

Abstract
Plant immune responses are triggered during the interaction with pathogens. The fungus Botrytis cinerea has previously been reported to use small RNAs (sRNAs) as effector molecules capable of interfering with the host immune response. Conversely, a host plant produces sRNAs that may interfere with the infection mechanism of an intruder. We used high‐throughput sequencing to identify sRNAs produced by B. cinerea and Solanum lycopersicum (tomato) during early phases of interaction and to examine the expression of their predicted mRNA targets in the other organism. A total of 7042 B. cinerea sRNAs were predicted to target 3185 mRNAs in tomato. Of the predicted tomato target genes, 163 were indeed transcriptionally down‐regulated during the early phase of infection. Several experiments were performed to study a causal relation between the production of B. cinerea sRNAs and the down‐regulation of predicted target genes in tomato. We generated B. cinerea mutants in which a transposon region was deleted that is the source of c.10% of the fungal sRNAs. Furthermore, mutants were generated in which both Dicer‐like genes (Bcdcl1 and Bcdcl2) were deleted and these displayed a >99% reduction of transposon‐derived sRNA production. Neither of these mutants was significantly reduced in virulence on any plant species tested. Our results reveal no evidence for any detectable role of B. cinerea sRNAs in the virulence of the fungus.
Keywords: Botrytis cinerea, high‐throughput sequencing, host immunity, small RNA, tomato
Botrytis cinerea mutants lacking both Dicer‐like genes displayed a >99% reduction of transposable element‐derived small RNA (sRNA) production, but were not reduced in virulence, indicating redundancy of sRNAs in fungal virulence.
1. INTRODUCTION
Grey mould caused by the fungus Botrytis cinerea is a disease that affects a wide range of hosts (Fillinger & Elad, 2016; van Kan et al., 2014). For many years, infection by B. cinerea was considered to mainly require production of phytotoxic metabolites and plant cell wall‐degrading enzymes (van Kan, 2006). In the past decade it has become clear that B. cinerea also produces effector proteins with cell death‐inducing capacity that may contribute to its virulence towards a broad range of host plants (Choquer et al., 2007; Noda et al., 2010; Zhu et al., 2017). An additional layer of complexity was revealed by the report that small RNAs (sRNAs) can act as a novel class of fungal effectors, which can be translocated into host cells and suppress the expression of host target genes by RNA interference (RNAi). A subset of these host genes is involved in immune responses, and their silencing by fungal sRNAs might thus facilitate invasion of the pathogen (Weiberg et al., 2013). It has been known for years that plant transgene‐derived double‐stranded RNAs (dsRNAs) could induce gene silencing in invading pathogens and pests (Baum et al., 2007; Iqbal et al., 2020; Nowara et al., 2010), a process referred to as host‐induced gene silencing (HIGS). However, Weiberg et al. (2013) reported that a pathogen can also produce sRNAs that actively suppress host immune responses, indicative of a true RNA information warfare between B. cinerea and its host (Chaloner et al., 2016; Weiberg et al., 2014). This concept was later also demonstrated in other pathosystems. For instance, the parasitic plant Cuscuta campestris secretes microRNAs to silence plant genes involved in defence signalling to promote parasitism (Shahid et al., 2018). The pathogenic oomycete Hyaloperonospora arabidopsidis was also reported to deliver sRNAs to silence Arabidopsis thaliana defence genes and these sRNAs are important for virulence (Dunker et al., 2020). Moreover, Wong‐Bajracharya et al. (2022) recently reported that sRNAs from the ectomycorrhizal fungus Pisolithus microcarpus can enter a host plant and regulate host transcripts in a symbiotic interaction. The first report that plant sRNAs are translocated to an invading fungal pathogen was in the cotton–Verticillium dahliae interaction (Zhang et al., 2016). Cai et al. (2018) demonstrated that a host plant can secrete exosome‐like vesicles containing sRNAs that can subsequently be taken up by B. cinerea hyphae at the infection site. Another study by Hou et al. (2019) described how a plant can produce small interfering RNAs (siRNAs) to confer resistance to Phytophthora capsici through cross‐kingdom RNAi, while inversely a Phytophthora effector can specifically inhibit the biogenesis of host siRNAs to facilitate infection. The delivery of host sRNAs can subsequently mediate silencing of genes in pathogens (Cai et al., 2018; Hou et al., 2019), indicating that sRNA translocation and RNAi induction between plants and microbial pathogens (fungi, oomycetes) is bidirectional. By contrast, Kettles et al. (2019) reported that RNAi‐deficient Zymoseptoria tritici mutants were fully pathogenic to wheat and Z. tritici was unable to take up external dsRNAs that were derived from transgenic wheat plants or artificially synthesized, suggesting that cross‐kingdom RNAi may not occur in the wheat–Z. tritici pathosystem. Cross‐kingdom RNAi seems to be confined to certain host–pathogen interactions and may not be a ubiquitous phenomenon.
sRNAs are mostly cleaved products from noncoding dsRNAs and single‐stranded RNAs with hairpin structures, which are processed by the endoribonuclease activity of Dicer‐like (DCL) proteins (Baulcombe, 2004; Ghildiyal & Zamore, 2009; Torres‐Martínez & Ruiz‐Vázquez, 2017). Complexes composed of sRNAs, Argonaute (AGO) proteins and auxiliary proteins conduct silencing of target mRNAs when the sRNAs are fully or partially complementary to their target (Novina & Sharp, 2004; Silvestri et al., 2019). Cross‐kingdom RNAi caused by sRNAs generated by host and pathogens is considered a well‐documented phenomenon that may have a significant effect on resistance or susceptibility during host–pathogen interactions (Dubey et al., 2019). For the first time, Weiberg et al. (2013) reported that the majority of sRNAs produced by B. cinerea during infection of A. thaliana are derived from long terminal repeat (LTR) retrotransposons in the fungal genome. Interestingly, a recent study reported that retrotransposons can promote virulence of B. cinerea, as a fungal strain that carried only silenced transposon relicts and produced few sRNAs became more virulent by introduction of an exogenous active retrotransposon (Porquier et al., 2021). Retrotransposon‐derived dsRNA molecules in B. cinerea are predominantly processed into sRNAs by two DCL proteins, BcDCL1 and BcDCL2 (Wang et al., 2016; Weiberg et al., 2013). It was reported that removal of both Bcdcl1 and Bcdcl2 genes in B. cinerea almost abolished the production of sRNA species with a size range of 20–26 nucleotides (nt) (Wang et al., 2016), and significantly reduced the virulence of B. cinerea on A. thaliana and Solanum lycopersicum (tomato) leaves (Weiberg et al., 2013). However, the ∆dcl1/∆dcl2 double mutant in the studies of Weiberg et al. (2013) and Wang et al. (2016) unexpectedly displayed reduced growth and aberrant sporulation phenotypes.
We aimed to establish in more detail the role of sRNAs in the early interaction between B. cinerea and tomato. Considering that the decisive processes in the interaction between B. cinerea and its host plants would occur within the first 24 h postinoculation (hpi) (Veloso & van Kan, 2018), we generated a dataset from samples taken at 12 hpi (when penetration is just accomplished), 16 hpi (occurrence of first signs of cell death), and 24 hpi (onset of lesion expansion). Samples were sequenced at sufficient read depth to also analyse B. cinerea sRNAs and mRNAs at these early timepoints, when fungal biomass is low. We examined the data for occurrence of inverse correlations in transcript levels between sRNAs and their corresponding (predicted) target transcripts from the pathogen and host. Subsequently, we performed functional analyses to evaluate whether the observed down‐regulation of mRNAs could have been mediated by cross‐kingdom RNAi. Neither the deletion of a retrotransposon region producing approx. 10% of the total sRNAs from the B. cinerea genome nor the deletion of both Dicer‐like genes impaired virulence on tomato and three other hosts. Our results suggest that natural production of sRNAs by B. cinerea may not contribute as much to virulence as previously reported.
2. RESULTS
We extracted sRNA and mRNA samples from B. cinerea‐infected tomato leaves harvested at 12, 16, and 24 hpi. Mock‐inoculated tomato leaves at the same time points as well as a B. cinerea liquid culture were included as controls. All samples were used for generating strand‐specific libraries and sequenced at read depths varying from 4.2 to 89 M per sample (Table S1). For infected leaf samples, sRNA reads were mapped to the B. cinerea genome (van Kan et al., 2017) and the tomato genome (Hosmani et al., 2019) to identify whether they were of fungal or host‐plant origin. sRNA reads that were mapped perfectly to both genomes (100% identity over the entire length) were eliminated from further analysis.
2.1. Many B. cinerea and tomato sRNAs and their predicted targets are expressed during early infection
The sRNA read length distribution in B. cinerea and tomato samples, grown separately, were different between the organisms (Figure 1a,b). The B. cinerea sRNA molecules showed a somewhat ragged distribution with two main peaks at 25 and 33 nt. Because it was reported that sRNAs derived from LTR retrotransposons can contribute to fungal pathogenesis by hijacking the host RNAi machinery (Weiberg et al., 2013; Weiberg & Jin, 2015), we further dissected the sRNA reads that were mapped to transposable elements (TEs) in the B. cinerea genome. The TE‐derived sRNA pool contained mainly 20–24 nt sRNA molecules with a peak at 22 nt (Figure 1a). Tomato sRNA sequences contained two sharp peaks, one at 24 nt and the other at 32 nt (Figure 1b). The peak at 32 nt consisted mainly of sequences representing half‐size tRNA molecules, cut near the anticodon loop. The 24 nt‐sRNAs are related to cis‐acting siRNAs (typically associated with RNA‐dependent DNA methylation) or natural antisense transcript‐derived siRNAs. The former is associated with transposon silencing and the latter with the regulation of stress‐response genes (Ghildiyal & Zamore, 2009). Further analyses only focused on sRNA reads with sizes 20–24 nt because these are considered important in gene silencing (Kamthan et al., 2015), either by transcriptional or posttranscriptional gene silencing (Sijen et al., 2001). The entire dataset of sRNAs from B. cinerea contained 27,918 unique sRNA molecules with lengths between 20 and 24 nt (Table S2). These sequences predominantly originated from the ribosomal RNA repeat region (13,258 sRNAs, 47.5%) and TE loci (9368 sRNAs, 33.5%). Other sources of sRNAs were tRNA loci, coding sequences as well as introns (Table S2). The entire dataset of B. cinerea sRNAs of 20–24 nt contains only 62 of the 73 sRNA sequences published by Weiberg et al. (2013), despite samples being sequenced at much greater read depths (Table S2). The 11 absent sRNA sequences do not map on the latest version of the B. cinerea genome (van Kan et al., 2017).
FIGURE 1.
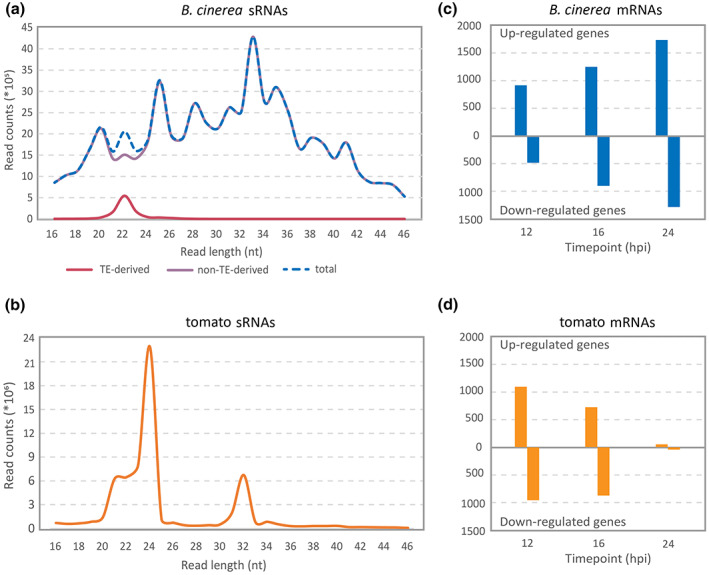
Small RNA (sRNA) profiles of tomato and Botrytis cinerea, and differential expression of plant and fungal mRNAs during their interaction. (a) Read length distribution of unique sRNA sequences in B. cinerea liquid cultures. The pink line represents the transposable elements (TE)‐derived B. cinerea sRNAs, the purple line shows the non‐TE‐derived B. cinerea sRNAs, and the blue‐dashed line represents the total B. cinerea sRNAs. (b) Read length distribution of unique sRNA sequences in the compiled tomato mock‐treated samples (orange line). The scale on the left in both (a) and (b) provides a read count for each read length of the samples. (c) Numbers of differentially expressed genes in B. cinerea at 12, 16, and 24 hours postinoculation (hpi). B. cinerea grown in liquid medium for 16 h was used as control for all three infection time points for the analysis of differentially expressed fungal genes (blue bars). (d) Numbers of differentially expressed genes in tomato. Mock‐treated leaves collected at 12, 16, and 24 hpi were used as the respective controls for B. cinerea‐inoculated leaves collected at 12, 16, and 24 hpi for the analysis of differentially expressed tomato genes (orange bars)
A total of 33 transposon regions in the B. cinerea genome produced >100,000 sRNA reads each, with two regions on Chr14 being predominant sources of sRNAs: the Gypsy‐type retrotransposon regions annotated as ms3003 (coordinates Chr14: 1,705,089–1,712,486) and ms3095‐3099 (a complex array of multiple elements, Chr14: 253,000–283,000) of which ms3095 and ms3097 are transcriptionally active. Each of these regions produced >500,000 sRNA reads and each contributed roughly 10% to the total sRNA read counts (Table S3) produced by TEs. Of the 27,918 unique sRNAs from B. cinerea with a length between 20 and 24 nt in the entire dataset, 5859 (21%) originated from these two regions. The total number of unique sRNA molecules produced by tomato with a length between 20 and 24 nt was 934,159 (Table S4), which predominantly originated from TE loci (573,727 sRNAs, 61%), annotated gene regions (324,294 sRNAs, 35%), and tRNA loci (2206 unique sRNAs, 0.2%). Due to the incomplete assembly and annotation of repeats in the tomato genome, these numbers are likely to be underestimates.
2.2. B. cinerea and tomato genes down‐regulated during interaction and potentially targeted by sRNAs from tomato and B. cinerea
After mapping mRNA reads on the fungal and tomato genomes, differential expression analyses were performed by comparing gene expression during B. cinerea infection of tomato with the respective controls, a B. cinerea liquid culture, and the mock‐inoculated tomato leaves. Differentially expressed genes of B. cinerea and tomato during the early interaction (12, 16, and 24 hpi) were listed for each time point (Figure 1c,d). Many B. cinerea genes were up‐ or down‐regulated during infection, as compared to growth in liquid culture, particularly at 24 hpi (>3000 genes, one quarter of the annotated genes) (Figure 1c). Among the most strongly up‐regulated genes of B. cinerea were genes encoding polygalacturonases and other Carbohydrate‐Active enZYmes (CAZymes), proteases and botcinic acid biosynthetic enzymes (Table S5). Tomato genes that were up‐ or down‐regulated during B. cinerea infection, as compared to the mock‐treated samples, were identified predominantly at 12 and 16 hpi (Figure 1d). Up‐regulated tomato genes included genes encoding chitinases, peroxidases, and ethylene‐responsive genes (Table S6). To analyse whether down‐regulation of tomato mRNAs could be mediated by B. cinerea sRNAs that participate in cross‐kingdom gene silencing, a target gene prediction was performed. Sequences of B. cinerea sRNAs (Table S2) were used as input to predict whether mRNAs of tomato could be targeted for silencing. Conversely, tomato sRNAs were used to predict target mRNAs in B. cinerea. Of the 27,918 unique sRNAs from B. cinerea with a length between 20 and 24 nt and coverage of at least 10 reads in the dataset, a quarter (7042 sRNAs) were predicted to potentially target 3185 distinct mRNAs in tomato. B. cinerea sRNAs with predicted target mRNAs in tomato originated from the ribosomal DNA repeat region on Chr4 (3279 sRNAs, 47%) and from TE loci on different chromosomes (2398 sRNAs, 34%) (Figure 2). Conversely, of 934,159 unique sRNAs molecules produced by tomato with a length between 20 and 24 nt, 114,011 had a predicted target mRNA in B. cinerea (11,434 distinct mRNAs, >97% of the fungal gene models).
FIGURE 2.
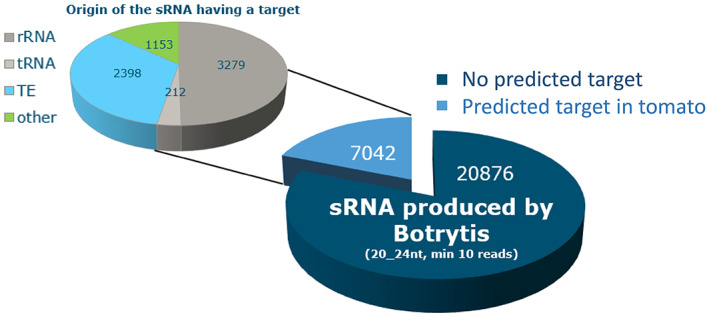
Number of unique small RNAs (sRNAs) produced by Botrytis cinerea with a length of 20–24 nucleotides. The genomic origin of sRNAs predicted to have a target in tomato is shown: ribosomal RNA (rRNA), transfer RNA (tRNA), transposable elements (TE), and other origins
A simulation was performed to evaluate the significance of the number of potential sRNA target transcripts. Three sets of 27,918 random sRNA sequences were generated with the same length distribution and GC content as the experimental B. cinerea sRNA dataset, and these random sequences were used as input for target prediction in tomato. The numbers of predicted target tomato transcripts for the three random sRNA sets were 6882, 6699, and 6798. Next, we generated a set of 934,159 random sRNA sequences with the same length distribution and GC content as the experimental tomato sRNA dataset, and used these random sequences as input for target site prediction in B. cinerea. The number of predicted target fungal transcripts was 11,331. These simulations indicate that the number of predicted target transcripts based on the biological samples was lower than or similar to the predicted targets based on randomly generated datasets.
Figure 3a illustrates relations between sRNAs originating from B. cinerea TE regions (fungal chromosomes in blue) and the genomic locations of their predicted target genes on tomato chromosomes (in orange). Predicted target mRNAs in tomato for each B. cinerea sRNA are listed in Table S7. Of the 3185 tomato genes predicted to be targeted by B. cinerea sRNAs, 163 indeed displayed significantly lower transcript levels during the course of infection (Table S8). Notably, 158 tomato genes that were predicted to be targeted by B. cinerea sRNAs also displayed significantly higher transcript levels (Table S8).
FIGURE 3.
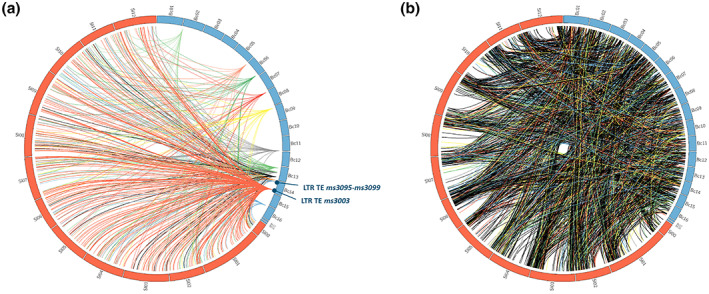
Genomic regions of small RNA (sRNA)‐producing loci in Botrytis cinerea with their predicted target genes in tomato (a) and of sRNA‐producing loci in Solanum lycopersicum (tomato) with their predicted target genes in B. cinerea (b). Blue boxes represent the 18 chromosomes of B. cinerea, whereas orange boxes represent the 12 chromosomes of tomato. B. cinerea chromosomes are oversized for illustration purposes. Only sRNAs produced by transposable element (TE) regions of B. cinerea (a) and of S. lycopersicum (b) are shown
To evaluate the significance of obtaining a certain number of potential target genes in tomato that indeed were down‐regulated during B. cinerea infection, a simulation was performed. From the entire set of tomato gene models, we selected random samples of 3185 genes (in 100 iterations) and recorded how many genes were down‐regulated or up‐regulated in the experimental dataset. The means of the number of up‐ and down‐regulated genes in these 100 iterations were 131 and 153, respectively. The experimental observation of 163 down‐regulated transcripts among 3185 predicted target genes did not significantly deviate from the number of down‐regulated transcripts that one would expect by chance among a random set of genes.
Most of the sRNAs produced by tomato originated from 215 TE regions (Table S9), with nine TEs producing more than 1 million sRNA read counts in the dataset. The total of 934,159 unique tomato sRNAs was predicted to target almost all 11,700 B. cinerea genes (Figure 3b; predicted targets of tomato sRNAs listed in Table S10). Most tomato TEs are located near the ends of chromosomes (Figure 3b). From the 114,011 tomato sRNAs that were predicted to have a target mRNA in B. cinerea, 70,189 were produced from TEs. The number of predicted B. cinerea target genes that displayed a significant down‐regulation in at least one of the infection time points studied (12, 16, and 24 hpi) as compared to the liquid culture was 1713, which is about 15% of the B. cinerea genes (Table S11).
2.3. Correlation between the down‐regulation of tomato mRNAs and levels of B. cinerea sRNAs that are predicted to target them
After analysing data from B. cinerea‐ and mock‐inoculated tomato leaves sampled at 12, 16, and 24 hpi, we aimed to validate the in silico prediction and establish more detailed sRNA–mRNA profiles during the early infection. New inoculations were performed and sampled at seven time points within the first 24 hpi for extraction of sRNA and mRNA. The expression levels of selected sRNAs and their matching target genes were quantified by reverse transcription‐quantitative PCR (RT‐qPCR). We selected 10 Bc‐sRNAs (sRNAs from B. cinerea) together with their predicted nine Sl‐mRNA targets (tomato transcripts) (Table S12) for molecular validation. The selection of sRNA–mRNA pairs was based on the following criteria: (i) both the sRNA and target mRNA showed sufficient reads in the data at all time points; (ii) the predicted target mRNA was significantly down‐regulated at one or more time point(s) compared to a previous time point; (iii) at most two unique sRNAs were predicted to target a single mRNA in tomato; and (iv) the target gene might participate in plant immune responses. Nine immune‐related genes in tomato were selected as candidate targets (Table S12), including three receptor‐like kinases (named LRR, RLK, and RLPK in this study), one mitogen‐activated protein kinase (MAPK3), a MAP kinase kinase kinase (MAPKKK56), a casein kinase (CK), and three transcription factors (WRKY23, WRKY46, and WRKY50).
Molecular quantification results from RT‐qPCR indicated correlations between most tested Bc‐sRNAs and their matching Sl‐mRNA targets (Figure 4). The only exception was the mRNA from the tomato CK gene, which displayed stable levels at all time points analysed (Figure S1). For the tomato genes analysed, transcript levels of LRR and RLPK decreased over time as infection progressed, RLK, MAPK3, MAPKKK56, WRKY23, and WRKY46 genes showed fluctuating expression profiles within 24 h after B. cinerea inoculation, and the mRNA level of WRKY50 strongly decreased at 16 and 20 hpi then recovered to a level similar to the mock control at 24 hpi (Figures 4a and S1). Despite various expression profiles of predicted target genes, two main correlations were observed between the sRNAs and their predicted target mRNAs. The first type was a synchronized down‐regulation of target mRNA with the high level of the corresponding sRNA in the other organism. Figure 4 shows the correlation between the tomato RLK transcript level and the Bc‐sRNA_RLK level at 10 and 20 hpi, and similarly for the MAPKKK56 transcript level and the Bc‐sRNA_MAPKKK56 level at 12 and 14 hpi. In the second type of correlation, suppression of mRNA level was slightly delayed as compared to the time when sRNA was highly produced. For example, mRNA levels of WRKY50 decreased between 12 and 20 hpi, while the expression level of Bc‐sRNA_WRKY50 was highest at 12 and 14 hpi (Figure 4a).
FIGURE 4.
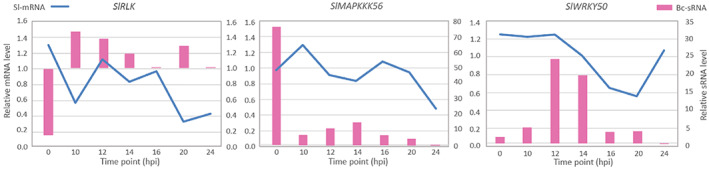
Correlations between production levels of small (RNAs) sRNAs and their predicted target mRNAs in Botrytis cinerea and tomato through seven sampling time points. Reverse transcription‐quantitative PCR results showing dynamics in the production levels of three chosen Bc‐sRNAs (pink bars) and the corresponding tomato transcripts (blue lines). The predicted target tomato genes are shown above each chart
2.4. Effects of deleting a retrotransposon in B. cinerea on virulence
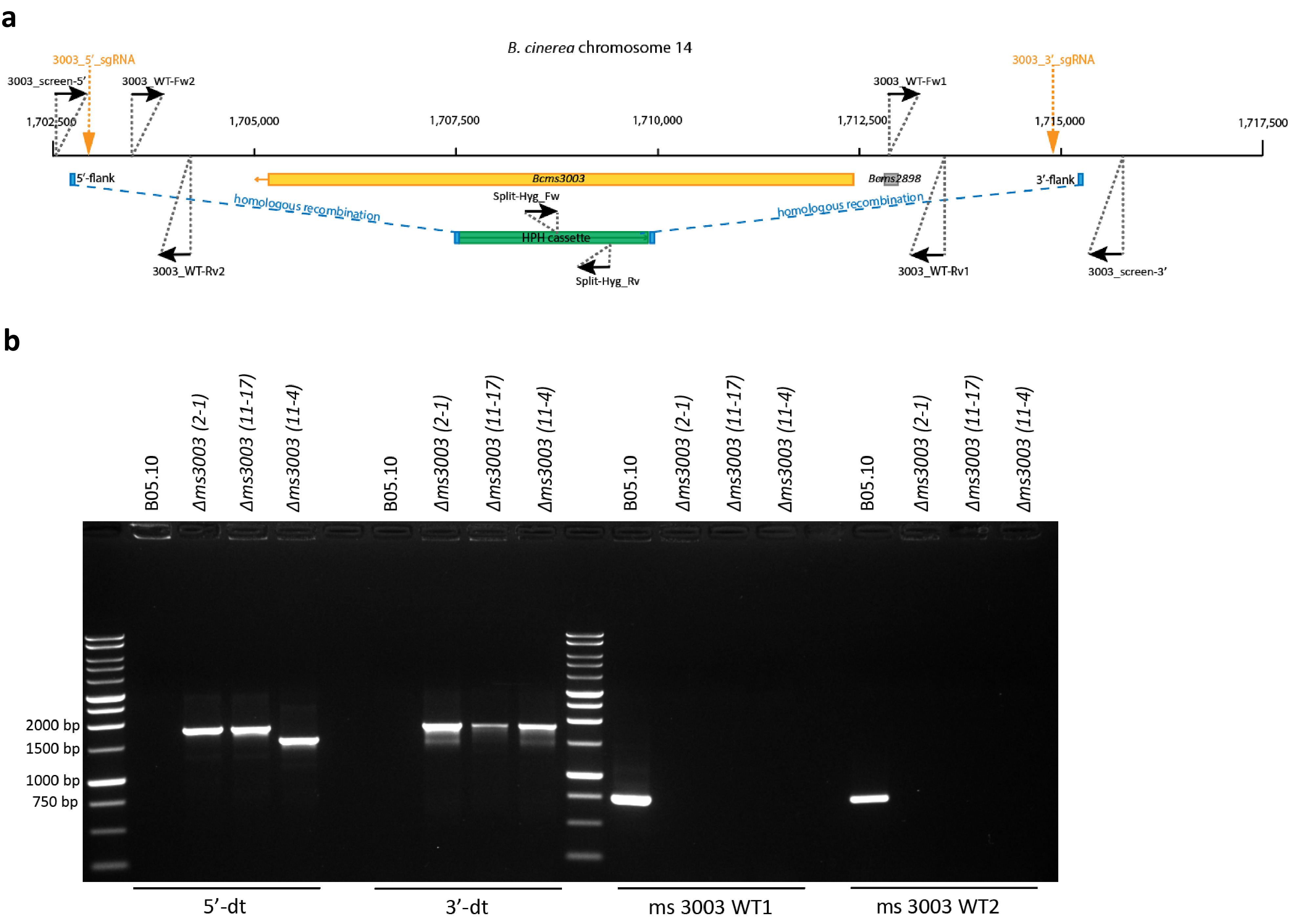
Approximately 10% of the total sRNAs produced by B. cinerea strain B05.10 originated from a retrotransposon region on Chr14 annotated as ms3003 (Figure 3a and Table S3). To verify whether sRNAs originating from ms3003 contribute to fungal virulence, the entire ms3003 region (c.12.5 kb) was deleted and virulence assays were performed with mutants. Three independent ∆ms3003 knockout transformants were obtained by a CRISPR‐Cas9 mediated system (Leisen et al., 2020) (Figure S2). Infection assays on tomato leaves indicated that the ∆ms3003 mutants caused lesions of equal sizes as the recipient B05.10 (Figure 5a,b). Furthermore, expression levels of three tomato genes, which were predicted targets of Bc‐sRNAs derived only from ms3003, were quantified by RT‐qPCR for six time points within the first 30 h after inoculation by B05.10 or ∆ms3003. As shown in Figure 5c, Sl05g014130, Sl06g074390, and Sl01g060030 displayed similar expression profiles in tomato leaves infected by B05.10 and ∆ms3003, although the levels of the corresponding Bc‐sRNAs targeting Sl05g014130 and Sl01g060030 seemed to be lower (with no statistical significance) in the ∆ms3003 mutant as compared to B05.10 (Figure 5c). We also attempted to delete a second TE region on Chr14 with a size of about 30 kb, designated ms3095‐3099 (Figure 3a), which has a complex tandem array of transposons and is also the source of about 10% of the total sRNA reads (Table S3). The deletion was partially successful: from multiple experiments, few transformants were obtained in which the 5′‐ or 3′‐end of the region was replaced by a donor template, while the other side was retained. Despite numerous attempts, we failed to obtain transformants lacking the entire ms3095‐3099 region.
FIGURE 5.
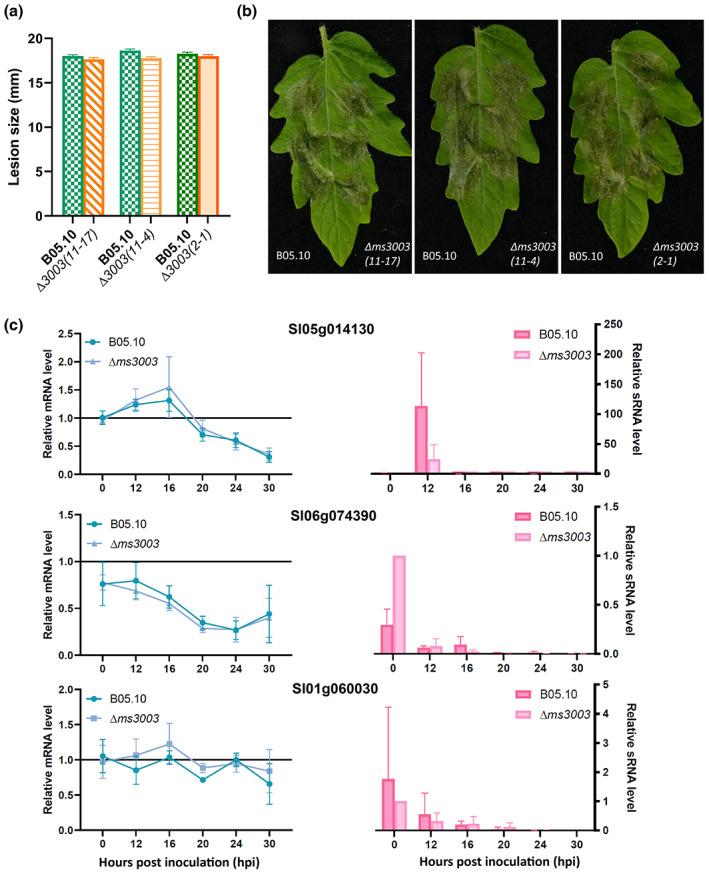
(a and b) The virulence of three ∆ms3003 independent mutants compared with the wild‐type B05.10 strain on tomato leaves, evaluated by lesion sizes in diameter (mm) at 3 days postinoculation (dpi). The measured data were plotted as a bar chart (a). Error bars represent standard error of 72 data points from three experiments; statistical analyses by Student's t test indicated no significant differences. Infected leaves were photographed at 3 dpi (b). The virulence assay was performed three times, each with similar results. (c) Transcript profiles of three tomato genes (predicted targets of Bc‐sRNAs derived from ms3003) during the infection of tomato leaves by B05.10 or a ∆ms3003 mutant. Error bars represent standard error from four biological replicates and two biological replicates in the plot of relative mRNA level and small (sRNA) level, respectively
2.5. Deleting B. cinerea Bcdcl1 and Bcdcl2 eliminated transposon‐derived sRNAs, but did not affect virulence
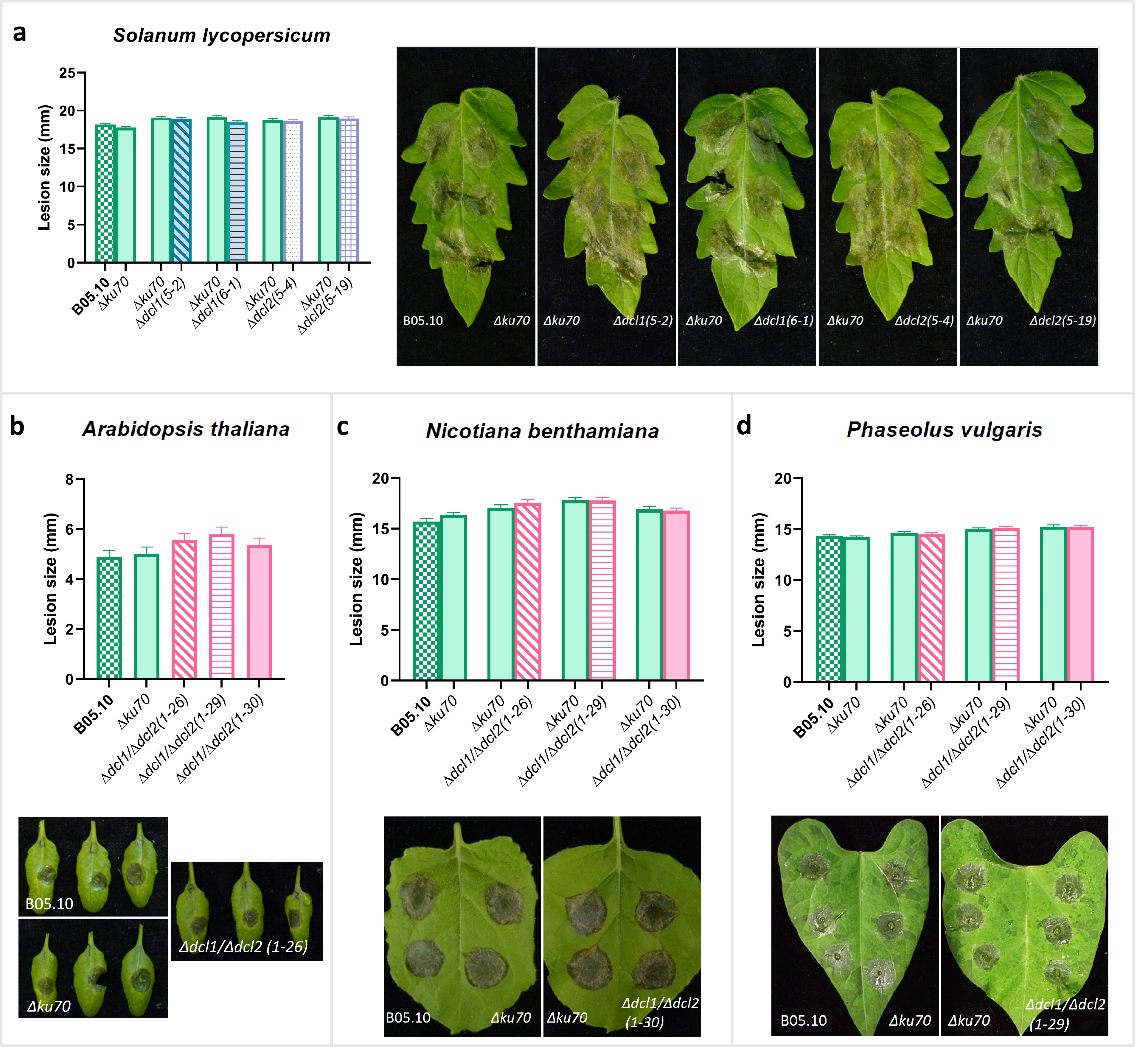
Generation of sRNAs in fungi requires the action of Dicer‐like proteins to cleave double‐stranded replication intermediates generated from retrotransposon transcripts into small interfering RNAs that can mediate gene silencing. Weiberg et al. (2013) reported that a B. cinerea double mutant that was defective in both Bcdcl1 and Bcdcl2 was reduced in virulence, presumably due to its inability to generate siRNAs with a size of 20–26 nt (Wang et al., 2016). However, the ∆Bcdcl1/∆Bcdcl2double mutant in the studies of Weiberg et al. (2013) and Wang et al. (2016) displayed reduced mycelial growth and abnormal sporulation. Such phenotypes were not necessarily expected for such mutants, and we suspected the occurrence of additional pleiotropic mutations in this double mutant. Thus, we generated novel single and double mutants in the Bcdcl1 and Bcdcl2 genes. Initial attempts to generate knockout mutants in strain B05.10 as recipient resulted in the generation of several independent ∆Bcdcl2 mutants. However, the ∆Bcdcl1 mutant could never be obtained in the homokaryotic state, despite applying a few rounds of single‐spore isolation. To overcome this issue, we chose as recipient mutant strain ∆ku70, in which nonhomologous end‐joining repair is disturbed (Choquer et al., 2008; Pinedo et al., 2008), to facilitate homokaryotic deletion of Bcdcl1 and Bcdcl2 genes. At least three independent knockout strains were obtained for each single or double mutant (Figure S3). To verify that the ∆Bcdcl1/∆Bcdcl2 double mutants were indeed defective in generating siRNAs, we sequenced the sRNA pool of recipient strain ∆ku70 and three independent double mutants, grown in liquid culture. Indeed ∆Bcdcl1/∆Bcdcl2 double mutants showed a reduced amount of sRNAs, specifically in the range between 20 and 24 nt (Figure 6a). The double mutants still contained a residual amount of sRNAs (Figure 6a). Mapping of the reads revealed that none of the unique sRNA sequences in the ∆Bcdcl1/∆Bcdcl2 double mutants was derived from TE regions (at a threshold of 10 reads) while in the recipient strain ∆ku70, 1409 of the 5996 unique sRNA sequences (23%) were derived from TE regions (Figure 6b). Two strains of each single mutant and three strains of the double mutant were tested for virulence and in vitro growth on two solid media. All ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 mutants displayed similar radial growth and developmental behaviour (sporulation and sclerotia formation) as the ∆ku70 recipient and the wild‐type strain B05.10 (Figure S4). In multiple biological repetitions, we never observed any significant reduction in lesion size between the wild type and mutants on tomato (Figure 6c), A. thaliana, Nicotiana benthamiana or Phaseolus vulgaris (Figure S5) despite testing multiple independent mutants for each gene, as well as multiple independent double mutants. These results indicated that TE‐derived 20–24 nt sRNAs from B. cinerea played an undetectable role in the developmental behaviour and aggressiveness on host plants of the fungus.
FIGURE 6.
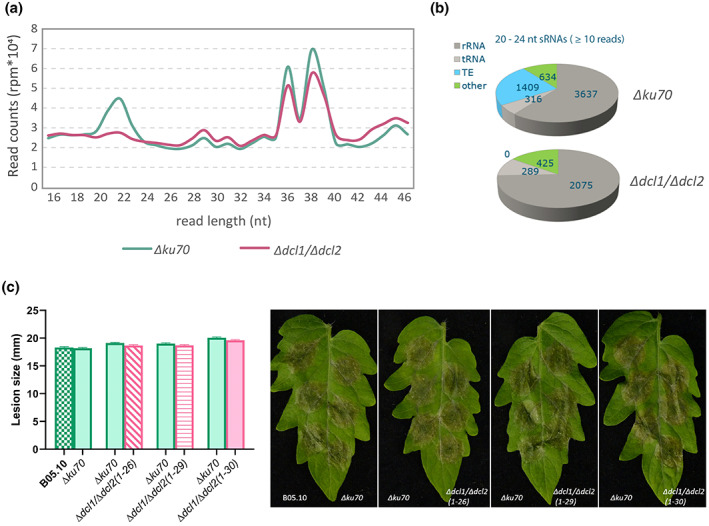
(a) Profile of normalized read counts (rpm) for total small RNA (sRNA) (length 16–46 nucleotides [nt]) produced in ∆ku70 and ∆Bcdcl1/∆Bcdcl2 mutants. (b) The numbers of unique sRNAs produced by Botrytis cinerea∆ku70 and ∆Bcdcl1/∆Bcdcl2 mutants with a length of 20–24 nt and their origin in the B. cinerea genome: ribosomal RNA (rRNA), transfer RNA (tRNA), transposable elements (TE), and other origins. The pie chart shows the results of three biological replicates. (c) The virulence of ∆Bcdcl1/∆Bcdcl2 compared with ∆ku70 and ∆ku70 with wild‐type B05.10 strain on tomato leaves. The diameter of lesions was measured at 3 days postinoculation (dpi), represented as a bar chart on the left side of panel (c). Error bars represent standard error of 72 inoculations from three independent experiments, and there were no significant differences between each comparison (statistical analyses were performed by Student's t test). Symptoms were photographed at 3 dpi, shown on the right side of panel (c)
3. DISCUSSION
Both plants and fungi produce various types of sRNAs through a number of pathways, and these sRNAs may have important regulatory functions in an organism by either mediating DNA methylation or RNA interference that result in transcriptional or posttranscriptional gene silencing, respectively. The studies by Weiberg et al. (2013) and Wang et al. (2017) reported that sRNAs can not only modulate transcript levels in the organism producing the sRNAs (Chang et al., 2012; Lax et al., 2020), but also reduce transcript levels in a different organism during a pathogenic interaction. On the one hand, sRNAs produced by B. cinerea could affect the transcript levels of A. thaliana genes involved in plant immunity (Weiberg et al., 2013), while on the other hand, sRNAs from a host plant could affect the transcript levels of fungal genes participating in the infection process (Cai et al., 2018; Wang et al., 2016). One crucial step in this RNA warfare obviously is the translocation of the sRNAs from the producing organism (either the plant or the fungus) to the opponent in the interaction. Extracellular vesicles were reported to be key players in the delivery of the sRNAs into the cells of the opponent. Studies by Cai et al. (2018) and He et al. (2021) revealed that not all sRNA molecules produced are effectively loaded into vesicles and thereby provided indications for the selectivity of sRNA translocation, although the mechanism of selectivity was not understood. Rutter and Innes (2020), however, have raised concerns about the conclusions of some recent studies on plant extracellular vesicles because the experimental approaches may insufficiently distinguish bona fide vesicle cargo from merely co‐purifying contaminants. Studies using a complementary approach (Wang et al., 2016) demonstrated that synthetic sRNAs that are topically applied to plant surfaces can be taken up in B. cinerea and lower the expression of target genes in the invading fungus, thereby reducing disease development.
The studies by Weiberg et al. (2013) and Wang et al. (2017) had a major shortcoming that we considered important from a biological perspective of the plant–pathogen interaction. The tissue samples analysed in these studies were taken from 24 hpi onwards, at a moment when the interaction between B. cinerea and its host is already well advanced and the fate of the interaction is largely decided (Veloso & van Kan, 2018). Under commonly used laboratory conditions for inoculation, B. cinerea spores germinate within 3–4 hpi and infection structures are formed around 6–8 hpi. Fungal penetration is completed around 10–12 hpi and first signs of cell death become evident at 16 hpi. Therefore, sampling tissues at 24–72 hpi provides a snapshot of sRNA and mRNA profiles that does not reflect the decisive moments in this plant–fungus interaction. This consideration stimulated us to sample at earlier time points to explore the sRNA and mRNA profiles at three decisive moments: 12 hpi (following penetration, during the early biotrophic phase of the interaction), 16 hpi (around the transition from the biotrophic to the necrotrophic phase), and 24 hpi (when the necrotic lesion in a host is established and the fungus expands beyond the inoculation spot). The sRNA sequence data that we obtained were different from the data obtained by Weiberg et al. (2013), but difficult to compare. Not only were time points different, the read depth was also much deeper and the reads could be mapped to more completely assembled and better annotated versions of the B. cinerea and tomato genomes (Hosmani et al., 2019; van Kan et al., 2017). The sRNA dataset from B. cinerea contained almost 28,000 unique sRNA sequences with a length of 20–24 nt, yet only 62 sRNAs from the 73 sequences described by Weiberg et al. (2013) were present in our data with a coverage of at least four reads. The 11 missing sRNAs of Weiberg et al. (2013) did not map to the recent version of the B. cinerea genome or were not detected in our dataset. The 28,000 B. cinerea sRNAs were predicted to target >3000 genes in the tomato genome, including hundreds of genes that could potentially participate in plant defence or immunity. Yet only 163 of the predicted tomato target mRNAs indeed showed significant down‐regulation in the same tissue samples, while 158 potential tomato target mRNAs showed up‐regulation.
We identified a set of genes related to plant immunity that were predicted to be targeted by B. cinerea sRNAs and did indeed display a reduced mRNA level during infection, as compared to mock controls. A detailed RT‐qPCR analysis was performed with sampling at seven time points between 0 and 24 hpi to establish an association between the level of the Bc‐sRNA and its target Sl‐mRNA. The results in Figures 4 and S1 show that in some cases there was an inverse correlation between the level of a Bc‐sRNA and its predicted target in tomato, which might indicate that cross‐kingdom RNAi was indeed operative to achieve this down‐regulation. However, whether the production of a unique Bc‐sRNA that is predicted to target a Sl‐mRNA actually causes the down‐regulation of its target is difficult to establish. First, many sRNAs are derived from transposons, some with multiple closely related, but nonidentical, copies and it is genetically impossible to dissect the function of individual sRNAs. Second, many of the Bc‐sRNAs are predicted to target multiple distinct Sl‐mRNAs. Conversely, many Sl‐mRNAs are predicted to be targeted by multiple distinct Bc‐sRNA molecules from various genomic origins. Whether a predicted sRNA–mRNA interaction actually occurs in the plant–fungus interaction depends on multiple factors: the concentration of the sRNA within the (plant or fungal) tissue, the effective translocation of sRNA between fungus and plant, through vesicles or otherwise, the efficacy of sRNA–mRNA hybrid formation, dependent on the free energy, and the efficacy of the RNAi process that destroys the target mRNA. All these processes will have an impact on the outcome of the plant–fungus interaction only if they occur sufficiently rapidly and efficiently, and only if the reduction of mRNA results in a notable reduction of the encoded protein that has a meaningful impact on the interaction. The reduction of transcript levels by, for example, 50% does not necessarily result in an equivalent reduction of the protein. Even if the protein levels were reduced to a similar extent as its transcript, this would only lead to physiological problems if the protein catalyses a rate‐limiting step in the biological process of interest. Furthermore, the overall consequences of differential expression of plant immunity genes, mediated by fungal sRNAs, are hard to predict and do not necessarily lead to increased susceptibility of the infected plant.
During the B. cinerea–tomato interaction, numerous fungal and plant genes were down‐regulated over the course of the infection process (Figure 1c,d). Based on the observations of Weiberg et al. (2013) and Wang et al. (2016), and the identification of about 95,000 potential sRNA–mRNA interactions in our dataset (7042 Bc sRNA–Sl mRNA interactions and 88,196 Sl sRNA–Bc mRNA interactions), it was tempting to consider that many changes in transcript levels were indeed caused by cross‐kingdom RNAi. However, simulations indicated that similar, or even higher, numbers of predicted sRNA–mRNA interactions occur with randomly generated sRNA datasets, and the 163 down‐regulated tomato mRNAs in the experimental dataset did not deviate from the number of down‐regulated transcripts that one may expect in a random subset of 3185 tomato genes. Other explanations for down‐regulation, besides cross‐kingdom RNA interference, should therefore be considered. B. cinerea undergoes developmental transitions during early phases of infection that are associated with transcriptional reprogramming. To penetrate the host tissue, fungal germ tubes develop infection structures, with their specific developmental and transcriptional programme (Choquer et al., 2021; Leroch et al., 2013). Once infection structures have completed host surface penetration (10–14 hpi), they are redundant and the fungus switches to intercellular hyphal growth while suppressing host cell death (Veloso & van Kan, 2018). From about 16 hpi, host cells are triggered to undergo programmed cell death and the fungus is exposed to oxidative stress in dying host tissue (Choquer et al., 2007; Torres et al., 2006). On the plant side, tomato genes that are down‐regulated on B. cinerea inoculation could be regulated by numerous physiological processes that are associated with defence responses and/or disease development, rather than by fungal sRNAs.
The majority of sRNAs produced by B. cinerea is derived from the rRNA repeat and a number of very active TEs of the Gypsy family. Two regions in chromosome 14 (ms3003 and ms3095–ms3097) stood out as producers of massive amounts of sRNAs, with each of these regions producing c.10% of the total read counts (Figure 3a and Table S3). These elements not only produced many reads, but also a great diversity of sRNAs, from a range of lengths and often overlapping in sequence. Production of such molecules is a random “disposal process” to eradicate the replication intermediate and thereby modulate the activity of the retrotransposon. The fact that sRNAs are produced that may, in addition, silence transcripts in a host plant should thus be considered accidental and a “collateral benefit”. The sheer number and diversity of sRNAs will inevitably have numerous (predicted) target genes within any given host plant, just by coincidence. If silencing of host immunity confers advantage to the fungus, B. cinerea genotypes that contain active retrotransposon copies are likely to be more successful than genotypes devoid of active transposons. Evolutionary selection will not operate at specific sequence level, but on the possession of such (active) elements. Studies on genetic diversity in the B. cinerea population revealed that a subset of isolates were considered to lack active transposons and were referred to as “vacuma” isolates (Giraud et al., 1999; Martinez et al., 2003; Samuel et al., 2012). Such isolates were often reported to be less virulent on particular hosts (Martinez et al., 2003), but it was difficult to experimentally validate this correlation because of differences in the origin and genetic backgrounds of isolates. In a study by Porquier et al. (2021), a vacuma isolate was transformed with an LTR‐type TE and became slightly but significantly more virulent than the recipient strain. We applied an inverse strategy and eliminated one TE region (ms3003, c.12.5 kb) that was responsible for the generation of ~c.10% of the sRNA population from the genome of the highly virulent strain B05.10 (Figure S2). This is to our knowledge the first successful targeted deletion of a TE region from the B. cinerea genome and perhaps from any other fungus. We also attempted to delete a second TE region, designated ms3095–3099, with a size of c.30 kb, which also generates c.10% of the sRNA population. This locus has a complex tandem array of multiple transposons in direct or inverted repeat orientation, and we attempted to delete it by using CRISPR guide RNAs that would cut in the unique sequences flanking the TE region, and transforming with a donor template that would merge the two flanking regions by a selection marker cassette. In multiple experiments a few transformants were obtained in which either the 5′ part or the 3′ part of the region was recombined with the donor template, but the recombination target site at the other end was retained. These mutants were not further analysed as they did not contain the complete desired deletion. Despite the successful knockout of a c.12.5 kb TE region ms3003, the virulence of the mutants was not notably reduced (Figure 5). This was probably due to the presence of additional TEs, including the ms3095–3099 region on Chr14, that compensate for the loss of the ms3003 region. An alternative explanation would be that the cross‐kingdom RNAi as such does not have a significant impact on the infection process to affect the virulence of mutants. This hypothesis was tested by generating double mutants that were defective in both Dicer‐like genes Bcdcl1 and Bcdcl2. The study by Weiberg et al. (2013) reported an almost absolute loss of virulence of such a mutant, but their mutant displayed serious growth retardation and abnormal sporulation. By contrast, the ∆Bcdcl1/∆Bcdcl2 mutants that we generated independently displayed a >99% reduction in TE‐derived sRNA production (of 20–24 nt length molecules), while the rDNA‐derived sRNAs were largely unaffected by deletion of Bcdcl1 and Bcdcl2. Three independent ∆Bcdcl1/∆Bcdcl2 mutants did not show any pleiotropic phenotypes as reported by Weiberg et al. (2013), nor did they show reduced virulence on any of four distinct host plants (Figure 6c). This indicates that, in our view, the role of cross‐kingdom RNAi at the level of single sRNA–mRNA interactions appears too small to be detected in the infection on leaves of several plant species under controlled laboratory conditions. These results are difficult to reconcile with reports that plant protection from B. cinerea infection can be achieved by applying onto plant surfaces sRNAs that can silence B. cinerea Dicer‐like genes as reported by Wang et al. (2016). This study made use of single‐ and double‐stranded RNAs that target the B. cinerea Dicer‐like genes, with the reasoning that silencing of Bcdcl1 and Bcdcl2 would prevent the fungus from producing sRNAs that can suppress plant immune responses and thereby trigger resistance. Our observation that ∆Bcdcl1/∆Bcdcl2 double mutants were equally virulent as the wild‐type strain (Figures 6c and S5), despite depletion of an immense proportion of its sRNAs (Figure 6a,b), suggests that the protection conferred by sRNAs that target B. cinerea Dicer‐like genes might operate through a different mechanism than merely the disruption of the fungal sRNA producing capacity. More recent publications have reported the topical application of a plethora of distinct sRNAs that confer resistance to B. cinerea (McLoughlin et al., 2018; Nerva et al., 2020; Spada et al., 2021). In these studies, the sRNAs were designed to target fungal genes with a role in growth (lanosterol C‐14‐α demethylase, chitin synthase, elongation factor EF2) or signal transduction required for virulence (MAP kinase BMP3). A very extensive study on Sclerotinia sclerotiorum (McLoughlin et al., 2018; Nerva et al., 2020; Spada et al., 2021) showed that 20 out of 59 sRNAs (targeting a range of physiological functions) applied on Brassica napus leaves could reduce S. sclerotiorum lesion development. Subsequent tests with sRNAs targeting the orthologs of five of these 20 genes from B. cinerea also conferred reduction of B. cinerea lesion development (McLoughlin et al., 2018; Nerva et al., 2020; Spada et al., 2021). Technically, the topical application of sRNAs to confer plant protection from disease falls under the definition of spray‐induced gene silencing (SIGS) (Wang & Jin, 2017) and offers an attractive perspective for future crop protection strategies. However, the success of this method does not necessarily provide evidence for the importance of natural cross‐kingdom RNA interference in the B. cinerea–host interaction as earlier proposed by Weiberg et al. (2013).
4. EXPERIMENTAL PROCEDURES
4.1. Fungal strains, plant material, and growth conditions
The B. cinerea strains used in this study (Table 1) were grown and spores collected as described in File S1. Tomato (S. lycopersicum 'Moneymaker') and Nicotiana benthamiana were grown in a greenhouse at 20°C. A. thaliana and French bean (P. vulgaris) were grown in a climate chamber at 21 and 19°C day and night temperatures, respectively. The photoperiod was 12 h of light per day and the chamber was kept at 70% relative humidity.
TABLE 1.
Botrytis cinerea strains used in this study
| B. cinerea strain | Description | Origin/reference |
|---|---|---|
| B05.10 | Wild‐type B. cinerea | van Kan et al. (2017) |
| ∆ms3003 (#2–1, #11–4, #11–17) | Bcms3003 knockout mutant generated from B05.10 background, hygromycin resistant | This study |
| ∆ku70 | Bcku70 knockout mutant generated from B05.10 background, hygromycin resistant | Choquer et al. (2008) |
| ∆dcl1 (#5–2, #6–1) | Bcdcl1 knockout mutant generated from ∆ku70 background, hygromycin and fenhexamid resistant | This study |
| ∆dcl2 (#5–4, #5–19) | Bcdcl2 knockout mutant generated from ∆ku70 background, hygromycin and nourseothricin resistant | This study |
| ∆dcl1/∆dcl2 (#1–26, #1–29, #1–30) | Bcdcl1 and Bcdcl2 double knockout mutant generated from ∆ku70 background, hygromycin, fenhexamid, and nourseothricin resistant | This study |
4.2. Tomato inoculations with B. cinerea
B. cinerea conidia were diluted in potato dextrose broth (PDB, 12 g/L) medium to 1000 conidia/μl and inoculated on detached tomato leaves essentially as described by Zhang and van Kan (2013). Details on the inoculation and sampling design are provided in File S1.
4.3. RNA extractions
Fungal mycelium or tomato leaf samples were frozen in liquid nitrogen and used for extraction of sRNA using the mirPremier microRNA Isolation Kit (Sigma‐Aldrich) while the mRNA fraction was isolated using the SV Total RNA Isolation System (Promega).
4.4. Generation and analyses of the RNA sequencing dataset
Single‐end Illumina sequencing was applied to all sRNA and mRNA samples by Vertis Biotechnologie AG (Martinsried, Germany) on a strand‐specific library with a read length of 75 nt. Sequence processing and bioinformatic analyses of data are described in detail in File S1.
4.5. RT‐qPCR quantification of mRNA and sRNA levels
Reverse transcription from mRNA was done using Superscript III reverse transcriptase (Invitrogen). For reverse transcription of sRNA, the qScript microRNA cDNA Synthesis kit (Quanta Bioscience) was used. Twenty nanograms of each cDNA sample was input for performing qPCR using SensiMix SYBR Hi‐ROX Kit (Bioline). The primer combinations shown in Table S13 were used to quantify levels of mRNAs and sRNAs. An actin gene from tomato was used to normalize plant mRNA levels, and the relative mRNA level of each time point was calculated by comparing with the mock‐inoculated sample at the same time point. An sRNA derived from B. cinerea 28S rRNA was used to normalize fungal sRNA levels. The threshold cycle (C t) values were determined by Bio‐Rad CFX Manager 3.1 and fold‐changes calculated using the ΔΔC t method (Dorak, 2007).
4.6. B. cinerea transformation by CRISPR‐Cas9 mediated approach
The B. cinerea mutant strains used in this study were generated by CRISPR‐Cas9 mediated transformation, as described by Leisen et al. (2020), with the modifications specified in File S1.
Supporting information
Figure S1 Correlations between production levels of Bc‐sRNAs and their predicted target mRNAs in tomato through seven sampling time points. Reverse transcription‐quantitative PCR results showing dynamics in the production levels of six chosen Bc‐sRNAs (pink bars) and the corresponding tomato transcripts (blue lines). The predicted target tomato genes are shown above each chart
{kind=link}
Figure S2 (a) Schematic representation of the generation of ∆ms3003 transformants via CRISPR‐Cas9‐mediated transformation with two sgRNAs and the primers used in genotyping the transformants. (b) Genotyping of ∆ms3003 transformants by PCR, representing the target region, was replaced by the donor template via homologous recombination on both 5′‐ and 3′‐flanks. The same PCRs were performed with wild‐type B05.10 as the nontransformed control
{kind=link}
Figure S3 (a) Schematic representation of the generation and characterization of ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 transformants. (b) Genotyping of ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 transformants by PCR with primers shown in (a) and File S1. For the Bcdcl1‐check reaction, a band of 6.7 kb in B05.10 and ∆ku70 indicates the wild‐type Bcdcl1 gene and a band of 2.7 kb in ∆Bcdcl1/∆Bcdcl2 mutants as the replacement of this gene by the donor template. Similarly, the Bcdcl2‐check resulted in a product of 5.6 kb for B05.10 and ∆ku70 and one of 2.0 kb for ∆Bcdcl1/∆Bcdcl2 mutants. For the Bcdcl1‐WT reaction, only the B05.10, ∆ku70, and ∆Bcdcl2 strains showed amplicons of the Bcdcl1 gene, while the ∆Bcdcl1 and ∆Bcdcl1/∆Bcdcl2 strains did not. Similarly, the Bcdcl2‐reaction could only amplify the Bcdcl2 fragment in the B05.10, ∆ku70, and ∆Bcdcl1 strains, but not in the ∆Bcdcl2 and ∆Bcdcl1/∆Bcdcl2 strains
{kind=link}
Figure S4 In vitro mycelial growth, conidia formation, and sclerotial development of B05.10 wild‐type (WT), ∆ku70, ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 mutant strains on MEA (a) and PDA (b) plates. The line charts below the photos represent the mycelial growth rate of B05.10 WT, ∆ku70, ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 mutant strains on MEA (a) and PDA (b) plates for 3 and 4 consecutive days, respectively
{kind=link}
Figure S5 (a) Virulence assays of ∆Bcdcl1 and ∆Bcdcl2 single mutants compared with ∆ku70, and ∆ku70 compared with B05.10, which were performed with tomato leaves. (b–d) Virulence assays of ∆Bcdcl1/∆Bcdcl2 double mutant strains compared with ∆ku70, and ∆ku70 compared with B05.10. The assays were performed with Arabidopsis thaliana (b), Nicotiana benthamiana (c), and Phaseolus vulgaris (d), represented as bar charts and photos. Error bars in the charts represent standard errors of 72, 72, and 120 data points from three experiments for A. thaliana, N. benthamiana, and P. vulgaris, respectively. No significant reduction in the virulence of ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 compared with ∆ku70, and the same for ∆ku70 compared with B05.10, statistically analysed by Student’s t test
{kind=link}
File S1 More detailed description of the experimental procedures
Table S1 Sequence read depth of the mRNA and small RNA samples
Table S2 Sheet 1: sequences of 27,918 unique small RNAs (sRNAs) from Botrytis cinerea with length between 20 and 24 nucleotides, and their genomic origin: chromosome coordinates, ribosomal RNA repeat, transposable element loci, tRNA loci, gene regions. Sheet 2: correspondence of 73 sRNAs described by Weiberg et al. (2013) to our data
Table S3 Transposable elements (TEs) in Botrytis cinerea as sources of small RNAs. The list provides the genomic location (chromosome, start and end coordinates), the TE identifier from REPET annotation, the read counts for the entire repeat region as well as the percentage of the total read counts. Only repeat elements that generated at least 100 read counts are included
Table S4 Sequences of 934,154 unique small RNAs from tomato with a length between 20 and 24 nucleotides, and their genomic origin: chromosome coordinates, gene region, transposable element loci
Table S5 Differentially expressed genes of Botrytis cinerea during tomato leaf infection at different hours postinoculation (hpi) as compared to in vitro liquid culture. Each sheet provides the gene ID, the log2FC, the adjusted p value and a gene description. Sheet 1: expression at 12 hpi; sheet 2: expression at 16 hpi; sheet 3: expression at 24 hpi
Table S6 Differentially expressed genes of tomato during Botrytis cinerea infection at different hours postinoculation (hpi) as compared to mock‐treated leaves at the same time point. Each sheet provides the gene ID, the log2FC, the adjusted p value and a gene description. Sheet 1: expression at 12 hpi; sheet 2: expression at 16 hpi; sheet 3: expression at 24 hpi
Table S7 Predicted target mRNAs in tomato for each of the 7042 Botrytis cinerea small RNAs (sRNAs). The sheet provides the sRNA sequence, its coordinates on the B. cinerea genome, the transposable element (TE) origin, the gene ID of the predicted target gene, and a description of this gene
Table S8 Tomato genes predicted to be targeted by Botrytis cinerea small RNAs that display significant differential expression during B. cinerea infection, as compared to mock‐inoculated leaves. Sheet contains both down‐regulated and up‐regulated genes
Table S9 Transposable elements in tomato as sources of small RNAs. The list provides the genomic location (chromosome, start and end coordinates), the transposable element (TE) identifier from REPET annotation, the read counts for the entire repeat region as well as the percentage of the total read counts. Only repeat elements that generated at least 30,000 read counts are included
Table S10 Predicted target mRNAs in Botrytis cinerea for each of 114,011 tomato small RNAs (sRNAs). The sheet provides the sRNA sequences, their coordinates on the tomato genome, the gene or transposable element (TE) origin, and the gene ID of the predicted target genes in B. cinerea
Table S11 Botrytis cinerea genes predicted to be targeted by tomato small RNAs that display significant down‐regulation during infection, as compared to liquid culture controls
Table S12 Target genes from tomato and Botrytis cinerea selected for detailed reverse transcription‐quantitative PCR validation. The table contains the functional annotation of target genes, gene names used in this study, the gene ID (in tomato or B. cinerea), and small RNA (from B. cinerea or tomato) that is predicted to target the gene
Table S13 All primers used in this study
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the assistance of Henriek Beenen in RNA isolations. The research of S.Q. was supported by a scholarship from the China Scholarship Council.
Qin, S. , Veloso, J. , Baak, M. , Boogmans, B. , Bosman, T. & Puccetti, G. et al. (2023) Molecular characterization reveals no functional evidence for naturally occurring cross‐kingdom RNA interference in the early stages of Botrytis cinerea–tomato interaction. Molecular Plant Pathology, 24, 3–15. Available from: 10.1111/mpp.13269
Si Qin and Javier Veloso contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data of this project have been deposited in the NCBI database under Bioproject at www.ncbi.nlm.nih.gov/bioproject/ with accession number PRJNA496584. Raw sequence reads are deposited in the Sequence Read Archive at www.ncbi.nlm.nih.gov/sra/ project SRP166089 under accession numbers SRX4902781‐SRX4902781 and SRX4902791‐SRX4902800 (sRNAs) SRX4902771‐SRX4902780 and SRX4902783‐SRX4902790 (mRNAs).
REFERENCES
- Baulcombe, D. (2004) RNA silencing in plants. Nature, 431, 356–363. [DOI] [PubMed] [Google Scholar]
- Baum, J.A. , Bogaert, T. , Clinton, W. , Heck, G.R. , Feldmann, P. , Ilagan, O. et al. (2007) Control of coleopteran insect pests through RNA interference. Nature Biotechnology, 25, 1322–1326. [DOI] [PubMed] [Google Scholar]
- Cai, Q. , Qiao, L. , Wang, M. , He, B. , Lin, F. , Palmquist, J. et al. (2018) Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science, 360, 1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaloner, T. , van Kan, J.A.L. & Grant‐Downton, R.T. (2016) RNA “information warfare” in pathogenic and mutualistic interactions. Trends in Plant Science, 21, 738–748. [DOI] [PubMed] [Google Scholar]
- Chang, S.S. , Zhang, Z. & Liu, Y. (2012) RNA interference pathways in fungi: mechanisms and functions. Annual Review of Microbiology, 66, 305–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquer, M. , Fournier, E. , Kunz, C. , Levis, C. , Pradier, J.‐M. , Simon, A. et al. (2007) Botrytis cinerea virulence factors: new insights into a necrotrophic and polyphageous pathogen. FEMS Microbiology Letters, 277, 1–10. [DOI] [PubMed] [Google Scholar]
- Choquer, M. , Robin, G. , Le Pêcheur, P. , Giraud, C. , Levis, C. & Viaud, M. (2008) Ku70 or Ku80 deficiencies in the fungus Botrytis cinerea facilitate targeting of genes that are hard to knock out in a wild‐type context. FEMS Microbiology Letters, 289, 225–232. [DOI] [PubMed] [Google Scholar]
- Choquer, M. , Rascle, C. , Gonçalves, I.R. , de Vallée, A. , Ribot, C. , Loisel, E. et al. (2021) The infection cushion of Botrytis cinerea: a fungal “weapon” of plant‐biomass destruction. Environmental Microbiology, 23, 2293–2314. [DOI] [PubMed] [Google Scholar]
- Dorak, M.T. (2006) Relative quantification. In: Real‐time PCR. London: Taylor & Francis, pp. 89–108. [Google Scholar]
- Dubey, H. , Kiran, K. , Jaswal, R. , Jain, P. , Kayastha, A.M. , Bhardwaj, S.C. et al. (2019) Discovery and profiling of small RNAs from Puccinia triticina by deep sequencing and identification of their potential targets in wheat. Functional & Integrative Genomics, 19, 391–407. [DOI] [PubMed] [Google Scholar]
- Dunker, F. , Trutzenberg, A. , Rothenpieler, J.S. , Kuhn, S. , Pröls, R. , Schreiber, T. et al. (2020) Oomycete small RNAs bind to the plant RNA‐induced silencing complex for virulence. eLife, 9, e56096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillinger, S. & Elad, Y. (2016) Botrytis – the fungus, the pathogen and its management in agricultural systems. Cham: Springer, pp. 1–486. [Google Scholar]
- Ghildiyal, M. & Zamore, P.D. (2009) Small silencing RNAs: an expanding universe. Nature Reviews Genetics, 10, 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud, T. , Fortini, D. , Levis, C. , Lamarque, C. , Leroux, P. , LoBuglio, K. et al. (1999) Two sibling species of the Botrytis cinerea complex, transposa and vacuma, are found in sympatry on numerous host plants. Phytopathology, 89, 967–973. [DOI] [PubMed] [Google Scholar]
- He, B. , Cai, Q. , Qiao, L. , Huang, C.Y. , Wang, S. , Miao, W. et al. (2021) RNA‐binding proteins contribute to small RNA loading in plant extracellular vesicles. Nature Plants, 7, 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosmani, P.S. , Flores‐Gonzalez, M. , van de Geest, H. , Maumus, F. , Bakker, L.V. , Schijlen, E. et al. (2019) An improved de novo assembly and annotation of the tomato reference genome using single‐molecule sequencing, hi‐C proximity ligation and optical maps. bioRxiv, 767764 [preprint], 10.1101/767764. [DOI] [Google Scholar]
- Hou, Y. , Zhai, Y. , Feng, L. , Karimi, H.Z. , Rutter, B.D. , Zeng, L. et al. (2019) A Phytophthora effector suppresses trans‐kingdom RNAi to promote disease susceptibility. Cell Host & Microbe, 25, 153–165.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal, S. , Fosu‐Nyarko, J. & Jones, M.G.K. (2020) Attempt to silence genes of the RNAi pathways of the root‐knot nematode, Meloidogyne incognita results in diverse responses including increase and no change in expression of some genes. Frontiers in Plant Science, 11, e00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamthan, A. , Chaudhuri, A. , Kamthan, M. & Datta, A. (2015) Small RNAs in plants: recent development and application for crop improvement. Frontiers in Plant Science, 6, e00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kan, J.A.L. (2006) Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends in Plant Science, 11, 247–253. [DOI] [PubMed] [Google Scholar]
- van Kan, J.A.L. , Shaw, M.W. & Grant‐Downton, R.T. (2014) Botrytis species: relentless necrotrophic thugs or endophytes gone rogue? Molecular Plant Pathology, 15, 957–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kan, J.A.L. , Stassen, J.H.M. , Mosbach, A. , v an der Lee, T.A.J. , Faino, L. , Farmer, A.D. et al. (2017) A gapless genome sequence of the fungus Botrytis cinerea . Molecular Plant Pathology, 18, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettles, G.J. , Hofinger, B.J. , Hu, P. , Bayon, C. , Rudd, J.J. , Balmer, D. et al. (2019) sRNA profiling combined with gene function analysis reveals a lack of evidence for cross‐kingdom RNAi in the wheat–Zymoseptoria tritici pathosystem. Frontiers in Plant Science, 10, e00892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax, C. , Tahiri, G. , Patiño‐Medina, J.A. , Cánovas‐Márquez, J.T. , Pérez‐Ruiz, J.A. , Osorio‐Concepción, M. et al. (2020) The evolutionary significance of RNAi in the fungal kingdom. International Journal of Molecular Sciences, 21, 9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisen, T. , Bietz, F. , Werner, J. , Wegner, A. , Schaffrath, U. , Scheuring, D. et al. (2020) CRISPR/Cas with ribonucleoprotein complexes and transiently selected telomere vectors allows highly efficient marker‐free and multiple genome editing in Botrytis cinerea . PLoS Pathogens, 16, e1008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroch, M. , Kleber, A. , Silva, E. , Coenen, T. , Koppenhöfer, D. , Shmaryahu, A. et al. (2013) Transcriptome profiling of Botrytis cinerea conidial germination reveals upregulation of infection‐related genes during the prepenetration stage. Eukaryotic Cell, 12, 614–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, F. , Blancard, D. , Lecomte, P. , Levis, C. , Dubos, B. & Fermaud, M. (2003) Phenotypic differences between vacuma and transposa subpopulations of Botrytis cinerea . European Journal of Plant Pathology, 109, 479–488. [Google Scholar]
- McLoughlin, A.G. , Wytinck, N. , Walker, P.L. , Girard, I.J. , Rashid, K.Y. , De Kievit, T. et al. (2018) Identification and application of exogenous dsRNA confers plant protection against Sclerotinia sclerotiorum and Botrytis cinerea . Scientific Reports, 8, 7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerva, L. , Sandrini, M. , Gambino, G. & Chitarra, W. (2020) Double‐stranded RNAs (dsRNAs) as a sustainable tool against gray mold (Botrytis cinerea) in grapevine: effectiveness of different application methods in an open‐air environment. Biomolecules, 10, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda, J. , Brito, N. & González, C. (2010) The Botrytis cinerea xylanase Xyn11A contributes to virulence with its necrotizing activity, not with its catalytic activity. BMC Plant Biology, 10, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novina, C.D. & Sharp, P.A. (2004) The RNAi revolution. Nature, 430, 161–164. [DOI] [PubMed] [Google Scholar]
- Nowara, D. , Schweizer, P. , Gay, A. , Lacomme, C. , Shaw, J. , Ridout, C. et al. (2010) HIGS: host‐induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis . The Plant Cell, 22, 3130–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinedo, C. , Wang, C. , Pradier, J. , Dalmais, B. , Choquer, M. , Pascal, L. et al. (2008) The sesquiterpene synthase from the botrydial biosynthetic gene cluster of the phytopathogen Botrytis cinerea . ACS Chemical Biology, 3, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porquier, A. , Tisserant, C. , Salinas, F. , Glassl, C. , Wange, L. , Enard, W. et al. (2021) Retrotransposons as pathogenicity factors of the plant pathogenic fungus Botrytis cinerea . Genome Biology, 22, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter, B.D. & Innes, R.W. (2020) Growing pains: addressing the pitfalls of plant extracellular vesicle research. New Phytologist, 228, 1505–1510. [DOI] [PubMed] [Google Scholar]
- Samuel, S. , Veloukas, T. , Papavasileiou, A. & Karaoglanidis, G.S. (2012) Differences in frequency of transposable elements presence in Botrytis cinerea populations from several hosts in Greece. Plant Disease, 96, 1286–1290. [DOI] [PubMed] [Google Scholar]
- Shahid, S. , Kim, G. , Johnson, N.R. , Wafula, E. , Wang, F. , Coruh, C. et al. (2018) MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature, 553, 82–85. [DOI] [PubMed] [Google Scholar]
- Sijen, T. , Vijn, I. , Rebocho, A. , Van Blokland, R. , Roelofs, D. , Mol, J.N.M. et al. (2001) Transcriptional and posttranscriptional gene silencing are mechanistically related. Current Biology, 11, 436–440. [DOI] [PubMed] [Google Scholar]
- Silvestri, A. , Fiorilli, V. , Miozzi, L. , Accotto, G.P. , Turina, M. & Lanfranco, L. (2019) In silico analysis of fungal small RNA accumulation reveals putative plant mRNA targets in the symbiosis between an arbuscular mycorrhizal fungus and its host plant. BMC Genomics, 20, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spada, M. , Pugliesi, C. , Fambrini, M. & Pecchia, S. (2021) Silencing of the Slt2‐type MAP kinase Bmp3 in Botrytis cinerea by application of exogenous dsRNA affects fungal growth and virulence on Lactuca sativa . International Journal of Molecular Sciences, 22, 5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, M.A. , Jones, J.D.G. & Dangl, J.L. (2006) Reactive oxygen species signaling in response to pathogens. Plant Physiology, 141, 373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres‐Martínez, S. & Ruiz‐Vázquez, R.M. (2017) The RNAi universe in fungi: a varied landscape of small RNAs and biological functions. Annual Review of Microbiology, 71, 371–391. [DOI] [PubMed] [Google Scholar]
- Veloso, J. & van Kan, J.A.L. (2018) Many shades of grey in Botrytis–host plant interactions. Trends in Plant Science, 23, 613–622. [DOI] [PubMed] [Google Scholar]
- Wang, M. & Jin, H. (2017) Spray‐induced gene silencing: a powerful innovative strategy for crop protection. Trends in Microbiology, 25, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Weiberg, A. , Lin, F.M. , Thomma, B.P.H.J. , Da Huang, H. & Jin, H. (2016) Bidirectional cross‐kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nature Plants, 2, 16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Weiberg, A. , Dellota, E. , Yamane, D. & Jin, H. (2017) Botrytis small RNA Bc‐siR37 suppresses plant defense genes by cross‐kingdom RNAi. RNA Biology, 14, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiberg, A. & Jin, H. (2015) Small RNAs – the secret agents in the plant–pathogen interactions. Current Opinion in Plant Biology, 26, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiberg, A. , Wang, M. , Lin, F.M. , Zhao, H. , Zhang, Z. , Kaloshian, I. et al. (2013) Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science, 342, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiberg, A. , Wang, M. , Bellinger, M. & Jin, H. (2014) Small RNAs: a new paradigm in plant‐microbe interactions. Annual Review of Phytopathology, 52, 495–516. [DOI] [PubMed] [Google Scholar]
- Wong‐Bajracharya, J. , Singan, V.R. , Monti, R. , Plett, K.L. , Ng, V. , Grigoriev, I.V. et al. (2022) The ectomycorrhizal fungus Pisolithus microcarpus encodes a microRNA involved in cross‐kingdom gene silencing during symbiosis. Proceedings of the National Academy of Sciences of the United States of America, 119, e2103527119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Zhao, Y.L. , Zhao, J.H. , Wang, S. , Jin, Y. , Chen, Z.Q. et al. (2016) Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nature Plants, 2, 10. [DOI] [PubMed] [Google Scholar]
- Zhu, W. , Ronen, M. , Gur, Y. , Minz‐Dub, A. , Masrati, G. , Ben‐Tal, N. et al. (2017) BcXYG1, a secreted xyloglucanase from Botrytis cinerea, triggers both cell death and plant immune responses. Plant Physiology, 175, 438–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. & van Kan, J.A.L. (2013) Botrytis cinerea mutants deficient in D‐galacturonic acid catabolism have a perturbed virulence on Nicotiana benthamiana and Arabidopsis, but not on tomato. Molecular Plant Pathology, 14, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Correlations between production levels of Bc‐sRNAs and their predicted target mRNAs in tomato through seven sampling time points. Reverse transcription‐quantitative PCR results showing dynamics in the production levels of six chosen Bc‐sRNAs (pink bars) and the corresponding tomato transcripts (blue lines). The predicted target tomato genes are shown above each chart
Figure S2 (a) Schematic representation of the generation of ∆ms3003 transformants via CRISPR‐Cas9‐mediated transformation with two sgRNAs and the primers used in genotyping the transformants. (b) Genotyping of ∆ms3003 transformants by PCR, representing the target region, was replaced by the donor template via homologous recombination on both 5′‐ and 3′‐flanks. The same PCRs were performed with wild‐type B05.10 as the nontransformed control
Figure S3 (a) Schematic representation of the generation and characterization of ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 transformants. (b) Genotyping of ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 transformants by PCR with primers shown in (a) and File S1. For the Bcdcl1‐check reaction, a band of 6.7 kb in B05.10 and ∆ku70 indicates the wild‐type Bcdcl1 gene and a band of 2.7 kb in ∆Bcdcl1/∆Bcdcl2 mutants as the replacement of this gene by the donor template. Similarly, the Bcdcl2‐check resulted in a product of 5.6 kb for B05.10 and ∆ku70 and one of 2.0 kb for ∆Bcdcl1/∆Bcdcl2 mutants. For the Bcdcl1‐WT reaction, only the B05.10, ∆ku70, and ∆Bcdcl2 strains showed amplicons of the Bcdcl1 gene, while the ∆Bcdcl1 and ∆Bcdcl1/∆Bcdcl2 strains did not. Similarly, the Bcdcl2‐reaction could only amplify the Bcdcl2 fragment in the B05.10, ∆ku70, and ∆Bcdcl1 strains, but not in the ∆Bcdcl2 and ∆Bcdcl1/∆Bcdcl2 strains
Figure S4 In vitro mycelial growth, conidia formation, and sclerotial development of B05.10 wild‐type (WT), ∆ku70, ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 mutant strains on MEA (a) and PDA (b) plates. The line charts below the photos represent the mycelial growth rate of B05.10 WT, ∆ku70, ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 mutant strains on MEA (a) and PDA (b) plates for 3 and 4 consecutive days, respectively
Figure S5 (a) Virulence assays of ∆Bcdcl1 and ∆Bcdcl2 single mutants compared with ∆ku70, and ∆ku70 compared with B05.10, which were performed with tomato leaves. (b–d) Virulence assays of ∆Bcdcl1/∆Bcdcl2 double mutant strains compared with ∆ku70, and ∆ku70 compared with B05.10. The assays were performed with Arabidopsis thaliana (b), Nicotiana benthamiana (c), and Phaseolus vulgaris (d), represented as bar charts and photos. Error bars in the charts represent standard errors of 72, 72, and 120 data points from three experiments for A. thaliana, N. benthamiana, and P. vulgaris, respectively. No significant reduction in the virulence of ∆Bcdcl1, ∆Bcdcl2, and ∆Bcdcl1/∆Bcdcl2 compared with ∆ku70, and the same for ∆ku70 compared with B05.10, statistically analysed by Student’s t test
File S1 More detailed description of the experimental procedures
Table S1 Sequence read depth of the mRNA and small RNA samples
Table S2 Sheet 1: sequences of 27,918 unique small RNAs (sRNAs) from Botrytis cinerea with length between 20 and 24 nucleotides, and their genomic origin: chromosome coordinates, ribosomal RNA repeat, transposable element loci, tRNA loci, gene regions. Sheet 2: correspondence of 73 sRNAs described by Weiberg et al. (2013) to our data
Table S3 Transposable elements (TEs) in Botrytis cinerea as sources of small RNAs. The list provides the genomic location (chromosome, start and end coordinates), the TE identifier from REPET annotation, the read counts for the entire repeat region as well as the percentage of the total read counts. Only repeat elements that generated at least 100 read counts are included
Table S4 Sequences of 934,154 unique small RNAs from tomato with a length between 20 and 24 nucleotides, and their genomic origin: chromosome coordinates, gene region, transposable element loci
Table S5 Differentially expressed genes of Botrytis cinerea during tomato leaf infection at different hours postinoculation (hpi) as compared to in vitro liquid culture. Each sheet provides the gene ID, the log2FC, the adjusted p value and a gene description. Sheet 1: expression at 12 hpi; sheet 2: expression at 16 hpi; sheet 3: expression at 24 hpi
Table S6 Differentially expressed genes of tomato during Botrytis cinerea infection at different hours postinoculation (hpi) as compared to mock‐treated leaves at the same time point. Each sheet provides the gene ID, the log2FC, the adjusted p value and a gene description. Sheet 1: expression at 12 hpi; sheet 2: expression at 16 hpi; sheet 3: expression at 24 hpi
Table S7 Predicted target mRNAs in tomato for each of the 7042 Botrytis cinerea small RNAs (sRNAs). The sheet provides the sRNA sequence, its coordinates on the B. cinerea genome, the transposable element (TE) origin, the gene ID of the predicted target gene, and a description of this gene
Table S8 Tomato genes predicted to be targeted by Botrytis cinerea small RNAs that display significant differential expression during B. cinerea infection, as compared to mock‐inoculated leaves. Sheet contains both down‐regulated and up‐regulated genes
Table S9 Transposable elements in tomato as sources of small RNAs. The list provides the genomic location (chromosome, start and end coordinates), the transposable element (TE) identifier from REPET annotation, the read counts for the entire repeat region as well as the percentage of the total read counts. Only repeat elements that generated at least 30,000 read counts are included
Table S10 Predicted target mRNAs in Botrytis cinerea for each of 114,011 tomato small RNAs (sRNAs). The sheet provides the sRNA sequences, their coordinates on the tomato genome, the gene or transposable element (TE) origin, and the gene ID of the predicted target genes in B. cinerea
Table S11 Botrytis cinerea genes predicted to be targeted by tomato small RNAs that display significant down‐regulation during infection, as compared to liquid culture controls
Table S12 Target genes from tomato and Botrytis cinerea selected for detailed reverse transcription‐quantitative PCR validation. The table contains the functional annotation of target genes, gene names used in this study, the gene ID (in tomato or B. cinerea), and small RNA (from B. cinerea or tomato) that is predicted to target the gene
Table S13 All primers used in this study
Data Availability Statement
The data of this project have been deposited in the NCBI database under Bioproject at www.ncbi.nlm.nih.gov/bioproject/ with accession number PRJNA496584. Raw sequence reads are deposited in the Sequence Read Archive at www.ncbi.nlm.nih.gov/sra/ project SRP166089 under accession numbers SRX4902781‐SRX4902781 and SRX4902791‐SRX4902800 (sRNAs) SRX4902771‐SRX4902780 and SRX4902783‐SRX4902790 (mRNAs).