

Abstract
Idiopathic pulmonary fibrosis (IPF) is a poorly understood, progressive lethal lung disease with no known cure. In addition to alveolar epithelial cell (AEC) injury and excessive deposition of extracellular matrix proteins, chronic inflammation is a hallmark of IPF. Literature suggests that the persistent inflammation seen in IPF primarily consists of monocytes and macrophages. Recent work demonstrates that monocyte-derived alveolar macrophages (moAMs) drive lung fibrosis, but further characterization of critical moAM cell attributes is necessary. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is an important epidermal growth factor receptor ligand that has essential roles in angiogenesis, wound healing, keratinocyte migration, and epithelial-mesenchymal transition. Our past work has shown HB-EGF is a primary marker of profibrotic M2 macrophages, and this study seeks to characterize myeloid-derived HB-EGF and its primary mechanism of action in bleomycin-induced lung fibrosis using Hbegff/f;Lyz2Cre+ mice. Here, we show that patients with IPF and mice with pulmonary fibrosis have increased expression of HB-EGF and that lung macrophages and transitional AECs of mice with pulmonary fibrosis and humans all express HB-EGF. We also show that Hbegff/f;Lyz2Cre+ mice are protected from bleomycin-induced fibrosis and that this protection is likely multifactorial, caused by decreased CCL2-dependent monocyte migration, decreased fibroblast migration, and decreased contribution of HB-EGF from AEC sources when HB-EGF is removed under the Lyz2Cre promoter.
Keywords: fibrosis, heparin-binding epidermal growth factor-like growth factor, monocyte-derived alveolar macrophage, alveolar epithelial cells, fibroblasts
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease with increasing incidence and high mortality (1). Although the etiology of IPF remains unclear, evidence suggests repetitive injuries to the epithelium, resulting inflammation, accumulation of fibroblasts and increased deposition of extracellular matrix (ECM) proteins by activated myofibroblasts lead to stiffening of the lung, impaired gas exchange, and ultimately respiratory failure (2–4).
Accumulating evidence implicates both monocytes and macrophages in the pathogenesis of lung fibrosis (1, 5, 6). Higher levels of circulating monocytes predict IPF disease progression (7–9) and in mice, depleting circulating monocytes or macrophages, or limiting their recruitment to the lung ameliorates fibrosis (5, 10–13). Typically, monocytes travel to sites of injury and differentiate into inflammatory macrophages (14). In the lung, recruited bone marrow–derived monocytes pass through an interstitial macrophage phenotype as they differentiate to monocyte-derived alveolar macrophages (moAMs) (12, 15). Recent work showed genetic deletion of moAMs protects from fibrosis development (12). Based on this work and others (11), there is renewed energy to understand how moAMs contribute to pathogenesis and to discover druggable targets (15).
First isolated from macrophage supernatant and expressed by profibrotic M2-like macrophages (16, 17), heparin-binding epidermal growth factor-like growth factor (HB-EGF) is one of four high-affinity ligands of EGFR (epidermal growth factor receptor) (18). HB-EGF is known for its role in cell maintenance, epithelialization, and wound healing (19–21). However, the role of HB-EGF in pulmonary diseases is complex with both profibrotic and antifibrotic effects (19, 22). For example, HB-EGF induces airway remodeling and epithelial mesenchymal transition in human bronchial epithelial cells in chronic obstructive pulmonary disease (COPD) and expression levels correlate with disease severity in COPD patients (19). Alternatively, in mouse models of emphysema, HB-EGF promotes survival, increases body weight, attenuates lung injury and inflammation, and prevents lung function decline (22).
No studies to date have examined the role of HB-EGF in patients with IPF nor used the bleomycin mouse model to investigate HB-EGF. Herein, we show HB-EGF is increased in patients with IPF and in mice with pulmonary fibrosis and that moAMs and alveolar epithelial cells (AECs) express HB-EGF. We also show Hbegff/f;Lyz2Cre+ mice are protected from bleomycin-induced fibrosis and that this protection is likely attributable to decreased monocyte recruitment and fibroblast migration, due to knockout of HB-EGF from both AEC and monocyte sources.
Methods
IPF Study
Plasma from patients in the COMET (Correlating Outcomes with biochemical Markers to Estimate Time-progression in IPF) study (NCT01071707) were analyzed by aptamer array (SomaLogics). Biomarker thresholds were chosen to maximize the combined sensitivity plus specificity (23).
Animals and Housing.
Six- to eight-week-old male and female Hbegff/f;Lyz2Cre+, Lyz2Cre+, and C57Bl/6J mice were bred at the University of Michigan. Animal experiments were approved by the Institutional Animal Committee on Use and Care.
Bleomycin Administration.
Mice were anesthetized with ketamine and xylazine. Bleomycin (1U/kg) was administered via oropharyngeal inoculation in a 50-μl volume.
Lung Homogenate, BAL, and Protein Assays.
Perfused mouse lungs were snap frozen and homogenized in Trizol (ThermoFisher) for RNA extraction. BAL fluid and cells were collected as described (11) in 1 ml of PBS/5 mM EDTA (Sigma-Aldrich). Cell-free supernatant was used for total protein assays or CCL2 and albumin ELISAs.
Flow Cytometry.
Lung cells were isolated by collagenase digestion (11), treated with Fc block followed by primary antibodies (30 min at 4°C). When necessary, cells were fix-permed and stained with intracellular antibodies (30 min at 25°C). See the data supplement for RNA PrimeFlow details.
Hydroxyproline Assays and Histology.
Descriptions can be found in (11) and the data supplement.
Pulmonary Function Tests.
Descriptions can be found in the data supplement.
Reverse Transcriptase (RT)-qPCR.
RNA was isolated using Trizol. qRT-PCR was performed on an ABI StepOnePlus real-time thermocycler (ThermoFisher) using a TaqMan RNA-to-Ct 1-Step Kit (ThermoFisher). Primers/probes are in Table 1.
Table 1.
Primers and Probes
| Gene | Direction | Sequence |
|---|---|---|
| GAPDH | Forward | 5′-GGGCCACGCTAATCTCATTT-3′ |
| Reverse | 5′-ATACGGCCAAATCCGTTCAC-3′ | |
| Probe | 5′-CTCCTCGAGCCTCGTCCCGT-3′ | |
| β-actin | Forward | 5′-CCGTGAAAAGATGACCCAGATC-3′ |
| Reverse | 5′-CACAGCCTGGATGGCTACGT-3′ | |
| Probe | 5′-TTTGAGACCTTCAACACCCCA-3′ | |
| Hbegf | Forward | 5′-CGGTGATGCTGAAGCTCTTT-3′ |
| Reverse | 5′-GCTGGTTTGTGGATCCAGTG-3′ | |
| Probe | 5′-ACGCGGACAACACTGCGGCC-3′ | |
| Collagen 1 | Forward | 5′-TGACTGGAAGAGCGGAGAGTACT-3′ |
| Reverse | 5′-GGTCTGACCTGTCTCCATGTTG-3′ | |
| Probe | 5′-CTGCAACCTGGACGCCATCAAGG-3′ | |
| Collagen 3 | Forward | 5′-GGATCTGTCCTTTGCGATGAC-3′ |
| Reverse | 5′-GCTGTGGGCATATTGCACAA-3′ | |
| Probe | 5′-TGCCCCAACCCAGAGATCCCATTT-3′ | |
| Fibronectin | Forward | 5′-TCGAGCCCTGAGGATGGA-3′ |
| Reverse | 5′-GTGCAAGGCAACCACACTGA-3′ | |
| Probe | 5′-CTGCAGGGCCTCAGGCCGG-3′ | |
| Survivin | Forward | 5′-GATCTGGCAGCTGTACCTCA-3′ |
| Reverse | 5′-ATCAGGCTCGTTCTCGGTAG-3′ | |
| Probe | 5′-CTGGAGGACTGCGCCTGCAC-3′ | |
| XIAP | Forward | 5′-ACCCTGCCATGTGTAGTGAA-3′ |
| Reverse | 5′-TCTCTGGGGGTTAAATGAGC-3′ | |
| Probe | 5′-TGAAGTCATTTCAGAACTGGCCGG-3′ | |
| BCL2 | Forward | 5′-GGAGTGTGAGGACCCAATCT-3′ |
| Reverse | 5′-CAACCACACCATCGATCTTC-3′ | |
| Probe | 5′-AGCCCCAGACCCCAACTCC-3′ | |
| Periostin | Forward | 5′-GGGGTTGTCACTGTGAACTG-3′ |
| Reverse | 5′-CGGCTGCTCTAAATGATGAA-3′ | |
| Probe | 5′-CGTGTCCTGACACAAATTGG-3′ | |
| VEGF | Forward | 5′-CTTGCAGATGTGACAAGCCA-3′ |
| Reverse | 5′-GAGAGGTCTGGTTCCCGAAA-3′ | |
| Probe | 5′-TGCAGCCTGGCTCACCGCCT-3′ | |
| CTGF | Forward | 5′-GAGTGTGCACTGCCAAAGAT-3′ |
| Reverse | 5′-GGCAAGTGCATTGGTATTTG-3′ | |
| Probe | 5′-CGCAGCGGTGAGTCCTTCCA-3′ | |
| PDGFa | Forward | 5′-CGAAGTCAGATCCACAGCAT-3′ |
| Reverse | 5′-GGGCTCTCAGACTTGTCTCC-3′ | |
| Probe | 5′-CCGGGACCTCCAGCGACTCT-3′ | |
| Cox2 | Forward | 5′-TGACCCCAAGGCTCAAAT-3′ |
| Reverse | 5′-GAACCCAGGTCCTCGCTTATG-3′ | |
| Probe | 5′-TTTGCCCAGCACTTCACCCATCAG-3′ | |
| PGE synthase | Forward | 5′-AACCTGGGCGAGTGGATCT-3′ |
| Reverse | 5′-CTGTAAGTGGCTCCAAATGGG-3′ | |
| Probe | 5′-ACATGTGTGTTTCTTAGCCTTTTG-3′ | |
| CCL2 | Forward | 5′-GGCTCAGCCAGATGCAGTTAAC-3′ |
| Reverse | 5′-CCTACTCATTGGGATCATCTTGCT-3′ | |
| Probe | 5′-CCCCACTCACCTGCTGCTACTCAT-3′ | |
| EGF | Forward | 5′-CAACACTGAAGGTGGCTACG-3′ |
| Reverse | 5′-GTGCGGTAGAGTCGAAACAG-3′ | |
| Probe | 5′-CCCGTCTCCTTCGTAGCCTTCTGAGC-3′ | |
| TGFα | Forward | 5′-CTGAAGGGAAGGACTGCTTG-3′ |
| Reverse | 5′-CCTGGCCAAATTCCTCCTCT-3′ | |
| Probe | 5′-CTGCCACTCTGAGACAGTGGTCTGA-3′ | |
| AREG | Forward | 5′-GCGAGGATGACAAGGACCTA-3′ |
| Reverse | 5′-TCGTTTCCAAAGGTGCACTG-3′ | |
| Probe | 5′-CCTCGCAGCTATTGGCATCGGCA-3′ | |
| EREG | Forward | 5′-GTGCATCTACCTGGTGGACA-3′ |
| Reverse | 5′-GAAGTGCTCACATCGCAGAC-3′ | |
| Probe | 5′-CAGTGTAGCCCACTTCACATCTGCAGA-3′ | |
| EPGN | Forward | 5′-AGCAGTCTGCCTCTTGTTCA-3′ |
| Reverse | 5′-TGTAGTCAGCTTCGGTGTTG-3′ | |
| Probe | 5′-TGCTTCTTCGCTCAGTGCTGCC-3′ | |
| BTC | Forward | 5′-GCCCTGGGTCTTGCAATTCT-3′ |
| Reverse | 5′-AGCTCCACAAAGAGAGCCAT-3′ | |
| Probe | 5′-TGTTCCCATCTGCTACCACACAGTGG-3′ |
Blood Cell Isolation.
Blood was collected via cardiac puncture and lysed twice with red blood cell lysis buffer for cell isolation.
Bone Marrow Transplantation (BMT).
Recipient mice received two 6.5 Gy irradiations 3 hours apart using a 137Cesium irradiator (24). 5 × 106 donor marrow cells/200 μl serum-free medium (SFM) were injected i.v. After BMT, mice were given acidified water (pH 3.3) for 3 weeks, normal water for 2 weeks, then treated with bleomycin.
AEC Isolation and Apoptosis Assay.
AECs were isolated as described (25) and plated at 500,000 cells/well on 24-well fibronectin-coated plates for RNA extraction or at 50,000 cells/well on opaque 96-well plates for apoptosis assays (17).
Fibroblast Assays.
Fibroblasts were isolated from lung mince cultures grown for 14 days. For proliferation assays, fibroblasts were plated at 10,000 cells/well in a 96-well plate in complete Dulbecco’s modified Eagle medium for 4 hours, followed by overnight treatment in SFM. Next day, medium was replaced with 90 μl of recombinant (r)-HB-EGF (Sigma-Aldrich) in SFM. MTT was added at 24 hours and the plate read at 48 hours. For migration, cells were plated at 20,000 cells/well in clear-bottomed 96-well plates (Essen Biosciences) in complete Dulbecco’s modified Eagle medium, then switched to SFM overnight. The next day, a scratch was made and media was replaced with rHB-EGF in SFM. The IncuCyte (Sartorius) acquired images every 4 hours and data were quantified using IncuCyte software.
Analysis of single cell (sc)-RNAseq datasets.
Single cell RNA sequencing datasets were accessed from (26) and (27). The transitional state was identified as the Krt8 + ADI cluster from the mouse bleomycin dataset (27) and the “KRT5-/KRT17+” cluster from the human IPF dataset (26).
Statistics.
Statistics were performed as described in the data supplement.
Results
Increased HB-EGF Is Associated with IPF Disease Progression and Fibrosis in Mice
The abundance of HB-EGF and its primary receptor, EGFR, was measured by aptamer array in plasma from IPF patients in the COMET trial (23). We noted that IPF patients who demonstrate disease progression have plasma levels of EGFR and HB-EGF above the identified thresholds compared with IPF patients who did not demonstrate progression over the course of the study (Figures 1A and 1B).
Figure 1.
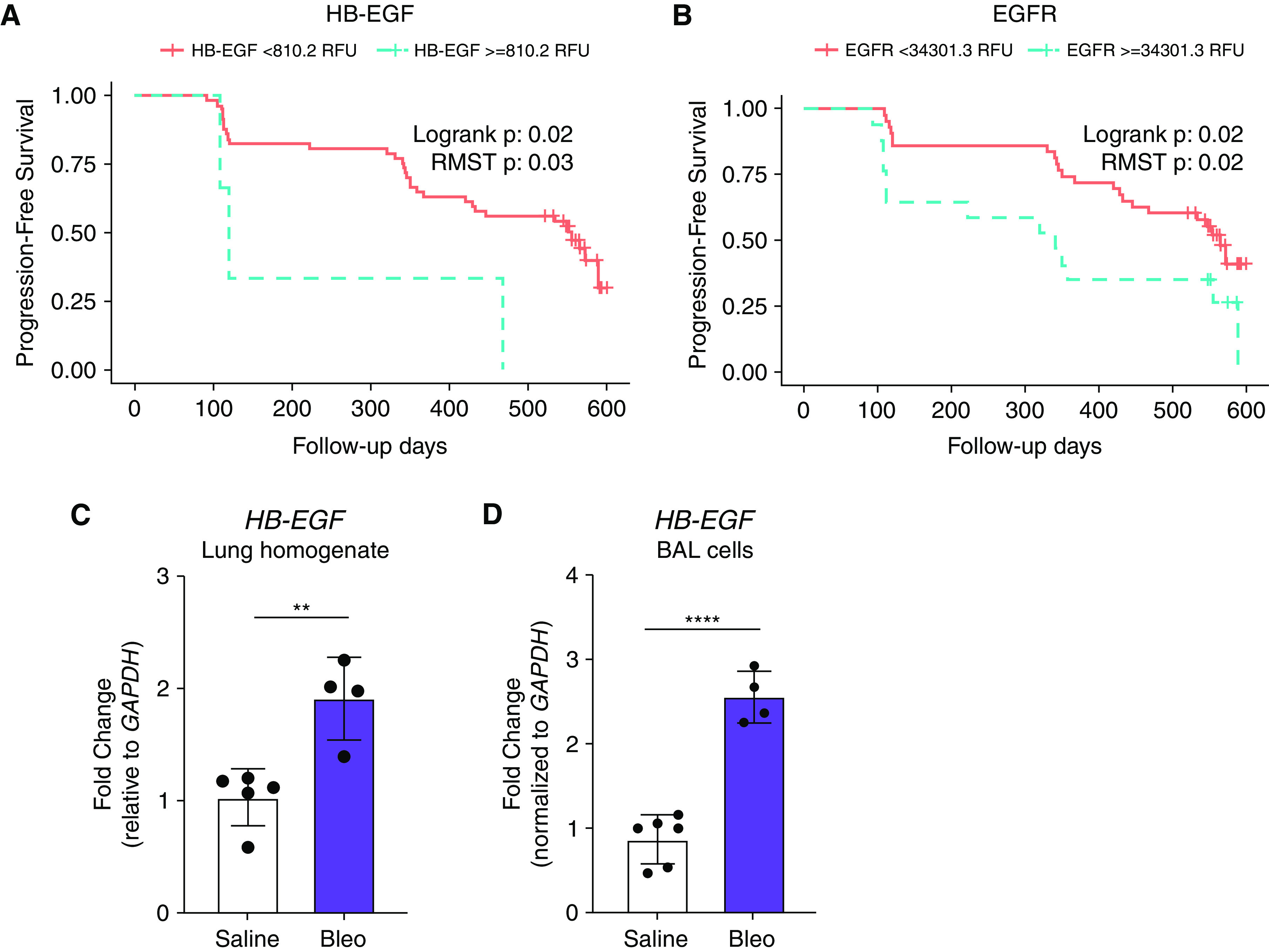
Increased levels of heparin-binding epidermal growth factor-like growth factor (HB-EGF) are associated with increased risk of idiopathic pulmonary fibrosis (IPF) disease progression and fibrosis in mice. IPF patients who demonstrated disease progression have higher levels of (A) HB-EGF and (B) its receptor, epidermal growth factor receptor (EGFR), in their bloodstream and worsened survival compared with IPF patients who did not demonstrate progression over the course of the study. Sensitivity and specificity was determined for different thresholds of the measured biomarker, where high HB-EGF relative fluorescence unit (RFU) values or high EGFR RFU values, respectively, were used to flag progression. The maximum sensitivity plus specificity of HB-EGF was achieved using a threshold of 810.2 RFU. Kaplan-Meier curves for groups above and below this threshold showed an increased risk for progression for those below 810.2 RFU (logrank P = 0.02, restricted means survival test [RMST] P = 0.03). The maximum sensitivity plus specificity of EGFR was achieved using a threshold of 34,301.3 RFU. Kaplan-Meier curves for groups above and below this threshold showed significantly increased risk of progression for those below 34,301.3 RFU (logrank P = 0.02, RMST P = 0.02). Orange lines indicate patients below threshold and blue lines indicate patients above the threshold. Wild-type mice treated with bleomycin-treated (bleo) mice have increased levels of HB-EGF mRNA expression in the lung homogenate (C) and in BAL cells compared with saline-treated mice. Representative reverse transcriptase (RT)-qPCR data in (C) (2 experiments, n = 4–6) and (D) (1 experiment, n = 4–6). **P < 0.01 and ****P < 0.0001.
To investigate the role of HB-EGF in pulmonary fibrosis, we used the bleomycin mouse model. Hbegf transcript was significantly upregulated 21 days post-bleomycin in whole lung (Figure 1C) as well as in the BAL cells (Figure 1D), a primarily myeloid population.
moAMs Express Hbegf
In the absence of validated antibodies for murine HB-EGF, we used RNA PrimeFlow, a technique that targets fluorophores to RNA transcripts rather than proteins, to measure Hbegf mRNA expression in myeloid cells along with a panel of cell surface markers on Day 21 post-saline or bleomycin instillation. Figure 2 shows a tSNE plot of HB-EGF-expressing myeloid cells. Lungs of both C57Bl/6J saline and bleomycin-treated animals contained HB-EGF-expressing alveolar macrophages (AMs, CD64+CD11c+SiglecFhi) and interstitial macrophages (IMs, CD64+CD11b+SiglecFlo). However, the total number of HB-EGF-expressing moAMs was much higher in bleomycin-treated mice (CD64+CD11c+CD11b+SiglecFint) (Figures 2A, 2B, and E2 in the data supplement) suggesting myeloid-derived HB-EGF may promote lung fibrosis.
Figure 2.
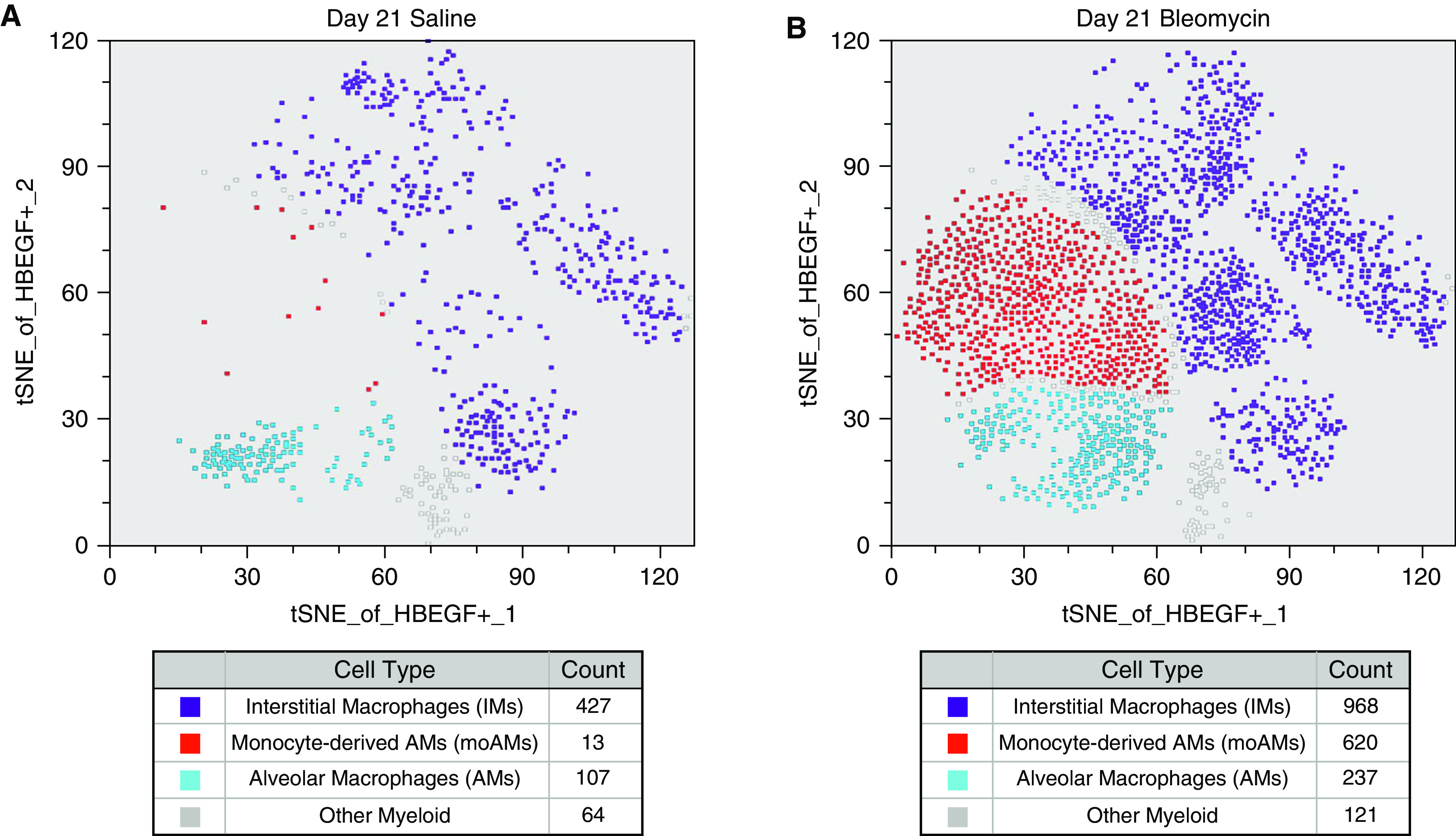
Monocyte-derived alveolar macrophages (moAMs) express HB-EGF. PrimeFlow RNA assays show the presence of HB-EGF + alveolar macrophages (AMs) (CD11c + SiglecFhi), HB-EGF + moAMs (CD11c + CD11b + SiglecFint), and HB-EGF + interstitial macrophages (IMs) (CD11b + SiglecFlo) in C57Bl/6J mice in control animals (A). Oral-pharyngeal administration of bleomycin induces an expansion of HB-EGF + moAMs and IMs after 21 days (B) compared with mice treated with a saline control (A) in C57Bl/6J mice. t-Distributed Stochastic Neighbor Embedding (tSNE) data is representative of one experiment with three mice per group. All data representative of 3 experiments, n = 3.
Hbegff/f;Lyz2Cre+ Mice Are Protected from Bleomycin-induced Fibrosis Despite Similar Levels of Acute Lung Injury
To assess the impact of myeloid-derived HB-EGF on lung fibrosis, we bred Hbegff/f;Lyz2Cre+ mice (28, 29). This mouse has HB-EGF genetically removed through a Cre-lox system using the Lyz2 promoter. Since myeloid cells express Lyz2, HB-EGF should be removed from monocytes, macrophages, dendritic cells, and neutrophils. Interestingly, Hbegff/f;Lyz2Cre+ mice were protected from bleomycin-induced fibrosis when compared with Lyz2Cre+ control mice. At day 21 post-bleomycin, Hbegff/f;Lyz2Cre+ mice showed significantly decreased amounts of hydroxyproline (Figure 3A), and decreased expression of collagen 1, collagen 3, and fibronectin in the lung (Figure 3B). The Hbegff/f;Lyz2Cre+ mice also showed improved histology with less immune cell infiltration, less interstitial thickening, and less collagen deposition compared with Lyz2Cre+ control animals (Figures 3C and E3). Further, Hbegff/f;Lyz2Cre+ mice showed the ability to inflate the lung to higher volume at lower pressure than could Lyz2Cre+ mice as measured by pressure–volume (P–V) loops (Figure 3D).
Figure 3.
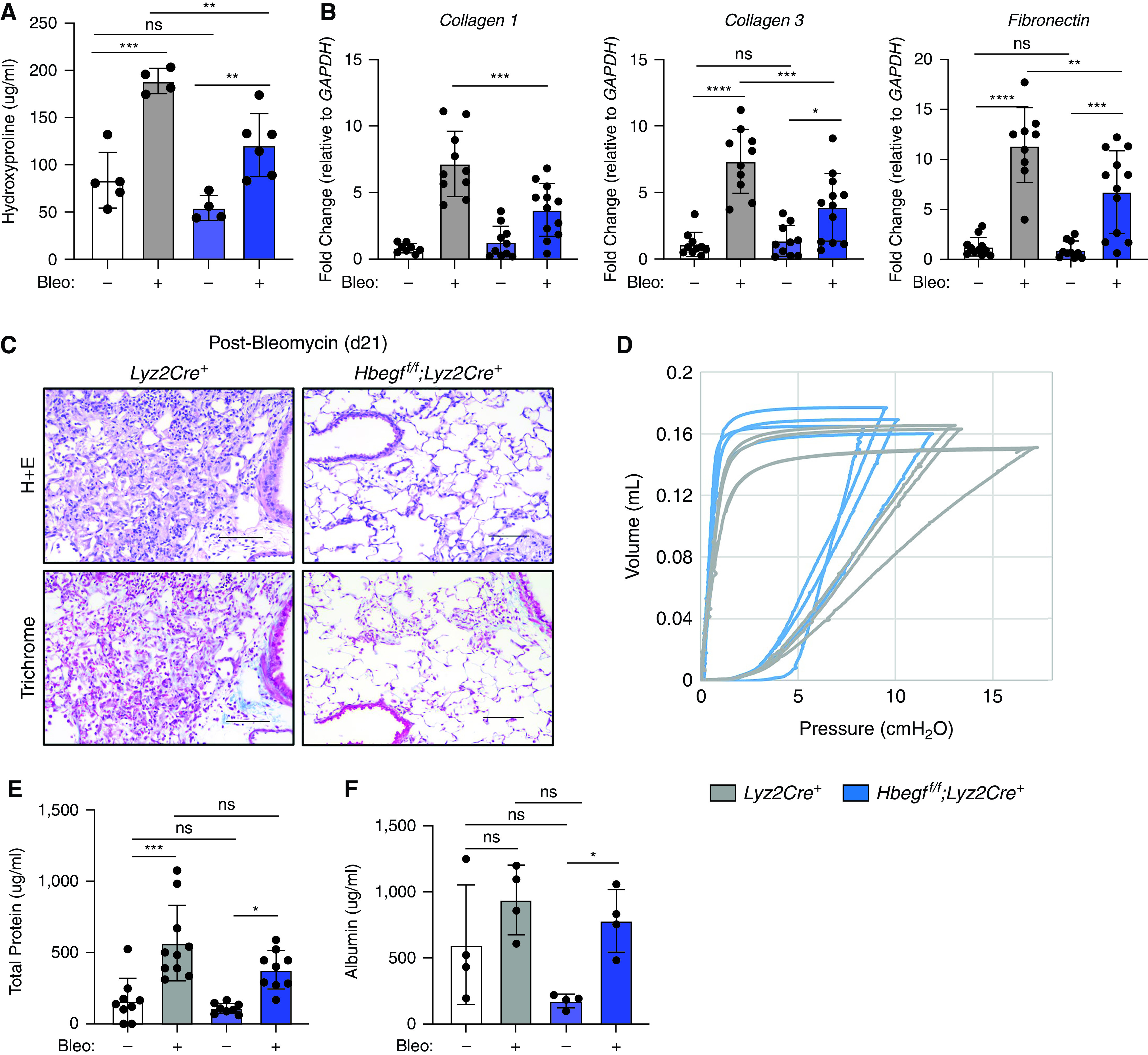
Hbegff/f;Lyz2Cre+ mice are protected from bleomycin-induced fibrosis despite similar levels of acute lung injury. Hbegff/f;Lyz2Cre+ mice have lower levels of hydroxyproline (A), decreased expression of profibrotic genes (B) Collagen 1, Collagen 3, and Fibronectin, improved lung histology (C), and improved pulmonary function as noted by the Hbegff/f;Lyz2Cre+ mice expanding to higher lung volumes at lower pressures than Lyz2Cre+ control mice measured by P-V loops 21 days post-bleomycin treatment (D). Additionally, levels of total protein (E) and albumin detected in the BAL fluid (BALF) (F) is not different between Lyz2Cre+ and Hbegff/f;Lyz2Cre+ mice three days post-bleomycin treatment, suggesting initial lung injury between genotypes is the same. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Lines in (C) are 100 μm length. All data are combined from 2–4 experiments or are representative of 2–4 experiments, n = 4–6. n.s. = not significant; P–V = pressure–volume.
Although the Hbegff/f;Lyz2Cre+ mice had decreased levels of key fibrosis markers at Day 21, it is possible that these differences result from reduced lung injury rather than ameliorated fibrosis development/progression. To assess initial lung injury, we performed BAL on mice at Day 3 post-bleomycin and analyzed the BAL fluid (BALF) for total protein. Our data showed Hbegff/f;Lyz2Cre+ mice have no difference in total protein nor in albumin concentration, indicative of alveolar or vascular leak, compared with control animals (Figures 3E and 3F). Thus, protection from lung fibrosis in Hbegff/f;Lyz2Cre+ mice cannot be explained by reduced lung injury.
Hbegff/f;Lyz2Cre+ Mice have Decreased Macrophage Numbers during Fibrosis Initiation
In the bleomycin mouse model, peak inflammation occurs around Day 7 post-treatment. While flow analysis of Day 7 lung cells showed no difference in total number of AMs after bleomycin, both moAMs and IMs had decreased numbers (Figure 4A) and decreased relative fold change between saline and bleomycin conditions by genotype (Figures E4A and E5A). At Day 21, AMs and IMs were not statistically different after bleomycin between Hbegff/f;Lyz2Cre+ mice and Lyz2Cre+ controls (Figures 4B, E4B, and E5B). MoAMs remained lower in Hbegff/f;Lyz2Cre+ mice compared with Lyz2Cre+ mice controls at Days 7 and 21 by total number (Figure 4B) and by relative fold change (Figures E4B, E5A, and E5B). These results suggest fewer moAMs are present during peak inflammation (Day 7) in Hbegff/f;Lyz2Cre+ mice and that this difference persists during the fibroproliferative stage.
Figure 4.
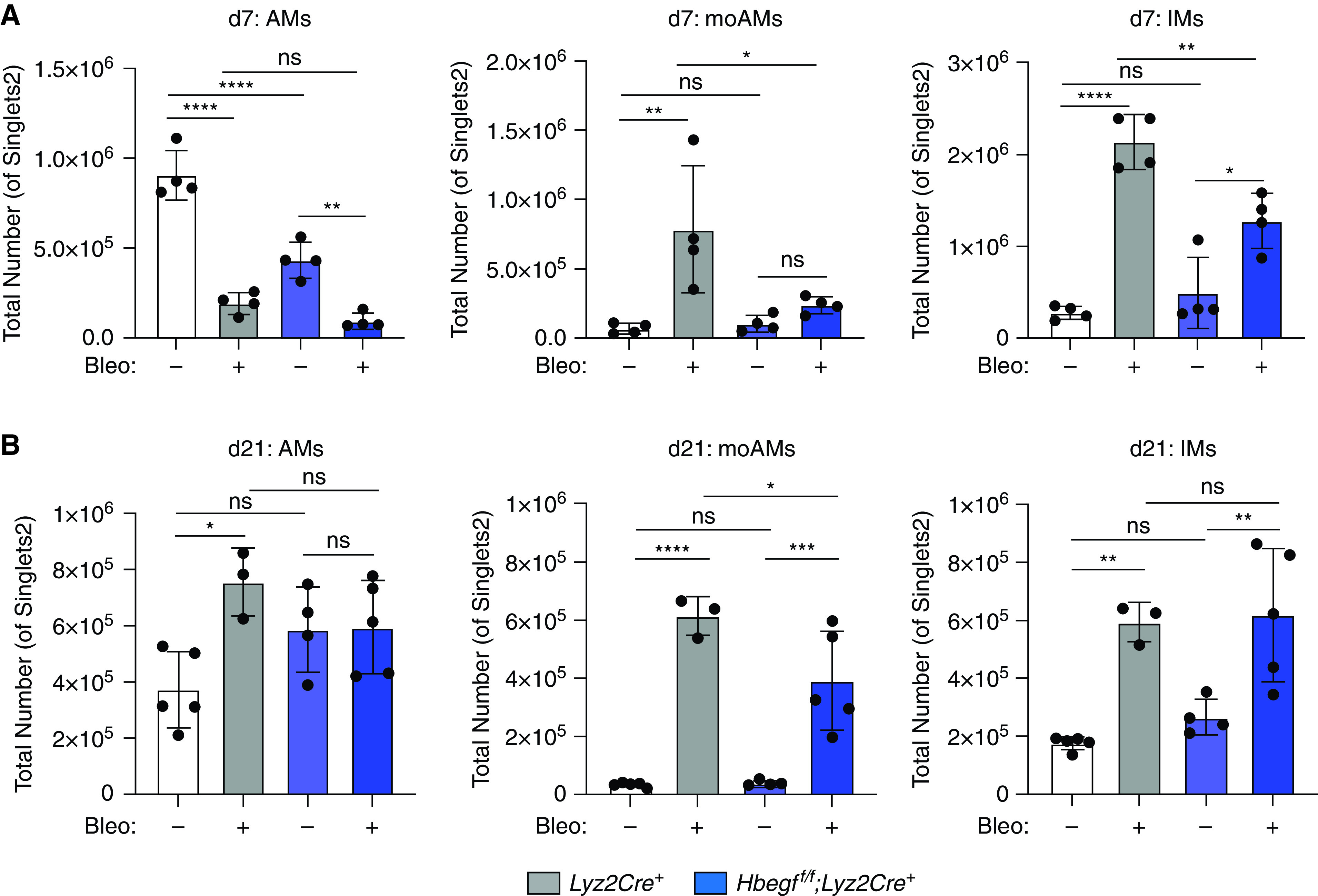
Hbegff/f;Lyz2Cre+ mice have decreased macrophage numbers during fibrosis initiation. (A) Hbegff/f;Lyz2Cre+ number at Day 7 post-bleomycin. (B) Hbegff/f;Lyz2Cre+ mice also have lower levels of moAMs 21 days post-bleomycin treatment by total cell number. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. All data representative of 3–5 experiments, n = 4–6.
Protection in Hbegff/f;Lyz2Cre+ Mice Correlates with Impaired Macrophage Migration and Decreased CCL2 Expression
Next, we wanted to determine why Hbegff/f;Lyz2Cre+ mice have fewer moAMs at Day 7 during fibrosis development. Decreased numbers of moAMs could be the result of decreased proliferation, increased apoptosis, decreased migration, or a combination. The relative changes in Ki67 staining for AMs, IMs, moAMs, or Ly6C+ monocytes at Day 7 was similar between Lyz2Cre+ and Hbegff/f;Lyz2Cre+ animals (Figures E6A and E6B), likely ruling out proliferative defects. Similarly, there were no statistical differences between Hbegff/f;Lyz2Cre+ and control mice in total numbers of dead moAMs or moAMs in early or late apoptosis (Figure E6C) ruling out increased apoptosis of these cells in Hbegff/f;Lyz2Cre+ mice.
As moAMs arise from inflammatory (Ly6C+) monocytes, we measured the total number of monocytes in the lung of wild type and Hbegff/f;Lyz2Cre+ mice. We found Hbegff/f;Lyz2Cre+ mice have fewer Ly6C+ monocytes at Day 7 post-bleomycin compared with controls (P = 0.054; Figure 5A), but there are no differences in Ly6C+ monocytes in blood (Figures 5B and E7). Thus, these data support a defect in monocyte recruitment to the lungs in Hbegff/f;Lyz2Cre+ mice post-bleomycin.
Figure 5.
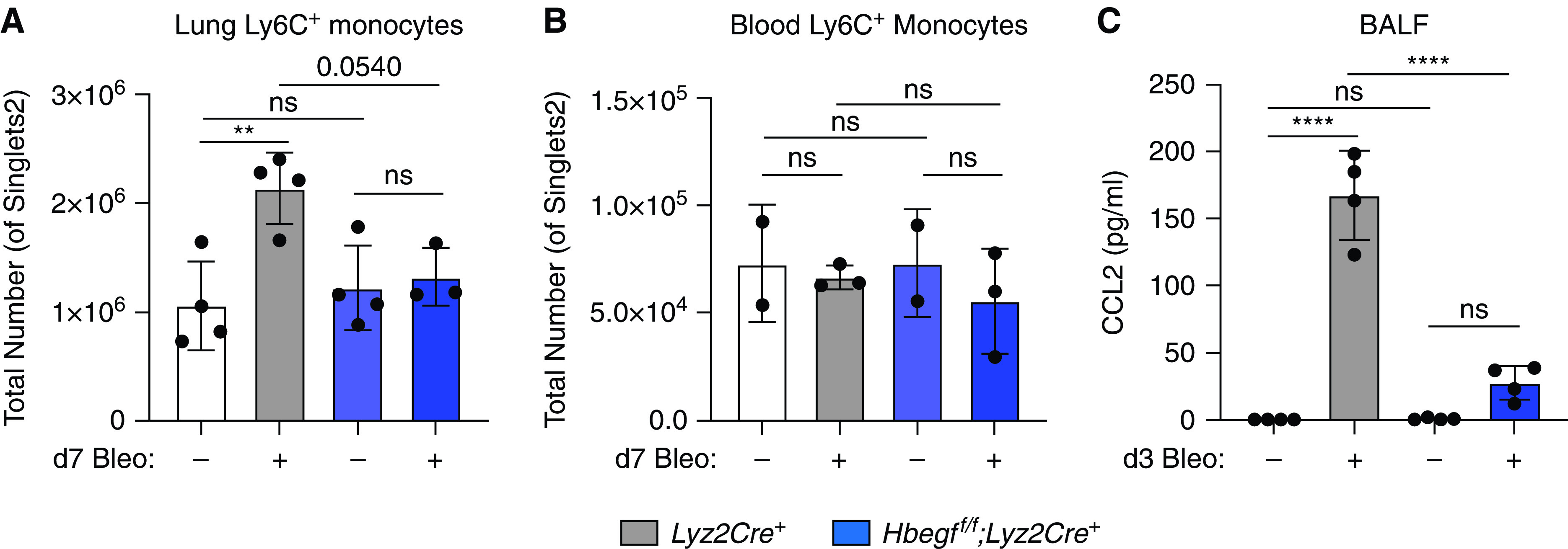
Protection in Hbegff/f;Lyz2Cre+ mice correlates with impaired macrophage migration and decreased CCL2 expression. Hbegff/f;Lyz2Cre+ mice have decreased numbers of Ly6C+ monocytes in the lungs compared with wild-type mice 7 days post-bleomycin treatment (A), but no difference in total Ly6C+monocytes in the blood at the same time point (B). Hbegff/f;Lyz2Cre+ mice have lower levels of CCL2 in their BALF (C) 3 days post-bleomycin compared with Lyz2Cre+ controls. **P < 0.01 and ****P < 0.0001. All data are representative from 2–3 experiments with n = 2–5. Combined data are shown in Figure E7 as fold change. BALF = BAL fluid.
To determine which chemokine(s) could be responsible for decreased monocyte recruitment, we performed a multigene qPCR array (not shown) and identified CCL2, a chemokine known for its ability to recruit monocytes and other immune cells. Elevated levels of CCL2 have been linked to acute respiratory distress syndrome (ARDS), COPD, animal models of fibrosis and human IPF (11, 30–32). BAL fluid from Lyz2Cre+ control and Hbegff/f;Lyz2Cre+ mice 3 days after bleomycin treatment showed levels of CCL2 protein are greatly diminished in Hbegff/f;Lyz2Cre mice (Figures 5C and E7C). Additionally, CCL2 mRNA expression is decreased in AECs (Figure E7D), and in bone marrow-derived macrophages (BMDMs) (Figure E7D) of Hbegff/f;Lyz2Cre+ mice 3 days post-bleomycin. Given the differences in CCL2 production between genotypes, we examined whether recombinant mouse (r)HB-EGF could directly stimulate cultured lung cells to produce CCL2. We did not see any induction of CCL2 mRNA expression in AECs, BMDMs, or fibroblasts treated with rHB-EGF (Figure E8), nor an increase in CCL2 mRNA expression in bleomycin-primed AECs treated with rHB-EGF ex vivo (Figure E8). Thus, HB-EGF alone cannot induce CCL2 expression.
Lyz2Cre Deletes HB-EGF in AECs Which May Also Contribute to Protection from Fibrosis
While the Lyz2Cre+ mouse was originally designed to target macrophages and granulocytes (33), studies have confirmed that Lyz2 is expressed in type 2 AECs (AT2s) and that Lyz2Cre+ removes floxed genes from both the myeloid lineage as well as from AT2s (34–36). We isolated AECs from Lyz2Cre+ and Hbegff/f;Lyz2Cre+ mice 21 days after bleomycin treatment. Although bleomycin decreases Hbegf expression in AECs from control mice, levels of Hbegf mRNA in Hbegff/f;Lyz2Cre+ AECs are similarly low in both saline and bleomycin-treated animals (Figure 6A) making it difficult to determine whether loss of Hbegf expression in AECs post-bleomycin contributed to the protection from fibrosis seen in Hbegff/f;Lyz2Cre+ mice. Interestingly, removal of HB-EGF from AECs did not result in compensation by any other ligands of the ErbB pathway in Hbegff/f;Lyz2Cre+ mice 21 days postbleomycin (Figure E9).
Figure 6.

Lyz2Cre deletes HB-EGF in alveolar epithelial cells (AECs) which may also contribute to protection from fibrosis. (A) AECs from Hbegff/f;Lyz2Cre+ mice have highly decreased levels of Hbegf at baseline (saline treatment) as well as 21 days postbleomycin treatment. Representative data of two experiments, n = 6. (B) Lethally irradiated C57Bl/6J mice reconstituted with Hbegff/f;Lyz2Cre+ bone marrow showed no difference in overall hydroxyproline content compared with lethally-irradiated C57Bl/6J mice reconstituted with C57Bl/6J bone marrow. Representative of three experiments, n = 3–7. (C) Lethally-irradiated Hbegff/f;Lyz2Cre+ mice reconstituted with C57Bl/6J bone marrow showed no difference in overall hydroxyproline compared with Hbegff/f;Lyz2Cre+ mice reconstituted with Hbegff/f;Lyz2Cre+ bone marrow, implying HB-EGF derived from the epithelium is more important in fibrosis protection than immune cell-derived HB-EGF. Representative of one experiment, n = 3–7. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Since HB-EGF is removed in both myeloid cells and in AECs in our Hbegff/f;Lyz2Cre+ mouse, we were interested to determine which cellular compartment was primarily responsible for protection against bleomycin-induced fibrosis. Thus, we designed a bone marrow transplant experiment where C57Bl/6J mice were lethally irradiated and given bone marrow from either C57Bl/6J mice or Hbegff/f;Lyz2Cre+ mice, and were then challenged with bleomycin after 5 weeks. Bleomycin-treated C57Bl/6J mice that received Hbegff/f;Lyz2Cre+ bone marrow (KO-WT) had high levels of hydroxyproline 21 days after bleomycin, and these levels were not different from those in the C57Bl/6J mice that received C57Bl/6J bone marrow (WT-WT) bleomycin-treated group (Figure 6B), implying that the protection noted in Figure 3 in the Hbegff/f;Lyz2Cre+ mice is related to HB-EGF removal from AECs.
To confirm this finding, we lethally irradiated Hbegff/f;Lyz2Cre+ mice and administered bone marrow from either C57Bl/6J mice (WT-KO) or Hbegff/f;Lyz2Cre+ donor mice (KO-KO). At Day 21, hydroxyproline levels of KO-KO bleomycin-treated mice were significantly decreased compared with WT-WT mice (Figure 6C). There was a strong trend toward protection from fibrosis in the WT-KO treated mice (P = 0.1190) and no difference in hydroxyproline levels between KO-KO and WT-KO bleomycin-treated mice (Figure 6C). Together, these data suggest that removal of HB-EGF from AECs in Hbegff/f;Lyz2Cre+ mice is critical for protection of Hbegff/f;Lyz2Cre+ mice from bleomycin-induced fibrosis. Given that Hbegf mRNA expression levels are similar in Lyz2Cre+ and Hbegff/f;Lyz2Cre+ AECs at Day 21 post-bleomycin, we speculate that the effects of AEC-derived HB-EGF to promote fibrosis may occur earlier than Day 21 in this model.
We and others recently identified a transitional AEC state that arises from AT2s during regeneration after injury and persists in IPF (26, 27, 37–40). This transitional state is characterized by activation of profibrotic processes including integrin αvβ6-mediated TGF-β activation (37), ER stress (41), and senescence (26, 27, 37–40), suggesting that the persistence of the transitional state drives fibrosis. Since our new data suggest that AECs contribute to the pathogenesis of fibrosis via HB-EGF production, we hypothesized that these profibrotic transitional AECs rather than mature AT2s may be the key source of HB-EGF in fibrosis. To explore this, we examined existing scRNAseq datasets from mouse and human fibrosis for HB-EGF expression by mature AT2s and transitional AECs, as well as myeloid cells and all other lung cells. We found that HB-EGF is upregulated in transitional AECs in both the bleomycin mouse model of fibrosis and in human IPF (Figure E10). Interestingly, in mouse but not human fibrosis, transitional AECs express more HB-EGF than macrophages. Taken together, these data suggest that HB-EGF is profibrotic, that transitional AECs are a major source of HB-EGF, and that production of HB-EGF is yet another mechanism by which transitional AECs drive fibrosis.
As the data show that transitional AEC-derived HB-EGF may be particularly potent in causing fibrosis, we next tested whether rHB-EGF induces profibrotic changes in AECs. Interestingly, rHB-EGF does not promote increased expression of key profibrotic genes periostin, Pdgfα, Vegf, and Ctgf (Figure E11). Further, the addition of rHB-EGF to AECs did not cause a change in anti-apoptotic genes BCL2, XIAP, or survivin (Figure E11), nor did it increase caspase 3/7 activation like positive control TGF-β (4 ng/ml) in an apoptosis assay (Figure E11). We conclude that rHB-EGF does not directly induce these profibrotic effects in AECs.
rHB-EGF Induces Fibroblast Migration with Little Impact on Proliferation or Matrix Gene Expression
To determine if rHB-EGF prompts a profibrotic response in fibroblasts in vitro, we measured fibroblast proliferation, ECM production, and migration. rHB-EGF did not induce fibroblast proliferation as measured by MTT assay (Figure 7A) or flow cytometry (data not shown). Similarly, addition of rHB-EGF did not cause an increase in expression of profibrotic genes collagen 1, collagen 3, or fibronectin (Figure 7B).
Figure 7.
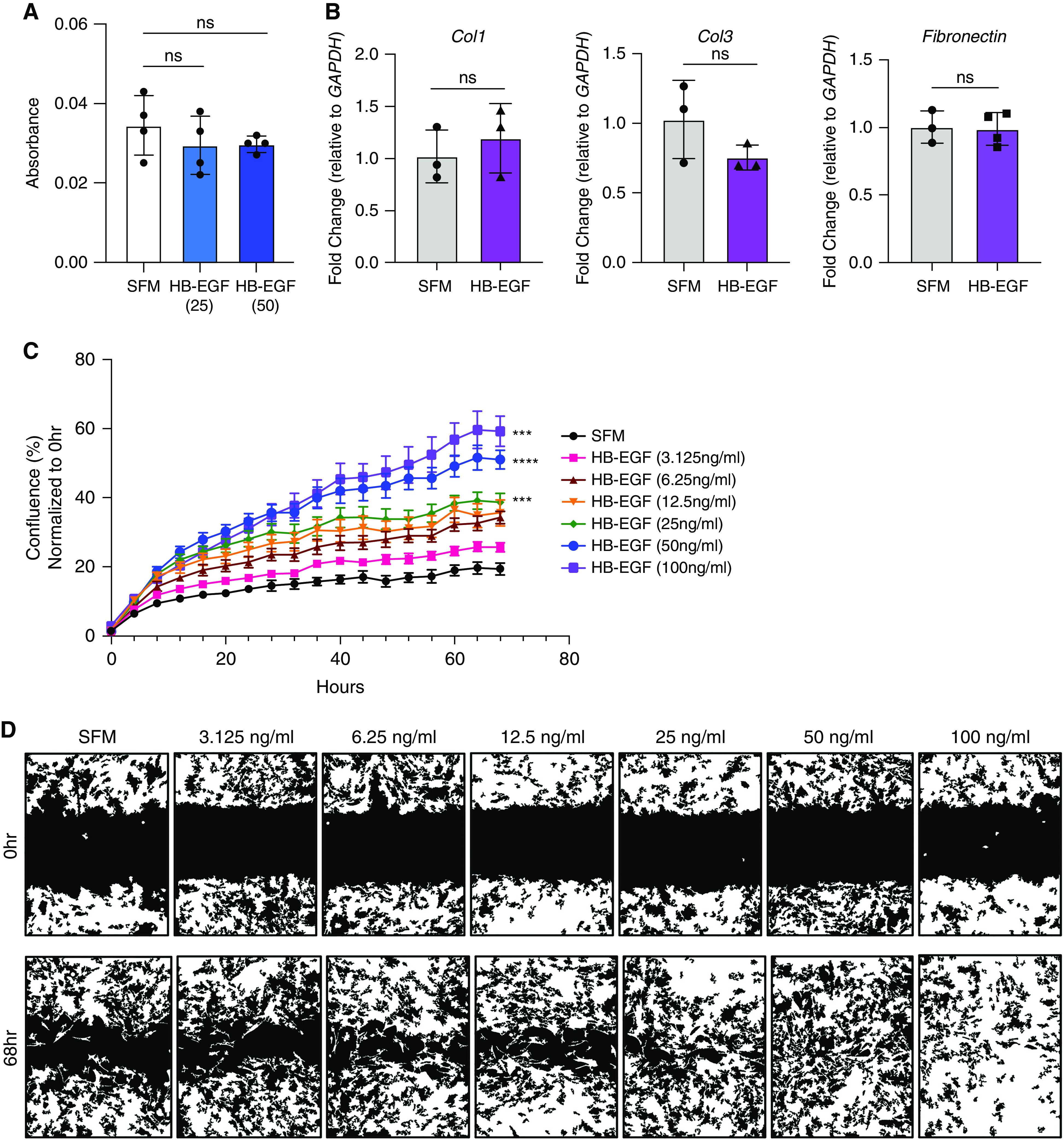
rHB-EGF induces fibroblast migration with little impact on proliferation or matrix gene expression. (A) Fibroblasts treated with rHB-EGF (25 and 50 ng/ml) do not have increased proliferation as measured by MTT. (B) qPCR fold change of key profibrotic genes collagen1, collagen3, and fibronectin is not induced in fibroblasts after rHB-EGF (50 ng/ml) administration. (C and D) rHB-EGF promotes fibroblast migration. Representative data shown of at least three experiments, n = 3–6. In (C), all doses of HB-EGF from 6.25 to 100 ng/ml are significantly different from SFM control starting at 12 hours. P values are given for HB-EGF (25 ng/ml), HB-EGF (50 ng/ml), and HB-EGF (100 ng/ml) and represent differences between treatment group and SFM at final time point (68 h). ***P < 0.001 and ****P < 0.0001. SFM = serum-free medium.
To measure fibroblast migration, we performed a scratch assay where primary lung fibroblasts were grown to confluence, evenly scratched with a WoundMaker device, then treated with concentrations of rHB-EGF (3.125–100 ng/ml), and the ability of the fibroblasts to migrate and close the wound was measured over time. As fibroblasts showed no increased proliferation in response to rHB-EGF (Figure 7A) and all cells were treated with rHB-EGF in SFM, we consider the scratch wound assay to be an accurate measure of migration. Intriguingly, rHB-EGF accelerated fibroblast migration in a dose-dependent fashion (Figures 7C and 7D).
Discussion
There is no work to date directly examining HB-EGF’s role in IPF or in a murine model of bleomycin-induced fibrosis. EGFR signaling may be important in IPF progression, as patients with disease progression as measured by a composite end point have higher levels of HB-EGF and EGFR compared with IPF patients who did not demonstrate progression over the course of the study (Figure 1). These data align with previous work showing lung fibrosis patients have heightened expression of EGFR (42) and that overexpression of TGFα (a different EGFR ligand) can promote pulmonary fibrosis in murine models (43).
Hbegf expression was robustly increased in our bleomycin-treated mice (Figure 1). Interestingly, both lung homogenate and BAL cells showed this Hbegf increase at Days 14 and 21 post-bleomycin. Murine and human scRNAseq studies reveal that Hbegf expression is upregulated in AT2s as they assume the transitional state (Figure E10), which in this murine dataset is shown at Day 7 post-bleomycin. Interestingly, despite this upregulation in the transitional AT2s, we don’t see elevated HB-EGF in whole lung RNA at Days 3 or 7 post-bleomycin (data not shown); presumably because the transitional AT2s are a relatively small percentage of total lung cells. This suggests that increased Hbegf expression in the whole lung during the later time points could be evident due to an influx of HB-EGF-producing monocytes/macrophages to the lung. Previous studies report that Ly6C+ monocytes migrate to sites of tissue injury and differentiate into macrophages upon arrival (14), and in the lungs, monocytes transit through an IM phenotypic stage during their transition to moAMs (12, 15). While all macrophage subtypes express Hbegf at baseline (Figures 2 and E2), there are twice as many Hbegf-expressing IMs and 47 times as many Hbegf-expressing moAMs in the lung at day 21 postbleomycin treatment compared with saline lungs (Figure 2). While our BMT chimeras suggest that AEC-derived HB-EGF may be more important than myeloid-derived HB-EGF in the mouse model, it is still possible that myeloid-derived HB-EGF may play important local roles in stimulating fibroblast migration. Furthermore, the scRNAseq data from human IPF patients suggests that in people, myeloid cells show similarly high levels of HB-EGF as transitional AECs, which may explain why elevated myeloid cells are well correlated with IPF progression (26).
Hbegff/f;Lyz2Cre+ mice were protected from fibrosis as measured by decreased hydroxyproline content, decreased profibrotic gene expression, improved histology, and improved pulmonary function compared with Lyz2Cre+ animals (Figure 3). While our dose of bleomycin (1 U/kg) causes < 25% mortality in wild-type mice, Hbegff/f;Lyz2Cre+ mice had decreased mortality and higher body weight at 21 days compared with wild-type mice (data not shown), further supporting the protected phenotype.
Given the importance of moAMs in fibrosis development and the protection phenotype of the Hbegff/f;Lyz2Cre+ mice, it is interesting that Hbegff/f;Lyz2Cre+ mice have fewer moAMs present during the inflammatory stage of fibrosis development (Figures 4, E4, and E5). Hbegff/f;Lyz2Cre+ mice routinely had lower numbers of moAMs at both Day 7 and Day 21 compared with Lyz2Cre+ mice. Considering these moAM differences at Day 21, however, it is interesting that total numbers of IMs are not different between genotypes at the same time point. This could be caused by impaired IM-moAM differentiation in Hbegff/f;Lyz2Cre+ mice, but if true, one might have predicted increased IM populations accumulating in the Hbegff/f;Lyz2Cre+ mice which we do not see. It could also relate to the difficulty of accurately identifying the heterogenous populations of recruited and inflammatory cells with dynamic changes in cell surface marker expression (2, 44, 45). We also noted total numbers of neutrophils are decreased at Day 7 post-bleomycin in Hbegff/f;Lyz2Cre+ mice (Figure E12). Neutrophils produce neutrophil elastase (NE), which promotes fibroblast migration and myofibroblast differentiation in vitro (46). A lack of neutrophils in these mice during peak inflammation is also consistent with a decreased fibrosis phenotype and it is possible that decreases in moAMs and neutrophils together contribute to protection of Hbegff/f;Lyz2Cre+ mice.
Our data show that the decreased numbers of moAMs in Hbegff/f;Lyz2Cre+ mice at Day 7 are the result of decreased monocyte migration (Figures 5, E6, and E7). As such, we add further evidence to the current theory that depleting monocytes in the bleomycin mouse model reduces fibrosis severity (5, 11, 44). In addition, our evidence suggests lower levels of monocyte migration to the lung is caused by decreased amounts of CCL2 in the BALF, BAL cells, and AECs of Hbegff/f;Lyz2Cre+ mice during initial lung injury (Figure 5). BMDMs of mice also have decreased levels of CCL2, suggesting that bleomycin induces a systemic effect that is likely regulated by a soluble factor (Figure 5) in Hbegff/f;Lyz2Cre+ mice. CCL2 binds its receptor, CCR2, found predominantly on monocytes (11). CCL2 is present in increased levels in IPF patients and is associated with increased lung injury scores in patients with ARDS (47). While rHB-EGF alone does not induce CCL2 expression in macrophages, fibroblasts, or AECs alone (Figure E8), it is possible that rHB-EGF requires a second stimulus such as LPS, cytokines (IL-4 and IL-13), or that the regulation of CCL2 involves cellular crosstalk, similar to what has been suggested for HB-EGF, causing fibroblast migration via induction of epithelial cell-derived IL-8 (48). HB-EGF is biologically active in both a soluble and membrane-bound (pro-HB-EGF) form. It is possible that CCL2 induction requires pro-HB-EGF for juxtacrine signaling, rather than paracrine or autocrine signaling alone. Furthermore, since monocytes and macrophages can recruit themselves to sites of injury, reduced numbers of both cell types could relate to the presence of fewer moAMs in the lung that produce CCL2.
Our chimeric BMT studies point to AEC-derived HB-EGF as a prominent reason for protection in Hbegff/f;Lyz2Cre+ animals. Interrogation of existing murine and human fibrosis scRNAseq datasets revealed that the transitional cells that arise from AT2s after injury express high levels of Hbegf. It is hard to know for sure how mRNA levels correlate to HB-EGF protein in the absence of good antibodies for murine HB-EGF; thus, another possibility is that the threshold level of HB-EGF in the lung is more critical than the cellular source and in mice, AECs may be a more prominent source of HB-EGF. Future experiments will be needed to address this more fully. Recent studies suggest that Lyz2Cre targets the alveolar epithelium, primarily in the AT2 cells (35, 36). AT2s are the surfactant-producing stem cells of the epithelium that assume the transitional state before differentiating into AT1 s to fill in gaps after injury (49, 50), and as such, Lyz2Cre-based removal of HB-EGF from AT2s should persist in daughter transitional cells and could also be deleted from AT1 cells which arise from gene-deleted AT2s. Given accumulating evidence that persistence of the transitional state in AECs contributes to fibrosis; we now identify HB-EGF as a possible mechanism by which transitional cells promote fibrosis.
We sought to determine how soluble HB-EGF (from any potential cell source) might induce profibrotic outcomes. Interestingly, rHB-EGF addition to AECs did not increase levels of profibrotic gene expression, decrease levels of anti-apoptotic gene expression, or increase apoptosis (Figure E11), all expected hallmarks of a profibrotic response. In addition, previous work suggested that HB-EGF regulates AT2 proliferation in vivo and thus it could be expected that HB-EGF would promote epithelial repair and improve fibrotic outcome (36). However, Hbegff/f;Lyz2Cre+ mice are protected; thus, it does not appear that loss of HB-EGF in the Lyz2Cre+ AECs limited lung repair. In examining the impacts on mesenchymal cells, rHB-EGF did not increase proliferation or expression of profibrotic genes in fibroblasts, but rHB-EGF did dramatically increase migration in a scratch wound assay (Figure 7). HB-EGF is also known to increase keratinocyte migration rather than proliferation (21) highlighting the multi-dimensional role of HB-EGF in various contexts.
This study has several caveats. First, this study was conducted in a murine model of fibrosis. Although the bleomycin model is the best characterized and most widely used animal model of pulmonary fibrosis, it does not fully recapitulate IPF (51). A second caveat is the technical limitations of the Lyz2Cre system and that lack of an appropriate inducible system. The conditional Lyz2Cre Cre-loxP system was chosen for this study because of its ability to remove HB-EGF from macrophage populations (AMs, IMs, and moAMs) with nearly 100% efficiency (35). We chose Lyz2Cre over other Cre-loxP systems we deemed less appropriate: Csf1r-Cre targets all leukocytes in addition to macrophages, and inducible Cre-loxP systems CX3CR1-ERCre and CD68-rtTA do not target every lung macrophage population (no resident AMs) (35). Like most Cre systems, however, Lyz2Cre also depletes off-target cell types. Previous work by McCubbrey and colleagues and Desai and colleagues have shown that Lyz2Cre successfully targets at least a subset of AECs for recombination, including the AT2 subtype (34–36, 52). However, RNAseq data suggests all AT2 cells express Lyz2; thus, deletion could be extensive. Additionally, HB-EGF is highly expressed in AT1 cells, and while AT1 cells do not express Lyz2, any AT1 cells derived from gene-deleted AT2 precursors would lose expression as well. Without an antibody specific to murine HB-EGF we cannot address this question at the protein level in AT1 and AT2 cells in vivo.
In addition to AT2s, Lyz2Cre targets other innate immune cells including neutrophils (100%), Ly6C+ monocytes (68%), dendritic cells (DCs) (50%), and eosinophils (<20%) (35). Although recent work has shown that all subtypes of peripheral blood dendritic cells are severely depleted in IPF patients, the role of DCs in fibrosis is not fully understood (53). However, neutrophils, dendritic cells, and eosinophils do not appear to express Hbegf in the bleomycin model (Figure E10). Additionally, we were unable to determine if the protection in the Hbegff/f;Lyz2Cre+ mice is simply a threshold effect. Finally, HB-EGF likely acts in profibrotic ways on certain cell types (e.g., fibroblasts) but in an antifibrotic manner for others (e.g., AECs). Ideally, the eventual availability of validated murine HB-EGF antibodies (e.g., ELISA kits) and murine HB-EGF inhibitors will allow this to be a more feasible future research direction.
Overall, we demonstrated that HB-EGF is expressed in IPF patients as well as in transitional AECs and all populations of lung macrophages (AMs, IMs, and moAMs) in mice with pulmonary fibrosis. We also showed that Hbegff/f;Lyz2Cre+ mice are protected from bleomycin-induced fibrosis and that this protection is multifactorial, caused by decreased CCL2-dependent monocyte migration and decreased fibroblast migration. The predominant source of the HB-EGF is likely the transitional AEC, although macrophage-derived HB-EGF may also contribute, given that the KO-WT mice had a trend toward protection in the bleomycin model (Figure 6B). Together, the data from this study provide evidence that HB-EGF is heavily involved in the development of pulmonary fibrosis.
Acknowledgments
Acknowledgment
The authors would like to thank the flow cytometry core at the University of Michigan for expertise in processing flow cytometry samples and Dr. Eliza Tsou for her assistance in the analysis of Incucyte images.
Footnotes
Supported by the National Heart, Lung, and Blood Institute grants F31 HL149150 (E.M.H.), R35 HL144481 (B.B.M.), and R01 HL147920 (R.L.Z); the Center for Strategic Scientific Initiatives, National Cancer Institute grants R01 CA247516 and U01 CA224145 (H.C.C.); the Division of Intramural Research, National Institute of Allergy and Infectious Diseases grant T32 AI007413 (E.M.H.); Francis Family Foundation (S.J.G.); Michigan Institute for Clinical and Health Research (S.J.G.); and Boehringer Ingelheim (S.J.G.).
Author Contributions: E.M.H., H.C.C., and B.B.M. conceived and designed research. E.M.H. and A.R. performed experiments. E.M.H. and S.J.G. analyzed data. E.M.H. and B.B.M. interpreted results of experiments. E.M.H., S.J.G., Y.W., D.N.O’D., and R.L.Z. prepared figures. S.M. conducted SOMAmer statistics. E.M.H. drafted manuscript. E.M.H. and B.B.M. edited and revised manuscript. E.M.H., S.J.G., D.N.O’D., S.M., R.L.Z., H.C.C., and B.B.M. approved the final version of manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0174OC on August 29, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax . 2015;70:1189–1196. doi: 10.1136/thoraxjnl-2015-207020. [DOI] [PubMed] [Google Scholar]
- 2. Zhang L, Wang Y, Wu G, Xiong W, Gu W, Wang CY. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res . 2018;19:170. doi: 10.1186/s12931-018-0864-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moore MW, Herzog EL. Regulation and relevance of myofibroblast responses in idiopathic pulmonary fibrosis. Curr Pathobiol Rep . 2013;1:199–208. doi: 10.1007/s40139-013-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ryu C, Homer RJ, Herzog EL. The airway in idiopathic pulmonary fibrosis: protecting the lung or promoting disease? Am J Respir Crit Care Med . 2016;193:1081–1082. doi: 10.1164/rccm.201601-0055ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wynn TA, Vannella KM. Review macrophages in tissue repair, regeneration, and fibrosis. Immunity . 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Joshi N, Walter JM, Misharin AV. Alveolar macrophages. Cell Immunol . 2018;330:86–90. doi: 10.1016/j.cellimm.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 7. Moore BB, Fry C, Zhou Y, Murray S, Han MK, Martinez FJ, et al. The COMET Investigators Inflammatory leukocyte phenotypes correlate with disease progression in idiopathic pulmonary fibrosis. Front Med . 2014;1:56. doi: 10.3389/fmed.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scott MKD, Quinn K, Li Q, Carroll R, Warsinske H, Vallania F, et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir Med . 2019;7:497–508. doi: 10.1016/S2213-2600(18)30508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kreuter M, Lee JS, Tzouvelekis A, Oldham JM, Molyneaux PL, Weycker D, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2021;204:74–81. doi: 10.1164/rccm.202003-0669OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med . 2011;184:569–581. doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 11. Moore BB, Paine R, III, Christensen PJ, Moore TA, Sitterding S, Ngan R, et al. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol . 2001;167:4368–4377. doi: 10.4049/jimmunol.167.8.4368. [DOI] [PubMed] [Google Scholar]
- 12. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med . 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCubbrey AL, Barthel L, Mould KJ, Mohning MP, Redente EF, Janssen WJ. Selective and inducible targeting of CD11b+ mononuclear phagocytes in the murine lung with hCD68-rtTA transgenic systems. Am J Physiol Lung Cell Mol Physiol . 2016;311:L87–L100. doi: 10.1152/ajplung.00141.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol . 2015;185:2596–2606. doi: 10.1016/j.ajpath.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J . 2020;55:1900646. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Besner G, Higashiyama S, Klagsbrun M. Isolation and characterization of a macrophage-derived heparin-binding growth factor. Cell Regul . 1990;1:811–819. doi: 10.1091/mbc.1.11.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hult EM, Gurczynski SJ, Moore BB. M2 macrophages have unique transcriptomes but conditioned media does not promote profibrotic responses in lung fibroblasts or alveolar epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol . 2021;321:L518–L532. doi: 10.1152/ajplung.00107.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Singh B, Carpenter G, Coffey RJ. EGF receptor ligands: recent advances [version 1; peer review: 3 approved] F1000 Res . 2016;5:2270. doi: 10.12688/f1000research.9025.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eapen MS, Sharma P, Thompson IE, Lu W, Myers S, Hansbro PM, et al. Heparin-binding epidermal growth factor (HB-EGF) drives EMT in patients with COPD: implications for disease pathogenesis and novel therapies. Lab Invest . 2019;99:150–157. doi: 10.1038/s41374-018-0146-0. [DOI] [PubMed] [Google Scholar]
- 20. Hirabayashi M, Asano Y, Yamashita T, Miura S, Nakamura K, Taniguchi T, et al. Possible pro-inflammatory role of heparin-binding epidermal growth factor-like growth factor in the active phase of systemic sclerosis. J Dermatol . 2018;45:182–188. doi: 10.1111/1346-8138.14088. [DOI] [PubMed] [Google Scholar]
- 21. Taylor SR, Markesbery MG, Harding PA. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) and proteolytic processing by a disintegrin and metalloproteinases (ADAM): a regulator of several pathways. Semin Cell Dev Biol . 2014;28:22–30. doi: 10.1016/j.semcdb.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 22. Su Y, Luo H, Yang J. Heparin-binding EGF-like growth factor attenuates lung inflammation and injury in a murine model of pulmonary emphysema. Growth Factors . 2018;36:246–262. doi: 10.1080/08977194.2018.1552270. [DOI] [PubMed] [Google Scholar]
- 23. Ashley SL, Xia M, Murray S, O’Dwyer DN, Grant E, White ES, et al. Six-SOMAmer index relating to immune, protease and angiogenic functions predicts progression in IPF. PLoS One . 2016;11:e0159878. doi: 10.1371/journal.pone.0159878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gurczynski SJ, Zhou X, Flaherty M, Wilke CA, Moore BB. Bone marrow transplant-induced alterations in Notch signaling promote pathologic Th17 responses to γ-herpesvirus infection. Mucosal Immunol . 2018;11:881–893. doi: 10.1038/mi.2017.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bauman KA, Wettlaufer SH, Okunishi K, Vannella KM, Stoolman JS, Huang SK, et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest . 2010;120:1950–1960. doi: 10.1172/JCI38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv . 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strunz M, Lukas MS, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun . 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wen HJ, Gao S, Wang Y, Ray M, Magnuson MA, Wright CVE, et al. Myeloid cell-derived HB-EGF drives tissue recovery after pancreatitis. Cell Mol Gastroenterol Hepatol . 2019;8:173–192. doi: 10.1016/j.jcmgh.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, et al. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci USA . 2003;100:3221–3226. doi: 10.1073/pnas.0537588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cui TX, Brady AE, Fulton CT, Zhang YJ, Rosenbloom LM, Goldsmith AM, et al. CCR2 mediates chronic LPS-induced pulmonary inflammation and hypoalveolarization in a murine model of bronchopulmonary dysplasia. Front Immunol . 2020;11:579628. doi: 10.3389/fimmu.2020.579628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Henrot P, Prevel R, Berger P, Dupin I. Chemokines in COPD: from implication to therapeutic use. Int J Mol Sci . 2019;20:2785. doi: 10.3390/ijms20112785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Belchamber KBR, Donnelly LE. Macrophage dysfunction in respiratory disease. Results Probl Cell Differ . 2017;62:299–313. doi: 10.1007/978-3-319-54090-0_12. [DOI] [PubMed] [Google Scholar]
- 33. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res . 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 34. Miyake Y, Kaise H, Isono K, Koseki H, Kohno K, Tanaka M. Protective role of macrophages in noninflammatory lung injury caused by selective ablation of alveolar epithelial type II cells. J Immunol . 2007;178:5001–5009. doi: 10.4049/jimmunol.178.8.5001. [DOI] [PubMed] [Google Scholar]
- 35. McCubbrey AL, Allison KC, Lee-Sherick AB, Jakubzick CV, Janssen WJ. Promoter specificity and efficacy in conditional and inducible transgenic targeting of lung macrophages. Front Immunol . 2017;8:1618. doi: 10.3389/fimmu.2017.01618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature . 2014;507:190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, et al. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight . 2019;5:e123637. doi: 10.1172/jci.insight.123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, et al. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med . 2020;201:1443–1447. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv . 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ting C, Aspal M, Vaishampayan N, Huang SK, Riemondy KA, Wang F, et al. Fatal COVID-19 and non-COVID-19 acute respiratory distress syndrome is associated with incomplete alveolar type 1 epithelial cell differentiation from the transitional state without fibrosis. Am J Pathol . 2022;192:454–467. doi: 10.1016/j.ajpath.2021.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watanabe S, Markov NS, Lu Z, Piseaux Aillon R, Soberanes S, Runyan CE, et al. Resetting proteostasis with ISRIB promotes epithelial differentiation to attenuate pulmonary fibrosis. Proc Natl Acad Sci USA . 2021;118:e2101100118. doi: 10.1073/pnas.2101100118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Braga TT, Agudelo JSH, Camara NOS. Macrophages during the fibrotic process: M2 as friend and foe. Front Immunol . 2015;6:602. doi: 10.3389/fimmu.2015.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-α in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2004;286:L741–L749. doi: 10.1152/ajplung.00208.2003. [DOI] [PubMed] [Google Scholar]
- 44. Koch CM, Chiu SF, Misharin AV, Ridge KM. Lung interstitial macrophages: establishing identity and uncovering heterogeneity. Am J Respir Cell Mol Biol . 2017;57:7–9. doi: 10.1165/rcmb.2017-0150ED. [DOI] [PubMed] [Google Scholar]
- 45. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med . 2016;22:303–316. doi: 10.1016/j.molmed.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 46. O’Dwyer DN, Ashley SL, Moore BB. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am J Physiol Lung Cell Mol Physiol . 2016;311:L590–L601. doi: 10.1152/ajplung.00221.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Antoniades HN, Neville-Golden J, Galanopoulos T, Kradin RL, Valente AJ, Graves DT. Expression of monocyte chemoattractant protein 1 mRNA in human idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA . 1992;89:5371–5375. doi: 10.1073/pnas.89.12.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y, Su G, Zhong Y, Xiong Z, Huang T, Quan J, et al. HB-EGF-induced IL-8 secretion from airway epithelium leads to lung fibroblast proliferation and migration. BMC Pulm Med . 2021;21:347. doi: 10.1186/s12890-021-01726-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc . 2006;3:364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 50. Parimon T, Yao C, Stripp BR, Noble PW, Chen P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci . 2020;21:2269. doi: 10.3390/ijms21072269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2008;294:L152–L160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 52. Radigan KA, Morales-Nebreda L, Soberanes S, Nicholson T, Nigdelioglu R, Cho T, et al. Impaired clearance of influenza A virus in obese, leptin receptor deficient mice is independent of leptin signaling in the lung epithelium and macrophages. PLoS One . 2014;9:e108138. doi: 10.1371/journal.pone.0108138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bocchino M, Zanotta S, Capitelli L, Galati D. Dendritic cells are the intriguing players in the puzzle of idiopathic pulmonary fibrosis pathogenesis. Front Immunol . 2021;12:664109. doi: 10.3389/fimmu.2021.664109. [DOI] [PMC free article] [PubMed] [Google Scholar]