

Abstract
Next generations sequencing (NGS)/deep sequencing has become an important tool in the study of viruses. The use of unique molecular identifiers/UMI can overcome the limitations of PCR errors and PCR-mediated recombination, and reveal the true sampling depth of viral population being sequenced in an NGS experiment. This approach of enhanced sequence data represents an ideal tool to study both high and low abundance drug resistance mutations, and more generally to explore the genetic structure of viral populations. Central to the use of the UMI/Primer ID approach is the creation of a template consensus sequence (TCS) for each genome sequenced. Here we describe a series of experiments to validate several aspects of the Mupltiplexed Primer ID (MPID) sequencing approach using the MiSeq platform. We have evaluated how multiplexing of cDNA synthesis and amplicons affects the sampling depth of the viral population for each individual cDNA/amplicon to understand the relationship between broader genome coverage versus maximal sequencing depth. We have validated reproducibility of the MPID assay in the detection of minority mutations in viral genomes. We have also examined the determinants that allow sequencing reads of PCR recombinants to contaminate the final TCS data set and show how such contamination can be limited. Finally, we provide several examples where we have applied MPID to analyze features of minority variants, and describe limits on their detection, in viral populations of HIV-1 and SARS-CoV-2 to demonstrate the generalizable utility of this approach with any RNA virus.
Keywords: next generations sequencing, unique molecular identifier, drug resistance mutation, viral diversity, Primer ID, template consensus sequence
Graphical Abstract
This graph shows how UMI-based sequencing can accurately measure the population diversity in RNA viruses.
RNA viruses, including human immunodeficiency virus type-1 (HIV-1), can display extensive genetic diversity arising from multiple factors, including the error-prone viral polymerase, rapid turnover of the viral population, selective pressures from the host immune system or from antiviral drugs, and viral recombination when two or more viruses co-infect the same cell.1–5 Genetic diversity can often be seen in the context of disease progression, pathogenesis, evolution of increased virulence, and the evolution of drug resistance. Information about viral populations can be used to look for drug resistant variants present as minor variants, or to track immune escape variants that may be escaping using several distinct pathways. Such information gives insight into the biology of a virus, but can also be useful in choosing the proper active antiviral regimen and potentially assist in vaccine design.
The key to understanding the genetic structure of a viral population is through accurate sequencing of individual viral genomes and accurate quantification of the number of genomes sequenced/sampled in the target viral population (i.e. sampling depth). There are two very different approaches to achieving such high quality information about a viral population. The first approach developed was template end-point dilution PCR (also known as single genome amplification [SGA] or single genome sequencing [SGS]) to generate amplicons from individual viral RNA templates combined with Sanger sequencing of the PCR product.6–10 This approach gives high quality data but is time-consuming, labor-intensive, and expensive with usually no more than a few dozen sequences generated to characterize the viral population.11–13 The low throughput of this approach limits the sampling depth of the viral population. Another approach to study the within-host viral population is to use next generation sequencing (NGS) technology to conveniently increase the sampling depth of the viral population. Unlike Sanger sequencing, which generates one consensus sequence, NGS generates many sequences in parallel. These deep sequencing technologies have the potential to greatly extend the sampling depths of viral populations but the direct application of these platforms suffers from two serious flaws. The first problem is that NGS platforms are error-prone, making the search for minor variants something of a futile act14, 15; and second, the preceding PCR amplification of the initial cDNA product completely obscures the number of viral genome templates being sequenced and breaks the link between specific sequences and specific viral RNA genomes. Thus, artifactual heterogeneity is introduced into the sequence data set due to PCR mis-incorporation/recombination16, 17 and platform sequencing error, while artifactual homogeneity is introduced by repetitive sequencing of PCR copies of the original templates (PCR resampling).18 Thus, simple applications of NGS approaches are seriously flawed in their ability to accurately probe and quantify diversity in viral populations.
We and others have resolved the limitations of conventional NGS approaches by including a block of degenerate nucleotides (unique molecular identifier [UMI] or Primer ID) in the initial cDNA primer which adds a unique sequence tag to each cDNA product. This tag is carried along through the entire PCR/sequencing process.19–25 Sequence reads in the final data set with the same Primer ID can now be identified as coming from the same original viral RNA template and thus can be collapsed to create a template consensus sequence (TCS) for each individual starting template. Fig. 1 shows the principle of using the Primer ID sequencing approach. Using the approach of creating a TCS for each starting template allows for the elimination of most of the errors generated by PCR mis-incorporation, PCR recombination and platform sequencing error. As important, the sampling depth (number of genomes sequenced) from the initial viral population is revealed by the total number of TCS (i.e., the number of different Primer IDs). We have directly measured the residual error rate of the Primer ID-guided sequencing to be around 1 in 10,000 nucleotides of TCSs from an RNA template control, potentially just the residual error of incorporation by reverse transcriptase during reverse transcription to generate the cDNA. Thus the Primer ID approach combined with NGS can be used to detect viral diversity with the advantage of revealing the true sampling depth of the viral population while also greatly suppressing the method-introduced sequencing errors. The addition of a Primer ID/UMI at the cDNA synthesis step represents a small modification to what is otherwise the widely used experimental design of sequencing viral RNA. The pooling of Primer IDs in the raw read output to create the error-corrected TCS for each starting RNA template requires a modest additional algorithm.
Figure 1.
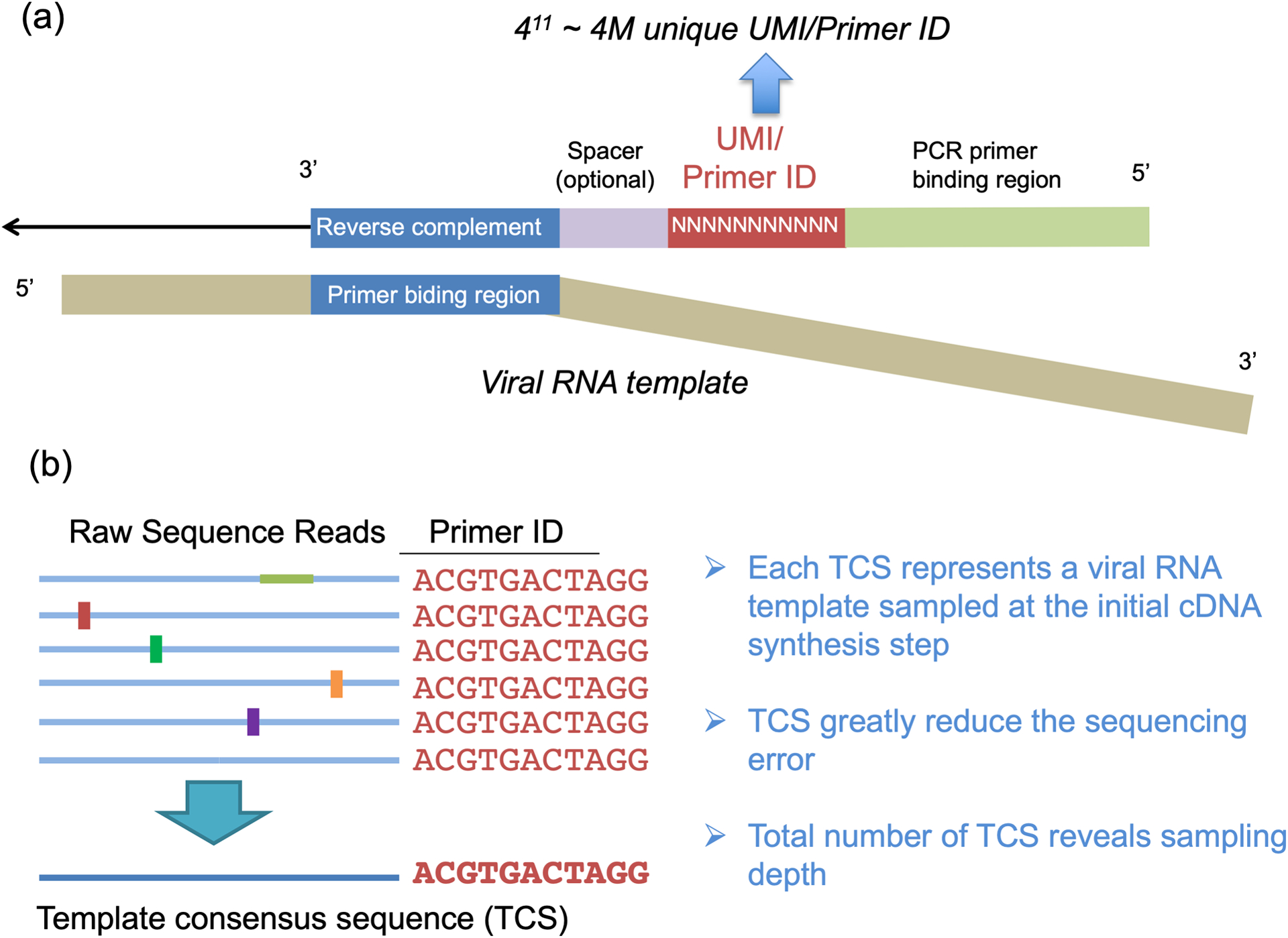
Primer ID Approach. a) The structure of the Primer ID primer and its binding to the viral RNA template. b) An example of creating the template consensus sequence from raw sequence reads.
In this manuscript we use Primer ID-guided NGS for deep sequencing RNA virus populations to detect overall diversity of a viral population and also to detect low level drug resistance mutations (DRMs) in viral populations. In addition, we describe our use of multiplexed Primer ID (MPID) with the MiSeq platform to extend the length of the viral genome sequenced within a single reaction. Along with these approaches we present data showing validation of the MPID MiSeq assay reproducibility. Finally, we present several examples where we have applied MPID MiSeq sequencing to the study of HIV-1 and SARS-CoV-2 viral populations.
RESULTS AND DISCUSSION
Impact Of Multiplexing cDNA and Forward Primers On Viral Population Sampling Depth.
The use of multiple cDNA primers in a single RT reaction and the subsequent PCR amplification of multiple amplicons, each with a distinct upstream/forward primer, increases the amount of sequence information from across the viral genome. However, this approach has the potential to reduce the number of viral genomes sequenced in each region within the viral population, which would limit the sampling depth of the population when evaluating the existence of minor variants. While the different amplicons have different upstream PCR primers they share a common downstream PCR primer included at the 5′ end of the cDNA primers. To examine the magnitude of the effect of multiplexing on sampling depth and the impact of cDNA primer placement, we used cDNA primers (with Primer ID) and PCR in a combinatorial design and determined the number of TCS recovered (a measure of sampling depth) in parallel sequencing runs (Multiplex Primer ID/MPID). The combination of amplicons is described in Fig. 2a. Each combination had two replicates and we used the same amount of input HIV-1 RNA (approximately 5,000 copies) from 8E5 cell supernatants for the library prep in each replicate. The multiple reads of a single Primer ID sequence were used to create a TCS for each sequenced template, thus the data in Fig. 2b are recorded as the number of TCS for each region sequenced as a function of the number of cDNA primers included in the initial cDNA synthesis reaction and the combination of regions tested.
Figure 2.
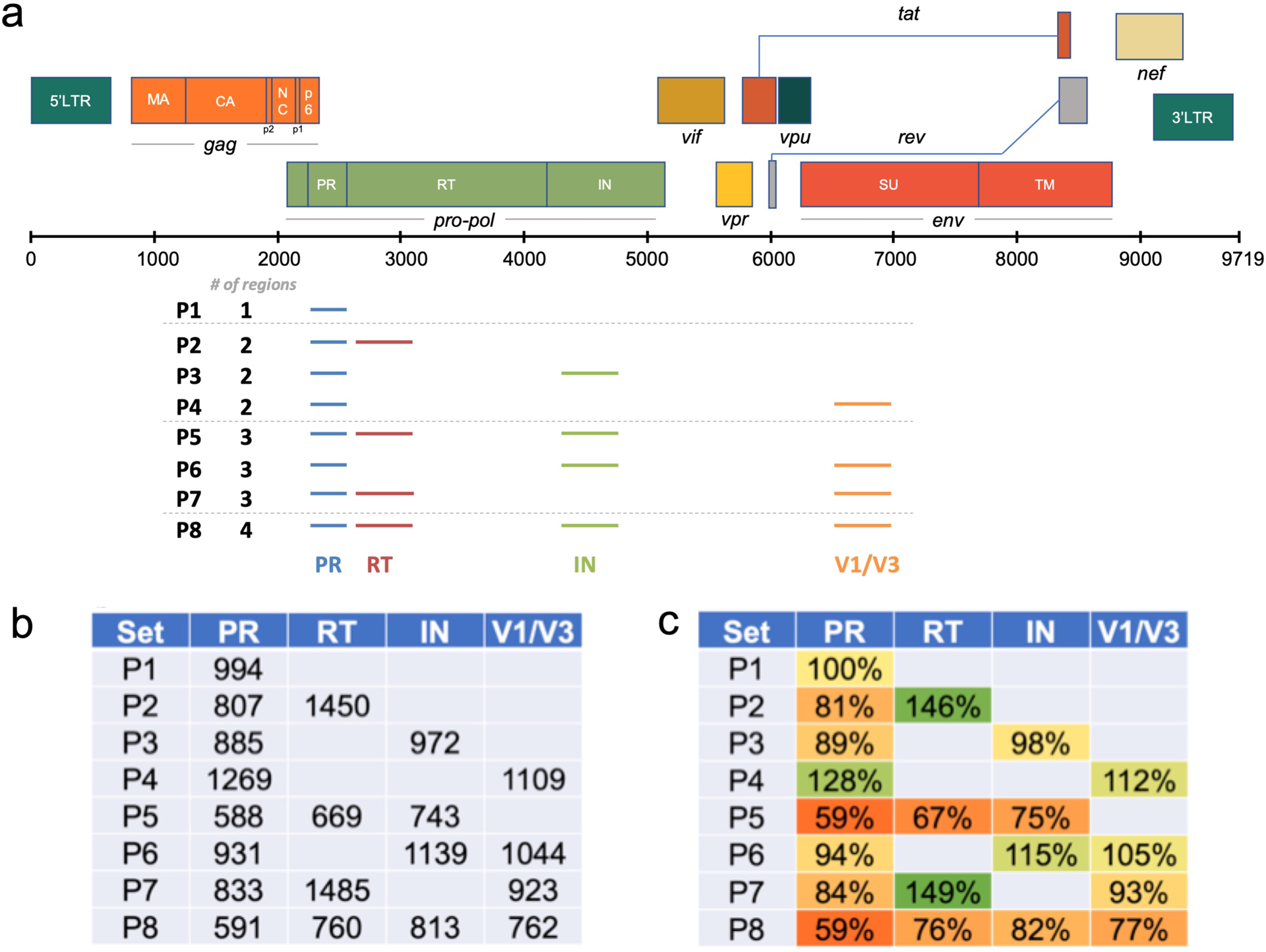
Multiplexing of regions impacts the recovery of TCS. In this experiment we used 8 different combination of HIV-1 amplicons. a) List of 8 combinations of regions in the experiment and their locations on the HIV-1 reference genome map. b) Average number of TCS at each region in 8 combinations. c) Normalized TCS numbers to P1 (PR only).
As can be seen in Fig. 2b, the different cDNA/forward primers vary in their efficiency of priming RT synthesis and PCR leading to different numbers of TCS recovered for each region even though they were all included in a single reaction. This difference for each region could be due to differing efficiencies of priming at the different regions of the genome, or differences between the complementary region of the primer and the viral genome for individual viral RNA populations. What is also apparent in the data in Fig. 2 is that the presence of multiple cDNA/forward primers can reduce the recovery of TCS. In Fig. 2c we normalized the TCS number with the P1 (protease/PR region primers alone). Three trends are apparent. First, including an additional amplicon in the pol region (RT or IN) reduced the sequencing depth in PR by 10–20% but this did not have an effect on the distal env amplicon V1/V2 (P2, P3, P6, P7). Second, when all three proximal amplicons were included (PR, RT, IN) then all amplicons were suppressed by 20–40% (P5, P8). Third, only when all four amplicons were included together did the distal amplicon (V1/V2) now show about a 25% decrease (P8). Thus there is a trade-off when using a multiplexing strategy in that overall sequence sampling depth of the viral population for each region can be reduced, especially for those regions in close proximity on the viral genome, and a higher number of amplicons can have a generally suppressive effect. However, these effects were all less than two-fold, and in many cases the number of templates sequenced is more than sufficient for the question under study making the multiplex design advantageous.
Validation of the Detection of Minority Variants Using Repeat Testing.
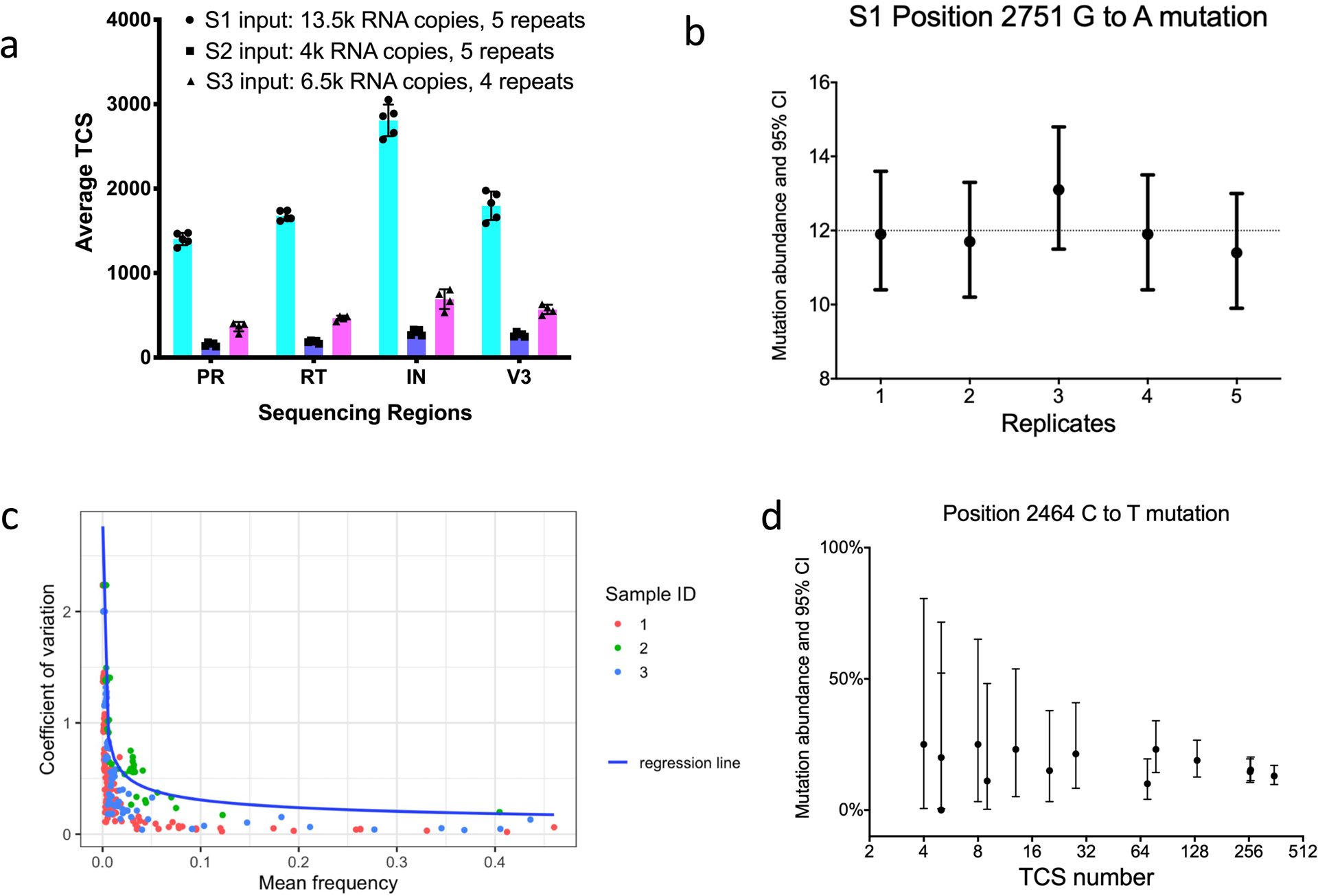
For a method to be useful in interpreting the sequence diversity of a viral population it must be reproducible. We tested the reproducibility of MPID MiSeq by repeatedly sequencing the same sample, using a total of 3 different HIV+ plasma specimens described in a previously published study.26 Fig. 3a shows the reproducibility of TCS obtained. The variations of TCS obtained among duplicates are all within 10% of the average of TCS obtained. It is worth noting that although similar numbers of RNA templates were present for each sample, the number of TCS recovered differed both by region and by clinical specimen. These data highlight the variability in the number of RNA templates actually sequenced between different clinical specimens, again, a feature that is obvious only when using a UMI to count the individual templates sequenced; in this case template utilization ranged from approximately 5% to 25% between the specimens and regions sequenced. The extreme example of poor template utilization comes when there is a failure to amplify anything and template utilization is 0%; in general, 25% represents the upper limit of what we have been able to obtain, and 5% to 10% template utilization from clinical specimens is a common outcome. Thus assuming a high percentage of RNA template utilization in an NGS experiment that does not include a UMI likely over-estimates the population sampling quality/depth by 10-fold or more.
Figure 3.
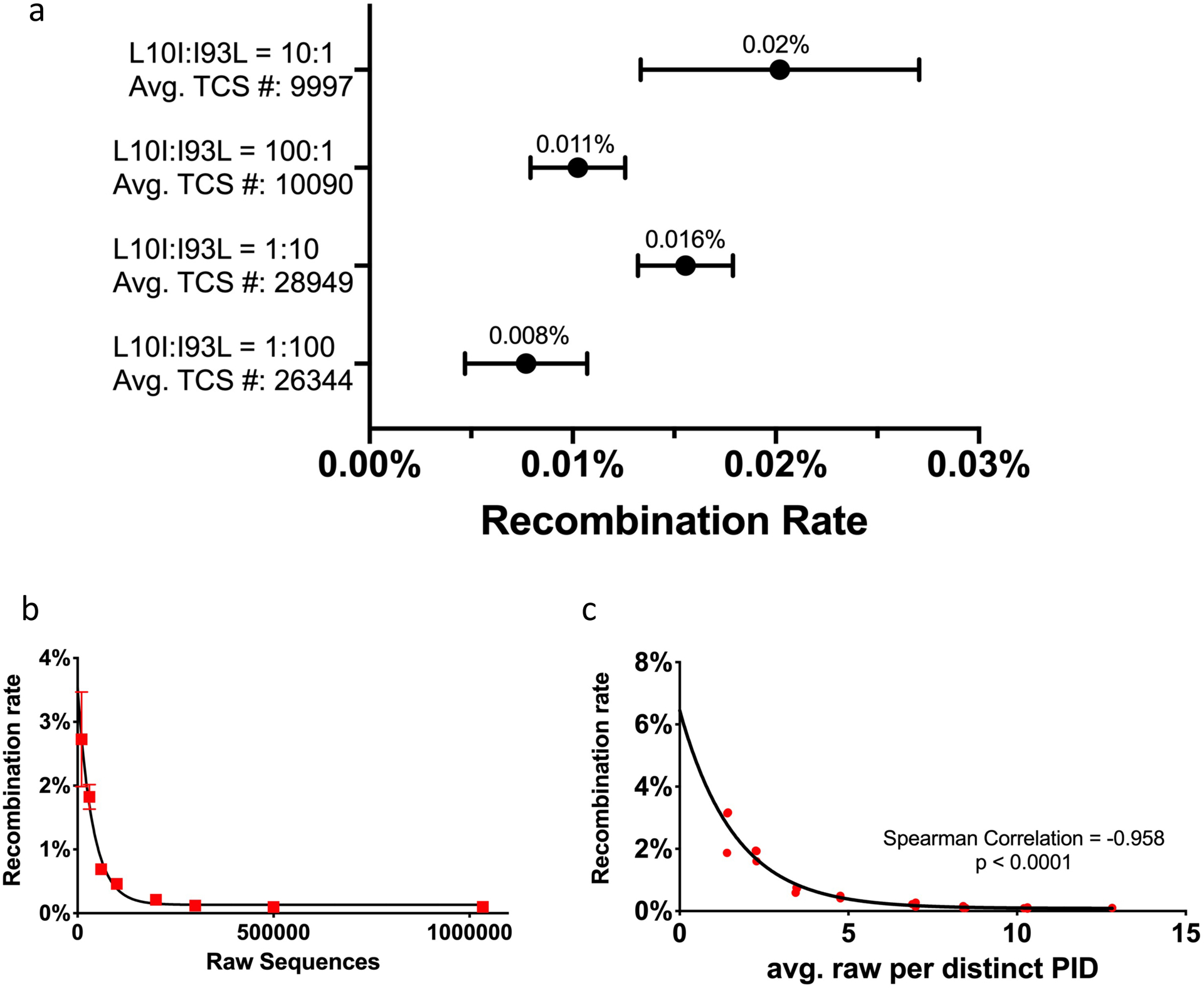
Recombination rate measure by Primer ID NGS. a) Recombination rates of 4 types of mutant mixtures after MPID MiSeq sequencing. The average recombination rates are labeled on top of the bars. b) Simulations to explore the factors correlated to residual recombination from one of the L10I:I93L=10:1 replicate, correlation of the number of raw sequences and recombination rate. c) Correlation of the average number per distinct PID and recombination rate. PID, Primer ID.
We next looked at the reproducibility of detection of minor variants in these viral populations. The abundances of minority variants (1% to 30%) were within 10% difference of the average abundance of all replicates. As shown in Fig. 3b, position 2751 (RT amino acid codon position 68) had a minority mutation (GGT to AGT, Gly to Ser), with an average abundance of 12% in specimen S1 (with sampling depth of approximately 1,800 TCS). The detection of mutations at this position is consistent throughout the 5 replicates. We also plotted the coefficient of variation (CV) for each minority mutation detected in these 3 specimens against their mean frequencies (Fig. 3c). It can be seen that the CV remains low (less than 0.5) for mutations greater than 1% abundance, but higher for mutations at lower abundance. The data suggest that MPID NGS protocol is consistent in the detection of minority mutations but can have higher variance in the mutation frequencies when the true abundance of a mutation is low, especially when the abundance gets closer to the lower detection sensitivity. This is the expected outcome based on the rules of random sampling. If greater accuracy is needed for low abundance variants then a larger number of TCS is required to increase sampling depth.
To demonstrate this feature of the detection of minor variants as a function of changing input template number, we varied template input. Higher numbers of templates (giving rise to higher numbers of TCS) should give similar values for the abundance of a minor variant but as templates becoming limiting the estimate of abundance should be compromised by sampling variability. This phenomenon can be seen in Fig. 3d. In this experiment we used different numbers of input templates from one specimen and sequenced them using the MPID MiSeq protocol. We plotted the observed frequencies and 95% CI of a position 2464 C to T mutation as a function of the TCS number. The mutation frequencies detected are generally consistent across the replicates but more variability is observed with the decrease in TCS number; the sum of the data suggests the true abundance of this variant in the population is in the range of 20%. This exercise highlights the difference between detecting a minor variant (with one or a few TCS with the mutation) as opposed to establishing an accurate estimate of its true abundance (with the accuracy going up with the number of TCS with that mutation recorded).
Evaluation of the Impact of PCR-Mediated Recombination.
During the PCR any prematurely terminated transcripts can serve as primers during a subsequent cycle creating artifactual recombinants between different templates, a well-known phenomenon during the many cycles used for PCR. For recombination events that happen after the first few cycles of PCR, the creation of a TCS for each RNA genome sequenced should obscure the presence of the recombinants since they will represent a minority of the sequences for that original template. To test the impact of PCR-mediated recombination on the structure of the viral population as detected by MPID, we carried out the following experiment. We used viral RNA isolated from tissue culture supernatants after separately growing two strains of HIV-1 in tissue culture. These two strains were generated from molecular clones that were identical except for two drug resistance mutations (DRM) positions placed near the ends of the PR region (L10I and I93L, referred as the L10I mutant and the I93L mutant, respectively). We mixed these two mutants as extracted viral RNA at ratios of 1:100, 1:10, 100:1 and 10:1, each with 4 replicates, and used the Primer ID MiSeq protocol to deep sequence the viral genomes at the PR region (examining between 10,000 and 29,000 TCS at each ratio). After sequencing, we examined each TCS for its sequences at PR positions 10 and 93. TCS with 10I and 93L (both mutations), as well as 10L and 93I (no mutations) on the same sequence, were classified as recombinants. We calculated the rate of recombination for each ratio of mutant mixture. Fig. 4a is the plot for the recombination rates for the 4 types of mutant mixtures. The average frequency of TCS that included recombination across replicates are labeled on top of the bars. Overall the recombination rates detected after TCS formation were extremely low across all types of mutant mixtures, ranging from 0.008% to 0.02%. The 10:1 and 1:10 mixtures had slightly higher recombination rate but not significantly different from 100:1 or 1:100 mixtures. This experiment demonstrated that the MPID protocol can eliminate the vast majority of the sequences generated by PCR-mediated recombination through the formation of TCS; in other words, when looking at 1000 TCS from a sample, it is likely that none of them represents an artifactual recombinant.
Figure 4.
Reproducibility of MPID NGS in the detection of minority mutations. A) Number of template consensus sequences (TCS) at all sequenced regions of multiplexed drug resistance testing for three specimens. b) Reproducibility of minority mutation detection in 3 specimen; mutation frequencies and confidence interval of position 2751 G to A mutation (RT amino acid codon position 68) in sample S1 across 5 replicates. c) Mean frequency and coefficient of variation of each minority mutations detected by repeated MPID-NGS of 3 different samples. d) Frequencies and confidence intervals of minority mutations detected from repeated sequencing using different number of RNA templates from the same specimen. TCS, template consensus sequence. CI, confidence interval. PR, protease, RT, reverse transcriptase, IN, integrase.
To further understand the reason for the residual TCS recombinants after MPID sequencing, we performed simulations to explore the correlation of the number of raw sequence reads per TCS and the recombination rate. The simulations were based on sequence data from one of the mutant mixtures (L10I:I93L=10:1). The total number of raw sequence reads for this library was 1,033,854, from which we created 10,017 TCS, and only 1 of the TCS was a recombinant. We randomly selected 10,000 to 500,000 raw sequence reads and processed the data using the same pipeline (tcs pipeline and variant analysis at PR position 10 and 93). We performed the process independently 3 times and plotted the recombination rate against the number of raw sequences on Fig. 4b. There is a strong inverse correlation between the number of raw sequences and the apparent recombination rate. When we used only 10,000 raw sequence reads, the average recovery of recombinant TCS was 2.7%. The recovery of recombinant TCS dropped quickly when more raw sequence reads were used to create TCS, and it dropped below 0.1% when at least 300,000 raw sequence reads were used to generate TCS. This question can also be interpreted as the number of raw sequence reads for each unique Primer ID determining the chance of fixing a PCR-mediated recombination event after data processing. Fig. 4c is the correlation of average number of raw sequence reads per distinct Primer ID/TCS and the observed recombination rate from the same simulated data. It is clear that with fewer raw sequence reads per distinct Primer ID/TCS, the more likely the chance to observe PCR-mediated recombination events that are present in the raw read data. This analysis shows that with at least 10 raw sequences per distinct Primer ID/TCS the artifactual recombination detection rate falls below 0.1%. This experiment and simulation emphasizes the importance of the careful design of MPID NGS protocol to ensure that a sufficient number of raw sequences can be obtained to construct TCS of the highest quality.
Having at least 10 raw reads per Primer ID (coverage) largely eliminates PCR recombinants from a typical data set. We have previously examined several other issues of Primer ID coverage that can be included in this discussion. In a separate simulation the number of raw sequence reads per Primer ID/TCS to adequately sample the sequences within raw sequence output. In that analysis19 we found that when the average number of raw sequences per Primer ID was 30 then over 90% of the genomes that were actually sequenced would appear in the analyzed dataset. Having an average number of raw reads per Primer ID/TCS greater than 30 does not cause any problems but rather represents diminishing returns on enhancing data quality for the available sequencing capacity. At the low end of the “reads per Primer ID” distribution, the raw sequence reads are actually dominated by a large number of apparent single Primer ID reads. These (and other low copy Primer ID reads) are the result of sequencing errors within the Primer ID (which can’t be corrected since they define the pooling for TCS formation); we have referred to these errors as “offspring” Primer IDs. The tcs pipeline includes a calculation based on the MiSeq error rate that generates a “minimum read number” cut-off based on the mean number of reads for each Primer ID. In this way offspring Primer IDs are excluded as these represent artifactual counting of raw reads that belong to an actual TCS. When the average number of raw reads per TCS is 30 then the minimum number of raw reads allowed to create a TCS will be 5 or greater. Finally, fortuitous resampling of a single Primer ID sequence to two different templates results in the loss of one of the template sequences when a TCS is created from the more abundantly read sequence with the same Primer ID (a modest loss when 100s or 1,000s of TCS are created). Thus, the final TCS pool represents a highly accurate dataset of sequences as they appeared in the original specimen minimally contaminated with sequencing errors and PCR-mediated recombinants.
MPID NGS and the Detection of Diversity of Viral Genome.
A key measurement of a viral population is the pairwise nucleotide diversity (Pi). An accurate assessment of Pi relies on accurate sequencing of the viral population since higher levels of sequencing error in the sequence data artifactually inflate the observed diversity. Since MPID NGS greatly reduces the sequencing method error rate and reveals the true sampling depth of the viral population, this makes it an ideal tool for accurately measuring pairwise diversity within the viral population.
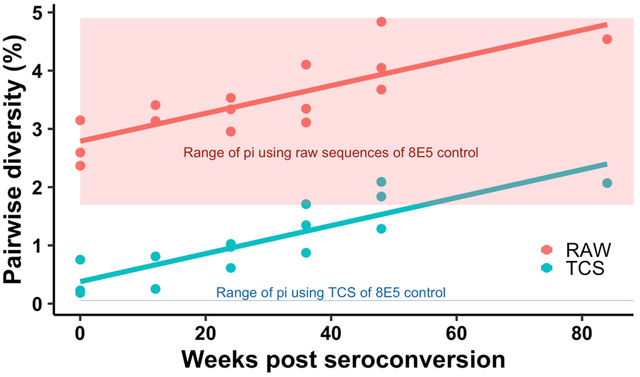
In Fig. 5a we show the change in the combined Pi values for a portion of the HIV-1 RT coding region in pol and V1/V3 in env using TCS (blue dots and line) compared to the raw MiSeq sequences of the same run (red dots and line) examining longitudinal specimens post seroconversion from 3 individuals27. It is clear that a majority of the signal for Pi is contributed by sequencing errors in the raw reads for samples within the first year of infection; by the middle of the second year true viral diversity makes up only one-half of the signal for Pi. This point is further emphasized when looking at apparent sequence diversity of a homogeneous RNA sequence. Fig. 5b is the the range of Pi values of the same regions obtained using the homogenous 8E5 HIV-1 genomic RNA28 from 11 individual MiSeq runs and processed using either raw reads (red) or TCS (blue). The data show that the raw reads values for Pi are dominated by the diversity generated by PCR/sequencing error (1.7–4.9%), and using Primer ID greatly reduced the method error with the residual error only contributing 0.05–0.06% to pairwise diversity.
Figure 5.
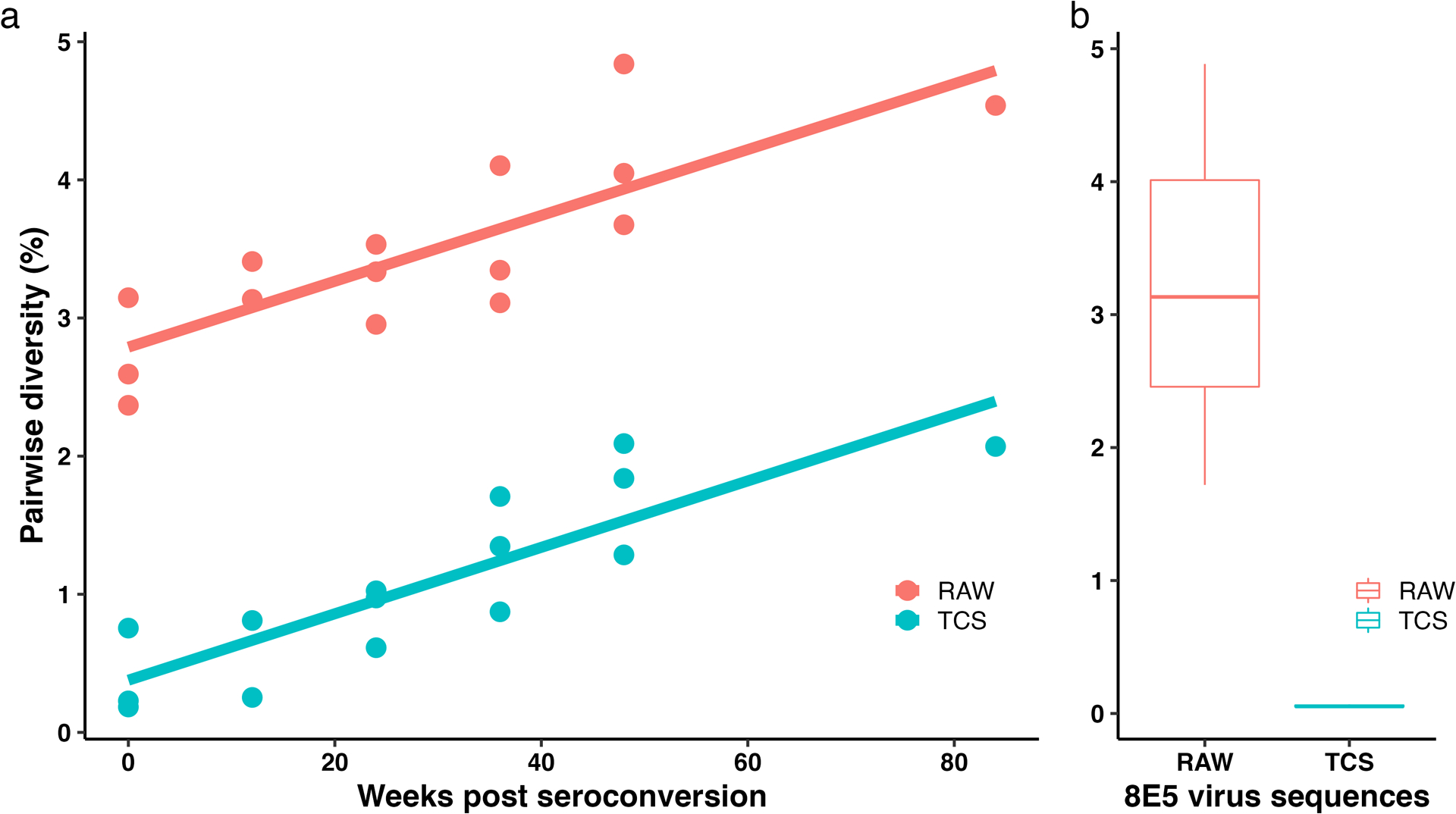
Pairwise diversity (Pi) of 3 CHAVI subjects after seroconversion in the background of Pi of the 8E5 control from 11 MiSeq runs. a) The change of Pi in the first two years post seroconversion of 3 subjects. Red line and dots show the Pi calculated using raw sequences, and the blue line and dots show the Pi calculated using TCS. b) Boxplot of the background of Pi of 8E5 control from 11 MiSeq runs. TCS, template consensus sequence.
Distinct Patterns Of Residual Background Sequencing Errors.
While the creation of a TCS for each sequenced template greatly reduces the errors from the error-prone sequencing platform and masks mutations introduced during PCR, there is still a small amount of residual error in the data set. This error comes from the host RNA polymerase used to synthesize viral RNA and/or from RT during the cDNA synthesis step. The first round of PCR also has the potential to introduce errors into the TCS. We have evaluated this error by sequencing viral RNA produced from the 8E5 cell line which carries a defective copy of viral DNA produces homogeneous viral RNA in noninfectious virions.
We sequenced 23,510 copies of HIV-1 RNA (i.e. TCS) collected from the supernatant of 8E5 cells and cataloged the frequency of different types of mutations (i.e. transitions and transversions) in the data set. These data are shown in Fig. 6. The overall mutation rate was 0.006% across the 519 positions sequenced. Transition substitutions were much higher than transversion substitutions, with C to T mutations the highest at 0.008%. In our estimates of residual errors in a dataset we have chosen to treat all mutations equally (see above). However, a more sophisticated matrix could be employed to adjust the false discovery rate estimate for individual observed mutations by scaling it to these observed differences in mutation frequency.
Figure 6.
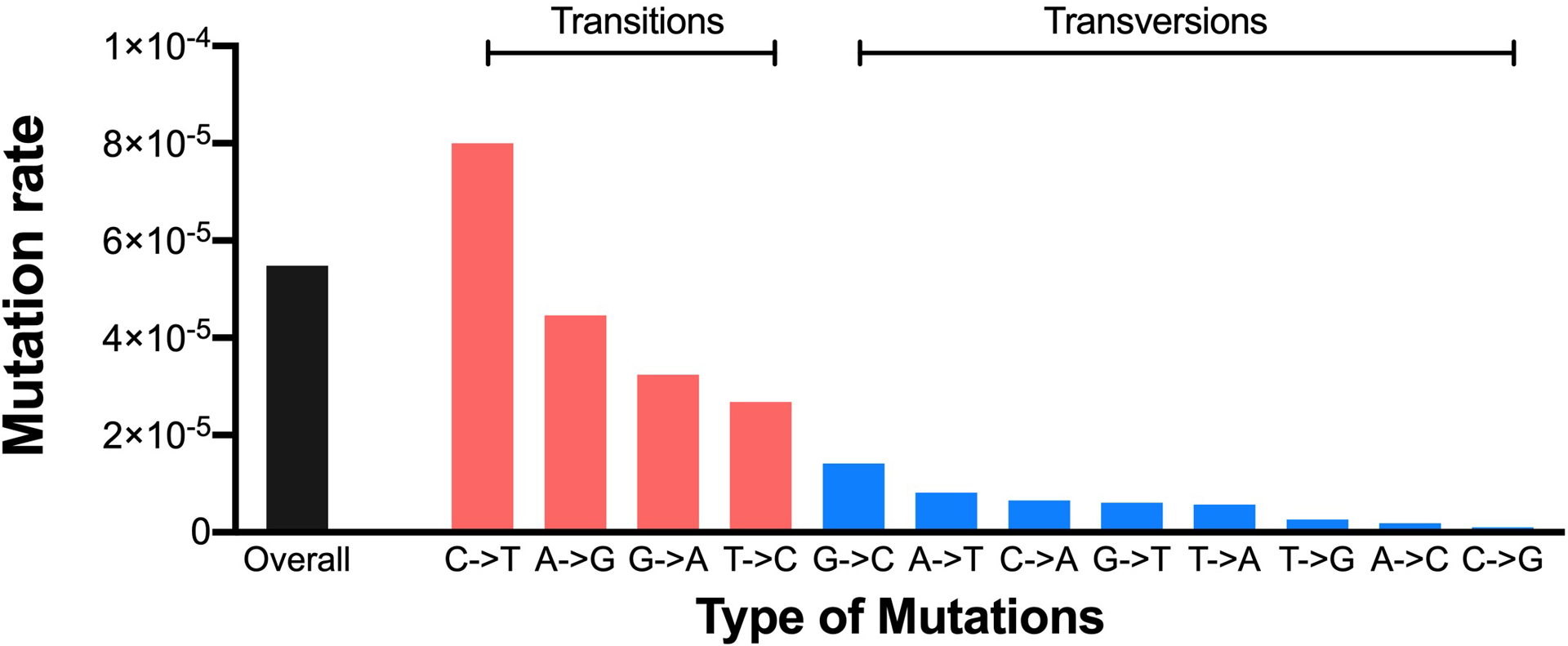
Types of mutations from homogenous HIV-1 RNA control isolated from supernatants of 8E5 cells.
Detection of Minor Variants In HIV-1 Viral Populations Approaching Equilibrium.
We applied the parameters of deep sequencing using TCS creation for each sequenced template of HIV-1 populations at approximately one year post infection, a time chosen to allow the large viral population to undergo mutation and selection to an extent that it might be approaching a steady-state sequence population. We did not attempt to test this assumption, which is an over-simplification since viral populations may undergo periodic bottlenecks due to immune selection. Our goal was to detect minor variants, especially DRMs, in the population whose existence could be validated with highly accurate sequence analysis. We examined virus in blood plasma from four participants from a cohort who were each originally infected with a single variant and who had untreated infections for 36 to 48 weeks.27 The chosen plasma sample was sequenced in all four regions using MPID with an attempt to achieve deeper sampling than typically obtained by adding more RNA templates and/or pooling separate sequencing runs of the same sample. For this analysis to be valid, it is necessary to account for residual method error (e.g. RT errors during the cDNA synthesis step). As described in the Methods, we calculated the false discovery rate (FDR) for each mutation detected to distinguish real DRMs from mutations likely appearing due to residual method error. Mutations with FDR no more than 5% were considered as detection of real DRMs. The results of this analysis are shown in Table 1. The median TCS number (sampling depth) in the pol region was 1,134, with which we had 95% confidence to detect minority DRMs as present as low as 0.32% of the population, calculated using a Binomial distribution. The median sampling depth at the V1/V3 env region was 427. We did not detect any notable DRMs in the RT region at this level of sampling. In the PR region, one participant had L90M as a majority mutation, which we consider to be a transmitted DRM. We found two participants with a detectable M46I mutation (in PR) in the viral population, and one participant with a detectable D30N mutation (in PR), all of which were below 1% abundance. At the IN region, one subject had an L74M mutation as a majority mutation. However, L74M can be found in some ART-naïve individuals as a polymorphism. Two other DRMs, S147G and V151I in IN, were found in two individuals as minority mutations with abundances below 0.5%. It should be noted that M46I and D30N in the PR region and V151I in the IN region could be generated as APOBEC3G/F mutations, representing three of the four low level DRMs detected. These results suggest that when viral populations are approaching equilibrium, minority DRMs can be observed at below 1% abundance, and APOBEC3G/F activity may contribute to the presence of minority DRMs in the viral populations.
Table 1.
Number of Template Consensus Sequences (TCS) and drug resistance mutations (DRMs) for HIV-1 identified in 4 CHAVI participants who had untreated infections for 36 to 48 weeks.
| ID | TCS and detection sensitivity | DRMs, percentage and 95% CI | |||||
|---|---|---|---|---|---|---|---|
| PR | RT | IN | V1V3 | PR | RT | IN | |
| A | 1424 (0.3%) | 1239 (0.3%) | 860 (0.4%) | 172 (2.1%) | M46I:0.56(0.24–1.1); L90M:99.86(99.49–99.98); |
S147G:0.35(0.07–1.02); | |
| B | 909 (0.4%) | 730 (0.5%) | 1198 (0.3%) | 616 (0.6%) | L74M:99.83(99.4–99.98); | ||
| C | 251 (1.5%) | 783 (0.5%) | 1070 (0.3%) | 237 (1.5%) | |||
| D | 2985 (0.1%) | 2057 (0.2%) | 5111 (0.1%) | 3020 (0.1%) | D30N:0.23(0.09–0.48); M46I:0.7(0.44–1.07); |
V151I:0.14(0.06–0.28); | |
Detection of Minority Mutations In the SARS-CoV-2 S Gene and nsp12.
New variants of SARS-CoV-2 are threats to efforts to control the ongoing pandemic. We have developed a MPID MiSeq protocol to accurately sequence most of the ectodomain of the SARS-CoV-2 S/Spike gene to enable a search for minority variants within a viral population within a person. In this MPID protocol, we multiplexed two sets of cDNAs/amplicons including part of nsp12 (for putative remdesivir DRMs detection) and the first two thirds of the S gene, for a total 3,658 bp sequenced.
We used this MPID MiSeq protocol to sequence SARS-CoV-2 RNA isolated from a nasal swab of a COVID-19 patient with a persistent infection. The specimen was collected under a SARS-CoV-2 study approved by the UNC IRB, and we sequenced de-identified specimens. We obtained significant sampling depth in all regions (median ~30,000 TCS per region, in part owing to the abundant amount of viral RNA in the specimen). The overall pairwise diversity was very low (0.02–0.03%). No potential resistance mutations to remdesivir in the nsp12 region sequences were detected, but we did find a minority mutation C464F at 0.12% abundance in nsp12. In the S gene RBD region, we found 2 positions with minority mutations at 0.06% abundance (I312V and D428E), while the rest of the sequence was identical to the circulating epidemic strain at that time. All three minor variants had FDR values of less than 5% and were considered as true variants. This experiment demonstrates that MPID MiSeq was able to detect low level minority variants in a SARS-CoV-2 viral population. Thus this approach has the potential to be a useful tool for monitoring the development of variants among infected individuals, especially for those with persistent infections and those exposed to non-clearing therapies that could select for resistance.
CONCLUSION
Here we have explored important and relevant features of the use of Primer ID, a form of UMI. The pairing of Primer ID and the MiSeq sequencing platform has provided a robust approach to studying viral sequence diversity, although limited by amplicon lengths in the range of 500 bp. We have overcome this limitation by multiplexing multiple cDNAs/amplicons in a single reaction and show only modest reductions in sequencing efficiency at up to four amplicons. PCR-mediated recombination is a well known phenomenon, but we showed that using the approach of TCS and having coverage in the range of 10-fold largely obscures the observation of recombinants in the processed data set. Using repeated sequencing runs we showed that the Primer ID/UMI protocol is highly reproducible and that, with the ability to know the number of templates actually sequenced, the sequence data is fully amenable to the application of standard statistical analysis for population sampling. The ability to suppress the method error rate allowed us to define the pattern of misincorporation/sequencing error that remain, to obtain highly accurate values for pairwise diversity in the viral population, and to search for DRMs within the unselected viral population. Finally, we show that all of these features are completely portable to another virus system, in this case SARS-CoV-2. Collectively this work validates MPID as a powerful approach to studying viral diversity within RNA virus populations.
METHODS
Cells and Virus.
The 8E5 cell clone was obtained from the HIV Reagent Program. Each 8E5 cell contains one copy of defective HIV-1 proviral DNA, and these cells produce virus particles with homogeneous viral RNA. Viral RNA was extracted from supernatant virus particles to generate a control viral RNA population. The NL4–3 clone of HIV-1 was modified to include point mutations in the protease coding region which were used in the PCR recombination experiment. We used HIV+ plasma specimens from previously published studies26, 27 for the validation of MPID-NGS in the detection of minority variants and the measurement of the pairwise diversity. We also used this MPID NGS protocol to sequence SARS-CoV-2 RNA isolated from a nasal swab of a person with a persistent infection.
Library Preparation.
The principle of the MPID NGS approach has been previously described29. In brief, we used Primer ID-tagged cDNA primers in the initial cDNA synthesis after viral RNA extraction to label each viral RNA template with a UMI. The multiplexing of the cDNA allowed us to sequence multiple regions of the viral genome in one reaction. After two rounds of PCR amplification of the cDNA, purified PCR products from different sequencing/amplification reactions were normalized for concentration and pooled for sequencing using the Illumina MiSeq. The primers used in the library prep can be found in Supplemental Table 1 (for HIV-1) and Supplemental Table 2 (for SARS-CoV-2).
Data Processing and Analysis.
The sequencing data were processed with tcs pipeline v.2.5.0 (https://github.com/ViralSeq/viral_seq) to construct the template consensus sequences (TCS). The number of TCS revealed the sampling depth of sequencing as the number of initial templates actually queried. Knowing the sampling depth/sample size allowed the calculation of the detection sensitivity based on the upper 95% confidence limits for the Binomial proportion when no mutation has been observed for a given the number of TCS, and knowing the number of TCS allowed the reporting of the observed frequencies of DRMs and their 95% confidence intervals for their true abundance (CIs). We did not use an arbitrary cut-off for the minority variants (as is often done when the sample size of the number of genomes sequenced is not known so a guess on sensitivity is made), rather we calculated the false discovery rate (FDR) adjusted p-value for each mutation detected using the Benjamini-Hochberg procedure to distinguish true DRMs from apparent substitutions likely caused by residual method error. In the FDR calculation, we used a previously measured residual error rate of 0.0001 for Primer ID sequencing19 as the substitution rate from the residual method error.
Supplementary Material
Acknowledgements.
This work was supported by grants from NIGMS (P01-GM109767, R01-GM135919) and NIAID (R01-AI140970, U19-AI142759) of the NIH. This research received infrastructure support from the UNC CFAR (P30-AI050410), and the UNC Lineberger Comprehensive Cancer Center (P30-CA016086). The support of the UNC High Throughput Sequencing Facility is also acknowledged.
Footnotes
Supporting Information: Two supplementary tables of sequences of the primers used in this study.
UNC is pursuing IP protection for Primer ID sequencing and RS has received nominal royalties from licensing.
REFERENCES
- 1.Holmes EC; Zhang LQ; Simmonds P; Ludlam CA; Brown AJ, Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci U S A 1992, 89 (11), 4835–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perelson AS; Neumann AU; Markowitz M; Leonard JM; Ho DD, HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 1996, 271 (5255), 1582–6. [DOI] [PubMed] [Google Scholar]
- 3.Preston BD; Poiesz BJ; Loeb LA, Fidelity of HIV-1 reverse transcriptase. Science 1988, 242 (4882), 1168–71. [DOI] [PubMed] [Google Scholar]
- 4.Burke DS, Recombination in HIV: an important viral evolutionary strategy. Emerg Infect Dis 1997, 3 (3), 253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung A; Maier R; Vartanian JP; Bocharov G; Jung V; Fischer U; Meese E; Wain-Hobson S; Meyerhans A, Recombination: Multiply infected spleen cells in HIV patients. Nature 2002, 418 (6894), 144. [DOI] [PubMed] [Google Scholar]
- 6.Salazar-Gonzalez JF; Bailes E; Pham KT; Salazar MG; Guffey MB; Keele BF; Derdeyn CA; Farmer P; Hunter E; Allen S; Manigart O; Mulenga J; Anderson JA; Swanstrom R; Haynes BF; Athreya GS; Korber BT; Sharp PM; Shaw GM; Hahn BH, Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 2008, 82 (8), 3952–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shankarappa R; Margolick JB; Gange SJ; Rodrigo AG; Upchurch D; Farzadegan H; Gupta P; Rinaldo CR; Learn GH; He X; Huang XL; Mullins JI, Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 1999, 73 (12), 10489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keele BF; Giorgi EE; Salazar-Gonzalez JF; Decker JM; Pham KT; Salazar MG; Sun C; Grayson T; Wang S; Li H; Wei X; Jiang C; Kirchherr JL; Gao F; Anderson JA; Ping LH; Swanstrom R; Tomaras GD; Blattner WA; Goepfert PA; Kilby JM; Saag MS; Delwart EL; Busch MP; Cohen MS; Montefiori DC; Haynes BF; Gaschen B; Athreya GS; Lee HY; Wood N; Seoighe C; Perelson AS; Bhattacharya T; Korber BT; Hahn BH; Shaw GM, Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 2008, 105 (21), 7552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer S; Kearney M; Maldarelli F; Halvas EK; Bixby CJ; Bazmi H; Rock D; Falloon J; Davey RT Jr.; Dewar RL; Metcalf JA; Hammer S; Mellors JW; Coffin JM, Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J Clin Microbiol 2005, 43 (1), 406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu SL; Rodrigo AG; Shankarappa R; Learn GH; Hsu L; Davidov O; Zhao LP; Mullins JI, HIV quasispecies and resampling. Science 1996, 273 (5274), 415–6. [DOI] [PubMed] [Google Scholar]
- 11.Ping LH; Joseph SB; Anderson JA; Abrahams MR; Salazar-Gonzalez JF; Kincer LP; Treurnicht FK; Arney L; Ojeda S; Zhang M; Keys J; Potter EL; Chu H; Moore P; Salazar MG; Iyer S; Jabara C; Kirchherr J; Mapanje C; Ngandu N; Seoighe C; Hoffman I; Gao F; Tang Y; Labranche C; Lee B; Saville A; Vermeulen M; Fiscus S; Morris L; Karim SA; Haynes BF; Shaw GM; Korber BT; Hahn BH; Cohen MS; Montefiori D; Williamson C; Swanstrom R; Study CAI; the Center for, H. I. V. A. V. I. C., Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. J Virol 2013, 87 (13), 7218–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sturdevant CB; Dow A; Jabara CB; Joseph SB; Schnell G; Takamune N; Mallewa M; Heyderman RS; Van Rie A; Swanstrom R, Central nervous system compartmentalization of HIV-1 subtype C variants early and late in infection in young children. PLoS Pathog 2012, 8 (12), e1003094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnell G; Joseph S; Spudich S; Price RW; Swanstrom R, HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog 2011, 7 (10), e1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huse SM; Huber JA; Morrison HG; Sogin ML; Welch DM, Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 2007, 8 (7), R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quail MA; Smith M; Coupland P; Otto TD; Harris SR; Connor TR; Bertoni A; Swerdlow HP; Gu Y, A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012, 13, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyerhans A; Vartanian JP; Wain-Hobson S, DNA recombination during PCR. Nucleic Acids Res 1990, 18 (7), 1687–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorzer I; Guelly C; Trajanoski S; Puchhammer-Stockl E, The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. J Virol Methods 2010, 169 (1), 248–52. [DOI] [PubMed] [Google Scholar]
- 18.Archer J; Rambaut A; Taillon BE; Harrigan PR; Lewis M; Robertson DL, The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time--an ultra-deep approach. PLoS Comput Biol 2010, 6 (12), e1001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou S; Jones C; Mieczkowski P; Swanstrom R, Primer ID Validates Template Sampling Depth and Greatly Reduces the Error Rate of Next-Generation Sequencing of HIV-1 Genomic RNA Populations. J Virol 2015, 89 (16), 8540–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jabara CB; Jones CD; Roach J; Anderson JA; Swanstrom R, Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A 2011, 108 (50), 20166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boltz VF; Rausch J; Shao W; Hattori J; Luke B; Maldarelli F; Mellors JW; Kearney MF; Coffin JM, Ultrasensitive single-genome sequencing: accurate, targeted, next generation sequencing of HIV-1 RNA. Retrovirology 2016, 13 (1), 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu GK; Hu J; Wang PH; Fodor SP, Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc Natl Acad Sci U S A 2011, 108 (22), 9026–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinde I; Wu J; Papadopoulos N; Kinzler KW; Vogelstein B, Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 2011, 108 (23), 9530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang RH; Mo T; Dong W; Lee GQ; Swenson LC; McCloskey RM; Woods CK; Brumme CJ; Ho CK; Schinkel J; Joy JB; Harrigan PR; Poon AF, Theoretical and experimental assessment of degenerate primer tagging in ultra-deep applications of next-generation sequencing. Nucleic Acids Res 2014, 42 (12), e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brodin J; Hedskog C; Heddini A; Benard E; Neher RA; Mild M; Albert J, Challenges with using primer IDs to improve accuracy of next generation sequencing. PLoS One 2015, 10 (3), e0119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clutter DS; Zhou S; Varghese V; Rhee SY; Pinsky BA; Jeffrey Fessel W; Klein DB; Spielvogel E; Holmes SP; Hurley LB; Silverberg MJ; Swanstrom R; Shafer RW, Prevalence of Drug-Resistant Minority Variants in Untreated HIV-1-Infected Individuals With and Those Without Transmitted Drug Resistance Detected by Sanger Sequencing. J Infect Dis 2017, 216 (3), 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dennis AM; Zhou S; Sellers CJ; Learner E; Potempa M; Cohen MS; Miller WC; Eron JJ; Swanstrom R, Using Primer-ID Deep Sequencing to Detect Recent Human Immunodeficiency Virus Type 1 Infection. J Infect Dis 2018, 218 (11), 1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Folks TM; Powell D; Lightfoote M; Koenig S; Fauci AS; Benn S; Rabson A; Daugherty D; Gendelman HE; Hoggan MD, Biological and biochemical characterization of a cloned Leu-3- cell surviving infection with the acquired immune deficiency syndrome retrovirus. J Exp Med 1986, 164 (1), 280–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou S; Hill CS; Clark MU; Sheahan TP; Baric R; Swanstrom R, Primer ID Next-Generation Sequencing for the Analysis of a Broad Spectrum Antiviral Induced Transition Mutations and Errors Rates in a Coronavirus Genome. Bio Protoc 2021, 11 (5), e3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.