uni-IRI, the total number of (myo)fibroblasts peaked at Day2 and was then decreased moderately with time (Figure S7C, left panel), whereas in UUO, myofibroblasts accumulated across the time course accounting for over 30% of the total kidney cells at D14 (Figure S7C, right panel).
Figure 7. Heterogeneity of kidney stromal cells and cell-cell communications in kidney fibrogenesis.
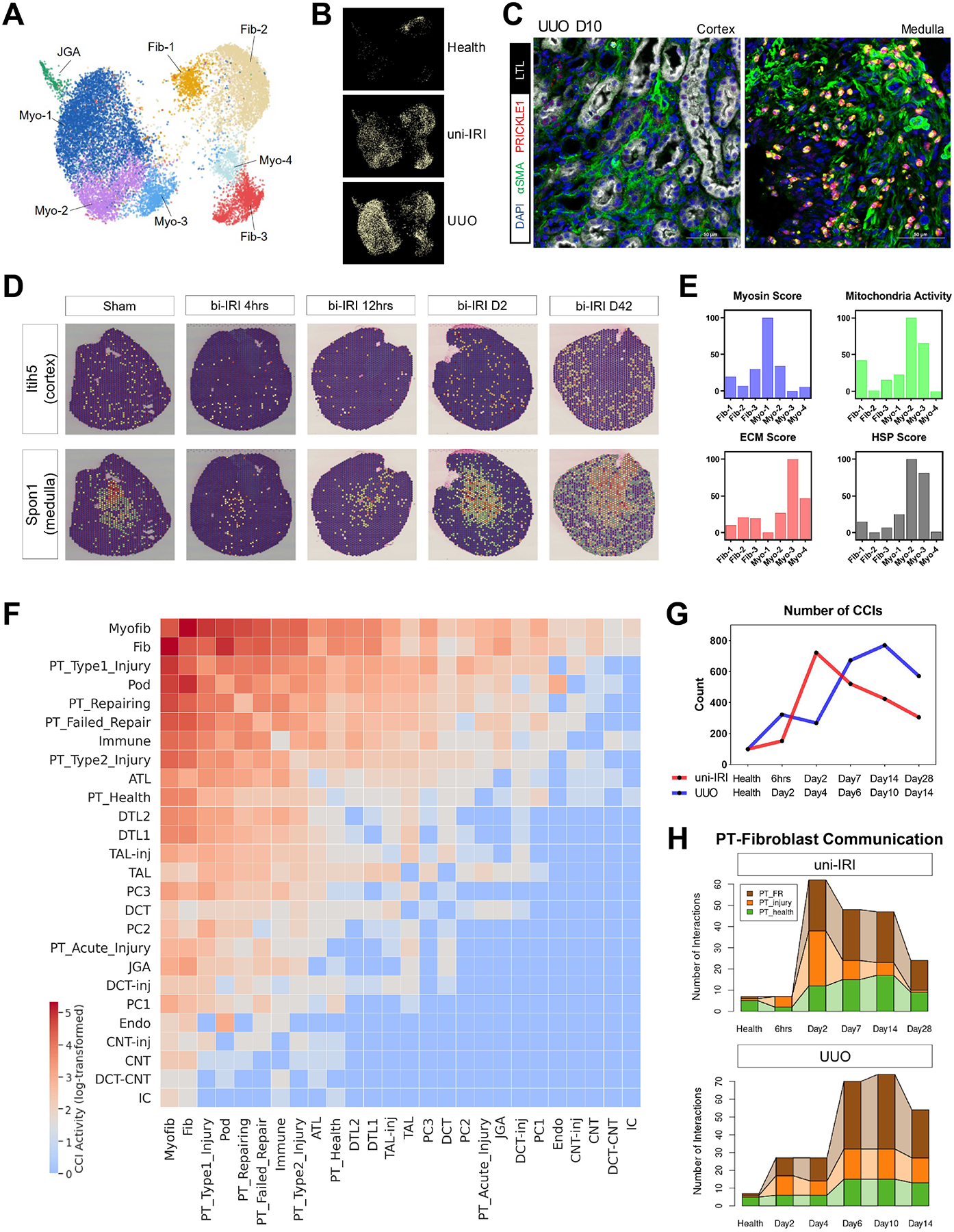
(A) UMAP plot of all stromal subtypes identified in subclustering analysis. Fib, fibroblast; Myo, myofibroblast; JGA, juxtaglomerular apparatus. See also Figure S7A.
(B) Condition map showing unique distribution of stromal cells in different experimental groups.
(C) Immunofluorescence staining of DAPI (blue), α-SMA (i.e., ACTA2) (green), PRICKLE1 (red) and LTL (white) on a tissue section collected from UUO D10 identifying PRICKLE1 expression on nuclear membranes of myofibroblasts in kidney medulla. Scale bars: 50 μm.
(D) Expression of two region-specific genes in a spatial transcriptomics dataset on female bi-IRI kidneys. Each spot of a tissue section is colored by gene expression. See also Figure S7D.
(E) Gene module activities on myosin, mitochondrial respiratory chain reactions, extracellular matrix (ECM) and heat-shock proteins (HSP) in each stromal subtype. Gene module scores are shown as means. For the convenience of data visualization, normalization is performed by adjusting the lowest score of each module as 0.
(F) Heat map showing the number of significant ligand-receptor pairs in cell-cell interaction (CCIs) analysis, predicted by CellPhoneDB, between major kidney cell types. Log-transformed data are shown. Populations with similar transcriptomics are combined for the convenience of data visualization.
(G) Numbers of significant CCIs identified by CellPhoneDB across the time courses of uni-IRI and UUO.
(H) Connected bar plots displaying the number of significant CCIs between (myo)fibroblasts and PT cells in each group condition. Fibroblast and myofibroblast are combined to increase robustness of data analysis. PT_injury combines PT-AcInj, PT-R and Type1/2 injured PT cells.
Kidney stromal heterogeneity included differences in regional localization. We identified Fib-1/2 as cortical fibroblasts (Itih5 high) and Fib-3 and Myo-4 as medullary (Spon1/Bmpr1b high) (Figure S7A). We found that Myo-4 specifically expressed Prickle1 (Figure S7A), which encodes a nuclear receptor that regulates cell polarity and is involved in Wnt signaling (Yang et al., 2013). Immunofluorescence analysis on UUO D10 tissues confirmed that PRICKLE1 was specifically expressed on nuclear membranes of α-SMA+ myofibroblasts in the inner medulla, but not in cortical regions (Figure 7C). The regional heterogeneity of stromal cells was further confirmed by spatial transcriptomic analysis of an existing dataset (Dixon et al., 2021), which indicated a higher expression of Itih5 in cortex than medulla and medulla-specific expression of Spon1 and Bmpr1b (Figure 7D and S7D).
Next, we assessed functional differences between myofibroblast subtypes. In addition to high expression of myosin genes (Figure 7E and S7B), Myo-1 cells showed increased expression of Il34 and components of the ERK/MAPK pathway (Figure S7A and S7E), indicating their inflammatory properties (Boström and Lundberg, 2013; Pat et al., 2003; Shoji et al., 2016). For Myo-2 cells, we observed upregulation of Il31ra (Figure S7A), a gene that is crucial for IL-31 signaling and has been found overexpressed in dermal fibroblasts of patients with systemic sclerosis (Kuzumi et al., 2021). Importantly, Myo-2 cells exhibited significantly enhanced activity of the mitochondrial respiratory chain as indicated by gene module scoring analysis (Mann-Whitney-U test) (Figure 7E), including subunits of NADH:ubiquinone oxidoreductase, ubiquinol-cytochrome c (CYC) reductase, CYC oxidase and ATP synthase (Figure S7F). A majority of heat shock protein-encoding genes were also upregulated in Myo-2 (Figure 7E and S7G), indicating that these cells performed highly active metabolic activities and stress response. In addition, even though all these myofibroblast subtypes had increased expression of Acta2 and Col1a1 compared with the other kidney cell types, we noticed that one myofibroblast cluster, Myo-3, exhibited the highest expression of these genes and extracellular matrix deposition score (Figure 7E), including elevated expression of various glycoproteins, collagens and proteoglycans (Figure S7H). Therefore, we annotated myofibroblast (Myo-3) as the major population responsible for ECM synthesis in kidney fibrosis. Although highly abundant collagens (i.e., collagen types I and III) were mostly detected in Myo-3, we found that genes encoding rare collagens, such as Col6a3, Col6a4, Col7a1 and Col9a1, were produced in myofibroblast group Myo-4 (Figure S7I), highlighting potential functional differences within myofibroblast subtypes.
Dynamics of Cell-cell Interactions in Kidney Fibrogenesis
Intercellular communication drives kidney fibrosis. We analyzed cell-cell interaction (CCI) activity across all major cell types based on their ligand-receptor transcriptomic signature and identified that fibroblast and myofibroblast displayed the strongest capacity to interact with other cell types (Figure 7F). We also observed higher CCI activity in diseased PT cells (e.g., Type1/2 injured PT and FR-PTC) compared to healthy PT (Figure 7F). We calculated CCI scores across the uni-IRI and UUO time courses and found that the total number of significant CCIs was low in health but increased after injury (Figure 7G). Specifically, in uni-IRI, the number of interactions peaked at Day2 and then gradually decreased, and in UUO, we observed an increasing activity of CCI which reached highest level at around Day10 (Figure 7G).
We next characterized proximal tubule and myofibroblast cross-talk. Fibroblasts and myofibroblasts were grouped together and CCI scores were calculated with healthy PT, FR-PTC and injured PT. We found that interactions between FR-PTC and fibroblasts (FR-fibroblast) had the most robust CCI score (Figure 7H). In uni-IRI, we observed a strong FR-fibroblast interaction beginning at Day2 when FR-PTC started to expand, and interestingly, the interaction was still active at Day28 (Figure 7H), even though FR-PTC only constituted < 5% of the total proximal tubule cells at this point (Figure 2E). In UUO, a similar pattern was evident beginning at Day 6 (Figure 7H). The importance of FR-fibroblast interactions encouraged us to identify molecule pairs responsible for the communication between the two cell types. In addition to strong interactions between fibroblast integrins and VCAM1/COL18A1/SPP1 expressed by FR-PTC (Figure S7J), we identified CD44-FGFR2 as a significantly dysregulated receptor-receptor pair in both uni-IRI and UUO (Figure S7J). The CD44-FGFR2 interaction was highly specific to the FR-fibroblast CCI as its activity was not statistically significant in interactions between fibroblasts and other PT subtypes (Figure S7K). CD44 is a receptor for hyaluronic acid and its upregulation in injured PT cells has been well characterized (Lewington et al., 2000; Schiessl et al., 2018), and FGFR2 is known to be essential for kidney development and its ablation ameliorates kidney fibrosis (Hains et al., 2008; Xu and Dai, 2017). In our dataset, Cd44 was specifically expressed in FR-PTC and Fgfr2 could be detected in multiple (myo)fibroblast subtypes with highest expression in Myo-1 (Figure S7L), which reinforced the critical role of CD44-FGFR2 interaction in FR-fibroblast intercellular communication. In addition, we also examined communications between fibroblasts and loop of Henle cells, identifying enhanced activity of EPHB2-EFNA5 interaction in kidney fibrogenesis (Figure S7M). The expression of Ephb2 (encoding Ephrin Type-B Receptor 2) was specific to loop of Henle cells and was upregulated in TAL-inj compared with its healthy state (Figure S11F), which was supported by several previous studies (Huang et al., 2021; Ogawa et al., 2006), suggesting that Eph/Ephrin signaling axis may be a mediator of kidney fibrogenesis.
DISCUSSION
Our dataset has been deposited into an online interactive scRNA-seq data analyzer (http://humphreyslab.com/SingleCell/), which allows researchers to visualize expression of any gene of interest among different cell types or disease groups. We specifically profiled samples of uni-IRI and UUO, two well-characterized models of kidney injury and fibrosis, and present a computational workflow (see Methods) for integrating our dataset with other scRNA-seq atlases so comparative and joint analysis can be performed with batch effects removed. For example, we integrated our previous scRNA-seq dataset on bi-IRI mouse kidneys (Kirita et al., 2020) with this uni-IRI subset, and found that all major cell states could be identified in both models (Figure S1E).
Our scRNA-seq library was generated with the sci-RNA-seq3 protocol, a technology based on single-cell combinatorial indexing (sci; also known as split-pool barcoding). sci-RNA-seq3 differs from widely adopted droplet microfluidic solutions, such as 10X Chromium, by marking each cell with a unique combination of several barcodes (instead of one barcode). Though still early in development, sci-based approaches have been applied to a growing number of studies in recent years, due to its high throughput capabilities, sample multiplexing capacity and utilization of common laboratory equipment (Li and Humphreys, 2021). Here, we demonstrated its applicability in solid tissues collected from disease models. The high-throughput and highly multiplexed dataset enables identification of rare cell types in the time course of disease progression, such as the Type1/2 injured PT cells described here. sci-based methods also provide a cost-effective solution to constructing comprehensive human cell atlases by profiling multiple samples in parallel to minimize batch effects, and recent improvements have been made to achieve higher gene detection sensitivity and co-measurement of multiple modalities (Ma et al., 2020; Martin et al., 2021).
Our results have been comprehensively validated through reanalyzing existing mouse and human datasets on relevant disease models. For example, we observed upregulation of Plin2 (or human PLIN2) in the folic acid-induced mouse nephropathy model (Craciun et al., 2016) (Figure S4A) and in a human renal IRI model (Park et al., 2020) (Mendeley Data). Our characterization of Type2 injured PT cells was supported by a previous dataset which profiled PT-enriched transcripts in UUO mice (Wu et al., 2020) (Figure S5B). We also surveyed a prior work on biopsy samples of patients with CKD (Nakagawa et al., 2015) and validated increased expression of Type2, but not Type1, injured PT marker genes in patients with CKD compared to control (Figure 4D–E and Mendeley Data). An interesting and open question is whether the abundance of either injured PT state in the early stages of human kidney disease correlates with long-term patient outcomes.
PT cells have high baseline energy demands and preferentially utilize lipids to generate ATP. Accumulation of lipids in PT is dependent on uptake of serum free fatty acids (Zeng et al., 2017) and defects in lipid metabolism are a well-recognized defect of CKD (Kang et al., 2014; Stadler et al., 2015; Tran et al., 2016). A recent study demonstrated that long-term fatty acid uptake (10-day palmitic acid administration) promoted inflammation and fibrogenesis of mouse PT cells (Mori et al., 2021). Here, we identified an unexpected, transient lipid accumulation and enhanced expression of FAO-related genes in Type1 injured PT cells (Figure 3A–F). Three experimental observations led to the conclusion that the increased expression of FAO genes contributed to an increased FAO phenotype: 1) Cells had very low content intracellular lipids at uni-IRI D2 (Figure 3D), implying the deposited lipids in the first 6 hours were utilized over the next day; 2) In vitro modeling of lipid accumulation, in combination with the use of a lipolysis inhibitor revealed that clearance of lipid droplets was through FAO (Figure 3H); 3) Direct metabolic measurement identified an increased OCR after 6-hour fatty acid treatment (Figure 3I and S3L). Interestingly, in our bulk RNA-seq analysis of cells harvested at 2 days after 6-hour oleic acid treatment, we found upregulation of genes involved in DNA replication, cell cycle regulation and cell proliferation (Figure 3J), which are high energy-demand cellular events. This was also consistent with our observation that Mki67-expressing Repairing PT cells were most abundant at uni-IRI D2 (Figure 2E). Therefore, the deposited lipids in 6 hours may serve as an essential energy source for injured epithelia following injury, promoting tubular repair through proliferative expansion.
Lipid droplets, also known as lipid vacuoles, are organelles whose phospholipid monolayer is decorated with lipid binding proteins and containing a hydrophobic core consisting of neutral lipids. Here, we identified a ~10-fold increased PLIN2 expression after a 6-hour fatty acid stimulus in vitro, with resolution of expression two days after removal of fatty acids from the media (Figure 4G), implying that PLIN2 plays a key role in how the cell responds to intracellular lipid accumulation. Further, PLIN2 gene knockdown caused a decrease in OCR and ECAR activities (Figure 4I and S4F), suggesting that PLIN2 regulates cellular metabolism. Although a reduced OCR may be expected to reflect a downregulation of FAO genes, we did not identify decreased expression of genes encoding mitochondrial FAO components such as CPT1A and CPT2. Instead, we observed significantly decreased expression of ACSL3, ACSL4 and ACSL5, which encode cytosolic proteins that convert lipolysis-derived free fatty acids into fatty acyl CoA (Li et al., 2010). These results indicate that PLIN2 regulates acyl CoA generation by lipolysis but does not directly affect mitochondrial β-oxidation. Overall, we propose the model presented in Figure 4K: after IRI, Type1 injured PT cells rapidly accumulate lipid droplets, inducing PLIN2 expression, leading to enhanced PLIN2-dependent FAO activity with subsequent consumption of these lipids, promoting epithelial proliferation and tubule regeneration. Why this lipid accumulation and consumption process does not occur in Type2 injured PT (or in UUO), and whether lack of lipid acquisition in early stages is responsible for the poor fate outcome of kidney fibrogenesis, requires further investigation.
We highlighted two dysregulated pathways, lipid and amino acid metabolism, in diseased PT cells, but we also acknowledge that kidney fibrogenesis affects many other metabolic networks. For example, SLC5A2 (the target of SGLT2 inhibitors) is responsible for ~90% of tubular glucose transport (Wen et al., 2021). As a consequence of cell dedifferentiation in both uni-IRI and UUO, we observed a remarkable reduction in expression of Slc5a2 in PT. We also identified decreased expression of genes encoding phosphofructokinase, glucose-6-phosphatase, and isocitrate dehydrogenase, which could be recovered in late timepoints of uni-IRI but remained at low levels in UUO, suggesting disrupted glucose metabolism in kidney fibrogenesis. In addition, we found that the two genes encoding subunits of lactate dehydrogenase, Ldha and Ldhb, were dysregulated with patterns consistent with a recent report (Osis et al., 2021) (Mendeley Data), indicating off-balance interconversion between lactate and pyruvate. Interestingly, we identified Hmox1, which encodes heme oxygenase-1 (HO-1, an essential modulator of glucose metabolism), and its transcription factor Nfe2l2 as two upregulated markers of early IRI injury (i.e., uni-IRI 6hrs) distributed in Type1 Injury and Acute Injury PT cells (Mendeley Data). Previous studies have demonstrated a protective role of HO-1 against kidney injury and exhibited significant elimination of tubular injury and interstitial fibrosis in UUO mice following treatment with an HO-1 inducer (Bolisetty et al., 2017; Kim et al., 2006). Understanding the specific role of HO-1 in maintaining renal glucose metabolism in disease states will require further investigation.
This high-throughput dataset enabled us to discover a shared injury response of all major TEC structures including PT, TAL, DCT, and CNT. Although TAL-inj, DCT-inj and CNT-inj were described as populations covering injured cells from both uni-IRI and UUO in this analysis, we could not exclude the possibility that they were heterogenous groups composed of multiple injury states, as characterized in injured PT. For example, we found that Gcnt2, whose deficiency can cause abnormal morphology of tubule epithelium (Chen et al., 2005), was upregulated in TAL-inj specifically at uni-IRI 6hrs, but not in UUO (Figure S6F). Higher detection resolution will be needed for this additional subclustering analysis.
This single-cell atlas of kidney fibrogenesis also serves as a unique resource to study fibrotic responses of other non-epithelial cells such as stromal cells (Figure 7A–E), immune cells (Figure S1C) and endothelial cells (ECs) (Figure S1D). We have performed subclustering analysis on all these populations to illustrate the complexity of the dataset. For example, we identified a group of macrophages (Mφ–2) marked by elevated expression of a lysozyme gene Lyz2, Tgfbi, and various genes encoding heat-shock proteins (Figure S1C and Mendeley Data). Mφ–2 showed high abundance at uni-IRI 6hrs/D2, but was low abundance in UUO (Mendeley Data). It has been reported that TGFBI+ macrophages can capture apoptotic cells and induce fibrotic responses (Nacu et al., 2008), and our results indicate that Mφ–2 could be an essential population initiating immune response against kidney injury. In the subclustering of endothelial cells, we found that a subgroup of EC (Activated EC) exhibited upregulated expression of Rapgef5 and Magi1, genes involved in abnormal angiogenesis and endothelial activation (Abe et al., 2019; Hong et al., 2007) (Figure S1D). This cell type was rarely observed in healthy, but could proliferate rapidly in disease, particularly after UUO D6 (Mendeley Data), and it would be interesting to learn its lineage progenitors and functional importance in kidney fibrogenesis in future studies.
In summary, we leveraged sci-RNA-seq3 to generate a high-throughput single-cell transcriptomic landscape of kidney fibrogenesis. PT cell dedifferentiation was a shared injury response in both uni-IRI and UUO models, but unique cell states existed in each model, such as the Type1 and Type2 injured PT, characterized by dysregulated lipid and amino acid metabolism, respectively. We also identified both shared and unique injury and repair responses in epithelial cells across nephron segments and demonstrated the heterogeneity of kidney stromal cells. Since kidney fibrosis affects nearly all renal cell types encompassing epithelia, stroma, endothelia and the immune system, it is critical to construct a comprehensive network of cell-cell communications for translational studies. Our work highlights the utility of analyzing detailed time courses of kidney fibrogenesis and validates sci-RNA-seq3 as a powerful method for analyzing multiple samples at once.
Limitations of study
Our work employs two widely adopted mouse kidney fibrogenesis models, uni-IRI and UUO, but how generalizable our findings are to other forms of kidney injury is unresolved. This version of sci-RNA-seq3 is technically limited in gene detection sensitivity. Also, it is challenging to assess the significance of Type1 injured PT in humans because this cell state only transiently appears ~ 6 hours after AKI, and few if any such early AKI human samples are available. Finally, our transcriptomic characterization is unimodal. Future multi-modality measurements such as combined transcriptomic and epigenomic readouts will be needed to depict a complete cell atlas of kidney fibrosis.
STAR METHODS
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Benjamin D. Humphreys (humphreysbd@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw (fastq) and pre-processed data (count matrix) and metadata of the sci-RNA-seq3 dataset have been deposited in NCBI’s Gene Expression Omnibus and are available through GEO Series accession number GSE190887. Raw and processed bulk RNA-seq data on RPTECs are available through GEO Series accession number GSE206084. All additional data are available at Mendeley Data (http://dx.doi.org/10.17632/hd3j7mdm2p.1).
Scripts for a pipeline of single-cell clustering and visualization and generation of all major figures in this study were written mostly in Python and R with code available at https://github.com/TheHumphreysLab/sci-RNA-seq-kidney. Data S1 - Source Data, containing values used to generate graphs related to Figures 3–5, 7 and S3–7, is also presented.
Experimental model and subject details
All mouse experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at Washington University in St. Louis. C57BL/6J mice were obtained from The Jackson Laboratory (000664, JAX). Both uni-IRI and UUO surgeries were performed as previously described (Clef et al., 2016; Wu et al., 2019). Briefly, 8- to 9-week-old male mice were anesthetized with isoflurane and buprenorphine Sustained-Release was administered for analgesia. Body temperature was monitored and maintained at 36.5–37.5°C throughout both procedures. After flank incision, in uni-IRI, ischemia was induced on the left kidney by clamping the renal pedicle with a nontraumatic micro aneurysm clamp (RS-5420, Roboz) for 22 minutes, and the clamp was subsequently removed. The 22-min clamping time was determined with a titration test before the experiment where we confirmed successful induction of tissue injury, fibrosis and repair. In UUO, irreversible obstruction was induced by ligating the left ureter twice with nonabsorbable silk suture (468782, McKesson) between the bladder and renal pelvis.
Mouse kidney sample processing
Mice were euthanized with isoflurane and the left ventricle was perfused with phosphate-buffered saline (PBS). Left kidneys were subsequently harvested. For tissues used in sci-RNA-seq3 library generation, a piece of cortical tissue was dissected from each kidney for subsequent qPCR analysis and the remaining tissue were snap-frozen with liquid nitrogen for nuclei preparation. For immunofluorescence, kidneys were fixed with 4% paraformaldehyde (PFA) (15713, Electron Microscopy Sciences) at 4°C for 2 hours, immersed in 30% sucrose at 4°C overnight and embedded in optimum cutting temperature compound (4583, Sakura) to cut sections.
Cell culture
HEK-293T cells (CRL-3216, ATCC) and C3H/10T1/2 cells (CCL-226, ATCC) were cultured in DMEM (11965, Gibco) supplemented with 10% Fetal Bovine Serum (F4135, Sigma) and 1× penicillin–streptomycin (15140122, Gibco). Primary human renal proximal tubule epithelial cells (RPTECs) (CC-2553, Lonza) were cultured in Renal Epithelial Cell Growth Medium (CC-3190, Lonza) with supplements provided in the kit. Primary RPTECs were used in early passage. All cells were maintained in a humidified 5% CO2 atmosphere at 37°C unless otherwise specified.
Quantitative polymerase chain reaction (qPCR) analysis
For each kidney sample, tissue was homogenized in TRIzol reagents (15596026, Invitrogen) on ice and RNA was extracted with the Direct-zol RNA Miniprep Kit (R2072, Zymo) following the manufacturer’s instructions. Complementary DNA (cDNA) was obtained by reverse transcribing the extracted RNA (~1.5 μg) with the High-Capacity cDNA Reverse Transcription Kit (4368813, Life Technologies), and then processed for qPCR using the iTaq Universal SYBR Green Supermix (1725125, BioRad). Gene expression was normalized to Gapdh or Actb expression and quantified with the 2−ΔΔCt method. For cultured human RPTECs, we performed cell lysis in TRIzol reagents and followed a similar procedure as mentioned above. Primer sequences can be found in Mendeley Data.
Immunofluorescence
6-μm tissue sections were fixed with 4% PFA for 5 minutes, washed with PBS and blocked with blocking buffer (1% Bovine Serum Albumin (BSA), 1% goat serum, 0.3% Triton X-100 in PBS) for 20–40 minutes at room temperature. For each target, sections were stained with the desired primary antibody for 45–90 minutes at room temperature or overnight at 4°C. Sections were washed with PBS (three times; 5 minutes each) and stained with the desired secondary antibody for 45–60 minutes at room temperature. After three washes with PBS for 5 minutes each, sections were counterstained with DAPI and mounted with Prolong Gold. Images were captured and processed with a confocal microscope (Eclipse Ti, Nikon) and examined in a blinded fashion.
Lipid staining
For Oil Red O staining, Oil Red O Stain Kit (ab150678, Abcam) was used. We incubated 6-μm fixed frozen tissue sections (formalin-fixed paraffin-embedded sections are not recommended) or fixed cells on a slide with Oil Red O Solution overnight at room temperature and followed the other procedures with the manufacturer’s instructions. Images were captured with a Zeiss Axio Scan Z1 light microscopy and examined in a blinded fashion.
BODIPY 493/503 (25892, Cayman Chemical) was used to label cellular lipid contents. Briefly, 6-μm fixed frozen tissue sections or fixed cells on a slide were incubated with 7.5–15 μM BODIPY 493/503 for 15 minutes at room temperature and washed with PBS. The sample was counterstained with DAPI and then processed for confocal imaging.
RNA in situ hybridization (ISH)
RNA ISH was performed as previously described (Chang-Panesso et al., 2019; Liu et al., 2017). Briefly, the cDNA used in qPCR was amplified by PCR with primer sequences described in Mendeley Data. Probes were transcribed in vitro by T7 (for antisense probes) (10881767001, Roche) or SP6 (for sense probes) (10810274001, Roche) RNA polymerases in the presence of DIG RNA Labeling Mix (11277073910, Roche). The reaction was performed at 37°C for 2 hours. Then, the reaction was supplemented with DNase (79254, Qiagen), incubated at 37°C for 15 minutes and stopped by adding EDTA pH 8.0. Probes were purified and eluted in hybridization buffer (50% formamide, 5× SSC, 1% SDS, 50 μg/mL yeast tRNA (15401011, Invitrogen) and 50 μg/mL heparin (H3393, Sigma)). Next, 15-μm sections were fixed with 4% PFA overnight, washed with PBS and incubated with 10 μg/mL Proteinase-K (25530049, Invitrogen) in PBS for 20 minutes. Sections were washed with PBS and incubated with acetylation solution (533 μl triethanolamine (T58300, Sigma), 70 μl 37% HCl and 150 μl acetic anhydride (320102, Sigma) in 40 mL H2O) for 10 minutes. Sections were then washed with PBS, H2O, 70% ethanol and 95% ethanol. Probes were added at a final concentration of 500–1000 ng/mL in hybridization buffer and the reaction was performed at 68°C overnight. After hybridization, sections were washed with 5× SSC, 1× SSC supplemented with 50% formamide, 2× SSC and 0.2× SSC at 68°C, and then washed with TBST buffer at room temperature. Sections were blocked with 2% blocking reagent (11096176001, Roche) for 1–2 hours at room temperature and 1:4000 diluted anti-DIG-AP Fab fragments antibody (11093274910, Roche) was added and incubated at 4°C overnight. Then, sections were washed with TBST and NTMT (100 mM NaCl, 100 mM Tris pH 9.5, 50 mM MgCl2, 0.1% Tween 20 and 2 mM Tetramisole (L9756, Sigma)) buffers and incubated with BM-Purple solution (11442074001, Roche) at room temperature. Sections were fixed again, washed with PBS and mounted with Prolong Gold. Images were captured with a Zeiss Axio Scan Z1 light microscopy and examined in a blinded fashion. To validate successful development of RNA ISH, we stained Slc22a7, a well-described gene expressed in S3 segment of PT in health (Chang-Panesso et al., 2019), and confirmed that it was highly expressed in healthy kidney tissues and was not observed at UUO D2 in antisense probe hybridization, and could not be detected in both conditions by sense probes (Mendeley Data).
For dual RNA-ISH, the aforementioned procedure was used with minor changes. Briefly, probes were transcribed in either DIG RNA Labeling Mix or Fluorescein RNA Labeling Mix (11685619910, Roche). The two probes were mixed and incubated on the slide at 68°C overnight. After hybridization, the Fluorescein-labelled probe was used to develop a blue color with anti-Fluorescein-AP Fab fragments (11426338910, Roche) coupled with NBT/BCIP solution (11681451001, Roche). The AP substrate was removed by incubating the slide at 68°C for 30–60 minutes. Then, the section was blocked again and the DIG-labelled probed was used to develop a brown color with anti-DIG-AP Fab fragments coupled with INT/BCIP solution (11681460001, Roche).
In vitro fatty acid exposure, ER stress induction and lipolysis inhibition
When human primary RPTECs reached 70%–80% confluence, cells were starved overnight by culturing in Renal Epithelial Cell Growth Medium without growth supplements. Then, cells were switched to complete growth medium and treated with BSA-conjugated oleate fatty acid (29557, Cayman Chemical), palmitate fatty acid (29558, Cayman Chemical) or ER stress inducers, Tunicamycin (SML1287, Sigma) or Thapsigargin (10 mM stock concentration) (T9033, Sigma) at desired concentrations, where BSA or DMSO treatments were used as control groups. To analyze the long-term effect of 6-hour fatty acid stimulus, cells were washed to ensure complete removal of fatty acids and incubated in complete growth medium with or without 10 μM Atglistatin (15284, Cayman Chemical), a lipolysis inhibitor, for 40–48 hours. Cells were then harvested for downstream analysis.
Fatty acid internalization assay
Human primary RPTECs were starved overnight and treated with green fluorescent fatty acid BODIPY FL C16 (D3821, Thermo Scientific) in complete growth medium for 6 hours. Cells were then washed with PBS and fixed with 4% PFA for 15 minutes. Cells were washed again, counterstained with DAPI and processed for confocal imaging.
Mitochondria staining on cultured cells
Human primary RPTECs were starved overnight and then exposed to complete growth medium with or without fatty acid treatment. Cells were incubated with MitoTracker Red CMXRos (9082S, Cell Signaling) at a final concentration of 200 nM for 30 minutes in the 37°C CO2 incubator before harvested. Then cells were washed with PBS and fixed with ice-cold methanol for 15 minutes at −20°C. Cells were washed again, counterstained with DAPI and processed for confocal imaging.
In vitro gene knockdown
PLIN2 expression in human primary RPTECs was knocked down with lipid-based siRNA transfection method. Briefly, Lipofectamine Reagent (13778150, Thermo Scientific) and PLIN2 Smartpool siRNA (L-019204-01-0010, Dharmacon) were mixed for 5 minutes and added to cells following the manufacturer’s instruction. ON-TARGETplus Non-targeting Control Pool (siNT) (D-001810-10-20, Dharmacon) was as control. Both siRNA molecules were used at a final concentration of 10 nM. Cells were examined or processed for subsequent treatments at 1–2 days after siRNA transfection.
ROS staining
2,7-Dichlorodihydrofluorescein diacetate (85155, Cayman Chemical) was added to cell culture medium at a final concentration of 50 μM and incubated with RPTECs for 45 minutes at 37°C. Cells were then washed and fixed with 4% PFA. Cells were counterstained with DAPI and processed for confocal imaging.
Metabolic measurement
Metabolic measurement of human primary RPTECs was performed as previously described (Kang et al., 2014) with minor changes. Briefly, a Seahorse XFe96 Analyzer (Agilent) was used to measure real-time oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). 10,000–20,000 cells were seeded into each well of a XF96 cell culture microplate (102416–100, Agilent). Cells were starved overnight in Renal Epithelial Cell Growth Medium without growth supplements. Cells were then treated with oleate or palmitate fatty acids in complete growth medium for 6 hours. The culture medium was switched into Seahorse XF DMEM assay medium (103680–100, Agilent) supplemented with 2 mM glucose and 0.5 mM L-carnitine (C0283, Sigma) at one hour before the real-time measurement. Oleate or palmitate fatty acids, etomoxir (11969, Cayman Chemical) and oligomycin (O4876, Sigma) were injected during the analysis at a final concentration of 100 μM, 40 μM and 1 μM, respectively.After the Seahorse measurement was completed, the number of cells per well was counted for data normalization. Briefly, cells were washed with PBS, fixed in ice-cold methanol at −20°C for 15 minutes and then stained with DAPI for 10 minutes. Cell counting was performed on the Lionheart FX Automated Microscope (Agilent) and the estimated mean number of cells was used to normalize the OCR and ECAR readouts. The four cell energy states (quiescent, aerobic, glycolytic, energetic) were defined according to existing publications (Hocaoglu et al., 2021) and the manufacturer’s instruction.
Nuclei isolation and fixation (mouse kidney)
Nuclei isolation from mouse kidney tissues was performed as previously described (Kirita et al., 2020) with minimal changes. Briefly, Nuclei EZ Lysis Buffer (NUC101, Sigma) was used and supplemented with EDTA-free protease inhibitor tablets (5892791001, Roche) and RNase inhibitors (N2615, Promega; AM2696, Thermo Scientific). Tissues were minced with a razor blade and homogenized with Dounce Tissue Grinders (885303–0002, Kimble) in ice-cold lysis buffer. The homogenate was filtered through a 200-μm strainer (43-50200-03, pluriSelect), incubated in the buffer for 5 minutes and then filtered again through a 40-μm strainer (43-50040-51, pluriSelect). The homogenate was centrifuged at 500×g for 4 minutes and the pellet was resuspended in the lysis buffer with gentle pipetting. After 5-minute incubation, the suspension was centrifuged at 500×g for 4 minutes and the nuclei pellet was resuspended with Nuclei Suspension Buffer (NSB), which was freshly prepared by supplementing nuclei buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3mM MgCl2) with 1% RNase inhibitor (AM2696, Thermo Scientific) and 2% BSA (B9000S, NEB). All procedures were performed at 4°C to protect RNA integrity.
Then, the nuclei were fixed with PFA (15713, Electron Microscopy Sciences) at a final concentration of 2.4% and incubated for 10 minutes on ice. The suspension was centrifuged at 500×g for 5 minutes at 4°C to remove supernatant and washed with NSB for 1–2 times. The nuclei were resuspended in NSB, snap-frozen in a cryotube and stored in liquid nitrogen for the sci-RNA-seq3 experiment. All centrifugation steps were performed using a centrifuge with a swinging bucket to reduce cell loss. In total, we generated 25 fixed frozen single-nuclei suspensions from 24 mouse kidney tissues and one mixture of cultured human and mouse cell lines (see below).
Nuclei isolation and fixation (cultured cells)
HEK-293T cells and C3H/10T1/2 cells were trypsinized at approximately 70% confluence, centrifuged at 300×g for 5 minutes at 4°C and washed twice with ice-cold PBS. Cell concentrations were measured, and equal number of cells from each cell line were pooled into a total of 5 million cells. The cell mixture was centrifuged at 300×g for 5 minutes at 4°C and the pellet was resuspended with 1 mL ice-cold cell line lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 1% SUPERaseIn RNase Inhibitor (AM2696, Thermo Scientific) and 2% BSA (B9000S, NEB)). Cells were incubated on ice for 4 minutes. Then, nuclei were filtered through a 40-μm Flowmi cell strainer (H13680–0040, Bel-Art), centrifuged at 500×g for 5 minutes at 4°C and washed with 1mL cell line lysis buffer. The nuclei pellet was resuspended with 1 mL PBS and PFA (15713, Electron Microscopy Sciences) was added to a final concentration of 3.2%. Fixation was performed on ice for 10 minutes. The fixed nuclei were centrifuged at 500×g for 5 minutes at 4°C, washed twice with cell line wash buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 1% SUPERaseIn RNase Inhibitor and 2% BSA) and snap-frozen in a cryotube and stored in liquid nitrogen.
sci-RNA-seq3 library generation
Generation of the sci-RNA-seq3 library was performed following a previously described protocol (Cao et al., 2019) with minor changes. In sci-RNA-seq3, both indexed reverse transcription (RT) and hairpin ligation were performed on 384 wells. Each of these wells contains a uniquely indexed oligo (a poly-T or ligation primer) with sequences described before (Cao et al., 2019). The two types of oligo-containing wells are referred to RT wells and ligation wells, respectively. Each RT well contains 2 μl poly-T primers (100 μM) and each ligation well contains 8μl hairpin ligation primers (100 μM). All RT wells and ligation wells were pre-prepared and stored at −20°C before use. All oligos used in this study were ordered from IDT.
Fixed frozen nuclei suspensions were thawed in a 37°C-water bath and then centrifuged at 500×g for 5 minutes at 4°C. For each sample, the nuclei were resuspended with freshly prepared NSB, permeabilized with Triton X-100 at a final concentration of 0.2 % for 5 minutes on ice and washed with NSB. Then, sonication was performed with a Bioruptor Pico sonication device (Diagenode) lightly (10 seconds) to reduce nuclei clumps. For each sample, the nuclei suspension was filtered through a 40-μm Flowmi cell strainer and nuclei concentration was measured.
Approximately 80,000 nuclei in 22 μl NSB and 2 μl 10 mM dNTP (639125, Clontech) were added into each RT well. The well IDs into which each sample was deposited were recorded for downstream sample demultiplexing analysis. In total, we deposited the mixed human/mouse cells into 2 wells and mouse kidney cells from the 24 samples into the other 382 wells (15 or 16 wells per sample). Then, all RT wells were incubated at 55°C for 5 minutes and 14 μl RT reaction mix, containing 2 μl SuperScript IV reverse transcriptase (18090050, Thermo Scientific), 8 μl 5× RT buffer, 2 μl 100 mM DTT and 2 μl RNaseOUT RNase inhibitor (10777019, Thermo Scientific), was added into each well. The RT reaction was performed by incubating all RT wells at 4°C, 10°C, 20°C, 30°C, 40°C and 50°C (2 minutes each) and then 55°C for 15 minutes. Then 60 μl NBB (nuclei buffer supplemented with 1.5% BSA) was added into each well and the nuclei suspensions from all RT wells were pooled. The pooled nuclei suspension was centrifuged at 500×g for 10 minutes at 4°C. The nuclei pellet was resuspended with 4.3 mL NSB and 10 μl was added into each ligation well supplemented with 2 μl ligase (M2200L, NEB) and 20 μl 2× ligation buffer. The ligation reaction was performed by incubating all ligation wells at 25°C for 10 minutes. Then, 60 μl NBB was added to each well and the nuclei suspensions from all ligation wells were pooled. The pooled suspension was centrifuged at 600×g for 10 minutes at 4°C and washed with NBB at 600×g for 10 minutes again. The pellet was gently resuspended with 4 mL NBB and filtered through a 40-μm Flowmi cell strainer. Then, we measured the nuclei concentration and distributed approximately 4,000 nuclei in 5 μl NBB into each well of several 96-well plates, which were stored at −80°C for further use.
Before sublibrary generation, we first prepared the transposome needed in the tagmentation step. Two lyophilized adapter oligos, ME_R ([PHO]CTGTCTCTTATACACATCT) and ME_A (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG) were reconstituted to 100 μM with annealing buffer (40 mM Tris-HCl pH 8.0, 50 mM NaCl). Then, 5 μl ME_R and 10 μl ME_A were mixed and incubated in a thermocycler. The oligo mixture was incubated at 95°C for 5 minutes, cooled down to 65°C at −0.1°C/second, incubated at 65°C for 5 minutes and finally cooled down to 4°C at −0.1°C/second. Then, 12.5 μl annealed oligos were added into 10 μl naked (unloaded) Tn5 transposase, and the mixture was incubated at 23°C for 30 minutes. Then, 12.5 μl glycerol was added and the loaded Tn5 was stored at −20°C for further use.
For sublibrary generation, one of the aforementioned plates was thawed in room temperature and 5 μl reaction mix, containing 0.67 μl second strand synthesis enzyme (E6111L, NEB), 1.33 μl reaction buffer and 3μl elution buffer, was added into each well. The plate was incubated at 16°C for 3 hours. For tagmentation, 2× TD buffer, containing 20 mM Tris-HCl pH 7.5, 10 mM MgCl2 and 20% (v/v) dimethylformamide (20673, Thermo Scientific), was added to the pre-prepared loaded Tn5 to a final concentration of 20 nM. Then, 10 μl 20 nM transposase was added into each well. Tagmentation was performed at 55°C for 5 minutes. Then, we added 20 μl DNA binding buffer (D4004–1-L, Zymo) into each well and incubated the reaction mix at room temperature for 5 minutes. Then, 40 μl Ampure XP beads (A63881, Beckman Coulter) were added into each well and the mixture was incubated at room temperature for 5 minutes. The supernatants were removed by placing the 96-well plate onto a magnetic stand. After washing with 80% ethanol twice, beads of each well were resuspended with 10 μl USER reaction mix, containing 1 μl enzyme (M5505L, NEB), 1 μl 10× buffer and 8 μl H2O. The plate was sealed and incubated at 37°C for 15 minutes. Then, 7 μl elution buffer was added into each well. The plate was placed onto a magnetic stand and 16μl supernatant was transferred to a new 96-well plate.
For indexed PCR reaction, the plate was first incubated at 80°C for 10 minutes. Then, 20 μl NEBnext 2× master mix (M0541L, NEB), 2 μl 10 μM P5 primer and 2 μl 10 μM P7 primer were added into each well. Primer sequences can be found in Mendeley Data. PCR was performed by incubating the plate at 72°C for 5 minutes, 98°C for 30 seconds, 15 cycles of [98°C 10 seconds, 66°C 30 seconds, 72°C 30 seconds] and 72°C for 5 minutes. Finally, PCR products from all wells were pooled together, purified with 0.7× Ampure XP beads and eluted with 100 μl elution buffer. The library was further purified with 0.7× Size-Select beads (D4084, Zymo) and visualized on a Bioanalyzer 2100 instrument.
Next-generation Sequencing for sci-RNA-seq3
In this study, a total of 8 sci-RNA-seq3 sublibraries were generated and pooled for sequencing. The pooled library was sequenced on the NovaSeq 6000 platform (Illumina) using a paired-end 200-cycle S4 kit (Read1: 34bp; Index1: 10bp; Index2: 10bp; Read2: 100bp).
sci-RNA-seq3 data pre-processing
Pre-processing sci-RNA-seq3 sequencing data was performed as previously described (Cao et al., 2019). Briefly, sequencing .fastq files were demultiplexed based on their i5 and i7 barcodes using deML (Renaud et al., 2015) with default settings. Then, unique molecular identifiers (UMIs), RT and ligation barcodes were extracted from the demultiplexed reads. RT and ligation barcodes were filtered with an edit distance (ED) < 2 and adapter-clipped with TrimGalore v.0.6.6 (https://github.com/FelixKrueger/TrimGalore). Trimmed reads were split into two modules based on their RT barcodes: the mouse kidney nuclei module (382 barcodes) and HEK-293T and C3H/10T1/2 mixed nuclei module (2 barcodes). Each module was mapped to its corresponding reference genome, mm10 or combined mm10 and hg19, using STAR v.2.7.7a (Dobin et al., 2013) (GENCODE vM11 for mouse and v19 for human). Low-quality aligners were filtered using samtools (Li et al., 2009) and reads with identical UMI, RT and ligation barcodes (ED < 2) were annotated as duplicates and removed. As recommended in the prior study (Cao et al., 2019), we performed UMI error correction (ED < 2) on a subset of our data and observed that 96.1% of reads remained after UMI error correction compared to skipping this step, suggesting good data quality. Next, mapped reads were split based on their combinatorial indexing barcodes and barcodes with at least 200 reads identified were processed for the next step. Finally, HTSeq v.0.12.4 (Anders et al., 2015) was used to generate a gene count matrix by calculating the number of strand-specific UMIs for each cell mapping to the exonic and intronic regions of each gene.
For species-mixing analysis, cells with over 85% of UMIs assigned to one species were annotated as species-specific cells and the other cells were annotated as cell collisions. Predicted doublet rate was calculated by dividing the number of cell collisions by total number of human and mouse cells after quality control. We performed ambient RNA removal with CellBender (Fleming et al., 2019) on the species-mixing data subset and observed similar doublet rates before and after data correction.
Pseudobulk trajectory ordering
First, each cell in the cell-by-gene count matrix (413,681 cells × 48,795 genes) was assigned to its original mouse kidney sample (n=24) based on its RT barcode. Then, a sample-by-gene count matrix was generated from the cell-by-gene count matrix by aggregating cells that originate from the same sample. Then, gene counts were normalized by the total number of reads per sample.
Pseudobulk trajectory analysis was performed using Monocle2 (Qiu et al., 2017). Briefly, the sample-by-gene count matrix was converted into a CellDataSet data class with the newCellDataSet function and size factors and dispersions were estimated with the estimateSizeFactors and estimateDispersions functions. Then, genes expressed in at least 2 samples were selected. Differentially expressed genes across the 24 mouse kidneys were identified with the differentialGeneTest function and top 5000 significant genes were selected as the ordering genes. Samples were ordered along a pseudo-trajecotry using the reduceDimension function (method = ‘DDRTree’) and the orderCells function. Data visualization was enabled with the plot_cell_trajectory function.
Doublet estimation, quality control and cell clustering
The gene count matrix generated above was converted into a data format (AnnData) compatible with the Scanpy v1.7.2 (Wolf et al., 2018) platform. Protein-coding genes, pseudogenes and genes encoding lincRNAs were maintained in further analysis. Estimation of cell doublets was implemented on Scrublet v.0.2.1 (Wolock et al., 2019) using the scrublet.Scrublet function with the expected overall doublet rate set as 0.06 and the number of neighbors set as 30. Then, a doublet score was calculated for each cell with the scrub_doublets function (min_counts = 3, min_cells = 3, min_gene_variability_pctl = 85, n_prin_comps = 30). Cells with the doublet score over 0.2 were annotated as expected doublets.
Only cells with less than 5% of counts derived from mitochondrial genes were processed. Cells with less than 80 genes detected and genes present in less than 30 cells were removed from the gene count matrix. Then, data was normalized and log-transformed. The top 5000 genes were selected with the highest variance using the scanpy.pp.highly_variable_genes function. The data was scaled and Principal Component Analysis (PCA) was performed for dimensionality reduction using the scanpy.tl.pca function (svd_solver = ‘arpack’, n_comps = 50). Next, a neighborhood graph of cells was computed using the scanpy.pp.neighbors function with the number of neighbors set as 50 (metric = ‘cosine’). Next, the neighborhood graph was embedded in two dimensions using Uniform Manifold Approximation and Projection (UMAP) with the scanpy.tl.umap function, in which the effective minimum distance between embedded points set as 0.01. Leiden clustering was performed (resolution = 2) and marker genes of each Leiden cluster was identified using the scanpy.tl.rank_genes_groups function (method = ‘wilcoxon’). One cluster showed significantly elevated mitochondrial gene counts and no expression of other cluster-specific genes, and therefore, annotated as an artefact cluster. Two minor clusters showed co-expression of well-known marker genes of PT and LoH cells or PT and DCT cells, and therefore, annotated as doublet clusters. These clusters were not specific to a kidney sample or disease condition and therefore removed. The remaining 309,666 cells were re-analyzed following a similar approach as described above (see Code Availability). For customized UMAP visualization used in Figure 1, we utilized a computational framework (https://github.com/TheHumphreysLab/plot1cell) described in a recent study (Wu et al., 2022). Individual clusters were annotated by manually inspecting the expression of lineage-specific genes and comparison with existing cell atlas datasets. For cell type subclustering, cells annotated as the subtype in the above analysis were extracted and re-analyzed following the similar procedure. Quality control was performed again, and low-quality cells may be further removed, and the effects of total counts per cell may be regressed out using the scanpy.pp.regress_out function to improve single-cell visualization and cell type annotation (see Code Availability).
Due to the unique sample multiplexing capacity of sci-RNA-seq3, all samples were expected to have very minimal technical batch effects since they were processed in one experiment and sequenced in one flow cell concurrently, and therefore, all presented data were analyzed without the use of batch effect removal tools, in a similar approach as described before (Cao et al., 2019). However, we compared our clustering results before and after batch effect correction with Harmony (Korsunsky et al., 2019). Briefly, the RunHarmony function was used and the 24 mouse kidney samples were considered as batch variables. Downstream analysis was performed on Seurat (Stuart et al., 2019). The RunUMAP function was used with [n.neighbors=40,dims = 1:40,min.dist=0.01] for global cell clustering and [n.neighbors=40,dims = 1:15,min.dist=0.1] for PT cell subclustering. Overall, we found the use of Harmony on our dataset resulted in impaired identification of sample-specific cell populations such as the uni-IRI 6hrs-specific Type1 injured PT cells, suggesting batch overcorrection.
Gene module scoring
Cell cycle scoring was performed with the scanpy.tl.score_genes_cell_cycle function with a list of genes associated with S-phase and G2M-phase. In addition, marker genes of cell cycle arrest and senescence were manually checked to verify the proliferate state of cells with high cell cycle scores. Scoring analysis of the other gene modules was performed with the scanpy.tl.score_genes function. The gene set used for ECM scoring was obtained from the Matrisome Project (Shao et al., 2020). The gene sets used for scoring of lipid droplet, heat shock proteins, mitochondrial respiration and myosin were obtained from the database of Harmonizome (Rouillard et al., 2016). All gene lists and their references can be found in Mendeley Data.
Single-cell trajectory inference
Single-cell pseudotemporal ordering was implemented on Monocle2 (Qiu et al., 2017). After single-cell clustering and sample demultiplexing, PT cells were downsampled for the Monocle analysis due to significant computation time required. Briefly, after size factors and dispersions were estimated, expressed genes were defined as genes that were identified in at least 30 cells. Then, highly variable genes were selected, and dimensionality reduction was performed using the reduceDimension function and cells were ordered along a pseudotemporal trajectory. The aforementioned analysis was performed in a semi-supervised manner with a few marker genes of health and injury selected. A similar unsupervised analysis was also performed, and no significant difference was identified. We also performed the analysis with Monocle v.3.alpha and obtained similar results. To compare the difference of gene expression signature between successful repair and failed repair branches, the branched expression analysis modeling (BEAM) was used. Genes with q values < 10−30 were processed and the two clusters were cataloged in a pseudotime manner. For analysis where cell downsampling was performed, the scanpy.pp.subsample function was used.
Single-cell fate mapping on time-series datasets
To infer the temporal couplings of the injury and repairing process of PT cells, which were sampled from various independent timepoints, we performed single-cell fate prediction analysis on the Cellrank (Lange et al., 2022) (development version of 1.3.2) platform which incorporates the Waddington Optimal-Transport (Schiebinger et al., 2019) modeling approach. Briefly, a subset of PT cells was selected to accelerate computation, and dimensionality reduction and UMAP embedding were conducted. Initial growth rates were estimated with default settings. Then, a cell-cell transition matrix was calculated using the compute_transition_matrix function (growth_iters = 3, last_time_point = “connectivities”), which uses a birth-death prediction process based on the Markov chains model. Then, the mass flow probabilities of the Type1 and Type2 PT injury states were computed and visualized using the plot_single_flow function.
Gene enrichment analysis
All gene enrichment analysis and protein-protein interaction enrichment analysis were performed on the Metascape v3.5 (Zhou et al., 2019) platform with default settings. Top differentially expressed genes of a cell type were selected and q-values were log-transformed and used for data visualization.
Single-cell pathway and transcription factor (TF) activity prediction
Progeny (v1) (Holland et al., 2020; Schubert et al., 2018) was used to analyze activities of key biological pathways in individual cells. Briefly, gene weights for each pathway were defined which considered top 2000 significant genes per pathway. Then, the dataset was normalized and scaled, and 100 permutations were conducted to calculate p-values of random activities.
For TF activity prediction in scRNA-seq data, DoRothEA (v1) (Garcia-Alonso et al., 2019; Holland et al., 2020) was used. Briefly, a TF-target network matrix was first defined in the package with confidence levels set as [‘A’, ‘B’, ‘C’]. TFs with less than 5 targets identified in the dataset were ignored and 100 permutations were performed on normalized and scaled data. The rank_tfs_groups function was used to identify genes that showed differential activities in each cell type.
Cell-cell interaction analysis
Cell-cell interaction analysis was performed using CellPhoneDB (v2.1.7) (Efremova et al., 2020) on all cells used in single-cell clustering. Cell subtypes with high transcriptomics similarity (e.g. Type1 Injured S1/2 and S3 cells of PT) were combined for the convenience of data visualization. For the use of CellPhoneDB, all genes were converted to human orthologues according to the Homology database of Mouse Genome Informatics. The analysis was implemented with the statistical method of CellPhoneDB (--iterations = 2000). For analyzing the dynamics of cell-cell interactions across multiple timepoints, data was demultiplexed and the same analysis was performed on each condition. Significantly dysregulated ligand-receptor pairs were identified based on their p-values and activity scores.
Comparison and integration with other datasets
For integration with a previous scRNA-seq dataset (Kirita et al., 2020) (126,578 cells) generated by 10X Chromium technologies, we first extracted cells originated from healthy and uni-IRI kidney samples (206,681 cells) from our sci-RNA-seq3 data. Then, the two datasets were combined using the AnnData.concatenate function, and then normalized and log-transformed. The top 3000 genes were selected as the highly variable genes. The data was scaled and PCA was performed using the scanpy.tl.pca function (svd_solver = ‘arpack’, n_comps = 30). A neighborhood graph was calculated (n_neighbors = 20, metric = ‘cosine’) and further embedded into a 2D UMAP graph using the scanpy.tl.umap function. Then, Batch Balanced K Nearest Neighbours (BBKNN) (Polański et al., 2020) was used with default settings to correct batch difference. For visualization of the combined dataset, UMAP space was computed again with the effective minimum distance between embedded points set as 0.1.
Spatial transcriptomics analysis was performed by revisiting a recent dataset (Dixon et al., 2021) generated with the 10X Genomics Visium System (PN-1000185, Lot No. 155614, Rev D). Quality control of spatial sequencing libraries was performed according to manufacturer’s instructions. Resulting Visium libraries were processed and aligned using the count and agg function of 10X Genomics SpaceRanger (v1.2.1). Analysis files were loaded into Seurat or Giotto (Dries et al., 2021) for normalization and plotting of spatial expression for genes of interest.
In addition, we compared our results with a few bulk RNA-seq datasets on kidney injury and fibrosis, which include a human renal IRI model (Park et al., 2020), folic acid-induced mouse nephropathy (Craciun et al., 2016) and UUO induced on Slc34a1GCE-eGFPL10a mice (PT-enriched transcripts) (Wu et al., 2020). Previous datasets generated by microarray approaches were also examined by surveying publicly available database collections (http://v5.nephroseq.org/). Protein expression of genes of interest was examined in The Human Protein Atlas (Uhlén et al., 2015).
Bulk RNA-seq
Human primary RPTECs were harvested at five different conditions for bulk RNA-seq with three biological replicates per group: (1) siNT treatment (control); (2) siNT and 6-hour 100 μM oleic acid exposure (siNT+Ole6hrs); (3) PLIN2 siRNA treatment and 6-hour 100 μM oleic acid exposure (siPLIN2+Ole6hrs), (4) siNT and 2-day normal medium exposure after 6-hour 100 μM oleic acid exposure (siNT+Ole6hrs+2d); and (5) siPLIN2+Ole6hrs+2d. RNA was extracted with RNeasy Kits (74104, Qiagen) following the manufacturer’s instruction. Libraries were generated with the poly-A selection method (mRNA Direct kit, Life Technologies) and sequenced with the NovaSeq 6000 S4 platform (2×150 bp) at a target of 30 million reads per library. RNA-seq reads were aligned and quantitated to the human reference genome Ensembl GRCh38.101 with an Illumina DRAGEN Bio-IT on-premise server running version 3.9.3–8 software. Differential expression analysis was performed with the exactTest function of edgeR v3.34.1 (Robinson et al., 2010). Genes with FDR < 0.01 were processed for GO enrichment analysis.
Lipidomics analysis
Triacylglycerol (TAG), free fatty acid (FFA), and cholesterol in mouse kidney tissues were analyzed with mass spectrometry-based lipidomics. Briefly, the mouse kidney samples were homogenized in water (1:4, w/v) using Omni Bead Ruptor. A modified Bligh and Dyer method was used to extract TAG, FFA, and cholesterol from 50 μL of homogenate. Internal standards of TAG (Nu-Chek Prep #T404), FFA (Cambridge Isotopes #DLM-2893–0.5) and cholesterol (Toronto Research Company #C432503) were added to the samples before extraction. The FFA was derivatized with 4-aminomethylphenylpyridium to improve mass spectrometric sensitivity. Measurement of TAG, FFA and cholesterol was performed with a Shimadzu 20AD HPLC system coupled to a 4000QTRAP mass spectrometer operated in positive multiple reaction monitoring mode. The electrospray ionization was used for TAG and FFA, and air pressure chemical ionization was used for cholesterol. Data processing was conducted with Analyst 1.6.3. Quality control (QC) samples were prepared by pooling the aliquots of the study samples and were used to monitor the instrument stability. Only the lipid species with CV < 15% in QC sample were reported. The data were reported as the peak area ratios of the analytes to the corresponding internal standards.
Measurement of BCAA concentration
Branched Chain Amino Acid Colorimetric Kit (MAK003, Sigma) was used to determine the concentration of BCAA in mouse kidneys. Briefly, mouse kidneys were completely perfused with PBS before dissection. For each sample, 10–20 mg tissue was dissected from mouse kidney cortex, weighted and homogenized in cold BCAA Assay Buffer. After centrifugation, supernatant was obtained and processed for the colorimetric detection with the manufacturer’s instructions. Leucine standards were used to generate the standard curve.
Statistical analysis
Unless otherwise specified, p values presented in tissue culture and biochemical assays were generated by unpair two-tailed Student’s t-tests and FDR values presented in bulk RNA-seq differential expression analysis were generated by exact tests on two groups of negative binomial random variables. Unless otherwise specified, Mann-Whitney-U test with the Benjamini-Hochberg correction was used for differential expression analysis of scRNA-seq data.
Supplementary Material
Data S1 – Source Data. Raw data used for figure generation.
Table S1. sci-RNA-seq3 protocol optimization and dataset overview.
Table S2. Summary of all cell subtypes and cell states identified.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-KIM-1 | R&D Systems | MAB1817 |
| Rabbit anti-PLIN2 | Abcam | ab108323 |
| Rabbit anti-BCAT1 | Proteintech | 13640–1-AP |
| Rabbit anti-PRICKLE1 | BiCell Scientific | Cat#50621 |
| FITC-conjugated mouse anti-ACTA2 | Sigma | Cat#F3777 |
| FITC-conjugated Lotus Tetragonolobus Lectin (LTL) | Vector Laboratories | FL-1321–2 |
| Biotinylated LTL | Vector Laboratories | B-1325–2 |
| Rabbit anti-oxLDL | Bioss | bs-1698R |
| Anti-DIG-AP Fab fragments | Roche | Cat#11093274910 |
| Chemicals, peptides, and recombinant proteins | ||
| NaCl 5M | Invitrogen | AM9759 |
| MgCl2 1M | Invitrogen | AM9530G |
| Tris-HCl pH 7.5 1M | Invitrogen | Cat#15567–027 |
| Tris-HCl pH 8.0 1M | Invitrogen | Cat#15568–025 |
| Triton X-100 | Sigma | T8787 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15713 |
| DMEM | Gibco | Cat#11965 |
| Fetal Bovine Serum | Sigma | F4135 |
| Penicillin-streptomycin | Gibco | Cat#15140122 |
| Renal Epithelial Cell Growth Medium | Lonza | CC-3190 |
| TRIzol | Invitrogen | Cat#15596026 |
| iTaq Universal SYBR Green Supermix | BioRad | Cat#1725125 |
| BODIPY 493/503 | Cayman Chemical | Cat#25892 |
| DIG RNA Labeling Mix | Roche | Cat#11277073910 |
| Proteinase-K | Invitrogen | Cat#25530049 |
| BM-Purple solution | Roche | Cat#11442074001 |
| Oleate fatty acid | Cayman Chemical | Cat#29557 |
| Palmitate fatty acid | Cayman Chemical | Cat#29558 |
| Atglistatin | Cayman Chemical | Cat#15284 |
| BODIPY FL C16 | Thermo Scientific | D3821 |
| MitoTracker Red CMXRos | Cell Signaling | 9082S |
| Lipofectamine Reagent | Thermo Scientific | Cat#13778150 |
| 2,7-Dichlorodihydrofluorescein diacetate | Cayman Chemical | Cat#85155 |
| Seahorse XF DMEM assay medium | Agilent | Cat#103680–100 |
| Etomoxir | Cayman Chemical | Cat#11969 |
| Oligomycin | Sigma | O4876 |
| Nuclei EZ Lysis Buffer | Sigma | NUC101 |
| EDTA-free protease inhibitor tablets | Roche | Cat#5892791001 |
| RNasin Ribonuclease Inhibitor | Promega | N2615 |
| SUPERase•In RNase Inhibitor | Thermo Scientific | AM2696 |
| RNaseOUT RNase inhibitor | Thermo Scientific | Cat#10777019 |
| BSA | NEB | B9000S |
| dNTP | Clontech | Cat#639125 |
| SuperScript IV reverse transcriptase | Thermo Scientific | Cat#18090050 |
| Quick Ligase | NEB | M2200L |
| Second strand synthesis enzyme | NEB | E6111L |
| Dimethylformamide | Thermo Scientific | Cat#20673 |
| USER enzyme | NEB | M5505L |
| NEBnext 2× master mix | NEB | M0541L |
| DNA binding buffer | Zymo | D4004–1-L |
| Critical commercial assays | ||
| Oil Red O Stain Kit | Abcam | ab150678 |
| Branched Chain Amino Acid Colorimetric Kit | Sigma | MAK003 |
| Seahorse XFe96 FluxPaks | Agilent | Cat#102416–100 |
| Deposited data | ||
| Raw and processed sci-RNA-seq3 data | This study | GSE190887 |
| Raw and processed RNA-seq data | This study | GSE206084 |
| Kidney Interactive Transcriptomics | The Humphreys Lab | http://humphreyslab.com/SingleCell/ |
| Mendeley Data | This study | http://dx.doi.org/10.17632/hd3j7mdm2p.1 |
| Data S1 – Source Data | This study | N/A |
| Experimental models: Cell lines | ||
| HEK-293T | ATCC | CRL-3216 |
| C3H/10T1/2 | ATCC | CCL-226 |
| Primary human renal proximal tubule epithelial cells | Lonza | CC-2553 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Lab | Cat#000664 |
| Software and algorithms | ||
| Scanpy | (Wolf et al., 2018) | https://scanpy.readthedocs.io/en/stable/ |
| Seurat | (Stuart et al., 2019) | https://satijalab.org/seurat/ |
| sci-RNA-seq3 preprocessing | (Cao et al., 2019) | https://github.com/JunyueC/sci-RNA-seq3_pipeline |
| deML | (Renaud et al., 2015) | https://github.com/grenaud/deML |
| TrimGalore | N/A | https://github.com/FelixKrueger/TrimGalore |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| samtools | (Li et al., 2009) | http://www.htslib.org/ |
| HTSeq | (Anders et al., 2015) | https://htseq.readthedocs.io/en/master/index.html |
| CellBender | (Fleming et al., 2019) | https://github.com/broadinstitute/CellBender |
| Monocle2 | (Qiu et al., 2017) | http://cole-trapnell-lab.github.io/monocle-release/ |
| Scrublet | (Wolock et al., 2019) | https://github.com/swolock/scrublet |
| plot1cell | (Wu et al., 2022) | https://github.com/TheHumphreysLab/plot1cell |
| Harmony | (Korsunsky et al., 2019) | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
| Cellrank | (Lange et al., 2022) | https://cellrank.readthedocs.io/en/stable/ |
| Metascape | (Zhou et al., 2019) | https://metascape.org |
| Progeny | (Holland et al., 2020) | https://github.com/saezlab/progeny-py |
| DoRothEA | (Holland et al., 2020) | https://github.com/saezlab/dorothea-py |
| CellPhoneDB | (Efremova et al., 2020) | https://github.com/Teichlab/cellphonedb |
| BBKNN | (Polański et al., 2020) | https://github.com/Teichlab/bbknn |
| SpaceRanger | 10X Genomics | https://support.10xgenomics.com/spatial-gene-expression/software/pipelines/latest/installation |
| Giotto | (Dries et al., 2021) | https://rubd.github.io/Giotto_site/ |
| edgeR | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
Highlights.
sci-RNA-seq3 transcriptionally profiles 309,666 cells from 24 kidneys without batch effects
Two injured proximal tubule cell states with distinct metabolic profiles revealed
Transiently activated lipid metabolism and PLIN2+ lipid droplets appear in early IRI
Nephron epithelia possess both shared and segment-specific injury and repair responses
Acknowledgments
These experiments were funded by NIH grants DK103740 and UC2DK126024 to BDH. The authors acknowledge the Washington University Diabetes Research Center for providing training for Seahorse Analyzer applications. The authors also acknowledge the Washington University Genome Technology Access Center and Center for Genome Sciences & Systems Biology for sequencing support.
Declaration of interests
B.D.H. is a consultant for Janssen Research & Development, LLC, Pfizer and Chinook Therapeutics, holds equity in Chinook Therapeutics and grant funding from Chinook Therapeutics, Janssen Research & Development, LLC and Pfizer; all interests are unrelated to the current work.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe J. ichi, Ko KA, Kotla S, Wang Y, Paez-Mayorga J, Shin IJ, Imanishi M, Vu HT, Tao Y, Leiva-Juarez MM, Thomas TN, Medina JL, Won JH, Fujii Y, Giancursio CJ, McBeath E, Shin JH, Guzman L, Abe RJ, Taunton J, Mochizuki N, Faubion W, Cooke JP, Fujiwara K, Evans SE, Le NT, 2019. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI insight 4. 10.1172/JCI.INSIGHT.125570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeva MM, Calviño J, Souto G, Donapetry C, 2011. Insulin resistance and the metabolism of branched-chain amino acids in humans. Amin. Acids 2011 431 43, 171–181. 10.1007/S00726-011-1088-7 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W, 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166. 10.1093/BIOINFORMATICS/BTU638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzer MS, Rohacs T, Susztak K, 2021. How Many Cell Types Are in the Kidney and What Do They Do? 10.1146/annurev-physiol-052521-121841 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolisetty S, Zarjou A, Agarwal A, 2017. Heme Oxygenase 1 as a Therapeutic Target in Acute Kidney Injury. Am. J. Kidney Dis 69, 531–545. 10.1053/J.AJKD.2016.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boström EA, Lundberg P, 2013. The Newly Discovered Cytokine IL-34 Is Expressed in Gingival Fibroblasts, Shows Enhanced Expression by Pro-Inflammatory Cytokines, and Stimulates Osteoclast Differentiation. PLoS One 8, e81665. 10.1371/JOURNAL.PONE.0081665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, Zager MA, Aldinger KA, Blecher-Gonen R, Zhang F, Spielmann M, Palis J, Doherty D, Steemers FJ, Glass IA, Trapnell C, Shendure J, 2020. A human cell atlas of fetal gene expression. Science (80-.) 370. 10.1126/SCIENCE.ABA7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, Trapnell C, Shendure J, 2019. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. 10.1038/s41586-019-0969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang-Panesso M, Kadyrov FF, Lalli M, Wu H, Ikeda S, Kefaloyianni E, Abdelmageed MM, Herrlich A, Kobayashi A, Humphreys BD, 2019. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J. Clin. Invest 129, 5501–5517. 10.1172/JCI125519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E, Tsai TH, Li L, Saha P, Chan L, Chang BHJ, 2017. PLIN2 is a Key Regulator of the Unfolded Protein Response and Endoplasmic Reticulum Stress Resolution in Pancreatic β Cells. Sci. Reports 2017 71 7, 1–12. 10.1038/srep40855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G-Y, Muramatsu H, Kondo M, Kurosawa N, Miyake Y, Takeda N, Muramatsu T, 2005. Abnormalities caused by carbohydrate alterations in Ibeta6-N-acetylglucosaminyltransferase-deficient mice. Mol. Cell. Biol 25, 7828–7838. 10.1128/MCB.25.17.7828-7838.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, Palmer M, Susztak K, 2019. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 30, 784–799.e5. 10.1016/J.CMET.2019.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clef N. Le, Verhulst A, D’Haese PC, Vervaet BA, 2016. Unilateral Renal Ischemia-Reperfusion as a Robust Model for Acute to Chronic Kidney Injury in Mice. PLoS One 11. 10.1371/JOURNAL.PONE.0152153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun FL, Bijol V, Ajay AK, Rao P, Kumar RK, Hutchinson J, Hofmann O, Joshi N, Luyendyk JP, Kusebauch U, Moss CL, Srivastava A, Himmelfarb J, Waikar SS, Moritz RL, Vaidya VS, 2016. RNA Sequencing Identifies Novel Translational Biomarkers of Kidney Fibrosis. J. Am. Soc. Nephrol 27, 1702–1713. 10.1681/ASN.2015020225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalen KT, Ulven SM, Arntsen BM, Solaas K, Nebb HI, 2006. PPARalpha activators and fasting induce the expression of adipose differentiation-related protein in liver. J. Lipid Res 47, 931–943. 10.1194/JLR.M500459-JLR200 [DOI] [PubMed] [Google Scholar]
- De Groot T, Bindels RJM, Hoenderop JGJ, 2008. TRPV5: an ingeniously controlled calcium channel. Kidney Int. 74, 1241–1246. 10.1038/KI.2008.320 [DOI] [PubMed] [Google Scholar]
- Dhillon P, Park J, Hurtado del Pozo C, Li L, Doke T, Huang S, Zhao J, Kang HM, Shrestra R, Balzer MS, Chatterjee S, Prado P, Han SY, Liu H, Sheng X, Dierickx P, Batmanov K, Romero JP, Prósper F, Li M, Pei L, Kim J, Montserrat N, Susztak K, 2021. The Nuclear Receptor ESRRA Protects from Kidney Disease by Coupling Metabolism and Differentiation. Cell Metab. 33, 379–394.e8. 10.1016/J.CMET.2020.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon E, Wu H, Muto Y, Wilson P, Humphreys B, 2021. Spatially Resolved Transcriptomic Analysis of Acute Kidney Injury in a Female Murine Model. J. Am. Soc. Nephrol ASN.2021081150. 10.1681/ASN.2021081150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/BIOINFORMATICS/BTS635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dries R, Zhu Q, Dong R, Eng CHL, Li H, Liu K, Fu Y, Zhao T, Sarkar A, Bao F, George RE, Pierson N, Cai L, Yuan GC, 2021. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 1–31. 10.1186/S13059-021-02286-2/FIGURES/6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R, 2020. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc 2020 154 15, 1484–1506. 10.1038/s41596-020-0292-x [DOI] [PubMed] [Google Scholar]
- Evers TMJ, Hochane M, Tans SJ, Heeren RMA, Semrau S, Nemes P, Mashaghi A, 2019. Deciphering metabolic heterogeneity by single-cell analysis. Anal. Chem 91, 13314. 10.1021/ACS.ANALCHEM.9B02410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulski KS, De Freitas DG, Kreepala C, Chang J, Sellares J, Sis B, Einecke G, Mengel M, Reeve J, Halloran PF, 2012. Molecular phenotypes of acute kidney injury in kidney transplants. J. Am. Soc. Nephrol 23, 948–958. 10.1681/ASN.2011090887/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SJ, Marioni JC, Babadi M, 2019. CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv 791699. 10.1101/791699 [DOI] [Google Scholar]
- Gao J, Serrero G, 1999. Adipose differentiation related protein (ADRP) expressed in transfected COS-7 cells selectively stimulates long chain fatty acid uptake. J. Biol. Chem 274, 16825–16830. 10.1074/JBC.274.24.16825 [DOI] [PubMed] [Google Scholar]
- Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, Saez-Rodriguez J, 2019. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 29, 1363–1375. 10.1101/GR.240663.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibotto Giacomo G, Sofia A, Saffioti S, Bonanni A, Mannucci I, Verzola D, 2010. Amino acid and protein metabolism in the human kidney and in patients with chronic kidney disease. Clin. Nutr 29, 424–433. 10.1016/J.CLNU.2010.02.005 [DOI] [PubMed] [Google Scholar]
- Gerhardt LMS, Liu J, Koppitch K, Cippà PE, McMahon AP, 2021. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc. Natl. Acad. Sci. U. S. A 118. 10.1073/PNAS.2026684118/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains D, Sims-Lucas S, Kish K, Saha M, Mchugh K, Bates CM, 2008. Role of fibroblast growth factor receptor 2 in kidney mesenchyme. Pediatr. Res 64, 592. 10.1203/PDR.0B013E318187CC12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harskamp LR, Gansevoort RT, Van Goor H, Meijer E, 2016. The epidermal growth factor receptor pathway in chronic kidney diseases. Nat. Rev. Nephrol 2016 128 12, 496–506. 10.1038/nrneph.2016.91 [DOI] [PubMed] [Google Scholar]
- Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, Hobbs FDR, 2016. Global Prevalence of Chronic Kidney Disease – A Systematic Review and Meta-Analysis. PLoS One 11, e0158765. 10.1371/journal.pone.0158765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocaoglu H, Wang L, Yang M, Yue S, Sieber M, 2021. Heritable shifts in redox metabolites during mitochondrial quiescence reprogramme progeny metabolism. Nat. Metab 2021 39 3, 1259–1274. 10.1038/s42255-021-00450-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland CH, Tanevski J, Perales-Patón J, Gleixner J, Kumar MP, Mereu E, Joughin BA, Stegle O, Lauffenburger DA, Heyn H, Szalai B, Saez-Rodriguez J, 2020. Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 2020 211 21, 1–19. 10.1186/S13059-020-1949-Z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Doebele RC, Lingen MW, Quilliam LA, Tang WJ, Rosner MR, 2007. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem 282, 19781–19787. 10.1074/JBC.M700128200 [DOI] [PubMed] [Google Scholar]
- Huang Z, Liu S, Tang A, Al-Rabadi L, Henkemeyer M, Mimche PN, Huang Y, 2021. Key role for EphB2 receptor in kidney fibrosis. Clin. Sci 135, 2127–2142. 10.1042/CS20210644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, 2018. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol 80, 309–335. 10.1146/annurev-physiol-022516-034227 [DOI] [PubMed] [Google Scholar]
- Kalantar-Zadeh K, Jafar TH, Nitsch D, Neuen BL, Perkovic V, 2021. Chronic kidney disease. Lancet 398, 786–802. 10.1016/S0140-6736(21)00519-5 [DOI] [PubMed] [Google Scholar]
- Kaleta B, 2019. The role of osteopontin in kidney diseases. Inflamm. Res 68, 93–102. 10.1007/S00011-018-1200-5 [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Fujii M, Mizokami A, Kittler JT, Nabekura J, Moss SJ, Hirata M, 2007. Phospholipase C-related inactive protein is implicated in the constitutive internalization of GABAA receptors mediated by clathrin and AP2 adaptor complex. J. Neurochem 101, 898–905. 10.1111/J.1471-4159.2006.04399.X [DOI] [PubMed] [Google Scholar]
- Kang HM, Ahn SH, Choi P, Ko Y-A, Han SH, Chinga F, Park ASD, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K, 2014. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med 2014 211 21, 37–46. 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamissi FZ, Ning L, Kefaloyianni E, Dun H, Arthanarisami A, Keller A, Atkinson JJ, Li W, Wong B, Dietmann S, Lavine K, Kreisel D, Herrlich A, 2022. Identification of kidney injury–released circulating osteopontin as causal agent of respiratory failure. Sci. Adv 8, 5900. 10.1126/SCIADV.ABM5900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Yang JI, Jung MH, Hwa JS, Kang KR, Park DJ, Roh GS, Cho GJ, Choi WS, Chang SH, 2006. Heme Oxygenase-1 Protects Rat Kidney from Ureteral Obstruction via an Antiapoptotic Pathway. J. Am. Soc. Nephrol 17, 1373–1381. 10.1681/ASN.2005091001 [DOI] [PubMed] [Google Scholar]
- Kimmel AR, Sztalryd C, 2016. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr 36, 471–509. 10.1146/ANNUREV-NUTR-071813-105410 [DOI] [PubMed] [Google Scholar]
- Kirita Y, Wu H, Uchimura K, Wilson PC, Humphreys BD, 2020. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. U. S. A 117, 15874–15883. 10.1073/pnas.2005477117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P. ru, Raychaudhuri S, 2019. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019 1612 16, 1289–1296. 10.1038/s41592-019-0619-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Patón J, Jansen J, Reimer KC, Smith JR, Dobie R, Wilson-Kanamori JR, Halder M, Xu Y, Kabgani N, Kaesler N, Klaus M, Gernhold L, Puelles VG, Huber TB, Boor P, Menzel S, Hoogenboezem RM, Bindels EMJ, Steffens J, Floege J, Schneider RK, Saez-Rodriguez J, Henderson NC, Kramann R, 2021. Decoding myofibroblast origins in human kidney fibrosis. Nature 589. 10.1038/s41586-020-2941-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzumi A, Yoshizaki A, Matsuda KM, Kotani H, Norimatsu Y, Fukayama M, Ebata S, Fukasawa T, Yoshizaki-Ogawa A, Asano Y, Morikawa K, Kazoe Y, Mawatari K, Kitamori T, Sato S, 2021. Interleukin-31 promotes fibrosis and T helper 2 polarization in systemic sclerosis. Nat. Commun 2021 121 12, 1–18. 10.1038/s41467-021-26099-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange M, Bergen V, Klein M, Setty M, Reuter B, Bakhti M, Lickert H, Ansari M, Schniering J, Schiller HB, Pe’er D, Theis FJ, 2022. CellRank for directed single-cell fate mapping. Nat. Methods 2022 1–12. 10.1038/s41592-021-01346-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewington AJP, Padanilam BJ, Martin DR, Hammerman MR, 2000. Expression of CD44 in kidney after acute ischemic injury in rats. 10.1152/ajpregu.2000.278.1.R247 278. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup, 1000 Genome Project Data Processing, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/BIOINFORMATICS/BTP352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Humphreys BD, 2021. Single Cell Technologies: Beyond Microfluidics. Kidney 360 2, 1196–1204. 10.34067/KID.0001822021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LO, Klett EL, Coleman RA, 2010. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 1801, 246. 10.1016/J.BBALIP.2009.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, Chen Y, Li M, Dessing MC, Parvez RK, Cippà PE, Krautzberger AM, Saribekyan G, Smith AD, McMahon AP, 2017. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2. 10.1172/JCI.INSIGHT.94716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YA, Te Liao C, Raybould R, Talabani B, Grigorieva I, Szomolay B, Bowen T, Andrews R, Taylor PR, Fraser D, 2021. Single-nucleus RNA sequencing identifies new classes of proximal tubular epithelial cells in kidney fibrosis. J. Am. Soc. Nephrol 32, 2501–2516. 10.1681/ASN.2020081143/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Zhang B, LaFave L, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Law T, Lareau C, Hsu Y-C, Regev A, Buenrostro JD, 2020. Chromatin potential identified by shared single cell profiling of RNA and chromatin. bioRxiv 2020.06.17.156943. 10.1101/2020.06.17.156943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markó L, Vigolo E, Hinze C, Park JK, Roël G, Balogh A, Choi M, Wübken A, Cording J, Blasig IE, Luft FC, Scheidereit C, Schmidt-Ott KM, Schmidt-Ullrich R, Müller DN, 2016. Tubular epithelial NF-κB activity regulates ischemic AKI. J. Am. Soc. Nephrol 27, 2658–2669. 10.1681/ASN.2015070748/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marneros AG, 2021. Magnesium and Calcium Homeostasis Depend on KCTD1 Function in the Distal Nephron. Cell Rep. 34, 108616. 10.1016/J.CELREP.2020.108616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marneros AG, 2020. AP-2β/KCTD1 Control Distal Nephron Differentiation and Protect against Renal Fibrosis. Dev. Cell 54, 348–366.e5. 10.1016/J.DEVCEL.2020.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BK, Qiu C, Nichols E, Phung M, Green-Gladden R, Srivatsan S, Blecher-Gonen R, Beliveau BJ, Trapnell C, Cao J, Shendure J, 2021. An optimized protocol for single cell transcriptional profiling by combinatorial indexing. [DOI] [PMC free article] [PubMed]
- Meliambro K, Wong JS, Ray J, Calizo RC, Towne S, Cole B, El Salem F, Gordon RE, Kaufman L, He JC, Azeloglu EU, Campbell KN, 2017. The Hippo pathway regulator KIBRA promotes podocyte injury by inhibiting YAP signaling and disrupting actin cytoskeletal dynamics. 10.1074/jbc.M117.819029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Ajay AK, Chang JH, Mou S, Zhao H, Kishi S, Li J, Brooks CR, Xiao S, Woo HM, Sabbisetti VS, Palmer SC, Galichon P, Li L, Henderson JM, Kuchroo VK, Hawkins J, Ichimura T, Bonventre JV, 2021. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 33, 1042–1061.e7. 10.1016/J.CMET.2021.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffari MS, 2003. TAURINE MODULATION OF RENAL EXCRETORY FUNCTION. THAI J. Physiol. Sci 16. [Google Scholar]
- Muto Y, Wilson PC, Ledru N, Wu H, Dimke H, Waikar SS, Humphreys BD, 2021. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat. Commun 2021 121 12, 1–17. 10.1038/s41467-021-22368-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacu N, Luzina IG, Highsmith K, Lockatell V, Pochetuhen K, Cooper ZA, Gillmeister MP, Todd NW, Atamas SP, 2008. MACROPHAGES PRODUCE TGFBI (BIGH3) FOLLOWING INGESTION OF APOPTOTIC CELLS AND REGULATE MMP14 LEVELS AND COLLAGEN TURNOVER IN FIBROBLASTS. J. Immunol 180, 5036. 10.4049/JIMMUNOL.180.7.5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Nishihara K, Miyata H, Shinke H, Tomita E, Kajiwara M, Matsubara T, Iehara N, Igarashi Y, Yamada H, Fukatsu A, Yanagita M, Matsubara K, Masuda S, 2015. Molecular Markers of Tubulointerstitial Fibrosis and Tubular Cell Damage in Patients with Chronic Kidney Disease. PLoS One 10, e0136994. 10.1371/JOURNAL.PONE.0136994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Wada H, Okada N, Harada I, Nakajima T, Pasquale EB, Tsuyama S, 2006. EphB2 and ephrin-B1 expressed in the adult kidney regulate the cytoarchitecture of medullary tubule cells through Rho family GTPases. J. Cell Sci 119, 559–570. 10.1242/JCS.02777 [DOI] [PubMed] [Google Scholar]
- Osis G, Traylor AM, Black LM, Spangler D, George JF, Zarjou A, Verlander JW, Agarwal A, 2021. Expression of lactate dehydrogenase A and B isoforms in the mouse kidney. Am. J. Physiol. - Ren. Physiol 320, F706–F718. 10.1152/AJPRENAL.00628.2020/ASSET/IMAGES/LARGE/AJPRENAL.00628.2020_F006.JPEG [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Suszták K, 2018. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science (80-.) 360. 10.1126/science.aar2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Kwon CH, Ha HK, Han M, Song SH, 2020. RNA-Seq identifies condition-specific biological signatures of ischemia-reperfusion injury in the human kidney. BMC Nephrol. 2020 211 21, 1–12. 10.1186/S12882-020-02025-Y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pat BK, Cuttle L, Watters D, Yang T, Johnson DW, Gobe GC, 2003. Fibrogenic stresses activate different mitogen-activated protein kinase pathways in renal epithelial, endothelial or fibroblast cell populations. Nephrology (Carlton). 8, 196–204. 10.1046/J.1440-1797.2003.00162.X [DOI] [PubMed] [Google Scholar]
- Pei K, Gui T, Li C, Zhang Q, Feng H, Li Y, Wu J, Gai Z, 2020. Recent Progress on Lipid Intake and Chronic Kidney Disease. Biomed Res. Int 2020. 10.1155/2020/3680397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkov D, Ni R, Else C, Piñol-Roma S, Ramirez F, Tanaka S, 2000. Cloning of a human gene closely related to the genes coding for the c-myc single-strand binding proteins. Gene 243, 27–36. 10.1016/S0378-1119(99)00515-6 [DOI] [PubMed] [Google Scholar]
- Piret SE, Guo Y, Attallah AA, Horne SJ, Zollman A, Owusu D, Henein J, Sidorenko VS, Revelo MP, Hato T, Ma’ayan A, He JC, Mallipattu SK, 2021. Krüppel-like factor 6-mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. Proc. Natl. Acad. Sci. U. S. A 118. 10.1073/PNAS.2024414118/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polański K, Young MD, Miao Z, Meyer KB, Teichmann SA, Park J-E, 2020. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965. 10.1093/BIOINFORMATICS/BTZ625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, Trapnell C, 2017. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979. 10.1038/NMETH.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramseyer VD, Garvin JL, 2013. Tumor necrosis factor-α: regulation of renal function and blood pressure. Am. J. Physiol. - Ren. Physiol 304, F1231. 10.1152/AJPRENAL.00557.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashmi RN, Eckes B, Glöckner G, Groth M, Neumann S, Gloy J, Sellin L, Walz G, Schneider M, Karakesisoglou I, Eichinger L, Noegel AA, 2012. The nuclear envelope protein Nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus 3, 172. 10.4161/NUCL.19090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud G, Stenzel U, Maricic T, Wiebe V, Kelso J, 2015. DeML: Robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics 31, 770–772. 10.1093/bioinformatics/btu719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/BIOINFORMATICS/BTP616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma’ayan A, 2016. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016. 10.1093/DATABASE/BAW100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudman-Melnick V, Adam M, Potter A, Chokshi SM, Ma Q, Drake KA, Schuh MP, Matthew Kofron J, Devarajan P, Steven Potter S, 2020. Single-cell profiling of AKI in a murine model reveals novel transcriptional signatures, profibrotic phenotype, and epithelial-to-stromal crosstalk. J. Am. Soc. Nephrol 31, 2793–2814. 10.1681/ASN.2020010052/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Yanagita M, 2017. Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm. Regen 37, 17. 10.1186/s41232-017-0048-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebinger G, Shu J, Tabaka M, Cleary B, Subramanian V, Solomon A, Gould J, Liu S, Lin S, Berube P, Lee L, Chen J, Brumbaugh J, Rigollet P, Hochedlinger K, Jaenisch R, Regev A, Lander ES, 2019. Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 176, 928–943.e22. 10.1016/J.CELL.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiessl IM, Grill A, Fremter K, Steppan D, Hellmuth M-K, Castrop H, 2018. Renal Interstitial Platelet-Derived Growth Factor Receptor-b Cells Support Proximal Tubular Regeneration. 10.1681/ASN.2017101069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Klinger B, Klünemann M, Sieber A, Uhlitz F, Sauer S, Garnett MJ, Blüthgen N, Saez-Rodriguez J, 2018. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun 2017 91 9, 1–11. 10.1038/s41467-017-02391-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Taha IN, Clauser KR, Gao Y. (Tom), Naba A, 2020. MatrisomeDB: the ECM-protein knowledge database. Nucleic Acids Res. 48, D1136–D1144. 10.1093/NAR/GKZ849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Bolati D, Adijiang A, Muteliefu G, Enomoto A, Nishijima F, Dateki M, Niwa T, 2011. NF-κb plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am. J. Physiol. - Cell Physiol 301, 1201–1212. 10.1152/AJPCELL.00471.2010/ASSET/IMAGES/LARGE/ZH00111167530011.JPEG [DOI] [PubMed] [Google Scholar]
- Shoji H, Yoshio S, Mano Y, Kumagai E, Sugiyama M, Korenaga M, Arai T, Itokawa N, Atsukawa M, Aikata H, Hyogo H, Chayama K, Ohashi T, Ito K, Yoneda M, Nozaki Y, Kawaguchi T, Torimura T, Abe M, Hiasa Y, Fukai M, Kamiyama T, Taketomi A, Mizokami M, Kanto T, 2016. Interleukin-34 as a fibroblast-derived marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci. Reports 2016 61 6, 1–11. 10.1038/srep28814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee AD, 2012. Exploring metabolic dysfunction in chronic kidney disease. Nutr. Metab 2012 91 9, 1–16. 10.1186/1743-7075-9-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler K, Goldberg IJ, Susztak K, 2015. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diab. Rep 15. 10.1007/S11892-015-0611-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P, 2017. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. 10.1038/nmeth.4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, Smibert P, Satija R, 2018. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224. 10.1186/s13059-018-1603-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, 2019. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. 10.1016/J.CELL.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Satija R, 2019. Integrative single-cell analysis. Nat. Rev. Genet 10.1038/s41576-019-0093-7 [DOI] [PubMed] [Google Scholar]
- Suliman ME, Bárány P, Filho JCD, Bergström J, 2002. Accumulation of taurine in patients with renal failure. Nephrol. Dial. Transplant 17, 528–529. 10.1093/NDT/17.3.528 [DOI] [PubMed] [Google Scholar]
- Tang C, Livingston MJ, Liu Z, Dong Z, 2020. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol 2020 169 16, 489–508. 10.1038/s41581-020-0309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Liu N, Zhuang S, 2013. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 83, 804–810. 10.1038/KI.2012.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M, Chen J, 2020. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12. 10.1186/s13059-019-1850-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, Parikh SM, 2016. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nat. 2016 5317595 531, 528–532. 10.1038/nature17184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson IM, Edlund K, Lundberg E, Navani S, Szigyarto CAK, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, Von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, Von Heijne G, Nielsen J, Pontén F, 2015. Tissue-based map of the human proteome. Science (80-.) 347. 10.1126/SCIENCE.1260419/SUPPL_FILE/1260419_UHLEN.SM.PDF [DOI] [PubMed] [Google Scholar]
- Weinreb C, Wolock S, Tusi BK, Socolovsky M, Klein AM, 2018. Fundamental limits on dynamic inference from single-cell snapshots. Proc. Natl. Acad. Sci. U. S. A 115, E2467–E2476. 10.1073/PNAS.1714723115/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Li Y, Li S, Hu X, Wei Q, Dong Z, 2021. Glucose Metabolism in Acute Kidney Injury and Kidney Repair. Front. Med 8, 2183. 10.3389/FMED.2021.744122/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, Theis FJ, 2018. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018 191 19, 1–5. 10.1186/S13059-017-1382-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, Klein AM, 2019. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291.e9. 10.1016/J.CELS.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Kirita Y, Donnelly EL, Humphreys BD, 2019. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol 30, 23–32. 10.1681/ASN.2018090912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lai CF, Chang-Panesso M, Humphreys BD, 2020. Proximal tubule translational profiling during kidney fibrosis reveals proinflammatory and long noncoding RNA expression patterns with sexual dimorphism. J. Am. Soc. Nephrol 31, 23–38. 10.1681/ASN.2019040337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Villalobos RG, Yao X, Reilly D, Chen T, Rankin M, Myshkin E, Breyer MD, Humphreys BD, 2022. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. Cell Metab. 34, 1064–1078.e6. 10.1016/J.CMET.2022.05.010/ATTACHMENT/666C88DC-99A5-46E8-A7BF-B1E5E6FC5510/MMC3.ZIP [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Dai C, 2017. Ablation of FGFR2 in Fibroblasts Ameliorates Kidney Fibrosis after Ischemia/Reperfusion Injury in Mice. Kidney Dis. 3, 160–170. 10.1159/000484604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Bassuk AG, Fritzsch B, 2013. Prickle1 stunts limb growth through alteration of cell polarity and gene expression. Dev. Dyn 242, 1293–1306. 10.1002/DVDY.24025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zager RA, Johnson ACM, Hanson SY, Lund S, 2005. Ischemic proximal tubular injury primes mice to endotoxin-induced TNF-alpha generation and systemic release. Am. J. Physiol. Renal Physiol 289. 10.1152/AJPRENAL.00023.2005 [DOI] [PubMed] [Google Scholar]
- Zeng L, Sas KM, Cifarelli V, Schoiswohl G, Huggins L, Gumaste N, Hu Y, Pennathur S, Abumrad NA, Kershaw EE, Hussain MM, Susztak K, Goldberg IJ, Scerbo D, Son N-H, Sirwi A, Zeng Lixia, Sas Kelli M, Cifarelli Vincenza, Schoiswohl Gabriele, Huggins L-A, Gumaste Namrata, Hu Yunying, Pennathur Subramaniam, Abumrad Nada A, Kershaw Erin E, Mahmood Hussain M, Susztak Katalin, Goldberg Ira J, 2017. Kidney triglyceride accumulation in the fasted mouse is dependent upon serum free fatty acids[S]. J. Lipid Res 58, 1132–1142. 10.1194/jlr.M074427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan M, Brooks C, Liu F, Sun L, Dong Z, 2013. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 83, 568–581. 10.1038/KI.2012.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen Y, Chen T, Miao B, Tang Z, Hu X, Luo Y, Zheng T, Na N, Ning Na C, 2021. Single-cell transcriptomics provides new insights into the role of fibroblasts during peritoneal fibrosis. Clin. Transl. Med 11, e321. 10.1002/CTM2.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang S, Liu S, Li C, Wang J, 2015. Role of Smad signaling in kidney disease. Int. Urol. Nephrol 2015 4712 47, 1965–1975. 10.1007/S11255-015-1115-9 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK, 2019. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 2019 101 10, 1–10. 10.1038/s41467-019-09234-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Jiang L, Long M, Wei X, Hou Y, Du Y, 2021. Metabolic Reprogramming and Renal Fibrosis. Front. Med 8, 2198. 10.3389/FMED.2021.746920/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 – Source Data. Raw data used for figure generation.
Table S1. sci-RNA-seq3 protocol optimization and dataset overview.
Table S2. Summary of all cell subtypes and cell states identified.
Data Availability Statement
Raw (fastq) and pre-processed data (count matrix) and metadata of the sci-RNA-seq3 dataset have been deposited in NCBI’s Gene Expression Omnibus and are available through GEO Series accession number GSE190887. Raw and processed bulk RNA-seq data on RPTECs are available through GEO Series accession number GSE206084. All additional data are available at Mendeley Data (http://dx.doi.org/10.17632/hd3j7mdm2p.1).
Scripts for a pipeline of single-cell clustering and visualization and generation of all major figures in this study were written mostly in Python and R with code available at https://github.com/TheHumphreysLab/sci-RNA-seq-kidney. Data S1 - Source Data, containing values used to generate graphs related to Figures 3–5, 7 and S3–7, is also presented.