

Short abstract
Content available: Audio Recording
Listen to an audio presentation of this article.

Roger W. Chapman
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a rare, progressive, cholestatic, immune‐mediated hepatobiliary disease. It is characterized by inflammation, stricturing, and concentric, obliterative fibrosis of the biliary system, ultimately leading to biliary cirrhosis, portal hypertension, and eventually hepatic failure in the majority of patients. 1 , 2 The pathogenesis of PSC remains unknown and, as yet, there is no proven medical or interventional therapy. Thus, with good reason, PSC has been called by Professor Michael Manns “… the last Black Box * remaining in Hepatology”.*
In 1976, as a young gastroenterology resident learning to perform Endoscopic Retrograde Cholangio Pancreatography (ERCP), I saw my first patient with PSC. The patient was a 32‐year‐old jaundiced man with ulcerative colitis and cholestatic serum liver chemical tests. We obtained the now‐characteristic cholangiogram showing multiple strictures and dilatations of the whole biliary tree (Figure 1A). A review of the world literature then did not provide much enlightenment. The first authentic case of PSC was published in 1924 3 by the Parisian vascular surgeon Paul Delbet (1866–1924)—seemingly in the year of his death—followed in 1925 with a report by Lafourcade, 4 also in Paris. However, as far back as 1867, a much earlier observation is credited to C.E.E. Hoffman, 5 who described biliary obstruction due to thickening of the bile duct wall. The existence of PSC confined within the liver, namely small duct PSC—see later—was heralded by Klemperer's 1937 description of chronic intrahepatic fibro‐obliterating cholangitis. 6 Sclerosing cholangitis had previously been labelled as both obliterative and stenosing cholangitis, which are consequences of the thickening of the bile duct wall, 5 whereas sclerosing cholangitis is the more fitting designation as it accurately reflects the overall disease process. Incidentally, it is intriguing that coining of the term sclerosing cholangitis in 1954 should have been as ascribed to Castleman, 7 because of the confounding similarities between immunoglobulin G4 (IgG4) ‐related disorders (which include a sclerosing cholangitis‐like entity), and bona fide multicentric plasma cell variant Castleman disease. 8
FIGURE 1.

(A) Endoscopic cholangiogram (ERCP) showing the typical changes of PSC, stricturing and dilatation of the biliary tree involving intrahepatic and extrahepatic bile ducts. (B) Magnetic Resonance Cholangiogram (MRCP) showing the same typical ductal changes of PSC. These radiographs are from the author's personal collection at the center in Oxford.
From the mid‐20s to the mid‐70s of the 20th century, fewer than 150 cases were described, the majority of which were diagnosed at postmortem after presenting with cholestatic jaundice. The breakthrough in the diagnosis of PSC occurred with the introduction of diagnostic ERCP in the early 1970s.† This allowed premortem and, importantly, pre‐operative cholangiograms to be obtained. It is tempting to speculate why so few cases of PSC were published during the more than half a century that elapsed between Delbet's description and the Anglo‐American revelations of 1980. 9 , 10 In those days, biliary obstruction was decidedly in the bailiwick of the surgeons, in whose literature most of the reports appeared. The surgical focus then was understandably on biliary strictures, mostly following inadvertent operative trauma—a euphemism for operative misadventure during cholecystectomy—or due to injury and inflammation caused by common duct gallstones, or the occasional congenital cases. In surgical practice the concept of operative correction was initially uppermost. Primary (i.e., idiopathic) diffuse, extensive, and multifocal stricturing of the extrabiliary tree (i.e., PSC with its usual co‐morbidities) and even intrahepatic PSC, were indeed recognized in the surgical literature, 11 , 12 , 13 , 14 , 15 , 16 including a well‐reasoned clinicopathology justification of the term PSC itself 15 —even before reliable ERCP replaced the near‐invisible foggy images of intravenous cholangiography, cholangiograms obtained intraoperatively and, by now, the largely obsolescent percutaneous transhepatic route. Surprisingly, PSC was not mentioned by the illustrious British surgeon of his day, Lord Edwin Rodney Smith KBE, FRCS, Baron of Marlow, ‡ in his 1964 definitive textbook on biliary surgery. 17 But, whereas various possible pathogenic explanations for idiopathic PSC were readily dismissed,—and parenthetically the precise pathogenesis of idiopathic PSC remains enigmatic—these alternative, secondary etiologies of biliary strictures were often competing diagnostic considerations, nonetheless. In this context, the importance of recognizing prior abdominal surgery was emphasized often, especially in the very early reports.† ‡
In high profile dedicated hepatobiliary surgery units, fully corrective operations were once shown to be feasible 18 and, in fact, seem to have been the desired intent from the surgical perspective in those distant days. Yet, surely, such complex and admittedly aggressive surgical prowess represented “the triumph of hope over experience”, as one of the greatest figures of 18th century life and letters, Dr. Samuel Johnson (1709–1794), ironically observed about second marriages. § The same criticism might have been levelled in its early days against liver transplantation (LT) for PSC. As we will see later, liver replacement may even be the treatment of choice for appropriate PSC patients, including highly selected cases of cholangiocarcinoma (CCA). Surgical drainage and bypass of the obstructed biliary tree have long since fallen from grace. 20 §
Diagnostic biliary imaging in PSC, by the noninvasive technique of Magnetic Resonance Cholangio Pancreatography (MRCP, Figure 1B) has by and large, replaced ERCP, 21 , 22 which is now mainly reserved as a prelude to therapeutic maneuvers. MRCP does not use radiation and is cheaper than ERCP, and although visualization of peripheral intrahepatic branches may be inferior, MRI can provide information about the hepatic parenchyma, complications of liver disease, and the opportunity to add MR elastography.
Three months after my initial encounter with PSC, I found myself at the Royal Free Hospital Liver Unit, in Hampstead in London, trying to persuade the formidable Dame Sheila Sherlock (Figure 2), one of the founding greats of Hepatology, to let me pursue a 3‐year research project on PSC. She was not impressed, “… there's nothing in that; you'll be studying iron metabolism”, which I duly did. However, I also gathered together the case records of the 29 known PSC patients who had been seen over the preceding decade at the Royal Free Hospital (RFH), initially at the Grays Inn Road site, and then at the RFH in Hampstead from 1974 onward. By chance, Nick LaRusso (Figure 3) from Mayo Clinic in Rochester MN visited The Royal Free Liver Unit in 1978, when he was also beginning to develop an interest in PSC; naturally, we discussed our findings. This led to the independent publication in 1980 of the results from our two retrospective observational studies, respectively, in which we and they described the characteristics and natural history of PSC; the Royal Free data appeared in Gut, 9 and the Mayo paper was published in Gastroenterology. 10
FIGURE 2.

(A) Dame Sheila Patricia Violet Sherlock DBE, FRCPE, FRS, HFRSE, FMGA, FCRGA. photographed in 1970, from Wikipedia. (B) Dame Sheila's London residence with memorial plaque (inset), From London Remembers—londonremembers.com.
FIGURE 3.

(A) Photograph of Dr Nicholas LaRusso in the mid‐1970s: a personal gift. (B) Current photograph of Dr. LaRusso, Credit: Mayo Clinic College of Medicine and Science.
After his return to Mayo, Nick further developed his stellar scientific career in biliary physiology, which began in the laboratory of the late legendary colorful Alan Hofmann (1931–2021) 23 at Mayo (Figure 4A), and in cholangiocyte biology that he pursued when he was a guest investigator at Rockefeller University in Manhattan New York City (1975–1977), in the laboratory of the late Nobel Laureate Christian René Marie Joseph, Viscount de Duve (1917–2013, Figure 4B). 24 This famed University of Louvain‐educated Belgian who solidified his global scientific reputation in New York, had actually been born in Thames‐Ditton, near London, whither his Belgian‐German parents had fled as refugees during the First World War. And yet, despite his demanding leadership and administrative roles over the years, ¶ Nick's engagement in the clinical and scientific aspects of PSC continues unabated. 26 ¶
FIGURE 4.

(A) Alan F Hofmann MD, PhD. Copyright @ 2022 AGA Institute.21 (B) Christian René Marie Joseph, Viscount de Duve lecturing at the age of 95 in 2012, on the origin of the eukaryotic cell reproduced from Reference 25 with permission.
The findings of both landmark PSC studies 9 , 10 were remarkably similar. There was a 2:1 male predominance with a mean age at presentation of around 35 years, and a strong association with ulcerative colitis in over 70% of the patients. The prognosis in this young group of patients was poor; 38% had died with a mean survival time of 7 years from diagnosis. CCA was the cause of death in 3 of 11 patients.
PATHOGENESIS: GENETIC STUDIES
The next tentative step forward came from our data 27 and a Norwegian group led by Erik Schrumpf, 28 namely that PSC had strong HLA associations on chromosome 6 (according to the nomenclature of the time) with the haplotype HLA B8 DR3 (Table 1). This haplotype is now known to be strongly associated with particular organ‐specific autoimmune diseases such as celiac disease and type 1 diabetes mellitus. Thus emerged the first evidence that PSC may be an autoimmune disease or at least immune‐mediated. 29
TABLE 1.
HLA types in PSC.
| Haplotype | Significance in PSC |
|---|---|
| A1B8‐TNF*2‐DR3*0101‐DRB1*0301‐DQ*0501‐DQB1*0201 | Strong association; auto‐immune haplotype |
| DR3 | |
| DRB3*0101‐DRB1*1301‐DQA1*0103‐DQB1*0603 | Strong association |
| DR6 | |
| DRB5*0101‐DRB1*1501‐DQA1*0102‐DQB1*0602 | Weak association |
| DR2 | |
| DRB4*0103‐DRB1*0401‐DQA1*03‐DQB1*0302 | Strong association with disease protection |
| DR4 | NB cholangoca/worse prognosis |
Over the next decade this immunological suggestion proved to be controversial, not least because PSC is male‐predominant and, in general, not responsive to immunosuppression with corticosteroids, unlike other organ‐specific autoimmune diseases. However, results of recent genetic studies using large cohorts of PSC patients, including the application of Genome‐Wide Association Study (GWAS) 30 and the use of Immunochip methodology 31 have shown that, in addition to confirming the HLA association, PSC is associated with a number of other immune response genes, in common with a number of other organ‐specific autoimmune diseases.
On this basis, it is now established that PSC is indeed an immune‐mediated disease and makes up one of the group of four autoimmune liver diseases—together with autoimmune hepatitis (AIH), primary biliary cholangitis (PBC)** and IgG4‐related disease. PSC is known to overlap with AIH and possibly with IgG4 disease but not with PBC, which has proven to be a completely distinct disorder. 32 The male predominance in PSC, which is not characteristic of the autoimmune diseases in general, including AIH and PBC remains unexplained, although intriguingly IgG4‐related biliary disease is associated with male predominance.**
PATHOGENESIS: ASSOCIATION WITH INFLAMMATORY BOWEL DISEASE
Results of our original RFH/Mayo studies 9 , 10 showed that approximately two thirds of PSC patients of Northern European Caucasian origin have inflammatory bowel disease (IBD), the majority of whom (approximately 85%) have an ulcerative colitis (UC) phenotype, whereas the remainder largely have Crohn colitis and to a lsser extent indeterminate colitis. It was initially thought that the prevalence of PSC in IBD was low, at 3%–5% of patients with UC. However, in more recent studies the prevalence of PSC is higher and occurs in approximately 8%–12% of patients with extensive UC. 33 In my opinion, all patients with extensive colitis should undergo MRCP at the time of the diagnosis of UC.
It was apparent from the early experience that the clinical phenotype of the colitis was significantly different in UC patients with PSC (PSC‐IBD), compared with UC patients without PSC. The differences are shown in Table 2. This led to the concept that PSC‐IBD may represent a distinct and separate disease from IBD without PSC. This concept has been confirmed by data from recent genetic studies that showed that patients with PSC‐IBD have less than 50% of genes in common in patients with IBD alone. 30 , 31
TABLE 2.
Clinical features of PSC/UC patients compared with UC patients alone.
|
In other genetic studies of PSC, an association has been demonstrated with the FUT2 gene, 34 which affects the bacterial composition of the gastrointestinal tract. Moreover, PSC patients with and without IBD have a bacterial and fungal microbiome that is distinct from controls and from UC patients without PSC. 35 These genetic and microbiome results strongly suggest that PSC‐IBD is a separate and distinct disease entity from IBD without PSC.
Current evidence suggests that PSC‐IBD is an immunologically mediated disease that is probably triggered in genetically susceptible individuals with a specific microbiome, by acquired toxic or infectious agents, which may gain access through the leaky diseased colon (Figure 5, 36 ). Gut‐derived lymphocytes aberrantly “home” to ligands in the biliary tract, in addition to gut epithelium. 37 Herein may be the explanation for the association with colitis.
FIGURE 5.

Factors involved in the pathogenesis of PSC. Reproduced with permission from Reference. 36
PSC IS A PREMALIGNANT DISEASE
One of the unexplained features of PSC is the strong association with malignancy, despite the paradox that PSC with or without IBD is not associated with cigarette smoking. To‐date, genetic studies have not uncovered any markers of malignancy that could identify the particular subgroup of PSC patients at high risk of developing malignancy. 38 Patients with total UC have an increased risk of colorectal cancer, associated with a long duration of disease and the extent of colitis. The risk of colonic dysplasia and colorectal cancer is significantly enhanced in those with co‐existent PSC; this risk continues after LT and increases with time. Thus, approximately 50% of patients with PSC‐UC will develop colonic dysplasia after 20 years of disease. 39 PSC is likely to be the most important co‐factor in the development of dysplasia and colon cancer in UC and is the commonest cause of death in patients with total UC.
Next, the devastating development of CCA complicates the clinical course in 8%–30% of adult patients with PSC, and can develop in either the intra‐ or more commonly in the extrahepatic bile ducts. 40 The incidence of CCA is approximately 0.5%–1.5% per year in those with large‐duct PSC, 41 and especially higher in patients with dominant strictures in the biliary tree. 41 In one‐third of patients with PSC who develop CCA, the cancer is identified within 1 year of the diagnosis of their PSC, when an astonishing 900‐fold increased incidence compared to the general population, was reported from Finland. 42 In a systemic review and meta‐analysis of published data from retrospective and prospective studies, the approximate relative risks of CCA, hepatobiliary, liver, gastrointestinal, pancreatic, colorectal and total cancer, were 584, 30, 17, 8, 6, 4, and 4, respectively. 43 There is a small subgroup of PSC patients with a CCA under 3 cm in diameter, who in the absence of evidence of metastatic spread may be eligible for LT. 44 However, in general, once CCA has developed in PSC, the prognosis is extremely poor with less than 5% of patients surviving 5 years. 37
Unfortunately to‐date, there are no available proven radiological signs or biomarkers that can identify CCA at an early enough stage to allow curative therapy. The development of effective, early markers for CCA represents one of the major unmet needs expressed by hepatologists and PSC patients alike. It may be that regular endoscopic surveillance of the biliary tree by cholangioscopy, analogous to endoscopic colonic surveillance, particularly in those patients with dominant strictures who are at particularly high risk, may offer earlier detection of biliary dysplasia. A recent American Gastroenterological Association (AGA) expert review clinical practice update on surveillance for hepatobiliary cancers in patients with PSC, 45 advised against ERCP and brush cytology for the early detection of CCA; but this needs to be formally tested in a clinical trial. Untoward symptoms, worsening cholestatic serum liver chemistry results, and/or the development of a new rapidly progressive high‐grade stricture on imaging raises the suspicion for CCA, which warrants thorough investigation. The watchword here is clinical vigilance. In a recent European Association for the Study of the Liver (EASL) practice guidelines on sclerosing cholangitis, a decision tree (Figure 6) was proposed for the diagnosis in patients with a suspicion of CCA. 46
FIGURE 7.

Survival of PSC Patients in Holland until liver transplantation or death. The estimated median transplant‐free survival population‐based PSC cohort was 21.2 years (bold black upper curve). Estimated median transplant‐free survival of PSC cohort patients at LT centers was 12.8 years (lower gray curve). Reproduced from Boonstra et al., Hepatology 2013, 49 with permission.
Hepatobiliary malignancies occur more commonly (10%–20%) in PSC than in the general population, 40 particularly hepatocellular carcinoma (HCC), i.e., hepatocellular adenocarcinoma in cirrhotic patients, and also carcinoma of the gallbladder. Other gallbladder pathology in PSC comprises cholecystitis, gallstones, epithelial dysplasia and polyps—with prevalence of the latter at 6%–17%. 47 , 48 The strong correlation between gallbladder dysplasia/adenocarcinoma and bile duct dysplasia/CCA supports the concept of a neoplastic “field effect” along the intrahepatic and extrahepatic biliary tract in PSC. 47 In a study of patients with radiographically detected polyps, the larger the polyp the greater was the cancer risk; overall gallbladder cancer occurred at a rate of 8.8 per 1000 patient years (Figure 7). 48
FIGURE 6.

Suggested diagnostic decision tree for patients with PSC, in whom there is suspicion for CCA. A high‐quality MRI with contrast should be performed. When strong suspicion of cancer or high‐grade dysplasia is found at a brush sample the patient should be evaluated at a high‐volume centre/specialised unit. Severe inflammation and biliary stenting may confound the diagnosis of true high‐grade dysplasia and a repeated ERCP with biopsies (if not already performed in the first ERCP) is recommended for confirmation of high‐grade dysplasia and/or malignancy, and tumour mapping of tumour involvement and spread before making a treatment decision. In cases with low‐grade dysplasia or benign brush sample, a follow‐up within 3 months is recommended with a new ERCP or MRI. MDT, multidisciplinary team. Reproduced from reference, 59 with permission.
PROGNOSIS
PSC patients who are asymptomatic at diagnosis, the majority of whom will develop progressive disease, have a mean survival rate of 88% at 5 years and more than 70% at 16 years after diagnosis. 2 , 38 Those who are symptomatic at diagnosis have shorter survival. Twenty years ago, it was thought that 50% of PSC patient would die or require LT within 10–12 years after the diagnosis. However these data arose from tertiary, LT centers; subsequently, it has become clear that when all PSC patients are included from nontransplant centers, then the median overall survival from diagnosis to death or LT, is approximately 22 years 49 (Figure 8). The median survival is 23 years for all‐comers in our center in Oxford.
FIGURE 8.
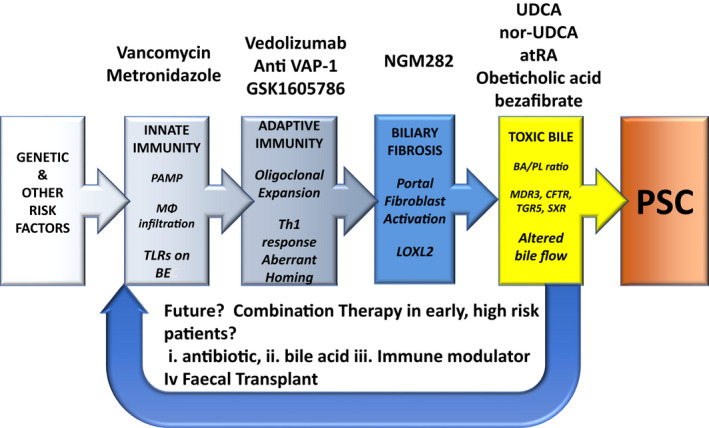
New therapeutic strategies for PSC. A schematic representation of the various elements contributing to pathophysiology of primary sclerosing cholangitis PSC (colored boxes), and the therapeutic agents (above), which may be directed against this particular element. Agents in italics are still in development and are not licensed. PAMP, pathogen‐associated molecular patterns; MΦ, monocyte; TLR, toll‐like receptors; BE, biliary epithelium; Th1, T‐helper 1; LOXL2, lysyl oxidase‐like 2; BA, bile acid; PL, phospholipid; MDR3, multidrug resistance 3 gene; CFTR, cystic fibrosis transmembrane conductance; TGR5, G‐protein coupled receptor; atRA, all‐trans retinoic acid. The figure shown was significantly altered from an original, courtesy of Chris Bowlus, University of California, Davis, CA, and published in Reference. 56
There is a distinct group of PSC patients whose serum liver chemical test results have a cholestatic pattern and who have features consistent with a diagnosis of sclerosing cholangitis on liver biopsy, but who have normal cholangiograms. These patients are described as having small duct disease, which accounts for 6%–16% of the PSC population. 50 Small duct PSC occurs more commonly in female patients and in those with Crohn colitis. For the most part, the course of their disease is milder and has a much better prognosis than large duct PSC. 50 Less than 20% of patients with small duct PSC will progress to large duct disease. 50 To‐date, and of importance, no cases of cholangiocarcinoma have been reported in small duct disease.
Findings in recent multicenter studies, utilizing data from over 7000 PSC patients, have revealed that the clinical phenotype may determine the prognosis; male gender, the presence of extrahepatic strictures, dominant strictures, an association with UC and high serum alkaline phosphatase (ALP), are all independent predictors of poor prognosis. 51 Persistently raised serum bilirubin for more than 3 months from diagnosis is an independent risk factor correlating with poor outcome and high risk of CCA. In contrast, when serum ALP is either normal or less than 1.5 times the upper limit of the normal range after 1 year of follow‐up, it has been shown to be a surrogate marker of a good prognosis. 52
Because of the great variability of the disease, in the past models have been of less use in individual cases of PSC than in PBC. However, three prognostic models have been validated recently in the assessment of the short and long‐term prognosis in PSC. 53 Unfortunately, and as stated previously, none of these models is able to identify the patient at risk of developing CCA.
MEDICAL TREATMENT
Over the last 40 years, a number of medical therapies including immunosuppressive agents and anti‐fibrotic therapy, have proved to be disappointing in the treatment of PSC. Some antibiotics, such as metronidazole and vancomycin, have shown promise but further studies are required to confirm efficacy. Almost all the trials that have been performed in PSC over the last 30 years can be criticized for including only small numbers of patients and heterogeneous patient selection. There is little evidence that corticosteroids are beneficial in patients with PSC, except for the minority of patients with a so‐called” overlap syndrome”, i.e., who have features of both PSC and AIH. 1
Ursodeoxycholic acid (UDCA) is a naturally occurring bile acid that improves survival in PBC in a dose of 13‐15 mg/kg/day. Whereas UDCA improves abnormalities of serum liver tests in PSC, its effect on PSC patient survival remains controversial. 54 A randomized controlled study employing high doses (25–30 mg/kg/day) in patients with advanced PSC was halted prematurely because of greater need for LT and death in the UDCA group than in the placebo recipients. 55 In my opinion, UDCA in a dose of 17‐20 mg/kg/day may be beneficial in a minority of PSC patients, but it has not been proven to be efficacious in the majority of patients and new strategies and therapies are urgently required.
There is currently major interest from Pharma in finding effective therapy for PSC. New therapeutic strategies have emerged, 56 , 57 and a number of therapeutic agents are currently being trialled including antibiotics, novel bile acids, agonists of the Farnesoid X (FXR) and other nuclear receptors, and of the FXR‐downstream target fibroblast growth factor‐19 (FGF19). These tactics are well‐detailed in the recent EASL clinical practice guidelines on PSC. 46 In addition, trials of faecal microbiota transplantation are in progress. 58 These agents are aimed to target different stages of the long disease process (Figures 5 and 8). It is disappointing that, despite the immune‐related foundation for PSC, an inventory of upward of a dozen immunosuppressive remedies has failed. 59 , 60 To‐date, positive results are still awaited for other anti‐tumor necrosis factor and comparable biologicals.
The ability to perform accurate patient stratification, which has recently become available, 51 will allow the most appropriate patients at the highest risk of disease progression to be included in trials. It is likely that over the next decade effective therapy either with single or, more likely, combination therapy, will become available for the treatment of PSC.
ENDOSCOPIC THERAPY IN PSC
As noted above, operative correction of biliary obstruction has been all but abandoned 20 and supplanted by LT for advanced disease, intractable complications and even hilar CCA, 44 , 61 except perhaps for the occasional non‐cirrhotic in whom endoscopic therapy has failed. 62 Regarding endoscopic therapy of PSC, and particularly the dilatation of high‐grade strictures in symptomatic patients using bougies or balloon catheters, it is salutary to accept that these skillful manipulations, which have gratifying short‐term success, 63 , 64 have yet to be sanctioned by randomized controlled trials but, nonetheless, are recommended 59 , 65 in patients with, what are now termed, clinically relevant 66 strictures.
LT IN PSC
Patents with advanced PSC have been candidates for LT ever since the early years of the procedure in Denver CO and Pittsburgh PA in the US, where it became the third commonest indication for LT after “post‐necrotic cirrhosis” and PBC. Accordingly, outcomes are comparably successful to those in other major indications. 67 The adverse effects of CCA and prior palliative biliary surgery, the need for elective and not moribund listing, the possibility of inflammatory bowel disease exacerbation and recurrence of PSC (rPSC) in the graft were all recognized. 67 These key features of LT for PSC are as germane today as they were then. 68 , 69 Meanwhile experience grows with LT and adjuvant therapy for selected cases of CCA complicating PSC.
CONCLUSION
The major interest in PSC among hepatologists and Pharma that has developed over the last 15 years, i.e., The long night's journey into day, has provided the stimulus for facilitating significant progress in our understanding of the pathogenesis of PSC—especially the immune and microbiome‐ related facets—its clinical phenotypes, the common association with IBD, and the prognosis. Yet, to date in the light of day, we afficianados and investigators of PSC have failed, both with regard to the early diagnosis and effective treatment of its most lethal complication, i.e., CCA, and development of effective medical therapy. The closed “Black Box” of PSC, the flight recorder so to speak, has been retrieved and begun to be prised open, but major challenges remain to be solved.
SERIES EDITOR'S POSTSCRIPT
In 1976, in the Liver Unit at the RFH in Hampstead in London, not far from ancient and iconic Hampstead Heath, †† an astute young Welsh lecturer noticed the unusual biliary anatomical irregularities in a patient with colitis and jaundice. As Louis Pasteur remarked, in a lecture at the University of Lille in 1854, “In the fields of observation chance favours only the prepared mind”. In Roger's case, his mind was prepared for a breakthrough (re)discovery, shared independently with Mayo, that fostered upward of 40 years ongoing clinical and basic research into a new cholestatic autoimmune liver disease, namely PSC.††
Roger W G Chapman was born, raised and schooled in Cardiff, (Caerdydd in Welsh, meaning the fort on the Taff, the river that flows by Cardiff), which is located in south‐east Wales and was proclaimed its capital as recently as 1955. After graduating in medicine in 1974 at St Bartholomew's Hospital (that was established together with the Priory of St Bartholomew in 1123, by a courtier of King Henry I) and completing his residency in medicine, Roger began training in liver disease in Southampton, with the renowned hepatologist Ralph Wright. Next, as a Lecturer and Research Felllow on the Liver Unit at RFH, from 1976 to 1981 under the supervision of Professor “Prof” Sheila Sherlock, he completed his thesis on “Iron Metabolism in Liver Disease” in 1981. That year, he moved to Oxford University and the John Radcliffe (University) Hospital, where he is now an Emeritus Consultant in Gastroenterology/Hepatology and member of Oxford University attached to the Oxford University Translational Gastroenterology Unit. Of course, he is much published, and a member of many learned societies and editorial boards. He was a founding member (in 1992) of the International Autoimmune Hepatitis Group (IAHG) producing position papers in the field of Autoimmune Hepatitis, and most recently he was involved in the foundation of the International PSC Study Group (2009).
He was awarded a Fellowship of American Association for the Study of Liver Disease (AASLD) in 2014, and a lifetime achievement award (2016) and Honorary Life Membership from the British Association for the Study of Liver Disease (BASL) in 2017. Whereas he appears most genial, it is well to remember that he is an accomplished examiner for the Universities of London, Cambridge, Oxford, Birmingham, Manchester, Newcastle, Bristol, Dublin, Oslo, Nottingham and Otago. Appearances can be deceptive.
But to end by returning to 1976, when in the summer London was withering under an unprecedented heat wave and drought that evidently was not a patch on the hyperthermal crisis that is ravaging Britain, southern Europe and other regions of the world at the time of this writing. It was commonplace, in that blighted ‘76 British summer, to see spotter aircraft buzzing overhead, scanning the parched countryside for green lawns in the yards of citizens, who could be prosecuted for violating the hosepipe ban. MCMLXXVI was a leap year in the Gregorian Calendar, the 1976th year of the Common Era (CE) and Anno Domini (AD) designations of the 20th century, but 2729 Ab urbe condita, 132‐133 for the Bahá'i, 7484–85 in Byzantium, 5736–37 in the Hebrew Calendar, 11,976 Holocene, 976–77 for the Igbo, and 13 days earlier jn the Julian calendar.
Aside from the timing of putting PSC on the map, 1976 was a propitious year. The bicentary of the Declaration of American Independence from the UK was celebrated enthusiastically in Britain, Jimmy Carter won the November USA presidential election, the trial of the Baader‐Meinhof terrorists began and Ulrike Meinhof hanged herself in prison, the Pittsburgh Steelers defeated the Dallas Cowboys at Super Bowl X, the first commercial Concorde flights began, UK Prime Minister Harold Wilson mysteriously resigned precipitously, Patty Hearst was convicted of armed robbery, in Argentina a military junta deposes president Isabel Peron in a coup, the Apple Computer Company is formed by Steve Jobs and Steve Wozniak, UK won the European Song Contest and ended the Third Cod War with Iceland, and Israeli commandos freed the Entebbe hostages in a dazzling raid.
The title that Roger chose for his essay was chosen to express the efforts in the dark to fully investigate PSC, eventually culminating in the daylight of our current understanding of this puzzling and often devastating condition. No relationship is implied with Eugene O′Neill's Tyrone family.
And so to 1977.
CONFLICT OF INTEREST
Nothing to report.
Chapman RW. Primary sclerosing cholangitis—A long night's journey into day. Clin Liver Dis. 2022;20:21–32. 10.1002/cld.1264
Footnotes
The expression Black Box is most commonly used nowadays for aircraft flight information recorders, which are actually bright orange. Possibly first developed at the Aeronautical Research Laboratory, Melbourne, Australia, in the 1950s by research scientist David Waren out of his interest in recording music, the two devices now in use are Cockpit Voice and Flight Data Recorders that document thousands of variables. In engineering, a black box system is viewed in in terms of inputs and outputs, without knowledge of its internal workings, i.e., its implementation is “opaque” (black), as is the pathogenesis of PSC.
A history of ERCP will be soon published in this series, by David Carr‐Locke.
President of the Royal College of Surgeons (1973–1977) and subsequently of the Royal Society of Medicine (1978–1980).
As reported by James Boswell, most celebrated biographer of the distinguished poet, playwright, essayist, moralistic, critic, biographer, editor, lexicographer, and overall consummate man of letters—in his acclaimed 1791 Life of Samuel Johnson. 19
Including Chair of the Division of Gastroenterology and Hepatology, and of the Department of Medicine at Mayo, Presidencies of both the American Association for the Study of Liver Diseases (1997) and (the American Gastroenterological Association 2007–2008), 25 Director of the Center for Innovation at Mayo, and one‐time Editor of Gastroenterology.
Formerly termed Primary Biliary Cirrhosis and soon to be reviewed by Raoul Poupon.
Mentioned historically as early as 986 CE and again in the Doomsday Book in 1086, Hampstead Heath is a 320 hectare (790 acre) rambling grassy, hilly, and wooded public space and is one of the highest points in London, from which to appreciate an unparalleled view of the city.
REFERENCES
- 1. Chapman RW, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis (PSC). Hepatology. 2010;51:660–78. [DOI] [PubMed] [Google Scholar]
- 2. Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary Sclerosing Cholangitis‐a comprehensive review. J Hepatol. 2017;67:1298–323. [DOI] [PubMed] [Google Scholar]
- 3. Delbet P. Retrecessement du choledoque: cholecystoduodeostomie. Bull Mem Soc Nat Chir (Paris). 1924;50:1144. [Google Scholar]
- 4. Lafourcade J. Deux observations d'obliteration cicatricielle de choledoque. Bull Mem Soc Nat Chir (Paris). 1925;50:828. [Google Scholar]
- 5. Hoffman CEE. Verschluss der Gallenwege curch Verdickung der Wandunger. Arch Pathol Anat Physiol. 1867;49:206–15. [Google Scholar]
- 6. Klemperer P. Chronic intrahepatic obliterating cholangitis. J Mount Sinai Hospital. 1937;4:279–29. [Google Scholar]
- 7. Krige JEJ, Bornman PC, Terblanche J. Primary sclerosing cholangitis. In: Holzheimer RG, Mannick JA, editors. Surgical treatment: evidence‐based and problem‐oriented. Munich: Zuckschwerdt; 2001. [PubMed] [Google Scholar]
- 8. Cheuk W, Yuen HKL, Chu SYY, Chiu EKW, Lam LK, Chan JK. Lymphadenopathy of IgG4‐related sclerosing disease. Am J Surg Path. 2008;32:671–81. [DOI] [PubMed] [Google Scholar]
- 9. Chapman RW, Arborgh BÅM, Rhodes JM, Summerfield JA, Dick R, Scheuer PJ, et al. Primary sclerosing cholangitis; a review of its clinical features, cholangiography and hepatic histology. Gut. 1980;21:870–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiesner RH, LaRusso NF. Clinicopathologic features of the syndrome of primary sclerosing cholangitis. Gastroenterology. 1980;79:200–6. [PubMed] [Google Scholar]
- 11. Judd ES. Stricture of the common bile duct. Ann Surg. 1926;84:404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller RT Jr. Benign stricture of the bile ducts. An Surg. 1927;86:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elliot E Jr. Benign cicatricial strictures of the bile ducts. Ann Surg. 1936;104:668–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ransom HK, Malcolm KD. Obstructive jaundice due to diffuse contracture of the extrahepatic bile ducts. Arch Surg. 1934;28:713–26. [Google Scholar]
- 15. Schwartz SI, Dale WA. Primary sclerosing cholangitis: review and report of six cases. AMA Arch Surg. 1958;77:439–51. [PubMed] [Google Scholar]
- 16. Tinkler L. Primary sclerosing cholangitis. Postgrad Med J. 1971;47:666–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith R. Surgery of the gallbladder and bile ducts. London: Butterworth and Co.; 1964. [Google Scholar]
- 18. Pitt HA, Thompson HH, Tompkins RK, Longmire WP. Primary sclerosing cholangitis: results of an aggressive surgical approach. Ann Surg. 1982;196:259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boswell J, Life of Samuel Johnson, LLD . Comprending an account of his studies and numerous works, in chronological order; a series of his epistolary correspondence and conversations with many eminent persons; and various original pieces of his composition, never before published. The whole exhibiting a view of literature and literary men in Great‐Britain for near half a century during which he flourished. London: Henry Baldwin for Charles Dilly, in the Poultry; 1791. [Google Scholar]
- 20. Lemmer ER, Bornman PC, Krige JEJ. Primary sclerosing cholangitis—requiem for biliary drainage operations. Arch Surg. 1994;129:723–8. [DOI] [PubMed] [Google Scholar]
- 21. Talwalker JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost‐minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology. 2004;40:39–45. [DOI] [PubMed] [Google Scholar]
- 22. Meagher S, Yusoff I, Kennedy W, Martel M, Adam V, Barkun A. The roles of magnetic resonance and endoscopic retrograde cholangiopancreatography (MRCP and ERCP) in the diagnosis of patients with suspected sclerosing cholangitis: a cost‐effectiveness analysis. Endoscopy. 2007;39:222–8. [DOI] [PubMed] [Google Scholar]
- 23. Barrett KE, LaRusso NF. In memoriam: Alan F Hofmann, MD, PhD, May 17, 1931 to September, 2021. Gastroenterology. 2022;162:997–8. [Google Scholar]
- 24. Opperdoes F. A feeling for the cell: Christian de Duve (1917–2013). PLoS Biol. 2013;11:e1001671. [Google Scholar]
- 25. Lazaridis KN, Gores GJ. Our new president—Nicholas LaRusso. MD Gastroenterology. 2007;132:2005–11. [DOI] [PubMed] [Google Scholar]
- 26. Trussoni CE, O'hara SP, LaRusso NF. Cellular senescence in the cholangiopathies: a driver of immunopathology and a novel therapeutic target. DSem Immunopathol. 2022;44:527–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chapman R, Varghese Z, Gaul R, Kokkinon N, Sherlock S. Association of primary sclerosing cholangitis with HLA‐B8. Gut. 1983;24:38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schrumpf E, Fausa O, Førre O, Dobloug JH, Ritland S, Thorsby E. HLA antigens and immunoregulatory T cells in ulcerative colitis associated with hepatobiliary disease. Scand J Gastroenterol. 1982;17:187–91. [DOI] [PubMed] [Google Scholar]
- 29. Chapman RW, Jewell DP. Primary sclerosing cholangitis: an immunologically mediated disease? West J Med. 1985;143:193–5. [PMC free article] [PubMed] [Google Scholar]
- 30. Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome‐wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet. 2017;49:269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al. Dense genotyping of immune‐related disease region identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. 2013;45(6):670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boberg KM, Chapman RW, Hirschfield GM, Manns MP, Lohse AW, Schrumpf E, et al. Autoimmune liver disorders and Overlap Syndromes; Position paper. J Hepatol. 2011;54:374–85. [DOI] [PubMed] [Google Scholar]
- 33. Lunder AK, Hov JR, Borthne A, Gleditsch J, Johannesen G, Tveit K, et al. Prevalence of sclerosing cholangitis, detected by magnetic resonance cholangiography, in patients with long‐term inflammatory bowel disease. Gastroenterology. 2016;151:660–9. [DOI] [PubMed] [Google Scholar]
- 34. Maroni L, van de Graaf SFJ, Hohenester SD, Oude Elferink RPJ, Beuers U. Fucosyltransferase 2: a genetic risk factor for primary sclerosing cholangitis and Crohn's disease—a comprehensive review. Clinic Rev Allerg Immunol. 2015;48:182–91. [DOI] [PubMed] [Google Scholar]
- 35. Hov JR, Karlsen TH. The microbiome in Primary Sclerosing Cholangitis: current evidence and potential concepts. Semin Liver Disease. 2017;37:314–31. [DOI] [PubMed] [Google Scholar]
- 36. Chapman RW. Update on primary sclerosing cholangitis. Cli Liv Dis (Hoboken). 2017;9:107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eksteen B, Miles AE, Grant AJ, Adams DH. Lymphocyte homing in the pathogenesis of extraintestinal manifestations of inflammatory bowel disease. Clin Med (Lond). 2004;4:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Williamson KD, Chapman RW. Primary sclerosing cholangitis. Dig Dis Sci. 2014;32:438–45. [DOI] [PubMed] [Google Scholar]
- 39. Broome U, Lofberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulverative colitis: evidence for increased neoplastic potential. Hepatology. 1995;22:1404–8. [DOI] [PubMed] [Google Scholar]
- 40. Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Lööf L, Danielsson Å, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002;36:321–7. [DOI] [PubMed] [Google Scholar]
- 41. Chapman RW, Williamson KD. Are dominant strictures in PSC a risk factor for cholangiocarcinoma? Current Hepatol Rep. 2017;16:124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barner‐Rassmussen N, Pukkala E, Jussila A, Färkkilä M. Epidemiology, risk of malignancy and patient survival in primary sclerosing cholangitis: a population‐based in Finland. Scand J Gastroenterol. 2020;55:74–81. [DOI] [PubMed] [Google Scholar]
- 43. Aune B, Sen A, Norat T, Riboli E, Folseraras T. Primary sclerosing cholangitis and the risk of cancer, cardiovascular disease, and all‐cause mortality: a systematic review and meta‐analysis of cohort studies. Sci Rep. 2021;11:10646. 10.1038/s41598-021-90175-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rosen CB, Heimbach JK, Gores GJ. Liver transplantation for cholangiocarcinoma. Transpl Int. 2010;23:692–7. [DOI] [PubMed] [Google Scholar]
- 45. Bowlus CL, Kim JK, Lindor KD. AGA Clinical Practice Update on surveillance for hepatobiliary cancers in patients with Primary Sclerosing Cholangitis: expert review. Clin Gastroenterol Hepatol. 2019;17:2416–22. [DOI] [PubMed] [Google Scholar]
- 46. European Association for the Study of the Liver . EASL Clinical Practice Guidelines on sclerosing cholangitis. J Hepatol. 2022;77:761–806. 10.1016/j.jhep.2022.05.011 [DOI] [PubMed] [Google Scholar]
- 47. Lewis JT, Talwalkar JA, Rosen CB, Smyrk TC, Abraham SC. Prevalence and risk factors for gallbladder neoplasia in patients with primary sclerosing cholangitis: evidence for a metaplasia‐dysplasia‐carcinoma sequence. Am J Surg Pathol. 2007;31:907–13. [DOI] [PubMed] [Google Scholar]
- 48. van Erp L, Cunningham M, Narasimman M, Ale Ali H, Jhaveri K, Drenth JPH, et al. Risk of gallbladder cancer in patients with primary sclerosing cholangitis and radiographically detected gallbladder polyps. Liver Int. 2020;40:382–92. [DOI] [PubMed] [Google Scholar]
- 49. Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, BWM S, Poen AC, et al. Population‐based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013;58:2045–55. [DOI] [PubMed] [Google Scholar]
- 50. Björnsson E, Olsson R, Bergquist A, Lindgren S, Braden B, Chapman RW, et al. Long term follow up of patients with Small Duct Primary Sclerosing Cholangitis. Gastroenterology. 2008;134:975–80. [DOI] [PubMed] [Google Scholar]
- 51. Weismuller TJ, Trivedi PJ, Bergquist A, Imam M, Lenzen H, Ponsioen CY, et al. Patient age, sex and inflammatory bowel disease phenotype associate with course of primary Sclerosing Cholangitis. Gastroenterology. 2017;152:1975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Al‐Mamari S, Djordjevic J, Halliday J, Chapman RW. Improvement of serum alkaline phosphatase to < 1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2013;58:329–34. [DOI] [PubMed] [Google Scholar]
- 53. de Vries EM, de Krijger M, Farkkila M, Arola J, Schirmacher P, Gotthardt D, et al. Validation of the prognostic scoring systems in primary sclerosing cholangitis; An international cohort study. Hepatology. 2017;65:907–19. [DOI] [PubMed] [Google Scholar]
- 54. Chapman RW. Primary Sclerosing Cholangitis; Is there still a role for ursodeoxycholic acid in the treatment of PSC? Nat Rev Gastroenterol Hepatol. 2010;7:74–5. [DOI] [PubMed] [Google Scholar]
- 55. Lindor KD, Kowdley KV, Luketic VAC, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of Primary Sclerosing Cholangitis. Hepatology. 2009;50:808–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Williamson KD, Chapman RW. New therapeutic strategies in primary Sclerosing Cholangitis. Semin Liver Dis. 2016;36:5–14. [DOI] [PubMed] [Google Scholar]
- 57. Gallo C, Howardson BO, Cristoferi L, Carbone M, Gershwin ME, Invernizzi P. An update on novel pharmacological agents for primary sclerosing cholangitis. Expert Opin Ther Targets. 2022;26:69–77. [DOI] [PubMed] [Google Scholar]
- 58. Allegretti JR, Kassam Z, Carrellas M, Mullish BH, Marchesi JR, Pechlivanis A, et al. Fecal microbiota transplantation in patients with primary sclerosing cholangitis: a pilot clinical trial. Am J Gastroenterol. 2019;114:1071–9. [DOI] [PubMed] [Google Scholar]
- 59. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: management of cholestatic liver disease. J Hepatol. 2009;51:237–67. [DOI] [PubMed] [Google Scholar]
- 60. Lindor KD, Kowdely KV, Harrison ME, American College of G . ACG guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015;110:646–59. [DOI] [PubMed] [Google Scholar]
- 61. Rosen CB. Transplantation versus resection for hilar cholangiocarcinoma. An argument for shifting paradigms for resectable disease in annals of surgery 2018. Ann Surg. 2018;267:808–9. [DOI] [PubMed] [Google Scholar]
- 62. Ahrendt SA. Surgical approaches to strictures in primary sclerosing cholangitis. J Gastrointestinal Surg. 2008;12:423–5. [DOI] [PubMed] [Google Scholar]
- 63. Eaton JE, Haseeb A, Rupp C, Eusebi LH, Munster K, Voitl R, et al. Predictors of jaundice resolution and survival after endoscopic treatment of primary sclerosing cholangitis. Hepatol Commun. 2022;6:809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bhat P, AAbakken L. Role of endoscopy in primary sclerosing cholangitis. Clin Endo. 2021;54:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. European Society of Gastrointestinal Endoscopy, European Association for the Study of the lLver . Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline. J Hepatol. 2017;66:1265–81. [DOI] [PubMed] [Google Scholar]
- 66. Venkatesh SK, Welle CL, Miller FH, Jhaveri K, Ringe KI, Eaton JE, et al. Reporting standards for primary sclerosing cholangitis using MRI and MR cholangiopancreatography: guidelines from MR Working Group of the International Primary Sclerosing Cholangitis Study Group. Euro Radiol. 2022;32:923–37 ‐author list corrected later. [DOI] [PubMed] [Google Scholar]
- 67. Marsh JW, Iwatsuki S, Makowka L, Esquivel CO, Gordon RD, Todo SA, et al. Orthotopic liver transplantation for primary sclerosing cholangitis. Ann Surg. 1988;207:21–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gow PJ, Chapman RW. Liver transplantation for primary sclerosing cholangitis. Liver. 2000;20:97–103. [DOI] [PubMed] [Google Scholar]
- 69. Martin EF. Timing, management, and outcomes of liver transplantation in Primary Sclerosing Cholangitis. Semin Liver Dis. 2017;37:305–13. [DOI] [PubMed] [Google Scholar]