

Abstract
Retinitis punctata albescens (RPA) is generally diagnosed by the presence of numerous clusters of white punctate lesions in the retina that progress over time and are related to several gene variants. The multifocal variant of congenital hypertrophy of the retinal pigment epithelium (CHRPE) is characterized by multiple, grouped, sharply circumscribed, pigmented spots. The PRPH2 gene encodes a photoreceptor-specific glycoprotein, which is essential for the morphogenesis of rod and cone photoreceptor outer segments. A 39-year-old Chinese female with nyctalopia, complained about blurred vision, presented a unique co-existing feature of RPA and CHRPE. Dilated fundus exam demonstrated numerous porcelain white discrete dots in both eyes and multiple, small, flat clusters of round brown to black pigmented lesions in the left eye. The full field electroretinography (ERG) showed decreased responses after standard dark adaptation and normal b-wave amplitudes after a long (4-hour) dark-adapted period. A heterozygous PRPH2 splicing variant was detected in the proband. In addition, the same variant was found in her mother, her son, and her daughter. We describe a PRPH2 variant in a rare case of RPA associated with multifocal CHRPE of the same individual.
Keywords: Congenital and Stationary Retinal Disease, RETINA, genetics, Electrophysiology, Retinitis Pigmentosa, Techniques of Retinal Examination
Introduction
Retinitis punctata albescens (RPA) is characterized by numerous dull-white punctate lesions, throughout the retina with a greater concentration in the posterior pole and midperiphery 1. As an autosomal recessive form of congenital stationary night blindness, RPA is associated with variants in retinaldehyde-binding protein 1 (RLBP1), Rhodopsin (RHO), Retinol Dehydrogenase 5 (RDH5), Peripherin 2 (PRPH2), and Lecithin Retinol Acyltransferase (LRAT) genes 2. Multifocal Congenital hypertrophy of the retinal pigment epithelium (CHRPE), also referred to as bear tracks, is flat, darkly pigmented, variably round lesions typically located in the peripheral retinal pigment epithelium (RPE) 3.
The PRPH2 gene, also known as retinal degeneration slow (RDS), encodes a photoreceptor-specific transmembrane glycoprotein, which is necessary for the morphogenesis of both rod and cone photoreceptor outer segments (OS) 4. Variants of the human PRPH2 gene have been associated with a wide variety of disease phenotypes, including retinitis pigmentosa 7 (OMIM: 608133), Leber congenital amaurosis 18 (OMIM: 608133), RPA (OMIM: 136880), macular dystrophy (OMIM: 608161), central areolar choroidal dystrophy 2 (OMIM: 613105) and macular pattern dystrophy-1 (OMIM 169150) 5. These conditions have significant disparity in severity and time of onset 5. Here we report a PRPH2 splicing variant in an unusual case of RPA associated with multifocal CHRPE.
Case Description
The patients consent to publish all contents including images of this case. A 39-year-old Chinese female reported blurry vision and metamorphopsia in the right eye for one week. She reported nyctalopia for as long as she could remember. Best corrected visual acuity (BCVA) was 20/50 OD and 20/20 OS. The anterior segment of both eyes was normal. Dilated fundus exam demonstrated numerous porcelain white discrete dots throughout the posterior pole and mid-peripheral retina in both eyes. Multiple, small, flat clusters of round brown to black pigmented lesions at RPE level in the inferior mid-periphery were observed only in the left eye (Fig. 1 a1, a2). Fundus autofluorescence (FAF) (Fig. 1 b1, b2) demonstrated hypoautofluorescence correlating with the RPE hypertrophy lesions in the left eye and granular hyperautofluorescence along the superotemporal arcade that correspond to the albinotic spots. Fluorescein angiography (FA) (Fig. 1 c1, c2) demonstrated persistent hypofluorescent blockage inferotemporally correlating with grouped congenital hypertrophy of the retinal pigment epithelium OS along with a granular hyperfluorescence with sharply demarcated borders OU that does not correspond to the albinotic spots. OCT Angiography (OCTA) demonstrates normal retinal and choroidal vascular structures in different layers, but subfoveal and parafoveal oval-shaped lesions which originate from the RPE with extension up to and elevating the overlying ellipsoid layer of the photoreceptors (Fig. 1 d, f). Horizontal SD-OCT scan showed extrafoveal dome-shaped lesions located at the level of the RPE (Fig. 1 g) and partial loss of ellipsoid layer of the photoreceptors (Fig. 1 h). The rod-specific dark-adapted electroretinography (ERG) showed decreased responses after standard dark adaptation (Figure. 2 a1). The cone-specific light-adapted ERG showed reduced b-wave amplitudes (Figure. 2 a2). The combined rod and cone dark-adapted standard flash ERG showed markedly reduced b-wave amplitudes (Figure. 2 a3). However, after a long (4-hour) dark-adapted period, full field ERG showed normal b-wave amplitudes. The EOG record showed a decreased Arden values OU (Figure. 2 b). Multifocal ERG showed delayed and decreased response amplitudes in the corresponding area (Fig 2 c1 and c2). There were not significant changes in the fundus appearance with two-year follow-up.
Fig. 1.
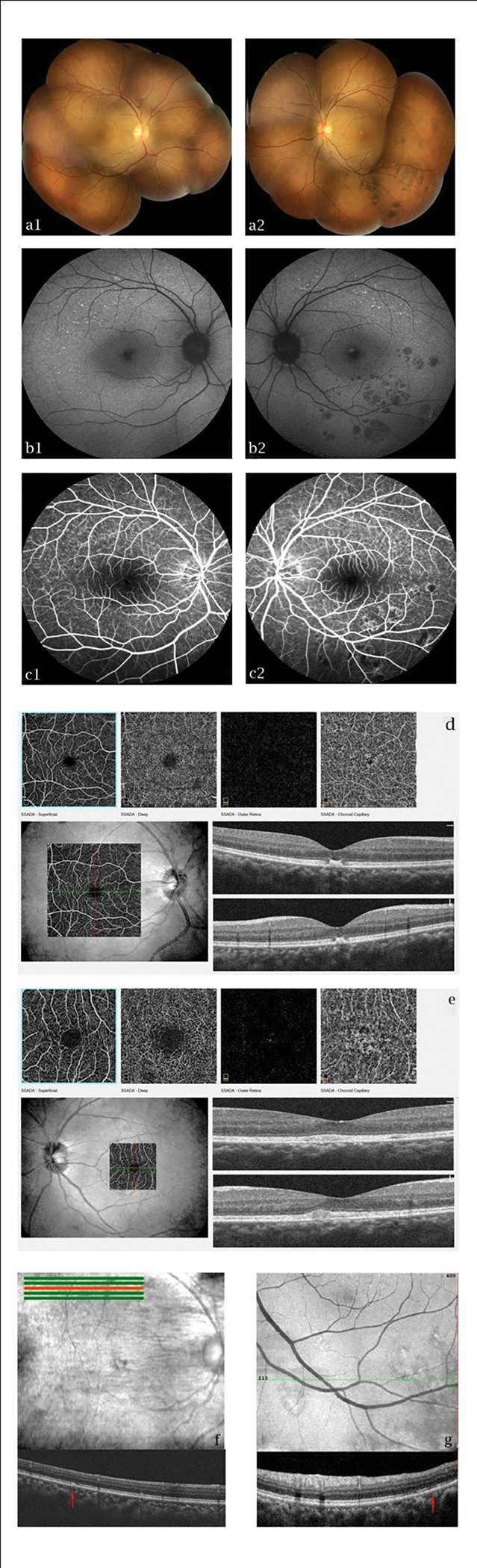
Fundus photographs (a1, a2), fundus autofluorescence (b1, b2), and fluorescein angiography (c1, c2) of the retina in the right (a1, b1 and c1) and the left (a2, b2 and c2) eye. A persistent hypofluorescence correlating with grouped congenital hypertrophy of the retinal pigment epithelium was showed in the left eye (a2, b2 and c2). Hypoautofluorescence correlating with the RPE hypertrophy lesions (b2) and granular hyperautofluorescence along the superotemporal arcade that correspond to the albinotic spots (b1, b2). A persistent hypofluorescent blockage inferotemporally correlating with grouped congenital hypertrophy of the retinal pigment epithelium (c2) along with a granular hyperfluorescence with sharply demarcated borders that does not correspond to the albinotic spots (c1, c2). OCT Angiography scan (d, e) shows subfoveal and parafoveal oval-shaped lesions which originate from the RPE with extension up to and elevating the overlying ellipsoid layer of the photoreceptors. Horizontal SD-OCT scan shows extrafoveal dome-shaped lesions located at the level of the RPE (red arrow in f) and partial loss of the ellipsoid layer of the photoreceptors (red arrow in g).
Fig. 2.

Electroretinography (ERG) test demonstrates reduced b-wave amplitudes in rod (a1), cone (a2) and combined (a3) responses after standard dark adaptation. The electroretinogram (EOG) record shows a decreased Arden values OU (b). Multifocal ERG shows delayed and decreased response amplitudes in the corresponding area (c1, c2).
DNA was extracted from peripheral blood before DNA sequencing performed. We tested 381 genes associated with ocular diseases, including RLBP1, RHO, RDH5 and LRAT, in the sample of the proband, however, only a splicing variant, the c.828+2T>C, was detected in exon 2 of the PRPH2 gene (transcript #NM_000322) in a heterozygous condition (Fig. 3 a). This is a +2 canonical splice variant that causes exon skipping and is predicted to disrupt the reading frame. Other family members also had genetic testing (Fig. 3 g), and the c.828+2T>C variant in PRPH2 gene was observed in her mother (Fig. 3 c), her son (Fig. 3 e), and her daughter (Fig. 3 f), in a heterozygous condition, while no variant in the PRPH2 gene was detected in her father (Fig. 3 b) or her husband (Fig. 3 d). The mother of the proband with a long-time visual impairment showed numerous punctate lesions and parafoveal structural disorder, especially on FAF (Fig. 3 h3, h4). The fundus appearance demonstrated numerous white discrete dots throughout the posterior pole and mid-peripheral retina in both eyes (Fig. 3 h1, h2). The father of the proband had a normal fundus appearance (Fig. 3 i1, i2). However, no obvious fundus lesions were present in the 15-year-old son of the proband (Fig. 3 j1, j2). The 11-month-old daughter was unable to cooperate with the exam.
Fig. 3.

Partial electropherograms of the PRPH2 exon 2 (a-f) and red arrows indicating the affected nucleotide. The heterozygous condition for c.828+2T>C variant was detected in the proband (a), the mother (c) the son (e) and the daughter (f) of the proband. The wild-type sequence was detected in the father (b) and the husband (d) of the proband. Familial pedigree (g) and arrow indicating the proband. Black shaded indicates affected by RPA, while red shaded indicates affected by CHRPE. The proband (39-year-old), the mother (67-year-old), the son (15-year-old) and the daughter (11-month-old) of the proband showed the heterozygous condition, while the father (67-year-old) and the husband (40-year-old) of the proband showed the wild-type condition. Fundus photographs (h1, h2) and fundus autofluorescence (h3, h4) of the mother of the proband demonstrate numerous punctate lesions and parafoveal structural disorder. There are a large number of hyperautofluorescence points corresponding to the white spots in the upper vascular arch and the nasal side of the optic disc. The macular area demonstrated mottled autofluorescence (h3, h4). Normal fundus images were captured in the eye of the father (i1, i2) and the son (j1, j2) of the proband.
Conclusions
RPA is characterized by multiple, variably sized, sharply circumscribed, white lesions with a greater concentration in the posterior pole and midperiphery, generally sparing the fovea 1. In this case report, subfoveal and parafoveal oval-shaped lesions extend from the RPE to the outer retinal layers were observed by OCT in both eyes. Elhannati et al. reported a patient with fundus albipunctatus showed a multitude of subtle, tiny yellow-white spots sparing the fovea 6. However, some patients with RPA demonstrate variable degrees of foveal cone death, even at an early stage 7. Therefore, foveal lesions are potentially involved in RPA. Moreover, the fundus changes of this patient were very similar to those observed in the two previous reports. A close relationship has been recognized between CHRPE and familial adenomatous polyposis (FAP) or Gardner syndrome, but the fundus lesions associated with tumor syndromes are usually bilateral and have more irregular or jagged borders, compared with the typical forms of multifocal CHRPE 3. All the family members denied the family history of FAP or other systemic diseases. Typical multifocal CHRPE was only observed in the left eye of the proband in this family. Turell et al. 8 described a case of CHRPE with nonpigmented, punctate lesions located within the macula. Meri et al. 9 described a presentation of concurrent congenital grouped pigmentation of the retina with albinotic spots. Although this is not the first report of RPA and CHRPE occurring simultaneously in the same individual, this is the first report of a variant of the PRPH2 gene associated with RPA and CHRPE.
Variants of RLBP1 and RDH5 gene in a homozygous state are commonly detected in RPA patients with typical fundus changes 2, while heterozygous variants in the PRPH2 gene are rarely seen in patients with RPA. It is remarkable that four members in this family, including the proband, presented heterozygous variant of PRPH2 gene, however, only the proband and her mother were observed with typical albinotic spots. According to a previous report, the presentation of retinal disease related to PRPH2 gene variant mainly occurs in the third to fifth decades of life 10. As mentioned before, the PRPH2 gene encodes a photoreceptor-specific transmembrane glycoprotein to mainly regulate the cone photoreceptor OS membranes 5. This missplicing of the PRPH2 we reported resulting in a truncated protein product, which could be the cause of macular dystrophy in a Spanish population 11. In addition, exon 2 mutations in PRPH2 gene were found previously in patients with retinitis pigmentosa and macular dystrophy 12,13. This suggests that the resulting protein lacking the region encoded by exon 2 is most likely non-functional and further studies would be required to confirm this. However, a mutation, the deletion of PRPH2 codon 153 or 154, has been reported to cause retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus all within the same family 14. Researchers found the most striking feature in families with the PRPH2 c.828+3A>T splice site mutation was the marked clinical diversity related to kinds of rod or cone dystrophy diseases, including retinitis pigmentosa, macular dystrophy and pattern dystrophy 15. Because RPA belong to the group of rod-cone dystrophies 2, we suppose this variant of PRPH2 we reported could present phenotypes in varied disease spectrum. According to the guidelines made by the American College of Medical Genetics and Genomics 16, this variant would be classified as likely pathogenic. Therefore, long-term follow-up is necessary for the children of the proband to focus on possible progression of disease even though currently there are no distinct lesions in the fundus. In this case, whether the PRPH2 gene contributed to CHRPE is still unclear.
We report a case with RPA associated with multifocal CHRPE of the same individual caused by the variant, the c.828+2T>C, in PRPH2 gene in a heterozygous condition. Because our current findings were based on a single case, additional cases and further genetic examination are required to characterize these rare and unusual associations between RPA and multifocal CHRPE.
Contributor Information
Aowang Qiu, Jiangsu Province Hospital and Nanjing Medical University First Affiliated Hospital, Ophthalmology.
Yan Yu, Jiangsu Province Hospital and Nanjing Medical University First Affiliated Hospital, Ophthalmology.
Junlong Huang, Jiangsu Province Hospital and Nanjing Medical University First Affiliated Hospital, Ophthalmology.
Qinghuai Liu, Jiangsu Province Hospital and Nanjing Medical University First Affiliated Hospital, Ophthalmology.
Yannis Paulus, University of Michigan, Ophthalmology and Visual Sciences.
wen fan, Jiangsu Province Hospital and Nanjing Medical University First Affiliated Hospital, Ophthalmology; University of Michigan, Ophthalmology and Visual Sciences.
References
- 1.Marmor MF. Long-term follow-up of the physiologic abnormalities and funduschanges in fundus albipunctatus. Ophthalmology 1990;97(3):380–4. [DOI] [PubMed] [Google Scholar]
- 2.Scimone C, Donato L, Esposito T, et al. A novel RLBP1 gene geographicalarea-related mutation present in a young patient with retinitis punctata albescens. Hum Genomics 2017;11(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shields JA, Shields CL. Tumors and Related Lesions of the Pigmented Epithelium.Asia Pac J Ophthalmol (Phila) 2017;6(2):215–23. [DOI] [PubMed] [Google Scholar]
- 4.Cheng T, al Ubaidi MR, Naash MI. Structural and developmental analysis of themouse peripherin/rds gene. Somat Cell Mol Genet 1997;23(3):165–83. [DOI] [PubMed] [Google Scholar]
- 5.Stuck MW, Conley SM, Naash MI. PRPH2/RDS and ROM-1: Historical context,current views and future considerations. Prog Retin Eye Res 2016;52:47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elhannati R, Tahri H. Fundus albipunctatus. Pan Afr Med J 2016;23:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song H, Latchney L, Williams D, et al. Fluorescence adaptive optics scanning laserophthalmoscope for detection of reduced cones and hypoautofluorescent spots in fundus albipunctatus. JAMA Ophthalmol 2014;132(9):1099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turell ME, Leonardy NJ, Singh AD A unique presentation of grouped congenitalhypertrophy of the retinal pigment epithelium. Ophthalmic Genet 2011;32(3):162–4. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe M, Makino S, Tampo H. An unusual presentation of concurrentcongenital grouped pigmentation of the retina with albinotic spots. Case Rep Ophthalmol 2015;6(1):39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boon CJ, den Hollander AI, Hoyng CB, et al. The spectrum of retinal dystrophiescaused by mutations in the peripherin/RDS gene. Prog Retin Eye Res 2008;27(2):213–35. [DOI] [PubMed] [Google Scholar]
- 11.Gamundi MJ, Hernan I, Muntanyola M, et al. High prevalence of mutations inperipherin/RDS in autosomal dominant macular dystrophies in a Spanish population. Mol Vis. 2007;13:1031–1037. [PMC free article] [PubMed] [Google Scholar]
- 12.Ekström U, Ponjavic V, Andréasson S, Ehinger B, Nilsson-Ehle P, Abrahamson M. Detection of alterations in all three exons of the peripherin/RDS gene in Swedish patients with retinitis pigmentosa using an efficient DGGE system. Mol Pathol. 1998;51(5):287–291. doi: 10.1136/mp.51.5.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Passerini I, Sodi A, Giambene B, Menchini U, Torricelli F. Phenotypicintrafamilial variability associated with S212G mutation in the RDS/peripherin gene. Eur J Ophthalmol. 2007;17(6):1000–1003. [DOI] [PubMed] [Google Scholar]
- 14.Weleber RG, Carr RE, Murphey WH, Sheffield VC, Stone EM. Phenotypicvariation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993;111(11):1531–1542. [DOI] [PubMed] [Google Scholar]
- 15.Shankar SP, Birch DG, Ruiz RS, et al. Founder Effect of a c.828+3A>T SpliceSite Mutation in Peripherin 2 (PRPH2) Causing Autosomal Dominant Retinal Dystrophies. JAMA Ophthalmol. 2015;133(5):511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations forinterpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39(11):1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]