

Abstract
Agonist antibodies that target immune checkpoints, such as those in the Tumor Necrosis Factor (TNF) receptor superfamily, are an important class of emerging therapeutics due to their ability to regulate immune cell activity, especially for treating cancer. Despite their great potential, to date they have shown limited clinical utility and further antibody optimization is urgently needed to improve their therapeutic potential. Here we discuss key antibody engineering approaches for improving the activity of antibody agonists by optimizing their valency, specificity for different receptors (e.g., bispecific antibodies) and epitopes (e.g., biepitopic or biparatopic antibodies), and Fc affinity for FcγR receptors. These powerful approaches are being used to develop the next generation of cancer immunotherapeutics with improved efficacy and safety.
Keywords: CD137, 4-1BB, OX40, CD40, mAb, immunotherapy
Optimizing agonist antibody properties to maximize therapeutic potential
In the last decade, therapeutic antibodies have seen exponential growth in treating many diseases including cancer, autoimmune diseases, and inflammatory disorders [1]. These molecules possess attractive biophysical properties such as high specificity, affinity, and solubility that are essential for their success [2]. The Food and Drug Administration (FDA) has approved over 100 antibodies, representing one-fifth of new drug approvals each year [3]. For example, antagonist antibodies that target immune checkpoint inhibitors such as programmed death-1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) have shown great promise in cancer treatment and several have been approved by the FDA [4]. On the other hand, agonist antibodies that activate immune receptors have been a subject of many ongoing clinical trials and have faced major roadblocks such as low efficacy and off-target effects despite their importance in mediating anti-tumor immunity [5]. Therefore, further optimization methods are needed to address these challenges in developing potent cancer immunotherapeutics.
The TNF receptor superfamily is a major player in a range of non-immune and immune functions such as cell differentiation and proliferation [6]. This diverse set of receptors consists of at least 29 members, of which several of them, including OX40, CD40, CD137, GITR and DR5, have been shown to play a significant role in cancer treatment. The expression profile of these TNF receptors is highly diverse on different types of immune and cancer cells. Antibodies specific for OX40, CD137 and GITR receptors mainly target T cells subtypes and mediate their activation, while antibodies specific for DR5 mainly target tumor cells and induce cellular apoptosis [7]. Antibodies specific for CD40 target both immune and cancer cells, which mediates activation of antigen-presenting and T cells as well as direct cytotoxic effects against cancer cells.
Several TNF-specific agonist antibodies have shown potent T-cell mediated immunity against cancer in preclinical and clinical trials [8]. However, the development of such antibodies is hindered by numerous and complex factors associated with activating the immunomodulatory receptors. Notably, conventional bivalent antibodies have limited ability to mediate higher-order receptor clustering, which is key for strong receptor activation [5]. While clustering can be achieved via FcγR-mediated crosslinking, there is scant evidence that clinically relevant antibodies are able to achieve sufficient crosslinking in vivo for therapeutic use. This issue is further compounded by the varying levels of FcγR expression on different immune cells, leading to a wide range of receptor agonism [9,10]. Therefore, the dependency on FcγR-mediated crosslinking remains a major pharmacologic barrier for clinical success. Finally, key agonist antibody properties include epitope, valency, specificity, Fc-mediated interactions, and isotype, all of which must be critically considered for developing potent agonist therapeutics (Figure 1). In this review, we highlight recent advancements in agonist antibody development for TNF receptors such as OX40, CD40, CD137, GITR, and Death Receptor 5 (DR5) to guide their development and optimization for clinical use (see Clinician’s Corner).
Figure 1.

Summary of antibody engineering approaches for optimizing agonist activity for therapeutic applications. Agonist activity can be optimized via engineering the Fab, hinge and Fc regions to enhance receptor clustering and activation.
Clinician’s Corner.
Agonist antibodies that activate TNF receptors have been shown to mediate potent anti-tumor responses in pre-clinical studies and clinical trials.
Mounting evidence suggests that antibody engineering may be needed to improve existing antibody therapeutics to enhance their agonist potential.
Recent clinical trial data suggest that agonists may be most beneficial in combination therapy with chemotherapy agents and inhibitory immune checkpoints (i.e., PD-1, CTLA-4).
Antibody Valency
Receptor clustering is a critical aspect of immune cell activation where monomeric or multimeric subunits are brought together to form higher-order receptor complexes to transduce intracellular signaling [11]. Mounting evidence has demonstrated that multivalent antibodies and Fc-fusion proteins elicit improved agonist function by forming higher-order receptor superclusters compared to their conventional bivalent counterparts (Figure 2). In one study, investigators engineered a novel hexameric Fc-fusion protein (MEDI1873) that targets glucocorticoid-induced TNFR-related protein (GITR, TNFR18) to improve anti-tumor immunity [12]. This novel Fc-fusion protein was constructed by attaching an IgG Fc region to a trimeric motif and the human GITR ligand ectodomain. In vitro studies demonstrated a >2.5-fold increase in T cell proliferation. Next, MEDI1873 was evaluated in a primate model where it induced increased T cell proliferation and elevated levels of immunoglobulin (IgG) circulating antibodies, indicating both strong cellular and humoral immune responses. Given GITR is highly expressed on both human effector and regulatory T cells (Tregs), it represents an important target for developing effective therapeutics [12].
Figure 2.

Multivalent Fc-fusion protein potently activates GITR receptor. Schematic illustration of a hexavalent Fc-fusion protein, which is composed of a trimeric form of the human GITR ligand ectodomain fused on the Fc region of each heavy chain, shows potent receptor clustering and enhanced T cell activation. Adapted from [12].
The role of valency is further bolstered by a similar study where multivalent nanobodies were designed to improve DR5 receptor signaling [13]. To do this, researchers engineered nanobodies that were fused together with a flexible glycine-serine linker to create trivalent and tetravalent constructs. These formats significantly reduced tumor cell viability by 40%, demonstrating a greater apoptotic response compared to the conventional bivalent counterpart. Interestingly, the agonist activity of the trivalent nanobody was comparable to the natural ligand as measured by caspase activity. Other studies on TNF receptors, including OX40, further support the claim that higher-order valency mediates strong receptor activation [10,14].
The success of multivalent antibodies in preclinical settings has garnered great interest for their use in early-stage clinical trials. This is exemplified by IGM-8444, an anti-DR5 multivalent antibody, which is currently undergoing a phase 1 study (Table 1, NCT04553692I) for relapsed and refractory cancer [15]. Another multivalent protein against OX40 known as MEDI6383 has recently completed phase 1 clinical testing. In this trial, the OX40 agonist is being assessed as a monotherapy and combination therapy with PD-1 antagonist MEDI4736, but study results have not yet been published (NCT02221960II) [16]. With an ever-increasing interest in the potency of multivalent antibodies, we expect to see considerable progress in designing these antibodies for therapeutic use.
Table 1.
Clinical trials.
| Title | Trial ID # | Treatment | Phase | Participants | Age eligibility (years) | Condition or disease |
|---|---|---|---|---|---|---|
| Phase I Study of IGM-8444 as a Single Agent and in Combination in Subjects with Relapsed and/or Refractory Solid Cancers | NCT04553692 I |
Agonist: IGM-8444 Other drugs: FOLFIRI, Bevacizumab (and approved biosimilars), Birinapant, Venetoclax |
1 | 320* | ≥ 18 | Solid Tumor, Colorectal Cancer, Gastric Cancer, Non-Hodgkin Lymphoma, Non-Small Cell Lung Cancer, Sarcoma, Chondrosarcoma, Small Lymphocytic Lymphoma, Chronic Lymphocytic Leukemia |
| A Phase 1 Study to Evaluate MEDI6383 Alone and in Combination with MEDI4736 in Adult Subjects With Select Advanced Solid Tumors | NCT02221960 II |
Agonist: MEDI6383 Other drugs: MEDI4736 |
1 | 39 | 18-99 | Recurrent or Metastatic Solid Tumors |
| First-in-human (FIH) phase I study of RO7122290 (RO), a novel FAP-targeted 4-1BB agonist, administered as single agent and in combination with atezolizumab (ATZ) to patients with advanced solid tumors | EUDRACT Number: 2017-003961-83; Protocol Number: BP40087III |
Agonist: RG7827 (also known as RO7122290) Other drugs: atezolizumab |
1 | 62 | Not provided | Advanced Solid Tumors |
| Phase 1 Study in Patients with Advanced and/or Refractory Solid Malignancies to Evaluate the Safety of ATOR-1015 | NCT03782467 IV | Agonist: ATOR-1015 | 1 | 33 | ≥ 18 | Solid Tumor, Neoplasms |
| A Study to Evaluate Safety, Pharmacokinetics and Anti-Tumor Activity of RO7300490, as Single Agent or in Combination with Atezolizumab in Participants with Advanced Solid Tumors | NCT04857138 V |
Agonist: RO7300490 Other drugs: Atezolizumab |
1 | 280* | ≥ 18 | Solid Tumors |
| FS120 First in Human Study in Patients with Advanced Malignancies | NCT04648202 VI | Agonist: FS120 | 1 | 277* | ≥ 18 | Advanced Cancer, Metastatic Cancer |
| A Study Of PF-05082566 As A Single Agent and In Combination with Rituximab | NCT01307267 VII |
Agonist: PF-05082566 Other drugs: rituximab |
1 | 190 | ≥ 18 | Lymphoma (Non-Hodgkin, Follicular, Large B-Cell (Diffuse)) Carcinoma (Non-Small-Cell Lung, Renal Cell, Squamous Cell of Head and Neck), Malignant Melanoma |
| Dose Finding Study Of CP-870,893, An Immune System Stimulating Antibody, In Combination with Paclitaxel And Carboplatin For Patients With Metastatic Solid Tumors | NCT00607048 VIII |
Agonist: CP-870,893 Other drugs: Paclitaxel, Carboplatin |
1 | 34 | 18-85 | Neoplasms |
| Phase 1 Trial of LVGN6051 as Single Agent and in Combination with Keytruda (MK-3475-A31/KEYNOTE-A31) in Advanced or Metastatic Malignancy | NCT04130542 IX | Agonist: LVGN6051 | 1 | 276* | ≥ 18 | Cancer |
| GEN1029 (HexaBody®-DR5/DR5) Safety Trial in Patients with Malignant Solid Tumors | NCT03576131 X | Agonist: GEN1029 | 1/2 | 48 | ≥ 18 | Colorectal Cancer, Non-small Cell Lung Cancer, Triple Negative Breast Cancer, Renal Cell Carcinoma, Gastric Cancer, Pancreatic Cancer, Urothelial Cancer |
| Safety, Tolerability, Pharmacokinetics, and Immunoregulatory Study of Urelumab (BMS-663513) in Subjects with Advanced and/or Metastatic Solid Tumors and Relapsed/Refractory B-cell Non-Hodgkin’s Lymphoma | NCT01471210 XI | Agonist: Urelumab (BMS-663513) | 1 | 124 | ≥ 18 | Cancer - Solid Tumors and B-Cell Non-Hodgkin’s Lymphoma |
| Combination Study of Urelumab and Rituximab in Patients With B-cell Non-Hodgkins Lymphoma | NCT01775631 XII |
Agonist: Urelumab Other drugs: Rituximab |
1 | 47 | ≥ 18 | B-Cell Malignancies |
| Phase I-II Study of Intratumoral Urelumab Combined with Nivolumab in Patients With Solid Tumors (INTRUST) | NCT03792724 XIII |
Agonist: Urelumab Other drugs: Nivolumab |
1/2 | 32* | ≥ 18 | Neoplasms |
| An Investigational Immuno-Therapy Study of Experimental Medication BMS-986178 by Itself or in Combination with Nivolumab and/or Ipilimumab in Participants with Solid Cancers That Are Advanced or Have Spread | NCT02737475 XIV |
Agonist: BMS-986178 Other drugs: Nivolumab, Ipilimumab, Tetanus vaccine Biological: DPV-001, vaccine Drug: Cyclophosphamide |
1/2 | 166 | ≥ 18 | Advanced Cancer |
Abbreviations:
=estimated
Multispecific Antibody Targeting
Bispecific antibodies that target both tumor-associated antigens and T cell receptors provide a useful approach for improving the function of agonist antibodies [17]. The key advantage of using antibodies targeting tumor-associated antigens is the ability to minimize off-target effects by directing the therapeutics to the tumor microenvironment. In a recent study, investigators engineered bispecific antibodies that target both DR5 and fibroblast-activation protein (FAP), the latter of which is highly expressed in the tumor microenvironment (Figure 3) [18]. The researchers reasoned that the FAP binding domain would lead to avidity-driven receptor clustering to induce strong antitumor activity. This avidity-driven clustering would also remove the need for Fc-mediated crosslinking, which is a critical limitation of agonist antibodies. In an in vitro assay, the bispecific antibody elicited a >2-fold improved apoptotic response compared to its parent DR5-targeting antibody. Additionally, human xenograft mouse models showed that an antibody-induced >4-fold decrease in tumor volume relative to a clinical antibody in an Fc-independent manner. Additional studies on bispecific antibodies targeting CD40 and extracellular matrix proteins have shown similar improvements in drug localization and anti-tumor efficacy [19].
Figure 3.
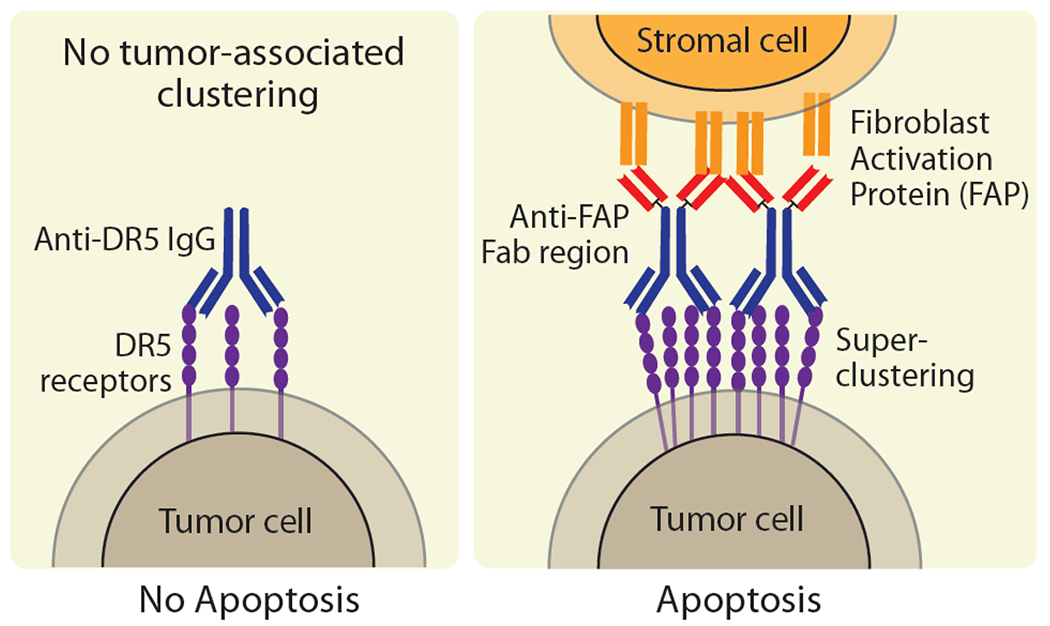
DR5-FAP bispecific antibodies promote Fc-independent DR5 receptor clustering and activation. Schematic illustration of tumor-specific antibodies targeting (left) only the DR5 receptor and (right) also the fibroblast activation protein (FAP) expressed on surrounding stromal cells to enhance DR5 receptor clustering and activation. Adapted from [18].
Bispecific antibodies have also been designed to target two different TNF receptors, namely CD137 and OX40, to improve antitumor immunity [20]. To achieve this, the bispecific antibody FS120 was engineered to bind CD137 via the Fab region and the OX40 receptor via the Fc region. In vitro studies showed that FS120 mediates strong IL-2 production with >20-fold improvement compared to monospecific antibodies. Furthermore, murine studies demonstrated that this bispecific antibody elicited >1.5-fold reduction in tumor volume and resulted in >3-fold higher CD4+ T cell proliferation. These findings are also supported by other studies [21,22] including one that reports a bispecific antibody targeting EGFR and CD137 with potent agonist function [23].
Similarly, biepitopic antibodies that target two different epitopes on the same receptor provide a useful avenue to induce Fc-independent receptor clustering. For example, researchers engineered biepitopic dual-variable domain (DVD) antibodies targeting the OX40 receptor to understand their effect on T-cell proliferation [10]. This unique tetravalent antibody format was constructed by placing an additional set of variable heavy and light domains, which together bind to a distinct receptor epitope, onto the N-termini of the heavy and light chains of the parental antibody. Interestingly, these antibodies demonstrated a >3-fold improvement in CD4+ T cell proliferation compared to their bivalent counterparts. These findings were further corroborated by murine studies that showed comparable results of >2-fold proliferation of effector CD4+ T cells and enhanced IFN-γ production. To understand the mechanism, the investigators conducted structure-based studies on the biepitopic antibodies, which revealed their ability to cluster nearby receptors through daisy-chaining and promote improved agonist function.
One bispecific antibody being evaluated in phase 1 clinical trials is RG7827 (also known as RO7122290), which binds to the CD137 receptor and FAP [24]. It is also being evaluated in a combination trial with atezolizumab (PD-L1 inhibitor) for advanced solid tumors (EUDRACT Number: 2017-003961-83; Protocol Number: BP40087III). The tumor biopsies revealed an increased level of proliferative CD8+ T-cells as measured by the Ki67+ proliferative marker, suggesting improved T cell response. Another bispecific antibody (ATOR-1015) that targets CTLA-4 and OX40 has been shown to improve anti-tumor immunity in a phase 1 clinical trial (NCT03782467IV) [25]. The preliminary results indicate that the treatment is well tolerated at doses of <200 mg and further dose escalation is currently being explored. Finally, bispecific antibody RO7300490, which targets CD40 and FAP domains (NCT04857138V), and FS120, which targets OX40 and CD137 (NCT04648202VI), are currently being evaluated in phase 1 clinical settings. Collectively, these clinical trials represent a burgeoning interest in developing bispecific antibodies for diverse cancer applications.
Antibody Isotype
Antibody isotypes, comprising four major subclasses, have been implicated in mediating receptor activation [26]. One major theme in isotype selection is the inverse relationship between hinge region flexibility and improved agonist function [27] (Figure 4). A recent study constructed a panel of anti-mouse CD40 antibodies with different human constant domains (i.e., IgG1-4) to evaluate the impact of the CH1-hinge region on agonist function mediated by antigen-presenting cells. Using a murine model, each human antibody isotype exhibited differential levels of CD8+ T cell activation, including a >6-fold improved response for IgG2 compared to the other isotypes. Next, the researchers sought to investigate the biophysical properties of each isotype in mediating divergent agonist activities. Structural analysis revealed that the IgG2 isotype had the most rigid hinge region whereas the least active isotype (IgG3) was the most flexible. These and other findings demonstrate that the rigidity of the human IgG2 hinge region is responsible for improved clustering of CD40 receptors compared to other isotypes [28].
Figure 4.
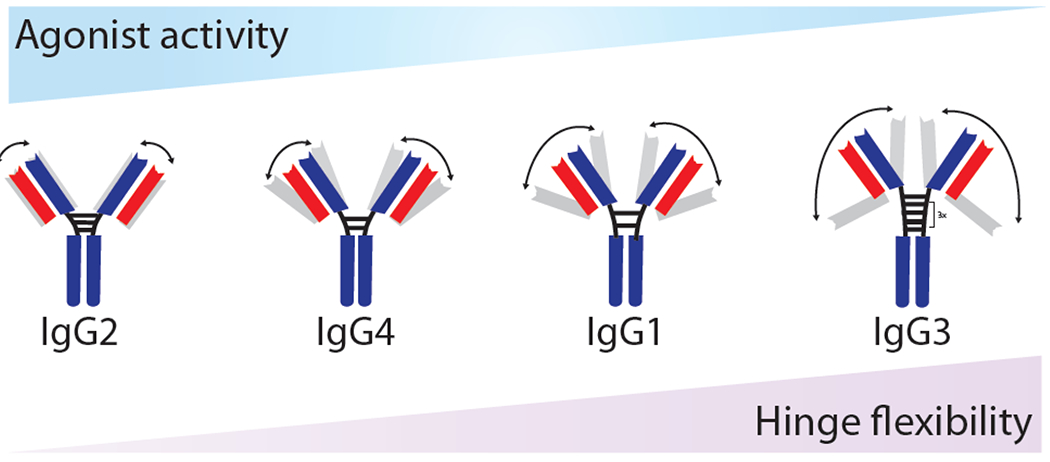
Inverse relationship between hinge region flexibility and receptor activation for anti-mouse CD40 agonist antibodies with human constant domains. Schematic illustration of the negative correlation between CD40 agonist activity and hinge flexibility of human IgG isotypes. Adapted from [27].
A unique phenomenon of isotype switching has been observed in a study where the human CD40 IgG4 antagonist antibody (bleselumab) was generated in human IgG1 and IgG2 formats [28]. Interestingly, the IgG1 isotype retained its antagonism as expected whereas the IgG2 isotype was converted to a superagonist. In vitro studies illustrated that the IgG2 isotype induced >3-fold improvement in B cell proliferation compared to the most effective clinical CD40 antibody (CP-870,893). These findings were further corroborated in an in vivo model, where switching to IgG2 isotype induced > 4-fold CD8+ T cell expansion This phenomenon was also generalized to other CD40 antagonists, which showed improved agonistic activity with isotype switching. Although the exact mechanism of the superagonist properties of IgG2 remains unknown, it is possible that the rigidity of the IgG2 hinge overrides the antagonist nature of the molecule through enhanced receptor clustering.
In a recent clinical trial, a CD137 IgG2 antibody (utomilumab) has been shown to be well tolerated in patients with advanced solid tumors (NCT01307267VII) [29]. Thus far, monotherapy treatment has only shown a mild improvement in mediating antitumor immunity. In terms of a combination trial with rituximab, for instance, 20% of patients with advanced solid tumors including colorectal and pancreatic cancer demonstrated complete or partial tumor reduction while >42% maintained stable disease [30]. The tumor biopsy studies showed amplified T cell activation and tumor cytotoxicity, which is consistent with an improved antitumor response. Finally, similar antibodies targeting CD40 in a combination trial have yielded comparable results in early phase clinical trials [31,32].
Antibody Fc Receptor Interactions
The interactions between IgG Fc regions and activating/inhibitory Fcγ receptors critically impact agonist activity [33]. One major antibody engineering approach in this research area is the introduction of favorable mutations in the Fc regions that selectively increase affinity to the inhibitory receptor FcyRIIB (Figure 5). This approach is effective because FcyRIIB is solely used as a scaffold for clustering monomeric receptors to meditate strong agonist activity [34]. To demonstrate this point, researchers constructed human anti-CD40 antibodies with Fc mutations such as S267E (SE) and S267E/L328F (SELF), which increased binding to both inhibitory FcγRIIB and activating FcγRIIA receptors [35]. Compared to wild type, these mutants demonstrated >2-fold improvement in CD8 T cell activation, suggesting improved T cell immunity. Given SE and SELF mutations increase affinity to both receptors, the investigators reasoned that selective engagement to FcyRIIB could further improve agonist function. To test this hypothesis, additional Fc mutants were developed to selectively optimize Fc engagement of FcγRIIB over FcγRIIA. The results demonstrated that one variant (V11, contained five mutations) resulted in a >97-fold improvement in affinity to FcyRIIB and 3-fold reduction in affinity to FcγRIIA. This mutant displayed significant improvement in CD8+ T cell activation compared to the wild-type antibody and >5-fold improvement over the SELF variant. In vivo tumor studies demonstrated that while the SELF variant reduced tumor volume by 65%, the V11 variant completely abrogated tumor growth. These findings indicated that engineering antibody Fc regions with high FcγRIIB/FcγRIIA binding affinity ratios represents a powerful approach to improve the efficacy of agonist antibodies. Other studies on CD137 and OX40 antibodies highlight comparable results where selective point mutations in the Fc region can dramatically improve agonist activity [36,37].
Figure 5.
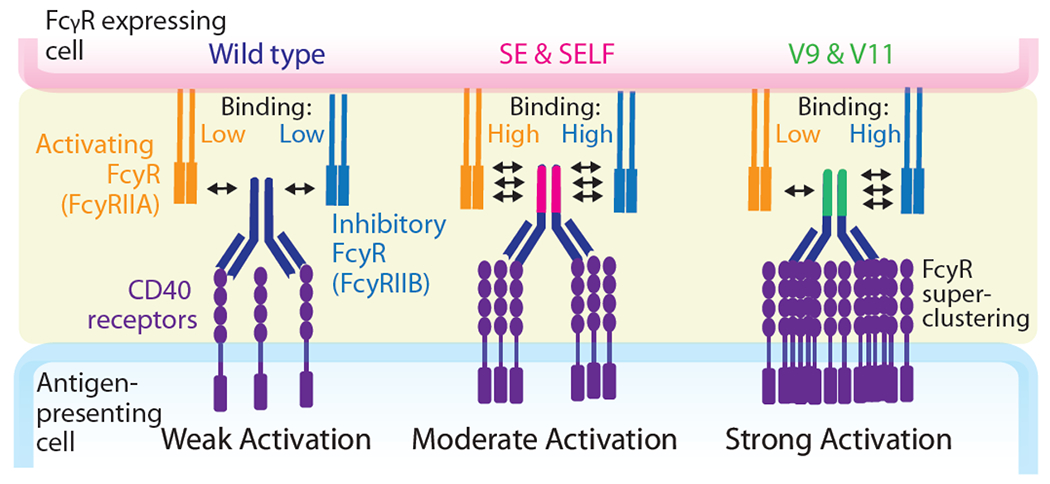
Selective FcγR engagement is required for optimal human CD40 antibody agonism and anti-tumor activity. Schematic illustration of the impact of activating and inhibitory FcγRs on CD40 receptor agonism. Selective antibody binding to the inhibitory receptor FcγRIIB is positively correlated with receptor activation. Adapted from [35].
Although improving affinity to Fcγ receptors is a viable option, this approach inherently depends on the availability of Fc receptors on antigen-presenting cells (APCs). The use of Fc mutations that promote Fc-Fc self-interactions is a unique method for improving agonist activity in an FcγR-independent manner. This is highlighted in an OX40 study where a set of Fc mutations (E345R, E430G, S440Y) promote hexamerization of IgGs [38]. Compared to wild type, the double mutant (E345R/E430G) showed the highest multimerization of receptor clusters in solution followed by the sets of triple and single mutations. An in vitro NF-kB assay was used to assess their biological response, which revealed that a single Fc mutation (E345R) induced the highest dose-dependent response compared to the sets of double and triple mutations. The authors hypothesized that the discrepancy between receptor multimerization in solution and agonist activity was partly due to the E345R mutation forming a favorable hexameric configuration upon antigen binding on the cell surface. Similar studies for other TNF receptors including DR5 demonstrate these favorable Fc mutations led to enhanced receptor agonism [39,40].
While the function of the inhibitory Fcγ receptor is well understood, activating Fcγ receptors are equally as important in mediating anti-tumor immunity. This is because activating FcγRs can induce antibody-dependent cellular cytotoxicity (ADCC) to deplete Tregs, which is a critical component of the anti-tumor response. To demonstrate this point, investigators highlighted the role of activating FcγRs on OX40-mediated Treg depletion [41]. The engagement of an OX40 mAb with activating FcγRs significantly depleted tumor infiltrating Treg cells while maintaining the CD8+ T cell subpopulation in a murine model, indicating the importance of activating receptors for mediating effective anti-tumor response. This observation is further supported by CD137 studies where binding to activating receptors resulted in amplified anti-tumor responses [42].
In terms of Fc-enhanced antibodies, a CD40 agonist mAb (known as CP-870,893 or selicrelumab) engineered with the V11 Fc mutations is currently being evaluated in a phase I combination trial with carboplatin and paclitaxel (NCT00607048VIII) [32]. This treatment has shown promise against melanoma cancer, where two patients displayed an increase in T-cell response. In terms of advanced solid tumors, 20% of patients exhibited a moderate decrease in target lesions, suggesting mild improvement in anti-tumor efficacy. Additionally, a CD137 antibody (LVGN6051) containing Fc mutations is also undergoing phase I clinical trials for advanced/metastatic cancer as a single agent and in combination with Keytruda, a humanized PD-1 inhibitor (NCT04130542IX) [43]. Thus far, preliminary data indicates a lack of adverse effects in monotherapy and only mild effects in combination therapy. In particular, a patient with metastatic head and neck squamous cell carcinoma has experienced 50% tumor reduction lasting for more than six months in combination therapy. Finally, a DR5 antibody (GEN1029) containing Fc hexamerization mutations is currently being evaluated in a phase I clinical trial (NCT03576131X) [44]. Thus, Fc-engineered antibodies have shown great promise in preclinical and clinical settings, strengthening their potential as cancer immunotherapeutics.
Receptor Epitope and Occupancy
The structure of TNF receptors (i.e., OX40, CD40, and CD137) consists of cysteine rich domain (CRD) subunits that can be selectively targeted by antibodies to impart superior agonist activity. Mounting evidence suggests that antibodies targeting the CRD regions outside the ligand-binding domain mediate improved agonist function (Figure 6) [45,46]. In the case of CD40 and CD137 receptors, antibodies that target the membrane distal CRD1 domain have significantly higher agonist activity compared to ligand-blocking antibodies that bind to CRD3 and CRD4. This is because antibodies that target distal domains allow for access to Fcγ receptors with reduced steric hindrance compared to antibodies targeting proximal domains. In a recent CD137 study, the epitope selection of clinical antibodies, namely urelumab and utomilumab, proved influential in mediating potent agonist activity. Structural analysis revealed that urelumab binds to CRD1 while utomilumab binds near the ligand-binding site in CRD3 and CRD4. Functional studies in CD8+ T cells showed that the non-ligand blocking antibody urelumab elicited >2 fold greater IL-2 and IFN-γ cytokine secretion than ligand-blocking utomilumab. The synergistic impact of the native ligand and clinical antibodies were evaluated for their ability to mediate clustering, which resulted in >4 times more receptor clusters for urelumab compared to utomilumab, suggesting a greater enhancement in agonist function [46]. Although urelumab shows superior agonist potential, recent studies have noted increased toxicity profiles in some patients at doses >1 mg/kg, which is much less of a problem for utomilumab for doses up to at least 10 mg/kg [46–48]. Finally, studies of other TNF receptors such as OX40 showed that non-blocking antibodies exhibit potent anti-tumor response [49,50].
Figure 6.
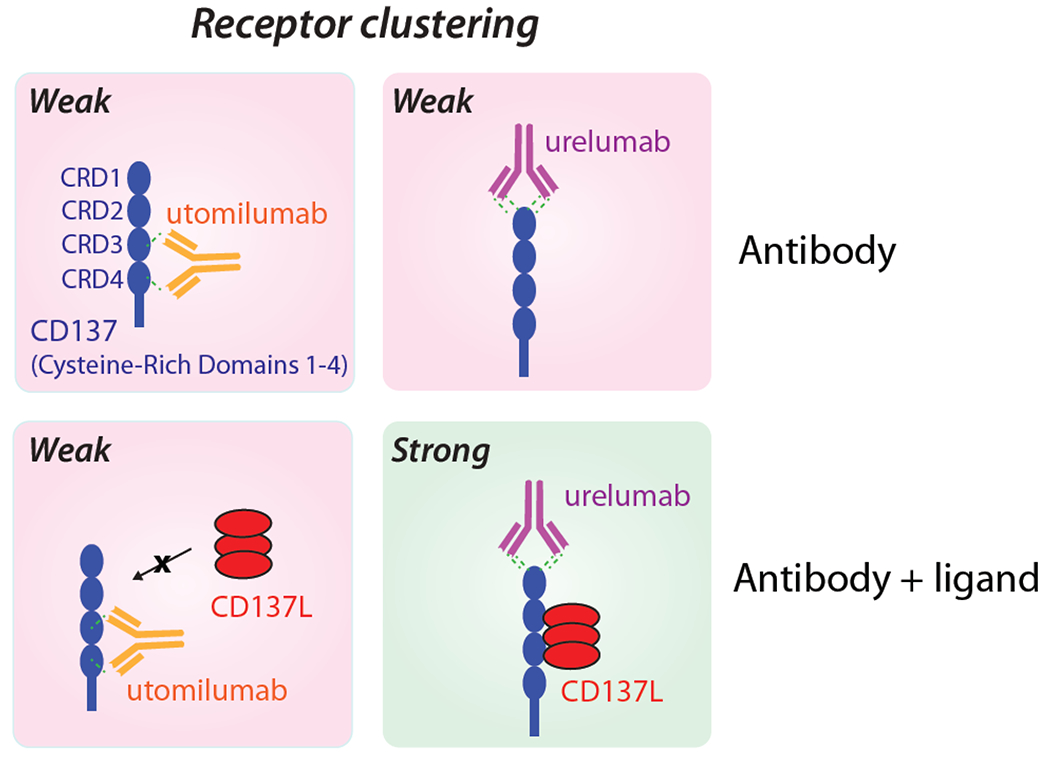
CD137 epitope strongly influences the activity of agonist antibodies. Schematic illustration of the impact of CD137 receptor epitopes on the activity of two agonist antibodies, namely urelumab and utomilumab. The non-ligand blocking antibody (urelumab) targets membrane-distal cysteine-rich domain 1 (CRD1) and displays enhanced agonist activity. In contrast, the ligand-blocking antibody (utomilumab) binds to CRDs 3 and 4 and displays reduced agonist function. Adapted from [46].
The importance of receptor epitope selection when designing effective antibodies for cancer therapeutics is highlighted in the case of urelumab (BMS-663513), which binds CD137 via CRD1, and has been the focus of multiple phase 1 clinical trials. In a combination trial (NCT01471210XI and NCT01775631XII) with rituximab (CD20 mAb), participants with refractory B-cell lymphoma and follicular lymphoma were treated with urelumab monotherapy and combination therapy. The combination trials demonstrated promising results where 35% of follicular lymphoma patients experienced a >30% tumor decrease, and 71% had stable disease, suggesting improved disease outcome. As a result, urelumab is proceeding to further clinical testing in combination with other drugs. The most notable example is an ongoing combination trial (NCT03792724XIII) with PD-1 inhibitor nivolumab for patients with B-cell lymphoma.
Receptor occupancy is also an important criterion for establishing optimal agonist function. Unlike antagonist antibodies that show sigmoidal dose-response, agonist antibodies exhibit a bell-shaped dose-response curve, which means that maximum activity occurs at intermediate concentrations [51,52]. To highlight this phenomenon, investigators demonstrated that OX40 antibodies mediated maximal agonist activity at intermediate dosing concentrations using a murine model [53]. To further test this hypothesis, the researchers examined antibody-induced proliferation of human T cells and found that ~40% receptor occupancy led to maximum CD4+ T cell proliferation. Notably, loss in receptor expression was observed at >40% receptor occupancy. Similarly, this general phenomenon has been observed for other T cell receptors, including for CD28, where occupying half of the receptors on the cell surface leads to maximal receptor activation [54,55].
In terms of receptor occupancy, an OX40 antibody (BMS-986178) displays peak agonist function at ~40% receptor occupancy in preclinical settings and is the subject of multiple clinical trials [56]. In a phase I trial (NCT02737475XIV), BMS-986178 has been tested as a monotherapy and combination therapy with nivolumab and ipilimumab (CTLA-4 antagonist) in 165 patients with advanced solid tumors. Amongst all treatment cohorts, only a small percentage (<15%) of participants experienced moderate to high-grade adverse effects, and no toxicity was observed even at the highest dose of 320 mg BMS-986178, indicating that the maximum tolerable dosage was not reached in this study. In terms of treatment efficacy, 12% of patients experienced a >30% tumor shrinkage, and one bladder cancer patient achieved complete eradication of cancerous tumors in nivolumab combination therapy. In combination cohorts with nivolumab and ipilimumab, the investigators also observed an increase in proliferating CD8+ T cells along with a decreasing percentage of Fox3+ regulatory T cells associated with an anti-tumor response. More clinical trials are warranted to examine receptor occupancy due to its critical role in enhancing agonist function.
Concluding Remarks
Agonist antibodies have shown great promise in mediating anti-tumor efficacy as cancer therapeutics. Despite their potential, these antibodies have faced many roadblocks that have hindered their progress in clinical settings. Recently, designing antibodies with unique binding epitopes, valency, specificity, Fc-mediated interactions, and isotypes has enabled the optimization of their biological function. These approaches have served a critical role in improving receptor activation while mitigating the current limitations of conventional mAb therapy including poor receptor activation using IgG bivalent antibodies. Their success in a preclinical setting has led to the evaluation of these antibodies in numerous early phase clinical trials. Further research and development are needed to fully uncover their potential as cancer therapeutics.
To develop the next generation of cancer therapeutics, novel engineering approaches highlighted in this review are required for their success in clinical trials (see Outstanding Questions). For example, the receptor structure and biology must be at the forefront of agonist antibody development. A greater understanding of the mechanism of receptor activation may open new avenues for generating optimized agonist antibodies. The trimeric formation of TNF receptors is critical for their intracellular receptor signaling and, therefore, molecular formats such as multivalency are favorable for receptor clustering. Additionally, isotype selection should be an important criterion because the rigid conformation of the IgG2 isotype allows for improved agonist function. Another key challenge for clinical antibodies is the dependency on FcγR-crosslinking, which is inherently variable for in vivo applications. This issue is compounded by varying levels of FcγR expression on different immune cells, which leads to diverse levels of receptor agonism. To overcome these challenges, bispecific antibodies have been developed to mediate potent agonist function in an Fc-independent manner. Beyond engineering the Fab region, optimization to the Fc region to improve its interaction with FcγR is a viable approach to generate improved agonist antibodies. Collectively, these methods can significantly improve T cell immunity to mediate potent anti-tumor response.
Outstanding Questions.
Most antibody engineering approaches are geared toward cancer therapy; to what extent are these approaches useful for other diseases, including autoimmune or neurological disorders?
Given the complexity of receptor signaling to what degree can antibody engineering approaches such as multivalency and bispecificity be generalized across the TNF receptor superfamily? Or across T cell receptors where receptor signaling is dependent on receptor clustering?
The clinical translation of most agonist antibodies has been modest for cancer therapy. Is this due to poor receptor expression in the tumor microenvironment? Or the lack of tumor-infiltrating T cells? Should adjuvants be considered to boost receptor expression and therefore improve clinical efficacy?
Factors including receptor epitope and occupancy have been shown to be influential in animal models; how well do they translate to clinical trials? Should dosing be critically considered given that partial receptor occupancy promotes optimal receptor signaling?
To what degree do different antibody engineering approaches work in concert? For example, would bispecific antibodies that target multiple receptors with enhanced Fc regions work well together to increase efficacy in pre-clinical studies and clinical trials?
What is the future of cancer immunotherapy with respect to agonist antibody therapeutics? Should we have a more personalized approach using bioinformatics to better understand differences in the tumor microenvironment? Are combination therapies the best way to move forward?
Finally, bispecific antibodies that can target tumor-associated antigens can improve tumor site localization and reduce off-target effects. Dosing regimens should also be considered given that partial receptor occupancy has yielded maximum agonist function. We anticipate that the combination of one or more of these optimization approaches can improve their agonist function and therapeutic efficacy.
Highlights.
Agonist antibodies that activate the tumor necrosis factor (TNF) receptor superfamily on T cells are being broadly pursued for cancer therapy. However, clinical translation is stymied by poor safety and efficacy.
Clustering of TNF receptors is critical for mediating potent receptor activation and bivalent antibodies have shown limited capacity to mediate receptor clustering on the cell surface. Thus, antibody engineering approaches for improving certain properties (i.e., multivalency and/or biepitopic targeting) are needed to enhance receptor clustering and agonist function.
Beyond antigen-binding fragment (Fab) engineering, antibody isotype selection and improving FcγR interactions are influential in improving anti-tumor immunity in pre-clinical studies and clinical trials.
Receptor binding epitopes and occupancy levels must be considered to mediate optimal receptor signaling and anti-tumor immunity.
Acknowledgements
We thank members of the Tessier lab for their helpful suggestions. This work was supported by the National Institutes of Health (R35GM136300 and RF1AG059723 to P.M.T.), National Science Foundation (CBET 1813963, 1605266 and 1804313 to P.M.T.), Department of Defense (W81XWH-21-1-0040), and the Albert M. Mattocks Chair (to P.M.T).
Glossary
- Receptor clustering
bringing together receptor subunits (monomers or multimers) to form higher-order receptor complexes on the cell surface.
- Multivalent antibodies
antibody possessing more than two antigen-binding sites.
- Nanobody
a type of antibody that consists of only variable heavy domains, primarily found in sharks and camelids.
- Bispecific antibodies
a class of antibodies that engage two distinct receptor epitopes or antigens.
- Tumor-associating antigens
antigens that are typically elevated on tumor cells relative to healthy cells.
- Isotype
also known as antibody class, which categorizes antibodies based on the heavy chain.
- Fcγ receptors
a group of receptors present on immune cells that bind to the Fc region of IgGs to mediate receptor signaling.
- Antibody-dependent cellular cytotoxicity (ADCC)
an immune mechanism in which an immune cell lyses a target cell (e.g., tumor cell).
- Cysteine-rich domain (CRD) subunits
structural regions within TNF receptors defined by the presence of repeated cysteine-rich sequence patterns.
- Receptor occupancy
the fraction of antibody-bound receptors on the cell surface
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
References
- 1.Grivennikov SI, Greten FR and Karin M (2010) Immunity, inflammation, and cancer. Cell 140, 883–899. 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rabia LA, Desai AA, Jhajj HS and Tessier PM (2018) Understanding and overcoming tradeoffs between antibody affinity, specificity, stability and solubility. Biochemical Engineering Journal 137, 365–374. 10.1016/j.bej.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullard A (2021) FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov 20, 491–495. 10.1038/d41573-021-00079-7. [DOI] [PubMed] [Google Scholar]
- 4.Mayes PA, Hance KW and Hoos A (2018) The promise and challenges of immune agonist antibody development in cancer. Nature Reviews Drug Discovery 17, 509–527. 10.1038/nrd.2018.75 [DOI] [PubMed] [Google Scholar]
- 5.Schardt JS, Jhajj HS, O’Meara RL, Lwo TS, Smith MD and Tessier PM (2022) Agonist antibody discovery: Experimental, computational, and rational engineering approaches. Drug Discovery Today 27, 31–48. 10.1016/j.drudis.2021.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watts TH (2005) TNF/TNFR family members in costimulation of T cell responses Annual Review of Immunology 23, 23–68. 10.1146/annurev.immunol.23.021704.115839 [DOI] [PubMed] [Google Scholar]
- 7.Sedger LM and McDermott MF (2014) TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants – past, present and future. Cytokine & Growth Factor Reviews 25, 453–472. 10.1016/j.cytogfr.2014.07.016 [DOI] [PubMed] [Google Scholar]
- 8.Brenner D, Blaser H and Mak TW (2015) Regulation of tumour necrosis factor signalling: live or let die. Nature Reviews Immunology 15, 362–374. 10.1038/nri3834 [DOI] [PubMed] [Google Scholar]
- 9.Furness AJ, Vargas FA, Peggs KS and Quezada SA (2014) Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol 35, 290–298. 10.1016/j.it.2014.05.002 [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Yeh SH, Madireddi S, Matochko WL, Gu C, Pacheco Sanchez P, Ultsch M, De Leon Boenig G, Harris SF, Leonard B, Scales SJ, Zhu JW, Christensen E, Hang JQ, Brezski RJ, Marsters S, Ashkenazi A, Sukumaran S, Chiu H, Cubas R, Kim JM and Lazar GA (2019) Tetravalent biepitopic targeting enables intrinsic antibody agonism of tumor necrosis factor receptor superfamily members. MAbs 11, 996–1011. 10.1080/19420862.2019.1625662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kucka K and Wajant H (2020) Receptor Oligomerization and Its Relevance for Signaling by Receptors of the Tumor Necrosis Factor Receptor Superfamily. Front Cell Dev Biol 8, 615141. 10.3389/fcell.2020.615141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tigue NJ, Bamber L, Andrews J, Ireland S, Hair J, Carter E, Sridharan S, Jovanović J, Rees DG, Springall JS, Solier E, Li YM, Chodorge M, Perez-Martinez D, Higazi DR, Oberst M, Kennedy M, Black CM, Yan L, Schwickart M, Maguire S, Cann JA, de Haan L, Young LL, Vaughan T, Wilkinson RW and Stewart R (2017) MEDI1873, a potent, stabilized hexameric agonist of human GITR with regulatory T-cell targeting potential. Oncoimmunology 6, e1280645. 10.1080/2162402X.2017.1280645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sadeghnezhad G, Romão E, Bernedo-Navarro R, Massa S, Khajeh K, Muyldermans S and Hassania S (2019) Identification of New DR5 Agonistic Nanobodies and Generation of Multivalent Nanobody Constructs for Cancer Treatment. International Journal of Molecular Sciences 20, 4818. 10.3390/ijms20194818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greer YE, Gilbert SF, Gril B, Narwal R, Peacock Brooks DL, Tice DA, Steeg PS and Lipkowitz S (2019) MEDI3039, a novel highly potent tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 2 agonist, causes regression of orthotopic tumors and inhibits outgrowth of metastatic triple-negative breast cancer. Breast Cancer Research 21, 27. 10.l186/s13058-019-1116-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang BT, Kothambawala T, Wang L, Matthew TJ, Calhoun SE, Saini AK, Kotturi MF, Hernandez G, Humke EW, Peterson MS, Sinclair AM and Keyt BA (2021) Multimeric Anti-DR5 IgM Agonist Antibody IGM-8444 Is a Potent Inducer of Cancer Cell Apoptosis and Synergizes with Chemotherapy and BCL-2 Inhibitor ABT-199. Mol Cancer Ther 20, 2483–2494. 10.1158/1535-7163.MCT-20-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, Curiel TJ, Colon-Otero G, Hamid O, Sanborn RE, O’Donnell PH, Drakaki A, Tan W, Kurland JF, Rebelatto MC, Jin X, Blake-Haskins JA, Gupta A and Segal NH (2016) Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. Journal of Clinical Oncology 34, 3119–3125. 10.1200/JCO.2016.67.9761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang S, van Duijnhoven SMJ, Sijts AJAM and van Elsas A (2020) Bispecific antibodies targeting dual tumor-associated antigens in cancer therapy. Journal of Cancer Research and Clinical Oncology 146, 3111–3122. 10.1007/s00432-020-03404-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brünker P, Wartha K, Friess T, Grau-Richards S, Waldhauer I, Koller CF, Weiser B, Majety M, Runza V, Niu H, Packman K, Feng N, Daouti S, Hosse RJ, Mössner E, Weber TG, Herting F, Scheuer W, Sade H, Shao C, Liu B, Wang P, Xu G, Vega-Harring S, Klein C, Bosslet K and Umaña P (2016) RG7386, a Novel Tetravalent FAP-DR5 Antibody, Effectively Triggers FAP-Dependent, Avidity-Driven DR5 Hyperclustering and Tumor Cell Apoptosis. Mol Cancer Ther 15, 946–957. 10.1158/1535-7163.MCT-15-0647 [DOI] [PubMed] [Google Scholar]
- 19.Ishihara J, Ishihara A, Potin L, Hosseinchi P, Fukunaga K, Damo M, Gajewski TF, Swartz MA and Hubbell JA (2018) Improving Efficacy and Safety of Agonistic Anti-CD40 Antibody Through Extracellular Matrix Affinity. Mol Cancer Ther 17, 2399–2411. 10.1158/1535-7163.MCT-18-0091 [DOI] [PubMed] [Google Scholar]
- 20.Gaspar M, Pravin J, Rodrigues L, Uhlenbroich S, Everett KL, Wollerton F, Morrow M, Tuna M and Brewis N (2020) CD137/OX40 Bispecific Antibody Induces Potent Antitumor Activity that Is Dependent on Target Coengagement. Cancer Immunol Res 8, 781–793. 10.1158/2326-6066.CIR-19-0798 [DOI] [PubMed] [Google Scholar]
- 21.Warwas KM, Meyer M, Gonçalves M, Moldenhauer G, Bulbuc N, Knabe S, Luckner-Minden C, Ziegelmeier C, Heussel CP, Zörnig I, Jäger D and Momburg F (2021) Co-Stimulatory Bispecific Antibodies Induce Enhanced T Cell Activation and Tumor Cell Killing in Breast Cancer Models. Front Immunol 12, 719116. 10.3389/fimmu.2021.719116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan S, Belmar N, Ho S, Rogers B, Stickler M, Graham M, Lee E, Tran N, Zhang D, Gupta P, Sho M, MacDonough T, Woolley A, Kim H, Zhang H, Liu W, Zheng P, Dezso Z, Halliwill K, Ceccarelli M, Rhodes S, Thakur A, Forsyth CM, Xiong M, Tan SS, Iyer R, Lake M, Digiammarino E, Zhou L, Bigelow L, Longenecker K, Judge RA, Liu C, Trumble M, Remis JP, Fox M, Cairns B, Akamatsu Y, Hollenbaugh D, Harding F and Alvarez HM (2022) An anti-PD-1–GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy. Nature Cancer 3, 337–354. 10.1038/s43018-022-00334-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Compte M, Harwood SL, Erce-Llamazares A, Tapia-Galisteo A, Romero E, Ferrer I, Garrido-Martin EM, Enguita AB, Ochoa MC, Blanco B, Oteo M, Merino N, Nehme-Álvarez D, Hangiu O, Domínguez-Alonso C, Zonca M, Ramírez-Fernández A, Blanco FJ, Morcillo MA, Muñoz IG, Melero I, Rodriguez-Peralto JL, Paz-Ares L, Sanz L and Alvarez-Vallina L (2021) An Fc-free EGFR-specific 4-1BB-agonistic Trimerbody Displays Broad Antitumor Activity in Humanized Murine Cancer Models without Toxicity. Clin Cancer Res 27, 3167–3177. 10.1158/1078-0432.CCR-20-4625 [DOI] [PubMed] [Google Scholar]
- 24.Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, Albrecht R, Herter S, Schlenker R, Hüsser T, Diggelmann S, Challier J, Mössner E, Hosse RJ, Hofer T, Brünker P, Joseph C, Benz J, Ringler P, Stahlberg H, Lauer M, Perro M, Chen S, Küttel C, Bhavani Mohan PL, Nicolini V, Birk MC, Ongaro A, Prince C, Gianotti R, Dugan G, Whitlow CT, Solingapuram Sai KK, Caudell DL, Burgos-Rodriguez AG, Cline JM, Hettich M, Ceppi M, Giusti AM, Crameri F, Driessen W, Morcos PN, Freimoser-Grundschober A, Levitsky V, Amann M, Grau-Richards S, von Hirschheydt T, Tournaviti S, Mølhøj M, Fauti T, Heinzelmann-Schwarz V, Teichgräber V, Colombetti S, Bacac M, Zippelius A, Klein C and Umaña P (2019) Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci TranslMed 11. 10.1126/scitranslmed.aav5989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kvarnhammar AM, Veitonmäki N, Hägerbrand K, Dahlman A, Smith KE, Fritzell S, von Schantz L, Thagesson M, Werchau D, Smedenfors K, Johansson M, Rosén A, Åberg I, Winnerstam M, Nyblom E, Barchan K, Furebring C, Norlén P and Ellmark P (2019) The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J Immunother Cancer 7, 103. 10.1186/s40425-019-0570-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S, Vamum B, Wypych J, Balland A and Bondarenko PV (2008) Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem 283, 16206–16215. 10.1074/jbc.M709988200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Zhao Y, Shi H, Zhang Y, Yin X, Liu M, Zhang H, He Y, Lu B, Jin T and Li F (2019) Human immunoglobulin G hinge regulates agonistic anti-CD40 immunostimulatory and antitumour activities through biophysical flexibility. Nature Communications 10, 4206. 10.1038/s41467-019-12097-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu X, Chan HTC, Fisher H, Penfold CA, Kim J, Inzhelevskaya T, Mockridge CI, French RR, Duriez PJ, Douglas LR, English V, Verbeek JS, White AL, Tews I, Glennie MJ and Cragg MS (2020) Isotype Switching Converts Anti-CD40 Antagonism to Agonism to Elicit Potent Antitumor Activity. Cancer Cell 37, 850–866.e857. 10.1016/j.ccell.2020.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, Cesari R, Chen Y, Davis CB, Huang B, Thall AD and Gopal AK (2018) Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin Cancer Res 24, 1816–1823. 10.1158/1078-0432.CCR-17-1922 [DOI] [PubMed] [Google Scholar]
- 30.Gopal AK, Levy R, Houot R, Patel SP, Popplewell L, Jacobson C, Mu XJ, Deng S, Ching KA, Chen Y, Davis CB, Huang B, Fly KD, Thall A, Woolfson A and Bartlett NL (2020) First-in-Human Study of Utomilumab, a 4-1BB/CD137 Agonist, in Combination with Rituximab in Patients with Follicular and Other CD20(+) Non-Hodgkin Lymphomas. Clin Cancer Res 26, 2524–2534. 10.1158/1078-0432.CCR-19-2973 [DOI] [PubMed] [Google Scholar]
- 31.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, Green SJ, O’Dwyer PJ, Running KL, Huhn RD and Antonia SJ (2007) Clinical Activity and Immune Modulation in Cancer Patients Treated With CP-870,893, a Novel CD40 Agonist Monoclonal Antibody. Journal of Clinical Oncology 25, 876–883. 10.1200/JCO.2006.08.3311 [DOI] [PubMed] [Google Scholar]
- 32.Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, Tolcher AW and Hamid O (2013) Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2, e23033. 10.4161/onci.23033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junker F, Gordon J and Qureshi O (2020) Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front Immunol 11, 1393. 10.3389/fimmu.2020.01393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li F and Ravetch JV (2012) Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcγ receptor engagement. Proceedings of the National Academy of Sciences 109, 10966–10971. 10.1073/pnas.1208698109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ and Ravetch JV (2016) Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcγR Engagement. Cancer Cell 29, 820–831. 10.1016/j.ccell.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mimoto F, Katada H, Kadono S, Igawa T, Kuramochi T, Muraoka M, Wada Y, Haraya K, Miyazaki T and Hattori K (2013) Engineered antibody Fc variant with selectively enhanced FcγRIIb binding over both FcγRIIaR131 and FcγRIIaH131. Protein Engineering, Design and Selection 26, 589–598. 10.1093/protein/gzt022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campos Carrascosa L, van Beek AA, deRuiter V, Doukas M, Wei J, Fisher TS, Ching K, Yang W, van Loon K, Boor PPC, Rakké YS, Noordam L, Doornebosch P, Grünhagen D, Verhoef K, Polak WG, JNM IJ, Ni I, Yeung YA, Salek-Ardakani S, Sprengers D and Kwekkeboom J (2020) FcγRIIB engagement drives agonistic activity of Fc-engineered αOX40 antibody to stimulate human tumor-infiltrating T cells. J Immunother Cancer 8. 10.1136/jitc-2020-000816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang D, Goldberg MV and Chiu ML (2016) Fc Engineering Approaches to Enhance the Agonism and Effector Functions of an Anti-OX40 Antibody. J Biol Chem 291, 27134–27146. 10.1074/jbc.M116.757773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Overdijk MB, Strumane K, Beurskens FJ, Ortiz Buijsse A, Vermot-Desroches C, Vuillermoz BS, Kroes T, de Jong B, Hoevenaars N, Hibbert RG, Lingnau A, Forssmann U, Schuurman J, Parren P, de Jong RN and Breij ECW (2020) Dual Epitope Targeting and Enhanced Hexamerization by DR5 Antibodies as a Novel Approach to Induce Potent Antitumor Activity Through DR5 Agonism. Mol Cancer Ther 19, 2126–2138. 10.1158/1535-7163.MCT-20-0044 [DOI] [PubMed] [Google Scholar]
- 40.Zhang D, Armstrong AA, Tam SH, McCarthy SG, Luo J, Gilliland GL and Chiu ML (2017) Functional optimization of agonistic antibodies to OX40 receptor with novel Fc mutations to promote antibody multimerization. mAbs 9, 1129–1142. 10.1080/19420862.2017.1358838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS and Brogdon JL (2014) OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol 92, 475–480. 10.1038/icb.2014.26 [DOI] [PubMed] [Google Scholar]
- 42.Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, Semmrich M, Teige I, Mårtensson L, Penfold CA, Chan HTC, Willoughby JE, Mockridge CI, Dahal LN, Cleary KLS, James S, Rogel A, Kannisto P, Jernetz M, Williams EL, Healy E, Verbeek JS, Johnson PWM, Frendéus B, Cragg MS, Glennie MJ, Gray JC, Al-Shamkhani A and Beers SA (2018) Antibodies to Costimulatory Receptor 4-1BB Enhance Anti-tumor Immunity via T Regulatory Cell Depletion and Promotion of CD8 T Cell Effector Function. Immunity 49, 958–970.e957. 10.1016/j.immuni.2018.09.014 [DOI] [PubMed] [Google Scholar]
- 43.Qi X, Li F, Wu Y, Cheng C, Han P, Wang J and Yang X (2019) Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nature Communications 10, 2141. 10.1038/s41467-019-10088-l [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Horst HJ, Gelderloos AT, Chamuleau MED, Breij ECW, Zweegman S, Nijhof IS, Overdijk MB and Mutis T (2021) Potent preclinical activity of HexaBody-DR5/DR5 in relapsed and/or refractory multiple myeloma. Blood Adv 5, 2165–2172. 10.1182/bloodadvances.2020003731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu X, Chan HTC, Orr CM, Dadas O, Booth SG, Dahal LN, Penfold CA, O’Brien L, Mockridge CI, French RR, Duriez P, Douglas LR, Pearson AR, Cragg MS, Tews I, Glennie MJ and White AL (2018) Complex Interplay between Epitope Specificity and Isotype Dictates the Biological Activity of Anti-human CD40 Antibodies. Cancer Cell 33, 664–675.e664. 10.1016/j.ccell.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chin SM, Kimberlin CR, Roe-Zurz Z, Zhang P, Xu A, Liao-Chan S, Sen D, Nager AR, Oakdale NS, Brown C, Wang F, Yang Y, Lindquist K, Yeung YA, Salek-Ardakani S and Chaparro-Riggers J (2018) Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nature Communications 9, 4679. 10.1038/s41467-018-07136-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, Schmidt H, Robert C, Chiarion-Sileni V, Ascierto PA, Maio M, Urba WJ, Gangadhar TC, Suryawanshi S, Neely J, Jure-Kunkel M, Krishnan S, Kohrt H, Sznol M and Levy R (2017) Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin Cancer Res 23, 1929–1936. 10.1158/1078-0432.CCR-16-1272 [DOI] [PubMed] [Google Scholar]
- 48.Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, Di Gravio D, Huang B, Gambhire D, Chen Y, Thall AD, Pathan N, Schmidt EV and Chow LQM (2017) Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin Cancer Res 23, 5349–5357. 10.1158/1078-0432.CCR-17-1243 [DOI] [PubMed] [Google Scholar]
- 49.Griffiths J, Hussain K, Smith HL, Sanders T, Cox KL, Semmrich M, Mårtensson L, Kim J, Inzhelevskaya T, Penfold CA, Tutt AL, Mockridge CI, Chan HC, English V, French RF, Teige I, Al-Shamkhani A, Glennie MJ, Frendeus BL, Willoughby JE and Cragg MS (2020) Domain binding and isotype dictate the activity of anti-human OX40 antibodies. J Immunother Cancer 8. 10.1136/jitc-2020-001557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang P, Tu GH, Wei J, Santiago P, Larrabee LR, Liao-Chan S, Mistry T, Chu ML, Sai T, Lindquist K, Long H, Chaparro-Riggers J, Salek-Ardakani S and Yeung YA (2019) Ligand-Blocking and Membrane-Proximal Domain Targeting Anti-OX40 Antibodies Mediate Potent T Cell-Stimulatory and Anti-Tumor Activity. Cell Rep 27, 3117–3123.e3115. 10.1016/j.celrep.2019.05.027 [DOI] [PubMed] [Google Scholar]
- 51.Nordstrom JL, Gorlatov S, Zhang W, Yang Y, Huang L, Burke S, Li H, Ciccarone V, Zhang T, Stavenhagen J, Koenig S, Stewart SJ, Moore PA, Johnson S and Bonvini E (2011) Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Research 13, R123. 10.1186/bcr3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I and Topalian SL (2010) Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. Journal of Clinical Oncology 28, 3167–3175. 10.1200/JCO.2009.26.7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang R, Gao C, Raymond M, Dito G, Kabbabe D, Shao X, Hilt E, Sun Y, Pak I, Gutierrez M, Melero I, Spreafico A, Carvajal RD, Ong M, Olszanski AJ, Milbum C, Thudium K, Yang Z, Feng Y, Fracasso PM, Korman AJ, Aanur P, Huang SA and Quigley M (2019) An Integrative Approach to Inform Optimal Administration of OX40 Agonist Antibodies in Patients with Advanced Solid Tumors. Clin Cancer Res 25, 6709–6720. 10.1158/1078-0432.CCR-19-0526 [DOI] [PubMed] [Google Scholar]
- 54.Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I, Fox B, Tarrant G, Robinson J, Meager T, Dolman C, Thorpe SJ, Bristow A, Wadhwa M, Thorpe R and Poole S (2007) “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol 179, 3325–3331. 10.4049/jimmunol.179.5.3325 [DOI] [PubMed] [Google Scholar]
- 55.Waibler Z, Sender LY, Kamp C, Müller-Berghaus J, Liedert B, Schneider CK, Lower J and Kalinke U (2008) Toward experimental assessment of receptor occupancy: TGN1412 revisited. J Allergy Clin Immunol 122, 890–892. 10.1016/jjaci.2008.07.049 [DOI] [PubMed] [Google Scholar]
- 56.Gutierrez M, Moreno V, Heinhuis KM, Olszanski AJ, Spreafico A, Ong M, Chu Q, Carvajal RD, Trigo J, Ochoa de Olza M, Provencio M, DeVos FY, DeBraud F, Leong S, Lathers D, Wang R, Ravindran P, Feng Y, Aanur P and Melero I (2021) OX40 Agonist BMS-986178 Alone or in Combination With Nivolumab and/or Ipilimumab in Patients With Advanced Solid Tumors. Clin Cancer Res 27, 460–472. 10.1158/1078-0432.CCR-20-1830 [DOI] [PubMed] [Google Scholar]